

Abstract
Severe acute respiratory syndrome coronavirus is a newly emergent virus responsible for a recent outbreak of an atypical pneumonia. The coronavirus spike protein, an enveloped glycoprotein essential for viral entry, belongs to the class I fusion proteins and is characterized by the presence of two heptad repeat (HR) regions, HR1 and HR2. These two regions are understood to form a fusion-active conformation similar to those of other typical viral fusion proteins. This hairpin structure likely juxtaposes the viral and cellular membranes, thus facilitating membrane fusion and subsequent viral entry. The fusion core protein of severe acute respiratory syndrome coronavirus spike protein was crystallized, and the structure was determined at 2.8 Å of resolution. The fusion core is a six-helix bundle with three HR2 helices packed against the hydrophobic grooves on the surface of central coiled coil formed by three parallel HR1 helices in an oblique antiparallel manner. This structure shares significant similarity with the fusion core structure of mouse hepatitis virus spike protein and other viral fusion proteins, suggesting a conserved mechanism of membrane fusion. Drug discovery strategies aimed at inhibiting viral entry by blocking hairpin formation, which have been successfully used in human immunodeficiency virus 1 inhibitor development, may be applicable to the inhibition of severe acute respiratory syndrome coronavirus on the basis of structural information provided here. The relatively deep grooves on the surface of the central coiled coil will be a good target site for the design of viral fusion inhibitors.
Severe acute respiratory syndrome (SARS)1 is a new life-threatening form of atypical pneumonia (1, 2) caused by a novel coronavirus, SARS-CoV (3, 4, 5, 6, 7, 8, 9, 10). Phylogenetic analysis of SARS-CoV shows that it is not closely related to any of the previously characterized coronaviruses isolated from either humans or animals, and it has therefore been assigned to a new, distinct group within the genus (4, 5, 9, 10).
Coronaviruses are enveloped, positive-strand RNA viruses with the largest genomes of any RNA virus and are characterized by 3–4 envelope proteins embedded on the surface (11, 12). Both the receptor binding and the subsequent membrane fusion process of coronaviruses are mediated by the spike membrane glycoprotein (S protein) (13). Recent studies show that murine coronavirus (mouse hepatitis virus (MHV)) uses a spike-mediated membrane fusion mechanism similar to that of so-called class I virus fusion proteins (14, 15).
Class I viral fusion proteins, including the hemagglutinin protein of influenza virus, gp160 of human immunodeficiency virus (HIV-1), glycoprotein of Ebola virus, and fusion protein (F protein) of paramyxovirus (16, 17), are all type I transmembrane glycoproteins that are displayed on the surface of viral membrane as oligomers. Most of these glycoproteins are synthesized as single chain precursors containing a protease cleavage site, and these precursors are cleaved into two noncovalently associated subunits: S1 and S2 in coronavirus, hemagglutinin 1 and 2 in influenza virus, gp120 + gp41 in HIV/simian immunodeficiency virus, glycoprotein-1 and -2 in Ebola virus, and F1 and -2 in paramyxovirus. Class I viral fusion proteins also contain a fusion peptide and at least two heptad repeat regions, termed HR1 and HR2. After binding to the receptor or induced by low pH, the fusion proteins undergo a series of conformational changes to mediate membrane fusion. The first step involves exposure of the fusion peptide, a hydrophobic region in the membrane-anchored subunit, which then inserts into the cellular lipid bilayer. Subsequently, HR1 and HR2 peptides form a trimer-of-hairpins-like structure via a transient pre-hairpin intermediate to facilitate juxtaposition of the viral and cellular membranes followed by virus-cell membrane fusion and viral entry. Biochemical and structural analysis of these fusion cores from class I viral fusion proteins shows that these complexes of two heptad repeat regions form a stable six-helix bundle, which is designated as a fusion core in which three HR1 helices form a central coiled coil surrounded by three HR2 helices in an oblique, antiparallel manner (18, 19, 20, 21, 22, 23, 24, 25, 26).
The coronavirus spike protein shares many features with other class I viral fusion proteins. It is a type I membrane protein that associates into trimers on the surface of coronavirus membrane (27). The distal subunit (S1) of the spike protein contains the receptor binding domain (28, 29), and the membrane-anchored subunit (S2) contains a putative internal fusion peptide and two heptad repeat regions (HR1 and HR2) (14, 15).
Agents that prevent conformational changes in the fusion protein by stabilizing the intermediate state are expected to prevent fusion activation and, thus, inhibit viral entry. In the case of HIV-1 gp41, peptides derived from C peptides can effectively inhibit infection in a dominant-negative manner by binding to the transiently exposed hydrophobic grooves of central coiled coil in the intermediate state and consequently blocking the formation of the fusion-active six-helix bundle structure (30).
We have recently determined the three-dimensional structure of murine coronavirus MHV spike protein fusion core and proposed a model for coronavirus-mediated membrane fusion in which the S protein undergoes a series of conformational changes similar to those of influenza virus and HIV-1 (14). Recent studies show that HR1 and HR2 of SARS-CoV spike protein form a stable six-helix bundle, and synthesized peptides corresponding to the HR2 region have inhibitory activity for viral fusion (31, 32, 33, 34, 35, 36).2 These studies also propose the binding regions between HR1 and HR2 and identify the inhibition efficiency of peptides derived from the HR2 region. However, the detailed three-dimensional structure of the HR1/HR2 complex remains unknown.
To verify that the SARS-CoV spike protein indeed forms a trimer-of-hairpins structure and to provide a structural basis for the design of viral fusion inhibitors, we characterized the binding of the two HR regions of the SARS-CoV spike protein and solved the crystal structure of this fusion core complex to 2.8 Å of resolution. The structure shows a similar conformation to other class I viral fusion proteins, especially MHV spike protein fusion core, suggesting that a similar approach might be used in identifying inhibitors of SARS-CoV infection effectively. This structure also provides important detailed structural information and target site for structure based drug design.
EXPERIMENTAL PROCEDURES
Construction, Expression, and Purification—The SARS spike gene was cloned from SARS coronavirus GZ02 (GenBank™ accession number AY390556). Two peptides (HR1 and HR2 regions of SARS-CoV spike protein) interact, and their binding regions were characterized by recent biochemical studies and sequence alignment with MHV fusion core complex.2 The HR1 and HR2 regions of SARS-CoV spike protein consist of residues 900–948 and residues 1145–1184, respectively, corresponding to the HR1 and HR2 regions in new MHV structure (14). The fusion core of SARS-CoV spike protein was prepared as a single chain (termed SARS 2-Helix) by linking the HR1 and HR2 domains via a 22-amino acid linker (LVPRGSGGSGGSGGLEVLFQGP), which is flexible and long enough to facilitate a natural interaction between the HR1 and HR2 peptides and allows for easy expression and purification of the fusion core complex (Fig. 1A ). The PCR-directed gene was inserted into pET22b (Novagen) vector, and the SARS 2-Helix protein was expressed in LB culture medium in Escherichia coli strain BL21(DE3). The product was purified by nickel nitrilotriacetic acid affinity chromatography and was further purified by gel filtration chromatography (Superdex 75™, Amersham Biosciences).
Fig. 1.

Structure determination of the MHV spike protein fusion core trimer.A, schematic diagram of SARS-CoV spike protein indicating the location of structurally significant domains. S1 and S2 are formed after proteolytic cleavage (vertical arrow) and noncovalently linked. The enveloped protein has an N-terminal signal sequence (SS) and a transmembrane domain (TM) adjacent to the C terminus. S2 contains two HR (heptad repeat) regions (hatched bars), HR1 and HR2 as indicated. The HR1 (898–1005) and HR2 (1145–1184) used in this study were derived from the LearnCoil-VMF prediction program (37). The 2-Helix protein construct consists of HR2 and part of HR1, which is the major region binding HR2, connected by a 22-amino acid linker (LVPRGSGGSGGSGGLEVLFQGP) as indicated. B, sequence alignment of coronavirus spike protein HR1 and HR2 regions. Letters above the sequence indicate the predicted hydrophobic residues at the a and d positions in two heptad repeat regions, which are highly conserved. FP, feline panleukopenia; IBV, infectious bronchitis virus; FIPV, feline infectious peritonitis virus.
Crystallization and Structure Determination—The purified protein was dialyzed against 10 mm Tris-HCl, pH 8.0, 10 mm NaCl and then concentrated to 15 mg ml-1. Crystals with good diffracting quality could be obtained after 2 weeks by using the hanging drop method by equilibrating a 2-μl drop (protein solution mixed 1:1 with reservoir solution) against a reservoir containing 0.1 m citric acid, pH 2.5, 10–15% polyethylene glycol 4000, 0.02 m spermidine tetra-HCl. The SARS 2-Helix crystal was mounted on nylon loops and flash-frozen in cold nitrogengas stream at 100 K using an Oxford Cryosystems coldstream with 0.1 m citric acid, pH 2.5, 25% polyethylene glycol 4000 as the cryoprotectant. The crystals have unit-cell parameters a = 121.2 Å, b = 66.3 Å, c = 70.0 Å, α = γ = 90°, β = 107.4° and belong to space group C2. The crystals contain 6 SARS 2-Helix molecules in one asymmetric unit, and the diffraction pattern extends to 2.8 Å of resolution. Data collection was performed in-house on a Rigaku RU2000 rotating copper-anode x-ray generator operated at 48 kV and 98 mA (Cu Kα; λ = 1.5418 Å) with a MAR 345 image-plate detector. Data were indexed, integrated, and scaled using DENZO and SCALEPACK programs (38).
The structure of SARS 2-Helix was determined by molecular replacement with the MHV 2-Helix structure (PDB code 1WDF) as a search model. Rotation and translation function searches were performed in the program CNS (39). The model was improved further by cycles of manual building and refinement using the programs O (40) and CNS (39). The quality of coordinates was examined by PROCHECK (41). The figures were generated with the programs GRASP (42), SPDBView (43), and MOLSCRIPT (44).
RESULTS AND DISCUSSION
Structure Determination—The crystal structure of SARS 2-Helix was solved by molecular replacement using the program CNS (39) with the MHV 2-Helix structure (PDB code 1WDF) employed as a search model. Six molecules (two trimers of SARS 2-Helix) per asymmetric unit were located from crossrotation and translation function searches. The model was improved further by cycles of manual building and refinement using the programs O (40) and CNS (39). The structure was subsequently refined to a final resolution of 2.8 Å with an R value of 23.3% and R free value of 27.3%. No residue was in disallowed regions of the Ramachandran plot. The statistics for the data collection, structure determination, and refinement are summarized in Table I .
Table I.
Data collection and final refinement statistics Numbers in parentheses correspond to the highest resolution shell. Rmerge = ΣhΣI|Iih – 〈Ih〉|/ΣhΣI〈Ih〉, where 〈Ih〉 is the mean of the observations Iih of reflection h. Rwork = Σ (|Fobs| – |Fcalc||)/Σ|Fobs|; Rfree is the R factor for a subset (5%) of reflections that was selected before refinement calculations and not included in the refinement. r.m.s.d., root mean square deviation from ideal geometry.
| Data collection statistics |
|
| Space group | C2 |
| Unit cell parameters | a = 121.2 Å, b = 66.3 Å, c = 70.0 Å, β = 107.4° |
| Wavelength (Å) | 1.5418 |
| Resolution limit (Å) | 2.8 |
| Observed reflections | 27,312 |
| Unique reflections | 11,974 |
| Completeness (%) | 91.4 (80.8) |
| [I/σ(I)] | 5.7 (1.2) |
| Rmerge (%) | 13.9 (51.1) |
| Final refinement statistics | |
| Rwork (%) | 23.3 |
| Rfree (%) | 27.3 |
| Resolution range (Å) | 50–2.8 |
| Total reflections used | 10,718 |
| Number of reflections in working set | 10,166 |
| Number of reflections in test set | 552 |
| Average B (Å2) | 42.7 |
| r.m.s.d. bonds (Å) | 0.013 |
| r.m.s.d. angles(°) |
1.8 |
Description of the Structure—One asymmetric unit contains six SARS 2-Helix molecules, including residues 902–947 in HR1 and 1153–1175 in HR2 (A molecule), residues 902–947 in HR1 and 1150–1184 in HR2 (B molecule), residues 902–947 in HR1 and 1153–1184 in HR2 (C molecule), residues 901–947 in HR1 and 1152–1181 in HR2 (D molecule), residues 901–948 in HR1 and 1153–1178 in HR2 (E molecule), and residues 901–947 in HR1 and 1154–1181 in HR2 (F molecule), respectively. The linker and several terminal residues could not be traced in the electron density map due to their disordered nature.
Here, we chose A, B, and C molecules as the SARS spike protein fusion core for the following structure description. In the SARS 2-Helix three-dimensional structure, the fusion core has a rod-shaped structure with a length of ∼70 Å and a diameter of ∼28 Å. Similar to MHV 2-Helix and other class I viral fusion proteins, the SARS spike protein fusion core is a six-helix bundle comprising a trimer of 2-Helix molecules. The center of the fusion core consists of a parallel trimeric coiled coil of three HR1 helices surrounded by three HR2 helices in an oblique, antiparallel manner (Fig. 2, A and B ). Residues 902–947 in HR1 fold into a 12-turn α-helix stretching the entire length of the fusion core. As in MHV 2-Helix and other class I viral fusion proteins, the residues in positions a and d of HR1 are predominantly hydrophobic (Fig. 1B). Residues 1160–1177 in HR2 form a 5-turn α-helix, whereas residues 1150–1159 at the N terminus and residues 1178–1184 at the C terminus of HR2 form two extended conformations, respectively. Three HR2 helices pack against the grooves formed by the interface of the central three HR1 helices, and no interaction was observed between individual HR2 regions. The N terminus of HR2 starts with Ile1150, which is aligned with Gln947 of HR1. The C terminus of HR2 ends with Leu1184, which is aligned with Gln902 of HR1 (see Fig. 4 ).
Fig. 2.
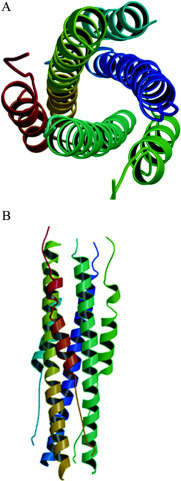
Overall views of the fusion core structure.A, top view of the SARS-CoV spike protein fusion core structure showing the 3-fold axis of the trimer. B, side view of the SARS-CoV spike protein fusion core structure showing the six-helix bundle.
Fig. 4.
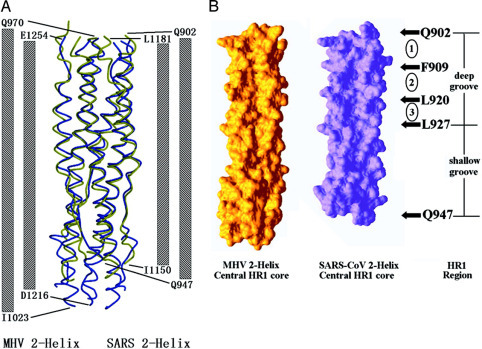
Comparison between fusion core structure of SARS-CoV and MHV.A, side view showing a structural comparison between SARS-CoV spike protein fusion core (colored in green) and MHV spike protein fusion core (colored in purple). The columns at both sides of the map represent two HR1 and HR2 regions of MHV and SARS fusion cores. The numbers at the end of these columns represent the specific boundaries of the HR1-HR2 interaction region in the two structures. B, surface map showing the comparison between hydrophobic grooves on the surface of three central HR1 regions of MHV (left side) and SARS-CoV (right side). The figure on the right side shows the deep and relatively shallow grooves on the surface of central HR1 coiled coil of SARS-CoV. Three numbers, 1–3, in circles represent three deep cavities, composing the deep grooves. The residues represent the boundaries of the grooves and cavities.
Interactions between HR1 and HR2—Three HR2 helices interact with HR1 helices mainly through hydrophobic interaction between hydrophobic residues in HR2 regions and the grooves on the surface of the central coiled coil. Similar to those in MHV 2-Helix, HR2 helices in SARS 2-Helix also contain OXO motifs, in which O represents a hydrophobic residue, and X represents any residue but is generally hydrophobic. 1153IN-ASVVNI1160 and 1178LIDL1181 in HR2 regions are both composed of OXO motifs; the side chains of the O residues inset into or align with the hydrophobic grooves of the central coiled coil, whereas the side chains of the X residues are directed into solvent (Fig. 3B ). As described in our previous paper (14), the OXO motifs are responsible for the partially extended conformation of HR2, and this pattern also makes the fusion core stable in solvent, as most of the hydrophobic residues in HR2 helices are packed against the central coiled coil, leaving the hydrophilic residues exposed to solvent.
Fig. 3.
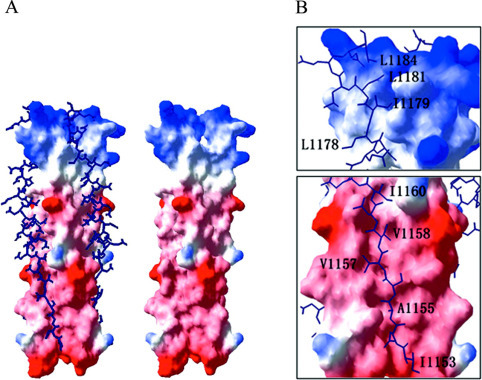
Detailed structure of the SARS-CoV spike protein fusion core and OXO motifs in HR2 regions.A, surface map showing the hydrophobic grooves on the surface of the central coiled coil (right side). Three HR2 helices pack against the hydrophobic grooves in an oblique antiparallel manner (left side). The helical regions and extended regions in HR2 helices could be observed clearly, and the boundaries of these regions are marked. B, OXO motifs in HR2 regions of SARS-CoV spike protein fusion core structure. The enlarged images show two regions containing OXO motifs. The hydrophobic residues in these motifs all pack against the hydrophobic grooves on the surface of three HR1 helices.
Comparison with MHV 2-Helix Structure—Among coronavirus spike proteins, only the structure of the MHV spike protein fusion core has been determined (14). In general, the fusion core from SARS-CoV and MHV adopt a similar fold, consistent with their high sequence identity and similarity between the two proteins. The structure of SARS spike protein fusion core was compared with that of MHV spike protein (Fig. 4). These structures can be superimposed with a root mean square difference of 0.91 Å for all Cα atoms. Alignments of the peptides derived from HR1 and HR2 regions of SARS-CoV spike protein and those of other coronaviruses reveal high sequence identity and similarity, suggesting that structures of spike protein fusion cores from other coronavirus might share significant similarity with those of MHV and SARS-CoV (Fig. 1B).
Although the main chains of the MHV 2-Helix and SARS-CoV 2-Helix structures can be superposed closely, the two complexes have significant differences in their hydrophobic grooves on the surface of the central coiled coil (Fig. 4B). As discussed in our previous paper detailing our SARS-CoV fusion core model, the central coiled coil had relatively deep grooves and relatively shallow grooves.2 The deep grooves, consisting of three hydrophobic deep pockets or cavities, were clearly deeper than the corresponding grooves on the surface of MHV 2-Helix control coiled coil. From the structure presented here, the helical regions of SARS-CoV HR2 segment (residues 1161–1184) pack exactly against the relatively deep grooves of the central coiled coil and the extended regions (residues 1150–1160) pack against the relatively shallow grooves. Based on our previous biochemical analysis and the crystal structure, we propose that the deep groove is an important target site for the design of viral fusion inhibitors (16, 30, 34, 45, 46, 47, 48).
Conformational Change and Membrane Fusion Mechanisms—Our previous structural study of the MHV spike protein fusion core led us to propose a model for coronavirus-mediated membrane fusion mechanism (14). The remarkable similarity between SARS-CoV and MHV spike protein fusion core structures as well as similar HR2 peptide inhibition phenomena and remarkable stability to both thermal denaturation and proteinase K digestion (31, 32, 33, 34, 35, 36) suggest a conserved mechanism of membrane fusion mediated by the spike protein. Similar to the MHV spike protein and HIV gp41, the SARS-CoV spike protein likely undergoes a series of conformational changes to become fusion-active. The fusion loop but not fusion peptide, which will insert into the cellular membrane, and distinct conformational states proposed for the MHV spike protein fusion core, including the native state, the pre-hairpin intermediate, and fusion-active hairpin state, may also apply to SARS-CoV spike protein. The existence of the pre-hairpin intermediate conformation of the SARS-CoV spike protein is strongly supported by the viral inhibition assay, in which the peptides corresponding to the HR2 regions can inhibit viral fusion in a dominant-negative manner (32, 34). A reasonable interpretation of these phenomena is that the HR2 peptide functions by binding to the transiently exposed hydrophobic groove on the surface of central coiled coil, thus blocking the conformational transition to the fusion active form and subsequent membrane fusion and viral entry.
Binding Regions of SARS-CoV Spike Protein Fusion Core— Recent studies on the fusion-active complex of SARS-CoV have confirmed that HR1 and HR2 associate into an antiparallel six-helix bundle, with structural features of other typical class I viral fusion proteins by means of CD, native PAGE, proteolysis protection analysis, and size-exclusion chromatography. In their biochemical analysis, Ingallinella et al. (31) mapped the specific boundaries of the key region of interaction between HR1 and HR2 peptides as residues 914 and 949 in HR1 region and residues 1148 and 1185 in HR2 region (31). The exact boundaries, residues 902 and 947 in HR1 region and residues 1150 and 1184 in HR2 region, can be observed from the crystal structure of SARS-CoV spike protein fusion core with a slight difference from previous results. The N terminus of the HR1 region (Gln902) aligns with the C terminus of the HR2 region (Leu1184), and the C terminus of the HR1 region (Gln947) aligns with the N terminus of the HR2 region (Ile1150). Although the residue numbers of the HR1 and HR2 regions are different (46 in HR1 and 35 in HR2), the two peptides are equivalent in length in the three-dimensional structure since HR1 forms a typical α-helix, whereas HR2 forms a partial helical conformation (Fig. 3A). This pattern of HR2 helices is also strongly supported by proteolysis protection experiments (31). The real boundaries of the fusion core might extend beyond those found in the structure studied here, whereas the major binding regions and the interactions between HR1 and HR2 peptides can be identified clearly from the SARS-CoV spike protein fusion core structure.
Inhibitory Molecules for SARS-CoV Infection—If small, bioavailable molecules that prevent hairpin formation can be identified, they may serve as useful drugs against SARS-CoV infection. In the case of HIV-1, several strategies to block hairpin formation have been successfully developed to identify viral entry inhibitors that bind to the hydrophobic pocket and grooves on the surface of the central coiled coil consisting of HIV-1 gp41 N peptides. These useful viral entry inhibitors include D peptides, 5-Helix, and synthetic peptides derived from N or C peptides (45, 46, 47). Successful viral entry inhibitors have also been identified for other viruses, such as T20 for HIV-1 (49, 50) and GP610 for Ebola virus (51). These strategies could also be used for the design of SARS fusion inhibitors. The well defined hydrophobic grooves on the surface of the central coiled coil of the SARS-CoV spike protein fusion core identified here may be a significant target for drug design.
Several peptides derived from the HR1 and HR2 regions of SARS-CoV spike proteins have been found to have inhibitory activity in recent studies (32, 33, 35) (Table II ). Analogous to the HIV-1 C peptides and MHV HR2 peptides, the HR2 peptides of SARS-CoV spike protein likely function in a dominant-negative manner by binding to the transiently exposed hydrophobic grooves in the pre-hairpin intermediate, thus, blocking viral entry. The efficacy of HR2 peptides of SARS-CoV spike protein is, however, significantly lower than corresponding HR2 peptides of murine coronavirus mouse hepatitis virus in inhibiting MHV infection (32). Synthetic peptides with the highest inhibitory efficacy encompass residues 1161–1187, derived from the HR2 region (33). This peptide is just the corresponding region that binds to the relatively deep grooves on the surface of central coiled coil. It is not clear why HR2 peptides of SARS-CoV have lower inhibitory efficacy. However, the structural information provided here will be useful for the design of antiviral compounds such as D peptides, 5-Helix, and some peptides (or mutants) derived from HR1 or HR2 peptides based on the crystal structure of SARS-CoV spike protein fusion core. The peptides encompassing residues 1161–1187 will be good targets for mutagenesis in the search for peptides with higher inhibitory efficacy. The exposed hydrophobic grooves in the intermediate state and, particularly, the relatively deep grooves on the surface of central coiled coil will be good targets for the discovery of viral entry inhibitors (Fig. 3A and 4B)..
Table II.
Amino acid sequences and EC50values of inhibitory peptides
| Peptide | EC50 | Sequence |
|---|---|---|
| μm | ||
| NP-1 (892–931)a | Marginal | GVTQNVLYENQKQIANQfnKAISQIQESLTTTSTALGKLQ |
| CP-1 (1153–1189)a | 19 | GINASVVNTQKEIDRLNEVAKNLNESLIDLQELGKYE |
| HR1 (889–926)b | 3.68 | NGIGVTQNVLYENQKQIANQfnKAISQIQESLTTTSTA |
| HR2 (1161–1187)b | 5.22 | IQKEIDRLNEVAKNLNESLIDLQELGK |
| HR2-1 (1126–1189)c | 43 ± 6.4 | ELDSPKEELDKYFKNHTSPDVDLGDISGINASVVNIQKEIDRLNEVAKNLNESLIDLQELGKYE |
| HR2-2 (1130–1189)c | 24 ± 2.8 | PKHELDKYFKNHTSPDVDLGDISGINASVVNIQKEIDRLNEVAKNLNESLIDLQELGKYE |
| HR2-8 (1126–1193)c | 17 ± 3.0 | ELDSFKEELDKYFKNHTSPDVDLGDISGINASVVNIQKEIDRLNEVAKNLNESLIDLQELGKYEQYIK |
| HR2-9 (1126–1184)c | 34 ± 4.0 | ELDSFKEELDKYFKNHTSPDVDLGDISGINASVVNIQKEIDRLNEVAKNLNESLIDLQEL |
| HR2-6 (1150–1189)c |
>50 |
DISGINASVVNIQKEIDRLNEVAKNLNESLIDLQELGKYE |
Acknowledgments
We thank Dr. Mark Bartlam for comments and critical reading.
Footnotes
The atomic coordinates and structure factors (code 1WNC) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The abbreviations used are: SARS, severe acute respiratory syndrome; HIV, human immunodeficiency virus; HR, heptad repeat; SARS-CoV, severe acute respiratory syndrome coronavirus; MHV, mouse hepatitis virus.
Y. Xu, J. Zhu, Y. Liu, Z. Lou, F. Yuan, Y. Liu, D. K. Cole, L. Ni, N. Su, L. Qin, X. Li, Z. Bai, J. I. Bell, H. Pang, P. Tien, Z. Rao, and G. F. Gao, submitted for publication.
References
- 1.Tsang K.W., Mok T.Y., Wong P.C., Ooi G.C. Respirology. 2003;8:259–265. doi: 10.1046/j.1440-1843.2003.00486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poutanen S.M., Low D.E., Henry B., Finkelstein S., Rose D., Green K., Tellier R., Draker R., Adachi D., Ayers M., Chan A.K., Skowronski D.M., Salit I., Simor A.E., Slutsky A.S., Doyle P.W., Krajden M., Petric M., Brunham R.C., McGeer A.J. N. Engl. J. Med. 2003;348:1995–2005. doi: 10.1056/NEJMoa030634. [DOI] [PubMed] [Google Scholar]
- 3.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 4.Eickmann M., Becker S., Klenk H.D., Doerr H.W., Stadler K., Censini S., Guidotti S., Masignani V., Scarselli M., Mora M., Donati C., Han J.H., Song H.C., Abrignani S., Covacci A., Rappuoli R. Science. 2003;302:1504–1505. doi: 10.1126/science.302.5650.1504b. [DOI] [PubMed] [Google Scholar]
- 5.Stadler K., Masignani V., Eickmann M., Becker S., Abrignani S., Klenk H.D., Rappuoli R. Nat. Rev. Microbiol. 2003;1:209–218. doi: 10.1038/nrmicro775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 7.Kuiken T., Fouchier R.A., Schutten M., Rimmelzwaan G.F., van Amerongen G., van Riel D., Laman J.D., de Jong T., van Doornum G., Lim W., Ling A.E., Chan P.K., Tam J.S., Zambon M.C., Gopal R., Drosten C., van der Werf S., Escriou N., Manuguerra J.C., Stohr K., Peiris J.S., Osterhaus A.D. Lancet. 2003;362:263–270. doi: 10.1016/S0140-6736(03)13967-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yung R.W., Ng T.K., Yuen K.Y. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 10.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 11.Siddell S., Wege H., Ter Meulen V. J. Gen. Virol. 1983;64:761–776. doi: 10.1099/0022-1317-64-4-761. [DOI] [PubMed] [Google Scholar]
- 12.Cavanagh D. J. Gen. Virol. 1983;64:2577–2583. doi: 10.1099/0022-1317-64-12-2577. [DOI] [PubMed] [Google Scholar]
- 13.Gallagher T.M., Buchmeier M.J. Virology. 2001;279:371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Y., Liu Y., Lou Z., Qin L., Li X., Bai Z., Pang H., Tien P., Gao G.F., Rao Z. J. Biol. Chem. 2004;279:30514–30522. doi: 10.1074/jbc.M403760200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bosch B.J., van der Zee R., de Haan C.A., Rottier P.J. J. Virol. 2003;77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckert D.M., Kim P.S. Annu. Rev. Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 17.Hernandez L.D., Hoffman L.R., Wolfsberg T.G., White J.M. Annu. Rev. Cell Dev. Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- 18.Bullough P.A., Hughson F.M., Skehel J.J., Wiley D.C. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 19.Lu M., Blacklow S.C., Kim P.S. Nat. Struct. Biol. 1995;2:1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 20.Weissenhorn W., Dessen A., Harrison S.C., Skehel J.J., Wiley D.C. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 21.Chan D.C., Fass D., Berger J.M., Kim P.S. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 22.Tan K., Liu J., Wang J., Shen S., Lu M. Proc. Natl. Acad. Sci. U. S. A. 1997;94:12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caffrey M., Cai M., Kaufman J., Stahl S.J., Wingfield P.T., Covell D.G., Gronenborn A.M., Clore G.M. EMBO J. 1998;17:4572–4584. doi: 10.1093/emboj/17.16.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weissenhorn W., Carfi A., Lee K.H., Skehel J.J., Wiley D.C. Mol. Cell. 1998;2:605–616. doi: 10.1016/s1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- 25.Weissenhorn W., Calder L.J., Wharton S.A., Skehel J.J., Wiley D.C. Proc. Natl. Acad. Sci. U. S. A. 1998;95:6032–6036. doi: 10.1073/pnas.95.11.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baker K.A., Dutch R.E., Lamb R.A., Jardetzky T.S. Mol. Cell. 1999;3:309–319. doi: 10.1016/s1097-2765(00)80458-x. [DOI] [PubMed] [Google Scholar]
- 27.Delmas B., Laude H. J. Virol. 1990;64:5367–5375. doi: 10.1128/jvi.64.11.5367-5375.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonavia A., Zelus B.D., Wentworth D.E., Talbot P.J., Holmes K.V. J. Virol. 2003;77:2530–2538. doi: 10.1128/JVI.77.4.2530-2538.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taguchi F. J. Virol. 1995;69:7260–7263. doi: 10.1128/jvi.69.11.7260-7263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan D.C., Kim P.S. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 31.Ingallinella P., Bianchi E., Finotto M., Cantoni G., Eckert D.M., Supekar V.M., Bruckmann C., Carfi A., Pessi A. Proc. Natl. Acad. Sci. U. S. A. 2004;101:8709–8714. doi: 10.1073/pnas.0402753101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bosch B.J., Martina B.E., Van Der Zee R., Lepault J., Haijema B.J., Versluis C., Heck A.J., De Groot R., Osterhaus A.D., Rottier P.J. Proc. Natl. Acad. Sci. U. S. A. 2004;101:8455–8460. doi: 10.1073/pnas.0400576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan K., Yi L., Chen J., Qu X., Qing T., Rao X., Jiang P., Hu J., Xiong Z., Nie Y., Shi X., Wang W., Ling C., Yin X., Fan K., Lai L., Ding M., Deng H. Biochem. Biophys. Res. Commun. 2004;319:746–752. doi: 10.1016/j.bbrc.2004.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu J., Xiao G., Xu Y., Yuan F., Zheng C., Liu Y., Yan H., Cole D.K., Bell J.I., Rao Z., Tien P., Gao G.F. Biochem. Biophys. Res. Commun. 2004;319:283–288. doi: 10.1016/j.bbrc.2004.04.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu S., Xiao G., Chen Y., He Y., Niu J., Escalante C.R., Xiong H., Farmar J., Debnath A.K., Tien P., Jiang S. Lancet. 2004;363:938–947. doi: 10.1016/S0140-6736(04)15788-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tripet B., Howard M.W., Jobling M., Holmes R.K., Holmes K.V., Hodges R.S. J. Biol. Chem. 2004;279:20836–20849. doi: 10.1074/jbc.M400759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh M., Berger B., Kim P.S. J. Mol. Biol. 1999;290:1031–1041. doi: 10.1006/jmbi.1999.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Otwinowski Z., Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 39.Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S., Read R.J., Rice L.M., Simonson T., Warren G.L. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 40.Jones T.A., Zou J.Y., Cowan S.W., Kjeldgaard M. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 41.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 42.Nicholls A., Sharp K.A., Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 43.Guex N., Peitsch M.C. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 44.Kraulis P.J. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- 45.Eckert D.M., Kim P.S. Proc. Natl. Acad. Sci. U. S. A. 2001;98:11187–11192. doi: 10.1073/pnas.201392898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Root M.J., Kay M.S., Kim P.S. Science. 2001;291:884–888. doi: 10.1126/science.1057453. [DOI] [PubMed] [Google Scholar]
- 47.Eckert D.M., Malashkevich V.N., Hong L.H., Carr P.A., Kim P.S. Cell. 1999;99:103–115. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- 48.Wild C.T., Shugars D.C., Greenwell T.K., McDanal C.B., Matthews T.J. Proc. Natl. Acad. Sci. U. S. A. 1994;91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kilby J.M., Hopkins S., Venetta T.M., DiMassimo B., Cloud G.A., Lee J.Y., Alldredge L., Hunter E., Lambert D., Bolognesi D., Matthews T., Johnson M.R., Nowak M.A., Shaw G.M., Saag M.S. Nat. Med. 1998;4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 50.Imai M., Okada N., Okada H. Microbiol. Immunol. 2000;44:205–212. doi: 10.1111/j.1348-0421.2000.tb02485.x. [DOI] [PubMed] [Google Scholar]
- 51.Watanabe S., Takada A., Watanabe T., Ito H., Kida H., Kawaoka Y. J. Virol. 2000;74:10194–10201. doi: 10.1128/jvi.74.21.10194-10201.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]