

Abstract
In solid malignancies, including head and neck squamous cell carcinoma (HNSCC), the immunosuppressive molecule adenosine, which accumulates in the tumor, suppresses cytotoxic CD8+ T cell functions including chemotaxis and tumor infiltration. Adenosine functions through binding to the adenosine A2A receptor (A2AR) present on T cells. In order to increase T cell migration into the tumor, the negative effect of adenosine must be abrogated. Systemic drug treatments targeting A2AR are available; however, they could lead to negative toxicities due to the broad expression of this receptor. Herein, we developed a lipid nanoparticle (NP)-based targeted delivery approach to knock down A2AR in T cells in order to increase their chemotaxis in the presence of adenosine. By using flow cytometry, immunofluorescence, qRT-PCR, and 3D-chemotaxis, we demonstrated that CD45RO-labeled nanoparticles delivering ADORA2A gene-silencing-RNAs decreased ADORA2A mRNA expression and rescued the chemotaxis of HNSCC CD8+ memory T cells. Overall, the data indicate that targeting the adenosine signaling pathway with lipid NPs is successful at suppressing the inhibitory effect of adenosine on the chemotaxis of HNSCC memory T cells, which could ultimately help increase T cell infiltration into the tumor.
Graphical abstract
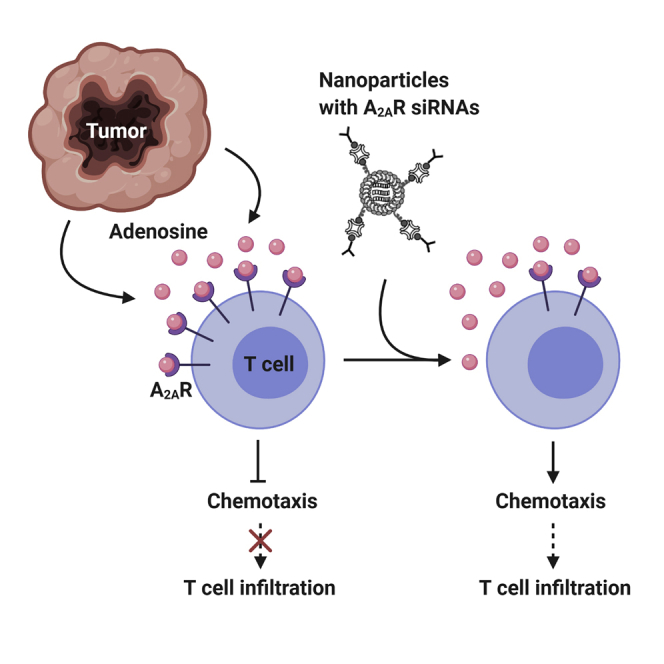
Adenosine in the tumor microenvironment inhibits cytotoxic T cell chemotaxis through adenosine A2A receptor signaling. Conforti and colleagues developed a lipid nanoparticle-based therapeutic, which delivered adenosine A2A receptor-silencing RNAs, knocked down the adenosine A2A receptor, and rescued the chemotaxis of cytotoxic memory T cells in the presence of adenosine.
Introduction
The immune system plays a critical role in the control of cancer development. CD8+ cytotoxic memory T cells in particular are key in eliminating cancer cells.1 High CD8+ T cell infiltration into the tumor is, in fact, associated with a favorable prognosis and response to immunotherapy.2,3 However, in many solid malignancies, such as head and neck squamous cell carcinoma (HNSCC), the immune system fails in part due to the limited ability of cytotoxic T cells to infiltrate the tumor microenvironment (TME).4, 5, 6 Thus, it is important to develop new therapeutic approaches that improve cytotoxic T cell tumor infiltration.
Adenosine, an immunosuppressive purine nucleoside, which accumulates in the TME, suppresses T cell function including chemotaxis.7, 8, 9, 10 Indeed, the presence of adenosine-generating ecto-nucleosidase CD73 (indicative of adenosine accumulation) in the TME is associated with a poor prognosis.11 Adenosine binds to an array of adenosine receptors (A1, A2A, A2B, A3) including the adenosine A2A receptor (A2AR), which initiates a signaling cascade that culminates with the inhibition of KCa3.1, a Ca2+-dependent K+ channel that controls human T cell migration.9,12 In addition to inhibiting chemotaxis, adenosine suppresses other anti-tumor functions of T cells such as the production of interferon-γ (IFN-γ).10,13, 14, 15 Importantly, the inhibitory effect of adenosine is particularly potent in circulating CD8+ T cells of HNSCC patients due to an increased sensitivity to adenosine, in part, conferred by a defect in KCa3.1, which ultimately limits tumor infiltration.6,13
One approach that could improve T cell migration and function in the TME, and ultimately enhance the response to immune checkpoint inhibitors and adoptive T cell therapies, is to target the adenosine signaling pathway.11,16, 17, 18, 19, 20 Many studies have shown that targeting A2AR (through either pharmacological inhibition or genetic deletion) increases cytotoxic T cell function and decreases tumor burden.4,11,14,18,21 A2AR blockade has also been shown to enhance the efficacy of immune checkpoint inhibitors—immunotherapies that are associated with a high degree of resistance.19,22 The efficacy of a therapeutic approach targeting A2AR has been established in a phase I clinical trial where renal cell carcinoma patients were treated with an A2AR antagonist (ciforadenant) alone and in combination with the immune checkpoint inhibitor programmed cell death ligand 1 (anti-PD-L1) antibody.22 In this study, clinical responses (decreased tumor burden and increased survival) were reported for treatment combinations with some degree of associated toxicity.22 Durable responses were also associated with increased CD8+ T cell tumor infiltration.22
Despite these great successes of targeting A2AR in cancer, systemic pharmacological approaches come with limitations. The adenosine signaling pathway is important in many cellular processes including the prevention of overactivation of the immune system in inflammatory settings and thus A2AR is expressed in many different immune cells.9,23 Additionally, A2AR is expressed in other tissues including vasculature, platelets, and brain striatum.24,25 Thus, A2AR signaling is also necessary for proper blood circulation, angiogenesis, and inhibition of platelet aggregation, as well as central nervous system regulation of motor activity and behavior.23, 24, 25, 26, 27 In fact, adenosine and A2AR agonists have been used in many clinical trials; examples include protection from post-operative liver ischemia, treatment of sickle cell anemia, and reduction of reperfusion injury due to coronary stenting.24,25 Therefore, a targeted delivery approach may allow for increased targeted dosage and resultant efficacy without increased toxicity compared to systemic pharmacological A2AR blockers.
One type of targeted delivery approach that has shown great clinical promise in different disease settings is the use of lipid nanoparticles (NPs).28, 29, 30 Lipid NPs are small particles constructed with different phospholipids that can be functionalized to target specific cell types and utilized for targeted drug delivery and gene therapy.31 In vivo murine studies with drugs and small interfering/silencing RNAs (siRNAs) delivered via lipid NPs have emphasized treatment specificity and reduction in off-target effects.28,29,32,33 Furthermore, we have developed lipid NPs that target CD45RO+ memory T cells and successfully deliver siRNAs against the target gene.34,35
Thus, in this study we investigated whether lipid NPs functionalized with either anti-CD8 or anti-CD45RO antibodies could effectively deliver siRNAs against ADORA2A (the gene encoding the A2AR) to the targeted T cells, block the adenosine signaling pathway, and rescue the chemotactic ability of these cells. CD45RO-labeled NPs are a reasonable alternative to CD8-labeled NPs as CD8+ CD45RO+ T cells make up about 90% of T cells in the tumor.5 We determined that, although CD8-labeled NPs were specific for CD8+ T cells, these NPs were unsuccessful at knocking down ADORA2A mRNA expression because they were more quickly internalized into the lysosome. Instead, CD45RO-labeled NPs escaped lysosome internalization and successfully knocked down ADORA2A mRNA expression and restored memory T cell chemotaxis in an adenosine-rich tumor-like microenvironment.
Results
ADORA2A siRNAs knocked down A2AR expression in healthy donor T cells
The goal of this investigation was to knock down the A2AR in specific T cell subsets by targeted delivery of ADORA2A siRNAs via functionalized lipid NPs. The composition and structure of the lipid NPs that were loaded with ADORA2A siRNAs are shown in Figure 1A. Experiments were thus performed to verify that the ADORA2A siRNAs were incorporated into the NPs could knock down A2AR in treated cells. Activated healthy donor (HD) T cells were transfected with 10 nM ADORA2A siRNAs; scramble sequence RNAs (scr-RNAs) served as controls. ADORA2A mRNA levels were determined via quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR). 24 h post-transfection, ADORA2A mRNA levels decreased by 65% in the ADORA2A siRNA-treated cells as compared to the scr control (Figure 1B). Additionally, A2AR protein levels were measured via flow cytometry 24–72 h post-transfection (Figures 1C and 1D). The degree of A2AR knockdown (fold change of normalized A2AR MFI in ADORA2A siRNA-treated cells to scr-RNA-treated cells) varied greatly in time among different donors with some individuals reaching the greatest degree of knockdown at 24 h post-transfection while others had the greatest degree of knockdown at 48 and 72 h (Figure 1C, left). Overall, when greatest degrees of A2AR knockdown (fold change less than one) were compiled for each sample within the 24–72 h time frame, the A2AR protein expression was significantly decreased in the ADORA2A siRNA-treated cells (Figure 1C, right). The gating strategy for the flow cytometry experiments and representative histograms are shown in Figure 1D; the specificity of the A2AR antibody is shown in Figure 1E. In this set of experiments, we tested both CD8+ T cells and a mixed population of CD3+ T cells. We observed a comparable inhibition of ADORA2A mRNA expression by ADORA2A siRNAs in these two groups (67.8% ± 2.0% for CD3+ T cells, n = 3, and 61.6% ± 0.1% in CD8+ T cells, n = 2). The average data shown in Figure 1B comprises both cell types. For the effect on A2AR protein levels, we observed a time-dependent effect in both groups. The two experiments with CD8+ T cells in Figure 1C are reported in yellow and green. Overall, these data showed that ADORA2A siRNAs were effective at knocking down A2AR expression.
Figure 1.

ADORA2A siRNAs decrease ADORA2A mRNA and A2AR protein expression
(A) Representation of the structure of the lipid NPs labeled with fluorescent streptavidin and biotinylated targeting antibody and loaded with siRNAs. PE: phosphoethanolamine; PEG: polyethylene glycol (B) Relative quantity (RQ) of ADORA2A mRNA expression in activated HD CD3+ or CD8+ T cells 24 h post-transfection with 10 nM scr or ADORA2A siRNAs (n = 5; 3 CD3+ T cell samples, 2 CD8+ T cell samples). GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) was used as the reference gene. Data are shown as mean ± standard deviation and significance is determined by a paired Student’s t test. (C) (Left) Fold change of normalized A2AR protein expression in activated HD CD3+ or CD8+ T cells 24 h (n = 5; 3 CD3+ T cell samples, 2 CD8+ T cell samples), 48 h (n = 5; 3 CD3+ T cell samples, 2 CD8+ T cell samples), or 72 h (n = 4; 2 CD3+ T cell samples, 2 CD8+ T cell samples) post-transfection with 10 nM ADORA2A siRNAs as compared to scr-RNAs. (Right) The normalized MFI of A2AR expression showing the highest degree of A2AR protein knockdown post-transfection with 10 nM ADORA2A siRNAs as compared to corresponding scr-RNAs for each individual sample regardless of time point at which knockdown occurred. Data are shown as mean ± standard error of the mean (n = 5). Data are compared using a paired Student’s t test. (D) Flow cytometry gating strategy for determining A2AR expression in transfected cells. The cells were first gated on T lymphocytes and then on GFP fluorescence (indicating that the cells were successfully transfected with siRNAs) to determine A2AR expression. The unstained sample is indicated in orange in the histogram while the A2AR expression of cells treated with scr-RNAs is indicated in blue and the A2AR expression of cells treated with ADORA2A siRNAs is indicated in red. (E) Representative histogram showing A2AR antibody specificity where CD3+ T cells were stained with A2AR antibody ± peptide followed by secondary antibody. Unstained cells are indicated in gray, A2AR antibody + peptide is indicated in red and A2AR antibody alone is indicated in blue. Shown here is a representative experiment from two identical experiments performed in HDs.
CD8-targeting NPs are trapped in the lysosome and fail to deliver siRNAs to T cells
Once the ADORA2A siRNAs were validated, we proceeded to determine whether CD8-NPs specifically bound to and entered CD8+ T cells, and then delivered the intact siRNA content capable of ultimately decreasing ADORA2A expression. In order to determine the specificity of the CD8-NPs for CD8+ T cells, CD3+ T cells isolated from the blood of HDs via negative selection were incubated with fluorescently tagged empty (no siRNAs) CD8-NPs overnight, stained with an anti-CD4 antibody, and analyzed via flow cytometry (Figure 2A). Overall, it was determined that 94% of the CD8-NPs were bound to the CD4– T cells, i.e., CD8+ cytotoxic T cells, as seen in the comparison of the percentage of fluorescent NPs attached to CD4– T cells in quadrant 1 versus the percentage of fluorescent NPs attached to CD4+ T cells in quadrant 2 (Figure 2B). Furthermore, about 86% of CD4– T cells had CD8-NPs either bound to their membrane or incorporated into the cells while only 1.7% of CD4+ T cells were positive for CD8-NPs (Figure 2C). Therefore, we determined that not only were the CD8-NPs specific for CD4– T cells but they also were bound to/incorporated into the majority of the CD4– T cells. In order to ensure that the CD8-NPs bound to CD8+ T cells due to the CD8+ label rather than non-specific binding of the NPs to the cells due to their liposomal structure, fluorescent NPs were labeled with an isotype-specific immunoglobulin G (IgG) antibody (IgG-NPs; thus having no cellular target) and were incubated with CD3+ T cells. It was determined that IgG-NPs did not bind to any cells in the CD3+ mixed T cell population (Figure 2D) indicating that CD8-NPs needed the CD8 antibody label in order to bind to CD8+ T cells. This indicated that the CD8-NPs were specific for CD8+ T cells and that the majority of CD8+ T cells had bound CD8-NPs.
Figure 2.

CD8-NPs are specific toward CD8+ T cells
(A) Flow cytometry gating strategy for determining specificity of CD8-NPs for CD8+ T cells. Cells were first gated based on the CD3+ lymphocyte population (left). The unstained sample was then used to determine the quadrant gate placement for the CD4 stain and NP fluorescence (middle). A representative plot for the distribution of CD8-NPs in CD4– and CD4+ T cells is shown on the right. Quadrant 1 represents the CD4– T cells that are positive for CD8-NPs. Quadrant 2 represents the CD4+ T cells that are positive for CD8-NPs. Quadrant 3 represents CD4+ T cells that are negative for CD8-NPs. Quadrant 4 represents CD4– T cells that are negative for CD8-NPs. Representative plots for n = 4 independent experiments. (B) Distribution of CD8-NPs in HD CD4– and CD4+ T cells within a CD3+ T cell population (the percent of CD8-NPs bound to CD4– T cells versus the percent of CD8-NPs bound to the CD4+ T cells; n = 4). (C) Percentage of HD CD4– and CD4+ T cells within a CD3+ T cell population that are positive for CD8-NPs (n = 4). (B and C) Data are bar graphs representing the mean ± standard deviation. Comparisons are made using paired Student’s t test. (D) Flow cytometry gating strategy for determining non-specific binding of NPs (labeled with IgG antibody) for CD8+ T cells. Cells were first gated based on the CD3+ lymphocyte population (left). The unstained sample was then used to determine the quadrant gate placement for the CD4 stain and NP fluorescence (middle). A representative plot for the distribution of IgG-NPs in CD4– and CD4+ T cells is shown on the right. Quadrant 1 represents the CD4– T cells that are positive for IgG-NPs. Quadrant 2 represents the CD4+ T cells that are positive for IgG-NPs. Quadrant 3 represents CD4+ T cells that are negative for IgG-NPs. Quadrant 4 represents CD4– T cells that are negative for IgG-NPs. Representative plots for n = 3 independent experiments.
We then tested whether the CD8-NPs could effectively deliver intact ADORA2A siRNAs to CD8+ T cells and knock down ADORA2A mRNA expression. To this effect, HD CD8+ T cells were incubated with CD8-NPs loaded with either scr-RNAs or ADORA2A siRNAs: CD8(scr)-NPs or CD8(A2AR)-NPs, respectively, and activated for 48–72 h. In parallel, we conducted experiments using CD45RO-NPs loaded with ADORA2A siRNAs (CD45RO(A2AR)-NPs) as a positive control. Our laboratory has previously shown that CD45RO-NPs are specific to CD45RO+ T cells, are internalized by the cells and release intact siRNA contents capable of knocking down the target gene.34,35 CD8(A2AR)-NPs did not reduce ADORA2A mRNA expression as compared to CD8(scr)-NPs (Figure 3A). Thus, CD8-NPs do not serve as good delivery system for siRNAs. However, CD45RO(A2AR)-NPs reducedADORA2A mRNA expression by 57% as compared to CD45RO(scr)-NPs (Figure 3B). This effect was comparable to the 65% knockdown obtained by the transfection of naked siRNAs (Figure 1B) indicating that the CD45RO(A2AR)-NPs were successful at knocking down ADORA2A mRNA expression.
Figure 3.

ADORA2A siRNAs delivered via CD45RO-NPs knock down ADORA2A mRNA expression in CD8+ memory T cells
(A) RQ of ADORA2A mRNA expression in activated CD8+ T cells 48–72 h post-incubation with CD8-NPs loaded with 200 pmol scr or ADORA2A siRNAs (n = 2 for both time points). (B) RQ of ADORA2A mRNA expression in activated CD8 memory T cells 72 h post-incubation with CD45RO-NPs loaded with 200 pmol scramble or ADORA2A siRNAs (n = 2). (A and B) 18S rRNA was used as reference gene. Data are represented as mean ± standard deviation. Significance determined by paired Student’s t test.
We then determined the reason for the inability of the CD8-NPs to successfully deliver intercellular siRNAs compared to CD45RO-NPs. Immunofluorescence was used to determine the localization of the CD8-NPs and CD45RO-NPs in the lysosome at 24 h. We determined that CD8-NPs had a higher percentage of localization in the lysosome than the CD45RO-NPs (Figures 4A and 4B). This indicated that at 24 h, a larger percentage of CD8-NPs were already degraded in the lysosome as compared to CD45RO-NPs and therefore CD8-NPs likely did not have time to release the ADORA2A siRNA contents into the cell. Therefore, CD45RO-NPs serve as better delivery systems for siRNAs and were utilized for subsequent experiments.
Figure 4.

CD8-NPs co-localize with the lysosome
(A) Representative confocal images of an activated CD3+ T cell incubated with CD8-NPs (top panel) and CD45RO-NPs (bottom panel) for 24 h. NPs visualized in green. Lysosomal marker LAMP-1 is visualized in red. Merge of NPs with LAMP-1 is shown with co-localization indicated by the presence of yellow. All images shown are shown at the same scale. (B) Single cell percentage of NP raw integrated density (NP intensity) in the lysosome as compared to total NP intensity in the cell. Approximately 20–45 cells were visualized from a total of two donors. Data represented as boxplots: line indicates the median; lower box is the 25th percentile; upper box is the 75th percentile; and whiskers represent the 10th and 90th percentiles. Each dot represents an individual cell. Significance determined by Mann-Whitney rank sum test.
CD45RO-targeting NPs loaded with ADORA2AsiRNAs rescue adenosine-mediated inhibition of HNSCC T cell chemotaxis
Because CD45RO(A2AR)-NPs were successful at knocking down ADORA2A mRNA expression, we proceeded to determine whether these NPs could rescue the chemotactic ability of CD8+ memory T cells from HNSCC patients in the presence of adenosine. Circulating CD8+ T cells in HNSCC patients are highly sensitive to adenosine that blocks their chemotaxis by 80%.6 This process reduces the number of CD8+ T cells that infiltrate the tumor.6 CD8+ CD45RO+ T cells make up about 90% of the cells that infiltrate solid tumors; therefore, targeting memory T cells with CD45RO(A2AR)-NPs could ultimately facilitate tumor infiltration with this cytotoxic cell population.5
In order to determine the effect of CD45RO(A2AR)-NPs, activated HNSCC CD8+ memory T cells were incubated with either CD45RO(A2AR)-NPs or CD45RO(scr)-NPs for 72–96 h and then were resuspended in a collagen gel that mimics the TME. A chemokine gradient was then formed over the cells with either CXCL10 only or CXCL10 and adenosine. Time lapse microscopy was then used to record the migration of these cells. Two point trajectories of the CD8+ memory T cells treated with either CD45RO(scr)-NPs or CD45RO(A2AR)-NPs are depicted in Figure 5A and Figure 5B, respectively. Each trajectory represents the distance traveled by a single cell along the chemokine gradient in the y-direction and the Y-center of mass (Y-COM) reports the average distance traveled within a certain time frame.6 We have previously shown that this parameter is a good indicator to quantify the effect of adenosine on T cell chemotaxis.6 Furthermore, we have shown that HNSCC CD8+ T cells are very sensitive to adenosine; more than their healthy counterparts.6 This difference in adenosine sensitivity was also observed in CD8+ memory T cells where the inhibition of chemotaxis by adenosine was higher in CD8+ memory T cells from HNSCC (70.9% ± 26.6%, n = 2 patients) than from HD patients (16.2% ± 2.6%, n = 2 patients; Figures S1A and S1B). We thus tested the effect of CD45RO(A2AR)-NPs on HNSCC CD8+ memory T cell chemotaxis. In control experiments, adenosine inhibited the chemotaxis of CD8+ memory T cells treated with CD45RO(scr)-NPs by 117%, as expected (Figure 5C). This inhibition of chemotaxis was comparable to the inhibition seen in untreated HNSCC CD8+ memory T cells in the presence of CXCL10 + adenosine and comparable to the inhibition seen in HNSCC T cells in the presence of CXCL12 + adenosine, thus indicating that the inhibitory effect of adenosine on chemotaxis of HNSCC T cells does not differ because of the presence of NPs or the chemokine used (Figure S2).6,36 In contrast to CD45RO(scr)-NPs, the inhibitory effect of adenosine was abrogated by CD45RO(A2AR)-NPs (Figures 5B and 5C). Overall, the percent inhibition of the Y-COM in the presence of adenosine for cells treated with CD45RO(A2AR)-NPs (−35.0%) was significantly lower than cells treated with CD45RO(scr)-NPs (117%; Figure 5D). These data indicated that the CD45RO(A2AR)-NPs effectively reduced the sensitivity of HNSCC CD8+ memory T cells to adenosine ultimately restoring their ability to chemotax in an adenosine-rich immunosuppressive microenvironment.
Figure 5.

CD45RO(A2AR)-NPs rescue the chemotactic ability of HNSCC CD8+ memory T cells in the presence of adenosine
(A and B) Representative two point trajectories of T cells migrating along the CXCL10 gradient (green) or CXCL10 + adenosine (ADO) gradient (blue) in HNSCC CD8+ memory T cells treated with (A) CD45RO(scr)-NPs or (B) CD45RO(A2AR)-NPs. Trajectories artificially set to start at the origin with the red triangle representing the Y-COM. (C) Y-COM of HNSCC CD8+ memory T cells treated with (left) CD45RO(scr)-NPs (n = 6) or (right) CD45RO(A2AR)-NPs (n = 6) in the presence of CXCL10 or CXCL10 + adenosine. Significance was determined using (left) a Wilcoxon signed rank test or (right) paired Student’s t test. (D) Percent inhibition of Y-COM of HNSCC CD8+ memory T cells in the presence of CXCL10 + adenosine compared to CXCL10 alone when treated with CD45RO(scr)-NPs (n = 6) compared to CD45RO(A2AR)-NPs (n = 6). Data are represented single dot plots (colored dots; wherein each dot represents a single donor) and as mean ± standard deviation (shown in black). Significance was determined using Student’s t test.
Discussion
The infiltration of CD8+ T cells into solid tumors is important for a favorable prognosis and response to immunotherapies.2,3,5 However, immunosuppressive components in the TME, such as adenosine, inhibit CD8+ T cell motility and tumor infiltration.6,7 Herein, we provide evidence that functionalized lipid NPs are able to effectively knock down A2AR in selective T cells subsets from HNSCC patients and increase the chemotactic ability of this subpopulation of cells in the presence of adenosine. These data show the therapeutic potentials of targeted NP delivery to block the inhibitory effect of the adenosine signaling pathway in cancer.
Immunotherapies targeting the adenosine signaling pathway have been thoroughly studied and show great clinical promise.11,14,17,18,22 However, a targeted therapeutic approach could limit the side effects caused by the broad expression A2AR in a wide variety of cells and tissues.24, 25, 26, 27 While lipid NP-based drug-delivery formulations have been clinically approved for use in cancer therapy and viral vaccines, the use of lipid NPs has been associated with side effects such as complement activation, which can induce complement associated-pseudoallergy.30,37 However, the formulation of lipid NPs that we used here is advantageous because the phospholipids are biocompatible, as they are present in the cell membrane.31 Furthermore, NPs of similar lipid composition have been used in vivo in animal models with no reported side effects.32 Still, the NPs we have used may present with limitations associated with the presence of polyethylene glycol (PEG) (used to stabilize the NPs and prolong their half-life in vivo) and streptavidin (SAV; used to facilitate the binding to biotinylated targeting antibodies). PEG can be recognized by anti-PEG antibodies that individuals may have developed by exposure to other pharmaceutical or cosmetic preparations.38 Additionally, while the SAV-biotin non-covalent interaction is a widely used method for functionalization of nanomaterial, SAV has immunogenic properties and has been shown to induce inflammatory cytokine expression in vitro.39,40 Further in vivo toxicity studies will indicate if there is a need for chemical modifications of these NPs; however, there are viable avidin analogs, such as neutravidin and bradavidin II, that have less immunogenicity as compared to SAV.39,40
Herein, we established the basic initial requisites that functionalized NPs have to have in order to be considered as potential therapeutic options: specifically target a cell subset and be able to deliver intact contents in order to achieve the desired effects. Our results showed that CD45RO-NPs do indeed fulfil these requirements; CD8-NPs do not. While CD8-NPs were specific to CD8+ T cells, they failed to knock down the A2AR. The inability of CD8-NPs to effectively decrease ADORA2A mRNA expression resulted from their rapid internalization in the lysosome, which does not allow enough time for NP disassembly in the endosome and endosomal escape of the siRNAs.41,42 Furthermore, it has been shown that ligand-bound CD8 is poorly internalized both in unstimulated and activated T cells and is restricted to the membrane, which could add to the inability of CD8-NPs to deliver their siRNA contents.43,44 Therefore, although there have been some accounts of using CD8 as a target for NPs, CD8 may not be the most effective targeting moiety when the encapsulated material needs to be delivered into the cells.45,46
While the CD8 receptor was ultimately a poor target, CD45RO-NPs fulfilled our intent. Targeting the CD45RO+ T cells would be an effective alternative approach to targeting CD8+ T cells since 90% of CD8+ T cells that infiltrate tumors, especially in HNSCC, are CD8+ memory T cells.5 Additionally, CD45RO-NPs have already been characterized and validated.34 We previously utilized these NPs for targeted knockdown of Kv1.3 channels in memory T cells and demonstrated that they specifically target memory T cells, are internalized by the cells, and exert their desired effects.34,35 In the current study, when CD45RO-NPs were used instead of CD8-NPs, we showed that CD8+ memory T cells treated with CD45RO(A2AR)-NPs decreased ADORA2A mRNA expression and, ultimately, blocked the inhibitory effect of adenosine on chemotaxis. The degree of chemotaxis inhibition observed in vitro in circulating CD8+ T cells is a good marker of the ability of these cells to infiltrate the tumor as the two parameters are inversely correlated; i.e., patients whose circulating CD8+ T cell chemotactic ability were most severely inhibited by adenosine in vitro are also those who had the lowest infiltration of CD8+ T cells into the tumor.6 Therefore, since the CD45RO(A2AR)-NPs rescued the chemotactic ability of CD8+ memory T cells in the presence of adenosine, these cells may be better suited and better able to infiltrate the TME and, possibly, allows them to maintain their function in the TME. Overall, this study introduces a novel targeted therapy aimed to oppose the immunosuppressive nucleoside adenosine and restore T cell infiltration and function in the TME. Ultimately, further in vivo murine models will determine the efficacy of these NPs against cancerous tumors.
Materials and methods
Human subjects
HD whole blood was obtained from consenting donors or from discarded blood units obtained from Hoxworth Blood Center at the University of Cincinnati. HNSCC whole blood was obtained from 8 patients whose demographics and clinical features are summarized in Table 1. The eligibility for HNSCC patients for this study was determined by a tissue-biopsy-confirmed diagnosis of HNSCC and the absence of any treatment prior to the blood draw. Informed consent was obtained for both the HDs and HNSCC patients included in this study, and the study was approved by the University of Cincinnati Institutional Review Board (IRB # 2014-4755). Sample collection protocols were conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. The data for HNSCC patients was managed using Research Electronic Data Capture tools (University of Cincinnati). Patient information from discarded blood units is not available.
Table 1.
Demographics and clinical table for HNSCC patients
| Age (years) | Value (%) |
|---|---|
| Range | 48 to 75 |
| Mean | 60.6 |
| Gender | |
| Male | 6 (75) |
| Female | 1 (12.5) |
| Unknown | 1 (12.5) |
| Tumor Site | |
| Oral cavity | 3 (37.5) |
| Oropharynx | 4 (50) |
| Unknown | 1 (12.5) |
| Clinical stage | |
| T1 | 3 (37.5) |
| T2 | 3 (37.5) |
| T3 | 1 (12.5) |
| Unknown | 1 (12.5) |
| Nodal status | |
| N0 | 1 (12.5) |
| N1 | 4 (50) |
| N2 | 2 (25) |
| Unknown | 1 (12.5) |
| HPV status/P16 status | |
| Negative | 3 (37.5) |
| Positive | 3 (37.5) |
| Unknown | 2 (25) |
| Smoking | |
| Yes (≥10 pack years) | 4 (50) |
| No (<10 pack years) | 3 (37.5) |
| Unknown | 1 (12.5) |
| Alcohol use | |
| Yes (≥5 drinks/week) | 2 (25) |
| No (<5 drinks/week) | 4 (50) |
| Unknown | 2 (25) |
HNSCC patients (n = 8) were enrolled in the study. Clinical stage from T1 to T3 refers to the size/extent of the tumors. Nodal status depicts the involvement of lymph nodes depending on their number and location.
Reagents
Phosphate-buffered saline (PBS), RPMI-1640, penicillin, streptomycin, L-glutamine, fetal bovine serum, and HEPES were all obtained from Thermo Fisher. Human serum and adenosine were obtained from Millipore Sigma and the adenosine solution was prepared in sterile H2O. CXCL10 and CXCL12 were obtained from R&D systems and the stock solutions were prepared in PBS with 0.1% bovine serum albumin.
Cell isolation
Whole blood from HNSCC patients and HDs was obtained and peripheral blood mononuclear cells were isolated using Ficoll-paque PLUS density gradient (GE Healthcare Biosciences) as described previously.6 CD3+, CD8+, and CD8+ memory T cells were each isolated by negative selection using commercially available kits (EasySep Enrichment kits, StemCell Technologies). The purities of these enriched T cell populations were determined using flow cytometry as indicated in Figure S3. T cells were maintained in T cell medium (RPMI-1640 medium containing L-glutamine supplemented with 200 U/mL penicillin, 200 mg/mL streptomycin, 1 mM L-glutamine, 10 mM HEPES, and 10% human serum) and then either remained unstimulated for 12–24 h (±NPs) or were activated in cell culture plates coated with 10 μg/mL anti-human CD3/CD28 antibodies (BioLegend) or 25 μL/mL ImmunoCult Human CD3/CD28 T Cell Activator (StemCell Technologies) for 72–96 h (±NPs) in the 37°C/5% CO2 incubator as indicated in the following protocols.
Transfection
Activated HD CD3+ or CD8+ T cells (either by plate bound antibodies or soluble activators; 1–6 × 106 cells per condition) were transfected using the Amaxa P3 Primary Cell 4D-Nucleofector X Kit L (Lonza Cologne GmbH) as indicated by the manufacturer’s instructions. Briefly, 10 nM ADORA2A siRNAs (Ambion ADORA2A Silencer Select siRNA; ID: S1088; Thermo Fisher) or scr-RNAs (Ambion Silencer Select Negative Control #2 siRNA; Thermo Fisher) and pMaxGFP vector (2 μg; from Lonza Nucleofector kit) were added to the T cells and the samples were transfected in the 4-Nucleofector X Unit (Lonza) using the program “T cell, human, stimulated” (E0–E115). The transfected T cells were then incubated in T cell medium in the 37°C/5% CO2 incubator for the indicated time points (24–72 h). Transfected cells were then used to determine A2AR mRNA and protein expression.
Lipid NP production and functionalization
Lipid NPs (100 nm) were developed as previously described.34 In brief, L-a-phosphatidylcholine, 1,2-distearoyl-snglycero-3-phosphoethanolamine-N-[biotinyl(polyethylene glycol)-2000] and cholesterol (Avanti Lipids) were mixed together, dried with nitrogen gas, and rehydrated with PBS. The mixture was the shaken for 2 h at 37°C to form multilamellar vesicles (Barnstead Model: Max Q 4000, Thermo Fisher). The multilamellar vesicles were then sonicated (Fisherbrand Sonic Dismembrator Ultra Sonic liquid Processor Model: FB120) and extruded through a 100 nm filter (LIPEX Thermobarrel Extruder, Northern Lipids) to form unilamellar vesicles (ULVs). ULV size was then determined using the Nanoseries Zetasizer (Malvern Instruments) and labeled with appropriate antibodies for both fluorescence and cell targeting. ULVs were labeled with 10 μg/mL SAV conjugated with Alexa Fluor-488 or Alexa Fluor-647 (BioLegend) and 10 μg/mL biotin anti-human CD8 antibody (clone: MEM31, Genetex), 10 μg/mL biotin anti-human CD45RO antibody (clone: UCHL1; BioLegend), or 10 μg/mL biotin anti-human IgG Fc antibody (clone: HP6017; BioLegend). Once the ULVs were labeled, the unbound antibodies were removed using Sephrose CL-4B columns (GE Healthcare Life Sciences). The ULVs were then frozen at −80°C, lyophilized overnight (FreeZone2.5 lyophilizer, LabConco) and then stored at −80°C. ULVs are referred to as CD8-NPs, CD45RO-NPs, or IgG-NPs based upon antibody labeling.
Insertion of siRNAs into NPs and NP treatment of cells
For CD8-NP specificity and internalization experiments, at the time of experimentation, lyophilized CD8-NPs or IgG-NPs were reconstituted in 100 μL sterile H2O. HD CD8+ T cells (0.3–1 × 106 cells at 2 × 106 cells/mL per condition) were resuspended in T cell medium and incubated with 50 μL of empty NPs (no siRNAs) and left unstimulated for 12–24 h in the 37°C/5% CO2 incubator.
Alternatively, for experiments determining ADORA2A mRNA expression and T cell chemotaxis, the lypophilized CD8-NPs or CD45RO-NPs were reconstituted in 100 μL sterile H2O containing protamine-sulfate (20 μM; Sigma) and 200 pmol of ADORA2A siRNAs or scr-RNAs. HD CD8+ or HD/HNSCC CD8+ memory T cells (∼0.3 × 106 cells at 2 × 106 cells/mL per condition) were resuspended in T cell medium and incubated with 50 μL of scr-RNA or ADORA2A siRNA-loaded CD8-NPs or CD45RO-NPs. CD8+ T cells were treated with siRNA-loaded CD8-NPs and CD8+ memory T cells were treated with siRNA-loaded CD45RO-NPs. Samples were then activated in cell culture plates coated with 10 μg/mL anti-CD3/CD28 antibodies in the 37°C/5% CO2 incubator for up to 96 h.
Flow cytometry
Flow cytometry was used to determine both A2AR protein expression post-transfection and CD8-NP specificity. A2AR expression post-transfection is as follows: activated HD CD3+ or CD8+ T cells transfected with scr or A2AR siRNAs were fixed with 1%–4% paraformaldehyde (Affymetrix, Thermo Fisher Scientific), permeablized with BD Perm/Wash buffer (Fixation/Permeabilization Solution kit; BD Cytofix/Cytoperm Plus, BD Biosciences) and then stained with anti-human anti-adenosine A2AR rabbit polyclonal antibody (Alomone labs) in BD Perm/Wash buffer followed by staining with a secondary anti-rabbit antibody—either Alexa Fluor-647 conjugated donkey anti-rabbit antibody (Thermo Fisher) or Brilliant Violet 421 conjugated donkey anti-rabbit antibody (BioLegend). Specificity of the anti-adenosine A2AR rabbit polyclonal antibody was determined by peptide adsorption. Anti-human anti-adenosine A2AR rabbit polyclonal antibody (0.75 μg) was incubated with 1.5 μg A2AR blocking peptide (Alomone) at a 1:2 ratio (1.5 μg peptide was added to 0.75 μg antibody). CD3+ T cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 (Millipore Sigma) and incubated with either the antibody alone or the antibody + peptide solution followed by incubation with Alexa Fluor 488 conjugated donkey anti-rabbit antibody.
CD8-NP specificity is as follows: resting HD CD3+ T cells were incubated with Alexa Fluor 488-labeled empty CD8-NPs (or Alexa Fluor 647-labeled empty IgG-NPs as control) for 12–24 h as indicated above. The samples were then fixed with 1% paraformaldehyde and stained with an anti-human CD4 antibody (Alexa Fluor 700 conjugated anti-human CD4 antibody [clone: RPA-T4, BioLegend], PerCPCy5.5 anti-human CD4 antibody [clone: OKT4, BioLegend], or PE anti-human CD4 antibody (clone: A161A1; BioLegend; used with IgG-NPs]). All flow cytometry samples were run on a LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo software (FlowJo).
qRT-PCR
qRT-PCR was used to determine ADORA2A mRNA expression in activated HD CD3+/CD8+ T cells after transfection and in activated HD CD8+ and CD8+ memory T cells after incubation with either siRNA-loaded CD8-NPs or CD45RO-NPs, respectively. The GFP-positive cell population (which indicated transfected cells) was not separated from non-transfected cells when measuring mRNA expression. Therefore, gene expression is a result from both transfected and non-transfected cells combined. The E.Z.N.A. total RNA isolation kit (Omega Biotek) was used to isolate total RNA from activated HD CD3+, CD8+, and CD8+ memory T cells after transfection or after incubation with either siRNA-loaded CD8-NPs or CD45RO-NPs as described above. Complementary DNA (cDNA) was synthesized from at least 200 ng of total RNA per condition using the Maxima First Strand cDNA Synthesis Kit for qRT-PCR (Thermo Fisher) as per the manufacturer’s instructions. qRT-PCR was then performed (as described in the manufacturer’s instructions) in order to detect the gene expression of ADORA2A, GAPDH, and 18S rRNA. In brief, the qRT-PCR was assembled in a 96-well plate with each well having 10 μL of 2× TaqMan Gene Expression Master Mix (Applied Biosystems), 1 μL of predesigned TaqMan Gene Expression Assay primer (A2AR: ADORA2A [assay ID: Hs00169123_m1], GAPDH [assay ID: HS03929097_g1], or 18S rRNA [assay ID:Hs99999901_s1]; all primers from Applied Biosystems), and 9 μL of diluted cDNA samples (diluted from 20 μL to 170 μL in sterile H2O). Each sample was prepared in technical replicates of four and the plate was run in Applied Biosystems StepOne Real-Time PCR System (Thermo Fisher) to obtain the CT values (StepOne software version 2.1). The ΔCT value for each ADORA2A technical replicate was calculated by subtracting the average CT value of GAPDH or 18S rRNA from the CT value of each corresponding ADORA2A technical replicate. The difference between the ΔCT value of the scr-RNA-treated sample and the ΔCT value of the corresponding ADORA2A siRNA-treated sample was then used to calculate the ΔΔCT values. Finally, the relative quantity (RQ) or fold change of ADORA2A mRNA expression in ADORA2A siRNA versus scr-RNA-treated samples was calculated as the 2-ΔΔCT value as described previously.6
Immunofluorescence
Immunofluorescence was used to determine CD8-NP and CD45RO-NP localization in the lysosome. HD CD3+ T cells were incubated with either Alexa Fluor 488 labeled empty CD8-NPs or CD45RO-NPs and activated for 24 h with soluble anti-CD3/CD28 antibodies. Cells were then plated on poly-L-lysine (Millipore Sigma) coated coverslips for at least 30 min in the 37°C/5% CO2 incubator. Cells were fixed with 4% paraformaldehyde at room temperature for 30 min, permeabilized for 20 min at room temperature with PBS + 0.2% Triton-100X, and blocked with a solution of PBS containing 10% FBS for 1 h at room temperature, and lysosomes were stained with a monoclonal rabbit anti-human LAMP-1 antibody (clone D2D11, Cell Signaling Technology) diluted in the blocking solution for 2 h at room temperature. Cells were then washed with PBS and stained with Alexa Fluor 568 conjugated donkey anti-rabbit secondary antibody diluted in the blocking solution for 1 h at room temperature. All coverslips were washed and labeled with Alexa Fluor 647-conjugated rabbit anti-human CD8 alpha antibody (clone EP1150, Abcam) diluted in blocking solution for 2 h at room temperature. The coverslips were then washed and mounted on slides with Fluoromount G (Thermo Fisher). Images were then obtained with an inverted Zeiss LSM 710 microscope (Carl Zeiss Microscopy GmbH). Images were taken at 100× oil immersion lens at room temperature and the pinhole was set at 1 airy unit. Approximately 20–45 cells were obtained per condition. Percent of NP localization in the lysosome was determined using ImageJ (National Institutes of Health). A region of interest (ROI) was drawn around the entirety of the cell. The raw integrated density of NP signal within the total cell ROI was measured. A ROI was then drawn around the lysosome and the raw integrated density of NP signal within the lysosome ROI was measured. The percent of NP localization in the lysosome was determined by dividing raw integrated density of NP signal found inside the lysosome by the total cell raw integrated density of NP signal multiplied by 100.
Chemotaxis
The effect of CD45RO(A2AR)-NPs on CD8+ memory T cell chemotaxis was determined using u-Slide Chemotaxis assays (ibidi GmbH) as previously described.6 This technique allows for 3D chemotaxis of T cells in a collagen matrix. In brief, 0.2–0.6 × 106 activated HNSCC CD8+ memory T cells were incubated with either scr or ADORA2A siRNA-loaded Alexa Fluor 488 labeled CD45RO-NPs overnight as indicated above. Each cell suspension was then centrifuged and washed to remove any unbound NPs and the samples were reactivated for a total activation time of 72–96 h. The cell samples were then resuspended in a gel containing rat tail collagen type I (Corning) and inserted into the center chamber portion of the u-Slide as per the manufacturer’s instructions. Migration medium was then inserted into the reservoir to the right of each center chamber. Migration medium plus 8 μg/mL CXCL10 was added to the left reservoir of one chamber forming a concentration gradient over the cells. In a separate chamber, in order to determine the effects of adenosine on migration, 1 μM adenosine (diluted in migration medium) plus 8 μg/mL CXCL10 was added to the left reservoir as previously indicated.6 The inverted Zeiss LSM 710 microscope with a 37°C incubator was then used to perform time lapse microscopy with the u-Slide capturing images every 3 s up to a total of 1,000 images in both brightfield and 488 channels as described previously.6 Two point manual cell tracking was performed using ImageJ software and was analyzed using the Chemotaxis and Migration Tool (ibidi GmbH). 10–20 cells were tracked per condition regardless of the presence of NP (Alexa Fluor 488) signal. The Y-COM, which is the average movement of the cells toward the chemokine in the y-direction, was measured as the chemotactic effect.6 We also performed control chemotaxis experiments in non-nanoparticle treated HD and HNSCC CD8+ memory T cells (Figure S1).
Statistical analysis
Statistical analysis was performed using SigmaPlot 13.0 (Systat Software). Student’s t tests, paired Student’s t tests, or Mann-Whitney rank sum tests or Wilcoxon signed rank tests (when samples failed normality or had unequal variance) were used to compare samples as indicated in the figure legends. Statistical significance was defined by a p value of 0.05 or less.
Acknowledgments
We would like to acknowledge Dr. Heather Duncan for her aid with IRB regulatory affairs (Division of Nephrology, Department of Internal Medicine, University of Cincinnati) and the clinical coordinators for their assistance in patient sample collection (Clinical Trials Office, University of Cincinnati Cancer Center). We would like to thank Dr. Nancy Ratner for use of the 4-Nucleofector X Unit and Drs. Giovanni Pauletti and Gerald Kasting for use of the Zetasizer. Flow cytometry was performed at Shriner’s Hospital for Children Flow Cytometry Core. 3D Chemotaxis and immunofluorescence were performed at the Live Microscopy Core in the Department of Pharmacology and Systems Physiology at the University of Cincinnati. Funding was provided by the National Cancer Institute (CA095286; L.C.) and the Brandon C. Gromada Head & Neck Cancer Foundation (L.C.). H.S.N. was supported by NCI T32CA117846 and University of Cincinnati Graduate Student URC Grant.
Author contributions
H.S.N. designed and performed experiments, analyzed and interpreted the data, and wrote the manuscript. A.A.C. performed experiments and contributed to data analysis and manuscript preparation. M.J.A. performed experiments and analyzed data. T.M.W.-D. supervised specimen procurement and provided expertise in HNSCC. L.C. supervised the design, analysis, and interpretation of the experiments, and wrote the manuscript. All the authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.03.001.
Supplemental information
References
- 1.Weninger W., Biro M., Jain R. Leukocyte migration in the interstitial space of non-lymphoid organs. Nat. Rev. Immunol. 2014;14:232–246. doi: 10.1038/nri3641. [DOI] [PubMed] [Google Scholar]
- 2.Leffers N., Gooden M.J., de Jong R.A., Hoogeboom B.N., ten Hoor K.A., Hollema H., Boezen H.M., van der Zee A.G., Daemen T., Nijman H.W. Prognostic significance of tumor-infiltrating T-lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer. Cancer Immunol. Immunother. 2009;58:449–459. doi: 10.1007/s00262-008-0583-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gooden M.J., de Bock G.H., Leffers N., Daemen T., Nijman H.W. The prognostic influence of tumour-infiltrating lymphocytes in cancer: a systematic review with meta-analysis. Br. J. Cancer. 2011;105:93–103. doi: 10.1038/bjc.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohta A., Gorelik E., Prasad S.J., Ronchese F., Lukashev D., Wong M.K., Huang X., Caldwell S., Liu K., Smith P. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA. 2006;103:13132–13137. doi: 10.1073/pnas.0605251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chimote A.A., Hajdu P., Sfyris A.M., Gleich B.N., Wise-Draper T., Casper K.A., Conforti L. Kv1. 3 channels mark functionally competent CD8+ tumor-infiltrating lymphocytes in head and neck cancer. Cancer Res. 2017;77:53–61. doi: 10.1158/0008-5472.CAN-16-2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chimote A.A., Balajthy A., Arnold M.J., Newton H.S., Hajdu P., Qualtieri J., Wise-Draper T., Conforti L. A defect in KCa3. 1 channel activity limits the ability of CD8+ T cells from cancer patients to infiltrate an adenosine-rich microenvironment. Sci. Signal. 2018;11:eaaq1616. doi: 10.1126/scisignal.aaq1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chimote A.A., Hajdu P., Kucher V., Boiko N., Kuras Z., Szilagyi O., Yun Y.H., Conforti L. Selective inhibition of KCa3.1 channels mediates adenosine regulation of the motility of human T cells. J. Immunol. 2013;191:6273–6280. doi: 10.4049/jimmunol.1300702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blay J., White T.D., Hoskin D.W. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. 1997;57:2602–2605. [PubMed] [Google Scholar]
- 9.Sitkovsky M.V., Lukashev D., Apasov S., Kojima H., Koshiba M., Caldwell C., Ohta A., Thiel M. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu. Rev. Immunol. 2004;22:657–682. doi: 10.1146/annurev.immunol.22.012703.104731. [DOI] [PubMed] [Google Scholar]
- 10.Mandapathil M. Adenosine-mediated immunosuppression in patients with squamous cell carcinoma of the head and neck. HNO. 2016;64:303–309. doi: 10.1007/s00106-016-0137-7. [DOI] [PubMed] [Google Scholar]
- 11.Leone R.D., Emens L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer. 2018;6:57. doi: 10.1186/s40425-018-0360-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuras Z., Yun Y.H., Chimote A.A., Neumeier L., Conforti L. KCa3.1 and TRPM7 channels at the uropod regulate migration of activated human T cells. PLoS ONE. 2012;7:e43859. doi: 10.1371/journal.pone.0043859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mandapathil M., Szczepanski M., Harasymczuk M., Ren J., Cheng D., Jackson E.K., Gorelik E., Johnson J., Lang S., Whiteside T.L. CD26 expression and adenosine deaminase activity in regulatory T cells (Treg) and CD4(+) T effector cells in patients with head and neck squamous cell carcinoma. OncoImmunology. 2012;1:659–669. doi: 10.4161/onci.20387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whiteside T.L. Targeting adenosine in cancer immunotherapy: a review of recent progress. Expert Rev. Anticancer Ther. 2017;17:527–535. doi: 10.1080/14737140.2017.1316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mastelic-Gavillet B., Navarro Rodrigo B., Décombaz L., Wang H., Ercolano G., Ahmed R., Lozano L.E., Ianaro A., Derré L., Valerio M. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8+ T cells. J. Immunother. Cancer. 2019;7:257. doi: 10.1186/s40425-019-0719-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sek K., Kats L.M., Darcy P.K., Beavis P.A. Pharmacological and genetic strategies for targeting adenosine to enhance adoptive T cell therapy of cancer. Curr. Opin. Pharmacol. 2020;53:91–97. doi: 10.1016/j.coph.2020.07.002. [DOI] [PubMed] [Google Scholar]
- 17.Sitkovsky M.V. Lessons from the A2A Adenosine Receptor Antagonist-Enabled Tumor Regression and Survival in Patients with Treatment-Refractory Renal Cell Cancer. Cancer Discov. 2020;10:16–19. doi: 10.1158/2159-8290.CD-19-1280. [DOI] [PubMed] [Google Scholar]
- 18.Vigano S., Alatzoglou D., Irving M., Ménétrier-Caux C., Caux C., Romero P., Coukos G. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front. Immunol. 2019;10:925. doi: 10.3389/fimmu.2019.00925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beavis P.A., Milenkovski N., Henderson M.A., John L.B., Allard B., Loi S., Kershaw M.H., Stagg J., Darcy P.K. Adenosine receptor 2A blockade increases the efficacy of anti–PD-1 through enhanced antitumor T-cell responses. Cancer Immunol. Res. 2015;3:506–517. doi: 10.1158/2326-6066.CIR-14-0211. [DOI] [PubMed] [Google Scholar]
- 20.Allard B., Allard D., Buisseret L., Stagg J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 2020;17:611–629. doi: 10.1038/s41571-020-0382-2. [DOI] [PubMed] [Google Scholar]
- 21.Ma S.R., Deng W.W., Liu J.F., Mao L., Yu G.T., Bu L.L., Kulkarni A.B., Zhang W.F., Sun Z.J. Blockade of adenosine A2A receptor enhances CD8+ T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol. Cancer. 2017;16:99. doi: 10.1186/s12943-017-0665-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fong L., Hotson A., Powderly J.D., Sznol M., Heist R.S., Choueiri T.K., George S., Hughes B.G.M., Hellmann M.D., Shepard D.R. Adenosine 2A receptor blockade as an immunotherapy for treatment-refractory renal cell cancer. Cancer Discov. 2020;10:40–53. doi: 10.1158/2159-8290.CD-19-0980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lukashev D.E., Smith P.T., Caldwell C.C., Ohta A., Apasov S.G., Sitkovsky M.V. Analysis of A2a receptor-deficient mice reveals no significant compensatory increases in the expression of A2b, A1, and A3 adenosine receptors in lymphoid organs. Biochem. Pharmacol. 2003;65:2081–2090. doi: 10.1016/s0006-2952(03)00158-8. [DOI] [PubMed] [Google Scholar]
- 24.Fredholm B.B., IJzerman A.P., Jacobson K.A., Linden J., Müller C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors--an update. Pharmacol. Rev. 2011;63:1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J.F., Eltzschig H.K., Fredholm B.B. Adenosine receptors as drug targets--what are the challenges? Nat. Rev. Drug Discov. 2013;12:265–286. doi: 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montesinos M.C., Desai A., Chen J.-F., Yee H., Schwarzschild M.A., Fink J.S., Cronstein B.N. Adenosine promotes wound healing and mediates angiogenesis in response to tissue injury via occupancy of A(2A) receptors. Am. J. Pathol. 2002;160:2009–2018. doi: 10.1016/S0002-9440(10)61151-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Varani K., Portaluppi F., Merighi S., Ongini E., Belardinelli L., Borea P.A. Caffeine alters A2A adenosine receptors and their function in human platelets. Circulation. 1999;99:2499–2502. doi: 10.1161/01.cir.99.19.2499. [DOI] [PubMed] [Google Scholar]
- 28.Dolina J.S., Sung S.S., Novobrantseva T.I., Nguyen T.M., Hahn Y.S. Lipidoid Nanoparticles Containing PD-L1 siRNA Delivered In Vivo Enter Kupffer Cells and Enhance NK and CD8(+) T Cell-mediated Hepatic Antiviral Immunity. Mol. Ther. Nucleic Acids. 2013;2:e72. doi: 10.1038/mtna.2012.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stephan M.T., Stephan S.B., Bak P., Chen J., Irvine D.J. Synapse-directed delivery of immunomodulators using T-cell-conjugated nanoparticles. Biomaterials. 2012;33:5776–5787. doi: 10.1016/j.biomaterials.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bulbake U., Doppalapudi S., Kommineni N., Khan W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics. 2017;9:E12. doi: 10.3390/pharmaceutics9020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grimaldi N., Andrade F., Segovia N., Ferrer-Tasies L., Sala S., Veciana J., Ventosa N. Lipid-based nanovesicles for nanomedicine. Chem. Soc. Rev. 2016;45:6520–6545. doi: 10.1039/c6cs00409a. [DOI] [PubMed] [Google Scholar]
- 32.Peer D., Park E.J., Morishita Y., Carman C.V., Shimaoka M. Systemic leukocyte-directed siRNA delivery revealing cyclin D1 as an anti-inflammatory target. Science. 2008;319:627–630. doi: 10.1126/science.1149859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khodoun M., Chimote A.A., Ilyas F.Z., Duncan H.J., Moncrieffe H., Kant K.S., Conforti L. Targeted knockdown of Kv1.3 channels in T lymphocytes corrects the disease manifestations associated with systemic lupus erythematosus. Sci. Adv. 2020;6:eabd1471. doi: 10.1126/sciadv.abd1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hajdu P., Chimote A.A., Thompson T.H., Koo Y., Yun Y., Conforti L. Functionalized liposomes loaded with siRNAs targeting ion channels in effector memory T cells as a potential therapy for autoimmunity. Biomaterials. 2013;34:10249–10257. doi: 10.1016/j.biomaterials.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chimote A.A., Hajdu P., Kottyan L.C., Harley J.B., Yun Y., Conforti L. Nanovesicle-targeted Kv1.3 knockdown in memory T cells suppresses CD40L expression and memory phenotype. J. Autoimmun. 2016;69:86–93. doi: 10.1016/j.jaut.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolff H.A., Rolke D., Rave-Fränk M., Schirmer M., Eicheler W., Doerfler A., Hille A., Hess C.F., Matthias C., Rödel R.M., Christiansen H. Analysis of chemokine and chemokine receptor expression in squamous cell carcinoma of the head and neck (SCCHN) cell lines. Radiat. Environ. Biophys. 2011;50:145–154. doi: 10.1007/s00411-010-0341-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sercombe L., Veerati T., Moheimani F., Wu S.Y., Sood A.K., Hua S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015;6:286. doi: 10.3389/fphar.2015.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lubich C., Allacher P., de la Rosa M., Bauer A., Prenninger T., Horling F.M., Siekmann J., Oldenburg J., Scheiflinger F., Reipert B.M. The mystery of antibodies against polyethylene glycol (PEG)-what do we know? Pharm. Res. 2016;33:2239–2249. doi: 10.1007/s11095-016-1961-x. [DOI] [PubMed] [Google Scholar]
- 39.Jain A., Cheng K. The principles and applications of avidin-based nanoparticles in drug delivery and diagnosis. J. Control. Release. 2017;245:27–40. doi: 10.1016/j.jconrel.2016.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jain A., Barve A., Zhao Z., Jin W., Cheng K. Comparison of avidin, neutravidin, and streptavidin as nanocarriers for efficient siRNA delivery. Mol. Pharm. 2017;14:1517–1527. doi: 10.1021/acs.molpharmaceut.6b00933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alshehri A., Grabowska A., Stolnik S. Pathways of cellular internalisation of liposomes delivered siRNA and effects on siRNA engagement with target mRNA and silencing in cancer cells. Sci. Rep. 2018;8:1–9. doi: 10.1038/s41598-018-22166-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dominska M., Dykxhoorn D.M. Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci. 2010;123:1183–1189. doi: 10.1242/jcs.066399. [DOI] [PubMed] [Google Scholar]
- 43.Boyer C., Auphan N., Gabert J., Blanc D., Malissen B., Schmitt-Verhulst A.-M. Comparison of phosphorylation and internalization of the antigen receptor/CD3 complex, CD8, and class I MHC-encoded proteins on T cells. Role of intracytoplasmic domains analyzed with hybrid CD8/class I molecules. J. Immunol. 1989;143:1905–1914. [PubMed] [Google Scholar]
- 44.Marsh M., Parsons I.J., Reid P., Pelchen-Matthews A. Endocytic regulation of the T lymphocyte co-receptor proteins CD4 and CD8. Biochem. Soc. Trans. 1993;21:703–706. doi: 10.1042/bst0210703. [DOI] [PubMed] [Google Scholar]
- 45.Bicho A., Peça I.N., Roque A.C., Cardoso M.M. Anti-CD8 conjugated nanoparticles to target mammalian cells expressing CD8. Int. J. Pharm. 2010;399:80–86. doi: 10.1016/j.ijpharm.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 46.Schmid D., Park C.G., Hartl C.A., Subedi N., Cartwright A.N., Puerto R.B., Zheng Y., Maiarana J., Freeman G.J., Wucherpfennig K.W. T cell-targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity. Nat. Commun. 2017;8:1747. doi: 10.1038/s41467-017-01830-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.