

Abstract
The molecular mechanisms underlying the cardiotoxicity associated with bevacizumab, a first-line immunotherapeutic agent used to treat lung cancer, are not fully understood. Here, we examined intracellular signal transduction in cardiomyocytes after exposure to different doses of bevacizumab in vitro. Our results demonstrated that bevacizumab significantly and dose-dependently reduces cardiomyocyte viability and increases cell apoptosis. Bevacizumab treatment also led to mitochondrial dysfunction in cardiomyocytes, as evidenced by the decreased ATP production, increased ROS production, attenuated antioxidative enzyme levels, and reduced respiratory complex function. In addition, bevacizumab induced intracellular calcium overload, ER stress, and caspase-12 activation. Finally, bevacizumab treatment inhibited the ERK signaling pathway, which, in turn, significantly reduced cardiomyocyte viability and contributed to mitochondrial dysfunction. Together, our results demonstrate that bevacizumab-mediated cardiotoxicity is associated with mitochondrial dysfunction, ER stress, and ERK pathway inactivation. These findings may provide potential treatment targets to attenuate myocardial injury during lung cancer immunotherapy.
1. Introduction
Bevacizumab is a first-line immunotherapeutic agent used for the treatment of lung cancer [1]. The cancer-suppressing effects of bevacizumab are associated with inhibition of vascular endothelial growth factor signaling, which leads to decreased tumor growth and impaired invasion [2, 3]. However, bevacizumab is also associated with cardiovascular toxicities, including decreased left ventricular ejection fraction, vasculitis, hypertension, arrhythmias, vascular bed degeneration, and limited angiogenesis response [4–8]. Several mechanisms, including accumulation of toxic metabolites, cardiac microvascular vasospasm, and excessive activation of the renin-angiotensin system, might underlie the adverse effects of bevacizumab on the heart [4–7]. However, the intracellular molecular mechanisms underlying bevacizumab-associated cardiotoxicity are not fully understood.
At the molecular level, cardiomyocyte viability and function are greatly affected by mitochondrial performance [9, 10]. Mitochondria regulate cardiomyocyte contraction and relaxation by controlling ATP production [11, 12]. As the primary site of protein manufacturing, the endoplasmic reticulum (ER) controls protein synthesis, folding, and release [13, 14]. Previous studies have reported that targeted cancer therapies, such as doxorubicin [15], anthracycline [16], and cantharidin [17], can result in cardiovascular toxicities. The adverse effects of chemotherapy drugs may result from impaired mitochondrial function, increased oxidative stress [15, 18], increased mitochondria-proteasome interactions [19], impaired mitochondrial autophagy [20, 21], activation of mitochondrial inflammation signaling pathways such as NF-κB [22], mitochondrial energy metabolic dysfunction [23], and mitochondrial apoptosis [24]. In addition, ER-mediated abnormalities in intracellular calcium signaling, protein misfolding as a result of ER stress, and ER-dependent cell apoptosis [25–27] can also contribute to cardiomyocyte damage during chemotherapy. Whether bevacizumab-mediated cardiovascular disorders are attributable to mitochondrial damage and/or ER stress remains to be determined.
The MAPK/ERK signaling pathway plays an important role in cardiomyocyte survival under stress conditions [28, 29]. Activated ERK attenuates oxidative stress in cardiomyocytes by promoting the transcription of antioxidative stress genes [30]. In addition, ERK alleviated chronic cardiac hypertrophy by improving mitochondrial metabolism [31]. As in cardiomyocytes, ERK promotes growth during tumor proliferation and invasion, and upregulation of the ERK pathway increased angiogenesis [32]. The ERK pathway also affects immune response in various tumors [33, 34]. In this study, we examined whether mitochondrial dysfunction, ER stress, and the ERK pathway are involved in bevacizumab-induced cardiotoxicity.
2. Materials and Methods
2.1. Cell Culture
H9C2 cell lines purchased from ATCC were grown in DMEM supplemented with 10% fetal bovine serum. The cells were maintained at 37°C and 5% carbon dioxide in a humidified environment. Cardiomyocytes were incubated with 0.1 or 5 mM bevacizumab as described in previous studies. Cardiomyocytes were also incubated with PD98059, an ERK pathway inhibitor, as described in previous reports to examine the influence of ERK inhibition on cardiomyocyte viability.
2.2. Cell Viability Assay
A total of 3000 treated cells per well were seeded in 96-well culture plates and incubated with or without bevacizumab for the indicated times; fresh media containing 50 μl of CCK8 solution (5 mg/ml) (Dojindo Laboratories, Kumamoto, Japan) was then added followed by incubation at 37°C for 3 hours according to the manufacturer's protocol [35]. Absorbance was measured at 450 nm using an enzyme e-linked immunosorbent assay reader [36].
2.3. Mitochondrial ROS Detection
Cardiomyocytes were treated with bevacizumab and then washed with cold PBS three times. Then, 0.5 ng/ml MitoSOX Red mitochondrial superoxide indicator (Molecular Probes, USA) was added to the cardiomyocyte medium and incubated for 30 minutes in the dark. Cells were then washed with cold PBS three times [37]. Mitochondrial ROS production was observed under a confocal laser scanning microscope (LSM780; Carl Zeiss, Oberkochen, Germany, or TCS SP8; Leica, Wetzlar, Germany) and an Axio Zoom V16 stereo microscope (Carl Zeiss) [38].
2.4. Intracellular Calcium Content
Intracellular calcium measurements were performed as previously described with minor modifications [39]. Briefly, H9C2 cells were loaded with 5 μM Fluo4-AM calcium probe for 30 minutes at 37°C [40]. Cells were then washed with PBS, and intracellular calcium was observed under a confocal laser scanning microscope (LSM780; Carl Zeiss, Oberkochen, Germany, or TCS SP8; Leica, Wetzlar, Germany) and an Axio Zoom V16 stereo microscope (Carl Zeiss) [41].
2.5. Immunofluorescence
For fluorescence microscopy-based detection of target proteins, 5 μm thick cryosections were deparaffinized in xylene and rehydrated through graded ethanol [42]. Antigen retrieval was performed for 20 min at 95°C with 0.1% sodium citrate buffer (pH 6.0). After endogenous peroxidase activity was quenched with 3% H2O2·dH2O and nonspecific binding was blocked with 1% bovine serum albumin buffer, sections were incubated overnight at 4°C with primary antibodies using an Alexa 594 TSA Kit (Invitrogen) according to the manufacturer's instructions [43]. Stained sections were observed under a confocal laser scanning microscope (LSM780; Carl Zeiss, Oberkochen, Germany, or TCS SP8; Leica, Wetzlar, Germany) and an Axio Zoom V16 stereo microscope (Carl Zeiss) [44].
2.6. Detection of GSH, SOD, and GPX Activities
GSH, SOD, and GPX activities were detected using a Zymography Assay Kit (Applygen Technologies, China) according to the manufacturer's protocol [45]. GSH, SOD, and GPX were separated by SDS-PAGE. SDS was then extracted from the gel by incubating with Triton X-100 for 48 hours at 37°C. Finally, the gels were stained with Coomassie Brilliant Blue G250 and decolorized. A bright band against the blue background indicated the activity of targeted proteins. A Gel Image System (image master 1D analysis software, Pharmacia) was used to image the band [46].
2.7. Western Blot Analysis
Cell lysate was boiled in a sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% sodium dodecyl sulfate, 20% glycerol, and 10% 2-mercaptoethanol), and protein concentration was determined using a Bradford protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA) with bovine serum albumin as the standard [47]. Following protein transfer, the membrane was blocked with 5% skim milk in PBS Tween- (PBST-) 20 for 2 h at room temperature and then incubated overnight with antibodies at 4°C (the primary antibodies include Bcl2, 1 : 1000, Cell Signaling Technology, #3498; Bax, 1 : 1000, Cell Signaling Technology, #2772; caspase-9, 1 : 1000, Cell Signaling Technology, #9504; c-IAP, 1 : 1000, Cell Signaling Technology, #4952). The membranes were then washed with PBST containing 0.1% Tween. After three washes in PBST, each blot was incubated with peroxidase-conjugated secondary antibody for 1 h at 37°C. Labeled proteins were visualized using the Odyssey infrared scanner (LI-COR, Lincoln, NB, USA) [48]. Signals were densitometrically assessed and normalized to the β-actin signals, and an enhanced chemiluminescence detection system (Amersham, Piscataway, NJ, USA) was used to visualize the antibody-specific proteins in accordance with the manufacturer's recommended protocol [49].
2.8. Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was isolated from samples according to miRNeasy Mini Kit (217004, Qiagen Company, Hilden, Germany) instructions [50]. All primers were synthesized by Takara Holdings Inc., Kyoto, Japan. RNA was then reverse transcribed into cDNA using the PrimeScript RT kit (RR036A, Takara). Next, fluorescence quantitative PCR was conducted using the SYBR® Premix ExTaq™ II kit (RR820A, Takara) on an ABI 7500 quantitative PCR instrument (7500, ABI Company, Oyster Bay, N.Y., USA). All samples were normalized to U6 and GAPDH using the 2- ΔΔCT method [51].
2.9. Mitochondrial Respiratory Chain Complex Activity Analysis
Mitochondrial respiratory chain activity was assessed using the Mitochondrial Respiratory Chain Complex Activity Assay Kit (Solarbio, Beijing, China) according to the manufacturer's instructions [52]. Briefly, the mitochondrial complex was extracted from cells and 10 μl of the extract was added to each well of a 96-well plate. Detection reagents were then added to the wells followed by gentle mixing and incubation at 37°C for 2 min. Absorbance values were measured before and after the reaction using a microplate reader (BioTek, Vermont, VT), and the difference was calculated [53]. Respiratory complex enzyme activity was then calculated using the formula provided in the kit manual [54].
2.10. Measurement of ATP Levels
ATP production was measured using the luminometric ATP Assay kit (AAT Bioquest, Sunnyvale, CA) according to the manufacturer's instructions [55]. Briefly, H9C2 cells were seeded in a 96-well white plate and 200 μl ATP assay solution was added. After mixing gently and incubating for 20 min at room temperature, luminescence intensity was measured using the luminometer mode on a plate reader (Tecan, Zurich, Switzerland) [56]. The readings were normalized to the total protein content.
2.11. Statistical Analysis
All results were confirmed in three independent experiments, and all quantitative data are expressed as the mean ± SD. Differences in quantitative variables between two groups were analyzed using Student's t-test, and differences in quantitative variables for three or more groups were analyzed by one-way ANOVA. p < 0.05 was considered statistically significant.
3. Results
3.1. Bevacizumab Reduces Cardiomyocyte Viability and Function
After H9C2 cells were treated with low and high concentrations of bevacizumab, cell viability was measured through a CCK-8 assay. As shown in Figure 1(a), compared to the control group, bevacizumab treatment significantly reduced cardiomyocyte viability in a dose-dependent manner. Moreover, an LDH release assay, which measures levels of LDH released into the culture medium as a result of cell membrane breakage, showed that bevacizumab-treated H9C2 cells released more LDH than control cells (Figure 1(b)). Because decreased cardiomyocyte viability is strongly associated with impaired cardiomyocyte function, we next examined single-cell contraction function in primary cardiomyocytes. While the average length of primary cardiomyocytes was not affected by bevacizumab (Figures 1(c)–1(h)), peak heights and maximal shortening velocity decreased after bevacizumab treatment, suggesting that bevacizumab disturbs cardiomyocyte contraction (Figures 1(c)–1(h)). In addition, maximal relengthening velocity and time-to-peak values increased after bevacizumab treatment (Figures 1(c)–1(h)), suggesting that bevacizumab also inhibits cardiomyocyte relaxation. Together, these results indicate that bevacizumab treatment dose-dependently reduces cardiomyocyte viability and impairs cardiomyocyte contraction/relaxation index.
Figure 1.
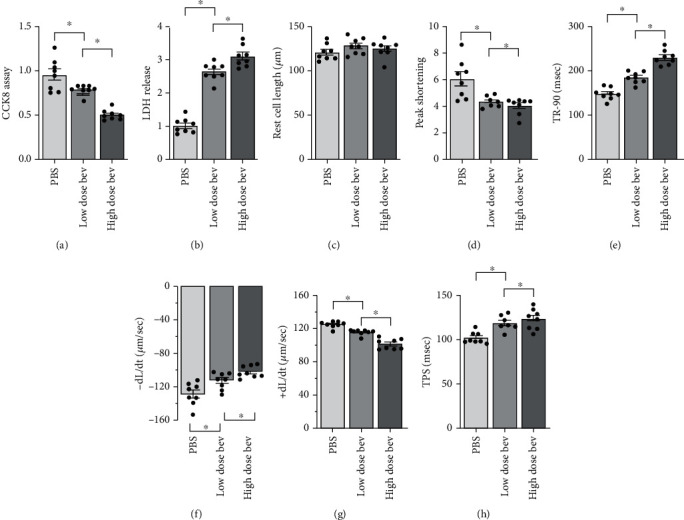
Bevacizumab reduces cardiomyocyte viability and function. (a) Cell viability was determined using a CCK-8 assay. (b) An LDH release assay was performed to examine whether bevacizumab induced cardiomyocyte damage. (c–h) The following single cardiomyocyte contractile properties were measured: peak height, maximal shortening velocity, maximal relengthening velocity, and time-to-peak. ∗p < 0.05.
3.2. Bevacizumab Induces Cell Apoptosis
Next, we examined alterations in cardiomyocyte apoptosis after bevacizumab treatment. First, TUNEL staining was used to observe cell apoptotic rate. As shown in Figures 2(a) and 2(b), compared to the control group, bevacizumab treatment significantly increased the number of TUNEL-positive cells in a dose-dependent manner; the low concentration of bevacizumab increased cardiomyocyte apoptosis rates to ~15%, while the high concentration increased rates to ~30%. Western blots were also performed to analyze changes in proapoptotic protein levels in cardiomyocytes. As shown in Figures 2(c)–2(e), compared to the control group, Bax, Bad, and caspase-9 protein levels increased dramatically in response to bevacizumab treatment in a dose-dependent manner (Figures 2(c)–2(e)). In contrast, Bcl-2 and c-IAP1 levels decreased significantly after bevacizumab treatment in a dose-dependent manner (Figures 2(f)–2(h)). Together, these results indicate that bevacizumab induces cardiomyocyte apoptosis.
Figure 2.
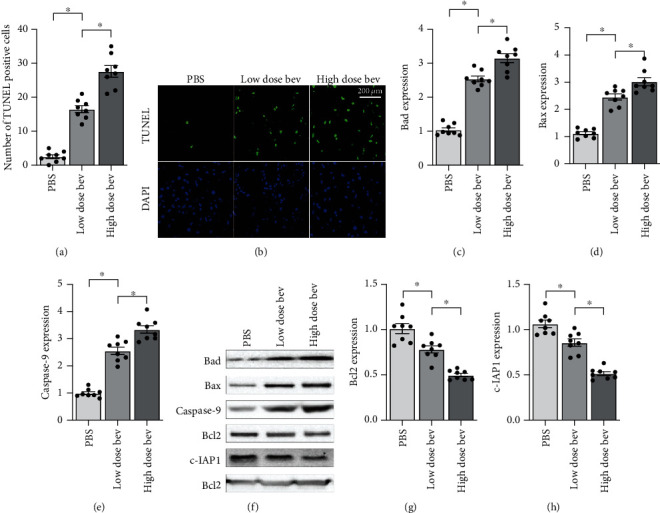
Bevacizumab induces cell apoptosis. (a, b) TUNEL staining was used to observe cell apoptosis. (c–h) Western blots were used to analyze alterations in expression of apoptosis-related proteins such as Bax, Bad, caspase-9, Bcl-2, and c-IAP1. ∗p < 0.05.
3.3. Bevacizumab Treatment Is Associated with Mitochondrial Dysfunction
Mitochondrial dysfunction is considered the primary mechanism underlying chemotherapy-mediated myocardial injury. The subsequent experiments were therefore performed to analyze the effects of bevacizumab on mitochondrial function. Mitochondrial metabolism was examined first due to the importance of ATP production for cardiomyocyte viability and function. Bevacizumab treatment significantly decreased ATP production in a dose-dependent manner (Figure 3(a)). Reduced mitochondrial ATP production can result from oxidative stress and respiration dysfunction. A mitochondrial ROS probe was therefore used to analyze changes in mitochondrial oxidative stress. As shown in Figures 3(b) and 3(c), compared to the control group, mitochondrial ROS levels were significantly increased after bevacizumab treatment in a dose-dependent manner. In contrast, concentrations of antioxidative enzymes such as GSH and SOD were significantly lower after bevacizumab treatment (Figures 3(d) and 3(e)), confirming increased oxidative stress within mitochondria. In addition to mitochondrial oxidative stress, an ELISA assay also demonstrated that mitochondrial respiration complex activity (including COX-I and COX-III) decreased significantly in cardiomyocytes treated with bevacizumab (Figures 3(f) and 3(g)). These findings indicate that bevacizumab induces mitochondrial dysfunction characterized by ROS production, respiration impairments, and reduced metabolism in cardiomyocytes.
Figure 3.
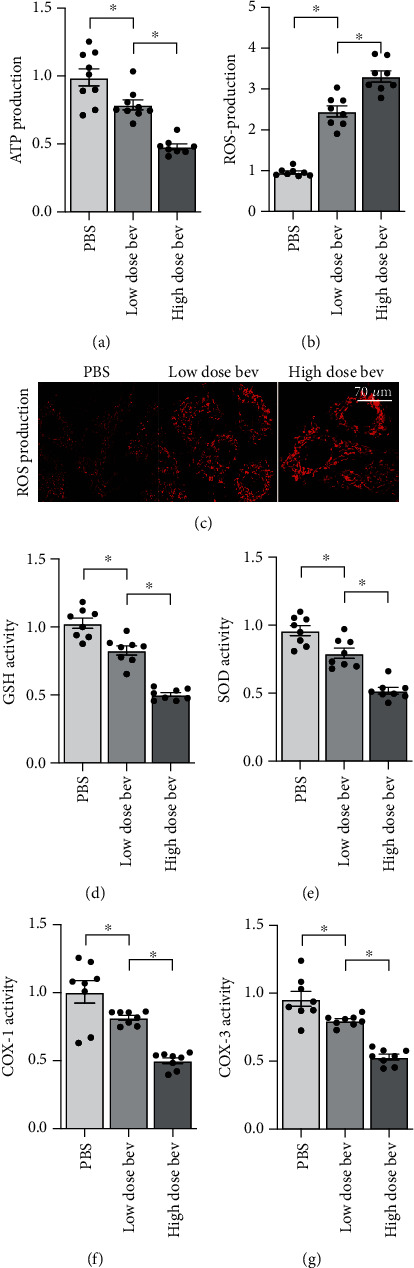
Bevacizumab treatment is associated with mitochondrial dysfunction. (a) ATP production was measured via ELISA in cardiomyocytes treated with bevacizumab. (b, c) Immunofluorescence staining was used to quantify mitochondrial ROS levels. (d, e) Changes in GSH, SOD, and GPX levels were analyzed in an ELISA. (f, g) Mitochondrial respiration complex activity was examined in an ELISA. ∗p < 0.05.
3.4. Bevacizumab Treatment Induces ER Stress in Cardiomyocytes
Next, we examined alterations in ER stress in cardiomyocytes treated with bevacizumab. ER stress is associated with intracellular calcium overload. An immunofluorescence assay was therefore used to examine alterations in intracellular calcium concentration. As shown in Figures 4(a) and 4(b), compared to the control group, bevacizumab treatment significantly increased intracellular calcium concentration in a dose-dependent manner. Abnormal calcium signaling activates ER stress, which is characterized by increased expression of CHOP and PERK. qPCR demonstrated that CHOP and PERK expression increased significantly in response to bevacizumab treatment (Figures 4(c) and 4(d)), indicative of ER stress activation. Excessive ER damage activates caspase-12, an upstream activator of caspase-3. An ELISA showed that caspase-12 activity increased dramatically after bevacizumab treatment in a dose-dependent manner (Figure 4(e)). Taken together, these results demonstrate that bevacizumab can induce ER stress in cardiomyocytes.
Figure 4.
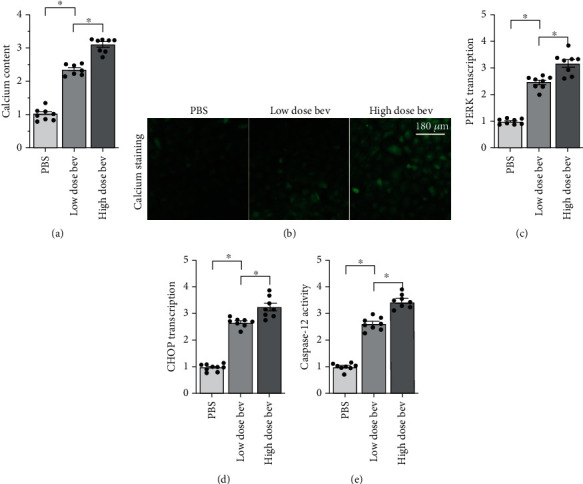
Bevacizumab treatment induces ER stress in cardiomyocytes. (a, b) Intracellular calcium levels were measured using immunofluorescence. (c, d) qPCR was used to analyze changes in CHOP and PERK levels. (e) ELISA was used to detect changes in caspase-12 activity. ∗p < 0.05.
3.5. Bevacizumab Inactivates the ERK Pathway in Cardiomyocytes
The ERK pathway, a classical signaling pathway responsible for cardiomyocyte survival, is a potential target of many chemotherapy drugs. We therefore examined whether ERK pathway activity was affected by bevacizumab. Western blots demonstrated that the ERK pathway was inactivated, as indicated by decreased ERK phosphorylation, in cardiomyocytes after bevacizumab treatment (Figures 5(a) and 5(b)). These results demonstrated that the ERK pathway was a downstream target of bevacizumab. To understand whether ERK inactivation contributed to bevacizumab-induced cardiomyocyte death and mitochondrial dysfunction, cardiomyocytes were incubated with an ERK inhibitor (PD98059). Cell viability was measured using an CCK-8 assay, and mitochondrial function was examined by measuring ATP production. As shown in Figure 5(c), compared to the control group, PD98059 treatment significantly reduced cardiomyocyte viability, and this effect was accompanied by a drop in mitochondrial ATP production (Figure 5(d)). This finding confirmed that ERK inhibition plays a role in cardiomyocyte damage associated with bevacizumab treatment.
Figure 5.
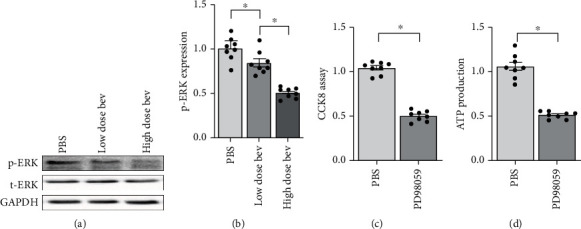
The ERK pathway is inactivated in cardiomyocytes treated with bevacizumab. (a, b) Western blots were used to analyze changes in ERK phosphorylation. (c) Cell viability was measured in a CCK-8 assay. Cardiomyocytes were treated with PD98059 to inhibit ERK activity. (d) ATP production was measured using an ELISA after PD98059 treatment. ∗p < 0.05.
4. Discussion
The cardiotoxic effects of chemotherapy have been well documented. Although chemotherapy and immunotherapy improve the prognoses of lung cancer patients, these treatments also significantly increase the risk of myocardial injury and heart failure. In the present study, we found that bevacizumab treatment induces cardiomyocyte damage as indicated by decreased cell viability, impaired contraction/relaxation, attenuated ATP production, elevated mitochondrial ROS production, decreased mitochondrial respiration complex transcription, increased intracellular calcium concentration, and activation of ER stress. These alterations are similar to pathological changes observed in various cardiovascular disorders such as myocardial infarction, heart failure, diabetic cardiomyopathy, and hypertension, suggesting that common molecular mechanisms and signaling pathways may be involved. Furthermore, we found that the ERK pathway is inactivated by bevacizumab, and decreased ERK activity promotes cardiomyocyte death and mitochondrial damage. Overall, this study helps to explain the intracellular molecular mechanisms, including mitochondrial dysfunction, ER stress, and inhibition of the ERK pathway, that underlie bevacizumab-induced cardiovascular toxicity.
Mitochondrial damage has been proposed as the primary cause of cardiomyocyte death and dysfunction [57]. Induction of mitochondrial fission promotes cardiomyocyte death by activating the caspase-9-related mitochondrial apoptotic pathway [58]. Additionally, inhibition of mitophagy leads to the accumulation of damaged mitochondria [59]. Chemotherapy drugs also contribute to cardiomyocyte dysfunction and death by affecting mitochondrial function. For example, doxorubicin-induced upregulation of p53 inhibits protective mechanisms in cardiac fibroblast mitochondria [20], and administration of mitochondria-associated protein LRPPRC protects against doxorubicin-induced cardiac injury by inhibiting ROS production [60]. Anthracycline-mediated cardiotoxicity is also associated with dysregulation of mitochondrial metabolism, although the detailed molecular mechanisms involved are not fully understood [23]. In the present study, bevacizumab treatment resulted in mitochondrial injury associated with metabolic dysregulation in cardiomyocytes. As far as we know, this is the first study to explore the influence of bevacizumab on mitochondrial homeostasis. While recent studies have examined the relationship between morphological alterations in mitochondria and chemotherapy-induced cardiovascular damage, additional studies are required to determine whether bevacizumab also alters mitochondrial morphology.
ER stress can act as an adaptive response in cardiomyocytes [61]. Mild ER stress is characterized by increased intracellular calcium levels, which accelerate calcium-dependent ATP metabolism [62]. However, excessive ER stress is associated with the activation of CHOP and PERK, transcription factors that promote expression of apoptosis-related genes such as Bax and Bad [63]. Irreversible ER stress promotes the activation of caspase-12, which in turn directly induces caspase-3 cleavage that ultimately leads to cell apoptosis [64]. ER stress has also been identified as a potential intracellular signaling transduction mechanism underlying cardiotoxicity associated with chemotherapy drugs [65, 66]. In the present study, we found that bevacizumab treatment is associated with intracellular calcium overload, ER stress, and caspase-12 activation, suggesting that ER stress contributes to bevacizumab-mediated cardiomyocyte damage. Notably, while our data do not establish a causal relationship between ER stress and mitochondrial dysfunction, many studies have reported that ER stress may contribute to mitochondrial damage [67]. We also did not explore the possibility of interactive effects between ER stress and mitochondrial dysfunction.
Taken together, our results demonstrate that bevacizumab-mediated myocardial injury is associated with mitochondrial damage, ER stress, and ERK pathway inactivation. However, some limitations of this study should be considered when interpreting these results. First, only in vitro experiments were performed, and in vivo studies are needed to confirm our findings on the cardiotoxic effects of bevacizumab. Second, although we found that ERK inactivation is associated with cardiomyocyte death and mitochondrial damage, it remains unclear whether bevacizumab induces mitochondrial damage and ER stress specifically by directly inhibiting the ERK pathway. Third, additional bevacizumab concentrations beyond those tested here may have different effects on cardiomyocyte viability and function, and further study is needed to determine the dose threshold beyond which bevacizumab triggers cardiomyocyte damage.
Data Availability
The analyzed datasets that were generated during the study are available from the corresponding author upon reasonable request.
Conflicts of Interest
All authors declare that there are no conflicts of interest associated with this study.
Authors' Contributions
Yue Li and Wei Tian contributed to this article equally.
References
- 1.Akamatsu H., Toi Y., Hayashi H., et al. Efficacy of osimertinib plus bevacizumab vs osimertinib in patients with EGFRT790M–mutated non-small cell lung cancer previously treated with epidermal growth factor receptor-tyrosine kinase inhibitor. JAMA Oncology. 2021;8 doi: 10.1001/jamaoncol.2020.6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamamoto N., Seto T., Nishio M., et al. Erlotinib plus bevacizumab vs erlotinib monotherapy as first-line treatment for advanced EGFR mutation-positive non-squamous non-small-cell lung cancer: survival follow-up results of the randomized JO25567 study. Lung Cancer. 2021;151:20–24. doi: 10.1016/j.lungcan.2020.11.020. [DOI] [PubMed] [Google Scholar]
- 3.di Somma M., Vliora M., Grillo E., et al. Role of VEGFs in metabolic disorders. Angiogenesis. 2020;23(2):119–130. doi: 10.1007/s10456-019-09700-1. [DOI] [PubMed] [Google Scholar]
- 4.van Leeuwen M. T., Luu S., Gurney H., et al. Cardiovascular toxicity of targeted therapies for cancer: an overview of systematic reviews. JNCI Cancer Spectrum. 2020;4(6) doi: 10.1093/jncics/pkaa076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yetkin-Arik B., Kastelein A. W., Klaassen I., et al. Angiogenesis in gynecological cancers and the options for anti-angiogenesis therapy. Biochimica Et Biophysica Acta. Reviews on Cancer. 2021;1875(1, article 188446) doi: 10.1016/j.bbcan.2020.188446. [DOI] [PubMed] [Google Scholar]
- 6.Huang H., Cao Y., Dong Y., et al. Cardiac hemodynamic response to the 6-minute walk test in patients with intestinal carcinoma undergoing bevacizumab treatment. Annals of Palliative Medicine. 2021;10(2):1362–1369. doi: 10.21037/apm-20-435. [DOI] [PubMed] [Google Scholar]
- 7.Winter M. P., Sharma S., Altmann J., et al. Interruption of vascular endothelial growth factor receptor 2 signaling induces a proliferative pulmonary vasculopathy and pulmonary hypertension. Basic Research in Cardiology. 2020;115(6):p. 58. doi: 10.1007/s00395-020-0811-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buglak D. B., Kushner E. J., Marvin A. P., Davis K. L., Bautch V. L. Excess centrosomes disrupt vascular lumenization and endothelial cell adherens junctions. Angiogenesis. 2020;23(4):567–575. doi: 10.1007/s10456-020-09737-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghaemi Kerahrodi J., Michal M. The fear-defense system, emotions, and oxidative stress. Redox Biology. 2020;37, article 101588 doi: 10.1016/j.redox.2020.101588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalyanaraman B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: have we been barking up the wrong tree? Redox Biology. 2020;29:p. 101394. doi: 10.1016/j.redox.2019.101394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou H., Toan S. Pathological roles of mitochondrial oxidative stress and mitochondrial dynamics in cardiac microvascular ischemia/reperfusion injury. Biomolecules. 2020;10(1):p. 85. doi: 10.3390/biom10010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J., Toan S., Zhou H. Mitochondrial quality control in cardiac microvascular ischemia-reperfusion injury: new insights into the mechanisms and therapeutic potentials. Pharmacological Research. 2020;156:p. 104771. doi: 10.1016/j.phrs.2020.104771. [DOI] [PubMed] [Google Scholar]
- 13.Yuan M., Gong M., Zhang Z., et al. Hyperglycemia induces endoplasmic reticulum stress in atrial cardiomyocytes, and mitofusin-2 downregulation prevents mitochondrial dysfunction and subsequent cell death. Oxidative Medicine and Cellular Longevity. 2020;2020:14. doi: 10.1155/2020/6569728.6569728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nie X., Tang W., Zhang Z., et al. Procyanidin B2 mitigates endothelial endoplasmic reticulum stress through a PPARδ-dependent mechanism. Redox Biology. 2020;37:p. 101728. doi: 10.1016/j.redox.2020.101728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmed A. Z., Satyam S. M., Shetty P., D’Souza M. R. Methyl gallate attenuates doxorubicin-induced cardiotoxicity in rats by suppressing oxidative stress. Scientifica. 2021;2021:12. doi: 10.1155/2021/6694340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia P., Chen J., Liu Y., Fletcher M., Jensen B. C., Cheng Z. Doxorubicin induces cardiomyocyte apoptosis and atrophy through cyclin- dependent kinase 2-mediated activation of forkhead box O1. The Journal of Biological Chemistry. 2020;295(13):4265–4276. doi: 10.1074/jbc.RA119.011571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shao H., Dong L., Feng Y., Wang C., Tong H. The protective effect of L-glutamine against acute cantharidin-induced cardiotoxicity in the mice. BMC Pharmacology and Toxicology. 2020;21(1):p. 71. doi: 10.1186/s40360-020-00449-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dieterich L. C., Tacconi C., Menzi F., et al. Lymphatic MAFB regulates vascular patterning during developmental and pathological lymphangiogenesis. Angiogenesis. 2020;23(3):411–423. doi: 10.1007/s10456-020-09721-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brandolini L., Antonosante A., Giorgio C., et al. NSAIDs-dependent adaption of the mitochondria-proteasome system in immortalized human cardiomyocytes. Scientific Reports. 2020;10(1, article 18337) doi: 10.1038/s41598-020-75394-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mancilla T. R., Davis L. R., Aune G. J. Doxorubicin-induced p53 interferes with mitophagy in cardiac fibroblasts. PLoS One. 2020;15(9, article e0238856) doi: 10.1371/journal.pone.0238856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao F., Maguire M. L., McAndrew D. J., et al. Overexpression of mitochondrial creatine kinase preserves cardiac energetics without ameliorating murine chronic heart failure. Basic Research in Cardiology. 2020;115(2):p. 12. doi: 10.1007/s00395-020-0777-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu A., Deng F., Chen Y., et al. NF-κB pathway activation during endothelial-to-mesenchymal transition in a rat model of doxorubicin-induced cardiotoxicity. Biomedicine & Pharmacotherapy. 2020;130, article 110525 doi: 10.1016/j.biopha.2020.110525. [DOI] [PubMed] [Google Scholar]
- 23.Murabito A., Hirsch E., Ghigo A. Mechanisms of anthracycline-induced cardiotoxicity: is mitochondrial dysfunction the answer? Frontiers in Cardiovascular Medicine. 2020;7:p. 35. doi: 10.3389/fcvm.2020.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang B., Li H., Qiao Y., et al. Tetramethylpyrazine attenuates the endotheliotoxicity and the mitochondrial dysfunction by doxorubicin via 14-3-3γ/Bcl-2. Oxidative Medicine and Cellular Longevity. 2019;2019:20. doi: 10.1155/2019/5820415.5820415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bae H., Lee J. Y., Yang C., Song G., Lim W. Fucoidan derived from Fucus vesiculosus inhibits the development of human ovarian cancer via the disturbance of calcium homeostasis, endoplasmic reticulum stress, and angiogenesis. Marine Drugs. 2020;18(1):p. 45. doi: 10.3390/md18010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lage R., Cebro-Márquez M., Rodríguez-Mañero M., González-Juanatey J. R., Moscoso I. Omentin protects H9c2 cells against docetaxel cardiotoxicity. PLoS One. 2019;14(2, article e0212782) doi: 10.1371/journal.pone.0212782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L., Chan J. Y., Zhou X., et al. A novel agent enhances the chemotherapeutic efficacy of doxorubicin in MCF-7 breast cancer cells. Frontiers in Pharmacology. 2016;7:p. 249. doi: 10.3389/fphar.2016.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lama S., Monda V., Rizzo M. R., et al. Cardioprotective effects of Taurisolo® in cardiomyoblast H9c2 cells under high-glucose and trimethylamine N-oxide treatment via de novo sphingolipid synthesis. Oxidative Medicine and Cellular Longevity. 2020;2020:11. doi: 10.1155/2020/2961406.2961406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xin T., Lv W., Liu D., Jing Y., Hu F. Opa1 reduces hypoxia-induced cardiomyocyte death by improving mitochondrial quality control. Frontiers in Cell and Development Biology. 2020;8:p. 853. doi: 10.3389/fcell.2020.00853. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Shi G., Wang Y., Yang J., et al. Effect of cryptotanshinone on measures of rat cardiomyocyte oxidative stress and gene activation associated with apoptosis. Cardiorenal Medicine. 2021;11(1):18–26. doi: 10.1159/000507184. [DOI] [PubMed] [Google Scholar]
- 31.Shen Y., Zhang X., Li C., et al. Pressure overload promotes cystatin C secretion of cardiomyocytes to regulate the MAPK signaling pathway and mediate cardiac hypertrophy. Annals of Translational Medicine. 2020;8(22):p. 1514. doi: 10.21037/atm-20-7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russignan A., Dal Collo G., Bagnato A., et al. Targeting the endothelin-1 receptors curtails tumor growth and angiogenesis in multiple myeloma. Frontiers in Oncology. 2021;10, article 600025 doi: 10.3389/fonc.2020.600025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang F., Wang H., Liu X., et al. Neuregulin-1 alleviate oxidative stress and mitigate inflammation by suppressing NOX4 and NLRP3/caspase-1 in myocardial ischaemia-reperfusion injury. Journal of Cellular and Molecular Medicine. 2021;25(3):1783–1795. doi: 10.1111/jcmm.16287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Z., Zhu L., Xing Y., Zhang Z. Praja2 suppresses the growth of gastric cancer by ubiquitylation of KSR1 and inhibiting MEK-ERK signal pathways. Aging. 2021;13:3886–3897. doi: 10.18632/aging.202356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J., Lin F. L., Leung J. Y. K., et al. A drug-tunable Flt23k gene therapy for controlled intervention in retinal neovascularization. Angiogenesis. 2021;24(1):97–110. doi: 10.1007/s10456-020-09745-7. [DOI] [PubMed] [Google Scholar]
- 36.Adapala R. K., Kanugula A. K., Paruchuri S., Chilian W. M., Thodeti C. K. TRPV4 deletion protects heart from myocardial infarction-induced adverse remodeling via modulation of cardiac fibroblast differentiation. Basic Research in Cardiology. 2020;115(2):p. 14. doi: 10.1007/s00395-020-0775-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou H., Toan S., Zhu P., Wang J., Ren J., Zhang Y. DNA-PKcs promotes cardiac ischemia reperfusion injury through mitigating BI-1-governed mitochondrial homeostasis. Basic Research in Cardiology. 2020;115(2):p. 11. doi: 10.1007/s00395-019-0773-7. [DOI] [PubMed] [Google Scholar]
- 38.Escoll M., Lastra D., Pajares M., et al. Transcription factor NRF2 uses the Hippo pathway effector TAZ to induce tumorigenesis in glioblastomas. Redox Biology. 2020;30, article 101425 doi: 10.1016/j.redox.2019.101425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lubos N., van der Gaag S., Gerçek M., Kant S., Leube R. E., Krusche C. A. Inflammation shapes pathogenesis of murine arrhythmogenic cardiomyopathy. Basic Research in Cardiology. 2020;115(4):p. 42. doi: 10.1007/s00395-020-0803-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan Y., Mui D., Toan S., Zhu P., Li R., Zhou H. SERCA overexpression improves mitochondrial quality control and attenuates cardiac microvascular ischemia-reperfusion injury. Molecular Therapy Nucleic Acids. 2020;22:696–707. doi: 10.1016/j.omtn.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lyu J., Wang M., Kang X., et al. Macrophage-mediated regulation of catecholamines in sympathetic neural remodeling after myocardial infarction. Basic Research in Cardiology. 2020;115(5):p. 56. doi: 10.1007/s00395-020-0813-3. [DOI] [PubMed] [Google Scholar]
- 42.Wang J., Toan S., Li R., Zhou H. Melatonin fine-tunes intracellular calcium signals and eliminates myocardial damage through the IP3R/MCU pathways in cardiorenal syndrome type 3. Biochemical Pharmacology. 2020;174:p. 113832. doi: 10.1016/j.bcp.2020.113832. [DOI] [PubMed] [Google Scholar]
- 43.Wang J., Zhu P., Toan S., Li R., Ren J., Zhou H. Pum2-Mff axis fine-tunes mitochondrial quality control in acute ischemic kidney injury. Cell Biology and Toxicology. 2020;36(4):365–378. doi: 10.1007/s10565-020-09513-9. [DOI] [PubMed] [Google Scholar]
- 44.García-Martínez E., Redondo A., Piulats J. M., Rodríguez A., Casado A. Are antiangiogenics a good ‘partner’ for immunotherapy in ovarian cancer? Angiogenesis. 2020;23(4):543–557. doi: 10.1007/s10456-020-09734-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuo K. L., Zhao J. F., Huang P. H., Guo B. C., Tarng D. C., Lee T. S. Indoxyl sulfate impairs valsartan-induced neovascularization. Redox Biology. 2020;30:p. 101433. doi: 10.1016/j.redox.2020.101433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H., Xia N. The role of oxidative stress in cardiovascular disease caused by social isolation and loneliness. Redox Biology. 2020;37, article 101585 doi: 10.1016/j.redox.2020.101585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma Y. S., Chu K. J., Ling C. C., et al. Long noncoding RNA OIP5-AS1 promotes the progression of liver hepatocellular carcinoma via regulating the hsa-miR-26a-3p/EPHA2 axis. Molecular Therapy Nucleic Acids. 2020;21:229–241. doi: 10.1016/j.omtn.2020.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Kong E., Kim H. D., Kim J. Deleting key autophagy elongation proteins induces acquirement of tumor- associated phenotypes via ISG15. Cell Death and Differentiation. 2020;27(8):2517–2530. doi: 10.1038/s41418-020-0519-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou H., Zhu P., Wang J., Toan S., Ren J. DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal Transduction and Targeted Therapy. 2019;4(1, article 56) doi: 10.1038/s41392-019-0094-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang Y., Ma L., Wang C., et al. Matrix metalloproteinase-7 in platelet-activated macrophages accounts for cardiac remodeling in uremic mice. Basic Research in Cardiology. 2020;115(3):p. 30. doi: 10.1007/s00395-020-0789-z. [DOI] [PubMed] [Google Scholar]
- 51.Nguyen Q. L., Okuno N., Hamashima T., et al. Vascular PDGFR-alpha protects against BBB dysfunction after stroke in mice. Angiogenesis. 2021;24(1):35–46. doi: 10.1007/s10456-020-09742-w. [DOI] [PubMed] [Google Scholar]
- 52.Wang J., Zhu P., Li R., Ren J., Zhang Y., Zhou H. Bax inhibitor 1 preserves mitochondrial homeostasis in acute kidney injury through promoting mitochondrial retention of PHB2. Theranostics. 2020;10(1):384–397. doi: 10.7150/thno.40098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu J., Cai Q., Wang W., et al. Ginsenoside Rh2 pretreatment and withdrawal reactivated the pentose phosphate pathway to ameliorate intracellular redox disturbance and promoted intratumoral penetration of adriamycin. Redox Biology. 2020;32:p. 101452. doi: 10.1016/j.redox.2020.101452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J., Chen Z., Dai Q., et al. Intravenously delivered mesenchymal stem cells prevent microvascular obstruction formation after myocardial ischemia/reperfusion injury. Basic Research in Cardiology. 2020;115(4):p. 40. doi: 10.1007/s00395-020-0800-8. [DOI] [PubMed] [Google Scholar]
- 55.Wang J., Zhu P., Li R., Ren J., Zhou H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biology. 2020;30:p. 101415. doi: 10.1016/j.redox.2019.101415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mulcahy Levy J. M., Thorburn A. Autophagy in cancer: moving from understanding mechanism to improving therapy responses in patients. Cell Death and Differentiation. 2020;27(3):843–857. doi: 10.1038/s41418-019-0474-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang J., Toan S., Zhou H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Angiogenesis. 2020;23(3):299–314. doi: 10.1007/s10456-020-09720-2. [DOI] [PubMed] [Google Scholar]
- 58.Jin Q., Li R., Hu N., et al. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff- required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biology. 2018;14:576–587. doi: 10.1016/j.redox.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou H., Zhu P., Wang J., Zhu H., Ren J., Chen Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death and Differentiation. 2018;25(6):1080–1093. doi: 10.1038/s41418-018-0086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang Q., Xiong W., Ke X., Zhang J., Xia Y., Liu D. Mitochondria-associated protein LRPPRC exerts cardioprotective effects against doxorubicin-induced toxicity, potentially via inhibition of ROS accumulation. Experimental and Therapeutic Medicine. 2020;20:3837–3845. doi: 10.3892/etm.2020.9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou H., Wang S., Hu S., Chen Y., Ren J. ER-mitochondria microdomains in cardiac ischemia-reperfusion injury: a fresh perspective. Frontiers in Physiology. 2018;9:p. 755. doi: 10.3389/fphys.2018.00755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li J., Xu C., Liu Y., et al. Fibroblast growth factor 21 inhibited ischemic arrhythmias via targeting miR-143/EGR1 axis. Basic Research in Cardiology. 2020;115(2):p. 9. doi: 10.1007/s00395-019-0768-4. [DOI] [PubMed] [Google Scholar]
- 63.Heusch G. Coronary microvascular obstruction: the new frontier in cardioprotection. Basic Research in Cardiology. 2019;114(6):p. 45. doi: 10.1007/s00395-019-0756-8. [DOI] [PubMed] [Google Scholar]
- 64.Hughes W. E., Beyer A. M., Gutterman D. D. Vascular autophagy in health and disease. Basic Research in Cardiology. 2020;115(4):p. 41. doi: 10.1007/s00395-020-0802-6. [DOI] [PubMed] [Google Scholar]
- 65.Kerkelä R., Grazette L., Yacobi R., et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nature Medicine. 2006;12(8):908–916. doi: 10.1038/nm1446. [DOI] [PubMed] [Google Scholar]
- 66.Lu M., Merali S., Gordon R., et al. Prevention of doxorubicin cardiopathic changes by a benzyl styryl sulfone in mice. Genes & Cancer. 2012;2(10):985–992. doi: 10.1177/1947601911436199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giamogante F., Barazzuol L., Brini M., Calì T. ER-mitochondria contact sites reporters: strengths and weaknesses of the available approaches. International Journal of Molecular Sciences. 2020;21(21):p. 8157. doi: 10.3390/ijms21218157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The analyzed datasets that were generated during the study are available from the corresponding author upon reasonable request.