

Abstract
During the past decade, many research groups have described catalytic methods for 1,2-carboboration, allowing access to structurally complex organoboronates from alkenes. Various transition metals, especially copper, palladium, and nickel, have been widely used in these reactions. This review summarizes advances in this field, with a special focus on the catalytic cycles involved in different metal-catalyzed carboboration reactions, as well as the regio- and stereochemical consequences of the underlying mechanisms. 1,2-Carboboration of other unsaturated systems, such as alkynes and allenes, are outside of the scope of this review.
Keywords: 1,2-Carboboration; Transition-metal; Alkene; Catalysis; Mechanism
1. Introduction
Boron-containing organic molecules are versatile synthetic intermediates due to the ability to transform C–B bonds into many other functional groups.[1] Because of the widespread availability of alkene substrates, conversion of alkenes into alkylboronates is a powerful synthetic strategy. One classical example of this type of transformation is alkene hydroboration, which has emerged as a central reactivity paradigm in organic synthesis since its initial discovery in the 1950s.[2] After decades of successfully developing alkene hydroboration chemistry, researchers have recently started to pursue alkene functionalization reactions that generate an even greater level of complexity, such as borylative alkene 1,2-difunctionalizations, where a second functional group is introduced instead of a hydrogen atom.[3] In this review, we will focus on a powerful subset of borylative 1,2-difunctionalization, namely 1,2-carboboration of alkenes, which is rapidly emerging as a powerful means of synthesizing organoboronates that complements existing methods.[4] By simultaneously introducing a boron and carbon moiety across a C=C bond, 1,2-carboboration forges a new C(sp3)–C bond and a new C(sp3)–B bond. Hence, this type of reaction offers a platform to access compounds with two contiguous stereogenic centers in a manner that quickly assembles molecular complexity from simple starting materials.
Starting from a common alkene substrate, 1,2-carboboration can be promoted by different transition metals (at various oxidation states). The unique mechanisms at play can result in divergent regio- and stereochemical outcomes, with the regio- and stereoselectivity/specificity dictated by the elementary steps involved (Scheme 1). This diversity of pathways presents an exciting opportunity in enabling divergent synthesis of organoboronates; however, it also represents a challenge in terms of developing predictable methods as well as a robust conceptual framework for synthetic planning. Among the results covered in this review are numerous examples of enantioselective transformations that have been achieved by adding appropriate non-racemic ligands. Elegant radical-based approaches, which are quickly emerging as a powerful complementary technology, are outside of the scope of this review.[5]
Scheme 1.
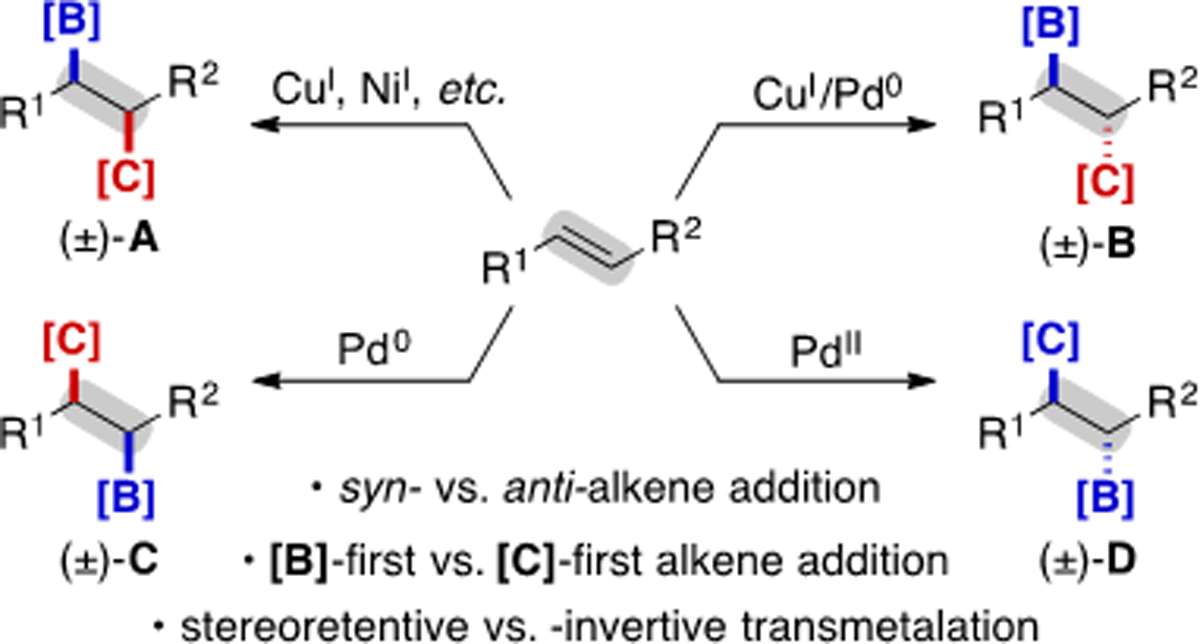
General depiction of catalyst-controlled 1,2-carboboration. In this scheme and throughout the manuscript, nucleophilic reaction partners are drawn in blue, and electrophilic reaction partners are drawn in red.
2. Transition-Metal-Catalyzed Alkene 1,2-Carboboration Reaction
2.1. Cu-catalyzed Alkene Carboboration
During the past decade, many examples of Cu-catalyzed alkene carboboration have been reported. Typically, a diboron reagent, such as bis(pinacolato)diboron (B2pin2), serves as the boron source, which undergoes transmetalation to transfer a boryl group onto the copper center. The resulting copper–boryl species then undergoes syn-migratory insertion to form the C(sp3)–B bond and concomitantly generate an alkylcopper intermediate.[6] Electrophilic trapping with a carbon-based electrophile then forges the C(sp3)–C bond and closes the catalytic cycle (Scheme 2). The mechanism for the final C–C-bond-forming step could either be SN2 or an oxidative addition/reductive elimination sequence, depending on the electrophile.[7]
Scheme 2.
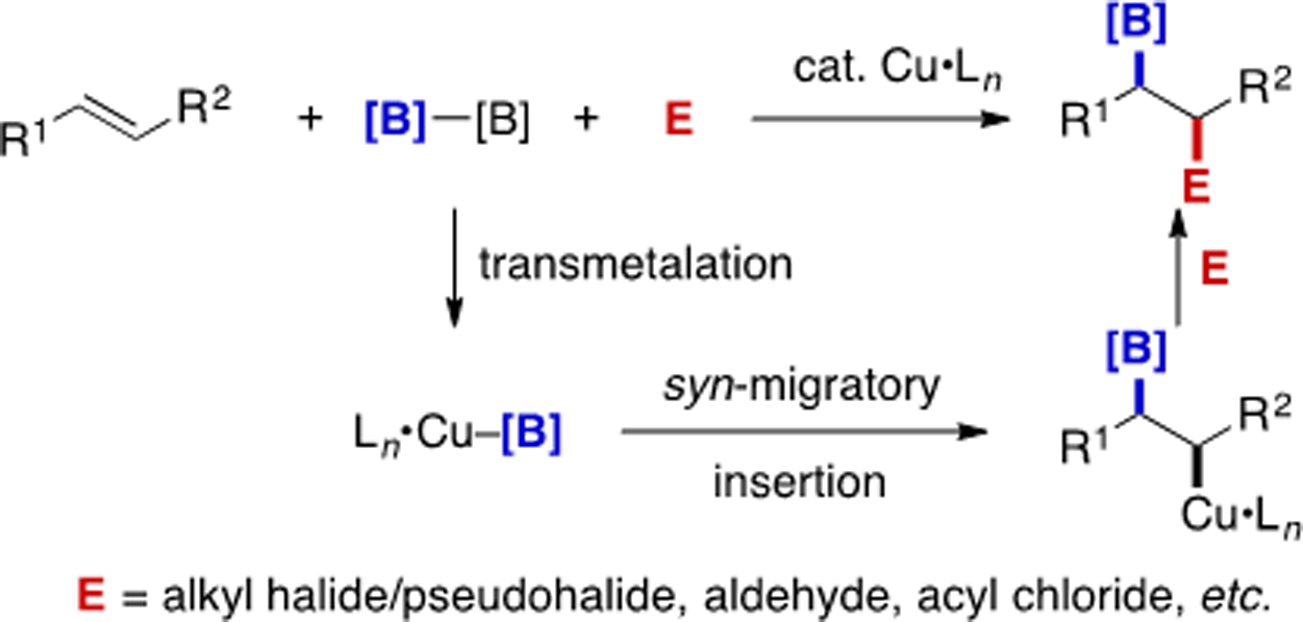
Mechanism of Cu-catalyzed alkene 1,2-carboboration.
2.1.1. Two-component Carboboration Involving an Intramolecular Cyclization
Since 2008, the groups of Ito and Sawamura have developed an elegant series of cyclization reactions triggered by borocupration.[8] Different sized rings can be constructed using this strategy, ranging from 3- to 5-membered ring systems. Compatible substrates include allylic carbonates, phosphates, alkyl sulfonates, and alkyl bromides. With chiral bisphospine ligands L1–L3, several transformations were rendered enantioselective.
In 2017, Ito and coworkers reported a Cu(I)-catalyzed borylative radical cyclization reaction (Scheme 4).[9] A radical clock experiment was designed to probe the mechanism, and the observation of ring opening is consistent with the intermediacy of a carbon-centered radical. This work is one of the few examples of Cu(I)-catalyzed carboboration that is believed to proceed by a radical mechanism.
Scheme 4.
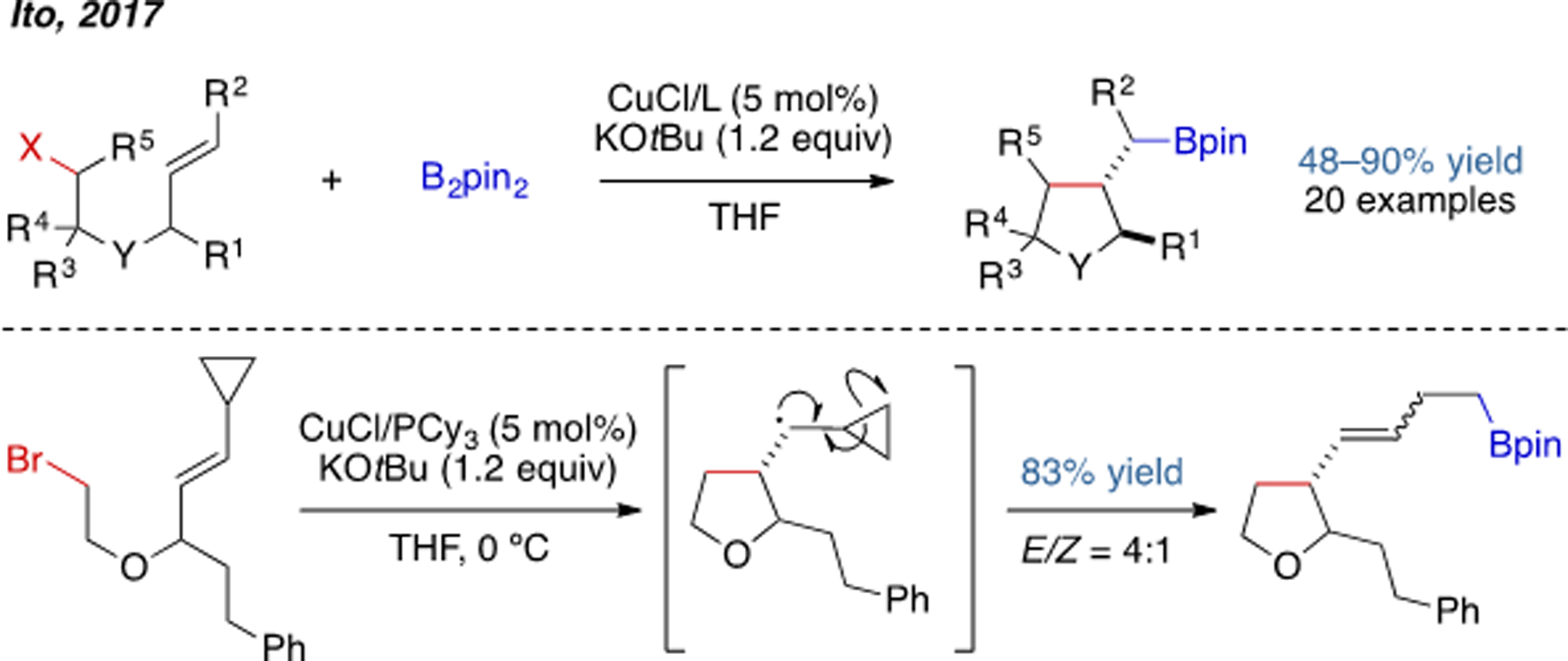
Cu(I)-catalyzed borylative radical cyclization.
In 2012, Lam and coworkers achieved an enantioselective Cu(I)-catalyzed cascade desymmetrization of α,β-unsaturated carbonyl compounds containing an intramolecularly tethered 1,3-diketone. Following conjugate borocupration, the resultant copper–enolate undergoes aldol cyclization to one of the ketone groups (Scheme 5).[10] With JosiPhos (L6) as the optimal ligand, an array of multifunctionalized bicyclic compounds could be generated in excellent enantioselectivity and moderate to high diastereoselectivity.
Scheme 5.
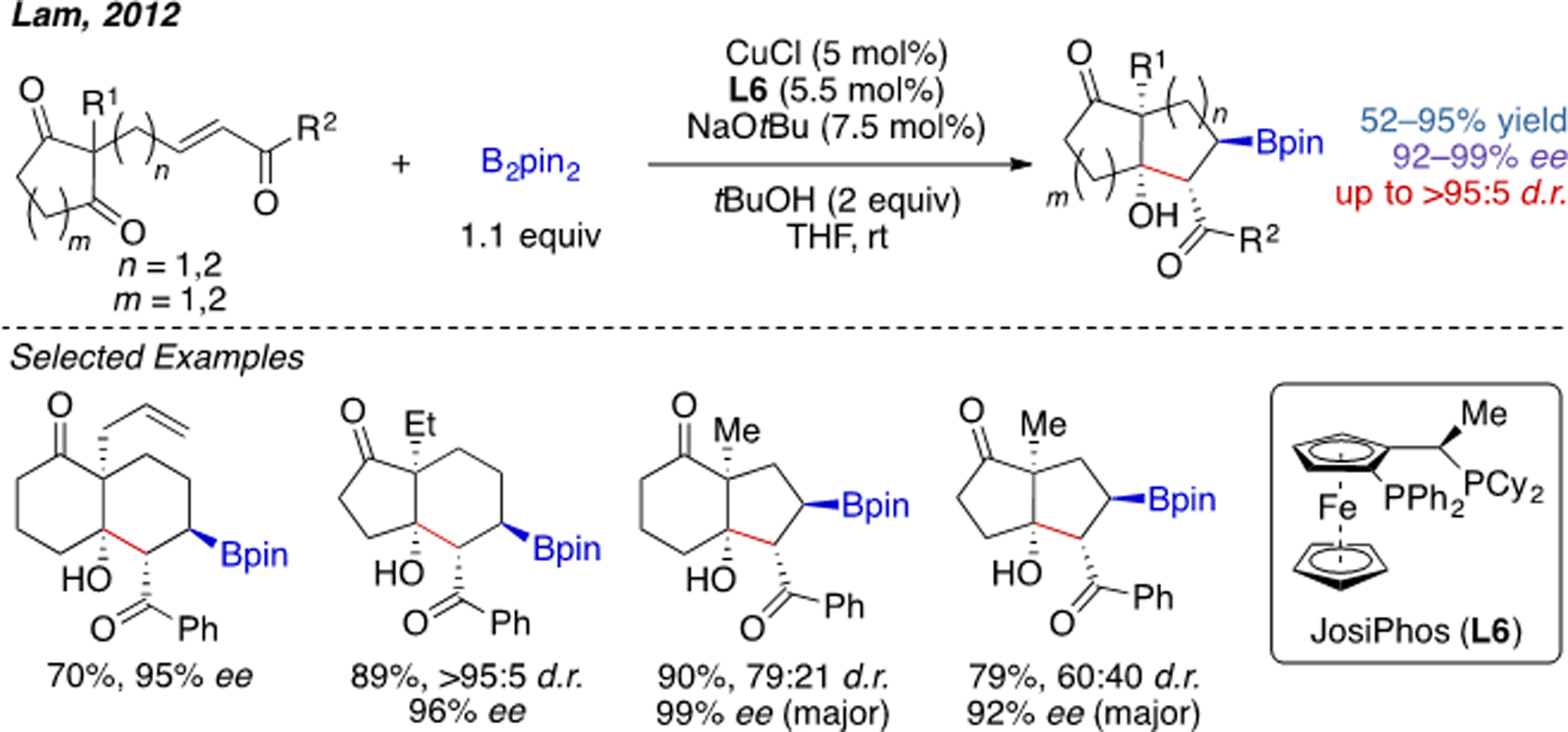
Cu(I)-catalyzed intramolecular carboboration involving an aldol cyclization.
Later, in 2018, Lautens and coworkers developed a Cu(I)-catalyzed intramolecular acylboration reaction using a tethered carbamoyl chloride as the electrophile (Scheme 6).[11] It is notable that the products are enantioenriched 3,3-disubstituted oxindoles, which are biologically interesting molecules.[12]
Scheme 6.
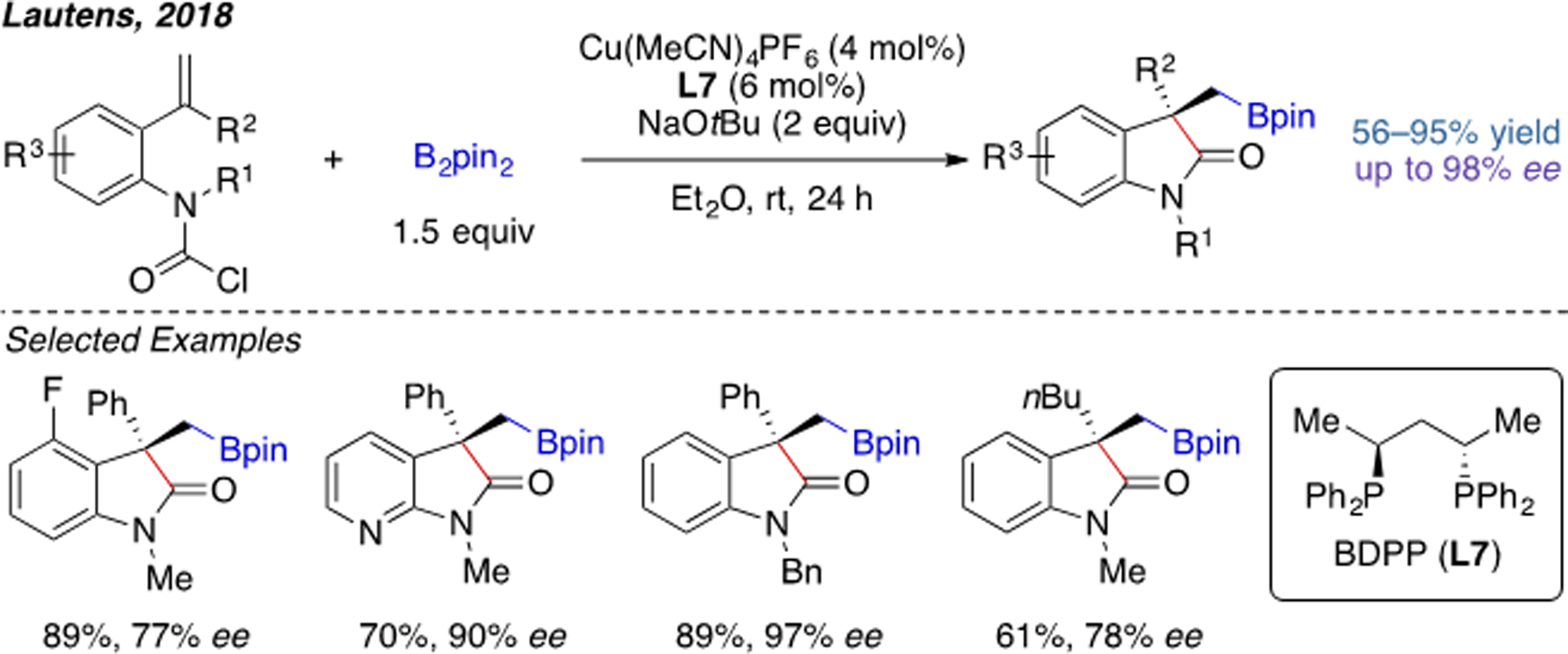
Cu(I)-catalyzed intramolecular acylboration.
2.1.2. Three-component Alkene Carboboration Reactions
Three-component carboboration is generally more challenging to achieve compared to its intramolecular counterpart, since the final electrophilic trapping step becomes slower and the side reaction of direct coupling between the boron nucleophile and carbon electrophile becomes significant. The first three-component alkene carboboration was described in a seminal report by Yoshida in 2013 using Cu catalysis (eq. 1).[13] Although only three examples of alkene benzylboration were disclosed with styrene, a vinyl boronate, and a vinyl silane, this work nevertheless demonstrated the viability of a fully intermolecular 1,2-carboboration, setting the stage for further development to unlock the full potential of this transformation in organic synthesis.
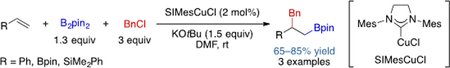 |
eq. 1 |
An extension of this alkene alkylboration system was published by Xiao, Fu, and coworkers in 2015 (Scheme 7).[14] The key alkylcopper intermediate generated by migratory insertion is resistant to β-H elimination compared to analogous intermediates derived from other transition metals, such palladium. Interestingly, in this work, the authors found that either regiochemical outcome could be obtained by adopting two structurally similar bisphosphine ligands (L5 and L8). DFT calculations of the relative Gibbs free energies of migratory insertion transition states revealed an opposite trend between these two ligands. With L5, anti-Markovnikov addition is favored because of a better d(Cu)-π(alkene) back donation. In contrast, with a bulkier ligand L6, the reaction favors Markovnikov addition due to steric effect. Notably, a weakly coordinating Lewis basic group on the alkene substrate is needed in this chemistry in order to overcome the direct boration of the alkyl halide.
Scheme 7.
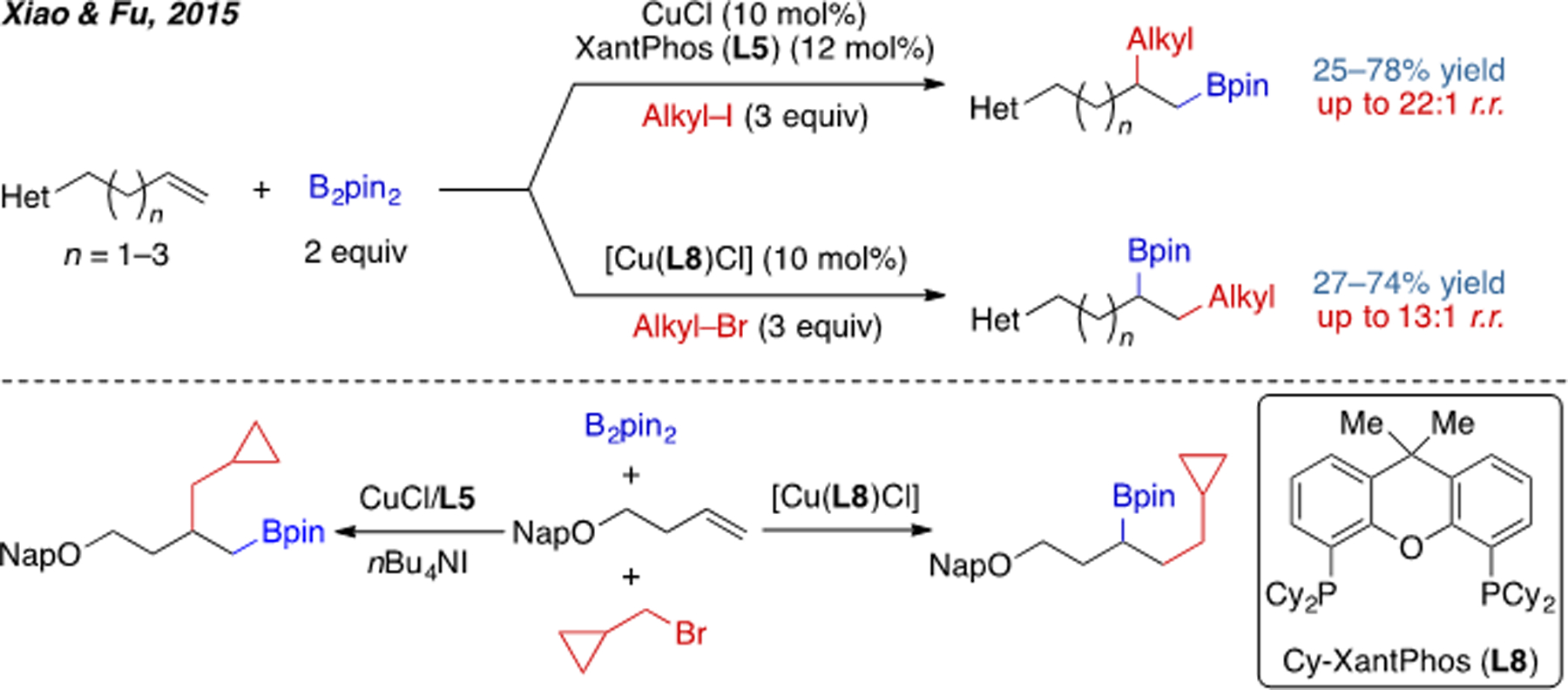
Cu(I)-catalyzed divergent alkylboration of alkenes.
In addition to alkyl halides, carbonyl-containing compounds have also been successfully employed as electrophiles in three-component alkene 1,2-carboboration. In 2014, Hoveyda and coworkers reported the first example of three-component carboboration using aldehydes as the coupling partner and 1,3-enynes as the substrate (Scheme 8).[15] Later, in 2017, Meek and coworkers achieved a similar transformation using vinyl boronate substrates.[16] These two reactions were rendered enantioselective through use of with chiral bisphosphine ligands L9 and L10, respectively. CO2 was also found to be a compatible electrophile in a so-called boracarboxylation reaction of styrenes reported by the group of Popp (eq. 2).[17] In all these cases, stabilized alkylcopper intermediates were generated from conjugated alkenes, which is a critical aspect of their success.
Scheme 8.
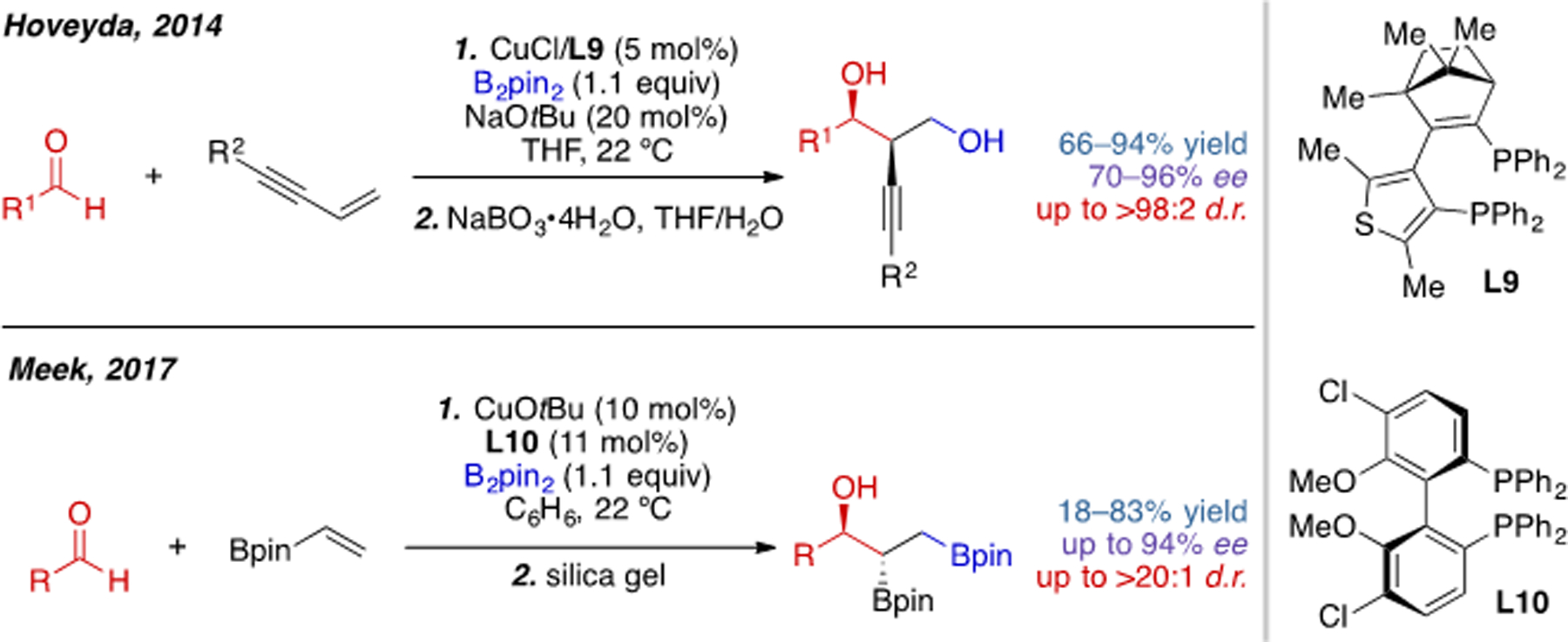
Cu(I)-catalyzed alkene carboboration with aldehydes as the electrophile.
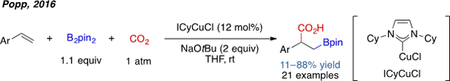 |
eq. 2 |
Cu(I)-catalyzed intramolecular acylboration has been introduced in the previous section. It should be noted that the three-component variant of this reaction was achieved even earlier by Brown and coworkers in 2017 (Scheme 9).[18] The compatible alkene substrates include vinyl arenes, 1,2-dienes, and strained alkenes. The authors also rendered this reaction enantioselective by preparing a chiral (NHC)Cu catalyst. In 2018, the group of Brown also reported a heteroarylboration of 1,3-dienes using pyridine-containing heterocycles as imine surrogates (Scheme 10).[19] The overall cine substitution of bromopyridines results from addition of the allylcopper intermediate via a six-membered transition state, in which C–C bond formation occurs at the position ortho to the bromide. Formal 1,5-elimination of the bromide and rearomatization through 1,2-hydride migration then delivers the final product. This work represents a novel protocol to access multisubstituted pyridines or quinolines. Though highly enabling in their own right, carboboration reactions involving copper–boryl intermediates have thus far been limited in terms of the scope of electrophiles that are compatible. This issue has been partially overcome by enlisting a second metal to promote the C–C bond forming step (see Section 2.4).
Scheme 9.
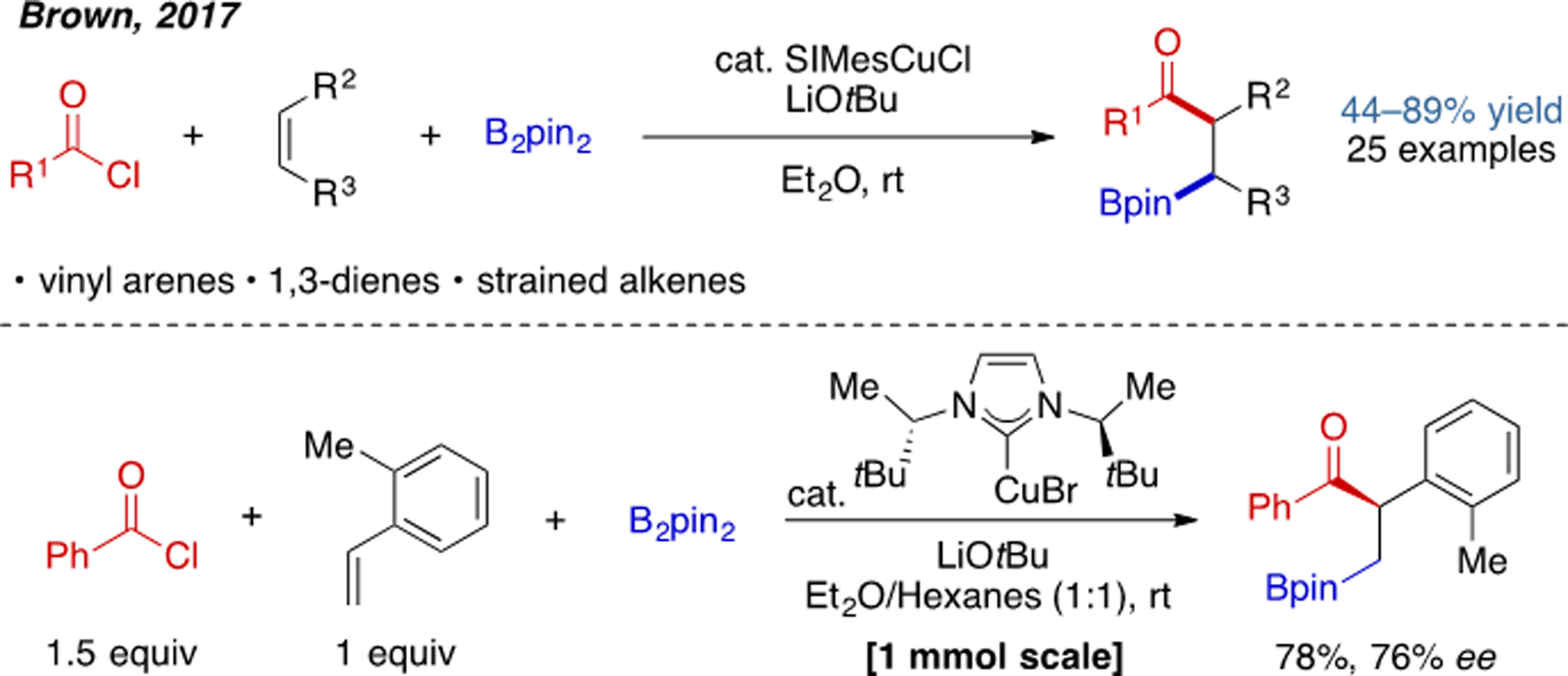
Cu(I)-catalyzed three-component acylboration.
Scheme 10.
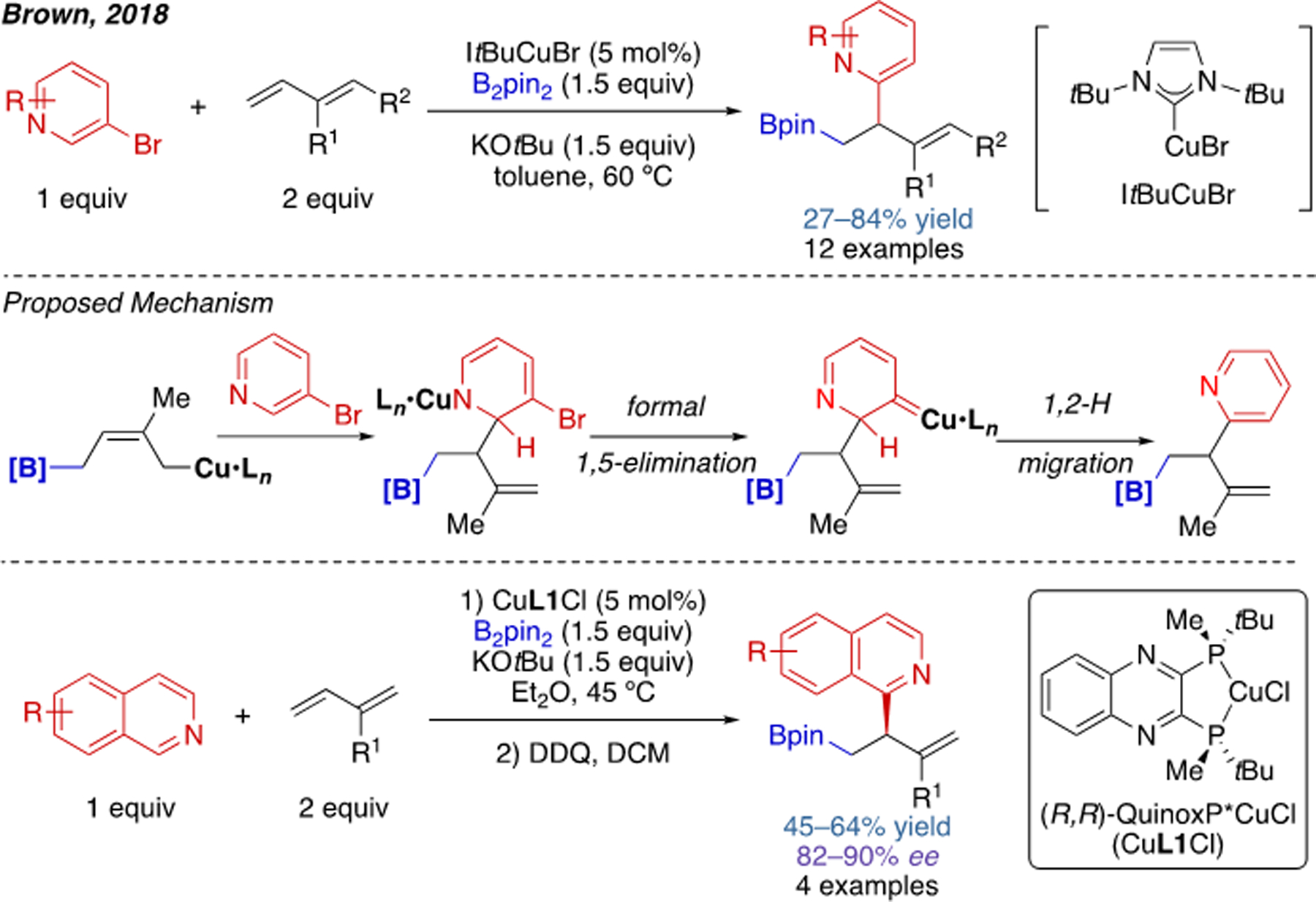
Cu(I)-catalyzed heteroarylboration of 1,2-dienes.
2.2. Pd-catalyzed Alkene Carboboration
Palladium catalysts are remarkably robust and versatile in traditional C(sp2)–C(sp2) and C(sp2)–heteroatom cross-coupling.[1b, 20] For example, Miyaura borylation typically uses a Pd(0) catalyst and is one of the most powerful C(sp2)–B-bond-forming methods in the modern synthetic repertoire.[21] Generally speaking alkene addition chemistry with palladium can take place through two different catalytic manifolds: Pd(0) catalysis via an inner-sphere Heck-type pathway or Pd(II) catalysis via an outer-sphere Wacker-type pathway. Indeed, several conceptually distinct Pd-catalyzed methods for alkene carboboration with different regio- and stereoselectivity have been successfully achieved. The first example of Pd-catalyzed alkene carboboration was reported by Suginome and coworkers in 2011 (Scheme 11).[22] The proposed reaction mechanism involves oxidative addition of Pd(0) into B–Cl bond and bora-Heck-type syn-migratory insertion of the tethered alkene. This mode of reactivity is unique in the context of alkene carboboration, presumably because boron halides are unstable in the present of many Lewis basic functional groups and are challenging to prepare. In general, “nucleophilic” boron reagents, as exemplified by as B2pin2, are far more common in alkene carboboration than “electrophilic” reagents, such as B–X (X=halide) compounds.
Scheme 11.
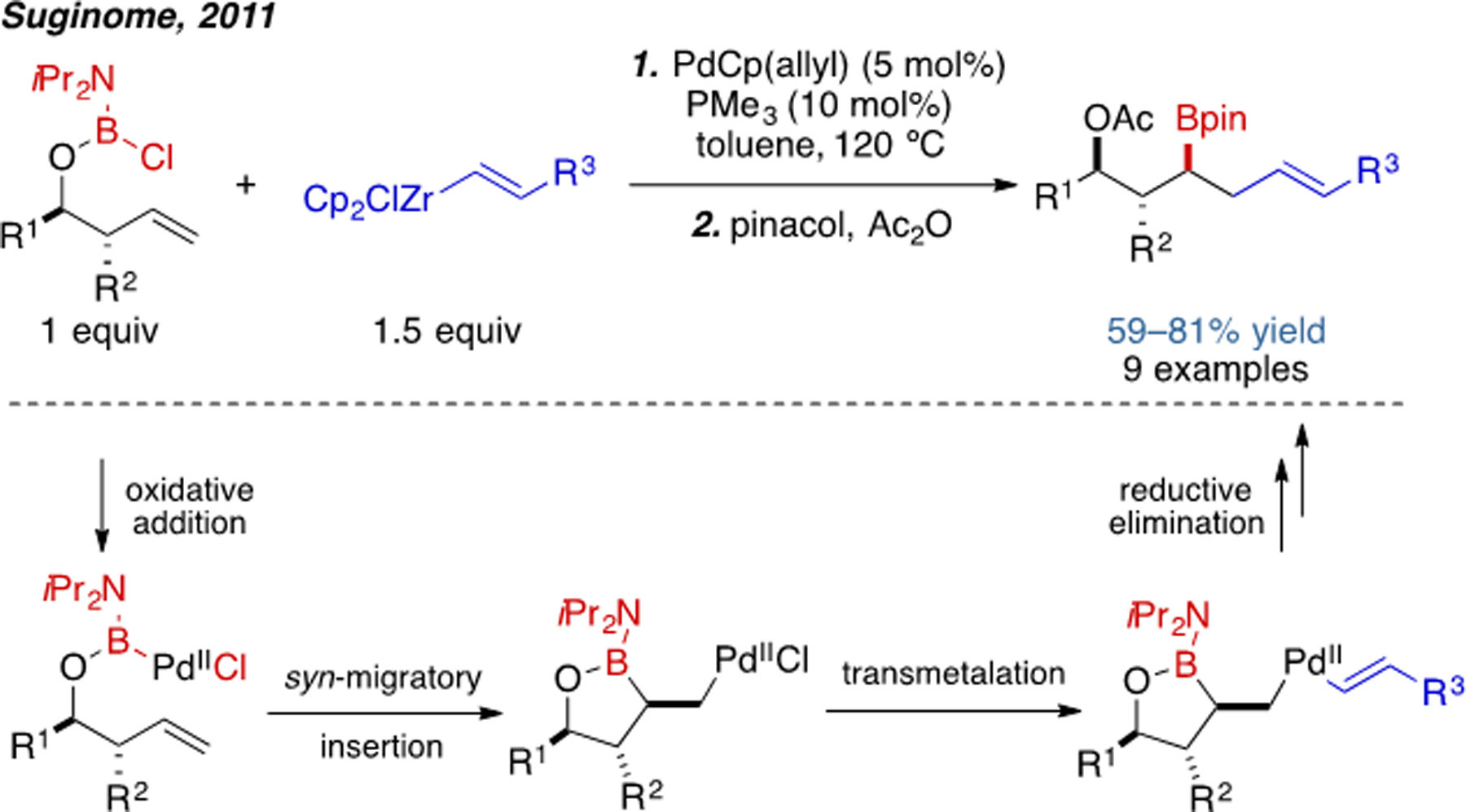
Pd(0)-catalyzed intramolecular vinylboration.
As mentioned above, the two canonical Pd-catalyzed alkene functionalization reactions are the Mizoroki–Heck reaction and Wacker reaction.[20b, 23] These two reaction modes proceed via different mechanisms, which can deliver stereoisomeric products (Scheme 12). In a Pd(0)/Pd(II) catalytic cycle, Heck-type migratory insertion across an alkene proceeds in syn-stereospecific manner to access an alkylpalladium(II) intermediate. In contrast, in a Pd(II)/Pd(0) catalytic cycle, Wacker-type nucleopalladation can take place in an anti-selective fashion via Pd(II) π-Lewis acid activation. In either case, the resulting alkylpalladium(II) species can then undergo transmetalation and C–B bond forming step proceed in a stereoretentive fashion. In contrast to Cu catalysis, where a Cu–Bpin species is first generated and reacts with alkene, the formation of C–B bond is generally the final step in a Pd-catalyzed carboboration reaction, Suginome’s 2011 precedent notwithstanding.
Scheme 12.
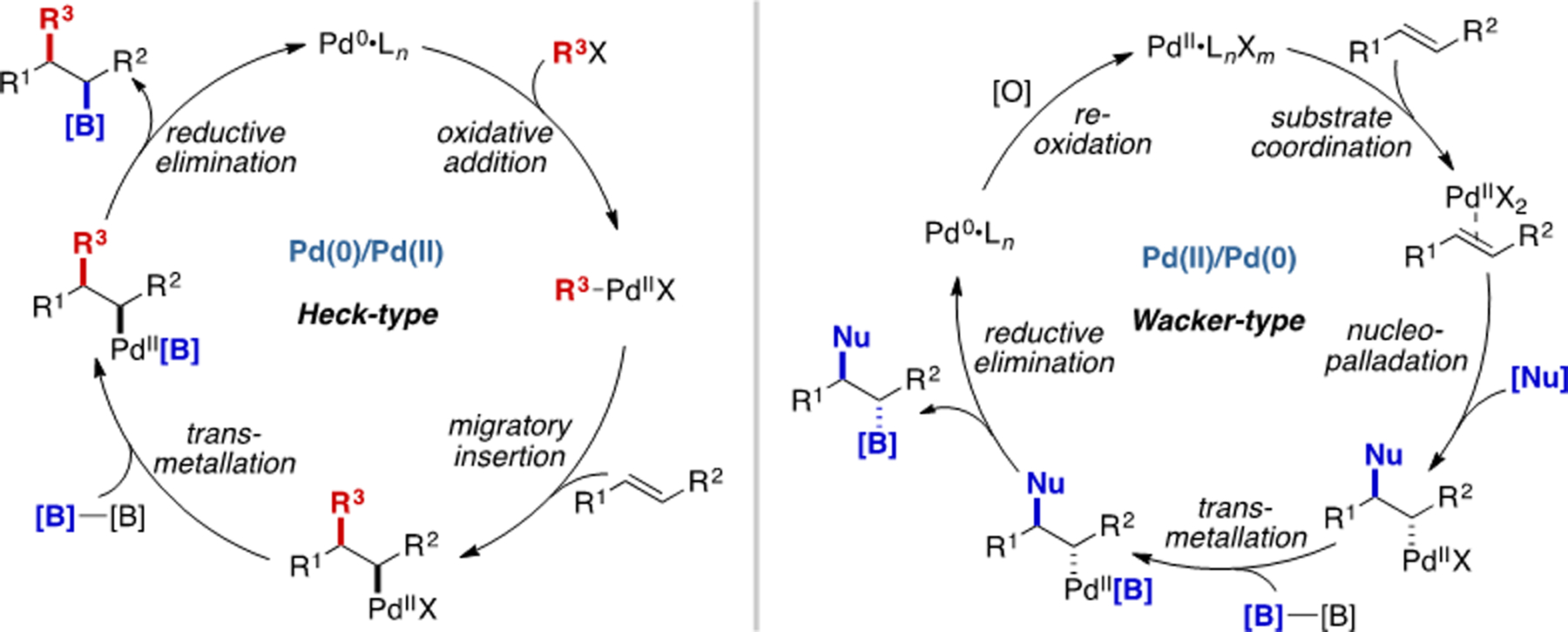
Two general modes of Pd-catalyzed alkene carboboration.
β-Hydride elimination of an alkylPd(II) complex is typically a rapid process[24] and is key elementary step in Mizoroki–Heck coupling and in the Wacker process. In the context of alkene carboboration, β-H elimination can complicate product selectivity. For example, in 2015 the group of Toste reported an example of alkene 1,1-arylboration by taking advantage of β-hydride elimination to generate a stabilized π-benzyl intermediate (Scheme 13).[25] To achieve high selectivity for 1,2-carboboration, β-hydride elimination is an undesired pathway and should be suppressed. Several strategies have been employed to prevent β-hydride elimination in alkene carboboration (vide infra). One is to use a geometrically constrained alkene substrate that does not have an appropriate β-hydrogen atom to eliminate. Another strategy is to use alkene substrates that can form electronically stabilized polyhaptic organopalladium(II) intermediates, such as allyl- or benzylpalladium(II) species. Lastly, substrates containing Lewis basic directing groups, which form palladacycle intermediates upon carbopalladation or migratory insertion, have been successfully employed.
Scheme 13.
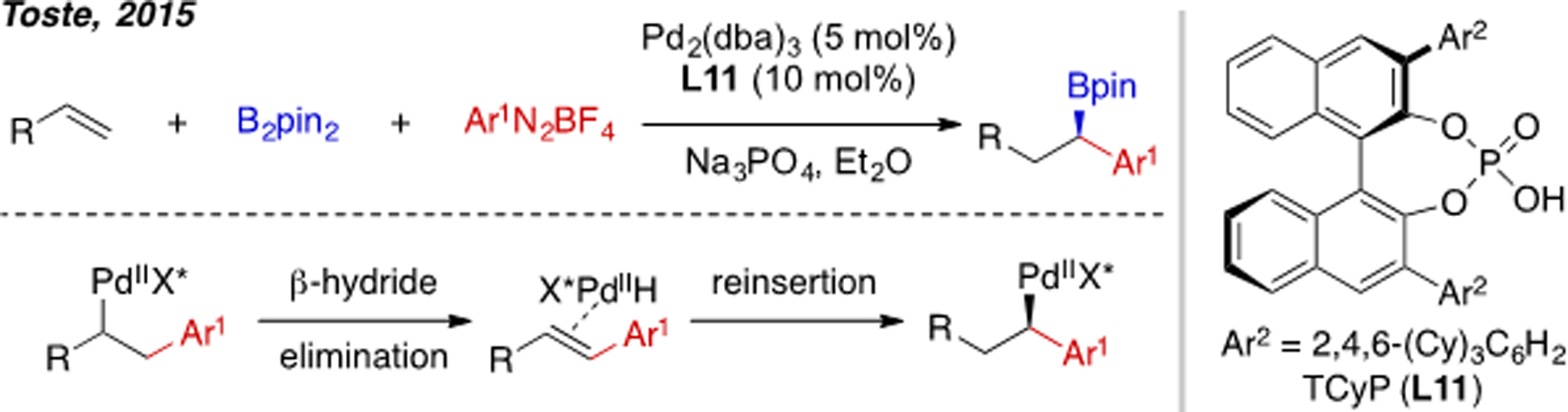
Pd(0)-catalyzed alkene 1,1-arylboration.
2.2.1. Carboboration Initiated by Heck-type Migratory Insertion
Heck-type migratory insertion is a robust means of delivering aryl and vinyl groups in a syn-specific manner. Several examples of 1,2-arylboration via Heck-type chemistry have been reported by the group of Song (eq. 4).[26] In these papers, regioselective 1,2-arylboration of vinyl arenes and strained alkenes was successfully achieved.
 |
eq. 4 |
In order to overcome problematic β-hydride elimination in alkene difunctionalization, our laboratory has developed a series of mechanistically diverse reactions using a strongly coordinating 8-aminoquinoline amide (AQ) as the directing group.[27] This directing group is beneficial for stabilizing the putative alkylPd(II) intermediates by forming a conformationally rigid palladacycle, as originally demonstrated by Daugulis in the context of C(sp3)–H activation.[28] By exploiting this strategy, our lab has recently developed a syn-carboboration reaction, where both aryl and alkenyl triflates are compatible coupling partners (Scheme 14).[29] The alkene scope is rather broad for this chemistry, including di- tri- and tetrasubstituted alkenes as suitable substrates. Interestingly, the C=C bonds of indole and benzofuran can be readily functionalized through this reaction with excellent diastereoselectivity, leading to highly substituted dearomatized heterocycles.
Scheme 14.
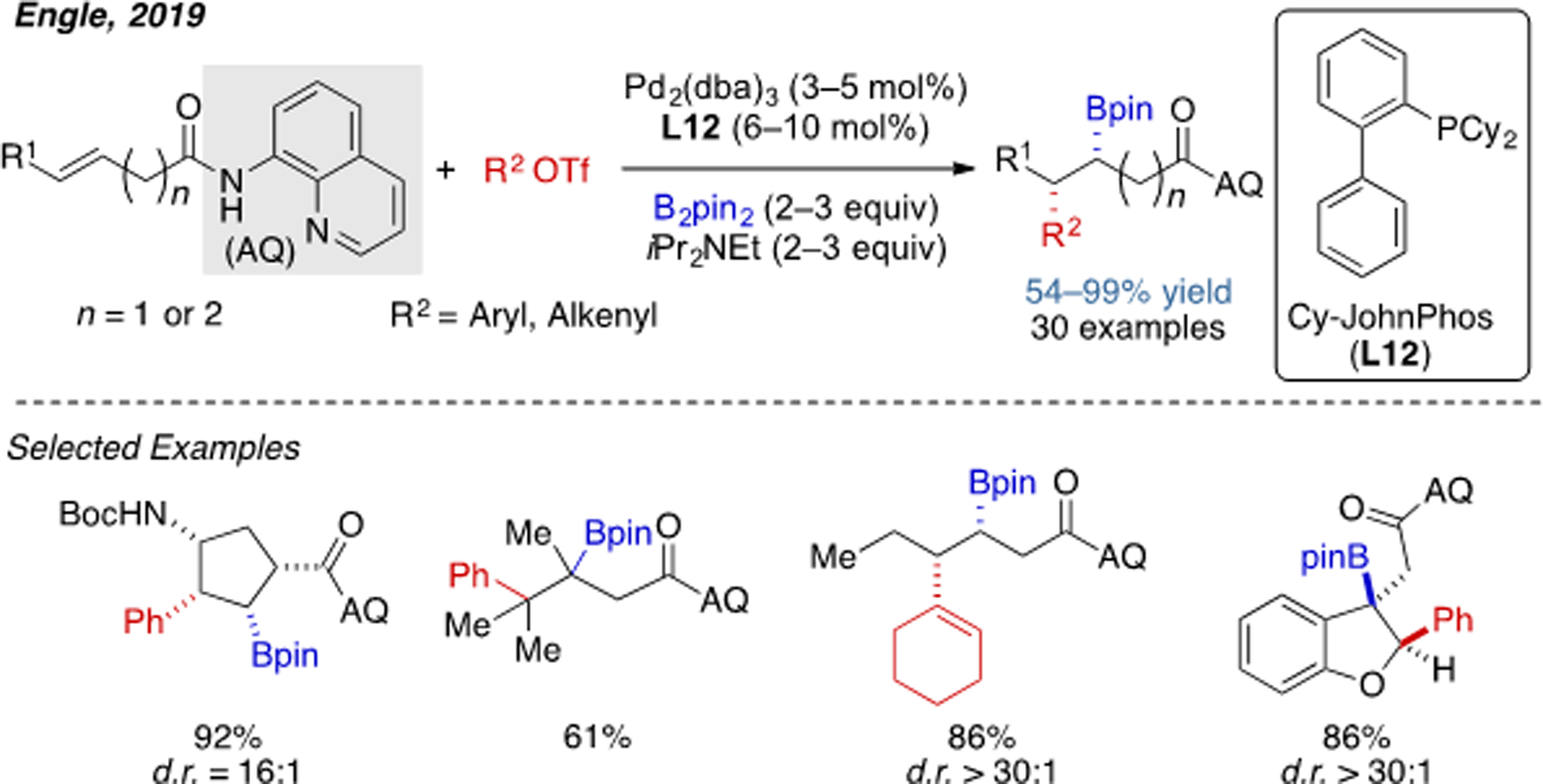
Pd(0)-catalyzed 1,2-carboboration of alkenes with AQ directing group.
As an alternative strategy to classical Heck-type chemistry through oxidative addition of an organohalide to Pd(0), Bäckvall and coworkers developed a mechanistically unique approach to access alkenylpalladium(II) intermediates by taking advantage of Pd(II)-mediated C(sp3)–H bond cleavage of substituted allenes (Scheme 15).[30] A tethered hydroxyl or carbonyl group is crucial for the desired cyclization, presumably by stabilizing the intermediates through weak coordination.
Scheme 15.
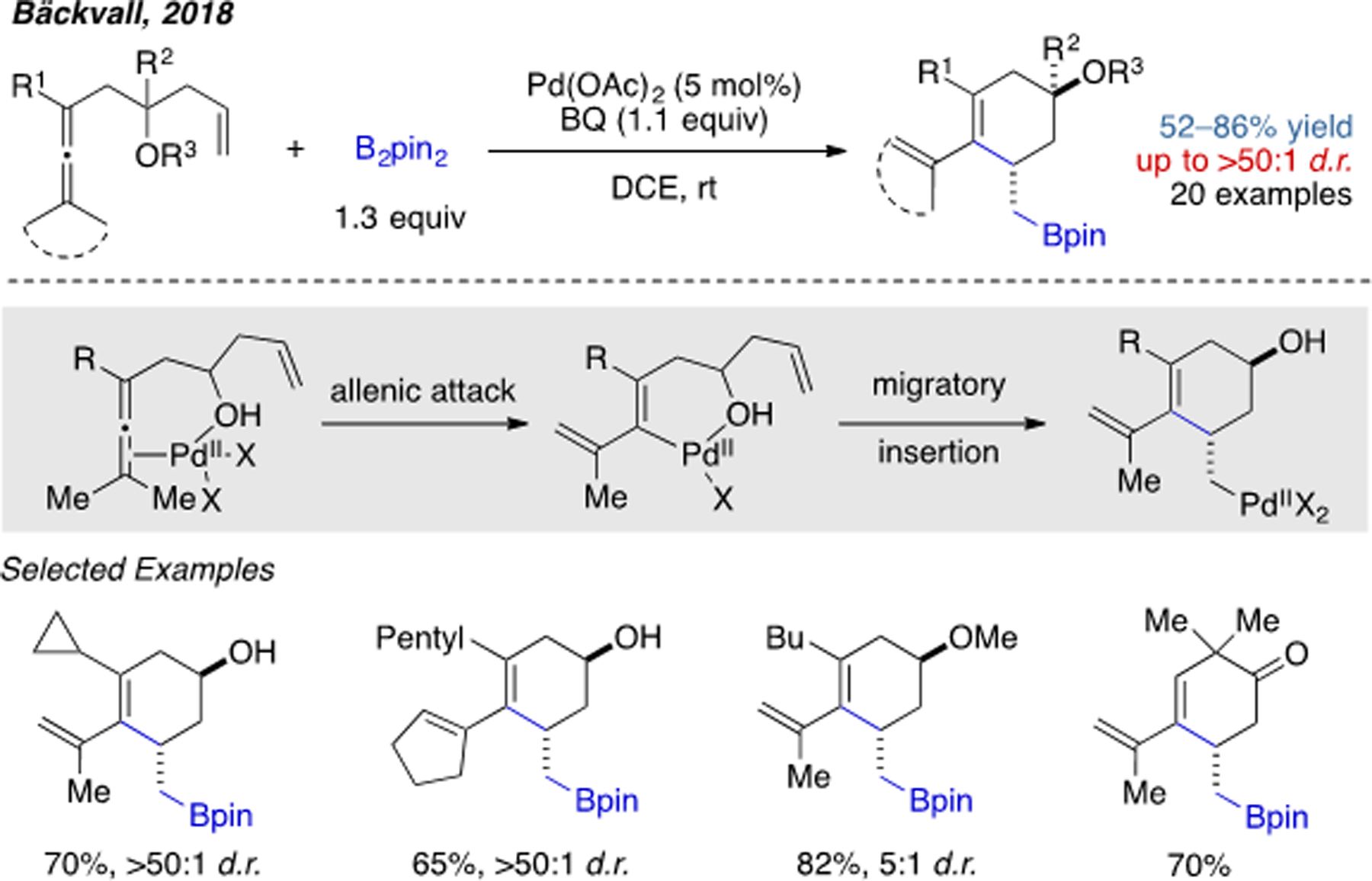
Pd(II)-catalyzed intramolecular 1,2-arylboration involving Heck-type migratory insertion.
2.2.2. Carboboration Initiated by Wacker-type Nucleopalladation
Alkene difunctionalization involving a Wacker-type mechanism has become an active research area in recent years.[23b] However, examples of Pd(II)-catalyzed 1,2-carboboration are very rare. As mentioned above, this is mainly because the undesired β-hydride elimination outcompetes the desired reaction in many cases. The first example of intermolecular (three-component) alkene carboboration using Pd(II) catalysis was reported by our research group (Scheme 16).[31] Again, a directing group strategy was key to successful execution of this chemistry. Upon anti-nucleopalladation, the resulting alkylpalladium(II) intermediate can either be a 5- or 6-membered palladacycle, which enables installation of a boron group at the β- and γ-positions with respect to the carbonyl group.
Scheme 16.
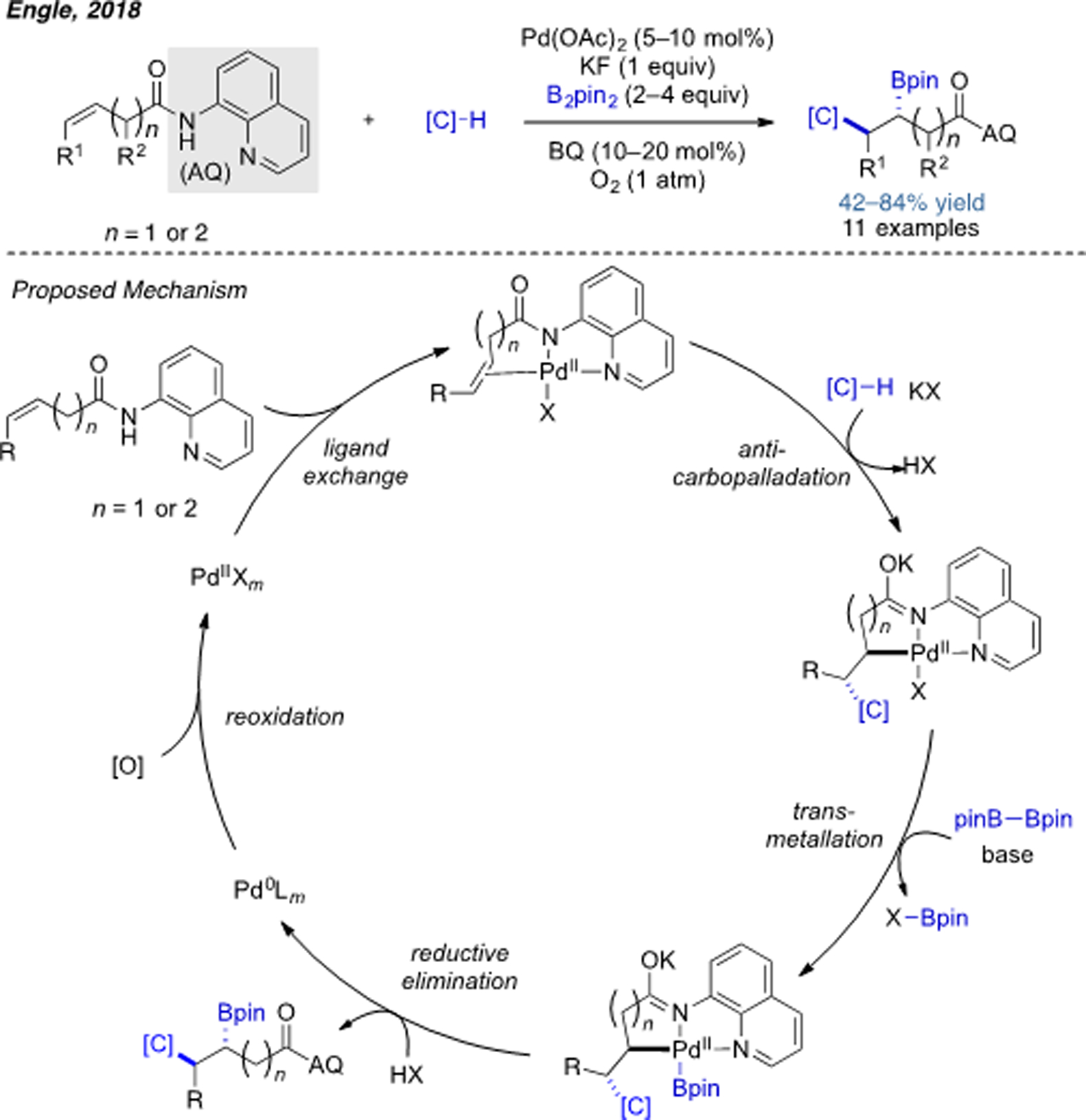
Pd(II)-catalyzed alkene 1,2-carboboration through Wacker-type mechanism.
After extensive screening of a variety of chiral ligands, we later rendered this transformation enantioselective[32a] by adopting a monodentate oxazoline ligand L13 (MOX) that had recently been described in a related report[32b] by He, Peng, and Chen (Scheme 17). Interestingly, the carboboration reaction was found to be stereoconvergent, providing the same major diastereomer from both the E- and Z-alkene substrates. This phenomenon is caused by the rapid E/Z isomerization prior to nucleopalladation, as supported by stoichiometric experiments. Shortly after our report, the group of Peng, Chen, He and coworkers also described an asymmetric carboboration method with a structurally similar ligand L14 that offered higher levels of enantioselectivity.[33] The origin of the effectiveness of this particular ligand was elucidated by DFT studies. Compared with L13, the larger aromatic ring in L14 could provide a better shield to differentiate the face selectivity of alkene.
Scheme 17.
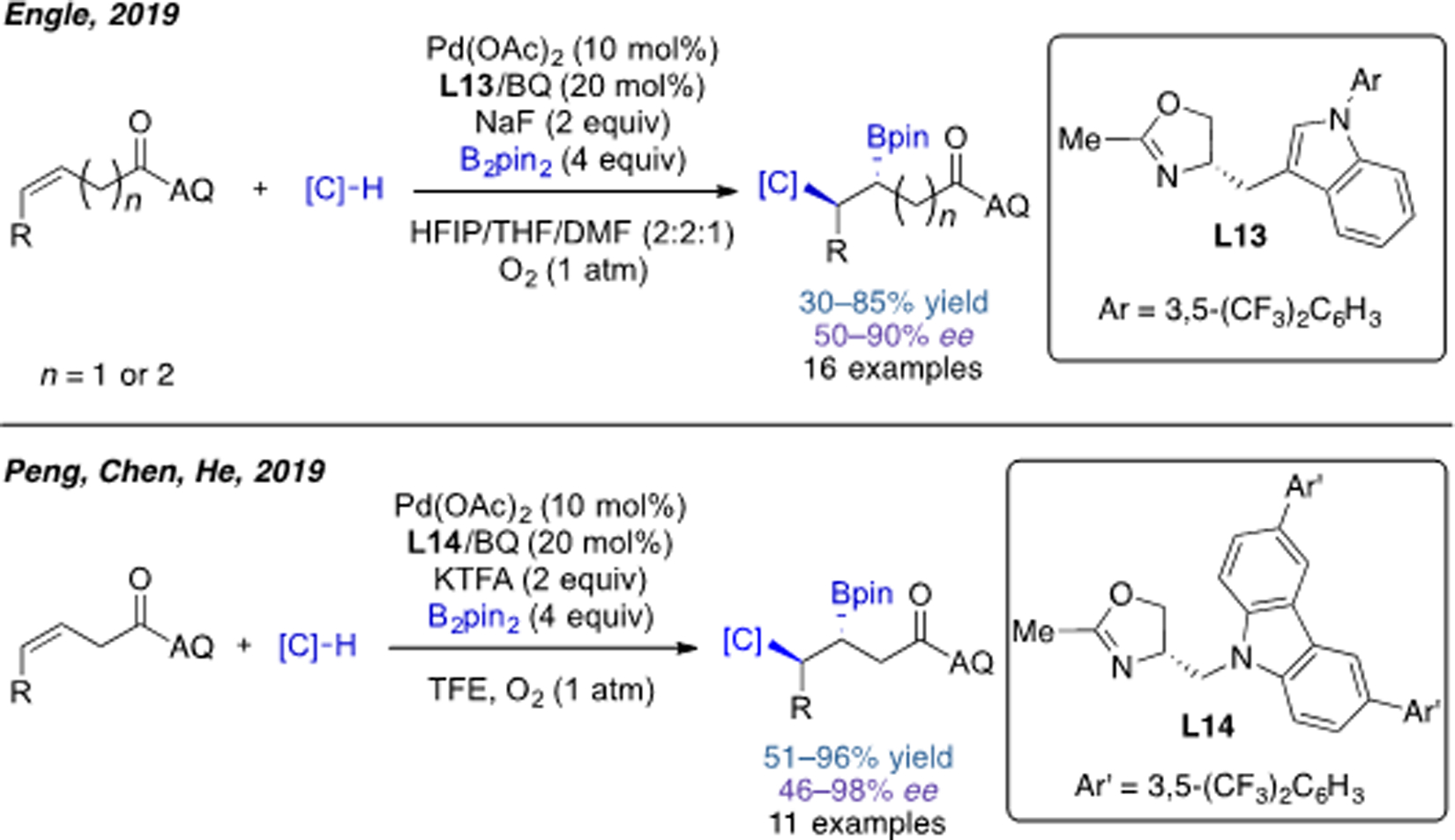
Pd(II)-catalyzed enantioselective alkene 1,2-carboboration reactions.
2.3. Ni-catalyzed Alkene Carboboration
Ni-catalyzed alkene carboboration is distinct from previously described approaches, presenting its own challenges and opportunities. In terms of mechanism, it is generally believed to be similar to Cu(I)-catalyzed carboboration. First, transmetalation of Ni catalyst with B2pin2 generates a Ni(I)–B complex, followed by syn-migratory insertion to form an alkylNi(I) species. This species can then be trapped by the electrophile, undergoing a sequence of oxidative addition/reductive elimination (Scheme 18). In terms of carbon electrophile scope, Ni-catalyzed carboboration has been shown to be capable of incorporating aryl or alkenyl groups, similar to a Pd-catalyzed Heck-type carboboration reaction. Although in principle nickel should be well suited to engage alkyl electrophiles,[34] nickel-catalyzed 1,2-alkylboration has not yet been described, likely due to the complications associated with competitive β-H elimination. Indeed, Yin has recently taken advantage of this phenomenon in a 1,1-alkylboration.[35]
Scheme 18.
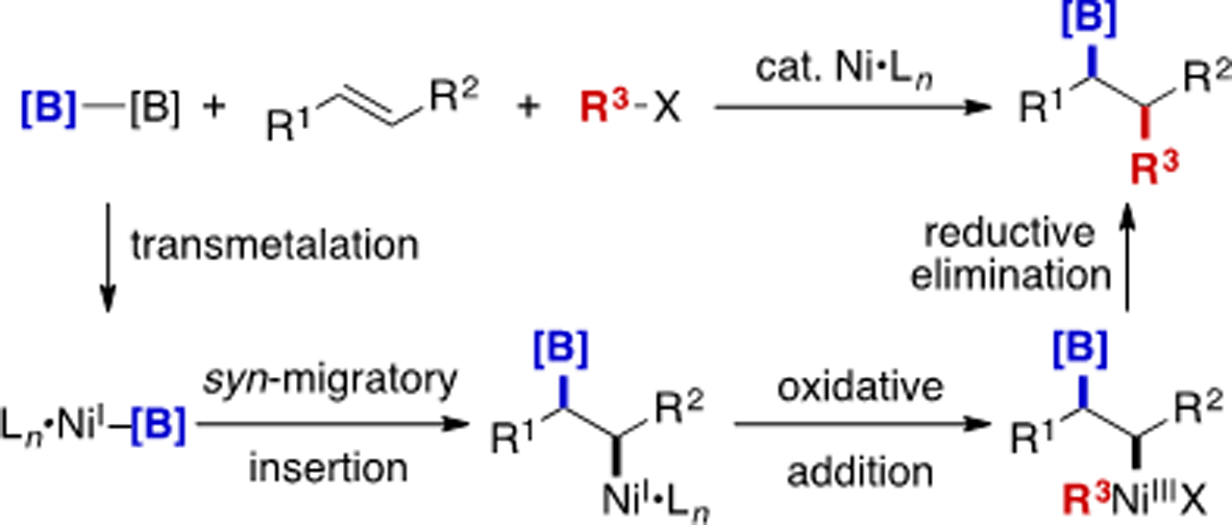
General depiction of a Ni-catalyzed alkene 1,2-carboboration.
In 2017, Brown and coworkers reported the first Ni-catalyzed alkene 1,2-arylboration reaction (Scheme 19).[36] Many simple alkene substrates are competent in this transformation, especially cyclic alkenes. Compared to an alkylPd(II) species, the key alkylNi(I) intermediate is more resistant to β-hydride elimination. Thus a directing group intended to mitigate β-hydride elimination is not required.[37] Recently, an improved set of reaction conditions with DMA (dimethylacetamide) as an additive was developed by the same group.[38] DMA serves as a weakly coordinating ligand to stabilize the alkylnickel(I) intermediate, further suppressing β-H elimination. Tri-substituted alkenes were able to participate in arylboration reaction. Detailed mechanistic studies were conducted showing that alkene insertion is the turnover-limiting step. With terminal non-conjugated alkenes, Yin has developed a 1,n-arylboration through alkylnickel chain-walking.[39]
Scheme 19.
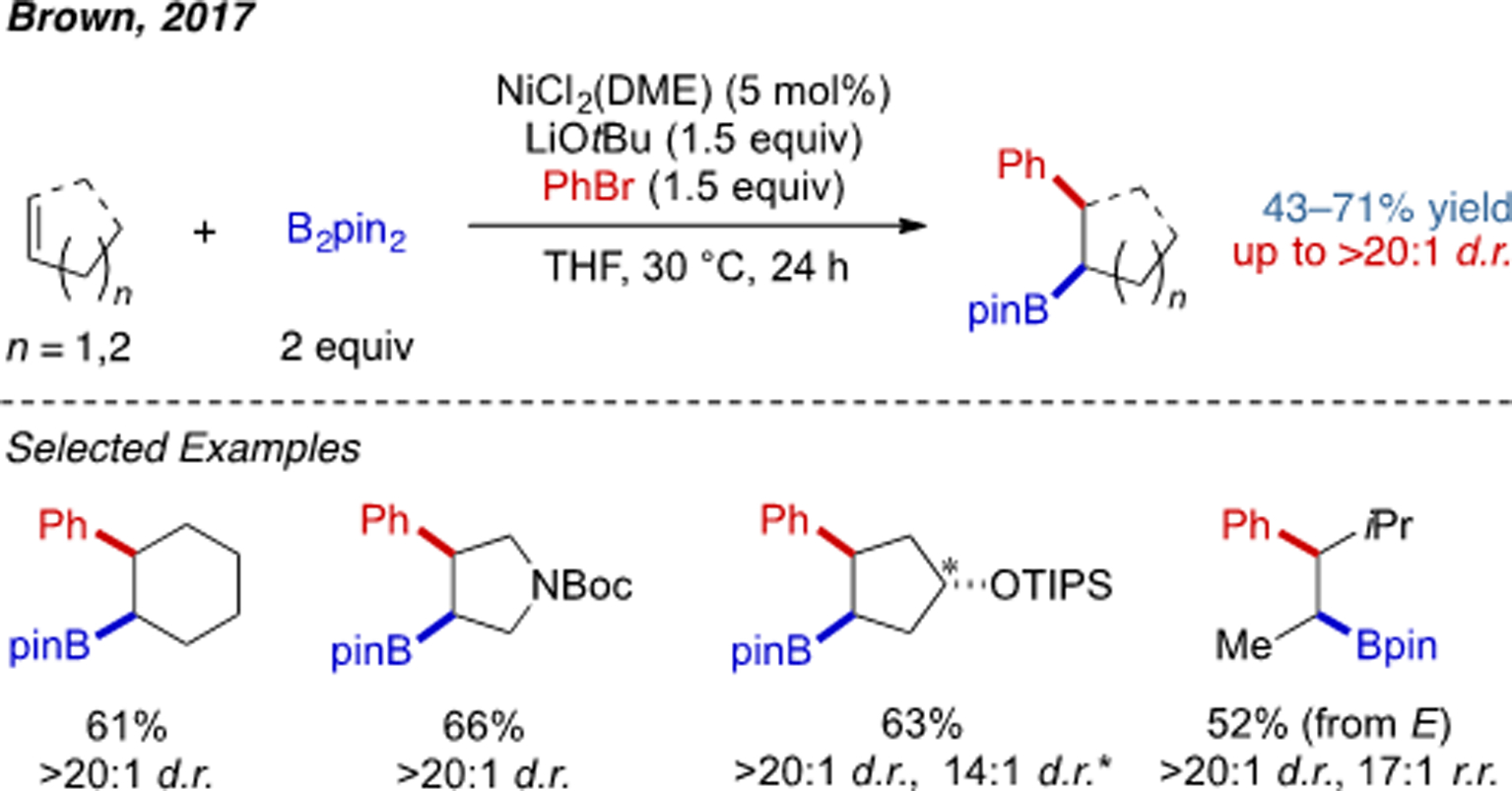
Ni-catalyzed alkene 1,2-arylboration reaction.
In 2019, Yin and coworkers developed a Ni-catalyzed 1,2-arylboration of vinyl arenes. A variety of aryl halides were compatible electrophiles in this transformation. Shortly afterwards, Brown and coworkers reported a method of 1,2-arylboration of tri- and disubstituted vinyl arenes (Scheme 20).[40,41]
Scheme 20.
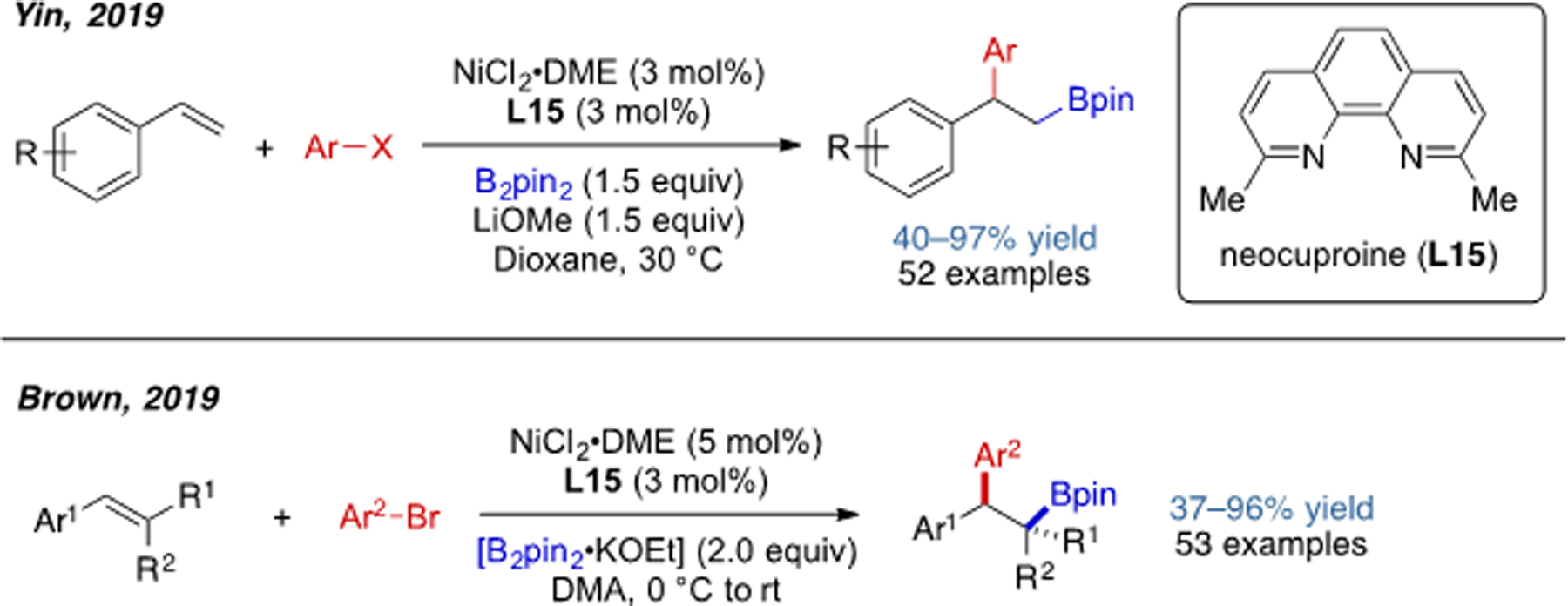
Ni-catalyzed 1,2-arylboration of vinyl arenes.
2.4. Alkene Carboboration by Cooperative Catalysis
Cooperative catalysis is a strategy to activate multiple reaction components simultaneously through use of more than one catalyst in order to achieve transformations that would otherwise be kinetically prohibited.[42] By combining multiple metal catalysts in one-pot, several historically challenging alkene 1,2-carboboration reactions have been realized. In 2014, the first examples of alkene 1,2-arylboration enabled by cooperative catalysis were published independently by the group of Semba/Nakao[43] and Brown[44] (Scheme 21). Interestingly, a Pd/Cu catalytic system with similar ligands was used in both cases. The plausible mechanism consists of two catalytic cycles. In the Pd cycle (Cycle I), an aryl Pd(II) intermediate is first formed through oxidative addition. In the meantime, Cu(I) catalyst undergoes a base-mediated transmetalation, followed by migratory insertion to generate an alkyl cuprate (Cycle II). Transmetalation between the organocuprate and Pd(II) complex, transfers the alkyl group from Cu to Pd center. Reductive elimination from the Pd(II) catalyst delivers the final product and regenerates a Pd(0) species. Through this mechanism, aryl electrophiles become suitable coupling partners in carboboration reaction, which is not trivial to achieve for Cu sole catalysis.[45] In addition, β-hydride elimination is no longer a significant issue for the putative alkyl Pd(II) intermediates, as the alkyl group is carried by Cu in the form of alkylcuprate for most of the time.
Scheme 21.
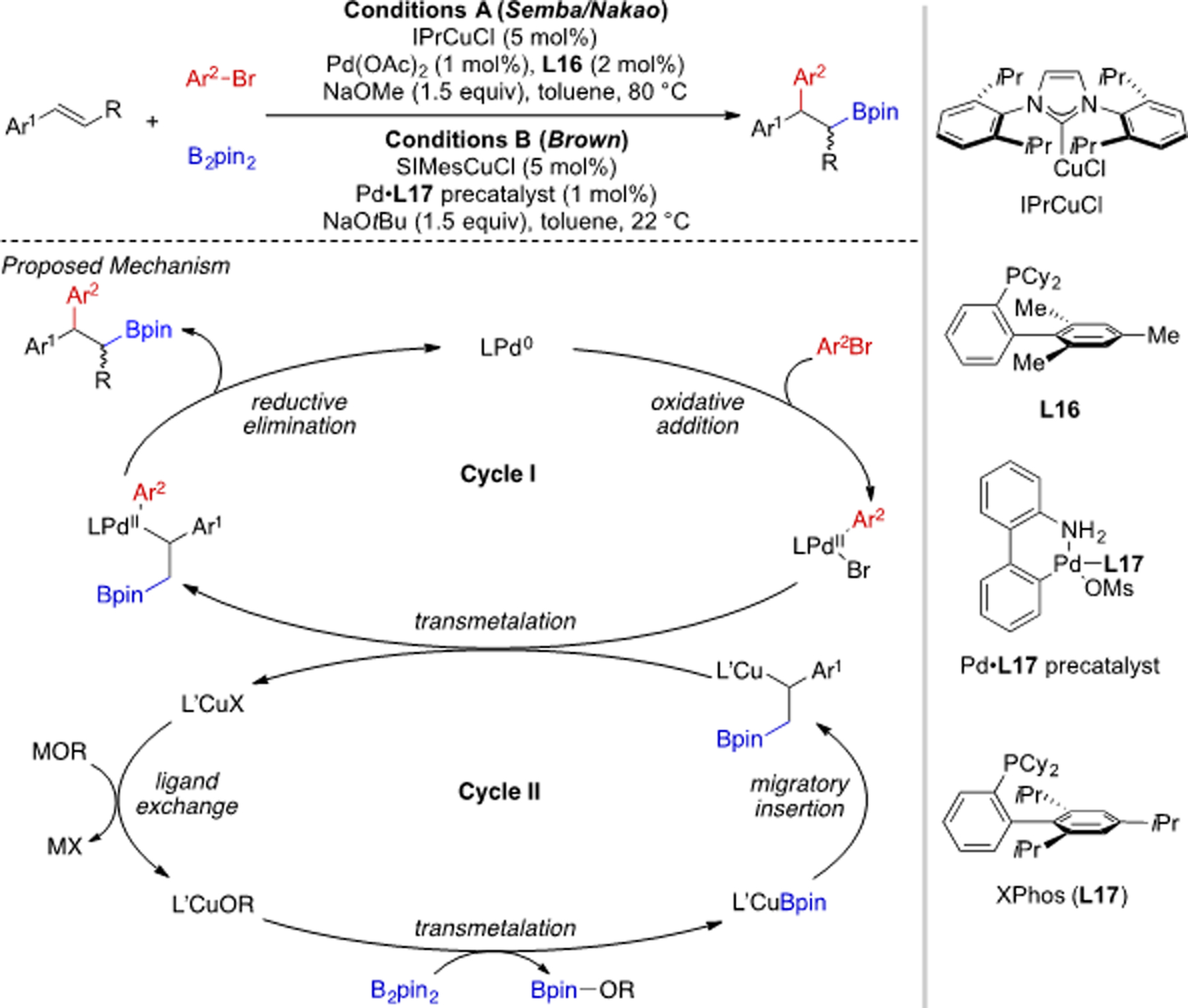
Alkene 1,2-arylboration reaction by Pd/Cu cooperative catalysis.
In 2015, Brown and coworkers found that the stereoselectivity of arylboration can be determined by the phosphine ligand (Scheme 22).[46] With Buchwald ligands, such as RuPhos (L17), the reaction is highly syn-selective. In contrast, with a trialkylphosine ligand (PiBu3), this reaction was optimized to be highly anti-selective. Different solvents also have a major impact on the stereochemical outcome. Since Cu–Bpin migratory insertion is syn-selective and Pd(II) reductive elimination is stereoretentive, a plausible mechanism is that RuPhos and THF facilitate a stereoretentive Cu/Pd transmetalation. While, PiBu3 and nonpolar solvents promote a stereoinvertive transmetalation. In terms of preparative chemistry, these complementary methods further enhance the ability to access structurally diverse alkyl boronates from common starting materials.
Scheme 22.
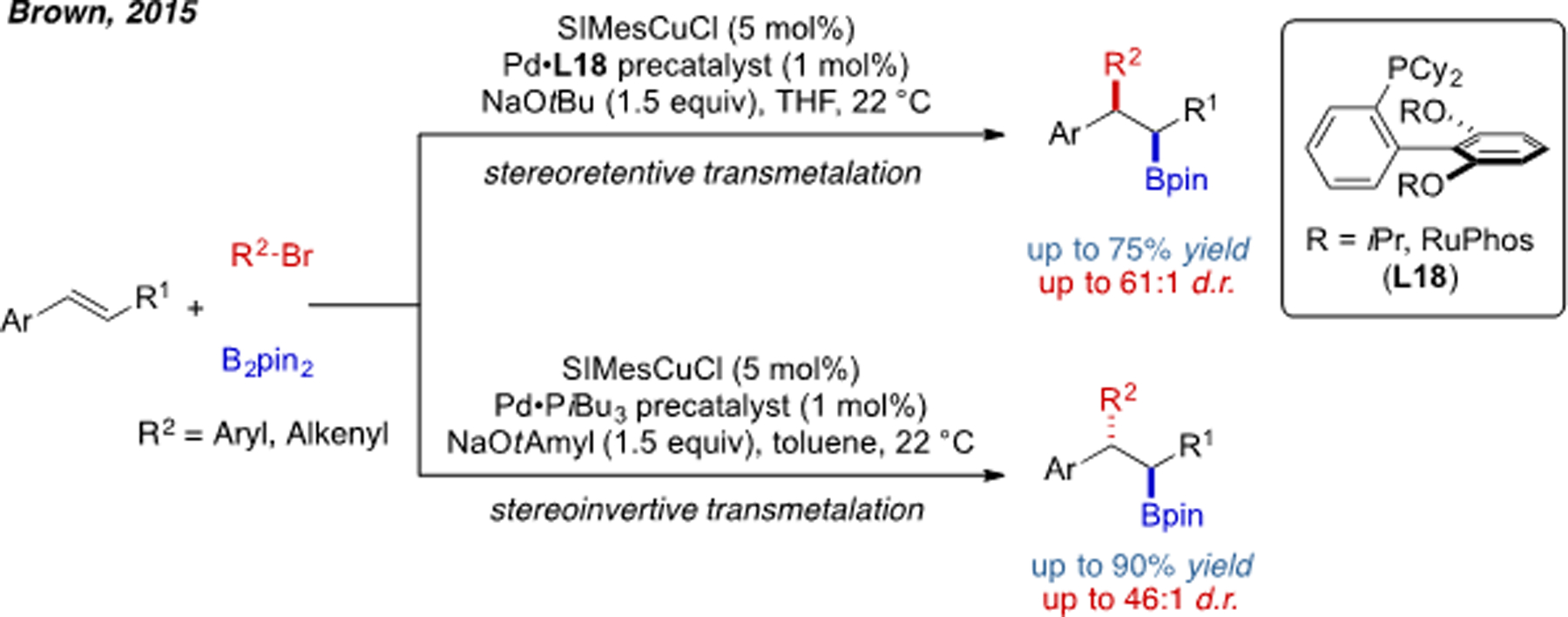
Stereodivergent alkene 1,2-arylboration reaction controlled by transmetalation.
Beyond arylboration, Pd/Cu cooperative catalysis has also been used in other types of carboboration reactions. For example, in 2015, Liao and coworkers developed the first enantioselective allylboration of vinyl arenes (eq. 5).[47] A dialkylarylphosphine containing a tethered chiral sulfoxide was found to be the optimal ligand, catalyzing the reaction in good to excellent ee.
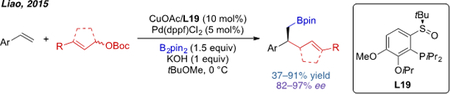 |
eq. 5 |
A different catalytic system was developed by the group of Nakao in 2016 (eq. 6).[48] They reported an arylboration of vinyl arenes via Ni/Cu cooperative catalysis. It is notable that weak electrophiles, such as aryl chlorides and tosylates were compatible, which had not been previously demonstrated in a carboboration reaction. The authors proposed that in this system the Ni catalyst plays a similar role as the Pd catalyst in the Cu/Pd systems. As discussed in the preceding section, it was later found that Ni is capable of carrying out both components of this dual system under different reaction conditions.
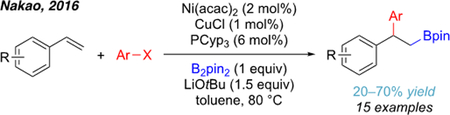 |
eq. 6 |
Dual catalytic systems are enabling in cases where each of the two metals is uniquely effective at some elementary steps but not others. However, cross-compatibility of the catalysts/ligands/reagents is a persistent challenge in such processes.
2.4. Alkene Carboboration Catalyzed by Other Transition Metals
Although Cu, Pd and Ni are the most explored transition-metal catalysts for alkene carboboration, other transition metals also hold promise to promote similar transformations. Recently, the group of Ge reported an example of cobalt-catalyzed intramolecular carboboration (Scheme 23).[49] Using Ph-BPE (L20) or JosiPhos (L6) as ligand, an excellent level of enantioselectivity was achieved. In terms of mechanism, the authors proposed that after generation of Co–H, an alkylcobalt species was formed by sequential insertion of the C≡C and C=C bonds, followed by σ-bond metathesis to install the C–B bond.
Scheme 23.
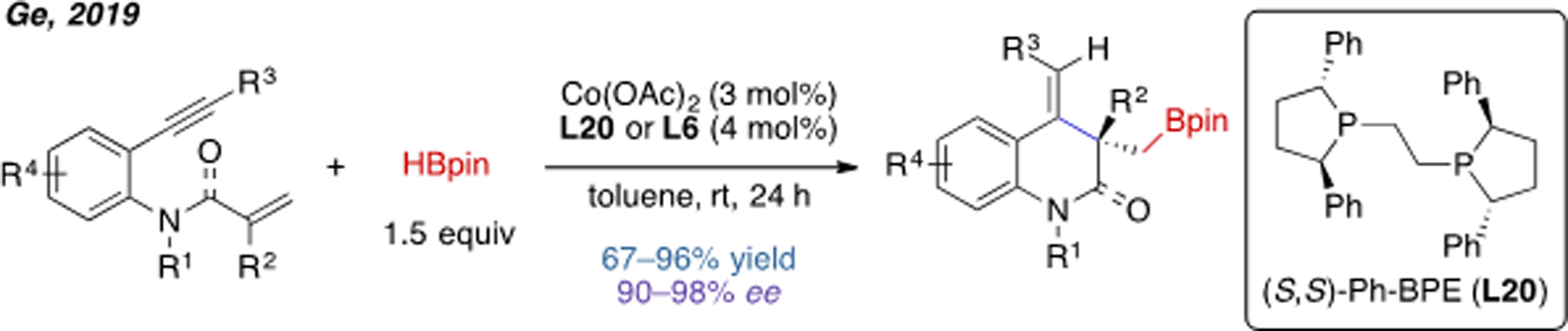
Co(II)-catalyzed intramolecular carboboration.
A Rh(III)-catalyzed intramolecular carboboration has also been achieved by Tian, Hong, Lin and coworkers (Scheme 24).[50] Mechanistically, this reaction involves initial formation of a bisboryl-Rh(III) intermediate. Next, syn-migratory insertion to the non-conjugated alkene leads to formation of an alkylrhodium intermediate that then undergoes conjugated addition to the tethered α,β-unsaturated alkene. This transformation was rendered highly enantioselective through the use of chiral NCN-pincer rhodium(III) catalysts (C1 and C2).
Scheme 24.
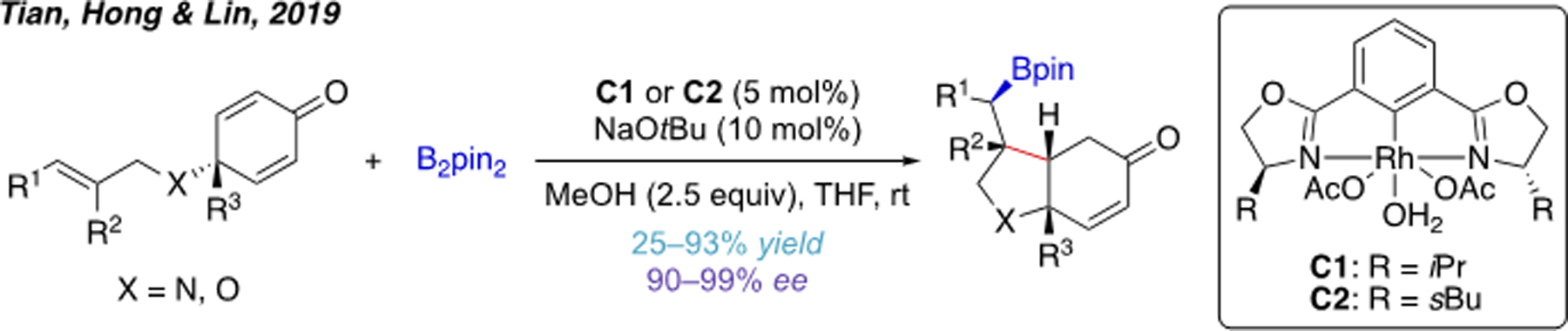
Rh(III)-catalyzed intramolecular carboboration.
3. Outlook
In this review, we highlighted representative examples to showcase the power of different approaches to 1,2-carboboration of alkenes catalyzed by Cu, Pd, and Ni. With different transition-metals as the catalyst, these reactions typically proceed by distinct mechanisms, leading to divergent regio- and stereochemical outcomes. Cu and Ni catalysts are believed to transmetalate with diboron reagents first to form a copper–boryl, which will then add across the C=C bond in a syn-fashion. In contrast to Cu and Ni catalysts, Pd catalyst typically react by first facilitating addition of the carbon moiety to alkene via syn-migratory insertion or anti-nucleopalladation depending on the conditions. On the other hand, cooperative catalysis systems have also been developed, offering more options to effect carboboration. With this expanding catalytic toolkit, a variety of carbon-based functional groups can now be installed, including alkyl, aryl, and alkenyl groups.
In order to realize increasingly general and practical alkene carboboration methods, there remain many outstanding problems to be addressed. For example, many existing reactions possess limited alkene scope. In Cu catalysis, there are almost no transformations that have been shown to be compatible with unactivated internal alkenes. In Pd catalysis, strained alkenes or styrene-type substrates have typically been required. More recent methods for non-conjugated alkenes require a directing group in proximity to the alkene to control regioselectivity and prevent the β-hydride elimination, but this directing group requires additional steps to install and remove. In Ni catalysis, although many unactivated alkenes are compatible in the reaction, the regioselectivity in this chemistry is dictated by the substitution pattern of the alkene substrate and can only be controllably switched in special cases, like with styrene substrates. In addition, enantioselective variants of Ni-catalyzed 1,2-carboboration remain unknown. While these are daunting challenges, the impressive progress of the field during the past ten years, augurs favorably for significant progress on these and other issues in the near future.
Scheme 3.
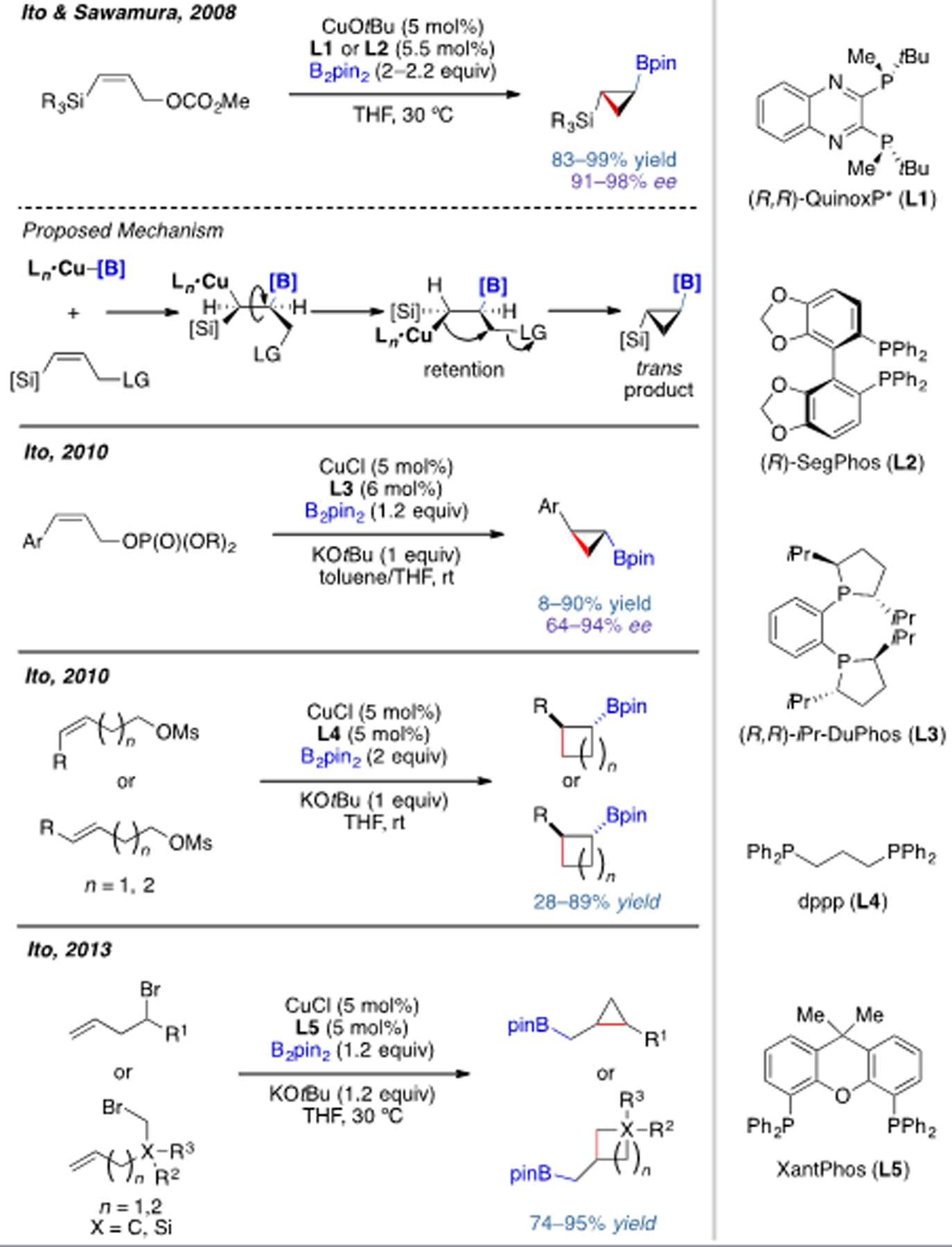
Cu(I)-catalyzed intramolecular carboboration with halides or pseudohalides as leaving groups.
Acknowledgements
We gratefully acknowledge financial support from the National Institutes of Health (5R35GM125052-02). We further thank Bristol-Myers Squibb (Graduate Fellowship to Z.L.; Unrestricted Grant to K.M.E.) and the Alfred P. Sloan Foundation (Fellowship to K.M.E.). We thank Joseph Derosa and Lucas J. Oxtoby for proofreading the manuscript.
Biographies
Zhen Liu was born in Xuzhou, Jiangsu Province, China. Zhen earned his B.S. degree from Peking University, where he carried out undergraduate research in the laboratory of Prof. Jianbo Wang. He is currently a Bristol-Myers Squibb Graduate Fellow in the Engle lab, working on transition-metal-catalyzed functionalization of alkenes and alkynes.

Dr. Yang Gao received his B.S. degree in Chemistry from Nanjing University. In 2018, he earned a Ph.D. in chemistry under the direction of Prof. Alan S. Goldman at Rutgers University, where he studied pincer iridium complexes and their applications in catalysis. He then joined the lab of Prof. Keary M. Engle as a postdoctoral associate. His current research interests are in transition-metal-catalyzed difunctionalization of alkenes and alkynes, and mechanistic interrogation of such reactions.

Tian Zeng was born and raised in Wuhan, China. She earned her B.S. degree in Chemistry from University of California, San Diego. During her college career she spent two years in the lab of Prof. Keary M. Engle studying Pd-catalyzed difunctionalization of alkenes. Tian is currently pursuing a doctorate degree at California Institute of Technology under the tutelage of Prof. Maxwell J. Robb.

Prof. Keary received his B.S. from the University of Michigan with Prof. Adam J. Matzger in 2007 and spent the following year as a Fulbright Scholar at the MPI für Kohlenforschung with Prof. Manfred T. Reetz. In 2013, he received a Ph.D. in Chemistry from Scripps Research with Prof. Jin-Quan Yu and a D.Phil. in Biochemistry from the University of Oxford with Profs. Véronique Gouverneur and John M. Brown. After a two-year appointment as an NIH Postdoctoral Fellow with Prof. Robert H. Grubbs at Caltech, Keary began his independent career at Scripps Research in 2015, where his research lab focuses on selective catalytic functionalization of alkenes and alkynes. He is a Bristol-Myers Squibb Unrestricted Grant recipient (2018), an Alfred P. Sloan Fellow (2019), and a Camille Dreyfus Teacher-Scholar (2019).

References
- [1].a) Pelter A, Smith K, Brown HC, in Borane Reagents, Academic Press, New York: 1988; [Google Scholar]; b) Miyaura N, Suzuki A, Chem. Rev 1995, 95, 2457–2483; [Google Scholar]; c) Sanford C, Aggarwal VK, Chem. Commun 2017, 53, 5481–5494. [DOI] [PubMed] [Google Scholar]
- [2].a) Brown HC, Zweifel G, J. Am. Chem. Soc 1959, 81, 247; [Google Scholar]; b) Burgess K, Ohlmeyer MJ, Chem. Rev 1991, 91, 1179–1191; [Google Scholar]; c) Thomas SP, Aggarwal VK, Angew. Chem 2009, 121, 1928–1930; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2009, 48, 1896–1898. [DOI] [PubMed] [Google Scholar]
- [3].a) Yin G, Mu X, Liu G, Acc. Chem. Res 2016, 49, 2413–2423. [DOI] [PubMed] [Google Scholar]; b) Lan X-W, Wang N-X, Xing Y, Eur. J. Org. Chem 2017, 5821–5851; [Google Scholar]; c) Derosa J, Tran VT, van der Puyl VA, Engle KM, Aldrichimica Acta 2018, 51, 21–32; [Google Scholar]; d) Zhang J-S, Liu L, Chen T, Han L-B, Chem. Asian J 2018, 13, 2277–2291. [DOI] [PubMed] [Google Scholar]
- [4].Shimizu Y, Kanai M, Tetrahedron Lett 2014, 55, 3727–3737. [Google Scholar]
- [5].For examples of transition-metal-free carboboration:; a) Ren S-C, Zhang F-L, Qi J, Huang Y-S, Xu A-Q, Yan H-Y, Wang Y-F, J. Am. Chem. Soc 2017, 139, 6050–6053; [DOI] [PubMed] [Google Scholar]; b) Sanzone JR, Hu CT, Woerpel KA, J. Am. Chem. Soc 2017, 139, 8404–8407; [DOI] [PubMed] [Google Scholar]; c) Cheng Y, Mück-Lichtenfeld C, Studer A, J. Am. Chem. Soc 2018, 140, 6221–6225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].For initial reports of catalytic hydroboration via Cu-Bpin species, see:; a) Ito H, Yamanaka H, Tateiwa J.-i., Hosomi A, Tetrahedron Lett 2000, 41, 6821–6825; [Google Scholar]; b) Takahashi K, Ishiyama T, Miyaura N, Chem. Lett 2000, 29, 982–983. [Google Scholar]
- [7].a) Tang C-T, Zhang Z-Q, Tajuddin H, Wu C-C, Liang J, Liu J-H, Fu Y, Czyzewska M, Steel PG, Marder TB, Liu L, Angew. Chem 2012, 124, 543–547; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2012, 51, 528–532; [DOI] [PubMed] [Google Scholar]; b) Zhou Y, You W, Smith KB, Brown MK, Angew. Chem 2014, 126, 3543–3547; [Google Scholar]; Angew. Chem. Int. Ed 2014, 53, 3475–3479. [DOI] [PubMed] [Google Scholar]
- [8].a) Ito H, Kosaka Y, Nonoyama K, Sasaki Y, Sawamura M, Angew. Chem 2008, 120, 7534–7537; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2008, 47, 7424–7427; [DOI] [PubMed] [Google Scholar]; b) Ito H, Toyoda T, Sawamura M, J. Am. Chem. Soc 2010, 132, 5990–5992; [DOI] [PubMed] [Google Scholar]; c) Zhong C, Kunii S, Kosaka Y, Sawamura M, Ito H, J. Am. Chem. Soc 2010, 132, 11440–11442; [DOI] [PubMed] [Google Scholar]; d) Kubota K, Yamamoto E, Ito H, J. Am. Chem. Soc 2013, 135, 2635–2640. [DOI] [PubMed] [Google Scholar]
- [9].Iwamoto H, Akiyama S, Hayama K, Ito H, Org. Lett 2017, 19, 2614–2617. [DOI] [PubMed] [Google Scholar]
- [10].a) Burns AR, González JS, Lam HW, Angew. Chem 2012, 124, 10985–10989; [Google Scholar]; Angew. Chem. Int. Ed 2012, 51, 10827–10831. [DOI] [PubMed] [Google Scholar]; Recently, a similar transformation that is initiated by borocupration of styrenes has been reported, see:; b) Zheng P, Han X, Hu J, Zhao X, Xu T, Org. Lett 2019, 21, 6040–6044. [DOI] [PubMed] [Google Scholar]
- [11].Whyte A, Burton KI, Zhang J, Lautens M, Angew. Chem 2018, 130, 14123–14126; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2018, 57, 13927–13930. [DOI] [PubMed] [Google Scholar]
- [12].Kaur M, Singh M, Chadha N, Silakari O, Eur. J. Med. Chem 2016, 123, 858–894. [DOI] [PubMed] [Google Scholar]
- [13].a) Yoshida H, Kageyuki I, Takaki K, Org. Lett 2013, 15, 952–955. [DOI] [PubMed] [Google Scholar]; For similar papers on Cu(I)-catalyzed alkylboration of vinyl arenes or strained alkenes, see:; b) Kageyuki I, Yoshida H, Takaki K, Synthesis 2014, 46, 1924–1932; [Google Scholar]; c) Parra A, López A, Díaz-Tendero S, Amenós L, Ruano JLG, Tortosa M, Synlett 2015, 26, 494–500; [Google Scholar]; d) Kageyuki I, Osaka I, Takaki K, Yoshida H, Org. Lett 2017, 19, 830–833; [DOI] [PubMed] [Google Scholar]; e) Kim N, Han JT, Ryu DH, Yun J, Org. Lett 2017, 19, 6144–6147. [DOI] [PubMed] [Google Scholar]
- [14].Su W, Gong T-J, Lu X, Xu M-Y, Yu C-G, Xu Z-Y, Yu H-Z, Xiao B, Fu Y, Angew. Chem 2015, 127, 13149–13153; [Google Scholar]; Angew. Chem. Int. Ed 2015, 54, 12957–12961. [DOI] [PubMed] [Google Scholar]
- [15].Meng F, Haeffner F, Hoveyda AH, J. Am. Chem. Soc 2014, 136, 11304–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Green JC, Joannou MV, Murray SA, Zanghi JM, Meek SJ, ACS Catal 2017, 7, 4441–4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Butcher TW, McClain EJ, Hamilton TG, Perrone TM, Kroner KM, Donohoe GC, Akhmedov NG, Petersen JL, Popp BV, Org. Lett 2016, 18, 6428–6431. [DOI] [PubMed] [Google Scholar]
- [18].a) Huang Y, Smith KB, Brown MK, Angew. Chem 2017, 129, 13499–13503; [Google Scholar]; Angew. Chem. Int. Ed 2017, 56, 13314–13318. [DOI] [PMC free article] [PubMed] [Google Scholar]; For an example of borocyanation of 1,3-dienes, see:; b) Jia T, He Q, Ruscoe RE, Pulis AP, Procter DJ, Angew. Chem 2018, 130, 11475–11479; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2018, 57, 11305–11309. [DOI] [PubMed] [Google Scholar]
- [19].Smith KB, Huang Y, Brown MK, Angew. Chem 2018, 130, 6254–6257; [Google Scholar]; Angew. Chem. Int. Ed 2018, 57, 6146–6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].For representative reviews, see:; a) Nicolaou KC, Bulger PG, Sarlah D, Angew. Chem 2005, 117, 4516–4563; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2005, 44, 4442–4489; [DOI] [PubMed] [Google Scholar]; b) Johansson Seechurn CCC, Kitching MO, Colacot TJ, Snieckus V, Angew. Chem 2012, 124, 5150–5174; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2012, 51, 5062–5085. [DOI] [PubMed] [Google Scholar]
- [21].For selected examples, see:; a) Ishiyama T, Murata M, Miyaura N, J. Org. Chem 1995, 60, 7508–7510; [Google Scholar]; b) Murata M, Oyama T, Watanabe S, Masuda Y, J. Org. Chem 2000, 65, 164–168; [DOI] [PubMed] [Google Scholar]; c) Joshi-Pangu A, Ma X, Diane M, Iqbal S, Kribs RJ, Huang R, Wang C-Y, Biscoe MR, J. Org. Chem 2012, 77, 6629–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Daini M, Suginome M, J. Am. Chem. Soc 2011, 133, 4758–4761. [DOI] [PubMed] [Google Scholar]
- [23].For a representative review on the Mizoroki-Heck reaction, see:; a) Beletskaya IP, Cheprakov AV, Chem. Rev 2000, 100, 3009–3066. [DOI] [PubMed] [Google Scholar]; For a review on Wacker-type alkene functionalization:; b) McDonald RI, Liu G, Stahl SS, Chem. Rev 2011, 111, 2981–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Lu X, Top. Catal 2005, 35, 73–86. [Google Scholar]; For a mechanistic study, see:; b) Shultz LH, Brookhart M, Organometallics 2011, 20, 3975–3982. [Google Scholar]
- [25].Nelson HM, Williams BD, Miró J, Toste FD, J. Am. Chem. Soc 2015, 137, 3213–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Yang K, Song Q, J. Org. Chem 2016, 81, 1000–1005; [DOI] [PubMed] [Google Scholar]; b) Yang K, Song Q, Org. Lett 2016, 18, 5460–5463. [DOI] [PubMed] [Google Scholar]
- [27].a) Gurak JA Jr., Yang KS, Liu Z, Engle KM, J. Am. Chem. Soc 2016, 138, 5805–5808; [DOI] [PubMed] [Google Scholar]; b) Yang KS, Gurak JA Jr., Liu Z, Engle KM, J. Am. Chem. Soc 2016, 138, 14705–14712; [DOI] [PubMed] [Google Scholar]; c) Liu Z, Zeng T, Yang KS, Engle KM, J. Am. Chem. Soc 2016, 138, 15122–15125; [DOI] [PubMed] [Google Scholar]; d) Derosa J, Tran VT, Boulous MN, Chen JS, Engle KM, J. Am. Chem. Soc 2017, 139, 10657–10660; [DOI] [PubMed] [Google Scholar]; e) Zeng T, Liu Z, Schmidt MA, Eastgate MD, Engle KM, Org. Lett 2018, 20, 3853–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].For a representative review, see:; Daugulis O, Roane J, Tran LD, Acc. Chem. Res 2015, 48, 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liu Z, Chen J, Li X, Gao Y, Coombs JR, Goldfogel MJ, Engle KM, ChemRxiv 2019, DOI: 10.26434/chemrxiv.8865143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Posevins D, Qiu Y, Bäckvall J-E, J. Am. Chem. Soc 2018, 140, 3210–3214. [DOI] [PubMed] [Google Scholar]
- [31].Liu Z, Ni H-Q, Zeng T, Engle KM, J. Am. Chem. Soc 2018, 140, 3223–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].a) Liu Z, Li X, Zeng T, Engle KM, ACS Catal 2019, 9, 3260–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]; For the first report using the MOX ligand scaffold in Pd(II)-catalyzed AQ-directed alkene functionalization, see:; b) Wang H, Bai Z, Jiao T, Deng Z, Tong H, He G, Peng Q, Chen G, J. Am. Chem. Soc 2018, 140, 3542–3546. [DOI] [PubMed] [Google Scholar]
- [33].Bai Z, Zheng S, Bai Z, Song F, Wang H, Peng Q, Chen G, He G, ACS Catal 2019, 9, 6502–6509. [Google Scholar]
- [34].For selected examples, see:; a) Saito B, Fu GC, J. Am. Chem. Soc 2007, 129, 9602–9603; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) García-Domínguez A, Li Z, Nevado C, J. Am. Chem. Soc 2017, 139, 6835–6838; [DOI] [PubMed] [Google Scholar]; c) Derosa J, van der Puyl VA, Tran VT, Liu M, Engle KM, Chem. Sci 2018, 9, 5278–5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Li Y, Pang H, Wu D, Li Z, Wang W, Wei H, Fu Y, Yin G, Angew. Chem 2019, 131, 8964–8968; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2019, 58, 8872–8876. [DOI] [PubMed] [Google Scholar]
- [36].Logan KM, Sardini SR, White SD, Brown MK, J. Am. Chem. Soc 2018, 140, 159–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Xu H, White PB, Hu C, Diao T, Angew. Chem 2017, 129, 1557–1560; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2017, 56, 1535–1538. [Google Scholar]
- [38].Sardini SR, Lambright AL, Trammel GL, Omer HM, Liu P, Brown MK, J. Am. Chem. Soc 2019, 141, 9391–9400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang W, Ding C, Li Y, Li Z, Li Y, Peng L, Yin G Angew. Chem 2019, 131, 4660–4664; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2019, 58, 4612–4616. [DOI] [PubMed] [Google Scholar]
- [40].Wang W, Ding C, Pang H, Yin G, Org. Lett 2019, 21, 3968–3971. [DOI] [PubMed] [Google Scholar]
- [41].Recently, Brown and coworkers reported a nickel-catalyzed arylboration of internal alkenylarenes:; Chen L-A, Lear AR, Gao P, Brown MK, Angew. Chem 2019, DOI: 10.1002/ange.20190486; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2019, DOI: 10.1002/anie.201904861. [DOI] [Google Scholar]
- [42].For selected reviews on cooperative catalysis, see:; a) Allen AE, MacMillan DWC, Chem. Sci 2012, 3, 633–658; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Inamdar SM, Shinde VS, Patil NT, Org. Biomol. Chem 2015, 13, 8116–8162. [DOI] [PubMed] [Google Scholar]
- [43].Semba K, Nakao Y, J. Am. Chem. Soc 2014, 136, 7567–7570. [DOI] [PubMed] [Google Scholar]
- [44].Smith KB, Logan KM, You W, Brown MK, Chem. Eur. J 2014, 20, 12032–12036. [DOI] [PubMed] [Google Scholar]
- [45].For examples of enantioselective 1,2-arylboration via Pd/Cu cooperative catalysis, see:; a) Logan KM, Brown MK, Angew. Chem 2017, 129, 869–873; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2017, 56, 851–855; [Google Scholar]; b) Chen B, Cao P, Yin X, Liao Y, Jiang L, Ye J, Wang M, Liao J, ACS Catal 2017, 7, 2425–2429. [Google Scholar]
- [46].Logan KM, Smith KB, Brown MK, Angew. Chem 2015, 127, 5317–5320; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2015, 54, 5228–5231. [DOI] [PubMed] [Google Scholar]
- [47].Jia T, Cao P, Wang B, Lou Y, Yin X, Wang M, Liao J, J. Am. Chem. Soc 2015, 137, 13760–13763. [DOI] [PubMed] [Google Scholar]
- [48].Semba K, Ohtagaki Y, Nakao Y, Org. Lett 2016, 18, 3956–3959. [DOI] [PubMed] [Google Scholar]
- [49].Wu C, Liao J, Ge S, Angew. Chem 2019, 131, 8974–8978; [Google Scholar]
- Angew. Chem. Int. Ed 2019, 58, 8882–8886. [DOI] [PubMed] [Google Scholar]
- [50].Tan Y-X, Zhang F, Xie P-P, Zhang S-Q, Wang Y-F, Li Q-H, Tian P, Hong X, Lin G-Q, J. Am. Chem. Soc 2019, 141, 12770–12779. [DOI] [PubMed] [Google Scholar]