

Abstract
Introduction
The treatment for glioblastoma (GBM) has remained unchanged for the past decade, with only minimal improvements in patient survival. As a result, novel treatments are needed to combat this devastating disease. Immunotherapies are treatments that stimulate the immune system to attack tumor cells and can be either local or systemically delivered. Viral treatments can lead to direct tumor cell death through their natural lifecycle or through the delivery of a suicide gene, with the potential to generate an anti-tumor immune response, making them interesting candidates for combinatorial treatment with immunotherapy.
Methods
We review the current literature surrounding the interactions between oncolytic viruses and the immune system as well as the use of oncolytic viruses combined with immunotherapies for the treatment of GBM.
Results
Viral therapies have exhibited preclinical efficacy as single-agents and are being investigated in that manner in clinical trials. Oncolytic viruses have significant interactions with the immune system, although this can also vary depending on the strain of virus. Combinatorial treatments using both oncolytic viruses and immunotherapies have demonstrated promising preclinical findings.
Conclusions
Studies combining viral and immunotherapeutic treatment modalities have provided exciting results thus far and hold great promise for patients with GBM. Additional studies assessing the clinical efficacy of these treatments as well as improved preclinical modeling systems, safety mechanisms, and the balance between treatment efficacy and immune-mediated viral clearance should be considered.
Keywords: Oncolytic viruses, Glioblastoma, Immunotherapy, Combination, Treatment
Introduction
Glioblastoma (GBM) is the most common primary malignant brain tumor, with a highly aggressive nature and dismal prognosis. The current treatment for GBM has remained largely unchanged for over a decade and consists of surgical resection, radiation, and chemotherapy [1]. Despite treatment, most patients succumb to the disease within 15 months of diagnosis, highlighting the need for novel treatments [2]. Indeed, GBM is a uniquely challenging cancer to treat and develop new treatments for, as highlighted by the lack of effective novel treatments [3]. Immunotherapy, which harnesses the immune system to eradicate cancers, has seen success in other cancer types and is the focus of a number of current preclinical and clinical studies in GBM [4–6]. Immunotherapies can be locally or systemically administered, and can also be generally categorized based on mechanism of treatment into monoclonal antibodies targeting tumor antigens, tumor agnostic-treatments that work against a variety of cancer types such as checkpoint inhibitors, viral therapies, T-cell therapies, and cancer vaccines [4]. While previously thought to be immune-privileged, studies in the past decade have highlighted the brain as accessible to the immune system, suggesting that immunotherapies may hold promise in treating CNS tumors, including GBM [7]. However, preceding investigations into the use of single-agent immunotherapies for GBM have been met with limited success [4, 5]
The failure of single-agent immunotherapies in GBM is likely in part due to the low immunogenicity of the tumor cells as well as the severe local and systemic immune suppression mediated by the cancer [4, 5]. Contributing to the low immunogenicity of the GBM is the downregulation of MHC I [9–11] and relatively low mutational burden (TMB) seen in most GBM tumors when compared to other cancers that respond well to immunotherapies [12]. Previous studies have shown a positive relationship between TMB and response to immunotherapies across cancer types [13]. However, even the more rare GBM with an elevated mutational burden does not follow this trend, highlighting the unique response of GBM to immunotherapy, relative to other malignancies [13]. GBM also causes significant local and systemic immune-suppression [14, 15]. While the detailed signaling pathways and mechanisms underlying the immune-suppression seen in GBM are outside the scope of this review, they include expression of immune-checkpoint molecules, TGF-B signaling, STAT3, and expression of additional immunosuppressive cytokines by the tumor [16]. In addition, GBM has a low number of tumor-infiltrating T cells, which can mediate tumor cell death, and severe exhaustion and dysfunction of the T cells that do infiltrate [8, 17]. In fact, recent studies have highlighted the sequestration of T cells in the bone marrow in the setting of a GBM or other intra-cranial tumors as contributing to the systemic immune-suppression seen in afflicted patients [15]. Myeloid cells, including tumor-associated macrophages (TAMs) and myeloid derived suppressor cells (MDSCs) similarly contribute to the immunosuppressive GBM tumor microenvironment in a number of ways and have also been associated with reduced survival [18], highlighting the multidimensional immune-suppression present in GBM [19]. These myeloid cells, and the mechanisms previously described, contribute to the designation of GBM as an immunogenically “cold” tumor in contrast to other cancers, such as melanoma, with abundant infiltrating immune cells and high tumor mutational burdens.
Viral treatments used for cancer are commonly replication competent viruses that are either specific to tumor cells or lack the ability to spread outside of the immune suppressed tumor microenvironment [20]. They can lead to the death of infected cells through cell lysis as a part of their natural life cycle or through the delivery of genes, such as suicide genes, causing host cell death [20]. The resulting cell death can lead to the release of tumor associated antigens (TAAs), damage associated molecular patterns (DAMPs), and pathogen associated molecular patterns (PAMPs), which can activate the immune system and provide immunogenic targets [20, 21]. In addition, the oncolytic viral lifecycle, or certain suicide genes, can lead to immunogenic cell death, which can also stimulate the innate immune system leading to increased dendritic cell recruitment and antigen uptake and presentation, contributing to the generation of a robust anti-tumor immune response. Viral antigens, such as envelope proteins, can also further trigger an immune response, which may initially target viral epitopes, but is thought to undergo epitope focusing, eventually targeting tumor-specific epitopes as the virus is cleared by the immune system [20–22]. These mechanisms contribute to the view of some oncolytic viruses as a form of tumor specific vaccination in which TAAs are released in conjunction with immune stimulation, although this is dependent on the type of viral therapy and is likely reduced if replication-defective vectors or more immunogenically silent viruses are used. In fact, the anti-tumor response seen in some viral treatments is reduced or abrogated in immune deficient models, [21, 23, 24] highlighting the importance of the immune system in promoting the efficacy of many viral therapies. These findings also highlight the potential benefit of combining locally administered viral therapies with immunotherapy. In this review, we discuss the use of viral treatments for the treatment of GBM and the potential benefits that may be seen when combining them with immunotherapies.
Viral therapies for glioblastoma: evidence of interplay with the immune system
Several studies have highlighted the role of the immune system in facilitating an anti-tumor immune response following viral treatment in multiple cancer types [21, 23]. As an example, in a mouse model of melanoma being treated by vesicular stomatitis virus (VSV), an early study by Diaz et al. demonstrated a significant reduction in survival benefit in mice treated with VSV and depleted of CD8 T cells when compared to immunocompetent mice, highlighting the role of the immune system in mediating an anti-tumor response following viral treatment [23]. In clinical trials, patients injected with herpes simplex virus (HSV) for the treatment of metastatic melanoma demonstrated responses in both lesions that had been directly injected with virus and remote lesions not injected with the virus, further highlighting the role of the immune system in the clinical response to viral treatments [25].
In GBM specifically, a number of viral therapies have been utilized in the preclinical and early clinical settings. Some of the more commonly used viruses include retroviruses, measles virus, adenovirus, poliovirus, and HSV, which each have a unique set of advantages and disadvantages associated with their use [26]. Following promising preclinical studies, some viral therapies have reached clinical trials, with mixed results. A complete list of viral clinical trials for GBM can be found in Table 1. In the following section, we describe a select few viral therapies in later-stage clinical trials as well as specific evidence of each virus’ interaction with the immune system.
Table 1.
Recruiting, currently active, completed, and terminated clinical trials examining oncolytic viral therapy for GBM
| ClinicalTrials.gov Identifier | Experimental treatment | Control or comparator treatment | N | Primary endpoint or outcomes | Results for primary outcome | Study start date | Current status |
|---|---|---|---|---|---|---|---|
| Phase I Trials | |||||||
| NCT00805376 | DNX-2401, DNX-2401 plus surgical resection | None | 37 | Maximum Tolerated Dose (MTD) | Experimental dose of 3 × l010 vp in 1 mL was tolerated. A maximum dose was not identified | February 2019 | Completed |
| NCT03896568 | Ad5-DNX-2401, Ad5-DNX-2401 followed by surgery and an additional dose of Ad5-DNX-2401 | None | 36 | MTD and incidence of AEs | Not posted | February 12, 2019 | Recruiting |
| NCT03714334 | DNX-2440 | None | ~24 | Incidence of treatment associated AEs | Not posted | December 5, 2019 | Recruiting |
| NCT03072134 | NSC-CRAd-Survivin-pk7 (neural stem cells loaded with an oncolytic virus) plus chemoradiotherapy for patients with resectable tumors, NSC-CRAd-Survivin-pk7 plus chemoradiotherapy for patients with unresectable tumors | None | 13 | Determination of the maximum number of neural stem cells loaded with the oncolytic adenovirus | Not posted | April 24, 2017 | Completed |
| NCT01956734 | DNX2401 and Temozolomide | None | ~31 | Incidence of AEs | Not Posted | September 2013 | Completed |
| NCT00751270 | AdV-tk plus valacyclovir | None | 15 | Safety | No AEs related to viral treatment [27] | November 2005 | Completed |
| NCT03657576 | HSV-1 C134 | None | ~24 | Safety and tolerability assessed via incidence of AEs | Not posted | September 23, 2019 | Recruiting |
| NCT03911388 | HSV G207 | None | ~15 | Safety and tolerability assessed via incidence of ≥ grade 3 AEs | Not Posted | September 12, 2019 | Recruiting |
| NCT03152318 | rQNestin34.5v.2 3 + 3 dose escalation, rQNestin34.5v.2 with Cyclophosphamide (CPA) pre-treatment 3 + 3 dose escalation (for patients of previous arm who have met MTD or HTD) | None | ~108 | MTD | Not posted | July 18, 2017 | Recruiting |
| NCT02457845 | HSV G207 | None | ~18 | Safety and tolerability assessed via incidence of ≥ grade 3 AEs | Not posted | May 2016 | Recruiting |
| NCT02031965 | HSV-1716 plus dexamethasone | None | 2 | MTD | Not Posted | December 2013 | Terminated |
| NCT00157703 | HSV G207 | None | 9 | Incidence of AEs | Three patients with seizures potentially related to treatment, otherwise well tolerated [28] | May 2005 | Completed |
| NCT00028158 | HSV G207 | None | 65 | Safety and survival duration | Doses up to 3 × 109 p.f.u. can be inoculated safely. No complications or deaths have been unequivocally associated with G207 | December 2001 | Completed |
| NCT00002824 | HSV-TK with ganciclovir | None | ~18 | Assess overall response | Not Posted | February 1996 | Completed |
| NCT03294486 | TG6002/5-FC | None | ~78 | 6-month progression-free survival rate (PFS-6) and number of patients experiencing dose limiting toxicity | Not Posted | October 12, 2017 | Recruiting |
| NCT00390299 | MV-CEA administered into resection cavity, MV-CEA administered intratumorally followed by resection and cavity administration | None | 23 | MTD, incidence of ≥ grade 3 AEs | Not Posted | October 23, 2006 | Completed |
| NCT03043391 | Polio/rhinovirus recombinant (PVSRIPO) | None | ~12 | Percentage of participants with unacceptable toxicity over 14-day period | Not Posted | December 5, 2017 | Recruiting |
| NCT01491893 | Recombinant nonpathogenic polio-rhinovirus chimera (PVSRIPO) (several arms at escalating doses) | None | 61 | MTD, incidences of dose limiting toxicity, optimum dose | Dose level -1 (5.0 × 107 TCID50) was identified as phase 2 dose. One instance of dose limiting toxicity observed at 5 (1010 TCID50) | April 25, 2012 | Active, not recruiting |
| NCT02444546 | Sargramostim, Wild-type Reovirus | None | 6 | MTD | Not Posted | June 2015 | Active, not recruiting |
| NCT00528684 | REOLYSIN® | None | 18 | MTD/dose limiting toxicity, Objective Response Rate (ORR) | MTD not reached. No grade III or IV AEs [29] | July 2006 | Completed |
| NCT02576665 | Toca 511/Toca FC | None | 21 | Abnormal changes in immune activity in tumor/peripheral blood | Not Posted | July 2016 | Terminated |
| NCT01470794 | Toca 511/Toca FC | None | 58 | Safety and MTD | Serious adverse event rate of 7.1%. Durable response in 21.7% of patients [30] | February 2012 | Completed |
| NCT04327011 | Toca 511/Toca FC | None | 65 | Long term safety follow-up | Not Posted | February 2011 | Terminated |
| Phase II Trials | |||||||
| NCT00870181 | ADV-TK plus GCV | Control patients had surgery, systemic chemotherapy, or palliative care | 47 | PFS-6 | Experimental group had PFS-6 rate of 54.5% and median PFS of 29.6 weeks. Individuals in the control group had a PFS-6 rate of 13.6% and a median PFS of 8.4 weeks | January 2008 | Completed |
| NCT00589875 | AdV-tk plus Valacyclovir (GMCI) plus Standard of Care treatment (SOC) | SOC | 52 | OS and safety | No dose-limiting toxicities observed. Median OS in experimental group of 17.1 months versus 13.5 months in SOC group | March 2007 | Completed |
| NCT01301430 | H-1 Parvovirus (ParvO-ryx01) | None | 18 | Safety and tolerability | No MTD reached [31] | September 2011 | Completed |
| NCT01582516 | Dose escalation of Delta24-RGD | None | 20 | Incidence of AEs | 20% of patients survived > 3 years from treatment. MTD not reached [32] | June 2010 | Completed |
| Phase III Trials | |||||||
| NCT02414165 | Toca 511/Toca FC | Lomustine, Temozolomide, or Bevacizumab | 403 | Overall survival (OS) | OS for glioblastoma subjects was 11.6 months | November 30, 2015 | Terminated |
Results from ClinicalTrials.gov
Retroviral replicating vectors (RRV):
Replicating retroviruses (RRVs) are somewhat unique amongst replicating viral therapies in that they don’t lead to lytic cell death as a result of their lifecycle. Rather, the viruses integrate into host cell genomes and divide in a non-lytic manner, allowing for stealthy viral spread and persistence within tumor cells. As a result, RRVs have been utilized to deliver prodrug activator (“suicide”) genes that then lead to cancer cell death when a prodrug is given. Cancer cells that escape prodrug conversion-mediated cytotoxicity then act as ‘reservoirs’ of integrated retrovirus, which continues to be produced and re-infects cancer cells even as they recur, enabling efficacy of further prodrug treatment cycles. This is the concept behind Toca 511 (vocimagene amiretrorepvec), which delivers a yeast cytosine deaminase, which then converts the prodrug 5-fluorocytosine (5-FC) to the chemotherapeutic 5-fluorouracil (5-FU), leading to the death of infected cells [33]. Interestingly, preclinical investigations into Toca 511 demonstrated significant tumor growth in CD4 or CD4/CD8 depleted mice when rechallenged with tumor. This was in contrast to the lack of tumor cell growth seen in mice with just CD8 depletion, natural killer cell depletion, or no immune cell depletion at all, highlighting the value of the immune system in maintaining a memory of the tumor cells and rebuffing tumor rechallenge, or potentially recurrence in the clinical setting [34]. In addition, in a subcutaneous model of GBM, treatment with Toca 511 was shown to cause significant reductions in tumor infiltration of potentially immunosuppressive myeloid cells, including TAMs, MDSCs, and monocytes [35]. While a limitation of these findings is their discovery in a subcutaneous model of GBM, which has significant differences in immune cell infiltration and behavior relative to intracranial models, it was also demonstrated that T cells from these treated animals showed anti-tumor activity in vitro and when adoptively transferred to naïve animals bearing intracranial gliomas [36, 37]. The reduction of immunosuppressive cells, such as MDSCs, is also a promising finding with potential implications for the use of Toca511 in combination with immunotherapies that may otherwise be hampered by immunosuppressive myeloid cell populations.
As a result of the promising preclinical findings, Toca 511 was subsequently taken to clinical trials. Toca 511 was shown to be safe and provide a significant survival benefit in a phase 1 trial for recurrent high-grade glioma [38]. While a subsequent Phase III trial failed to meet its endpoints overall, likely due to lack of adequate prodrug cycles administered in the majority of patients, subsequent subgroup analysis revealed significant survival benefit in patients with 2 or more recurrences [39]. Further clinical trials of Toca 511 (now DB107) in specific patient populations that may benefit from this treatment are underway.
Poliovirus
Another viral therapy that has recently been studied in clinical trials is the modified poliovirus, known as PVSRIPO. PVSRIPO is genetically modified through the replacement of the native internal ribosome entry site (IRES) with that from rhinovirus, to reduce the neurovirulence of the virus and prevent viral replication in neurons. Virus particles infect cells expressing the poliovirus receptor (CD155), which is highly expressed on many solid tumors, including GBM cells, contributing to the specificity of the virus [40]. Preclinical studies of the virus were promising with significantly improved survival in treated mice using a subcutaneous GBM model [41]. Interestingly, the virus was also shown to sublethally infect antigen presenting cells (APCs) including macrophages and dendritic cells, leading to their activation, which then helped to drive an antitumor immune response. Similar to some of the preclinical immune-system findings in Toca511, the mechanisms of APC activation described in PVSRIPO should be interpreted with the caveat that this occurred in a subcutaneous model. Nevertheless, the results were promising and led to PVSRIPO being taken to clinical trials.
A phase I clinical trial has demonstrated the ability of the PVSRIPO to significantly extend patient survival, with 21% survival at 36 months in treated patients [40]. The safety of the virus was also demonstrated, although 69% of patients had a grade 1 or 2 adverse event attributed to the treatment [40]. PVSRIPO is now being evaluated in a multicenter Phase II trial for adult GBM, and in a Phase Ib trial for pediatric recurrent high-grade glioma. (Table 1).
Oncolytic adenovirus
Replicating oncolytic and replication-deficient adenoviruses have also been explored in the treatment of GBM. The most notable example of a replicating oncolytic adenovirus is Delta-24-RGD (now designated DNX-2401 (Tasadenoturev)), which carries specific mutations that confer tumor selectivity. These mutations include insertion of an ανβ3 / ανβ5 RGD sequence of the viral fiber to target redirect the virus to recognize surface integrins and enhance virus entry into tumor cells as well as a 24-bp deletion in the viral E1A gene to preclude viral replication in healthy cells that express a functional retinoblastoma protein but allow for viral replication in tumor cells with down-regulated retinoblastoma protein [32]. Preclinical studies have demonstrated that this virus can elicit anti-tumor immunity; mice injected with Delta-24-RGD had increased evidence of Th1 immunity, increased NK cells, and increased CD4 + lymphocytes in the tumor following virus injection. Treatment with the virus was also implicated in increased presentation of TAAs [42, 43]. A subsequent phase I clinical trial was promising, demonstrating the safety of the virus as well as a significant survival benefit and 20% of patients surviving for over three years [32, 42, 43]. Replication-deficient adenoviruses have also been used as a vehicle to carry suicide genes, most frequently thymidine kinase (TK), [27, 44, 45] directly to the GBM tumor mass. The killing of cells using TK from adenoviruses has been shown to increase costimulatory molecules on antigen presenting cells as well as infiltration of T cells and macrophages [27, 44, 45]. In addition, therapeutic responses seen have been higher in immune competent animals relative to immunodeficient models, emphasizing the role of the immune system in the response seen to these vectors [46].
As a result of the promising preclinical studies utilizing non-replicating adenoviruses carrying TK, subsequent clinical trials were initiated (Table 1). A phase I trial of a non-replicating adenovirus carrying TK were promising, demonstrating 25% survival at three years in newly diagnosed glioma [27]. Interestingly, significant CD3 + T cell infiltration was seen in 4/4 patient tumors analyzed following treatment, potentially suggesting some level of an immune response in these patients. Additional analysis of a single patient tumor revealed a large number of these CD3 + T cells to also be CD8 + , again potentially indicating cytotoxic T cells infiltrating as a response to the viral treatment. Significant increases in macrophage infiltration were also noted, further highlighting the immune response to the viral treatment and similarly corresponding to preclinical findings [27]. Two subsequent phase II clinical trials were completed, again demonstrating a survival benefit in patients with high grade glioma treated with an adenovirus carrying TK [47, 48].
Combining viral therapies with immunotherapy
As discussed in proceeding sections, viral therapies interact with the immune system in a number of ways, including the release of TAAs through tumor cell death, stimulation of the immune system through subsequent inflammatory pathways or direct infection of immune cells, and depletion of myeloid cells when specific suicide genes are utilized. Previous studies have demonstrated reduced efficacy of viral therapies in immunodeficient models, emphasizing the role of the immune system in viral treatment responses. Thus, combining oncolytic viruses with immunotherapies that increase the anti-tumor response of the immune system and potentially reduce tumor-mediated immune suppression is a subject of active investigation.
Reports of viral therapies combined with systemic immunotherapy have been successful in other cancers. In the B16 melanoma model, combination therapy with Newcastle disease virus (NDV) and checkpoint inhibition with CTLA-4 blockade led to the resolution of local and distant metastasis despite the resistance of tumor cells to NDV mediated lysis. This result was thought to be related to increased tumor T cell infiltration as a result of the viral infection, which subsequently increased tumor susceptibility to checkpoint inhibition [49]. Similarly, treatment with an oncolytic adenovirus has been shown to overcome PD-1 resistance in a mouse model of lung cancer by increasing the number of neoepitopes recognized by activated T cells [50]. The ability to increase the number of neoepitopes recognized by activated T cells may have implications for the treatment of GBM given the lower tumor mutational burden and limited number of TAAs available for the immune system to target.
As a result of these successes in other cancer types, the combination of viral therapies with systemic immunotherapies has also been investigated in GBM. Hardcastle et al. demonstrated an improved anti-tumor response in an orthotopic GL261 mouse model treated with oncolytic measles virus and anti-PD-1 compared to either anti-PD-1 or measles virus treatment alone [51]. Similar results were demonstrated when using VSV expressing TAAs in combination anti-PD-1 treatment [52] as well as in similar models using reovirus [53]. While these results are promising and recapitulate those seen in other cancers, they should be interpreted with caution considering the use of the GL261 model, which is overall significantly more immunogenic than human GBM and other mouse models, such as SB28 or Tu2449 [54, 55]. Interestingly, a study by Passaro et al. took a different approach to their combinatorial therapy; instead of delivering exogenous anti-PD-1 treatment, they designed an oncolytic HSV to express to express a single-chain fragment variable antibody against PD-1, leading to checkpoint inhibition in addition to tumor cell death via the virus’ oncolytic nature [56]. Using two different syngeneic mouse models, CT-2A and GL261, they demonstrated that mice treated with the HSV expressing PD-1 had a significant survival benefit relative to control mice, with some exhibiting long-term responses and resistance to rechallenge. However, this survival benefit was greater in GL261 tumors and there was no difference between the HSV expressing PD-1 and virus that did not express PD-1 [56]. In addition, there was a reduced therapeutic benefit of either virus in the CT-2A model, which is less immunogenic than GL261, highlighting the importance of model selection and the poor ability of GL261 to recapitulate the low immunogenicity of human GBM [56].
As seen in the aforementioned study by Passaro et al., an alternative approach to combining systemic immunotherapies with viral therapeutic modalities is by using the virus itself to deliver immunomodulatory genes, allowing the immune response to the virus and tumor cell death to be combined with the immune stimulating effects of the immunomodulatory gene. In the clinical setting, Talimogene laherparepvec (a herpes virus encoding for human granulocyte–macrophage colony stimulating factor) for the treatment of melanoma has also seen success in phase III trials, further demonstrating the potential of such treatments [57]. A similar approach has also been utilized in GBM. King et al. demonstrated the ability of two adenoviruses, one carrying TK, (leading to tumor cell death and the release of TAAs following prodrug administration) and one carrying FLT3L (which recruits dendritic cells to the tumor) to result in the long-term survival of rats with multifocal GBM [58]. In addition, Barret et al. used the GL261 model to evaluate the use of a replication incompetent adenovirus to deliver IL-12, which activates NK and T cells, to tumor cells in a regulatable manner that could be turned on or off using an activator (veledimex) [59]. Impressively, 65% of treated animals had a long term survival benefit, although again, this was in the GL261 model. These promising results prompted a subsequent phase I clinical trial. Using the same mechanism of regulation as the preceding preclinical trial, 31 patients were administered the adenovirus in their resection cavities following surgery with veledimex also given at varying doses, as tolerated. Interestingly, a patient requiring reresection of their tumor demonstrated increases in tumor infiltrating T cells, including CD3 + and CD3 + CD8 + T cells. A trend towards increased survival was also seen in patients who cumulatively received less than or equal to 20 mg of dexamethasone, suggesting the ability of the steroid to dampen potentially beneficial anti-tumor immune responses [60]. This system is currently being evaluated in a multicenter phase II study (Table 2). Similarly, HSV has been used to deliver IL-12 to the tumor microenvironment of 4C8 tumors, with an increase in survival and CD4 + , CD8 + , and NK cell tumor infiltration in treated mice when compared to controls [61]. Like it’s adenovirus counterpart, an HSV vector delivering IL-12 (M032) is now also in a clinical trial, with results pending (Table 2).
Table 2.
Recruiting, currently active, completed, and terminated clinical trials examining oncolytic viral and immunotherapeutic treatments for GBM
| ClinicalTrials.gov identifier | Experimental treatment | Control or comparator treatment | n | Primary endpoint or outcomes | Results for primary outcome | Study start date | Current status |
|---|---|---|---|---|---|---|---|
| Phase I trials | |||||||
| NCT03679754 | Ad-RTS-hIL-12 plus Veledimex | None | 36 | Safety and tolerability | Not Posted | September 5, 2018 | Active, not recruiting |
| NCT03636477 | Ad-RTS-hIL-12 plus veledimex in combination with nivolumab | None | 21 | Safety and Tolerability | Similar safety profile to Ad-RTS-hIL-12 plus Veledimex alone [60, 62] | June 18, 2018 | Active, not recruiting |
| NCT03330197 | Ad-RTS-hIL-12 plus Veledimex | None | ~ 24 | Safety and tolerability | Not Posted | September 26, 2017 | Active, not recruiting |
| NCT02026271 | Ad-RTS-hIL-12 plus Veledimex | None | ~ 48 | Safety and Tolerability | Frequency of AEs correlated with Veledimex dosing, reversed when veledimex was stopped. Median overall survival of 16.7 months in patients receiving ≤ 20 mg of dexamethasone [60] | June 2015 | Active, not recruiting |
| NCT01811992 | Dose escalation of Ad-hCMV-TK and Ad-hCMV-Flt3L | None | 19 | Tolerability and survival duration | MTD not reached [63] | April 2014 | Active, not recruiting |
| NCT02197169 | DNX-2401, DNX-2401 plus IFN-y | None | 37 | Objective Response Rate (ORR) | Not Posted | September 11, 2014 | Completed |
| NCT02062827 | HSV M032 producing IL-12 | None | 36 | MTD | Not Posted | February 14, 2014 | Recruiting |
| Phase II Trials | |||||||
| NCT02798406 | DNX-2401 plus pembrolizumab | None | 49 | ORR | Not Posted | June 2016 | Active, not recruiting |
| NCT04006119 | Ad-RTS-hIL-12 plus Veledimex and Cemipilimab-rwlc | None | 30 | Overall Survival (OS) and safety | Not Posted | August 1, 2019 | Recruiting |
Replication competent oncolytic adenoviruses have also been used to deliver immunomodulatory genes. Indeed, the oncolytic replication competent adenovirus Delta-24-RGD was used to deliver OX40L, an immune co-stimulator, to the GL261 mouse model of GBM. OX40L delivery resulted in a significant increase in survival relative to the unmodified Delta-24-RGD virus, demonstrating the benefit of combining an immunotherapy with the intrinsic immune-stimulating nature of an oncolytic virus [64]. A similar result was also demonstrated by the same group when they used the Delta-24-RDG virus to deliver another co-stimulatory ligand, GITRL (glucocorticoid-induced TNFR family-related gene) [65]. Compared to combining viral therapies with systemic immunotherapy, the advantage of local delivery of immunotherapy is avoiding the toxicity of systemic immunotherapy, while the disadvantage is that both therapies are localized, creating a potential risk for progression outside of the region of localized therapy.
Additional clinical trials involving the combination of viral therapies with immunotherapies can be found in Table 2. A schematic highlighting the interactions between oncolytic viral therapy and the immune system can be found in Fig. 1.
Fig. 1.
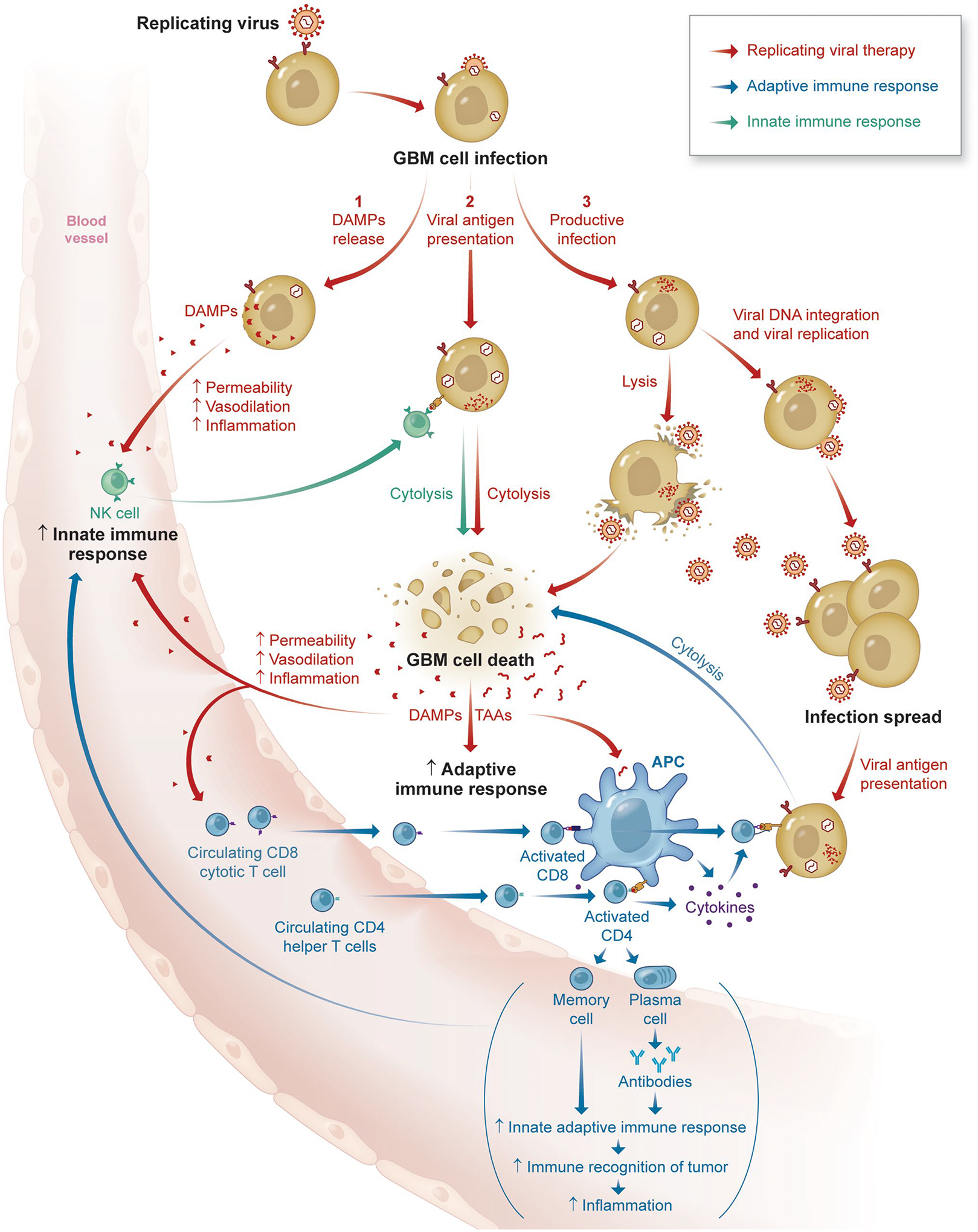
Schematic highlighting key interactions between oncolytic viral therapies and the immune system
Future directions
Using viral therapies in combination with immunotherapies for the treatment of GBM is an interesting treatment strategy with promising results to this point. Future treatments might combine viral therapies with immunotherapies that target multiple aspects of the immune system, such as T cell and myeloid compartments, in order to reverse the multidimensional immune suppression seen in GBM. Novel virus delivery mechanisms with increased payload abilities, tumor specificity, and safety will also continue to be explored and improved.
This review also highlights the challenges associated with testing and identifying effective viral treatments in the preclinical setting, highlighting the need for improved preclinical models in which to evaluate GBM viral and immunotherapies. As seen in multiple preclinical studies discussed in this review, the syngeneic GL261 model is commonly used to study viral treatments. However, a number of issues exist with the model. Primary amongst them is its high immunogenicity; GL261 has a much higher tumor mutational burden, MHC class I expression, and T cell infiltration than human GBM. The significant difference between the immunogenicity of GL261 and human GBM has likely contributed to the success of immunotherapies such as checkpoint inhibitors in GL261, but their subsequent failure in clinical trials [5, 54]. As a result, murine models induced by manipulation of tumor suppressor gene or oncogene expression, rather than induction through exposure to carcinogens, such as the SB28 model may be more biomimetic models of human GBM for studying immunotherapies [54]. Indeed, the immunogenicity of SB28 is more similar to that of human GBM as it has lower T cell infiltration, MHC Class I expression, and tumor mutational load than GL261. In fact, SB28 has 108 non-synonymous mutations, compared to 4978 in GL261, highlighting the reduced number of potential neoantigens in SB28 tumors [54]. In addition, murine models are frequently generated from cell lines and fail to replicate the intra-tumoral heterogeneity and other histologic/genetic characteristics seen in human GBM. As a result, a virus may replicate well through a murine model, but not in human GBM. The increased utilization of patient derived xenograft (PDX) models, which can more accurately recapitulate human GBM characteristics, presents an opportunity for investigators to model virus replication kinetics and/or transgene expression in a more clinically relevant tumor environment [66]. However, most PDX models also lack a functional immune system which similarly can influence viral replication and efficacy. While humanized PDX models that aim to recapitulate the human GBM tumor-immune system interface in a mouse model exist, they are technically challenging to create and expensive. Thus, careful consideration should be given to model selection with testing a viral vector in multiple model types likely providing the best insight into how a viral therapy will perform in human GBM.
Another critical consideration is the potential for concomitantly delivered immunotherapies to increase viral clearance, reducing the efficacy of the viral therapy. Anti-viral immune responses are largely mediated by type I interferons (IFNs), and additional components of the innate immune system, which can be downregulated in GBM, although this remains controversial [67, 68]. While some immunogenically silent viruses, such as replicating retroviruses [69] are adept at evading these anti-viral immune responses, others may be more immunogenic and susceptible to clearance by the immune system. As a result, the balance between stimulating the immune system against tumor cells, while still allowing for viral replication through the tumor is one that should be carefully considered and explored in future experiments. This may also highlight advantages and disadvantages between different viral treatments. For example, when attempting to deliver a therapeutic payload it may be beneficial to use an immunogenically silent virus that will be able to spread further through the tumor before clearance by the immune system rather than an immunogenic oncolytic virus. Nevertheless, combination treatments with viral treatments and immunotherapies will undoubtedly continue to see use in the treatment of GBM and are an exciting area of future research.
Footnotes
Conflict of interest The authors report no conflict of interest concerning materials or methods used in this study or the findings specified in this paper.
References
- 1.Stupp R, Mason WP, van den Bent MJ et al. (2005) Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med 352(10):987–996. 10.1056/NEJMoa043330 [DOI] [PubMed] [Google Scholar]
- 2.Ostrom QT, Cioffi G, Gittleman H et al. (2019) CBTRUS Statistical Report: primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro-Oncology 21(5):v1–v100. 10.1093/neuonc/noz150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jain KK (2018) A critical overview of targeted therapies for glioblastoma. Front Oncol 8:419. 10.3389/fonc.2018.00419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Young JS, Dayani F, Morshed RA, Okada H, Aghi MK (2019) Immunotherapy for high-grade gliomas: a clinical update and practical considerations for neurosurgeons. World Neurosurg 124:397–409. 10.1016/j.wneu.2018.12.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reardon DA, Brandes AA, Omuro A et al. (2020) Effect of Nivolumab vs Bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol 10.1001/jamaoncol.2020.1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Montoya ML, Kasahara N, Okada H (2020) Introduction to immunotherapy for brain tumor patients: challenges and future perspectives. Neuro-Oncology Pract. 10.1093/NOP/NPAA007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Louveau A, Smirnov I, Keyes TJ et al. (2015) Structural and functional features of central nervous system lymphatic vessels. Nature 523(7560):337–341. 10.1038/nature14432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE (2018) T-Cell dysfunction in glioblastoma: applying a new framework. Clin Cancer Res 24(16):3792–3802. 10.1158/1078-0432.CCR-18-0047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A (2015) Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol 129(6):829–848. 10.1007/s00401-015-1432-1 [DOI] [PubMed] [Google Scholar]
- 10.D’Alessio A, Proietti G, Sica G, Scicchitano BM (2019) Pathological and molecular features of glioblastoma and its peritumoral tissue. Cancers (Basel). 10.3390/cancers11040469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Facoetti A, Nano R, Zelini P et al. (2005) Human leukocyte antigen and antigen processing machinery component defects in astrocytic tumors. Clin Cancer Res 11(23):8304–8311. 10.1158/1078-0432.CCR-04-2588 [DOI] [PubMed] [Google Scholar]
- 12.Cristescu R, Mogg R, Ayers M et al. (2018) Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science (80-). 10.1126/science.aar3593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samstein RM, Lee C-H, Shoushtari AN et al. (2019) Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 51(2):202–206. 10.1038/s41588-018-0312-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nduom EK, Weller M, Heimberger AB (2015) Immunosuppressive mechanisms in glioblastoma. Neuro-Oncology 17 Suppl 7(Suppl 7):9–14. 10.1093/neuonc/nov151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chongsathidkiet P, Jackson C, Koyama S et al. (2018) Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med 24(9):1459–1468. 10.1038/s41591-018-0135-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim M, Xia Y, Bettegowda C, Weller M (2018) Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol 15(7):422–442. 10.1038/s41571-018-0003-5 [DOI] [PubMed] [Google Scholar]
- 17.Woroniecka K, Chongsathidkiet P, Rhodin K et al. (2018) T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res 24(17):4175–4186. 10.1158/1078-0432.CCR-17-1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alban TJ, Alvarado AG, Sorensen MD et al. (2018) Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI insight. 10.1172/jci.insight.122264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding AS, Routkevitch D, Jackson C, Lim M (2019) Targeting myeloid cells in combination treatments for glioma and other tumors. Front Immunol 10:1715. 10.3389/fimmu.2019.01715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Russell SJ, Barber GN (2018) Oncolytic viruses as antigen-agnostic cancer vaccines. Cancer Cell 33(4):599–605. 10.1016/j.ccell.2018.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC (2014) Going viral with cancer immunotherapy. Nat Rev Cancer 14(8):559–567. 10.1038/nrc3770 [DOI] [PubMed] [Google Scholar]
- 22.Ribas A, Dummer R, Puzanov I et al. (2017) Oncolytic virotherapy promotes intratumoral T cell infiltration and improves Anti-PD-1 immunotherapy. Cell 170(6):1109–1119.e10. 10.1016/j.cell.2017.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diaz RM, Galivo F, Kottke T et al. (2007) Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res 67(6):2840–2848. 10.1158/0008-5472.CAN-06-3974 [DOI] [PubMed] [Google Scholar]
- 24.Kim JW, Miska J, Young JS et al. (2017) A comparative study of replication-incompetent and -competent adenoviral therapy-mediated immune response in a Murine Glioma Model. Mol Ther - Oncolytics 5:97–104. 10.1016/j.omto.2017.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaufman HL, Bines SD (2010) OPTIM trial: a Phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future Oncol 6(6):941–949. 10.2217/fon.10.66 [DOI] [PubMed] [Google Scholar]
- 26.Lawler SE, Speranza MC, Cho CF, Chiocca EA (2017) Oncolytic viruses in cancer treatment a review. JAMA Oncol 3(6):841–849. 10.1001/jamaoncol.2016.2064 [DOI] [PubMed] [Google Scholar]
- 27.Chiocca EA, Aguilar LK, Bell SD et al. (2011) Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to upfront surgery and intensive timing radiation for malignant glioma. J Clin Oncol 29(27):3611–3619. 10.1200/JCO.2011.35.5222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Markert JM, Razdan SN, Kuo HC et al. (2014) A phase 1 trial of oncolytic HSV-1, g207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther 22(5):1048–1055. 10.1038/mt.2014.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forsyth P, Roldán G, George D et al. (2008) A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther 16(3):627–632. 10.1038/sj.mt.6300403 [DOI] [PubMed] [Google Scholar]
- 30.Cloughesy TF, Landolfi J, Vogelbaum MA et al. (2018) Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro Oncol 20(10):1383–1392. 10.1093/neuonc/noy075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geletneky K, Hajda J, Angelova AL et al. (2017) Oncolytic H-1 Parvovirus shows safety and signs of immunogenic activity in a first phase I/IIa Glioblastoma Trial. Mol Ther 25(12):2620–2634. 10.1016/j.ymthe.2017.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lang FF, Conrad C, Gomez-Manzano C et al. (2018) Phase I study of DNX-2401 (delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol 36(14):1419–1427. 10.1200/JCO.2017.75.8219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perez OD, Logg CR, Hiraoka K et al. (2012) Design and selection of toca 511 for clinical use: Modified retroviral replicating vector with improved stability and gene expression. Mol Ther 20(9):1689–1698. 10.1038/mt.2012.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hiraoka K, Inagaki A, Kato Y et al. (2017) Retroviral replicating vector-mediated gene therapy achieves long-term control of tumor recurrence and leads to durable anticancer immunity. Neuro Oncol 19(7):918–929. 10.1093/neuonc/nox038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitchell LA, Lopez Espinoza F, Mendoza D et al. (2017) Toca 511 gene transfer and treatment with the prodrug, 5-fluorocytosine, promotes durable antitumor immunity in a mouse glioma model. Neuro-Oncology 19(7):930–939. 10.1093/neuonc/nox037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okada H, Tsugawa T, Sato H et al. (2004) Delivery of interferon-α transfected dendritic cells into central nervous system tumors enhances the antitumor efficacy of peripheral peptide-based vaccines. Cancer Res 64(16):5830–5838. 10.1158/0008-5472.CAN-04-0130 [DOI] [PubMed] [Google Scholar]
- 37.Okada H, Thorne SH (2017) Is the immune response a friend or foe for viral therapy of glioma? Neuro-Oncology 19(7):882–883. 10.1093/neuonc/nox082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cloughesy TF, Landolfi J, Hogan DJ et al. (2016) Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci Transl Med 8(341):341ra75. 10.1126/scitranslmed.aad9784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cloughesy TF, Petrecca K, Walbert T et al. (2020) Effect of vocimagene amiretrorepvec in combination with flucytosine vs standard of care on survival following tumor resection in patients with recurrent high-grade glioma: a randomized clinical trial. JAMA Oncol. 10.1001/jamaoncol.2020.3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Desjardins A, Gromeier M, Herndon JE et al. (2018) Recurrent glioblastoma treated with recombinant poliovirus. NeuroOncol Pract 379(2):150–161. 10.1056/NEJMOA1716435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown MC, Holl EK, Boczkowski D et al. (2017) Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci Transl Med. 10.1126/scitranslmed.aan4220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang H, Clise-Dwyer K, Ruisaard KE et al. (2014) Delta-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. Castro MG, ed. PLoS ONE 9(5):e97407. 10.1371/journal.pone.0097407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kleijn A, Kloezeman J, Treffers-Westerlaken E et al. (2014) The in vivo therapeutic efficacy of the oncolytic adenovirus Delta24-RGD is mediated by tumor-specific immunity. Castro MG, ed. PLoS ONE 9(5):e97495. 10.1371/journal.pone.0097495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barba D, Hardin J, Sadelain M, Gage FH (1994) Development of anti-tumor immunity following thymidine kinase-mediated killing of experimental brain tumors. Proc Natl Acad Sci USA 91(10):4348–4352. 10.1073/pnas.91.10.4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vile RG, Castleden S, Marshall J, Camplejohn R, Upton C, Chong H (1997) Generation of an anti-tumour immune response in a non-immunogenic tumour: HSVtk killing in vivo stimulates a mononuclear cell infiltrate and a Th1-like profile of intratumoural cytokine expression. Int J Cancer. [DOI] [PubMed] [Google Scholar]
- 46.Gagandeep S, Brew R, Green B et al. (1996) Prodrug-activated gene therapy: involvement of an immunological component in the “bystander effect.” Cancer Gene Ther 3(2):83–88 [PubMed] [Google Scholar]
- 47.Ji N, Weng D, Liu C et al. (2016) Adenovirus-mediated delivery of herpes simplex virus thymidine kinase administration improves outcome of recurrent high-grade glioma. Oncotarget 7(4):4369–4378. 10.18632/oncotarget.6737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wheeler LA, Manzanera AG, Bell SD et al. (2016) Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro-Oncology 18(8):1137–1145. 10.1093/neuonc/now002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zamarin D, Holmgaard RB, Subudhi SK et al. (2014) Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 10.1126/scitranslmed.3008095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Woller N, Gürlevik E, Fleischmann-Mundt B et al. (2015) Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol Ther 23(10):1630–1640. 10.1038/mt.2015.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hardcastle J, Mills L, Malo CS et al. (2017) Immunovirotherapy with measles virus strains in combination with anti-PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro-Oncology 19(4):493–502. 10.1093/neuonc/now179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cockle JV, Rajani K, Zaidi S et al. (2016) Combination viroimmunotherapy with checkpoint inhibition to treat glioma, based on location-specific tumor profiling. Neuro-Oncology 18(4):518–527. 10.1093/neuonc/nov173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Samson A, Scott KJ, Taggart D et al. (2018) Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. 10.1126/scitranslmed.aam7577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Genoud V, Marinari E, Nikolaev SI et al. (2018) Responsiveness to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade in SB28 and GL261 mouse glioma models. Oncoimmunology 7(12):e1501137. 10.1080/2162402X.2018.1501137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smilowitz HM, Weissenberger J, Weis J, Brown JD, O’Neill RJ, Laissue JA (2007) Orthotopic transplantation of v-src-expressing glioma cell lines into immunocompetent mice: establishment of a new transplantable in vivo model for malignant glioma. J Neurosurg 106(4):652–659. 10.3171/jns.2007.106.4.652 [DOI] [PubMed] [Google Scholar]
- 56.Passaro C, Alayo Q, De Laura I et al. (2019) Arming an oncolytic herpes simplex virus type 1 with a single-chain fragment variable antibody against PD-1 for experimental glioblastoma therapy. Clin Cancer Res 25(1):290–299. 10.1158/1078-0432.CCR-18-2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kohlhapp FJ, Kaufman HL (2016) Molecular pathways: Mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res 22(5):1048–1054. 10.1158/1078-0432.CCR-15-2667 [DOI] [PubMed] [Google Scholar]
- 58.King GD, Muhammad AKMG, Curtin JF et al. (2008) Flt3L and TK gene therapy eradicate multifocal glioma in a syngeneic glioblastoma model. Neuro Oncol 10(1):19–31. 10.1215/15228517-2007-045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barrett JA, Cai H, Miao J et al. (2018) Regulated intratumoral expression of IL-12 using a RheoSwitch Therapeutic System® (RTS®) gene switch as gene therapy for the treatment of glioma. Cancer Gene Ther 25(5–6):106–116. 10.1038/s41417-018-0019-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chiocca EA, Yu JS, Lukas RV, et al. Regulatable Interleukin-12 Gene Therapy in Patients with Recurrent High-Grade Glioma: Results of a Phase 1 Trial. Vol 11.; 2019. http://stm.sciencemag.org/. Accessed June 10, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hellums EK, Markert JM, Parker JN et al. (2005) Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro Oncol 7(3):213–224. 10.1215/S1152851705000074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chiocca EA, Lukas RV, Rao G et al. (2019) Evaluation of controlled IL-12 in combination with a PD-1 inhibitor in subjects with recurrent glioblastoma. J Clin Oncol 37(15_Suppl):2020 [Google Scholar]
- 63.Lowenstein PR, Orringer DA, Sagher O et al. (2019) First-in-human phase I trial of the combination of two adenoviral vectors expressing HSV1-TK and FLT3L for the treatment of newly diagnosed resectable malignant glioma: Initial results from the therapeutic reprogramming of the brain immune system. J Clin Oncol 37(15_Suppl):2019 [Google Scholar]
- 64.Jiang H, Rivera-Molina Y, Gomez-Manzano C et al. (2017) Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous cancer vaccination. Cancer Res 77(14):3894–3907. 10.1158/0008-5472.CAN-17-0468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rivera-Molina Y, Jiang H, Fueyo J et al. (2019) GITRL-armed Delta-24-RGD oncolytic adenovirus prolongs survival and induces anti-glioma immune memory. NeuroOncol Adv 1(1):1–11. 10.1093/noajnl/vdz009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Patrizii M, Bartucci M, Pine SR, Sabaawy HE (2018) Utility of glioblastoma patient-derived orthotopic xenografts in drug discovery and personalized therapy. Front Oncol. 10.3389/fonc.2018.00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alain T, Lun XQ, Martineau Y et al. (2010) Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN production. Proc Natl Acad Sci USA 107(4):1576–1581. 10.1073/pnas.0912344107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Critchley-Thorne RJ, Simons DL, Yan N et al. (2009) Impaired interferon signaling is a common immune defect in human cancer. Proc Natl Acad Sci USA 106(22):9010–9015. 10.1073/pnas.0901329106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin AH, Burrascano C, Pettersson PL, Ibanez CE, Gruber HE, Jolly DJ (2014) Blockade of Type I Interferon (IFN) production by retroviral replicating vectors and reduced tumor cell responses to IFN likely contribute to tumor selectivity. J Virol 88(17):10066–10077. 10.1128/jvi.02300-13 [DOI] [PMC free article] [PubMed] [Google Scholar]