

Abstract
Prolonged ER stress has been known to be one of the major drivers of impaired lipid homeostasis during the pathogenesis of non-alcoholic liver disease (NAFLD). However, the downstream mediators of ER stress pathway in promoting lipid accumulation remain poorly understood. Here we present data showing the b-ZIP transcription factor E4BP4 in both hepatocytes and the mouse liver is potently induced by the chemical ER stress inducer tunicamycin or by high-fat, low-methionine and choline-deficient (HFLMCD) diet. We showed that such an induction is partially dependent on CHOP, a known mediator of ER stress and requires the E-box element of the E4bp4 promoter. Tunicamycin promotes lipid droplet formation and alters lipid metabolic gene expression in primary mouse hepatocytes from E4bp4flox/flox but not E4bp4 liver-specific KO (E4bp4-LKO) mice. Compared with E4bp4flox/flox mice, E4bp4-LKO female mice exhibit reduced liver lipid accumulation and partially improved liver function after 10-week HFLMCD diet feeding. Mechanistically, we observed elevated AMPK activity and the AMPKβ1 abundance in the liver of E4bp4-LKO mice. We have evidence supporting that E4BP4 may suppress the AMPK activity via promoting the AMPKβ1 ubiquitination and degradation. Furthermore, acute depletion of the Ampkβ1 subunit restores lipid droplet formation in E4bp4-LKO primary mouse hepatocytes. Our study highlighted hepatic E4BP4 as a key factor linking ER stress and lipid accumulation in the liver. Targeting E4BP4 in the liver may be a novel therapeutic avenue for treating NAFLD.
Keywords: lipid accumulation, lipid droplet, de novo lipogenesis, ER stress, high-fat, low-methionine, choline-deficient diet
Introduction
The ER system is an intracellular organelle responsible for the folding of membrane-bound and secreted proteins, lipid and sterol synthesis, and calcium storage in eukaryotes (1,2). When unfolded or misfolded proteins accumulate inside that ER lumen, ER stress triggers a group of signal transduction pathways namely “unfolded protein response (UPR)” to reprogram the transcriptional and translational processes for recovery or adaptation. A variety of pharmacological agents and dietary manipulations have been shown to cause ER stress and interrupt the normal ER function (3,4). One of metabolic consequences of ER stress in hepatocytes is lipid accumulation in the liver. Acute injection of the ER stress inducer tunicamycin in mice was shown to not only induce the UPR genes but also cause marked lipid droplet accumulation along with increased de novo lipogenesis and reduced fatty acid oxidation in the liver (5). Methionine and choline deficient (MCD) diet, which is known to induce liver steatosis and injury, induces potent ER stress in the liver (6). These findings highlighted that ER stress might be a critical regulator of hepatic lipid accumulataion, the early hallmark of non-alcoholic fatty liver disease (NAFLD).
The precise mechanisms of NAFLD remain poorly understood. Currently, the “multiple-hit hypothesis” has been recognized to address the development and progression of NAFLD (7,8). The initial hit leads to simple liver steatosis, while oxidative stress, insulin resistance, impairment in lipid metabolism, and Kupffer cell activation result in hepatic inflammation and apoptosis, prompting the progression from simple steatosis to non-alcoholic steatohepatitis (NASH) (9). Hepatocytes are rich in ER due to a high capacity for handling protein and lipid biosynthesis. It has been hypothesized that impaired ER homeostasis may be directly involved in the onset and progression of NAFLD (2,10). For example, the markers of ER stress were elevated in the liver of NAFLD (6,11,12). Chemical chaperons have been shown to not only reduce ER stress but also improve liver function (13,14). Deletion of the ER stress responsive chaperon p58IPK protects mice from diet-induced steatohepatitis (15). Several drugs including FXR agonists and GLP analogues have shown great promise in treating NAFLD/NASH through potent inhibition of the ER stress pathways in the liver (16,17).
There has been an intense interest in the role of transcription factors in the pathogenesis of NASH, particularly the ER-stress associated transcription factors, such as CHOP, ATF4, XBP1, CREBH, and C/EBP-β (8). The Friedman group showed that deletion of C/ebp-β attenuates inflammation and lipid accumulation in diet-induced NASH (18). In contrast, Chop deletion results in more severe NASH following diet high in fat, fructose, and cholesterol (18,19). Hepatic Xbp1 deficiency sensitizes mice to diet-induced NASH (20). Crebh deficiency also accelerated the development of NAFLD in mice fed atherogenic diet (21). These paradoxical results demonstrated the diverse functions of ER stress-associated transcription factors in the pathogenesis of NAFLD. Thus, a better understanding of the specific roles for individual components of the ER stress pathway may lead to effective treatment for NAFLD.
As a b-ZIP transcription factor, E4BP4 is ubiquitously expressed in various tissues, particularly abundant in the liver, spleen, and adipose tissue (22). Although E4BP4 is well known for its critical role in NK cell development and lymphocytes (23,24) and has been recently implicated in hepatic gluconeogenesis and the regulation of body composition (25,26), the involvement of E4BP4 in lipid metabolism remains largely unexplored. We previously reported that potent induction of E4BP4 by insulin contributes to the post-prandial de novo lipogenesis in an SREBP-1c-dependent manner (27). E4bp4-deficient hepatocytes exhibit a significant reduction in de novo lipogenesis, triglyceride content as well as the expression of lipogenic enzymes including Fasn and Scd1. The work from the Zhang group shows that E4BP4 could impact hepatic lipid metabolism via its interaction with CREBH, a liver-enriched ER-tethered transcription factor known to regulate hepatic lipid homeostasis (21). All these findings support a physiological role of E4BP4 in regulating hepatic lipid metabolism. Because dysregulated lipid metabolism is a major contributor to the pathogenesis of NAFLD, we set out to study the role of E4BP4 in this process. In the present study, we report that E4BP4 is highly induced by either chemical or diet inducers of ER stress in hepatocytes and the liver. E4bp4-deficient hepatocytes are resistant to tunicamycin-induced lipid accumulation. Moreover, the liver from E4bp4-LKO mice on NASH-inducing HFLMCD diet show decreased lipid accumulation, improved lipid metabolism, and reduced liver injury along with the enhanced AMPK signaling. Our data demonstrated that hepatic E4BP4 may drive the pathogenesis of diet-induced NAFLD likely through its inhibitory effects on the AMPK pathway.
Material and Methods
Animal and treatment
Animal experiments were conducted in accordance with the guidelines of the institutional Animal Care and Use Committee of University of Michigan Medical School. Male C57BL/6J mice and Albumin-Cre mice were purchased from the Jackson Laboratory. E4bp4flox/flox mice were generated via the conventional homologous recombination targeting exon 2 of the trimouse E4bp4 at Cyagen (Santa Clara, CA). Liver-specific E4bp4 knockout (E4bp4-LKO) mice were generated by crossing E4bp4flox/flox mice with Albumin-Cre mice. All mice were housed on a 12 hrs:12 hrs light/dark cycle at 25 °C with free access to water and regular chow (26.8% kcal from protein, 16.6% from fat, and 56.4% from starch). At the age of 2 months, E4bp4 flox/flox and E4bp4-LKO female mice was fed high-fat, low-methionine and choline-deficient (HFLMCD: 45% calories from fat, 0.1% methionine, and choline-deficient) diet for 10 weeks. For Ampk-β1 knockdown experiment, 2 months old WT mice were fed HFLMCD diet for 2 weeks, and then injected with adenoviral shRNA against LacZ, E4bp4, or E4bp4 plus Ampk β1 through tail vein. After injection, mice continued with HFLMCD diet for another 10 days prior to liver harvesting for metabolic assays.
Cell cultures
Hepa1c1c-7 cell was purchased from the ATCC and maintained according to the instructions. Isolation of primary mouse hepatocytes (PMHs) was described previously (Tong, Zhang et al. 2015). PMHs and Hepa1 cells were treated with DMSO and 5μM tunicamycin for 24 hrs and harvested for western blotting, RT-qPCR, and BODIPY staining.
Liver Histology and Sirius-Red staining
Liver tissues were immediately fixed in 10% formalin at room temperature overnight after paraffin embedding and H&E staining. While the H&E-stained slides were observed directly under microscope, the unstained slides were baked in an oven set at 58 °C for 1 hr. And the paraffin sections were rehydrated in ethanol solutions in various concentrations. Then, the slides were stained with Picro-Sirius Red Collagen Stain Kit (Sigma, Direct Red 80, 365548; FAST Green, 7252) or Masson’s Trichrome Stain Kit (Sigma, HT15-1KT) according to the user’s manuals and observed under microscope.
Transfection and Luciferase assay
293 AD cells were seeded in 24 wells plates. Transfection was performed with Opti-MEM (Gibco, 2021431) and polyethylenimine (PEI). The β-gal expression plasmid and Chop-HA over-expression plasmids were co-transfected with the E4bp4 WT promoter or the E4bp4 ΔE-box promoter-driven luciferase reporter construct plus the β-gal expression plasmid. 24 hrs later, the cells were harvest for luciferase and β-gal assays. Luciferase assay was performed with Luciferin (Gold Biotechnology, 103404-75-7) and normalized by the β-gal signal.
Protein extraction and immunoprecipitation
To prepare cytosolic and nuclear proteins, liver tissues were homogenized in hypotonic buffer, incubated on ice for 15-20 min, and centrifuged at 3000 rpm for 10 min at 4 °C. The supernatant was saved as the cytosolic fraction. The pellet was washed once with hypotonic buffer and re-suspended in RIPA buffer prior to sonication for 5 seconds. The nuclear fraction was then collected after centrifugation at 13000 x rpm for 10 min. Western blot analysis was performed using the following primary antibodies: anti-GAPDH (Santa Cruz, sc-25778), anti-Lamin A/C (Santa Cruz, sc-20681), anti-HSP90 (Santa Cruz, sc-13119), anti-CHOP (Santa Cruz, sc-575), anti-GRP78 (Santa Cruz, sc-13968), anti-E4BP4 (Santa Cruz, sc-28203; Cell Signaling, 14312S; DSHB, PCRP-NFIL3-2B5), anti-AMPK-pT172 (Cell Signaling, 2531S), anti-AMPK α1/α2 (Cell Signaling, 5831S), anti-AMPK-β1 (Santa Cruz, sc-100357), anti-AMPK-γ1 (Santa Cruz, sc-19138), anti-β-tubulin(Sigma,T5201), and anti-FLAG (Sigma, A8592).
cDNA Synthesis and qPCR
Total cellular RNA extraction was performed with TRIzol (Invitrogen) and chloroform. cDNA was synthesized with the Verso cDNA kit (ThermoFisher Scientific) and subjected to qPCR analysis with Radiant™ Green 2X qPCR Mix (Alkali Scientific) on an ABI 7900 HT thermal cycler (Applied Biosystems). The value of each cDNA was calculated using the ΔΔCT method and normalized to the values of the house-keeping gene control, the18s ribosomal RNA. The data were plotted as fold change. The primer sequences are listed below.
BODIPY staining
Primary mouse hepatocytes (PMHs) were isolated from E4bp4 flox/flox and E4bp4-LKO mice using a previously reported protocol. 8 x 104 cells were seeded per well of 12-well plates. Next day, cell culture medium was changed to serum-free DMEM (Gibco™) and the cells were treated with DMSO or tunicamycin (5 μg/mL). 24 hrs later, the cells were stained with BODIPY (Invitrogen, D3922, 493/503, 2 μM) using a protocol described by Bo Qiu (Bo Qiu et al. 2016).
Plasma and liver metabolite measurements
Serum cholesterol, triglycerides (TG), and alanine transaminase (ALT) were measured using the commercial kits (Pointe Scientific A7510, A7525 and T7532) according to the manufacturer’s instructions. For liver TG content, liver tissues were homogenized for total lipid extraction according to Bligh and Dyer (Bligh and Dyer 1959). Total lipids were re-suspended in 400 μL of 100% ethanol. 3 μL were used for the measurement of TG with the TG kit (Pointe Scientific T7532).
Statistical analysis
All data are reported as Mean ± S.D.. Differences between two groups were assessed by the two-tailed Student’s t-test. Difference between more than two groups was analyzed by ANOVA followed by the Tukey’s post-hoc testing. p value < 0.05 was deemed statistically different.
Results
E4BP4 is a novel target induced by ER stress in hepatocytes.
We previously reported that insulin induces the E4bp4 expression via an SREBP-1c-dependent manner and such an induction is critical for de novo lipogenesis in hepatocytes (27). Whether the E4bp4 expression is also sensitive to other lipogenic stimuli remains unknown. A recent study reported that the ER stress inducer tunicamycin increases the E4bp4 expression in pancreatic β-cells and impairs β-cell metabolism and insulin secretion (28). Given the sensitivity of hepatocytes to ER stress stimuli, we tested whether tunicamycin could induce E4bp4 by treating both primary mouse hepatocytes (PMHs) for indicated time points. Induction of the Chop mRNA confirmed the positive response to tunicamycin stimulation in hepatocytes. Meanwhile, the E4bp4 mRNA was induced as early as 2 hrs following tunicamycin treatment and peaked at 4 hrs with an increase by 3 fold (Figure 1A). Since the saturated fatty acid palmitate was shown to trigger ER stress in hepatocytes (29), we treated hepatocytes with palmitate and observed a similar elevation of E4bp4 expression in PMHs (Figure 1B). In contrast, oleic acid (OA), an unsaturated fatty acid, showed no effect on the expression of either Chop or E4bp4 mRNA in mouse hepatocytes (Figure 1C). Lastly, we observed a time-dependent induction of E4BP4 protein in PMHs (Figure 1D). Of note, similar results were observed in the mouse hepatoma Hepa1c1c7 cell line (Supplementary Figure 1A-D).
Figure 1. ER stress induces E4bp4 mRNA and protein in primary hepatocytes and mouse liver.
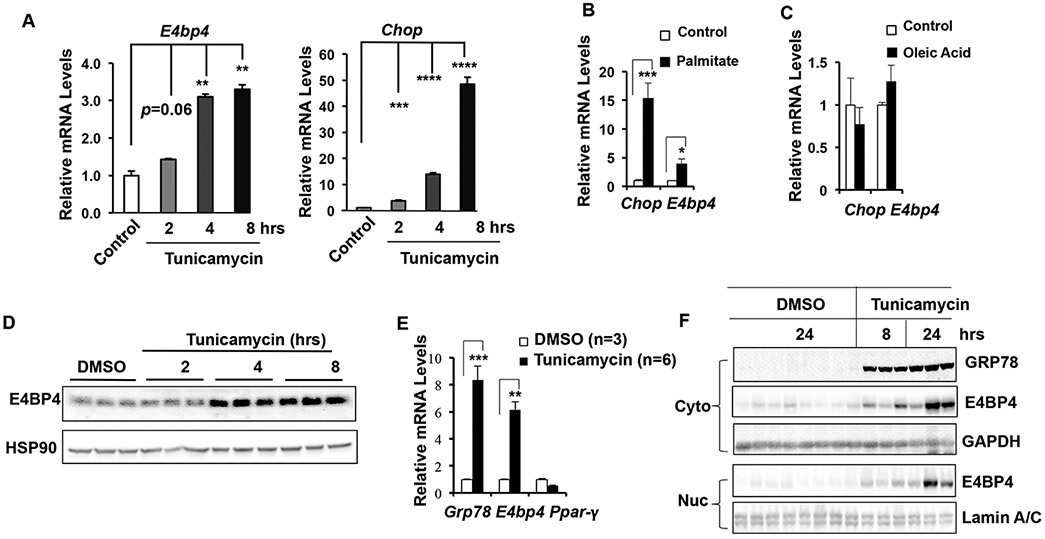
(A-C) Primary mouse hepatocytes were isolated from WT mice and then treated with tunicamycin at 5 μg/ml for 2, 4 8hr (A), or palmitate (400 μM) (B) or oleic acid (300 μM) (C) for 24 hr before harvest for mRNA analysis. The mRNA levels of Chop and E4bp4 were examined by RT-qPCR. (D) The protein levels of E4BP4 in primary hepatocytes were determined by immunblotting during a time-course study of tunicamycin treatment (E-F) WT male mice were acutely injected with either DMSO (n=3-7) or tunicamycin (n=3-6) and liver tissues were harvested at either 8 or 24 hrs for both mRNA and protein analysis. The mRNA abundance of Grp78, E4bp4, and Ppar-γ was analyzed by RT-qPCR, whereas the protein levels of E4BP4 and GRP78 were examined by immunoblotting. Data were presented as Mean ± S.D.. At least three independent experiments were performed with similar results.
To test whether this response is also conserved in the mouse liver, we challenged WT mice with tunicamycin through intraperitoneal injection. Eight and 24 hrs post injection, we examined both the protein and mRNA expression of E4bp4 in the liver. Indeed, liver E4bp4 mRNA and protein were potently induced by tunicamycin injection along with the induction of GRP78 (Figure 1E-F), a well-known target of acute ER stress response. Collectively, these data support that E4BP4 is a novel target of the ER stress responses in both hepatocytes and the liver.
Interestingly, E4BP4 induction as a novel target of the ER stress response is not limited to only hepatocytes. We also detected elevated levels of both E4bp4 mRNA and protein after tunicamycin treatment in the mouse macrophage RAW264.7 cells (Supplementary Figure 2A&B)
HFLMCD diet feeding increases liver E4BP4 expression in WT mice
A variety of diets enriched in saturated fat and cholesterol with certain nutrient depletion have been used to induce hepatic ER stress (6,11). In particular, high-fat, low methionine, and choline-deficient (HFLMCD) diet has been shown to stimulate hepatocyte ER stress, liver steatosis, and liver inflammation in a relatively short duration (30). In our hands, feeding with HFLMCD diet for 10 weeks led to elevated levels of serum ALT, liver TG, and liver cholesterol (Figure 2A-C), while liver histology demonstrated clear signs of lipid accumulation by H&E staining and fibrosis by Sirius Red staining (Figure 2D), mimicking human NASH. We also detected a significant induction of hepatic E4bp4 mRNA of female mice and a remarkable increase of hepatic E4BP4 protein in male mice along with the elevated ER stress marker Chop mRNA after 10-week HFLMCD diet (Figure 2E-F & Supplementary Figure 2C&D). Taken altogether, our data for the first time demonstrate that hepatic E4BP4 is elevated in the liver of mice with HFLMCD diet-induced steatosis and fibrosis.
Figure 2. HFLMCD diet induces E4BP4 expression in the mouse liver.

C57BL6 female mice were fed either regular chow (n=4) or HFLMCD diet (n=8) for 10 wks to induce NASH prior to the following assays: (A) serum ALT; (B-C) total liver TG & cholesterol; (D) liver histology by H&E staining and liver fibrosis by Sirius Red staining; (E-F) Liver E4bp4 mRNA and protein expression in regular chow vs. HFLMCD diet-fed females. Data were presented as Mean ± S.D.. *p<0.05, ***p<0.001 by unpaired Student t-test.
CHOP is partially required for tunicamycin-induced E4bp4 expression.
The transcriptional program upon ER stress has been well characterized in mammalian cells (1). CHOP acts downstream of the PERK-ATF4 pathway to promote cell death during prolonged ER stress (31). Hepatic induction of CHOP following tunicamycin injection is necessary for the suppression of genes critical for lipid metabolism (32). Interestingly, the global Chop KO mice fed a diet high in fat, fructose, and cholesterol for 16 wks developed more severe histological features of NASH compared with WT controls (19). We therefore examined whether Chop knockdown affects the tunicamycin-induced E4bp4 in both cultured hepatocytes and the mouse liver. Acute depletion of Chop in hepatocytes was achieved by transduction with Ad-shChop and confirmed by RT-qPCR (Figure 3A). In PMHs, tunicamycin induced a robust increase of E4bp4, Chop, and Grop78 in Ad-shLacZ-transduced controls, whereas Chop depletion largely abrogated the E4bp4 induction without affecting Grp78 (Figure 3A). A similar loss of E4bp4 induction was observed in the Ad-shChop-injected mouse liver after tunicamycin injection (Figure 3B), suggesting that CHOP is required for the maximal E4bp4 induction by tunicamycin in hepatocytes and the liver.
Figure 3. Chop is critical for the induction of E4bp4 by tunicamycin in PMHs and the mouse liver.
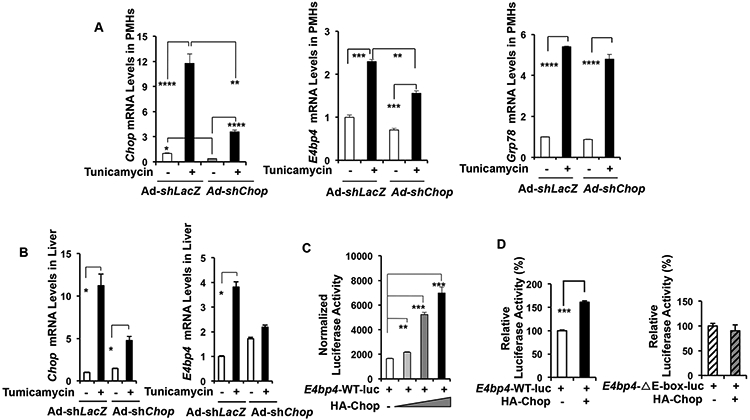
(A) PMHs were transduced with AdshlacZ or Ad-shChop before treatment of tunicamycin for 24 hr. The cells were then harvested for mRNA analysis of E4bp4, Chop, and Grp78. (B-C) WT mice were tail-vein injected with either AdshLacZ or AdshChop for 10 days before intra-peritoneal injection of DMSO or tunicamycin for 24 hrs. The mRNA and protein levels of E4BP4 were analyzed by TR-qPCR and immunoblotting, respectively. (C) Activation of the E4bp4 promoter activity by ectopic expression of Chop. 293AD cells were transfected with a luciferase reporter construct driven by the E4bp4-WT promoter plus an increasing amount of the HA-Chop expression construct. The luciferase activity was normalized by the β-gal activity. (D) The E-box element in the promoter is required for its induction by CHOP. 293AD cells were transfected with the E4bp4-WT-luc or the E4bp4-ΔE-box-luc mutant plus the HA-Chop expression construct. The luciferase activity was normalized by the β-gal activity. Data were presented as Mean ± SD. *p <0.05, ***p <0.001 by unpaired Student t-test. The in vitro experiments were repeated at least twice with similar results.
Given the role of CHOP as a transcription factor, we hypothesized that CHOP could directly activate the promoter of E4bp4 in response to tunicamycin. We did find that the ectopic expression of HA-Chop induced the luciferase activity driven by the mouse E4bp4 promoter in a dose-dependent manner in 293A cells (Figure 3C). We previously reported that the E-box element within the proximal region of the E4bp4 promoter is important for its activation by SREBP-1c (27). Thus, we tested whether this response element is also required for the CHOP-mediated induction by generating a mutant reporter construct with the deletion of the E-box element. As shown in Figure 3D, Chop overexpression was only able to activate the E4bp4-WT-luc but not the E4pb4-ΔE-box-luc mutant, suggesting that this E-box element is indeed critical for the transcriptional activation of E4bp4 by CHOP (Figure 3D). However, we were unable to detect direct binding of CHOP to the E4bp4 promoter by ChIP assay (data not shown), indicating that CHOP might stimulate other transcription factors to activate the E4bp4 promoter.
E4BP4 is required for tunicamycin-induced lipid droplet accumulation in cultured hepatocytes
Both acute and chronic ER stress potently impact hepatic lipid homeostasis (10). For example, administration of tunicamycin into WT mice was reported to influence lipid synthesis and breakdown, leading to massive liver steatosis (5). We previously reported that E4BP4 mediates de novo lipogenesis downstream of the insulin signaling in hepatocytes (27). To test whether E4BP4 is involved in the tunicamycin-induced lipid accumulation in hepatocytes and the liver, we generated E4bp4 liver-specific knockout (E4bp4-LKO) mice by crossing E4bp4flox/flox mice with Alb-Cre transgenic mice and validated the liver-specific deletion of E4BP4 (Figure 4A). Next, we challenged both E4bp4flox/flox and E4bp4-LKO PMHs with tunicamycin, and analyzed the impact of E4bp4 deficiency on lipid droplet formation and TG content in hepatocytes. As shown by BODIPY staining (33), overnight incubation of tunicamycin markedly increased both the number and size of lipid droplets in E4bp4flox/flox hepatocytes. However, tunicamycin-induced lipid droplets were nearly abolished in the E4bp4-LKO PMHs (Figure 4B), consistent with the reduced levels of TG content in tunicamycin-treated E4bp4-LKO vs. E4bp4flox/flox PMHs (Figure 4C). These observations support that E4BP4 is indeed required for lipid droplet accumulation in response to tunicamycin-induced ER stress in hepatocytes.
Figure 4. Hepatic E4bp4 is required for tunicamycin-induced accumulation of lipid droplets in PMHs.

(A) Validation of liver-specific E4bp4 deficiency in various tissues isolated from both E4bp4flox/flox (n=3) and E4bp4-LKO (n=3) mice by RT-qPCR. PMHs were isolated from E4bp4flox/flox and E4bp4-LKO mice and treated with tunicamycin overnight prior to BODIPY staining for lipid droplets and RNA extraction for RT-qPCR. (B) Image of lipid droplet formation after tunicamycin treatment; lipid droplets in 50 cells were countered per condition and presented in the bar graph; (C) Cellular triglycerides (TG) content in tunicamycin-treated E4bp4flox/flox or E4bp4-LKO PMHs; (D) Lipid metabolic genes assessed in PMHs of E4bp4-LKO mice following tunicamycin treatment.
Lipid homeostasis in hepatocytes is a dynamic process coordinated via lipid uptake, lipid synthesis, fatty acid oxidation, and lipid export (34). To explore the downstream targets of E4BP4 during lipid accumulation in hepatocytes, we measured the mRNA expression of common lipid metabolic pathways in PMHs. On one hand, tunicamycin elevated the expression of Fasn while repressing Cpt1a in E4bp4flox/flox but not E4bp4-LKO PMHs (Figure 4D). On the other hand, there were no differences in the expression of genes involved in autophagy, lipolysis, and lipid export between those two groups (Supplementary Figure 3). Chronic ER stress also has been reported to induce hepatocyte apoptosis (35,36). We therefore examined whether E4BP4 is required for this process. As shown in Supplementary Figure 5, tumicamycin treatment significantly induced pro-apoptotic genes including Noxa and Puma in E4bp4flox/flox but not E4bp4-LKO PMHs, indicating that E4BP4 induction may contribute to ER stress-induced apoptosis. Together, these findings reveal a broad role of E4BP4 in lipid metabolism and hepatocyte apoptosis following ER stress.
Loss of hepatic E4bp4 protects against HFLMCD diet-induced lipid accumulation
Given the important role of ER stress in hepatocytes during NAFLD (2) and the elevated E4BP4 expression in the liver of the HFLMCD-diet-induced ER stress mouse model (Figure 2), it was logical to examine whether manipulation of hepatic E4BP4 impacts lipid metabolism and NAFLD in vivo. We firstly compared E4bp4flox/flox vs. E4bp4-LKO mice on regular chow following fasting overnight and refeeding for 12 hrs. The reason we chose to refeed mice is because the E4BP4 mRNA and protein tend to be potently induced by refeeding (37). Body weight, triglycerides and cholesterol in serum/liver, as well as serum ALT all turned out to be comparable between the two groups of mice (Supplementary Figure 6), supporting that hepatic E4BP4 is dispensable in liver lipid metabolism under the normal physiological condition.
Next, we examined the impact of hepatic deletion of E4bp4 on lipid metabolism following HFLMCD diet. Upon 10-wk HFLMCD feeding, both Both E4bp4flox/flox and E4bp4-LKO mice showed similar body weight gain (Figure 5A). However, the ratio of liver weight/body weight was significantly reduced in E4bp4-LKO mice (Figure 5B). Serum ALT, the classical marker for liver injury, was about 20% lower in E4bp4-LKO mice (Figure 5C). Despite comparable levels of serum TG and cholesterol in both groups of mice (Figure 5D), E4bp4-LKO mice accumulated significantly less TG, total cholesterol, and lipid droplets in the liver by H&E staining (Figure 5D&E). In summary, all these data suggest that E4bp4-LKO mice are protected from HFLMCD-induced liver steatosis and cholesterol accumulation.
Figure 5. Loss of hepatic E4bp4 protects mice from HFLMCD diet-induced lipid accumulation in the liver.

Both E4bp4 flox/flox and E4bp4-LKO were fed HFLMCD diet for 10 weeks before sacrifice for the following assays: (A-B) body weight and liver weight/body weight ratio; (C) serum ALT, TG, and cholesterol; (D) liver TG and cholesterol; (E) Liver histology by H&E staining; (F) Quantification of lipid metabolic gene expression in the liver. (G) Immuoblotting of AKT-PS473, ATK, lipogenic enzymes, and SREBP-1c in the liver. *p <0.05; **p <0.01, compared with E4bp4flox/flox group. All data are expressed as Mean ± S.D..
To further explore how E4bp4 deficiency renders mice resistant to liver steatosis, we measured the genes of major lipid metabolic pathways in both groups of mice. In comparison with E4bp4flox/flox mice, E4bp4-LKO mice showed significantly lower expression of genes involved in de novo lipogenesis (Acc1 and Scd1) and lipid droplet formation (Plin4) in the liver (Figure 5F). At the protein levels, SCD1 protein levels were markedly reduced in the liver of E4bp4-LKO mice (Figure 5G). Of note, liver-specific Scd1 knockout mice showed less lipid accumulation in the liver in response to high-carbohydrate diet (38,39). In contrast, no significant differences were detected in the genes involved in lipolysis, cholesterol biosynthesis, fatty acid oxidation, and fatty acid uptake in the liver between those two groups of HFLMCD diet-fed mice (Supplementary Figure 7).
Since NASH is associated with increased liver inflammation and fibrosis, we then compared the expression of inflammatory and fibrotic markers in the liver between E4bp4flox/flox and E4bp4-LKO mice following HFLMCD. The mRNA levels of inflammatory cytokines (Tnfα, Il-1β, Mcp1, and F4-80) as well as fibrotic genes (Cola1, αSma, Tgfβ) in the liver were comparable between the two groups (Supplementary Figure 8A&B), in agreement with the similar staining pattern of the liver tissues by Masson’s Trichrome staining (Supplementary Figure 8C). These data suggest that under the diet-induced ER stress condition, hepatic E4bp4 deficiency protects mice from lipid accumulation without impacting inflammation and fibrosis in the liver.
The role of AMPK suppression in E4BP4-driven lipid accumulation in hepatocytes
To further uncover the molecular mechanisms underlying the reduced expression of de novo genes and lipid accumulation in the liver of HFLMCD diet-fed E4bp4-LKO mice, we focused on the energy-sensing kinase AMPK, a critical regulator of lipid homeostasis in the liver (40). It has been well recognized that the AMPK-dependent phosphorylation of ACC potently inhibits de novo lipogenesis in the liver (41), whereas the AMPK-dependent phosphorylation of ATGL promotes lipolysis (42). Several in vivo studies revealed the beneficial effects of chronic AMPK activation on diet-induced liver steatosis (43,44). Whether hepatic E4bp4 deficiency could affect the AMPK pathway in hepatocytes and subsequently diet-induced lipid accumulation in the liver is currently unknown.
AMPK activation is marked by AMPK phosphorylation at T172 (45). We first examined the effect of E4bp4 deficiency on AMPK-PT172 in the liver of HFLMCD diet-fed mice. Indeed, the AMPK-PT172 levels were elevated in the liver of E4bp4-LKO mice following HFLMCD diet feeding (Figure 6A). Similarly, we observed increased levels of AMPK-PT172 in E4bp4-LKO PMHs (Figure 6B), supporting that E4BP4 regulates AMPK-PT172 in a cell-autonomous manner in hepatocytes. Conversely, adenoviral over-expression of E4BP4 substantially reduced not only the basal but also the 2-Deoxy-D-glucose-induced AMPK-PT172 in WT mouse hepatocytes (Figure 6C). These findings collectively support that E4BP4 acts as a negative regulator of the AMPK pathway in hepatocytes.
Figure 6. E4BP4 targets the AMPK-β1 subunit to mediate the tunicamycin-induced lipid accumulation in hepatocytes.

(A) Comparison of AMPK activation by anti AMPK-phosphorylation at T172 in the liver E4bp4flox/flox and E4bp4-LKO mice following HFLMCD feeding. (B) The levels of AMPK-PT172 in PMHs from E4bp4flox/flox and E4bp4-LKO mice; (C) Adenoviral over-expression of E4BP4 in WT PMHs reduces the basal and 2-Deoxy-D-glucose-induced AMPK-PT172 level. (D) The protein abundance of AMPK subunits in E4bp4flox/flox and E4bp4-LKO PMHs by immunoblotting. (D) Effect of E4BP4 overexpression on the stability of AMPK-β1. (E) Effect of acute depletion of Ampk-β1 by adenoviral shAmpk-β1 on lipid droplets in E4bp4-LKO PMHs following tunicamycin treatment. The knockdown efficiency of Ad-shAmpk-1 was confirmed by immunblotting with anti-AMPK-β1. (F) The mRNA levels of candidate ubiquitin E3 ligases in the liver of E4bp4flox/flox and E4bp4-LKO mice following HFLMCD feeding. *p <0.05; **p <0.01, compared with E4bp4flox/flox group. All data are expressed as Mean ± S.D..
To test how hepatic E4BP4 regulates the AMPK pathway, we examined whether E4BP4, as a transcription factor, affects the expression of hepatic Ampk subunits. The mRNA levels of the major AMPK subunits were comparable in the liver of E4bp4flox/flox vs. E4bp4-LKO mice (Supplementary Figure 9A). However, at the protein level, AMPKβ1 was markedly elevated in both E4bp4-LKO PMHs (Figure 6D) and the liver of E4bp4-LKO mice (Supplemental Figure 9B). These data suggest that E4BP4 might control the proteolysis of AMPKβ1 in hepatocytes.
AMPKβ1 is the scaffolding subunit in the intact AMPK kinase complex in mouse hepatocytes (46). Interestingly, we found that, under the tunicamycin-induced ER stress, only the AMPKβ1 protein abundance was reduced (Supplementary Figure 9D). To directly test the impact of E4BP4 on the AMPK-β1 degradation, we measured the protein half-life of AMPKβ1 in Ad-E4bp4-transduced Hepa1 cells in cycloheximide chase analysis. In cells transduced with Ad-GFP, AMPKβ1 had a half-life of about 3 hrs. However, its half-life was shortened to less than 2 hrs in cells transduced with Ad-E4bp4 (Figure 6E). This observation was consistent with the formation of AMPKβ1-polyubiquitin conjugates in Ad-E4bp4-transduced PMHs (Supplementary Figure 10A). We previously reported that E4BP4 forms a complex with the nuclear SREBP-1c and promotes lipogenic gene expression in response to insulin signaling (27). We therefore checked whether SREBP-1c over-expression affects the AMPKβ1 protein abundance just like E4BP4. As shown in Supplementary Figure 10B, adenoviral SREBP-1c overexpression showed no effect on the AMPKβ1 abundance in cultured hepatocytes, suggesting that the E4BP4 action on the AMPKβ1 stability is likely to be independent of SREBP-1c in hepatocytes.
How does E4BP4 controls the AMPKβ1 proteolysis? We speculate that E4BP4 may control the expression of critical regulators that either promote or suppress the stability of AMPKβ1 in hepatocytes. It has been reported that the ubiquitin E3 ligase MKRN targets the AMPKα subunits for degradation, whereas the ubiquitin-specific protease USP10 protects the AMPK complex from proteolysis (47-49). We screened the expression of the reported regulators of the AMPK complex by RT-qPCR and found that several AMPK–specific E3 ligases including Rnf44, Ube2e, and, Celebron (49-51) were significantly down-regulated in the liver of E4bp4-LKO mice following HFLMCD diet feeding (Figure 6F). Thus, it is likely that E4BP4 could suppress the AMPKβ1 protein stability via the up-regulation of these E3 ligases.
Since AMPKβ1 is essential for the integrity of the AMPK protein complex (46), we reasoned that the depletion of Ampk-β1 by shRNA could lead to the disassociation of the AMPK kinase complex and subsequently suppress lipid droplet formation in E4bp4-LKO PMHs. To test this hypothesis, we challenged E4bp4-LKO PMHs with tunicamycin after depleting Ampkβ1 with Ad-shAmpk-β1 (validated by immunoblotting in Figure 6G) and then performed lipid droplet staining. As shown in Figure 6G, no increase of lipid droplets was detected in Ad-shLacZ-transduced E4bp4-LKO PMHs after tunicamycin treatment. However, knockdown of Ampk-β1 led to a drastic induction of lipid droplets in E4bp4-LKO PMHs in the presence or absence of tunicamycin treatment. In summary, our data support that E4BP4 is likely to target the AMPKβ1 subunit to promote lipid accumulation in hepatocytes in response to ER stress inducers.
Discussion
Previously, we reported that the b-ZIP transcription factor E4BP4 could be induced in the liver via actions of nutritional and hormonal signaling to regulate hepatic lipid metabolism (22,27). Here we demonstrate that ER stress potently induces E4BP4 in the liver and hepatocytes to cause lipid accumulation in the liver. Depletion of hepatic E4bp4 not only protects against tunicamycin-induced lipid droplet accumulation in cultured hepatocytes but also reduces liver steatosis in a diet-induced NAFLD mouse model. At the mechanistic level, we found that CHOP is required for the maximal induction of E4BP4 by ER stress in hepatocytes. We discovered that E4bp4 deletion impairs hepatic de novo lipogenesis and lipid accumulation likely through promoting the AMPK activity in hepatocytes following ER stress. Our findings point to E4BP4 as a novel target for treating liver diseases associated with ER stress (Supplementary Figure 10C).
It has been established that ER stress promotes NAFLD by altering hepatic lipid metabolism (4,10). Specific effectors of ER stress pathway and their downstream signaling molecules have been examined in cell cultures and animal models for their roles in the ER stress-induced lipid accumulation in hepatocytes. A PERK inhibitor was found to improve liver steatosis and insulin sensitivity in an animal model of NAFLD (52). In contrast, deletion of Ire1 leads to spontaneous liver steatosis (53), indicative of the unique functions of specific ER stress mediators in regulating lipid metabolism. In the current study, we obtained direct evidence from E4bp4-LKO PMHs to support the essential role of E4BP4 in tunicamycin-induced lipid accumulation in hepatocytes. A similar observation was also made in the liver of HFLMCD-challenged E4bp4-LKO mice. To investigate the impact of hepatic E4bp4 deficiency with a stronger relevance to advanced human NAFLD in the future, we intend to feed E4bp4-LKO mice a diet rich in saturated fat, cholesterol, and sugars (54). Recently, the Koo group observed reduced E4BP4 in the liver of either 27-week high-fat (60% calories from fat) diet-fed WT or ob/ob mice and provided evidence in support of E4BP4 as a negative regulator of hepatic gluconeogenic genes (25). In our study, both tunicamycin injection and 10-week HFLMCD (45% calories from fat) diet feeding potently induce the mRNA and protein levels of E4BP4 in the liver of E4bp4flox/flox mice. We speculate that genetic backgrounds, the type of nutritional stress, and feeding regimen may all contribute to the differences between the studies, highlighting the complexities in the E4BP4-regulated metabolic responses in the liver.
In our study, we examined lipid metabolism genes to assess how E4BP4 impacts lipid metabolism following ER stress. Consistent with our previous studies (27), several de novo lipogenic genes including Fasn and Acc1 were reduced in the absence of E4bp4 in hepatocytes. However, E4bp4 deficiency did not seem to impact fatty acid oxidation, lipid transport, and lipolysis gene expression. We speculated that hepatic E4bp4 deficiency might repress the activity of lipid metabolic enzymes via the AMPK pathway without significantly affecting the mRNA levels of these lipid metabolic enzymes. Our data indeed show that inhibition of the AMPK pathway by acute knockdown of Ampk-β1 restores lipid droplet formation in E4bp4-LKO PMHs. AMPK is a master regulator of multiple metabolic pathways and have been targeted in treating obesity, insulin resistance, type 2 diabetes, and NAFLD (40,55,56). One of our novel findings is that E4BP4 is both necessary and sufficient to inhibit the cellular AMPK activity in hepatocytes. E4bp4-deficiency in hepatocytes enhances the levels of AMPK-PT172, the hallmark of AMPK activation, whereas E4bp4 overexpression inhibits AMPK-PT172. The Sternberg’s group proposed three potential mechanisms by which AMPK activation may improve NAFLD: (1) the suppression of hepatic de novo lipogenesis; (2) the enhancement of fatty acid oxidation; and (3) the improvement of the mitochondrial function/integrity in adipose tissue (40,56,57). Surprisingly, despite the reduced ATP content in the livers of patients with NAFLD, hepatic AMPK activity was actually reduced, suggesting that AMPK activation could be regulated independently of ATP levels in the liver during NAFLD (56,58). Our data point out that E4BP4 could be a critical factor that links nutritional stress and AMPK suppression in hepatocytes during NAFLD. Therefore, the suppression of E4BP4 may be a valid avenue to activate hepatic AMPK activity for the treatment of NAFLD.
So, how does E4BP4 suppress the AMPK signaling in the liver? There has been extensive research on how the intracellular ADP:ATP ratio activates AMPK (57). However, very little is known about how the changes of the major components of the AMPK complex affect its activity. We found that the mRNA abundance of Ampk subunits was similar in the liver between E4bp4flox/flox and E4bp4-LKO mice (Supplementary Figure 9A). In contrast, the protein level of the AMPKβ1 subunit was elevated in both E4bp4-LKO PMHs and liver tissues. Moreover, E4BP4 overexpression enhances the ubiquitination of AMPKβ1 and accelerates the degradation of the AMPKβ1 subunit in hepatocytes. Therefore, E4BP4 is likely to inhibit hepatic AMPK pathway by enhancing the AMPKβ1 ubiquitination and degradation. So far, the AMPK subunits were shown to undergo the E3 ligase-mediated ubiquitination and degradation. A case in point, the lipid droplet-binding protein CIDEC was reported to interact with the AMPKα subunits and promote its ubiquitination-dependent degradation during adipogenesis (59). Recently, several E3 ligases were reported to modulate the protein stability of the AMPK complex, including MKRN, CRBN, RNF44, UBE2O (47,49-51). MKRN1 was found to directly target the AMPK-α subunits for ubiquitination and degradation (47). MKRN-null mice showed chronic AMPK activation in both liver and adipose tissues and were protected against diet-induced metabolic dysfunction. RNF44 promotes the degradation of AMPKα1 in melanoma cells in response to arginine deprivation (50). The E3 ligase CRBN binds to the AMPK complex and degrades the AMPKα subunits in multiple cell types (49). The Song group reported that UBE2O acts as a ubiquitin E3 ligase for the AMPK-α2 subunit in skeletal muscles (49). Intriguingly, we found that the mRNA levels of RNF44, CRBN, and Ube2O but not MKRN were significantly reduced in the liver of E4bp4-LKO mice. Our future study will determine whether these E3 ligases are involved in the regulation of the AMPKβ1 stability downstream of E4BP4 induction in hepatocytes in response to chronic ER stress.
ER stress in macrophages is important for inflammation and the inflammasome activation. Interestingly, we observed that, in the RAW264.1 macrophage cell line, tunicamycin also potently induces E4bp4 mRNA and protein expression (Supplementary Figure 2), suggesting that the ER stress-induced E4BP4 pathway could be conserved in multiple cell types. It has been reported that the ER stress pathway is activated in Kupffer cells during NAFLD, resulting in the M1-type polarization of macrophages and subsequently increased liver injury (60). Even though the essential role of E4BP4 in NK cell development was widely reported (23), so far, the contribution of E4BP4 in ER-stressed macrophages to NAFLD remains unknown. We will explore the role of E4BP4 in Kupffer cells during the pathogenesis of NAFLD by generating a mouse model with the macrophage-specific deletion of E4bp4 for future study.
One important finding in our study is the gender-specific protection against HFLMCD-induced NASH. Only female E4bp4-LKO mice were protected from TG and cholesterol accumulation in the liver even although hepatic E4BP4 was elevated in both female and male mice following 10-week HFLMCD diet. It has been previously recognized that NAFLD is a sexually dimorphic disease and the metabolic impact of sex hormones is likely to account for the differences between genders (61). In a human study, over 1000 genes display a sex bias in their expression in the liver with the top biological pathways related to lipid metabolism (62). Currently, there are no data suggesting a crosstalk between E4BP4 and the estrogen signaling in the liver. For future study, we will identify the gender-specific downstream targets of E4BP4 that might regulate lipid metabolism and the AMPK signaling in a sexually dimorphic manner.
In summary, we have shown that both the E4bp4 mRNA and protein are highly inducible by ER stress signals in cultured hepatocytes and the mouse liver. We also showed that hepatic E4BP4 is required for lipid accumulation in response to ER stress signals. E4bp4 liver-specific knockout mice are partially protected from hepatic lipid accumulation and injury induced by HFLMCD diet, likely due to the enhanced AMPK pathway. Furthermore, E4BP4 might reduce the AMPK-β1 stability in the liver to inhibit the AMPK pathway in response to chemical or diet-induced ER stress possibly by upregulating AMPK-specific E3 ligases. Taken together, our data reveal a novel molecular link between E4BP4 and ER stress-induced hepatic lipid accumulation during the pathogenesis of NAFLD and indicate that the suppression of hepatic E4BP4 activation might be a potential therapeutic avenue for treating NAFLD.
Supplementary Material
Table I.
qPCR primer sequences
| Forward | Reverse | |
|---|---|---|
| 18S | TTGACGGAAGGGCACCACCAG | GCACCACCACCCACGGAATCG |
| Acc1 | GAAGCCACAGTGAAATCTCG | GATGGTTTGGCCTTTCACAT |
| Acox1 | TGCTGCAGACGGCCAGGTTC | GGCCAGACTGCCACCTGCTG |
| Acox2 | CCTTTGCCCAACGACACTGGCA | ACCGGGAGGTACCAAGAACCTCTG |
| Acsl3 | GCAGCTGCGTCAGGGTCC | TAAGACCCGCGGGCTCCG |
| Atg12 | GGCCTCGGAACAGTTGTTTA | CAGCACCGAAATGTCTCTGA |
| Atg2a | CACTCTACGCCACTACAT | ATCCAGCACATCCAAGAA |
| Atg7 | CAGAAGAAGTTGAACGAGTA | CAGAGTCACCATTGTAGTAAT |
| Atgl | TTCACCATCCGCTTGTTGGAG | AGATGGTCACCCAATTTCCTC |
| Bax | GATCAGCTCGGGCACTTTAG | TTGCTGATGGCAACTTCAAC |
| Bcl-2l1 | CGGATTGCAAGTTGGATGGC | TCAGGAACCAGCGGTTGAAG |
| Beclin1 | GGCCAATAAGATGGGTCTGA | CACTGCCTCCAGTGTCTTCA |
| Bim | CGGTCCTCCAGTGGGTATTT | TATGGAAGCCATTGCACTGAGA |
| Cd36 | CCAAGCTATTGCGACATGATT | CCGAACCACAGCGTAGATAGACC |
| Cgi58 | CTTGCTTGGACACAACCTG | GAGGTGACTAACCCTTGATGG |
| Chop | CTGCCTTTCACCTTGGAGAC | CGTTTCCTGGGGATGAGATA |
| Cidea | ACAGAAATGGACACCGGGTAG | TGACATTGAGACAGCCGAGG |
| Col1a1 | GAGGCCTCCCCAGAACATCAC | CGATCTCGTTGGATCCCTGG |
| Col1a2 | AGTCGATGGCTGCTCCAAAA | AGCACCACCAATGTCCAGAG |
| Cpt1a | TCTGCATGTTTGACCCAAAA | TTGCTGGAGATGTGGAAGAA |
| Crbn | TCCTTTGCGGGTAAACAGACA | TCGGTTTTCTGGCTTCTTTACTA |
| Cyp4a10 | GGAGCTCCAATGTCTGAGAAGAGT | TCTCTGGAGTATTCTTCTGAAAAAGGT |
| Cyp4a14 | TCTCTGGCTTTTCTGTACTTTGCTT | CAGAAAGATGAGATGACAGGACACA |
| Dhcr7 | ATTGAGTTCAACCCCCGCAT | AACACGTAGATGGCCTGCAA |
| E4bp4 | ATGGGAAGGCTCTTTCTCCACT | TACCCGAGGTTCCATGTTTC |
| Elovl7 | GCCAAGAGCAATGAGGATGG | GGTCCACGGCATGATCGTAT |
| Fasn | TTGGCCCAGAACTCCTGTAG | CTCGCTTGTCGTCTGCCT |
| Fsp27α | GCCACGCGGTATTGCCAGGA | GGGTCTCCCGGCTGGGCTTA |
| Fsp27β | GTGACCACAGCTTGGGTCGGA | GGGTCTCCCGGCTGGGCTTA |
| Gadd45a | TGGTGACGAACCCACATTCA | CGGGAGATTAATCACGGGCA |
| Grp78 | GGTGCAGCAGGACATCAAGTT | CCCACCTCCAATATCAACTTGA |
| Hmgcr | CACAATAACTTCCCAGGGGT | GGCCTCCATTTAGATCCG |
| Hmgcs | AGAAATCCCTGGCTCGCTTG | AGCTTTAGACCCCTGAAGGC |
| Hrd1 | TTTTCGGCCTGTCAGATGGC | GGCCCAGAGACCTGTGAACG |
| Hsl | ACGCTACACAAAGGCTGCTT | TCGTTGCGTTTGTAGTGCTC |
| Idi1 | GACGTCACCCTTGTGCTAGA | TAGAACAGAGATTCCGGCTG |
| Il-1β | TGCAGCTGGAGAGTGTGGATCCC | TGTGCTCTGCTTGTGAGGTGCTG |
| Il-6 | GACAACTTTGGCATTGTGG | ATGCAGGGATGATGTTCTG |
| Lpl | AGAAGGGAAAGGACTCAGCAG | TCAAACACCCAAACAAGGGTA |
| Lss | ATCCAAGCACTGTTAGAGGCAGGT | TCCAGTGTGCTGAAGGAGAAACCA |
| Magl | GAACAAGTCGGAGGGTTCTGC | GAGGACGTGATAGGCACCTT |
| Mcp1 | ACTGAAGCCAGCTCTCTCTTCCTC | TTCCTTCTTGGGGTCAGCACAGAC |
| Mkrn1 | GCGAGAAAGGAGATCCGACC | 5‘-TTCGTATCTGCAGCGGTCTC |
| Mmp2 | AACGGTCGGGAATACAGCAG | CCACCCATGGTAAACAAGGC |
| Mttp1 | CTCCACAGTGCAGTTCTCACA | AGAGACATATCCCCTGCCTGT |
| Noxa | TGGAGTGCACCGGACATAAC | TCGTCCTTCAAGTCTGCTGG |
| Pcsk9 | CACAATGTAGGTTCCTGGCA | GAGGATGGCCTGGCTGAT |
| Plin2 | GTGGAAAGGACCAAGTCTGTG | GACTCCAGCCGTTCATAGTTG |
| Plin3 | TGTCCAGTGCTTACAACTCGG | CAGGGCACAGGTAGTCACAC |
| Plin4 | TCCTGCTCTGAGGGACCCTT | TCTTGCCTTTGGATTTGGGG |
| Plin5 | TGTCCAGTGCTTACAACTCGG | CAGGGCACAGGTAGTCACAC |
| Pnpla3 | CTCCCTCTCGGCCGTATAAT | AGTCGTGGATGCCCTGGTGT |
| Pparα | CCTTCTACGCTCCCGACCCA | CCATGTCCATAAATCGGCACCA |
| Puma | TACGAGCGGCGGAGACAAG | GTGTAGGCACCTAGTTGGGC |
| Rnf44 | AGAGCACAGCTGCGTCCTG | GGGCTCACAACCCGGCA |
| Scd1 | GCCGAGCCTTGTAAGTTCTG | CCTCCTGCAAGCTCTACACC |
| Sqle | GATGGGCATTGAGACCTTCT | TTTAAAAGAGCCCGACAGGA |
| Srebp-1c | AACGTCACTTCCAGCTAGAC | AACGTCACTTCCAGCTAGAC |
| Srebp2 | CCCTATTCCATTGACTCTGAGC | GAGTCCGGTTCATCCTTGAC |
| Syvn1 | GCGTCTAGGACCTTGTCCTTT | CCAGAGACCTGTGAACGCTAGG |
| Tgfβ | GCTGAACCAAGGAGACGG | ATGTCATGGATGGTGCCC |
| Timp1 | AGGTGGTCTCGTTGATTCGT | GTAAGGCCTGTAGCTGTGCC |
| Tnfα | ACTTCGGGGTGATCGGTCCCC | TGGTTTGCTACGACGTGGGCTAC |
| Ube2o | GCTCTATGGCCAAGAAGGTGA | CCCTATTTCACTCCCGGCTC |
| Ulk1 | ACCATTGTCTACCAGTGT | AGTGTCTTGTTCTTCTCTAA |
| Usp10 | CCAGTGCCTCCCAAACCCCG | GTCCTCCTGCCGGCCCTTTTC |
| Wipi1 | CACAGGATGGAGGAGAAT | GATGGAGGTAAGGAAGGT |
Acknowledgements
This work was supported by NIH R00 (DK077449) and R01 (DK099593) to L.Y., R01 (DK090313) to K.Z., and R01 (DK121170) to X.T.. Part of the work was also supported by pilot grants from the University of Michigan Medical School: Michigan Nutrition Obesity Research Center to L.Y. and X.T. (P30 DK089503), Michigan Diabetes Research Training Center to L.Y. and X.T. (P60-DK-020572), and Center for Gastrointestinal Research (P30 DK034933) to D.Z.. We thank Dr. Rutkowski for sharing the HA-Chop expression vector.
Abbreviations:
- E4BP4
E4 promoter–binding protein 4
- NAFLD
non-alcoholic fatty liver disease
- ER stress
endoplasmic reticulum stress
- NASH
nonalcoholic steatohepatitis
- AMPK signaling
AMP-activated protein kinase signaling
- PMHs
primary mouse hepatocytes
- PERK
protein kinase RNA-like endoplasmic reticulum kinase
- ATF4
activating transcription factor 4
- CHOP
C/EBP homologous protein
- UPR
unfolded protein response
- FXR
farnesoid X-activated receptor
- GLP
glucagon-like peptide
- XBP1
X-box binding protein 1
- CREBH
cyclic adenosine monophosphate (cAMP)-responsive element-binding protein H
- C/EBP β
CCAAT/enhancer-binding protein β
- FASN
fatty acid synthase
- SCD1
stearoyl-Coenzyme A desaturase 1
- ALT
alanine aminotransferase
- TG
triglycerides
- HFLMCD diet
high-fat, low-methionine, and choline-deficient diet
- ChIP
chromatin immunoprecipitation
- CPT1A
carnitine palmitoyltransferase 1A
- ACC1
acetyl-CoA carboxylase 1
- PLIN4
perilipin 4
- ATGL
adipose triglyceride lipase
- CIDEC
cell death-inducing DFFA-like effector C
- MKRN1
makorin ring finger protein 1
References
- 1.Grootjans J, Kaser A, Kaufman RJ, and Blumberg RS (2016) The unfolded protein response in immunity and inflammation. Nat Rev Immunol 16, 469–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henkel A, and Green RM (2013) The unfolded protein response in fatty liver disease. Semin Liver Dis 33, 321–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byun S, Kim YC, Zhang Y, Kong B, Guo G, Sadoshima J, Ma J, Kemper B, and Kemper JK (2017) A postprandial FGF19-SHP-LSD1 regulatory axis mediates epigenetic repression of hepatic autophagy. EMBO J 36, 1755–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han J, and Kaufman RJ (2016) The role of ER stress in lipid metabolism and lipotoxicity. J Lipid Res 57, 1329–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee JS, Mendez R, Heng HH, Yang ZQ, and Zhang K (2012) Pharmacological ER stress promotes hepatic lipogenesis and lipid droplet formation. Am J Transl Res 4, 102–113 [PMC free article] [PubMed] [Google Scholar]
- 6.Rinella ME, Siddiqui MS, Gardikiotes K, Gottstein J, Elias M, and Green RM (2011) Dysregulation of the unfolded protein response in db/db mice with diet-induced steatohepatitis. Hepatology 54, 1600–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samuel VT, and Shulman GI (2018) Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab 27, 22–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noureddin M, and Sanyal AJ (2018) Pathogenesis of NASH: The Impact of Multiple Pathways. Curr Hepatol Rep 17, 350–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedman SL, Neuschwander-Tetri BA, Rinella M, and Sanyal AJ (2018) Mechanisms of NAFLD development and therapeutic strategies. Nat Med 24, 908–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song MJ, and Malhi H (2019) The Unfolded Protein Response and Hepatic Lipid Metabolism in Non-alcoholic fatty liver disease. Pharmacol Ther, 107401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, and Hotamisligil GS (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 [DOI] [PubMed] [Google Scholar]
- 12.Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, and Sanyal AJ (2008) Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 134, 568–576 [DOI] [PubMed] [Google Scholar]
- 13.Ren LP, Song GY, Hu ZJ, Zhang M, Peng L, Chen SC, Wei L, Li F, and Sun W (2013) The chemical chaperon 4-phenylbutyric acid ameliorates hepatic steatosis through inhibition of de novo lipogenesis in high-fructose-fed rats. Int J Mol Med 32, 1029–1036 [DOI] [PubMed] [Google Scholar]
- 14.Cho EJ, Yoon JH, Kwak MS, Jang ES, Lee JH, Yu SJ, Kim YJ, Kim CY, and Lee HS (2014) Tauroursodeoxycholic acid attenuates progression of steatohepatitis in mice fed a methionine-choline-deficient diet. Dig Dis Sci 59, 1461–1474 [DOI] [PubMed] [Google Scholar]
- 15.Bandla H, Dasgupta D, Mauer AS, Nozickova B, Kumar S, Hirsova P, Graham RP, and Malhi H (2018) Deletion of endoplasmic reticulum stress-responsive co-chaperone p58(IPK) protects mice from diet-induced steatohepatitis. Hepatol Res 48, 479–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoo J, Cho IJ, Jeong IK, Ahn KJ, Chung HY, and Hwang YC (2018) Exendin-4, a glucagon-like peptide-1 receptor agonist, reduces hepatic steatosis and endoplasmic reticulum stress by inducing nuclear factor erythroid-derived 2-related factor 2 nuclear translocation. Toxicol Appl Pharmacol 360, 18–29 [DOI] [PubMed] [Google Scholar]
- 17.Han CY, Rho HS, Kim A, Kim TH, Jang K, Jun DW, Kim JW, Kim B, and Kim SG (2018) FXR Inhibits Endoplasmic Reticulum Stress-Induced NLRP3 Inflammasome in Hepatocytes and Ameliorates Liver Injury. Cell Rep 24, 2985–2999 [DOI] [PubMed] [Google Scholar]
- 18.Rahman SM, Schroeder-Gloeckler JM, Janssen RC, Jiang H, Qadri I, Maclean KN, and Friedman JE (2007) CCAAT/enhancing binding protein beta deletion in mice attenuates inflammation, endoplasmic reticulum stress, and lipid accumulation in diet-induced nonalcoholic steatohepatitis. Hepatology 45, 1108–1117 [DOI] [PubMed] [Google Scholar]
- 19.Rahman K, Liu Y, Kumar P, Smith T, Thorn NE, Farris AB, and Anania FA (2016) C/EBP homologous protein modulates liraglutide-mediated attenuation of non-alcoholic steatohepatitis. Lab Invest 96, 895–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Henkel AS, LeCuyer BE, Schipma MJ, Anderson KA, and Green RM (2015) Hepatocyte X-box binding protein 1 deficiency increases liver injury in mice fed a high-fat/sugar diet. Am J Physiol Gastrointest Liver Physiol 309, G965–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng Z, Kim H, Qiu Y, Chen X, Mendez R, Dandekar A, Zhang X, Zhang C, Liu AC, Yin L, Lin JD, Walker PD, Kapatos G, and Zhang K (2016) CREBH Couples Circadian Clock With Hepatic Lipid Metabolism. Diabetes 65, 3369–3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keniry M, Dearth RK, Persans M, and Parsons R (2014) New Frontiers for the NFIL3 bZIP Transcription Factor in Cancer, Metabolism and Beyond. Discoveries (Craiova) 2, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gascoyne DM, Long E, Veiga-Fernandes H, de Boer J, Williams O, Seddon B, Coles M, Kioussis D, and Brady HJ (2009) The basic leucine zipper transcription factor E4BP4 is essential for natural killer cell development. Nat Immunol 10, 1118–1124 [DOI] [PubMed] [Google Scholar]
- 24.Kostrzewski T, Borg AJ, Meng Y, Filipovic I, Male V, Wack A, DiMaggio PA, and Brady HJM (2018) Multiple Levels of Control Determine How E4bp4/Nfil3 Regulates NK Cell Development. J Immunol 200, 1370–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang G, Han HS, and Koo SH (2017) NFIL3 is a negative regulator of hepatic gluconeogenesis. Metabolism 77, 13–22 [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Kuang Z, Yu X, Ruhn KA, Kubo M, and Hooper LV (2017) The intestinal microbiota regulates body composition through NFIL3 and the circadian clock. Science 357, 912–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tong X, Li P, Zhang D, VanDommelen K, Gupta N, Rui L, Omary MB, and Yin L (2016) E4BP4 is an insulin-induced stabilizer of nuclear SREBP-1c and promotes SREBP-1c-mediated lipogenesis. J Lipid Res 57, 1219–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohta Y, Taguchi A, Matsumura T, Nakabayashi H, Akiyama M, Yamamoto K, Fujimoto R, Suetomi R, Yanai A, Shinoda K, and Tanizawa Y (2017) Clock Gene Dysregulation Induced by Chronic ER Stress Disrupts beta-cell Function. EBioMedicine 18, 146–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei Y, Wang D, Topczewski F, and Pagliassotti MJ (2006) Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab 291, E275–281 [DOI] [PubMed] [Google Scholar]
- 30.Khoury T, Ben Ya'acov A, Shabat Y, Zolotarovya L, Snir R, and Ilan Y (2015) Altered distribution of regulatory lymphocytes by oral administration of soy-extracts exerts a hepatoprotective effect alleviating immune mediated liver injury, non-alcoholic steatohepatitis and insulin resistance. World J Gastroenterol 21, 7443–7456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sano R, and Reed JC (2013) ER stress-induced cell death mechanisms. Biochim Biophys Acta 1833, 3460–3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chikka MR, McCabe DD, Tyra HM, and Rutkowski DT (2013) C/EBP homologous protein (CHOP) contributes to suppression of metabolic genes during endoplasmic reticulum stress in the liver. J Biol Chem 288, 4405–4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiu B, and Simon MC (2016) BODIPY 493/503 Staining of Neutral Lipid Droplets for Microscopy and Quantification by Flow Cytometry. Bio Protoc 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rui L (2014) Energy metabolism in the liver. Compr Physiol 4, 177–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brenner C, Galluzzi L, Kepp O, and Kroemer G (2013) Decoding cell death signals in liver inflammation. J Hepatol 59, 583–594 [DOI] [PubMed] [Google Scholar]
- 36.Iracheta-Vellve A, Petrasek J, Gyongyosi B, Satishchandran A, Lowe P, Kodys K, Catalano D, Calenda CD, Kurt-Jones EA, Fitzgerald KA, and Szabo G (2016) Endoplasmic Reticulum Stress-induced Hepatocellular Death Pathways Mediate Liver Injury and Fibrosis via Stimulator of Interferon Genes. J Biol Chem 291, 26794–26805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tong X, Muchnik M, Chen Z, Patel M, Wu N, Joshi S, Rui L, Lazar MA, and Yin L (2010) Transcriptional repressor E4-binding protein 4 (E4BP4) regulates metabolic hormone fibroblast growth factor 21 (FGF21) during circadian cycles and feeding. J Biol Chem 285, 36401–36409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flowers MT, and Ntambi JM (2009) Stearoyl-CoA desaturase and its relation to high-carbohydrate diets and obesity. Biochim Biophys Acta 1791, 85–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyazaki M, Sampath H, Liu X, Flowers MT, Chu K, Dobrzyn A, and Ntambi JM (2009) Stearoyl-CoA desaturase-1 deficiency attenuates obesity and insulin resistance in leptin-resistant obese mice. Biochem Biophys Res Commun 380, 818–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith BK, and Steinberg GR (2017) AMP-activated protein kinase, fatty acid metabolism, and insulin sensitivity. Curr Opin Clin Nutr Metab Care 20, 248–253 [DOI] [PubMed] [Google Scholar]
- 41.Winder WW, and Hardie DG (1996) Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol 270, E299–304 [DOI] [PubMed] [Google Scholar]
- 42.Kim SJ, Tang T, Abbott M, Viscarra JA, Wang Y, and Sul HS (2016) AMPK Phosphorylates Desnutrin/ATGL and Hormone-Sensitive Lipase To Regulate Lipolysis and Fatty Acid Oxidation within Adipose Tissue. Mol Cell Biol 36, 1961–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woods A, Williams JR, Muckett PJ, Mayer FV, Liljevald M, Bohlooly YM, and Carling D (2017) Liver-Specific Activation of AMPK Prevents Steatosis on a High-Fructose Diet. Cell Rep 18, 3043–3051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, Gao B, Wierzbicki M, Verbeuren TJ, Shaw RJ, Cohen RA, and Zang M (2011) AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 13, 376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hawley SA, Selbert MA, Goldstein EG, Edelman AM, Carling D, and Hardie DG (1995) 5'-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J Biol Chem 270, 27186–27191 [DOI] [PubMed] [Google Scholar]
- 46.Polekhina G, Gupta A, Michell BJ, van Denderen B, Murthy S, Feil SC, Jennings IG, Campbell DJ, Witters LA, Parker MW, Kemp BE, and Stapleton D (2003) AMPK beta subunit targets metabolic stress sensing to glycogen. Curr Biol 13, 867–871 [DOI] [PubMed] [Google Scholar]
- 47.Lee MS, Han HJ, Han SY, Kim IY, Chae S, Lee CS, Kim SE, Yoon SG, Park JW, Kim JH, Shin S, Jeong M, Ko A, Lee HY, Oh KJ, Lee YH, Bae KH, Koo SH, Kim JW, Seong JK, Hwang D, and Song J (2018) Loss of the E3 ubiquitin ligase MKRN1 represses diet-induced metabolic syndrome through AMPK activation. Nat Commun 9, 3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng M, Yang X, Qin B, Liu T, Zhang H, Guo W, Lee SB, Kim JJ, Yuan J, Pei H, Wang L, and Lou Z (2016) Deubiquitination and Activation of AMPK by USP10. Mol Cell 61, 614–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kwon E, Li X, Deng Y, Chang HW, and Kim DY (2019) AMPK is down-regulated by the CRL4A-CRBN axis through the polyubiquitination of AMPKalpha isoforms. FASEB J 33, 6539–6550 [DOI] [PubMed] [Google Scholar]
- 50.Li YY, Wu C, Shah SS, Chen SM, Wangpaichitr M, Kuo MT, Feun LG, Han X, Suarez M, Prince J, and Savaraj N (2017) Degradation of AMPK-alpha1 sensitizes BRAF inhibitor-resistant melanoma cells to arginine deprivation. Mol Oncol 11, 1806–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vila IK, Yao Y, Kim G, Xia W, Kim H, Kim SJ, Park MK, Hwang JP, Gonzalez-Billalabeitia E, Hung MC, Song SJ, and Song MS (2017) A UBE2O-AMPKalpha2 Axis that Promotes Tumor Initiation and Progression Offers Opportunities for Therapy. Cancer Cell 31, 208–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang W, Hietakangas V, Wee S, Lim SC, Gunaratne J, and Cohen SM (2013) ER stress potentiates insulin resistance through PERK-mediated FOXO phosphorylation. Genes Dev 27, 441–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.So JS, Hur KY, Tarrio M, Ruda V, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, Lichtman AH, Iwawaki T, Glimcher LH, and Lee AH (2012) Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab 16, 487–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Santhekadur PK, Kumar DP, and Sanyal AJ (2018) Preclinical models of non-alcoholic fatty liver disease. J Hepatol 68, 230–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu G, Huang K, and Zhou J (2018) Hepatic AMP Kinase as a Potential Target for Treating Nonalcoholic Fatty Liver Disease: Evidence from Studies of Natural Products. Curr Med Chem 25, 889–907 [DOI] [PubMed] [Google Scholar]
- 56.Smith BK, Marcinko K, Desjardins EM, Lally JS, Ford RJ, and Steinberg GR (2016) Treatment of nonalcoholic fatty liver disease: role of AMPK. Am J Physiol Endocrinol Metab 311, E730–E740 [DOI] [PubMed] [Google Scholar]
- 57.Lin SC, and Hardie DG (2018) AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab 27, 299–313 [DOI] [PubMed] [Google Scholar]
- 58.Boudaba N, Marion A, Huet C, Pierre R, Viollet B, and Foretz M (2018) AMPK Re-Activation Suppresses Hepatic Steatosis but its Downregulation Does Not Promote Fatty Liver Development. EBioMedicine 28, 194–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu Y, Gu Y, Liu G, Zhang F, Li J, Liu F, Zhang Z, Ye J, and Li Q (2015) Cidec promotes the differentiation of human adipocytes by degradation of AMPKalpha through ubiquitin-proteasome pathway. Biochim Biophys Acta 1850, 2552–2562 [DOI] [PubMed] [Google Scholar]
- 60.Yang F, Liu Y, Ren H, Zhou G, Yuan X, and Shi X (2019) ER-stress regulates macrophage polarization through pancreatic EIF-2alpha kinase. Cell Immunol 336, 40–47 [DOI] [PubMed] [Google Scholar]
- 61.Palmisano BT, Zhu L, Eckel RH, and Stafford JM (2018) Sex differences in lipid and lipoprotein metabolism. Mol Metab 15, 45–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu L, Shi J, Luu TN, Neuman JC, Trefts E, Yu S, Palmisano BT, Wasserman DH, Linton MF, and Stafford JM (2018) Hepatocyte estrogen receptor alpha mediates estrogen action to promote reverse cholesterol transport during Western-type diet feeding. Mol Metab 8, 106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.