

Abstract
Over the past decade, genome-wide association studies (GWAS) have emerged as a powerful tool to understand the genetic basis of complex traits in humans. The GWAS approach has been successfully applied to primary glomerular disorders, providing numerous novel insights into the genetic architecture of IgA nephropathy, membranous nephropathy, and steroid-sensitive nephrotic syndrome. IgA nephropathy appears to have a highly complex polygenic architecture, with nearly 20 genome-wide significant loci of small-to-moderate effects discovered to date. In contrast, the genetic susceptibility to membranous nephropathy and steroid-sensitive nephrotic syndrome appears to be driven by a small number of large-effect loci. The MHC locus on chromosome 6p21 is strongly associated with genetic susceptibility to all major types of immune-mediated glomerulopathies. However, a distinct set of classical HLA alleles is associated with each individual disease type, pinpointing to specific immune mechanisms underlying each of these conditions. Additional insights from the discovery of non-HLA risk loci reinforced the role of innate and adaptive immunity in the pathogenesis of these disorders, and highlighted important susceptibility overlaps between glomerular and other autoimmune and inflammatory conditions. Despite these initial successes, much larger GWAS and sequencing studies are still needed for each individual glomerular disease type. Increased power will be critical to comprehensively test for genetic effects across the full spectrum of allelic frequencies, to detect gene-gene and gene-environment interactions, and to potentially improve the performance of polygenic risk predictors. Moreover, the existing studies are limited mostly to European and East Asian populations, stressing the urgency to expand genetic discovery efforts to more diverse populations worldwide.
Keywords: glomerular disease, IgA nephropathy, membranous nephropathy, steroid sensitive nephrotic syndrome, GWAS, human genetics, kidney genomics series, nephrotic syndrome, glomerulonephritis
Introduction
Although individual histopathologic subtypes of glomerular disorders are relatively rare, cumulatively, glomerular diseases represent the third leading cause of end stage kidney failure in the United States. Most forms are thought to be immune mediated, but the underlying pathogenic mechanisms are likely heterogenous and may be unique to each individual glomerular disease type. An immune-mediated glomerular injury is complex and involves activation of innate and adaptive immune systems, resulting in diverse clinical and pathologic manifestations. Human genetic studies provide a powerful approach to uncover the earliest molecular disease drivers that could potentially be targeted therapeutically. Such knowledge can also facilitate improved disease classification based on molecular mechanisms, the first step in the development of targeted therapies and personalized care. The genome-wide association study (GWAS) approach has been greatly successful in uncovering the genetic underpinnings of other complex traits, especially autoimmune diseases and inflammatory conditions. In this review, we will summarize the current state of GWAS for the three most common types of glomerular disease: IgA nephropathy, membranous nephropathy, and steroid-sensitive nephrotic syndrome (Figure 1).
Figure 1.
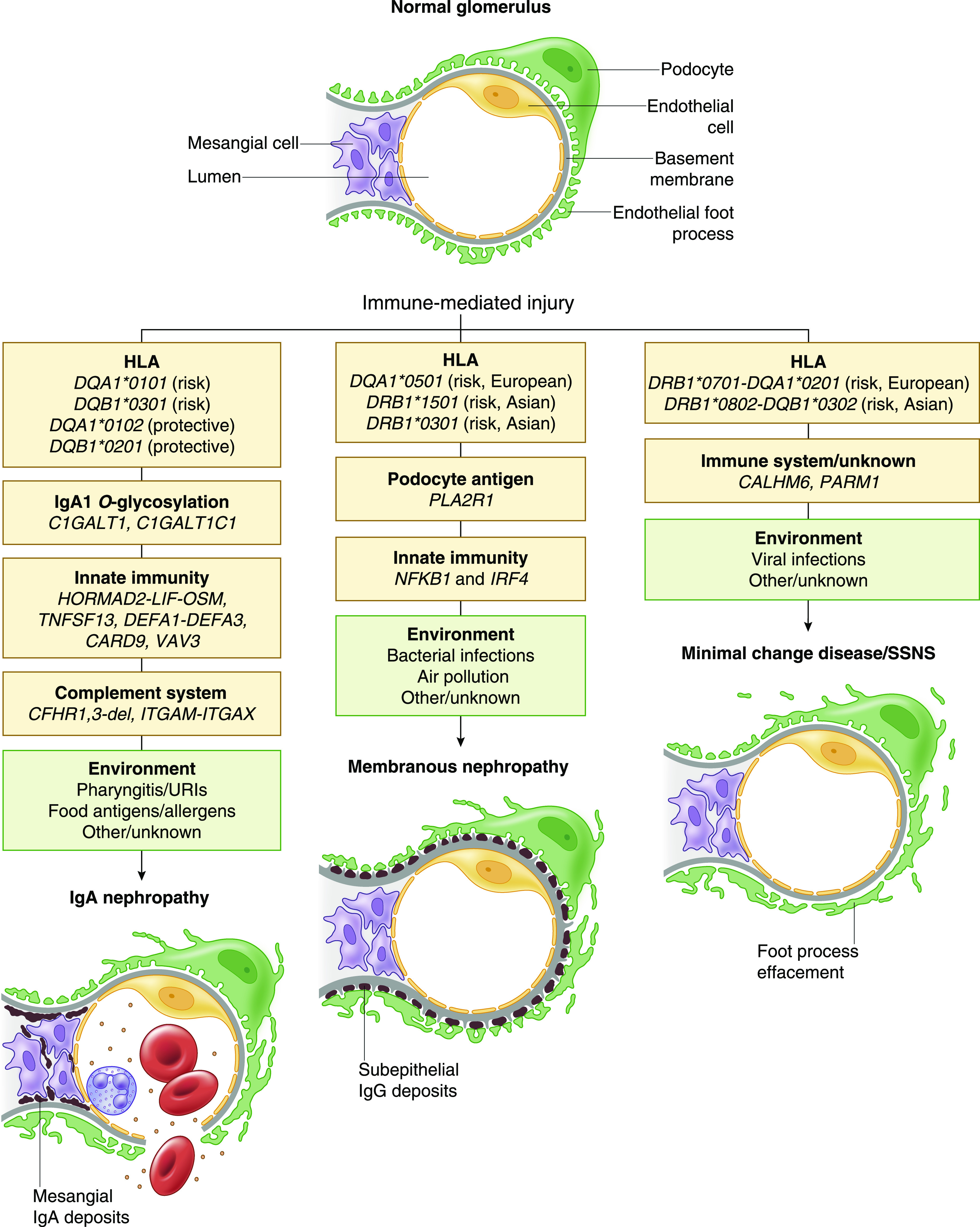
Complex determination of immune-mediated glomerular injury involves interactions of genetic risk loci with environmental exposures. The genetic factors are listed in yellow boxes and include high-priority candidate genes at genome-wide significant loci; potential environmental disease triggers are listed in green boxes. Schematic representations of histopathologic injury patterns for IgA nephropathy, membranous nephropathy, and steroid-sensitive nephrotic syndrome (SSNS) are also provided. URIs, upper respiratory infections.
Immunoglobulin A Nephropathy
The diagnosis of IgA nephropathy relies on the detection of mesangial IgA deposits in kidney biopsy specimens (1). The disease process is thought to be related to increased production of galactose-deficient IgA1 (Gd-IgA1) that triggers an abnormal anti-glycan antibody response in predisposed individuals, subsequently leading to the formation of circulating immune complexes and their trapping in the glomerular mesangium (2,3). Interestingly, IgA nephropathy is most common in East Asia and less frequent in Europe. The same trend is observed across immigrant populations in the United States, suggesting these differences are likely due to genetic factors rather than environmental exposures or disparities in clinical care (4). The prevalence of IgA nephropathy in native African populations is not well studied, but the disease appears to have lower prevalence in African American compared with European American populations (4). The role of inherited factors is further supported by numerous reports of extended pedigrees with familial IgA nephropathy, although no single gene causes have been identified to date.
Over the past decade, GWAS have provided numerous insights into the genetic architecture of IgA nephropathy. The first GWAS performed in cohorts of European ancestry (5) detected a strong and genome-wide significant signal in the MHC region (rs3115573; odds ratio [OR], 1.68; P=1.0×10−9). No association outside the MHC region were observed, but the study was underpowered to detect smaller effects.
The second GWAS in both East Asian and European cohorts confirmed the association signal in the MHC (rs9275596; OR, 1.32; P=1.6×10−26) and provided evidence for at least two independent signals within this region (6). Moreover, two non-MHC loci were also identified, including a variant on chromosome 1q32 at the CFH locus (rs6677604; OR, 0.68; P=3.0×10−10) subsequently fine mapped to a protective deletion of CFHR1 and CFHR3 genes (7). These genes encode factor H–related peptides that competitively inhibit factor H, the key regulator of the alternative pathway, and the deletion appears to enhance the protective effect of factor H (8). Similar protective effects of the CFHR1 and CFHR3 deletion were observed in GWAS of age-related macular degeneration, a retinal disease associated with overactivation of the alternative complement pathway (9). These findings provided the first genetic evidence for the involvement of the alternative pathway in IgA nephropathy, and prioritized this pathway as a potential therapeutic target (8).
The second non-MHC signal was detected on chromosome 22q12 (rs2412971; OR, 1.25; P=1.9×10−9). Due to extended linkage disequilibrium at this locus, the region of association was broad and spanned multiple genes. Among them were LIF and OSM, encoding two related cytokines previously implicated in mucosal immunity and inflammation. Notably, the IgA nephropathy risk allele at this locus has been reported to have a concordant effect on the risk of tonsillitis and tonsillectomy (10), but an opposed effect on the risk of early onset inflammatory bowel disease and Crohn’s disease (11). Although both LIF and OSM represent excellent positional candidates, the specific causal variant and its target gene have not yet been resolved for this locus.
The chromosome 22q12 association was subsequently replicated in the third GWAS for IgA nephropathy performed in Chinese cohorts (12). This study also identified two additional genome-wide significant loci encoding genes related to mucosal immunity: TNFSF13 locus (rs3803800; OR, 1.21; P=9.4×10−11) encoding APRIL, a powerful mucosal cytokine stimulating IgA production (13); and DEFA locus (rs2738048; OR, 1.21; P=3.2×10−14) encoding human antimicrobial peptides called α-defensins secreted at mucosal surfaces (14).
The identification of TNFSF13 locus was particularly exciting because APRIL is known to be directly involved in the regulation of IgA production. The inactivation of APRIL in mice produces partial IgA deficiency and reduced IgA antibody responses (15). Human mutations inactivating TACI, the main receptor for APRIL, produce IgA deficiency and common variable immunodeficiency syndrome. These findings pointed to APRIL-TACI signaling as a potential therapeutic target, motivating ongoing clinical trials of drugs targeting this ligand-receptor pair in IgA nephropathy.
The second locus discovered in this study encoded α-defensins 1 and 3 (DEFA1 and DEFA3), and was subsequently fine mapped in Chinese cohorts (16). DEFA1 and DEFA3 genes are subject to complex multicopy variation, and the number of gene copies correlated with the expression level of these peptides (17). Direct typing of copy number variants combined with conditional analyses of the locus demonstrated that higher copy numbers of DEFA1/A3 genes were significantly protective from IgA nephropathy (16). The analysis of clinical data also suggested the patients with fewer copy numbers of α-defensins were at a higher risk of disease progression.
A more recent GWAS published by the same group involved an expanded analysis of the previously reported Chinese cohorts (18) and described three additional genome-wide significant loci, including ST6GAL1 (rs7634389; OR, 1.13; P=7.3×10−10), ACCS (rs2074038; OR, 1.14; P=3.9×10−9), and ODF1-KLF10 (rs2033562; OR, 1.13; P=1.4×10−9). These loci are not supported by European GWAS and may be Asian specific, but still require independent replication in larger studies. The underlying causal genes and their pathogenic role in IgA nephropathy are not known.
The largest biethnic GWAS analyzed 7658 cases and 12,954 controls of European and East Asian ancestry (19). In addition to confirming associations at the MHC, HORMAD2, CFH, TNFSF13, and DEFA loci, the study identified several novel loci, including VAV3, ITGAM-ITGAX, and CARD9, as well as new independent signals at the HLA and DEFA loci.
The ITGAM-ITGAX locus represented the strongest signal outside the MHC region in European cohorts (rs11574637-T; OR, 1.32; P=8.1×10−13). The risk allele is fixed in East Asian populations, explaining why this signal was not detected in prior Chinese studies. ITGAM and ITGAX encode leukocyte-specific integrins αM and αX, components of complement receptors 3 and 4 involved in leukocyte cell adhesion, migration, and phagocytosis of complement-coated particles by macrophages.
The risk variant at the CARD9 locus (rs4077515-T; OR, 1.16; P=1.2×10−9) represented p.Ser12Asn substitution in CARD9 and had been associated with increased risk of inflammatory bowel disease in previous GWAS (11). This allele was also associated with higher mRNA expression of CARD9 in immune cells. Because CARD9 is a powerful proinflammatory molecule that promotes activation of NF-κB pathway, this locus provided the first genetic evidence for the involvement of NF-κB in the pathogenesis of IgA nephropathy.
Additional insights from this study were provided by investigations of a genetic risk score that captured cumulative effects of GWAS risk alleles. The geospatial distribution of GWAS risk alleles varied in a pattern that paralleled the geographic distribution of the disease prevalence, with strong west-to-east and south-to-north risk gradients (4). Simulation studies demonstrated this pattern is unlikely to occur as a result of genetic drift alone, suggesting a potential role of multilocus adaptation. Interestingly, the genetic risk correlated strongly with variation in local pathogens, particularly helminth diversity, suggesting a possible role for host-intestinal pathogen interactions in shaping the susceptibility landscape.
Moreover, the cumulative burden of risk alleles was strongly associated with age at disease onset. This implies that early onset or pediatric cases may have a stronger genetic determination, and genetic studies of this population could be most fruitful. To date, there are no GWAS or sequencing studies of pediatric IgA nephropathy, although a consortium named GIGA-kids (Genomics of IgA-related disorders in kids, www.gigakids.org) has been formed to specifically address this issue.
Another powerful GWAS approach involves mapping genetic loci for disease-related quantitative endophenotypes. In this approach, a continuous trait, such as serum level of a circulating biomarker causally related to the disease of interest, is regressed against each genotype genome wide. One of the most promising quantitative endophenotypes for IgA nephropathy is serum Gd-IgA1 level. This biomarker has been reproducibly associated with IgA nephropathy and its progression, experimental data supports its causal role, and it has high heritability in family-based studies ranging from 40% to 80% (20,21).
The first quantitative GWAS for serum levels of Gd-IgA1 was performed in 2633 subjects of European and East Asian ancestry and discovered two distinct genome-wide significant loci containing C1GALT1 (rs13226913-T; OR, 0.22; P=3.2×10−11) and C1GALT1C1 (rs5910940-T; OR, 0.14; P=2.7×10−8) genes. These genes encoded molecular partners essential for enzymatic O-glycosylation of IgA1. Jointly, these two loci explained approximately 7% of variance in circulating Gd-IgA1 in European cohorts, but only 2% in East Asian cohorts (22). The Gd-IgA1–increasing allele of rs13226913, a noncoding single-nucleotide polymorphism with a regulatory function, appears to have the largest effect and is common in European populations, but rare in East Asian populations. This allele exhibits a cis–expression quantitative trait locus (cis-eQTL) effect, reducing mRNA expression of C1GALT1 in blood and other tissues. Furthermore, in vitro small interfering RNA knockdown studies demonstrated that mRNA levels of both C1GALT1 and C1GALT1C1 determine the rate of secretion of Gd-IgA1 in IgA1-producing cells. The second independent GWAS for Gd-IgA1 (23) replicated the associations of C1GALT1 and C1GALT1C1 loci. This study also demonstrated that individuals of European ancestry exhibited significantly higher Gd-IgA1 levels compared with East Asian ancestry, and confirmed that high Gd-IgA1 levels correlated with kidney disease progression.
Taken together, several GWAS in predominantly European and East Asian cohorts identified nearly 20 independent risk alleles for IgA nephropathy. These alleles explained approximately 7% of the disease risk, and it is expected that a larger fraction of risk will be accounted for with expanded discovery studies. Although GWAS have already provided unbiased insights into pathogenic mechanisms, most loci still require follow-up to identify the underlying causal variants and related molecular mechanisms. Moreover, the contribution of rare variants remains understudied. Finally, there have been practically no genetic studies of IgA nephropathy in African, Central and Southwest Asian, or Native American populations, highlighting the need to expand genetic discovery efforts to more diverse populations worldwide.
Membranous Nephropathy
Membranous nephropathy is an adult-onset kidney disease, with a peak incidence between 30 and 50 years of age. Membranous nephropathy is considerably less prevalent compared with IgA nephropathy, affecting only one in 100,000 patients (24). Approximately 70% of cases have no apparent underlying systemic condition, and are thus labeled primary or idiopathic membranous nephropathy; whereas the remaining 30% cases are found in the setting of malignancy, infection, or autoimmune disease (25). The report of a rare form of neonatal membranous nephropathy caused by pathogenic antibodies against neutral endopeptidase suggested membranous nephropathy was a disease of anti-podocyte antibodies (26). Subsequent discovery of additional podocyte antigens targeted in primary membranous nephropathy, including PLA2R (70% of cases) (27) and THSD7A (2%–3% of cases) (28), further reinforced this model and allowed for a more meaningful antigen-based disease subclassification. More recently, antibodies against exostosin 1 and 2 and neural EGF-like 1 (NELL-1) protein have also been identified in a subgroup of patients with PLA2R1-negative membranous nephropathy (29,30). Additional, less common antigens are probably still unknown. Taken together, current studies support the pathogenesis model in which a kidney-specific genetic or environmental perturbation increases the immunogenicity of a podocyte antigen. When combined with an immune defect in self-recognition, this perturbation triggers an autoimmune humoral response characterized by anti-podocyte antibodies, leading to subsequent podocyte injury manifesting as nephrotic syndrome.
The first GWAS for membranous nephropathy involved European case-control cohorts with only 556 cases of primary disease (31). Despite small sample size, two highly significant loci were discovered: the HLA-DQA1 locus (rs2187668; OR, 4.32; P=8.0×10−93) and the PLA2R1 locus (rs4664308; OR, 2.28; P=8.6×10−29) encoding PLA2R, the most common antigen in primary membranous nephropathy. Notably, the effect estimates for these loci were high, and there was evidence for a remarkable genetic interaction between these loci, with homozygosity for both risk alleles increasing the odds of disease by nearly 80-fold. To date, similar interactions have only rarely been described in GWAS, highlighting the unusual genetic architecture of membranous nephropathy.
Several follow-up studies have replicated these loci and their interactions (32–35). Imputation analysis of the German CKD cohort that included 137 cases of idiopathic membranous nephropathy suggested DQA1*0501 as the most likely HLA risk allele in European cohorts (34). However, the studies originating from East Asia consistently implicated the nearby HLA-DRB1 gene, specifically the risk alleles DRB1*1501 (independent of DQA1*0501) and DRB1*0301 (in partial linkage disequilibrium with DQA1*0501) (36,37). These findings suggested the HLA associations may differ between East Asian and European populations.
The largest international genetic study of membranous nephropathy performed recently involved East Asian cohorts from China, Japan, and South Korea (1632 biopsy specimen–diagnosed cases and 3209 controls) and European-ancestry cohorts from the United States, United Kingdom, Italy, Poland, Germany, Belgium, Switzerland, and Turkey (2150 biopsy specimen–diagnosed cases and 5829 controls) (38). The joint transethnic meta-analysis of all 12,820 participants (3782 cases and 9038 controls) confirmed strong signals at the HLA and PLA2R1 loci. Importantly, large sample size and biethnic composition of the study permitted computational dissection of ethnicity-specific effects and interactions of the HLA locus. This analysis confirmed strong East Asian–specific risk effect of the DRB1*1501 allele (OR, 3.81; P=2.0×10−49), and demonstrated this allele had no disease associations in European cohorts. Moreover, although the association of DRB1*0301 was strongly supported by both ethnicities (OR, 3.50; P=9.2×10−23 in East Asian cohorts; OR, 3.39; P=5.2×10−82 in European cohorts), independent effects of DQA1*0501 were observed only in European cohorts (OR, 2.88; P=5.7×10−93). Imputation-based sequence analysis of HLA peptides further identified effects of individual amino acid residues within the DRβ antigen-binding groove, specifically at positions 13, 71, and 74. These findings provide data on the high-risk amino acid sequences of DRβ that can be used to identify specific T cell epitopes driving the disease process in different ancestral groups.
The PLA2R1 locus (rs17831251; OR, 2.25; P=4.7×10−103) represented the most strongly associated non-HLA signal in this study. Further fine mapping and annotation analyses of this locus demonstrated the risk allele intersected a regulatory element affecting PLA2R1 mRNA expression. Although the risk allele was associated with lower mRNA expression of PLA2R1 across multiple tissues, it correlated with higher PLA2R1 expression specifically in the kidney glomerular compartment. The same risk allele exhibited significant multiplicative interaction with HLA risk haplotypes, but the interactions were evident only for DRB1*1501 and DRB1*0301 alleles, and not for DQA1*0501 after accounting for the DRB1*0301 risk allele.
In addition, two novel genome-wide significant loci with genetic effects relevant to both ancestral groups were discovered. These include a locus on chromosome 4q24 encoding NFKB1 (rs230540; OR, 1.25; P=3.4×10−12) and another independent locus on chromosome 6p25.3 encoding IRF4 (rs9405192; OR, 1.29; P=1.4×10−14). NFKB1 gene encodes a subunit of the NF-κB transcription regulator that is activated by cytokines, stimulating an inflammatory response. IRF4 encodes the transcription factor that regulates IFN responses to viral infections, and downregulates Toll-like receptor signaling. These findings highlighted the role of transcriptional regulation of inflammatory response in the pathogenesis of membranous nephropathy, and raised the possibility of infectious disease triggers.
Remarkably, all of the above GWAS loci were highly predictive of the disease and jointly explained approximately one third of the overall disease risk. The polygenic risk score based on these GWAS loci correlated with the key parameters of disease severity, including proteinuria and anti-PLA2R antibody levels. When combined with serum ELISA for anti-PLA2R antibodies, the genetic risk score had superior diagnostic properties compared with serologic test alone, correctly reclassifying 20%–37% of antibody-negative cases in independent European validation cohorts. These findings indicate that genetic profiling could potentially be used to enhance a noninvasive diagnosis of membranous nephropathy. This can be especially useful in the clinical settings where kidney biopsy represents too great of a risk or is simply not available. One limitation is that the diagnostic properties of GWAS-based risk scores may be population specific. Thus, it would be important to direct future efforts at validation of these findings in more diverse populations that are not represented in current studies.
Steroid-Sensitive Nephrotic Syndrome
Steroid-sensitive nephrotic syndrome is the most common pediatric glomerular disease, affecting approximately one to 16 in 100,000 children (39). An infection, typically of the upper respiratory tract, often precedes onset of the disease. Kidney biopsy is not required to establish the diagnosis, but the most common underlying histologic lesion is that of minimal change disease. It is believed that this is an immune-mediated disease, but its precise etiology is uncertain.
Because steroid-sensitive nephrotic syndrome represents a relatively rare pediatric condition, the existing GWAS are small and generally underpowered. The first study was an exome chip-based study and involved only 214 pediatric cases of South Asian ancestry (40). The study described strong contribution of the HLA-DQA1 region (rs1129740; OR, 5.44; P=1.4×10−17), corroborating findings from earlier candidate gene studies (41,42). These associations were subsequently replicated in a cohort of European children, and then again in children of African American ancestry (43). Another study involved a collection of 132 European, 56 sub-Saharan African, and 85 North African cases, followed by an independent cohort of 112 European children. The transethnic meta-analysis confirmed three independent signals in the HLA-DQ/DR region (44). Moreover, higher burden of risk alleles was associated with younger age of disease onset and increased odds of complete remission. Imputation of classical HLA alleles identified the HLA-DQA1*0201-DRB1*0701 risk haplotype in European participants, but the small cohort size precluded further fine mapping of this locus. The third GWAS involved Japanese children (224 cases in the discovery and 216 in the replication) and confirmed the signal at the HLA-DQ/DR region (45). The analysis of classical HLA alleles pointed to a different risk haplotype, HLA-DRB1*0802-DQB1*0302, suggesting an ancestry-specific effect.
The latest and largest GWAS involved 422 pediatric patients of European ancestry (46). Consistent with prior studies, the strongest signal was found at the HLA-DR/DQ locus (rs9273542; OR, 3.39; P=1.6×10−43), but conditional analysis revealed a second independent genome-wide significant signal represented by rs2858317 (P=4.3×10−31). Imputation of classical HLA alleles confirmed HLA-DQA1*0201-HLA-DRB1*0701 as the most likely risk haplotype. Larger size compared with prior GWAS also enabled identification of two non-HLA risk loci, including a signal within an intronic region of the PARM1 gene on chromosome 4q13.3 (rs10518133; OR, 1.96; P=2.5×10−8) and another, stronger signal near the CALHM6 gene on chromosome 6q22.1 (rs2637678; OR, 1.96; P=1.3×10−17). Notably, this variant is associated with lower mRNA expression of CALHM6 in lymphocytes, and has previously been associated with increased risk of ulcerative colitis (47). The absence of expression of CALHM6 in the kidney is in contrast to the high expression observed in lymphocytes, and consistent with the notion that impaired immune regulation may represent one of the key factors in the development of steroid-sensitive nephrotic syndrome. Cumulatively, the independent genome-wide significant risk alleles had large effects, explaining approximately 14% of disease risk in the discovery cohort. Nevertheless, the new loci still require replication in independent cohorts.
In summary, steroid-sensitive nephrotic syndrome represents a rare pediatric condition, and practical implementation of large-scale genetic studies has been difficult. Similar to membranous nephropathy, GWAS findings suggest the existence of alleles with large effect sizes, including substantial contributions of the HLA locus and its potential ancestry-specific effects. The above studies have not yet been meta-analyzed to further increase power for new locus discovery. Nevertheless, these early studies provide support for the central role of adaptive immunity in steroid-sensitive nephrotic syndrome, and a strong rationale for further expanded discovery studies for this phenotype across diverse ancestries.
Conclusions and Future Directions
The GWAS approach has not been systematically applied to all types of glomerular diseases at a scale that is comparable to other complex traits, mainly due to the low overall prevalence of individual forms of glomerular disorders. Nevertheless, GWAS have already provided important insights into the pathogenesis of glomerular disorders.
GWAS findings highlighted the central role of the MHC class II locus in the susceptibility to common forms of immune-mediated glomerulonephropathy (GN). Importantly, each disease type was associated with a distinct set of classic HLA alleles, highlighting that the immune mechanisms are likely different for each of these conditions. These associations may provide insights into early events that trigger autoimmune response. For example, in rheumatoid arthritis, it had been known for decades that a specific amino acid sequence of HLA-DRB1 is strongly associated with the disease risk. Through GWAS and a technique of amino acid imputation, this sequence was fully defined, and the information was used to provide a molecular explanation for the propensity of peptides with citrullinated components to induce the disease (48). As exemplified by GWAS for membranous nephropathy (38), similar insights about the nature of immunogenic epitopes are likely to emerge from high-resolution HLA analysis in well-powered glomerular disease cohorts.
Additionally, GWAS have provided important insights into genetic architecture of glomerular disorders. GWAS have defined IgA nephropathy as a highly polygenic trait that involves a complex interplay of multiple risk alleles shaped by selective forces related to host-pathogen interactions. In contrast, the genetic risk of membranous nephropathy and steroid-sensitive nephrotic syndrome appears to be determined by a smaller number of large-effect loci. At the same time, single gene defects do not appear to be major contributors to IgA nephropathy, membranous nephropathy, or steroid-sensitive nephrotic syndrome in contrast to other glomerular disorders, such as FSGS. This could potentially be explained by the fact that the contributions of rare alleles have not yet been systematically explored in large sequenced cohorts. Another possibility is that some forms of disease may be due to rare mutations in noncoding regions missed by exome sequencing. This possibility will ultimately be addressed by whole-genome sequencing studies in large cohorts of GN patients, such as the CureGN study (49).
It is worth pointing out that the successes of GWAS are most apparent in the field of autoimmune disorders, allowing for identification of shared disease pathways and shedding light on the common underlying mechanisms that are now being pursued as therapeutic targets (50). This success is highly relevant to glomerular disease because many of the known GN alleles have overlapping effects on the risk of other autoimmune traits. The identification of shared causal pathways with GN could enable an effective genetics-driven approach for drug repurposing. Moreover, allelic effects picked up by GWAS of autoimmune disorders tend to be larger compared with cardiometabolic or neuropsychiatric traits. This has also been the case for GN, where several common alleles with unusually large effects have already been identified. Such findings can empower clinically important polygenic predictors that can be used for early diagnosis and risk stratification. The genetic risk models that incorporate the effects of glomerular risk alleles have already been shown to be clinically meaningful, as exemplified by the ability to enhance the diagnosis of membranous nephropathy. We anticipate that such approaches will only become more powerful when larger GWAS become available.
Finally, it is striking that each time a GWAS is performed for a primary glomerular disease, a new set of significant loci is being discovered. This success should motivate further investment into genetic studies of larger cohorts. Notably, for some forms of GN, such as poststreptococcal or membranoproliferative nephritis, no GWAS have been performed to date. Thus, although the pace of GWAS discoveries has been slower in glomerular diseases compared with other traits, there are still clear opportunities to substantially advance the field using this well-established approach.
In summary, GWAS have been remarkably successful for glomerular disorders, providing a compelling rationale to actively pursue expanded genetic discovery studies. The challenges are to systematically define disease contributions of common and rare alleles in larger and more diverse genetic cohorts, identify causal variants underlying each significant GWAS locus, and elucidate dysregulated molecular pathways downstream of risk alleles. Such insights can then be translated into clinical benefits, including genetically guided drug interventions and genetic strategies for screening and prevention. The successes of the existing studies strongly suggest that genetic approaches will continue to transform our field, illuminating the pathophysiology of glomerular diseases and paving the road to precision medicine in glomerular clinics.
Disclosures
K. Kiryluk reports employment by Columbia University during the conduct of the study. He also reports a relationship with AstraZeneca and Goldfinch Bio, outside the submitted work. All remaining authors have nothing to disclose.
Funding
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases through grants R01DK105124, RC2DK116690, and UH3DK114926 (to K. Kiryluk), and T35DK093430 (to C. Southard).
Acknowledgments
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Magistroni R, D’Agati VD, Appel GB, Kiryluk K: New developments in the genetics, pathogenesis, and therapy of IgA nephropathy. Kidney Int 88: 974–989, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kiryluk K, Novak J: The genetics and immunobiology of IgA nephropathy. J Clin Invest 124: 2325–2332, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suzuki H, Kiryluk K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, Wyatt RJ, Scolari F, Mestecky J, Gharavi AG, Julian BA: The pathophysiology of IgA nephropathy. J Am Soc Nephrol 22: 1795–1803, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kiryluk K, Li Y, Sanna-Cherchi S, Rohanizadegan M, Suzuki H, Eitner F, Snyder HJ, Choi M, Hou P, Scolari F, Izzi C, Gigante M, Gesualdo L, Savoldi S, Amoroso A, Cusi D, Zamboli P, Julian BA, Novak J, Wyatt RJ, Mucha K, Perola M, Kristiansson K, Viktorin A, Magnusson PK, Thorleifsson G, Thorsteinsdottir U, Stefansson K, Boland A, Metzger M, Thibaudin L, Wanner C, Jager KJ, Goto S, Maixnerova D, Karnib HH, Nagy J, Panzer U, Xie J, Chen N, Tesar V, Narita I, Berthoux F, Floege J, Stengel B, Zhang H, Lifton RP, Gharavi AG: Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS Genet 8: e1002765, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feehally J, Farrall M, Boland A, Gale DP, Gut I, Heath S, Kumar A, Peden JF, Maxwell PH, Morris DL, Padmanabhan S, Vyse TJ, Zawadzka A, Rees AJ, Lathrop M, Ratcliffe PJ: HLA has strongest association with IgA nephropathy in genome-wide analysis. J Am Soc Nephrol 21: 1791–1797, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, Sanna-Cherchi S, Men CJ, Julian BA, Wyatt RJ, Novak J, He JC, Wang H, Lv J, Zhu L, Wang W, Wang Z, Yasuno K, Gunel M, Mane S, Umlauf S, Tikhonova I, Beerman I, Savoldi S, Magistroni R, Ghiggeri GM, Bodria M, Lugani F, Ravani P, Ponticelli C, Allegri L, Boscutti G, Frasca G, Amore A, Peruzzi L, Coppo R, Izzi C, Viola BF, Prati E, Salvadori M, Mignani R, Gesualdo L, Bertinetto F, Mesiano P, Amoroso A, Scolari F, Chen N, Zhang H, Lifton RP: Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet 43: 321–327, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie J, Kiryluk K, Li Y, Mladkova N, Zhu L, Hou P, Ren H, Wang W, Zhang H, Chen N, Gharavi AG: Fine mapping implicates a deletion of CFHR1 and CFHR3 in protection from IgA nephropathy in Han Chinese. J Am Soc Nephrol 27: 3187–3194, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maillard N, Wyatt RJ, Julian BA, Kiryluk K, Gharavi A, Fremeaux-Bacchi V, Novak J: Current understanding of the role of complement in IgA nephropathy. J Am Soc Nephrol 26: 1503–1512, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raychaudhuri S, Ripke S, Li M, Neale BM, Fagerness J, Reynolds R, Sobrin L, Swaroop A, Abecasis G, Seddon JM, Daly MJ: Associations of CFHR1-CFHR3 deletion and a CFH SNP to age-related macular degeneration are not independent. Nat Genet 42: 553–555; author reply 555–556, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feenstra B, Bager P, Liu X, Hjalgrim H, Nohr EA, Hougaard DM, Geller F, Melbye M: Genome-wide association study identifies variants in HORMAD2 associated with tonsillectomy. J Med Genet 54: 358–364, 2017. [DOI] [PubMed] [Google Scholar]
- 11.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Büning C, Cohain A, Cichon S, D’Amato M, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L, Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW, Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, Newman W, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M, Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, Schreiber S, Simms LA, Sventoraityte J, Targan SR, Taylor KD, Tremelling M, Verspaget HW, De Vos M, Wijmenga C, Wilson DC, Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, Zhao H, Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford-Smith G, Mathew CG, Rioux JD, Schadt EE, Daly MJ, Franke A, Parkes M, Vermeire S, Barrett JC, Cho JH; International IBD Genetics Consortium (IIBDGC) : Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491: 119–124, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu XQ, Li M, Zhang H, Low HQ, Wei X, Wang JQ, Sun LD, Sim KS, Li Y, Foo JN, Wang W, Li ZJ, Yin XY, Tang XQ, Fan L, Chen J, Li RS, Wan JX, Liu ZS, Lou TQ, Zhu L, Huang XJ, Zhang XJ, Liu ZH, Liu JJ: A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat Genet 44: 178–182, 2011. [DOI] [PubMed] [Google Scholar]
- 13.He B, Xu W, Santini PA, Polydorides AD, Chiu A, Estrella J, Shan M, Chadburn A, Villanacci V, Plebani A, Knowles DM, Rescigno M, Cerutti A: Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity 26: 812–826, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Lehrer RI, Lu W: α-Defensins in human innate immunity. Immunol Rev 245: 84–112, 2012. [DOI] [PubMed] [Google Scholar]
- 15.Castigli E, Scott S, Dedeoglu F, Bryce P, Jabara H, Bhan AK, Mizoguchi E, Geha RS: Impaired IgA class switching in APRIL-deficient mice. Proc Natl Acad Sci U S A 101: 3903–3908, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ai Z, Li M, Liu W, Foo JN, Mansouri O, Yin P, Zhou Q, Tang X, Dong X, Feng S, Xu R, Zhong Z, Chen J, Wan J, Lou T, Yu J, Zhou Q, Fan J, Mao H, Gale D, Barratt J, Armour JA, Liu J, Yu X: Low α-defensin gene copy number increases the risk for IgA nephropathy and renal dysfunction. Sci Transl Med 8: 345ra88, 2016. [DOI] [PubMed] [Google Scholar]
- 17.Aldred PM, Hollox EJ, Armour JA: Copy number polymorphism and expression level variation of the human alpha-defensin genes DEFA1 and DEFA3. Hum Mol Genet 14: 2045–2052, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Li M, Foo JN, Wang JQ, Low HQ, Tang XQ, Toh KY, Yin PR, Khor CC, Goh YF, Irwan ID, Xu RC, Andiappan AK, Bei JX, Rotzschke O, Chen MH, Cheng CY, Sun LD, Jiang GR, Wong TY, Lin HL, Aung T, Liao YH, Saw SM, Ye K, Ebstein RP, Chen QK, Shi W, Chew SH, Chen J, Zhang FR, Li SP, Xu G, Shyong Tai E, Wang L, Chen N, Zhang XJ, Zeng YX, Zhang H, Liu ZH, Yu XQ, Liu JJ: Identification of new susceptibility loci for IgA nephropathy in Han Chinese. Nat Commun 6: 7270, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, Fasel D, Lata S, Prakash S, Shapiro S, Fischman C, Snyder HJ, Appel G, Izzi C, Viola BF, Dallera N, Del Vecchio L, Barlassina C, Salvi E, Bertinetto FE, Amoroso A, Savoldi S, Rocchietti M, Amore A, Peruzzi L, Coppo R, Salvadori M, Ravani P, Magistroni R, Ghiggeri GM, Caridi G, Bodria M, Lugani F, Allegri L, Delsante M, Maiorana M, Magnano A, Frasca G, Boer E, Boscutti G, Ponticelli C, Mignani R, Marcantoni C, Di Landro D, Santoro D, Pani A, Polci R, Feriozzi S, Chicca S, Galliani M, Gigante M, Gesualdo L, Zamboli P, Battaglia GG, Garozzo M, Maixnerová D, Tesar V, Eitner F, Rauen T, Floege J, Kovacs T, Nagy J, Mucha K, Pączek L, Zaniew M, Mizerska-Wasiak M, Roszkowska-Blaim M, Pawlaczyk K, Gale D, Barratt J, Thibaudin L, Berthoux F, Canaud G, Boland A, Metzger M, Panzer U, Suzuki H, Goto S, Narita I, Caliskan Y, Xie J, Hou P, Chen N, Zhang H, Wyatt RJ, Novak J, Julian BA, Feehally J, Stengel B, Cusi D, Lifton RP, Gharavi AG: Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet 46: 1187–1196, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gharavi AG, Moldoveanu Z, Wyatt RJ, Barker CV, Woodford SY, Lifton RP, Mestecky J, Novak J, Julian BA: Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J Am Soc Nephrol 19: 1008–1014, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiryluk K, Moldoveanu Z, Sanders JT, Eison TM, Suzuki H, Julian BA, Novak J, Gharavi AG, Wyatt RJ: Aberrant glycosylation of IgA1 is inherited in both pediatric IgA nephropathy and Henoch-Schönlein purpura nephritis. Kidney Int 80: 79–87, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kiryluk K, Li Y, Moldoveanu Z, Suzuki H, Reily C, Hou P, Xie J, Mladkova N, Prakash S, Fischman C, Shapiro S, LeDesma RA, Bradbury D, Ionita-Laza I, Eitner F, Rauen T, Maillard N, Berthoux F, Floege J, Chen N, Zhang H, Scolari F, Wyatt RJ, Julian BA, Gharavi AG, Novak J: GWAS for serum galactose-deficient IgA1 implicates critical genes of the O-glycosylation pathway. PLoS Genet 13: e1006609, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gale DP, Molyneux K, Wimbury D, Higgins P, Levine AP, Caplin B, Ferlin A, Yin P, Nelson CP, Stanescu H, Samani NJ, Kleta R, Yu X, Barratt J: Galactosylation of IgA1 is associated with common variation in C1GALT1. J Am Soc Nephrol 28: 2158–2166, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGrogan A, Franssen CF, de Vries CS: The incidence of primary glomerulonephritis worldwide: A systematic review of the literature. Nephrol Dial Transplant 26: 414–430, 2011. [DOI] [PubMed] [Google Scholar]
- 25.Gupta S, Köttgen A, Hoxha E, Brenchley P, Bockenhauer D, Stanescu HC, Kleta R: Genetics of membranous nephropathy. Nephrol Dial Transplant 33: 1493–1502, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Debiec H, Guigonis V, Mougenot B, Decobert F, Haymann JP, Bensman A, Deschênes G, Ronco PM: Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N Engl J Med 346: 2053–2060, 2002. [DOI] [PubMed] [Google Scholar]
- 27.Beck LH Jr., Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, Klein JB, Salant DJ: M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 361: 11–21, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomas NM, Beck LH Jr., Meyer-Schwesinger C, Seitz-Polski B, Ma H, Zahner G, Dolla G, Hoxha E, Helmchen U, Dabert-Gay AS, Debayle D, Merchant M, Klein J, Salant DJ, Stahl RAK, Lambeau G: Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med 371: 2277–2287, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sethi S, Madden BJ, Debiec H, Charlesworth MC, Gross L, Ravindran A, Hummel AM, Specks U, Fervenza FC, Ronco P: Exostosin 1/exostosin 2-associated membranous nephropathy. J Am Soc Nephrol 30: 1123–1136, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sethi S, Debiec H, Madden B, Charlesworth MC, Morelle J, Gross L, Ravindran A, Buob D, Jadoul M, Fervenza FC, Ronco P: Neural epidermal growth factor-like 1 protein (NELL-1) associated membranous nephropathy. Kidney Int 97: 163–174, 2020. [DOI] [PubMed] [Google Scholar]
- 31.Stanescu HC, Arcos-Burgos M, Medlar A, Bockenhauer D, Kottgen A, Dragomirescu L, Voinescu C, Patel N, Pearce K, Hubank M, Stephens HA, Laundy V, Padmanabhan S, Zawadzka A, Hofstra JM, Coenen MJ, den Heijer M, Kiemeney LA, Bacq-Daian D, Stengel B, Powis SH, Brenchley P, Feehally J, Rees AJ, Debiec H, Wetzels JF, Ronco P, Mathieson PW, Kleta R: Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med 364: 616–626, 2011. [DOI] [PubMed] [Google Scholar]
- 32.Lv J, Hou W, Zhou X, Liu G, Zhou F, Zhao N, Hou P, Zhao M, Zhang H: Interaction between PLA2R1 and HLA-DQA1 variants associates with anti-PLA2R antibodies and membranous nephropathy. J Am Soc Nephrol 24: 1323–1329, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bullich G, Ballarín J, Oliver A, Ayasreh N, Silva I, Santín S, Díaz-Encarnación MM, Torra R, Ars E: HLA-DQA1 and PLA2R1 polymorphisms and risk of idiopathic membranous nephropathy. Clin J Am Soc Nephrol 9: 335–343, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sekula P, Li Y, Stanescu HC, Wuttke M, Ekici AB, Bockenhauer D, Walz G, Powis SH, Kielstein JT, Brenchley P, Eckardt KU, Kronenberg F, Kleta R, Köttgen A; GCKD Investigators : Genetic risk variants for membranous nephropathy: Extension of and association with other chronic kidney disease aetiologies. Nephrol Dial Transplant 32: 325–332, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramachandran R, Kumar V, Kumar A, Yadav AK, Nada R, Kumar H, Kumar V, Rathi M, Kohli HS, Gupta KL, Sakhuja V, Jha V: PLA2R antibodies, glomerular PLA2R deposits and variations in PLA2R1 and HLA-DQA1 genes in primary membranous nephropathy in South Asians. Nephrol Dial Transplant 31: 1486–1493, 2016. [DOI] [PubMed] [Google Scholar]
- 36.Cui Z, Xie LJ, Chen FJ, Pei ZY, Zhang LJ, Qu Z, Huang J, Gu QH, Zhang YM, Wang X, Wang F, Meng LQ, Liu G, Zhou XJ, Zhu L, Lv JC, Liu F, Zhang H, Liao YH, Lai LH, Ronco P, Zhao MH: MHC class II risk alleles and amino acid residues in idiopathic membranous nephropathy. J Am Soc Nephrol 28: 1651–1664, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le WB, Shi JS, Zhang T, Liu L, Qin HZ, Liang S, Zhang YW, Zheng CX, Jiang S, Qin WS, Zhang HT, Liu ZH: HLA-DRB1*15:01 and HLA-DRB3*02:02 in PLA2R-related membranous nephropathy. J Am Soc Nephrol 28: 1642–1650, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie J, Liu L, Mladkova N, Li Y, Ren H, Wang W, Cui Z, Lin L, Hu X, Yu X, Xu J, Liu G, Caliskan Y, Sidore C, Balderes O, Rosen RJ, Bodria M, Zanoni F, Zhang JY, Krithivasan P, Mehl K, Marasa M, Khan A, Ozay F, Canetta PA, Bomback AS, Appel GB, Sanna-Cherchi S, Sampson MG, Mariani LH, Perkowska-Ptasinska A, Durlik M, Mucha K, Moszczuk B, Foroncewicz B, Pączek L, Habura I, Ars E, Ballarin J, Mani LY, Vogt B, Ozturk S, Yildiz A, Seyahi N, Arikan H, Koc M, Basturk T, Karahan G, Akgul SU, Sever MS, Zhang D, Santoro D, Bonomini M, Londrino F, Gesualdo L, Reiterova J, Tesar V, Izzi C, Savoldi S, Spotti D, Marcantoni C, Messa P, Galliani M, Roccatello D, Granata S, Zaza G, Lugani F, Ghiggeri G, Pisani I, Allegri L, Sprangers B, Park JH, Cho B, Kim YS, Kim DK, Suzuki H, Amoroso A, Cattran DC, Fervenza FC, Pani A, Hamilton P, Harris S, Gupta S, Cheshire C, Dufek S, Issler N, Pepper RJ, Connolly J, Powis S, Bockenhauer D, Stanescu HC, Ashman N, Loos RJF, Kenny EE, Wuttke M, Eckardt KU, Köttgen A, Hofstra JM, Coenen MJH, Kiemeney LA, Akilesh S, Kretzler M, Beck LH, Stengel B, Debiec H, Ronco P, Wetzels JFM, Zoledziewska M, Cucca F, Ionita-Laza I, Lee H, Hoxha E, Stahl RAK, Brenchley P, Scolari F, Zhao MH, Gharavi AG, Kleta R, Chen N, Kiryluk K: The genetic architecture of membranous nephropathy and its potential to improve non-invasive diagnosis. Nat Commun 11: 1600, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noone DG, Iijima K, Parekh R: Idiopathic nephrotic syndrome in children. Lancet 392: 61–74, 2018. [DOI] [PubMed] [Google Scholar]
- 40.Gbadegesin RA, Adeyemo A, Webb NJ, Greenbaum LA, Abeyagunawardena A, Thalgahagoda S, Kale A, Gipson D, Srivastava T, Lin JJ, Chand D, Hunley TE, Brophy PD, Bagga A, Sinha A, Rheault MN, Ghali J, Nicholls K, Abraham E, Janjua HS, Omoloja A, Barletta GM, Cai Y, Milford DD, O’Brien C, Awan A, Belostotsky V, Smoyer WE, Homstad A, Hall G, Wu G, Nagaraj S, Wigfall D, Foreman J, Winn MP; Mid-West Pediatric Nephrology Consortium : HLA-DQA1 and PLCG2 are candidate risk loci for childhood-onset steroid-sensitive nephrotic syndrome. J Am Soc Nephrol 26: 1701–1710, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kari JA, Sinnott P, Khan H, Trompeter RS, Snodgrass GJ: Familial steroid-responsive nephrotic syndrome and HLA antigens in Bengali children. Pediatr Nephrol 16: 346–349, 2001. [DOI] [PubMed] [Google Scholar]
- 42.Huang YY, Lin FJ, Fu LS, Lan JL: HLA-DR, -DQB typing of steroid-sensitive idiopathic nephrotic syndrome children in Taiwan. Nephron Clin Pract 112: c57–c64, 2009. [DOI] [PubMed] [Google Scholar]
- 43.Adeyemo A, Esezobor C, Solarin A, Abeyagunawardena A, Kari JA, El Desoky S, Greenbaum LA, Kamel M, Kallash M, Silva C, Young A, Hunley TE, de Jesus-Gonzalez N, Srivastava T, Gbadegesin R: HLA-DQA1 and APOL1 as risk loci for childhood-onset steroid-sensitive and steroid-resistant nephrotic syndrome. Am J Kidney Dis 71: 399–406, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Debiec H, Dossier C, Letouzé E, Gillies CE, Vivarelli M, Putler RK, Ars E, Jacqz-Aigrain E, Elie V, Colucci M, Debette S, Amouyel P, Elalaoui SC, Sefiani A, Dubois V, Simon T, Kretzler M, Ballarin J, Emma F, Sampson MG, Deschênes G, Ronco P: Transethnic, genome-wide analysis reveals immune-related risk alleles and phenotypic correlates in pediatric steroid-sensitive nephrotic syndrome. J Am Soc Nephrol 29: 2000–2013, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jia X, Horinouchi T, Hitomi Y, Shono A, Khor SS, Omae Y, Kojima K, Kawai Y, Nagasaki M, Kaku Y, Okamoto T, Ohwada Y, Ohta K, Okuda Y, Fujimaru R, Hatae K, Kumagai N, Sawanobori E, Nakazato H, Ohtsuka Y, Nakanishi K, Shima Y, Tanaka R, Ashida A, Kamei K, Ishikura K, Nozu K, Tokunaga K, Iijima K; Research Consortium on Genetics of Childhood Idiopathic Nephrotic Syndrome in Japan : Strong association of the HLA-DR/DQ locus with childhood steroid-sensitive nephrotic syndrome in the Japanese population. J Am Soc Nephrol 29: 2189–2199, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dufek S, Cheshire C, Levine AP, Trompeter RS, Issler N, Stubbs M, Mozere M, Gupta S, Klootwijk E, Patel V, Hothi D, Waters A, Webb H, Tullus K, Jenkins L, Godinho L, Levtchenko E, Wetzels J, Knoers N, Teeninga N, Nauta J, Shalaby M, Eldesoky S, Kari JA, Thalgahagoda S, Ranawaka R, Abeyagunawardena A, Adeyemo A, Kristiansen M, Gbadegesin R, Webb NJ, Gale DP, Stanescu HC, Kleta R, Bockenhauer D: Genetic identification of two novel loci associated with steroid-sensitive nephrotic syndrome. J Am Soc Nephrol 30: 1375–1384, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Julià A, Domènech E, Chaparro M, García-Sánchez V, Gomollón F, Panés J, Mañosa M, Barreiro-De Acosta M, Gutiérrez A, Garcia-Planella E, Aguas M, Muñoz F, Esteve M, Mendoza JL, Vera M, Márquez L, Tortosa R, López-Lasanta M, Alonso A, Gelpí JL, García-Montero AC, Bertranpetit J, Absher D, Myers RM, Gisbert JP, Marsal S: A genome-wide association study identifies a novel locus at 6q22.1 associated with ulcerative colitis. Hum Mol Genet 23: 6927–6934, 2014. [DOI] [PubMed] [Google Scholar]
- 48.Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J: 10 years of GWAS discovery: Biology, function, and translation. Am J Hum Genet 101: 5–22, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mariani LH, Bomback AS, Canetta PA, Flessner MF, Helmuth M, Hladunewich MA, Hogan JJ, Kiryluk K, Nachman PH, Nast CC, Rheault MN, Rizk DV, Trachtman H, Wenderfer SE, Bowers C, Hill-Callahan P, Marasa M, Poulton CJ, Revell A, Vento S, Barisoni L, Cattran D, D’Agati V, Jennette JC, Klein JB, Laurin LP, Twombley K, Falk RJ, Gharavi AG, Gillespie BW, Gipson DS, Greenbaum LA, Holzman LB, Kretzler M, Robinson B, Smoyer WE, Guay-Woodford LM; CureGN Consortium : CureGN study rationale, design, and methods: Establishing a large prospective observational study of glomerular disease. Am J Kidney Dis 73: 218–229, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Visscher PM, Brown MA, McCarthy MI, Yang J: Five years of GWAS discovery. Am J Hum Genet 90: 7–24, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]