

Abstract
Non-viral platforms can be applied rapidly and cost-effectively for chimeric antigen receptor (CAR)-T cell manufacturing. In the present paper, we describe in detail a clinically relevant manufacturing process for NKG2D CAR-T cells through electroporation of CAR-encoding piggyBac transposon plasmids and in vitro expansion with K562 artificial antigen-presenting cells. With an optimized protocol, we generated the final cell therapy products with 89.2% ± 10.2% NKG2D CAR-positive cells and achieved the corresponding antigen-dependent expansion between 50,000 and 60,000 folds within 4 weeks. To facilitate repeated CAR-T cell infusions, we evaluated the practicality of cryopreservation followed by post-thaw expansion and an extended manufacturing process for up to 9 rounds of weekly K562 cell stimulation. We found that neither compromised the in vitro anti-tumor activity of NKG2D CAR-T cells. Interestingly, the expression of T cell exhaustion markers TIGIT, TIM3, and LAG3 was reduced with extended manufacturing. To enhance the safety profile of the NKG2D CAR-T cells, we incorporated a full-length CD20 transgene in tandem with the CAR construct and demonstrated that autologous NK cells could mediate efficient antibody-dependent cell-mediated cytotoxicity to remove these CAR-T cells. Collectively, our study illustrates a protocol that generates large numbers of efficacious NKG2D CAR-T cells suitable for multiple rounds of infusions.
Keywords: piggyBac transposon, K562 aAPCs, NKG2D CAR-T cells
Graphical abstract
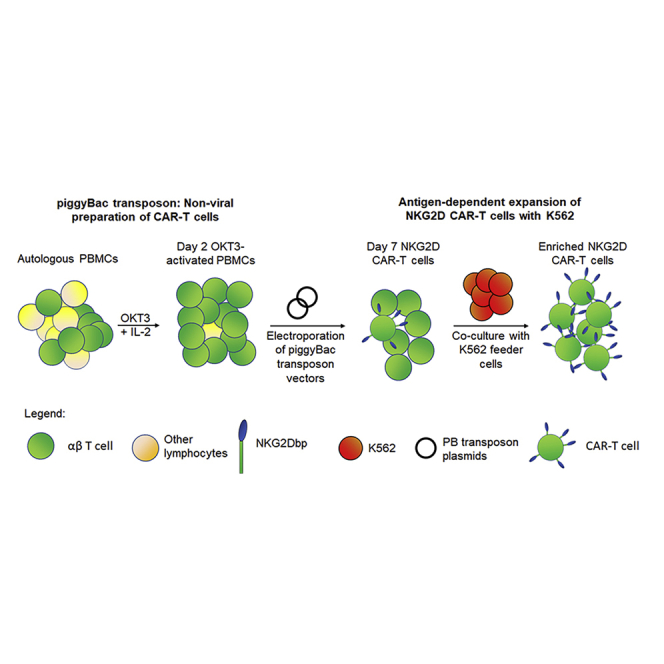
NKG2D CAR-T cells can be prepared in large numbers for clinical application through non-viral piggyBac transposon platform and antigen-dependent expansion by K562 feeder cells. Methods such as cryopreservation and extended expansion were explored to facilitate repeated CAR-T infusions. The CD20 transgene was also incorporated to facilitate CAR-T removal.
Introduction
NKG2D-based chimeric antigen receptor (CAR)-T cells have received increased attention over the past few years, because of their ability to recognize a panel of eight stress-associated ligands in the MIC and ULBP families. Within the MIC family, allelic polymorphism results in a wide panel of ∼105 protein variants encoded by the MICA and MICB genetic loci found on human chromosome 6, all of which are recognized by the cognate NKG2D receptor.1 Members of the ULBP family are glycoproteins encoded by the RAET1 genes found similarly on chromosome 6, and six functional proteins in this family have been identified so far: ULBP1 to ULBP6.1 Collectively, these NKG2D ligands are not known to be widely expressed on normal tissues but are upregulated in expression levels in response to malignant transformation, viral infection, and DNA damage.2 Various epidemiological studies have estimated that up to 95% of all solid tumors express at least one or a combination of two or more various NKG2D ligands.3 Therefore, a potential pan-tumor CAR-T approach can be developed with a NKG2D-based construct.
To generate T cells that stably express CAR, most studies utilize viral vectors such as lentiviral and retroviral vectors to integrate the CAR-expressing cassette randomly into the genome for persistent expression. Such transgene integration technologies have been reliably paired with other platform technologies to generate CAR-T cells, such as apheresis and gene modification integrated into the CliniMACS Prodigy system (Miltenyi Biotec). However, the use of viral vectors is associated with high manufacturing costs, cumbersome processes, and risks of residual viral elements.4 Thus, to overcome these limitations, non-viral methods such as mini-circles and transposon systems have been developed.4
The piggyBac (PB) transposon element was first discovered in the cabbage looper moth Trichoplusia ni TN-368 cell line and was later verified to cross-function in other species as well.5 The PB transposase mediates exchange of genetic materials between the vector and host genome through recognition of inverted terminal repeat sequences (ITRs) present on the transposon vector and the corresponding TTAA sequences present in the genome. Transgenes flanked by the ITRs can therefore be integrated stably into TTAA integration sites. The advantages afforded by PB systems include high cargo capacity of up to 100 kb DNA fragment for the delivery of multiple transgenes, significantly lower manufacturing costs, and potentially fewer regulatory restrictions in clinical translations. Considering these advantages, this non-viral platform has been suggested as an alternative gene delivery system to viral vectors for CAR-T cell production.6, 7, 8
In both preclinical and clinical settings, the use of NKG2D CAR-T cells has been explored experimentally by various research groups and clinically by Celyad Oncology mainly (Table S1). The reported manufacturing processes for NKG2D CAR-T cells start with peripheral blood mononuclear cells (PBMCs) prepared from apheresis or peripheral blood.9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 T cells in PBMCs are activated by either anti-CD3 antibody OKT3 or anti-CD3/CD28 beads, plus exogenous interleukin (IL)-2, followed by CAR gene transfer with efficient but costly retroviral or lentiviral vectors. Two NKG2D CAR-T research articles reported the use of non-viral platforms for CAR-T manufacturing, one involving mini-circles24 and another involving mRNA CAR electroporation.25 After the in vitro expansion of modified NKG2D CAR-T cells through exogenous IL-2 for several days to several weeks, CAR transgene expression efficiency between 40% and 90% can be achieved. However, NKG2D CAR-T cell expansion fold is commonly low, with 400-fold expansion being the highest value reported so far (Table S1).
The human K562 myelogenous leukemia cell line has been genetically modified in diverse ways for use as a feeder cell line to promote the expansion of NK cells26, 27, 28, 29, 30, 31, 32, 33, 34, 35 and, more recently, T lymphocytes.36, 37, 38 We have previously described the expression profile of NKG2D ligands on K562 cells and how they could be used as cell-based artificial antigen-presenting cells (aAPCs) to enrich and expand CAR-T cells.39 In this study, we reported an NKG2D CAR-T cell manufacturing protocol based on the combinatorial use of NKG2D CAR-encoding piggyBac transposon plasmids and K562 aAPCs genetically modified to express CD64, CD86, and CD137L.38 We showed that, starting with small volumes (60–80 mL) of peripheral blood sample, large numbers of NKG2D CAR-T cells could be produced and expanded to meet a clinical timeline designed for solid cancer therapy that often involves multiple cycles of infusion of this cell product.
Results
Optimization of transfection conditions to generate NKG2D CAR-T cells
Our group reported the generation of NKG2D CAR-T cells through electroporation with CAR-encoding piggyBac transposon plasmids.39 Briefly, the NKG2D CAR plasmid was constructed with the NKG2D ectodomain expressed in tandem with a streptavidin tag (Streptavidin II [STII] tag) for specific detection of NKG2D CAR, 4-1BB (CD137) as the costimulatory domain, and the single ITAM-containing DAP12 as the activation domain (Figure 1A). As an irrelevant CAR control, the NKG2D ectodomain was replaced by the anti-CD22 scFv (Figure 1A) derived from the m971 clone.40
Figure 1.

Optimization of electroporation conditions for NKG2D CAR-T cell generation
(A) NKG2D CAR and control CAR constructs used in the current study. (B) PBMC activation. Donor-derived PBMCs were activated with either 100 ng/mL or 500 ng/mL of OKT3 for either 2 or 3 days. Cell yield was measured with trypan blue exclusion assay after the activation and is presented as a percentage of total PBMCs initially used. (C) Electroporation efficiency. Activated PBMCs were transfected by electroporation of 5 μg of piggyBac transposase plasmid and 10 μg of NKG2D CAR donor plasmid in trial runs to evaluate cell survival. The percentage of CAR-T cells was measured by flow cytometry with an antibody against Streptavidin II (STII) tag on day 5 post-electroporation. (D) Cell yield post-electroporation. Electroporation was performed as described in (C). Cell yield as a percentage of total activated T cells electroporated was measured with trypan blue exclusion assay on day 5 post-electroporation. (E and F) Expression profile of NKG2D ligands on T cells at 2 days (E) and 5 days (F) post-electroporation. Cells were analyzed through staining with individual antibodies against MICA/MICB, ULBP1, ULBP2/5/6, ULBP3, and ULBP4. (G–I) Dose-dependent expression of NKG2D CAR on T cells 5 days post-electroporation. (G) Representative flow cytometry histograms from a single donor electroporated with 5 μg of piggyBac transposase plasmid and 5, 10, 20, or 30 μg of NKG2D CAR donor plasmid. (H) Electroporation efficiency increases with increasing amounts of the CAR plasmid used. (I) Cell yield as a percentage of total activated T cells electroporated decreases with increasing amounts of the CAR plasmid used. All data in (B)–(D), (H), and (I) are mean ± SD of three independent experiments with PBMC samples from three different donors. Data in (E) and (F) are mean ± SD of single measurements from five different donors.
We first looked at activation methods used by other research groups that studied human NKG2D CAR-T cells and found that a majority used either anti-CD3/CD28 beads or OKT3 (Table S1). OKT3 is a murine monoclonal antibody that targets the epsilon subunit of the CD3 complex. When used in combination with recombinant IL-2, OKT3 acts as a mitogenic agent that mediates T cell activation and proliferation.41 We thus evaluated both Dynabeads and OKT3 and found that OKT3 could generally provide purer CD3+ CD56− T cell populations (data not shown). Furthermore, the use of OKT3 will result in lower overall manufacturing costs compared with the use of Dynabeads. OKT3 activation resulted in increased cell granularity and an internalization of αβ-T cell receptor (TCR) but no upregulation of PD1 expression (Figure S1). We further optimized the dose and duration of OKT3 activation and found that the four conditions tested produced consistent cell yields post-activation (Figure 1B) and consistent electroporation efficiencies (Figure 1C). However, cell yield post-electroporation was higher in both groups treated with OKT3 for 2 days compared with those treated for 3 days (Figure 1D). In view of this, we focused on the use of 100 ng/mL OKT3 for 2 days as the standard activation protocol for subsequent sections.
NKG2D CAR-T cells have been shown to express NKG2D ligands after OKT3 activation and viral transduction, and this has led to the issue of T cell fratricide among NKG2D CAR-T cells.19 To understand whether our protocol of OKT3 activation followed by DNA electroporation would similarly upregulate these ligands, we characterized the surface expression of NKG2D ligands on the activated cells before electroporation and on electroporated cells at 2 and 5 days post-electroporation via flow cytometry. There was almost no expression of NKG2D ligands on the activated cells, with 2%–4% of cells expressing ULBP4 as the highest expression value (Figure S2). As shown in Figure 1E, both αCD22bp and NKG2Dbp CAR-T cells expressed very low levels of MICA/MICB, ULBP1, ULBP2/5/6, and ULBP3 (<2%), while ULBP4 expression was at 3.9% ± 1.2% and 5.7% ± 1.5% for αCD22bp and NKG2Dbp CAR-T cells, respectively. By 5 days post-electroporation, expression of all NKG2D ligands for both αCD22bp and NKG2Dbp CAR-T cells dropped to below 2% (Figure 1F).
To evaluate how different DNA amounts affect cell yield and electroporation efficiencies, we performed a dose escalation for the NKG2D CAR donor plasmid from 5 μg to 30 μg while keeping the piggyBac transposase plasmid constant at 5 μg. As shown in Figures 1G–1I, CAR expression levels increased from 8.2% ± 2.8% for 5 μg to 43.6% ± 4.9% for 30 μg, indicating that CAR expression could be enhanced through electroporation of higher doses. Conversely, cell yield on day 5 post-electroporation dropped from 52.0% ± 2.6% for 5 μg to 10.3% ± 0.6% for 30 μg, indicating that cell viability was inversely proportionate to the amount of DNA plasmids electroporated. Between 5 μg and 10 μg of plasmid used in electroporation, we also observed no significant difference in the expression levels of T cell exhaustion markers (Figure S3A). As the low expression of PD1 on OKT3-activated T cells from all four donors was unexpected, we validated the MIH1 αPD1 antibody against PD1-expressing PMA/ionomycin-treated Jurkat cells as a quality control (Figure S3B).42 In view of the need to produce both CAR-T cells at high numbers and a sufficient level of surface CAR expression for subsequent CAR-T cell expansion, we focused on the starting amounts of 5 μg of piggyBac transposase plasmid and 10 μg of NKG2D CAR plasmid for the purpose of electroporation in subsequent sections.
Antigen-dependent enrichment and expansion of NKG2D CAR-T population with K562 feeder cells
To generate large numbers of CAR-T cells, we used a human K562 myelogenous leukemia cell line33 that expresses NKG2D ligands (Figure S4) to stimulate the NKG2D CAR-T cell expansion in an antigen-dependent manner. As shown in Figure S4, K562 cells have a basal expression level of NKG2D ligands, which could be further upregulated after gamma irradiation. In preparation for a potential clinical application of the NKG2D CAR-T product, we outlined a 28-day manufacturing protocol that includes peripheral blood withdrawal from patients on day 0 and the subsequent re-introduction of autologous CAR-T cell products on day 28 (Figure 2A). This 28-day protocol consists of the 7-day activation protocol described in Figure 1 and three consecutive 7-day manufacturing phases that utilize gamma-irradiated K562 cells as feeder cells. As shown with the representative data in Figure 2B, we observed an antigen-dependent enrichment of NKG2D CAR-T cells from day 7 to day 28. During this period, the proportion of NKG2D CAR-T cells increased from 18.4% ± 8.1% on day 7 to 87.7% ± 8.2% on day 28 (Figure 2C). The corresponding expansion folds were 58,740 ± 31,730 by day 28 (Figure 2D).
Figure 2.

Antigen-dependent enrichment and expansion of NKG2D CAR-T population with K562 feeder cells
(A) Timeline for OKT3 activation, piggyBac-mediated genetic modification, and K562-based antigen-dependent expansion of NKG2D CAR-T cells. (B) Representative flow cytometry quadrant plots to illustrate the time course of antigen-dependent K562-based enrichment of NKG2D CAR-T cells from a single donor. CAR-T cells were analyzed through co-staining with anti-CD3 and anti-Streptavidin II tag antibodies. (C) Antigen-dependent enrichment of NKG2D CAR-T cells from day 7 to day 28 was similarly analyzed by flow cytometry (n = 7). (D) Antigen-dependent expansion of NKG2D CAR-T cells with K562 feeder cells from day 14 to day 28 was analyzed by trypan blue exclusion assay (n = 7). (E) ELISpot-IFNγ assay against CAOV3 and HCT-116 demonstrates that the total number of IFNγ-secreting spots increased with increasing enrichment of NKG2D CAR-T cells from day 7 to day 28. Data from 28-day Ctrl CAR-T cells are included as a negative control. Data shown are mean ± SD from a single representative assay. All data were derived from healthy donors.
To validate the antigen-dependent anti-tumor activity of NKG2D CAR-T cells, we used the human ovarian adenocarcinoma CAOV3 cell line and the human colon colorectal HCT-116 cell line as target cell lines that express NKG2D ligands.39 Congruent with the incremental enrichment of CAR-expressing T cells, we observed an increase in the number of interferon (IFN)γ-secreting ELISpots against both CAOV3 and HCT-116 target cells (Figure 2E). While there was no statistically significant difference in the responses from day-21 and day-28 CAR-T cells, there was a significant increase in IFNγ-secreting ELISpots with each stepwise CAR-T enrichment from day 7 to day 21, indicating that IFNγ secretion response is directly proportional to the percentage of NKG2D CAR-T cells within the bulk population. As an irrelevant CAR control, αCD22 CAR-T cells were prepared in a similar manner and enriched with gamma-irradiated CD22-expressing Raji feeder cells (data not shown). These CAR-T cells, however, did not stimulate IFNγ secretion when co-culturing with both CAOV3 and HCT-116 cells (Figure 2E).
Characterization of day 28 ex vivo expanded NKG2D CAR-T cells
Having shown the efficacy of the ex vivo expanded NKG2D CAR-T cells, we proceeded to characterize day 28 CAR-T cells derived from seven donors in terms of CD3/CD56 purity, CD4/CD8 proportion, expression of T cell exhaustion markers, and memory T cell development. Flow cytometry gating strategy for CAR-T cell characterization is presented in Figure S5. In terms of purity, we found that day 28 CAR-T cells were mostly CD3+ CD56−, with a mean of 88.3% ± 5.5% (Figure 3A), while the split in CD4 and CD8 proportions was more varied among donors, with CD4+ CAR-T cells at 34.5% ± 22.2% and CD8+ CAR-T cells at 56.3% ± 22.3% (Figure 3B). To determine whether this divergent development of CD4+ and CD8+ CAR-T cells would affect antigen-dependent cytotoxicity, we tested day 28 CAR-T cells from two donors that differed significantly in their CD4/CD8 proportions: CD8majority donor 1 with 83.4% CD8+ and 7.8% CD4+ (Figure S6A) and CD4majority donor 2 with 38.4% CD8+ and 55% CD4+ (Figure S6B). Using the europium release assay, we tested their antigen-dependent cytotoxicity against CAOV3 and HCT-116 and did not find any difference in cytotoxicity that could be attributed to this variation in CD4/CD8 proportions (Figure S6C). Further to this observation, we had the opportunity to track the composition of CD4+ and CD8+ T cells on day 7 and day 28 for four of these seven donors. Interestingly, we observed that two demonstrated an increase in CD4+ and the other two an increase in CD8+ between day 7 and day 28 (Figures S7A and S7B).
Figure 3.

Characterization of day 28 NKG2D CAR-T cells expanded with a K562-based protocol
(A) Development of CD3+ CD56- NKG2D CAR-T cells. Purity of NKG2D CAR-T cells was analyzed through co-expression patterns of CD3 and CD56 as detected by flow cytometry. Flow cytometry data shown on the left are representative of 7 independent experiments. Data shown on the right in each bar are mean ± SD of 7 donors. (B) Development of CD4+ and CD8+ NKG2D CAR-T cells. CAR-T cells collected on day 28 post-activation were analyzed through flow cytometry. For each plot, a single dot denotes a single donor, while mean ± SD of 7 donors are also shown. (C) Expression of exhaustion markers differed widely among donors. Expression profiles of PD1, TIGIT, LAG3, and TIM3 were analyzed on 28-day NKG2D CAR-T cells. For each plot, a single dot denotes a single donor, while means ± SD of 7 donors are also shown. (D) Development of the TEM subset. Memory T subsets were analyzed through co-expression patterns of CCR7 and CD45RA as detected by flow cytometry: TSCM, CCR7+ CD45+; TCM, CCR7+ CD45RA−; TEM, CCR7− CD45−; TEffector, CCR7− CD45RA+. Flow cytometry data shown on the left are representative of 7 independent experiments. Data shown on the right in each bar are means ± SD of 7 donors. (E) Cytotoxicity against NKG2DL-expressing CAOV3 and HCT-116. αCD22 CAR-T cells were used as a negative control. Cytotoxicity assay was performed at effector-to-target ratios of 20:1, 10:1, 5:1, and 1:1 in a standard DELFIA time-resolved fluorescence assay. The results of 1 representative experiment out of 3 independent experiments with different donors are shown. The differences between NKG2D CAR-T and Ctrl CAR-T are statistically significant (p < 0.05) for the 2 tested tumor cell lines.
Next, we evaluated the expression profile of canonical T cell exhaustion markers (Figure S7). Interestingly, none of the seven healthy donors tested showed any elevation of PD1 expression throughout the 21-day expansion with K562 feeder cells (Figure S7C), with the eventual expression level of 3.8% ± 2.5% on day 28 (Figure 3C). However, the expression of TIGIT, LAG3, and TIM3 showed greater variation among the seven donors characterized, with expression levels of TIGIT, LAG3, and TIM3 on day 28 at 37.3% ± 12.9%, 41.9% ± 21.7%, and 38.9% ± 30.5%, respectively (Figure 3C). Besides the variation among donors, there was also fluctuation in their expression levels within the expansion period of each individual donor (Figures S7D–S7F).
We also tracked and analyzed the development of memory T cells during the 21-day expansion protocol. Development of the CCR7− CD45RA− effector memory subset (TEM) was a consistent observation across all seven donors, with the eventual 28-day population at 87.1% ± 15.0%, with six donors having at least 88% on day 28 (Figure 3D). Compared with 28-day αCD22 CAR-T cells, 28-day NKG2D CAR-T cells also demonstrated significantly higher cytotoxicity against both CAOV3 and HCT-116 (Figure 3E), thus demonstrating tumor killing activity in an antigen-dependent manner.
Cryopreservation of NKG2D CAR-T cells to facilitate multiple injections of CAR-T cells
Compared with other NKG2D CAR-T studies reported, a key difference in our approach is the use of K562 feeder cells to expand CAR-T cells. This approach allows us to potentially produce a large quantity of cellular therapeutics from one single blood sample to facilitate multiple infusions of CAR-T cells and multi-cycles of cell therapy treatment over a long period that could be crucial in treating solid tumors.38 For this, we tested the generation of age-matched 28-day NKG2D CAR-T cells as first described in Figure 2A for multiple infusions. We first evaluated the feasibility of cryopreserving 21-day CAR-T cells that have undergone two phases of expansion with K562 feeder cells. With reference to the clinical practices of Novartis in handling tisagenlecleucel (anti-CD19 CAR-T cells), we similarly adopted the use of CryoStor10 (STEMCELL Technologies) in CAR-T cell preservation. We investigated two key parameters, namely the cell freezing density and the potential to retain its cytotoxicity after a subsequent round of expansion to obtain 28-day CAR-T cells. First, we froze 21-day CAR-T cells at differing cell densities of 2.5 × 107/mL, 5 × 107/mL, and 10 × 107 cells/mL and kept them in liquid nitrogen storage for 2 weeks prior to thawing (Figure 4A). After thawing, we found no significant difference in the cell recovery rates between them (Figure 4B). We then expanded the recovered 21-day CAR-T cells frozen at 10 × 107 cells/mL with K562 feeder cells. Expansion folds from these thawed cells were like those grown continuously as described in Figure 1A (Figure S8). We similarly performed a cytotoxicity assay against both CAOV3 and HCT-116 and observed that thawed and expanded CAR-T cells retained the ability to lyse these two target cell lines in a dose-dependent manner, with 100% cell lysis at 20:1 effector-to-target (E:T) ratio (Figure 4C). We further tested the cryopreservation of NKG2D CAR-T cells derived from blood samples collected from cancer patients. Five patients with ovarian cancer were included for the test. As shown in Table 1, we confirmed that cryopreserved CAR-T cells from cancer patient blood samples could be further expanded after thawing and the expanded CAR-T cells preserved the tumor cell lysing activity. The volume of blood collected from these patients ranged from 60 mL to 80 mL (Table 1).
Figure 4.

Cryopreservation to facilitate multiple injections of NKG2D CAR-T cells
(A) Schematic timeline to illustrate time points where cryopreservation and thawing of NKG2D CAR-T cells are performed. Cryopreservation on either day 14 or day 21 and subsequent thawing and expansion 1 week before infusion. A possible clinical application timeline with 3 infusions separated by 2 weeks is shown. (B) Varying freezing densities did not affect cell recovery yields of cryopreserved day 21 NKG2D CAR-T cells. CAR-T cells, after freezing for 2 weeks, were thawed and enumerated with a trypan blue exclusion assay immediately after thawing. Duplicate readings from each single donor were obtained from 2 independent thaw/count procedures. Average values were then pooled from 3 independent donors to derive mean ± SD shown in the chart. (C) Cryopreservation and subsequent K562-based expansion did not compromise antigen-dependent cytotoxicity against both CAOV3 and HCT-116 target cell lines. Cytotoxicity assay was performed at effector-to-target ratios of 20:1, 10:1, 5:1, and 1:1 in a standard DELFIA time-resolved fluorescence assay. Data shown are mean ± SD from a single representative assay. (D) Experimental outline of an animal study using cryopreserved NKG2D CAR-T cells. Three groups of mice (5 mice per group) received i.p. injection of 2 × 106 HCT116-Luc cells (day 0) followed by i.p. injection of PBS, ctrl T cells, or NKG2D CAR-T cells on day 7 and day 32 (1 × 107 cells per mouse). Bioluminescent imaging of tumor signals was performed on day 7 and day 35. (E) Bioluminescent images on day 7 and day 35 are shown. (F) Bioluminescence flux values on day 7 and day 35. The values from each mice of respective groups are plotted. ∗∗∗∗p < 0.0001. (G) Kaplan-Meier analysis of survival of the in vivo animal experiment. Statistical analysis between groups was performed with the log-rank test.
Table 1.
NKG2D CAR-T cell production with blood samples collected from patients with ovarian and colorectal cancera
| Characterization | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Mean ± SD |
|---|---|---|---|---|---|---|---|
| Blood vol. collected (mL) | 80 | 80 | 60 | 60 | 80 | 16 | 62.67 ± 24.87 |
| Total PBMCs isolated | 7.6 × 107 | 6.3 × 107 | 2.4 × 107 | 13.6 × 107 | 2.1 × 107 | 1.09 × 107 | 5.52 × 107 ± 4.72 × 107 |
| PBMCs/mL | 0.95 × 106 | 0.79 × 106 | 0.40 × 106 | 2.27 × 106 | 0.26 × 106 | 0.68 × 106 | 9.34 × 105 ± 7.98 × 105 |
| PBMCs used for CAR-T production | 6 × 107 | 5.3 × 107 | 2.16 × 107 | 5 × 107 | 1.92 × 107 | 9.4 × 106 | 3.55 × 107 ± 2.12 × 107 |
| The initial CAR-T cellsb | |||||||
| % CD3+ | 99.8 | 94.4 | 98.1 | 97.3 | 87.7 | 96.3 | 95.6 ± 4.27 |
| % CD4+ in CD3+ | 9.1 | 39.8 | 22.4 | 55.3 | 19.6 | 55.6 | 33.63 ± 19.57 |
| % CD8+ in CD3+ | 88.2 | 54.2 | 76.3 | 42.4 | 77.1 | 42.1 | 63.38 ± 19.74 |
| % CAR+ cells | 66 | 64.4 | 56.5 | 94.2 | 73.7 | 95.8 | 75.1 ± 16.36 |
| % Naive in CD3+ | 3.7 | 3.3 | 7.7 | 0.5 | 0.6 | 0.1 | 2.65 ± 2.91 |
| % Central memory in CD3+ | 0.9 | 3.5 | 5.6 | 22 | 37.1 | 15.2 | 14.05 ± 13.79 |
| % Effector memory in CD3+ | 46.3 | 56.3 | 32 | 76.6 | 62 | 84.6 | 59.63 ± 19.34 |
| % Effector in CD3+ | 49 | 36.9 | 54.8 | 0.9 | 0.4 | 0.1 | 23.68 ± 26.08 |
| % PD1+ | 6.7 | 10.7 | 5.6 | 29.4 | 17.5 | 31 | 16.82 ± 11.19 |
| % LAG3+ | 36.2 | 60 | 44.3 | 15.3 | 33.7 | 43 | 38.75 ± 14.71 |
| % TIGIT | 16.9 | 43.2 | 23.7 | 15.5 | 42.6 | 50.7 | 32.1 ± 15.21 |
| % Cytotoxic killing∗∗∗ | 64.77 | 72.68 | 61.62 | 89.2 | 89.62 | 66.39 | 74.05 ± 12.43 |
| CAR-T cells after freezing & thawingc | |||||||
| % CD3+ | 92.3 | 93 | 98.9 | 96.8 | 86.2 | 95 | 93.7 ± 4.41 |
| % CD4+ in CD3+ | 17.8 | 22.4 | 24.8 | 65.9 | 27.4 | 48.3 | 34.43 ± 18.69 |
| % CD8+ in CD3+ | 72.1 | 72.6 | 69.6 | 30.5 | 67.7 | 49.4 | 60.3 ± 16.96 |
| % CAR+ | 94 | 81.7 | 87.3 | 98.5 | 91.6 | 97.6 | 91.78 ± 6.41 |
| % Naive in CD3+ | 0.2 | 0 | 0.2 | 0 | 0.1 | 0 | 0.08 ± 0.1 |
| % Central memory in CD3+ | 1.3 | 0.4 | 1.5 | 20 | 17.2 | 5.7 | 7.68 ± 8.7 |
| % Effector memory in CD3+ | 97.9 | 99 | 96.6 | 79.6 | 82.3 | 94.3 | 91.62 ± 8.4 |
| % Effector in CD3+ | 0.6 | 0.6 | 1.8 | 0.4 | 0.4 | 0 | 0.63 ± 0.61 |
| % PD1+ | 15.8 | 11.8 | 7.6 | 48.9 | 31 | 49.4 | 27.42 ± 18.6 |
| % LAG3+ | 55.5 | 70 | 53.9 | 32.4 | 43 | 16.6 | 45.23 ± 18.89 |
| % TIGIT | 27.8 | 75.4 | 26.9 | 34.3 | 50.7 | 36.4 | 41.92 ± 18.5 |
| % Cytotoxic killingd | 93.86 | 70.85 | 91.76 | 57.35 | 72.01 | 77.47 | 77.22 ± 13.79 |
Patients 1–5 are ovarian cancer patients, and patient 6 is a patient with colorectal cancer.
Initial CAR-T cells: CAR-T cells before cryopreservation as indicated in Figure 4A.
CAR-T cells after freezing & thawing: CAR-T cells after cryopreservation and then being expanded again for 7 days.
Cytotoxic killing: Percentage of killing against SKOV3 assessed with the xCELLigence RTCA system at E:T 1:1 for 24 h. For patient 6, percentage of killing against SKOV3 assessed with the xCELLigence RTCA system at E:T 1:1 for 48 h.
We previously showed the efficacy of freshly prepared NKG2D CAR-T cells in eradicating established HCT-116 and SKOV3 xenografts.39 In this study, we further validated the ability of cryopreserved CAR-T cells to eradicate established human tumor xenografts. Briefly, luciferase-expressing HCT-116 cells were inoculated into NSG mice 1 week before the administration of NKG2D CAR-T cells along with PBS and control CAR-T cells (Figure 4D). A second injection of CAR-T cells and PBS was given on day 32 after initial HCT-116 inoculation. As shown in Figures 4E and 4F, while tumor cell flux values increased from day 7 to day 35 in mice treated with either PBS or control CAR-T cells, the bioluminescence signals in mice treated with NKG2D CAR-T cells were completely eliminated by day 35. Mice treated with PBS control and control CAR-T cells had a median survival of 42 days and 61 days, respectively. Conversely, mice treated with NKG2D CAR-T had a 100% survival rate until they were euthanized on day 150 (Figure 4G).
K562 feeder cell line allows a long-term in vitro expansion of NKG2D CAR-T cells
Long-term in vitro expansion would allow us to consider the possibility of maintaining a parallel working cell stock, in addition to a frozen cell stock, that could potentially facilitate ad hoc infusions of CAR-T cells per clinical needs. To address this issue, we weekly stimulated NKG2D CAR-T cells with K562 feeder cells up to day 63 (Figure 5A; Figure S9). Consistent with our observation that the TEM subset is preferably developed by the end of the 28-day manufacturing phase (Figure 3D), NKG2D CAR-T cells evaluated at day 35 and day 63 were still predominantly CCR7− and CD45RA− (Figure 5B). Interestingly, however, we observed a general downregulation of T cell exhaustion marker expression in all three donors, albeit to varying degrees (Figure 5C). PD1 expression remained unelevated, while expression levels of TIGIT, LAG3, and TIM3 dropped consistently throughout the extended manufacturing phase.
Figure 5.

K562 feeder cell line allows a long-term in vitro expansion of NKG2D CAR-T cells.
(A) Schematic figure to illustrate a possible extended manufacturing period until day 63. (B) Characterization of memory T cell development from day 28 to day 63 as analyzed through co-expression patterns of CCR7 and CD45RA based on flow cytometry: TSCM, CCR7+ CD45+; TCM, CCR7+ CD45RA−; TEM, CCR7− CD45−; TEffector, CCR7− CD45RA+. Data shown in each bar are mean ± SD of 3 donors. (C) Extended CAR-T manufacturing period resulted in reduced expression of T cell exhaustion markers: PD1, TIGIT, LAG3, and TIM3. Data shown are from 3 different donors, with flow cytometry analysis performed on days 28, 35, and 63. (D and E) Extended CAR-T manufacturing did not compromise antigen-dependent cytotoxicity (D) or IFNγ secretion (E) against NKG2DL-expressing CAOV3. Cytotoxicity assay and ELISpot-IFNγ assay were performed on days 28, 35, and 63. Data shown are mean ± SD of each time point from a single representative donor.
To evaluate whether the long-term culture would negatively modulate the anti-tumor functions of CAR-T cells, we assessed the ability of continuously expanded NKG2D CAR-T cells to recognize and lyse CAOV3 tumor cells. As shown in Figure 5D, cytotoxicity exhibited against CAOV3 was relatively consistent on day 28, day 35, and day 63, with a slight statistically significant difference between day 63 and day 28 at the highest E:T ratio of 20:1 (p < 0.05). Similarly, there were no statistically significant differences in the number of IFNγ-secreting spots between the three CAR-T groups (Figure 5E). Thus, these results demonstrated that an extended phase of manufacturing with K562 feeder cells up to day 63 did not negatively modulate the in vitro anti-tumor functions of NKG2D CAR-T cells.
Antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity as safety mechanisms to remove NKG2D CAR-T cells
To enhance the in vivo safety profile of this NKG2D CAR-T cell product, we modified the NKG2D CAR construct by inserting a full-length CD20 transcript downstream of the CAR sequence, with the resulting transcript expressed in a bicistronic protein linked by a P2A self-cleaving linker peptide (Figure 6A). We adopted the same protocol as described in Figure 1A to produce NKG2D-CD20 CAR-T cells. We tested the production of NKG2D and NKG2D-CD20 CAR-T cells in parallel with three healthy donors and assessed CAR expression through flow cytometry analysis of STII and CD20 expression on the two different CAR-T cells, respectively (Figure 6B). Expansion folds by day 21 were consistent at 987 ± 297 for NKG2D and 969 ± 284 for NKG2D-CD20 (Figure 6C). To show that the co-expression of CD20 does not affect the cytotoxicity of NKG2D CAR-T cells, we performed a standard europium release assay against both CAOV3 and HCT-116 cancer cell lines at varying E:T ratios and found no significant differences between these two NKG2D CAR-T cell products (Figure 6D).
Figure 6.

CD20-expressing NKG2D CAR-T cells can be removed through antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity
(A) Schematic representation of CD20-expressing piggyBac transposon. NKG2Dbp CAR and full-length CD20 are expressed in a polycistronic transcript separated by the P2A self-cleaving peptide. (B) Expression of CAR and CD20 on 21-day NKG2D CAR-T and NKG2D-CD20 CAR-T cells. Flow cytometry data shown are representative of three independent experiments. (C) Expansion of NKG2D-CD20 CAR-T cells and NKG2D CAR-T cells is achieved with the same methodology described in Figure 2A. Expansion folds shown are single measurements from three healthy donors on day 21 post-activation. (D) The cytotoxicity of NKG2D-CD20 CAR-T cells was assessed in parallel with donor-matched NKG2D CAR-T cells against CAOV3 and HCT-116. Data shown are mean ± SD of triplicates from a representative experiment (n = 3). (E) ADCC assay was performed with 21-day CAR-T cells and 17-day NK cells. Data shown are representative of three independent experiments. (F) CDC assay was performed by incubating CAR-T cells with baby rabbit complement and anti-CD20 antibody (rituximab). Data shown are pooled from two independent experiments.
To facilitate antibody-dependent cell-mediated cytotoxicity (ADCC), we cultured autologous NK cells with an IL-15-expressing K562 feeder cell line (Figure S10A). The purity of these cells was verified through flow cytometry analysis of CD3− CD56+ expression, and these cells also expressed high levels of CD16, an Fc receptor necessary for ADCC (Figure S10B). To perform ADCC, we used the non-CD20-expressing NKG2D CAR-T cells as a negative control. As expected, there was no significant difference in the cytotoxicity exerted by autologous NK cells against NKG2D CAR-T cells (Figure 6E) when either the isotype control antibody or the anti-CD20 antibody was added (NS; p > 0.05). Conversely, NK cells co-incubated with anti-CD20 antibody elicited a statistically significant cytotoxic response against NKG2D-CD20 CAR-T cells compared with those incubated with the isotype control antibody (∗∗∗p < 0.001).
In parallel with ADCC, we also demonstrated the efficacy of complement-dependent cytotoxicity (CDC) in removing CD20-expressing NKG2D CAR-T cells. As shown in Figure 6F, NKG2Dbp-CD20 CAR-T cells treated with both baby rabbit complement and anti-CD20 antibody underwent a higher degree of cytotoxicity compared with cells treated in media alone, complement alone, or antibody alone (∗p < 0.05). Collectively, this showed that the incorporation of CD20 allows the possibility of ADCC and CDC to remove NKG2D CAR-T cells systematically.
Discussion
At present, the process of CAR-T cell manufacturing remains a bottleneck for most clinical applications, with no single manufacturing protocol having been recognized as the gold standard for good manufacturing practice (GMP) manufacturing. For the manufacturing of NKG2D CAR-T cells to meet clinical applications, the same issue exists.17
In a recent study by Baumeister and colleagues,15,17 they described the use of GMP-compliant G-REX cell culture vessels and recombinant IL-2 to expand NKG2D CAR-T cells for a clinical application (NCT02203825). Their method depends on the expression of NKG2D ligands on activated CAR-T cells. However, T cell fratricide will eventually result in elimination of these ligand-expressing T cells from the population, which makes long-term antigen-dependent expansion of these cells difficult. To that end, two other methods have recently been reported: the use of LY294002 phosphatidylinositol 3-kinase (PI3K) inhibitor (PIK3i) to block or reduce NKG2D CAR signaling and the use of anti-NKG2D antibody to prevent NKG2D-mediated fratricide.19 Compared with PI3K inhibition, blockade of NKG2D improved cell yield directly while also maintaining anti-tumor activity.
Interestingly, our work showed that OKT3 activation followed by DNA electroporation did not upregulate the expression level of any NKG2D ligands. This can be contrasted with the upregulated levels observed in NKG2D CAR-T cells that were prepared by OKT3 activation and viral transduction.19 In that study, the authors observed that MICA, ULBP2, and ULBP4 were predominantly expressed at the protein level, with expression level of ULBP4 as high as nearly 40% by day 6.19 In our present study, we observed single-digit expression levels for all NKG2D ligands at 2 days (Figure 1E) and 5 days (Figure 1F) after DNA electroporation. In addition, the highest expression level observed for ULBP4 for both time points was 5.7% ± 1.5% for NKG2Dbp CAR-T cells at the 2-day time point. While this observation for a preferential expression of ULBP4 is congruent with that observed by Breman et al.,19 our protocol did not appear to induce as significant an increase in NKG2D ligand expression as OKT3 activation coupled with viral transduction. This suggests that the early stage of our manufacturing protocol need not be concerned with a low cell yield arising from T cell fratricide.
A limitation of our study is that we did not extend the characterization of NKG2D ligand expression on NKG2D CAR-T cells to the later stages of manufacturing. This would have allowed us to investigate whether K562 cells served as a decoy target or as an additional source of antigens. Nonetheless, even if our approach did not address this issue of T cell fratricide arising from expression of NKG2D ligands on activated T cells observed in other studies, the use of K562 feeder cells does provide an alternative source of NKG2D ligand antigens. Importantly, the expansion of NKG2D CAR-T cells in our work leverages on the natural ability of K562 cells to express NKG2D ligands, which could further be upregulated by gamma irradiation (Figure S4). Regardless of T cell fratricide, our data have shown a steady and robust antigen-dependent expansion of NKG2D CAR-T cells (Figure 2D). This also enables a long-term culture to ensure timely availability of ready-to-use CAR-T cells, should a clinical need for more injections arise. Furthermore, the use of K562 cells allows us to bypass an immediate need to construct a new cell line for antigen-dependent enrichment and expansion. Thus, our K562-based approach is advantageous, as studies reported to date have focused on the sole use of recombinant IL-2 to expand NKG2D CAR-T cells (Table S1).
We have previously reported the use of our K562 feeder cell line engineered to express CD64, CD86, and CD137L.37,38 In expanding Zometa-activated Vγ9Vδ2T cells through a G-Rex platform, the use of K562 feeder cells resulted in the downregulation of PD1, TIGIT, BTLA, CTLA, TIM3, and LAG3 expression levels in a single expansion phase from day 7 to day 17.37 While not entirely similar, this is somewhat congruent with our observations in the present study, where the expression levels of T cell exhaustion markers were downregulated over the extended 63-day manufacturing period. In the context of tonic CAR signaling, the use of CD137 costimulation in CAR-T cells has been shown to ameliorate T cell exhaustion arising from constitutive CAR activity.43 While we did not examine the mechanistic reasons for this downregulation of exhaustion markers in both studies, this could possibly be attributed to the activity of CD137L expressed on the feeder cells.
Current clinical CAR-T manufacturing for blood cancer treatment has focused on the introduction of CAR-T cells in the TCM or TSCM subsets.44 This has led to the development of apheresis methods that harness a large number of naive T cells for genetic modification.45 In this aspect, we recognized that the preference for TEM development in our application may attract criticisms in that it may have limitations on the in vivo expansion and persistence of the infused NKG2D CAR-T cells. TEM development in our study is expected, as soluble cytokines identified to be crucial in ex vivo development of TCM or TSCM, such as IL-7 and IL-15,46 were not used in our current K562 expansion protocol. Improvements to this protocol can be made by exploring various combinations of cytokines, inhibition of signaling pathways, such as rapamycin-mediated mTOR inhibition,47 modulation of potassium efflux,48 or genetic modifications to express membrane-bound cytokines on K562 feeder cells. K562 may also be modified to express membrane-bound variants of IL-7 and IL-15 to bolster the development of TCM or TSCM subsets. While not entirely similar, Hurton and colleagues suggested the use of tethered IL-15, which consists of a full-length IL-15 expressed in tandem with an IL15Rα chain, joined by a Ser-Gly linker peptide.49 This method enabled IL 15 signaling even in the absence of antigenic stimulation and promoted the maintenance of long-lived TSCM cells. In this regard, K562 cells expressing membrane-bound variants of IL-7, IL-15, and IL-21 may also selectively promote the development of memory CAR-T cells.
Unlike CAR-T cell therapy for liquid cancer, in which leukemic cells are disseminated in the circulation and CAR-T cells can easily access malignant cells and readily expand after being introduced into the blood, CAR-T cells injected for solid tumor therapy face tough challenges to achieve in vivo expansion and persistence. This is related to the fact that solid tumor malignancies are regionally localized at specific tissue sites and only a limited number of infused CAR-T cells can reach there and interact with tumor antigen-expressing solid tumor cells that stimulate their proliferation. Thus, multiple injections of CAR-T cells, even multi-cycles of treatment, are usually necessary for managing solid malignancies.50,51 To accommodate multiple injections of age-matched CAR-T cells, we explored the feasibility of cryopreservation coupled with an additional round of K562 stimulation subsequent to thawing so that we could consistently introduce 28-day NKG2D CAR-T cells back into the patient (Figure 4). Antigen-dependent expansion of CAR-T cells after cryopreservation is a novel concept, as current anti-CD19 clinical works at Novartis (tisagenlecleucel) and Kite Pharma (axicabtagene ciloleucel) involve only a single injection of ex vivo manufactured CAR-T cells.52 Should a need for additional doses arise, the entire manufacturing process from apheresis through expansion may have to be repeated. With cryopreservation of initial CAR-T cell products at sufficient numbers, our protocol potentially allows multiple injections of age-matched CAR-T cells way after the first injection back into the patient. Subsequent injections could be performed on prearranged clinical approval or on an ad hoc basis subject to clinically evaluated needs. This has ramifications for quality control measures, as stringent assays performed on the first batch could give better albeit not complete assurances about the quality of subsequent batches.
One innovation that we explored with CAR-T manufacturing was the long-term ex vivo manufacturing of these NKG2D CAR-T cells up to day 63. This approach allowed us to have a working cell stock functioning in parallel with real-time clinical evaluation of patient’s response to the initial doses. Besides the availability of frozen initial CAR-T cell products, an ongoing working cell stock could be attractive in providing an immediate source of CAR-T cells. An anticipated criticism of this approach would be the potential T cell exhaustion and resultant loss of anti-tumor activity. At least until day 63, we have shown that in vitro anti-tumor activity was not lost with the extended manufacturing period (Figure 5). However, we note that the in vivo persistence of these CAR-T cells has not been validated and is an area of future work. Another criticism of this approach would be the manufacturing costs associated with maintaining a parallel GMP-grade cell culture for an extended period. While an ongoing cell culture may provide a readily available source of CAR-T cells, this advantage may be tempered by a realistic need to balance competing needs for logistics, equipment, and facilities between different patients. Furthermore, while we have investigated the expression profile of exhaustion markers and validated their in vitro cytotoxicity, it is possible that an extended ex vivo expansion may induce other cellular changes that we have not yet characterized. This further complicates the requisite quality control tests needed to be satisfied during release testing. Thus, future studies will involve deeper characterization of the cellular properties of these “extended” CAR-T cells.
Studies to enhance the safety profile of infused T cells have included the tandem expression of an elimination gene,53, 54, 55 including that of CD20.56, 57, 58, 59 The study by Griffioen et al.59 similarly utilized a full-length CD20 transgene as a suicide gene for virus-specific T cells and did not report any effect on T cell function. In the present study, we have shown that the incorporation of a native full-length CD20 protein expressed in tandem with the primary CAR construct via the P2A self-cleaving peptide could help facilitate ADCC mediated by autologous NK cells and complement-mediated cytotoxicity. Future in vivo evaluation of this ADCC/CDC approach is warranted before clinical adoption.
In conclusion, we have described a piggyBac-based gene transfer protocol and an accompanying K562-based expansion protocol that allows a reproducible and reliable manufacturing of large numbers of autologous NKG2D CAR-T cells over an extended manufacturing period. The method accommodates a clinical application timeline involving multiple infusions of CAR-T cells and, potentially, multi-cycles of cell therapy treatment that would be necessary for cure of solid tumors. Modifications to the K562 feeder cell line can be made to accommodate antigen-dependent enrichment and expansion of other CAR-T cells.
Materials and methods
Cancer cell culture
Human cancer cell lines CAOV3 and HCT-116 were cultured in DMEM (Lonza, Basel, Switzerland) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA, USA). All human cell cultures were maintained at 5% CO2 in a humidified 37°C incubator.
Preparation and expansion of CAR-T cells
PBMCs were isolated from buffy coat samples from healthy donors (National University of Singapore, NUS-IRB B-14-133E) or blood samples from patients with ovarian cancer (Cancer Hospital of the University of Chinese Academy of Sciences, IRB-2020-15) with Ficoll-Paque-based density gradient centrifugation. For optimization of T cell activation, PBMCs were activated with soluble OKT3 anti-CD3 monoclonal antibody (eBioscience, San Diego, CA, USA) at either 100 ng/mL or 500 ng/mL and cultured in AIM-V (Invitrogen, Carlsbad, CA, USA) supplemented with 5% AB serum (Gemini Bio, West Sacramento, CA, USA) and 300 IU/mL IL-2 (Miltenyi Biotec, Germany) for 2 or 3 days. OKT3-activated PBMCs were then harvested and washed thrice with OptiMEM (Invitrogen) before resuspension in P3 Primary Cell Nucleofector Solution (Lonza) at a cell density of 1 × 108 cells/mL. For each electroporation reaction with 100 μL of cell suspension, 5 μg of piggyBac (PB) transposase plasmid and one of the following transposon donor plasmids were added at the indicated amounts: 10 μg of αCD22bp, 10 μg of NKG2D, or 30 μg of NKG2D-CD20.37 Cells and DNA mixtures were transferred to a 100-μL single Nucleocuvette (Lonza) and electroporated with the EO-115 program of the 4D-Nucleofector system (Lonza).
After electroporation, 500 μL of AIM-V supplemented with 5% AB serum and 300 IU/mL IL-2 was added to each Nucleocuvette. Cells were then allowed to rest for 30 min in the humidified 37°C incubator. Electroporated T cells were then transferred to a 12-well plate and cocultured in AIM-V supplemented with 5% AB serum and 300 IU/mL IL-2 for a further 5 days. Culture medium was replenished with fresh 300 IU/mL IL-2 every 2 days.
NKG2D and NKG2D-CD20 CAR-T cells were expanded through co-culture with gamma-irradiated K562 feeder cells modified to express CD64, CD86, and CD137L.37 αCD22bp CAR-T cells were expanded through co-culture with gamma-irradiated parental Raji cells. Both K562 and Raji feeder cells were prepared by gamma irradiation at 100 gy (A∗Star Biological Resource Centre, Singapore). For each round of stimulation, CAR-T cells and their respective feeder cells were seeded at 1:2 E:T ratio and cultured in AIM-V supplemented with 5% AB serum and 300 IU/mL IL-2 for 7 days. Cell culture was initiated by seeding 1 × 106 CAR-T cells and 2 × 106 feeder cells in a T75 flask within a 10 mL volume. Subsequently, cell density was maintained at 1 × 106 to 2 × 106 per mL between day 4 and day 7 of each 7-day expansion phase.
Flow cytometric analysis, cytotoxicity assay, and ELISpot-IFNγ assay
Flow cytometric analysis was performed with the Accuri C6 cytometer (BD Biosciences, Franklin Lakes, NJ, USA) with conjugated anti-human antibodies listed in Table S2. Cytolytic activities of CAR-modified T cells were examined with a non-radioactive method (DELFIA EuTDA Cytotoxicity Reagents kit, PerkinElmer, Waltham, MA, USA). IFNγ ELISpot assays were performed according to the protocols of ELISpot kits (Mabtech, Nacka Strand, Sweden). The plates were analyzed by an ELISpot scanner (CTL, Cleveland, OH, USA).
Animal experiments
Animal experiments were performed according to protocols reviewed and approved by Institutional Animal Care and Use Committee (IACUC), the Biological Resource Centre (BRC), the Agency for Science, Technology and Research (A∗STAR), Singapore (Permit number BRC IACUC#181324). NSG mice were inoculated via intraperitoneal (i.p.) injection of 2 × 106 HCT116-Luc cells to generate tumor models. To investigate in vivo anti-tumor effects of CAR-T cells, human T cells expressing an NKG2D CAR (1 × 107) were i.p. injected into tumor-bearing mice. Mice treated with PBS and control T cells were used as controls. Tumor progression was monitored by live bioluminescence imaging. For in vivo imaging, animals were anesthetized with isoflurane in the presence of O2 and injected with 150 mg/kg body weight of d-luciferin. Fifteen minutes after injection, animals were scanned with an IVIS100 imaging platform. The imaging data were acquired with the following parameters: 10 s of exposure time, medium binning, and f/stop of 1. Acquired images were then analyzed with Living software 3.2.
Statistical analysis
Data are presented as mean ± standard deviation (SD). All statistics were performed with GraphPad Prism 7 (San Diego, CA, USA). p values <0.05 were considered significant.
For details of the materials and methods, see Supplemental information.
Acknowledgments
This work was supported by Singapore Ministry of Health’s National Medical Research Council (NMRC/CIRG/1406/2014, NMRC/OFLCG/003/2018, and MOH-000465-01).
Author contributions
Conceived and designed the experiments: J.Z., X.H.X., and S.W.; performed the experiments: J.C.K.T., J.W., Z.D., Y.Y.N., Z.L., Y.R., and C.Z.; analyzed the data: J.C.K.T., J.W., Z.D., and Y.Y.N.; wrote the manuscript: J.C.K.T., S.W., J.Z., and X.H.X.
Declaration of interests
S.W., J.C.K.T., Y.Y.N., and Z.L. have filed patent applications related to CAR technology and could potentially receive licensing royalties in future.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.02.023.
Contributor Information
Jianqing Zhu, Email: zjq-hz@126.com.
Xue Hu Xu, Email: xxh@gzhmu.edu.cn.
Shu Wang, Email: dbsws@nus.edu.sg.
Supplemental information
References
- 1.Lanier L.L. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol. Res. 2015;3:575–582. doi: 10.1158/2326-6066.CIR-15-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spear P., Wu M.R., Sentman M.L., Sentman C.L. NKG2D ligands as therapeutic targets. Cancer Immun. 2013;13:8. [PMC free article] [PubMed] [Google Scholar]
- 3.Sentman C.L., Meehan K.R. NKG2D CARs as cell therapy for cancer. Cancer J. 2014;20:156–159. doi: 10.1097/PPO.0000000000000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardee C.L., Arévalo-Soliz L.M., Hornstein B.D., Zechiedrich L. Advances in Non-Viral DNA Vectors for Gene Therapy. Genes (Basel) 2017;8:65. doi: 10.3390/genes8020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim A., Pyykko I. Size matters: versatile use of PiggyBac transposons as a genetic manipulation tool. Mol. Cell. Biochem. 2011;354:301–309. doi: 10.1007/s11010-011-0832-3. [DOI] [PubMed] [Google Scholar]
- 6.Wilson M.H. Consider Changing the Horse for Your CAR-T? Mol. Ther. 2018;26:1873–1874. doi: 10.1016/j.ymthe.2018.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Neil R.T., Saha S., Veach R.A., Welch R.C., Woodard L.E., Rooney C.M., Wilson M.H. Transposon-modified antigen-specific T lymphocytes for sustained therapeutic protein delivery in vivo. Nat. Commun. 2018;9:1325. doi: 10.1038/s41467-018-03787-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakazawa Y., Huye L.E., Dotti G., Foster A.E., Vera J.F., Manuri P.R., June C.H., Rooney C.M., Wilson M.H. Optimization of the PiggyBac transposon system for the sustained genetic modification of human T lymphocytes. J. Immunother. 2009;32:826–836. doi: 10.1097/CJI.0b013e3181ad762b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang T., Barber A., Sentman C.L. Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Cancer Res. 2006;66:5927–5933. doi: 10.1158/0008-5472.CAN-06-0130. [DOI] [PubMed] [Google Scholar]
- 10.Barber A., Zhang T., DeMars L.R., Conejo-Garcia J., Roby K.F., Sentman C.L. Chimeric NKG2D receptor-bearing T cells as immunotherapy for ovarian cancer. Cancer Res. 2007;67:5003–5008. doi: 10.1158/0008-5472.CAN-06-4047. [DOI] [PubMed] [Google Scholar]
- 11.Barber A., Zhang T., Megli C.J., Wu J., Meehan K.R., Sentman C.L. Chimeric NKG2D receptor-expressing T cells as an immunotherapy for multiple myeloma. Exp. Hematol. 2008;36:1318–1328. doi: 10.1016/j.exphem.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barber A., Sentman C.L. NKG2D receptor regulates human effector T-cell cytokine production. Blood. 2011;117:6571–6581. doi: 10.1182/blood-2011-01-329417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song D.G., Ye Q., Santoro S., Fang C., Best A., Powell D.J., Jr. Chimeric NKG2D CAR-expressing T cell-mediated attack of human ovarian cancer is enhanced by histone deacetylase inhibition. Hum. Gene Ther. 2013;24:295–305. doi: 10.1089/hum.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X., Sun M., Yu S., Liu K., Li X., Shi H. Potential therapeutic strategy for gastric cancer peritoneal metastasis by NKG2D ligands-specific T cells. OncoTargets Ther. 2015;8:3095–3104. doi: 10.2147/OTT.S91122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernández L., Metais J.Y., Escudero A., Vela M., Valentín J., Vallcorba I., Leivas A., Torres J., Valeri A., Patiño-García A. Memory T Cells Expressing an NKG2D-CAR Efficiently Target Osteosarcoma Cells. Clin. Cancer Res. 2017;23:5824–5835. doi: 10.1158/1078-0432.CCR-17-0075. [DOI] [PubMed] [Google Scholar]
- 16.Tao K., He M., Tao F., Xu G., Ye M., Zheng Y., Li Y. Development of NKG2D-based chimeric antigen receptor-T cells for gastric cancer treatment. Cancer Chemother. Pharmacol. 2018;82:815–827. doi: 10.1007/s00280-018-3670-0. [DOI] [PubMed] [Google Scholar]
- 17.Murad J.M., Baumeister S.H., Werner L., Daley H., Trébéden-Negre H., Reder J., Sentman C.L., Gilham D., Lehmann F., Snykers S. Manufacturing development and clinical production of NKG2D chimeric antigen receptor-expressing T cells for autologous adoptive cell therapy. Cytotherapy. 2018;20:952–963. doi: 10.1016/j.jcyt.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han Y., Xie W., Song D.G., Powell D.J., Jr. Control of triple-negative breast cancer using ex vivo self-enriched, costimulated NKG2D CAR T cells. J. Hematol. Oncol. 2018;11:92. doi: 10.1186/s13045-018-0635-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breman E., Demoulin B., Agaugué S., Mauën S., Michaux A., Springuel L., Houssa J., Huberty F., Jacques-Hespel C., Marchand C. Overcoming Target Driven Fratricide for T Cell Therapy. Front. Immunol. 2018;9:2940. doi: 10.3389/fimmu.2018.02940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baumeister S.H., Murad J., Werner L., Daley H., Trebeden-Negre H., Gicobi J.K., Schmucker A., Reder J., Sentman C.L., Gilham D.E. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol. Res. 2019;7:100–112. doi: 10.1158/2326-6066.CIR-18-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang D., Sun B., Dai H., Li W., Shi L., Zhang P., Li S., Zhao X. T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells. J. Immunother. Cancer. 2019;7:171. doi: 10.1186/s40425-019-0642-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernández L., Fernández A., Mirones I., Escudero A., Cardoso L., Vela M., Lanzarot D., de Paz R., Leivas A., Gallardo M. GMP-Compliant Manufacturing of NKG2D CAR Memory T Cells Using CliniMACS Prodigy. Front. Immunol. 2019;10:2361. doi: 10.3389/fimmu.2019.02361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun B., Yang D., Dai H., Liu X., Jia R., Cui X., Li W., Cai C., Xu J., Zhao X. Eradication of Hepatocellular Carcinoma by NKG2D-Based CAR-T Cells. Cancer Immunol. Res. 2019;7:1813–1823. doi: 10.1158/2326-6066.CIR-19-0026. [DOI] [PubMed] [Google Scholar]
- 24.Deng X., Gao F., Li N., Li Q., Zhou Y., Yang T., Cai Z., Du P., Chen F., Cai J. Antitumor activity of NKG2D CAR-T cells against human colorectal cancer cells in vitro and in vivo. Am. J. Cancer Res. 2019;9:945–958. [PMC free article] [PubMed] [Google Scholar]
- 25.Lehner M., Götz G., Proff J., Schaft N., Dörrie J., Full F., Ensser A., Muller Y.A., Cerwenka A., Abken H. Redirecting T cells to Ewing’s sarcoma family of tumors by a chimeric NKG2D receptor expressed by lentiviral transduction or mRNA transfection. PLoS ONE. 2012;7:e31210. doi: 10.1371/journal.pone.0031210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ojo E.O., Sharma A.A., Liu R., Moreton S., Checkley-Luttge M.A., Gupta K., Lee G., Lee D.A., Otegbeye F., Sekaly R.P. Membrane bound IL-21 based NK cell feeder cells drive robust expansion and metabolic activation of NK cells. Sci. Rep. 2019;9:14916. doi: 10.1038/s41598-019-51287-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kweon S., Phan M.T., Chun S., Yu H., Kim J., Kim S., Lee J., Ali A.K., Lee S.H., Kim S.K. Expansion of Human NK Cells Using K562 Cells Expressing OX40 Ligand and Short Exposure to IL-21. Front. Immunol. 2019;10:879. doi: 10.3389/fimmu.2019.00879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sutlu T., Alici E. Ex vivo expansion of natural killer cells: a question of function. Cytotherapy. 2011;13:767–768. doi: 10.3109/14653249.2011.563295. [DOI] [PubMed] [Google Scholar]
- 29.Baek H.J., Kim J.S., Yoon M., Lee J.J., Shin M.G., Ryang D.W., Kook H., Kim S.K., Cho D. Ex vivo expansion of natural killer cells using cryopreserved irradiated feeder cells. Anticancer Res. 2013;33:2011–2019. [PubMed] [Google Scholar]
- 30.Zeng J., Tang S.Y., Toh L.L., Wang S. Generation of “Off-the-Shelf” Natural Killer Cells from Peripheral Blood Cell-Derived Induced Pluripotent Stem Cells. Stem Cell Reports. 2017;9:1796–1812. doi: 10.1016/j.stemcr.2017.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao L., Cen D., Gan H., Sun Y., Huang N., Xiong H., Jin Q., Su L., Liu X., Wang K. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol. Ther. 2019;27:1114–1125. doi: 10.1016/j.ymthe.2019.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oberoi P., Kamenjarin K., Ossa J.F.V., Uherek B., Bönig H., Wels W.S. Directed Differentiation of Mobilized Hematopoietic Stem and Progenitor Cells into Functional NK cells with Enhanced Antitumor Activity. Cells. 2020;9:811. doi: 10.3390/cells9040811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujisaki H., Kakuda H., Shimasaki N., Imai C., Ma J., Lockey T., Eldridge P., Leung W.H., Campana D. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009;69:4010–4017. doi: 10.1158/0008-5472.CAN-08-3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imai C., Iwamoto S., Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106:376–383. doi: 10.1182/blood-2004-12-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamiya T., Chang Y.H., Campana D. Expanded and Activated Natural Killer Cells for Immunotherapy of Hepatocellular Carcinoma. Cancer Immunol. Res. 2016;4:574–581. doi: 10.1158/2326-6066.CIR-15-0229. [DOI] [PubMed] [Google Scholar]
- 36.Zha S., Li Z., Chen C., Du Z., Tay J.C., Wang S. Beta-2 microglobulin knockout K562 cell-based artificial antigen presenting cells for ex vivo expansion of T lymphocytes. Immunotherapy. 2019;11:967–982. doi: 10.2217/imt-2018-0211. [DOI] [PubMed] [Google Scholar]
- 37.Xiao L., Chen C., Li Z., Zhu S., Tay J.C., Zhang X., Zha S., Zeng J., Tan W.K., Liu X. Large-scale expansion of Vγ9Vδ2 T cells with engineered K562 feeder cells in G-Rex vessels and their use as chimeric antigen receptor-modified effector cells. Cytotherapy. 2018;20:420–435. doi: 10.1016/j.jcyt.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 38.Du S.H., Li Z., Chen C., Tan W.K., Chi Z., Kwang T.W., Xu X.H., Wang S. Co-Expansion of Cytokine-Induced Killer Cells and Vγ9Vδ2 T Cells for CAR T-Cell Therapy. PLoS ONE. 2016;11:e0161820. doi: 10.1371/journal.pone.0161820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ng Y.Y., Tay J.C.K., Li Z., Wang J., Zhu J., Wang S. T Cells Expressing NKG2D CAR with a DAP12 Signaling Domain Stimulate Lower Cytokine Production While Effective in Tumor Eradication. Mol. Ther. 2020;29:75–85. doi: 10.1016/j.ymthe.2020.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haso W., Lee D.W., Shah N.N., Stetler-Stevenson M., Yuan C.M., Pastan I.H., Dimitrov D.S., Morgan R.A., FitzGerald D.J., Barrett D.M. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang S.C., Owen-Schaub L.B., Roth J.A., Grimm E.A. Characterization of OKT3-initiated lymphokine-activated effectors expanded with interleukin 2 and tumor necrosis factor alpha. Cancer Res. 1990;50:3526–3532. [PubMed] [Google Scholar]
- 42.Zheng Y., Fang Y.C., Li J. PD-L1 expression levels on tumor cells affect their immunosuppressive activity. Oncol. Lett. 2019;18:5399–5407. doi: 10.3892/ol.2019.10903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Long A.H., Haso W.M., Shern J.F., Wanhainen K.M., Murgai M., Ingaramo M., Smith J.P., Walker A.J., Kohler M.E., Venkateshwara V.R. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu Y., Zhang M., Ramos C.A., Durett A., Liu E., Dakhova O., Liu H., Creighton C.J., Gee A.P., Heslop H.E. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123:3750–3759. doi: 10.1182/blood-2014-01-552174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sommermeyer D., Hudecek M., Kosasih P.L., Gogishvili T., Maloney D.G., Turtle C.J., Riddell S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cieri N., Camisa B., Cocchiarella F., Forcato M., Oliveira G., Provasi E., Bondanza A., Bordignon C., Peccatori J., Ciceri F. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121:573–584. doi: 10.1182/blood-2012-05-431718. [DOI] [PubMed] [Google Scholar]
- 47.Brestrich G., Zwinger S., Fischer A., Schmück M., Röhmhild A., Hammer M.H., Kurtz A., Uharek L., Knosalla C., Lehmkuhl H. Adoptive T-cell therapy of a lung transplanted patient with severe CMV disease and resistance to antiviral therapy. Am. J. Transplant. 2009;9:1679–1684. doi: 10.1111/j.1600-6143.2009.02672.x. [DOI] [PubMed] [Google Scholar]
- 48.Eil R., Vodnala S.K., Clever D., Klebanoff C.A., Sukumar M., Pan J.H., Palmer D.C., Gros A., Yamamoto T.N., Patel S.J. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature. 2016;537:539–543. doi: 10.1038/nature19364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hurton L.V., Singh H., Najjar A.M., Switzer K.C., Mi T., Maiti S., Olivares S., Rabinovich B., Huls H., Forget M.A. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA. 2016;113:E7788–E7797. doi: 10.1073/pnas.1610544113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown C.E., Alizadeh D., Starr R., Weng L., Wagner J.R., Naranjo A., Ostberg J.R., Blanchard M.S., Kilpatrick J., Simpson J. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weiss T., Weller M., Guckenberger M., Sentman C.L., Roth P. NKG2D-Based CAR T Cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res. 2018;78:1031–1043. doi: 10.1158/0008-5472.CAN-17-1788. [DOI] [PubMed] [Google Scholar]
- 52.Zheng P.P., Kros J.M., Li J. Approved CAR T cell therapies: ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov. Today. 2018;23:1175–1182. doi: 10.1016/j.drudis.2018.02.012. [DOI] [PubMed] [Google Scholar]
- 53.Brandt L.J.B., Barnkob M.B., Michaels Y.S., Heiselberg J., Barington T. Emerging Approaches for Regulation and Control of CAR T Cells: A Mini Review. Front. Immunol. 2020;11:326. doi: 10.3389/fimmu.2020.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang X., Chang W.C., Wong C.W., Colcher D., Sherman M., Ostberg J.R., Forman S.J., Riddell S.R., Jensen M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bonifant C.L., Jackson H.J., Brentjens R.J., Curran K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics. 2016;3:16011. doi: 10.1038/mto.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Philip B., Kokalaki E., Mekkaoui L., Thomas S., Straathof K., Flutter B., Marin V., Marafioti T., Chakraverty R., Linch D. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. 2014;124:1277–1287. doi: 10.1182/blood-2014-01-545020. [DOI] [PubMed] [Google Scholar]
- 57.Marin V., Cribioli E., Philip B., Tettamanti S., Pizzitola I., Biondi A., Biagi E., Pule M. Comparison of different suicide-gene strategies for the safety improvement of genetically manipulated T cells. Hum. Gene Ther. Methods. 2012;23:376–386. doi: 10.1089/hgtb.2012.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vogler I., Newrzela S., Hartmann S., Schneider N., von Laer D., Koehl U., Grez M. An improved bicistronic CD20/tCD34 vector for efficient purification and in vivo depletion of gene-modified T cells for adoptive immunotherapy. Mol. Ther. 2010;18:1330–1338. doi: 10.1038/mt.2010.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Griffioen M., van Egmond E.H., Kester M.G., Willemze R., Falkenburg J.H., Heemskerk M.H. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica. 2009;94:1316–1320. doi: 10.3324/haematol.2008.001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.