

Abstract
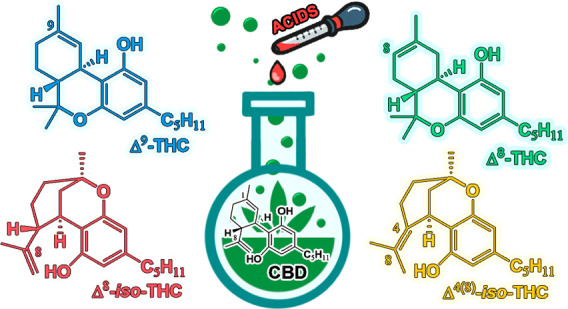
The chemical reactivity of cannabidiol is based on its ability to undergo intramolecular cyclization driven by the addition of a phenolic group to one of its two double bonds. The main products of this cyclization are Δ9-THC (trans-Δ-9-tetrahydrocannabinol) and Δ8-THC (trans-Δ-8-tetrahydrocannabinol). These two cannabinoids are isomers, and the first one is a frequently investigated psychoactive compound and pharmaceutical agent. The isomers Δ8-iso-THC (trans-Δ-8-iso-tetrahydrocannabinol) and Δ4(8)-iso-THC (trans-Δ-4,8-iso-tetrahydrocannabinol) have been identified as additional products of intramolecular cyclization. The use of Lewis and protic acids in different solvents has been studied to investigate the possible modulation of the reactivity of CBD (cannabidiol). The complete NMR spectroscopic characterizations of the four isomers are reported. High-performance liquid chromatography analysis and 1H NMR spectra of the reaction mixture were used to assess the percentage ratio of the compounds formed.
Recent years have seen a dramatically increasing interest in phytocannabinoids. Isolated from Cannabis in 1940,1,2 cannabidiol (CBD) is one of the most abundant phytocannabinoids in the species of Cannabis for textile uses.3,4 Despite the structural similarity between CBD and Δ9-THC (trans-Δ-9-tetrahydrocannabinol) (Figure 1), CBD has a low agonistic effect for cannabinoid receptors; in particular, it is considered an allosteric negative modulator of CB1 and CB2 receptors (cannabinoid receptor types 1 and 2).5,6 Current evidence shows that CBD exerts pharmacological effects via specific molecular targets such as adenosine, glycine, opioid, serotonin, nonendocannabinoid G protein-coupled, nicotinic acetylcholine, and proliferator-activated receptors.7 Moreover, CBD shows anticonvulsant, antispasmodic, anxiolytic, antinausea, antirheumatoid arthritis, and neuroprotective properties.5 Recently, it has been demonstrated that CBD is an inverse agonist for G protein-coupled orphan receptors, such as GPR3, GPR6, and GPR12, suggesting new therapeutic uses of CBD for Alzheimer’s disease, Parkinson’s disease, cancer, and infertility.8
Figure 1.

Structures of cannabidiol (CBD) and Δ-9-tetrahydrocannabinol (Δ9-THC).
Δ9-THC is the key compound of Cannabis sativa with major psychoactive effects.5 From a pharmacological perspective, Δ9-THC is a partial agonist at both cannabinoid receptors: CB1, a modulator of psychoactive effects, and CB2, a modulator of immunological and anti-inflammatory effects.5 The psychoactive effects of Δ9-THC include anxiety, paranoia, perceptual alterations, and cognitive deficits. All these CB1-mediated effects are caused by the perturbation of GABA (γ-aminobutyric acid)/glutamatergic neurotransmission and dopamine release, and above all, they are generally acute, transient, and self-limited.5 Moreover, a low Δ9-THC acute toxicity in murine models has also been observed. Lastly, after Δ9-THC administration, hypolocomotion, hypothermia, catalepsy, analgesia, and increased food intake have been reported.5
The possibility of inducing intramolecular cyclization of CBD to create the THC skeleton is well-known. Because of the remarkable difference in terms of the activity between CBD, Δ9-THC, and its isomers, we decided to study (a) the feasibility of this reaction, (b) its selectivity, and (c) the availability of an efficient and quick method for monitoring this conversion.
Thus, CBD was treated with Lewis and protic acids, and the composition of the resulting mixture was evaluated using high-performance liquid chromatography (HPLC) or direct NMR spectra analysis.
Results and Discussion
According to the literature, the cyclization reaction of CBD seems to occur following an acid-catalyzed activation of a specific double bond.9,10 A dihydrobenzopyran ring moiety is formed by internal ether formation of one of the phenolic groups with one of the double bonds. The two double bonds in the CBD structure are responsible for the formation of two different compounds (Scheme 1). If the activation occurs on the Δ8 double bond, the products show the THC scaffold (Δ9-THC, path b); otherwise, the Δ1 double bond activation leads to the formation of the iso-THC scaffold (Δ8-iso-THC, path a). The latter cyclization is much less frequent. However, acidic conditions are responsible for further isomerization toward the corresponding thermodynamically more stable compounds, Δ8-THC and Δ4(8)-iso-THC, respectively.11
Scheme 1. CBD Acid-Promoted Cyclization.
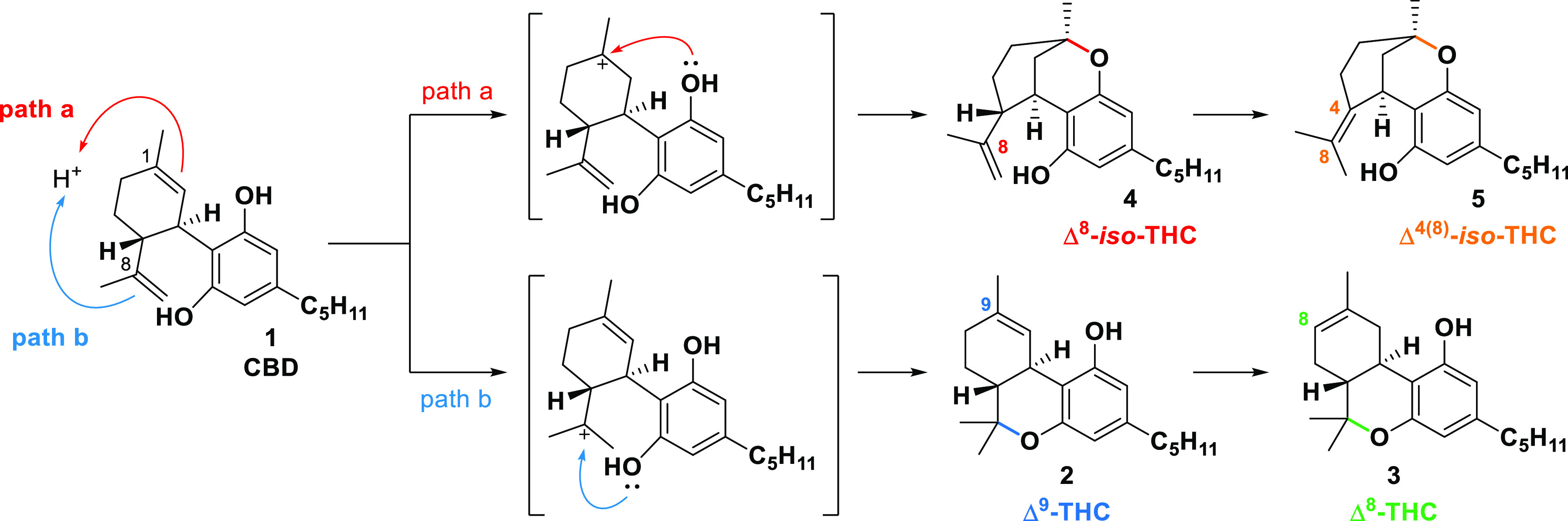
Although Δ9-THC and its derivatives have been widely explored and recognized as the major psychoactive Cannabis constituents, the iso-THC isomers have received little attention. For this reason, we wish to fill the literature gap, especially regarding the provision of full NMR data.
To investigate the susceptibility and selectivity of CBD cyclization, different reactions, including the use of Lewis and protic acids in different solvents and varying the temperature and reaction time, were performed (Scheme 2).
Scheme 2. CBD Conversions with Acids and the Structures of the Products.

The Lewis acids were evaluated first, starting with the recorded use of BF3·OEt2.12,13 The data suggest that performing the reaction with BF3·OEt2 in CH2Cl2 at a low temperature affords Δ9-THC as the main product, but increasing the temperature and reaction time results in preferential formation of the more stable Δ8-THC. The results support this assertion (Table 1, entries 1 and 2). Lowering the temperature also lowers the yields (Table 1, entry 3). Using other solvents gave different degrees of selectivity. In particular, toluene gave results similar to those in CH2Cl2, but iso-THCs always accompanied the Δ8- and Δ9-THCs (Table 1, entries 4 and 5). A reaction conducted in MeCN at −10 °C for 6 h yielded Δ8-iso-THC as the main product accompanied by trace amounts of Δ4(8)-iso-THC (Table 1, entry 7).
Table 1. Reaction Conditions Screening of Acid-Catalyzed Cyclization of CBD Using Lewis Acidsa.
| reaction
mixture composition (%)b |
||||||||
|---|---|---|---|---|---|---|---|---|
| entry | acid | solvent | T (°C) | time (h) | Δ9-THC | Δ8-THC | Δ8-iso-THC | Δ4(8)-iso-THC |
| 1 | BF3·OEt2 | CH2Cl2 | –10 | 4 | 44 | 1 | 3 | |
| 2 | BF3·OEt2 | CH2Cl2 | 0 | 6 | 2 | 52 | ||
| 3 | BF3·OEt2 | CH2Cl2 | –78 to −30 | 48 | 10 | 11 | 5 | |
| 4 | BF3·OEt2 | Tol | –10 | 3 | 41 | 2 | 29 | |
| 5 | BF3·OEt2 | Tol | 0 | 6 | 36 | 26 | ||
| 6 | BF3·OEt2 | THF | –10 | 6 | NR | NR | NR | NR |
| 7 | BF3·OEt2 | MeCN | –10 | 6 | 5 | 30 | 5 | |
| 8 | TMSOTf | CH2Cl2 | –10 | 6 | 93 | |||
| 9 | TMSOTf | Tol | –10 | 6 | 12 | 75 | ||
| 10 | In(OTf)3 | CH2Cl2 | –10 | 6 | 52 | 6 | 4 | |
| 11 | In(OTf)3 | CH2Cl2 | 0 to RT | 48 | 72 | |||
| 12 | In(OTf)3 | Tol | –10 | 4 | NR | NR | NR | NR |
| 13 | In(OTf)3 | Tol | 0 | 24 | 98 | |||
| 14 | ZnBr2 | CH2Cl2 | RT | 96 | NR | NR | NR | NR |
| 15 | TiCl4 | CH2Cl2 | –10 | 6 | 34 | 9 | ||
RT, room temperature; NR, no reaction.
Determined via HPLC and 1H NMR analysis.
To enhance the yields and the selectivity of the process, a series of tests with different acids were conducted, following the hypothesis that other Lewis acids could actively induce cyclization. Starting from the positive literature results regarding the use of TMSI (trimethylsilyl iodide), which showed a high yield of Δ9-THC formation without isomerization,14 TMSOTf (trimethylsilyl triflate) was tested as an acidic reagent. Contrary to the expectations, it displayed a high affinity for the formation of Δ8-THC when CH2Cl2 or toluene were used as solvents, even at a low temperature (Table 1, entries 8 and 9).
In(OTf)3 in CH2Cl2 converted CBD into Δ9-THC at a low temperature in a better yield than that of BF3·OEt2 (Table 1, entry 10). As in previous tests, higher temperatures caused the production of the thermodynamically more stable isomer in higher yields (Table 1, entry 11). Using toluene, the selectivity shifted to the formation of Δ8-THC in excellent yields (Table 1, entry 13). The use of ZnBr2 in CH2Cl2 did not promote the cyclization reaction even at room temperature (Table 1, entry 14), while TiCl4 showed a trend similar to that of BF3 (Table 1, entry 15). The activity of AlCl3, AgOTf, and Ti(OiPr)4 was also investigated, without any noteworthy results. Considering these outcomes, a unique preferential formation path for any of the possible isomers cannot be determined based on the characteristics of the Lewis acid used to induce the cyclization.
Subsequently, protic acid screening was performed (Table 2). The best results for CBD conversion were obtained with HCl, pTSA (p-toluenesulfonic acid), and CSA (camphorsulfonic acid). As reported,9 pTSA in CH2Cl2 led directly to the formation of Δ8-THC as the sole product (Table 2, entry 2). The nature of the solvent clearly affected the reaction outcome. The reaction in n-hexane afforded a mixture of Δ9-THC, Δ8-THC, and Δ8-iso-THC in a ratio of 1:5:1 (Table 2, entry 3), while the reaction in toluene gave a higher selectivity. pTSA gave different isomers in different percentages depending on the solvent and reaction time (Table 2, entries 2–5). The best selectivity of Δ9-THC and Δ8-THC formation was obtained with toluene and CH2Cl2, respectively. On the contrary, the use of 10% mmol catalytic amounts of acid in toluene resulted in almost complete isomerization of the double bond because of the increased reaction time that shifted the outcome to the thermodynamic isomer (Table 2, entry 6). Interestingly, CSA promoted the cyclization of CBD to Δ9-THC with complete selectivity and satisfactory yields regardless of the reaction time (Table 2, entry 7). Other protic acids gave worse results for the CBD conversion (Table 2, entries 8–15).
Table 2. Reaction Conditions Screening of Acid-Catalyzed Cyclization of CBD Using Protic Acidsa.
| reaction
mixture composition (%)b |
||||||||
|---|---|---|---|---|---|---|---|---|
| entry | acid | solvent | T (°C) | time (h) | Δ9-THC | Δ8-THC | Δ8-iso-THC | Δ4(8)-iso-THC |
| 1 | HCl | H2O | RT | 72 | 57 | |||
| 2 | pTSA | CH2Cl2 | RT | 36 | 94 | |||
| 3 | pTSA | n-Hex | RT | 36 | 13 | 66 | 13 | |
| 4 | pTSA | DMSO | RT | 18 | NR | |||
| 5 | pTSA | Tol | RT | 48 | 82 | 11 | ||
| 6 | pTSA catc | Tol | RT | 96 | 9 | 89 | ||
| 7 | CSA | Tol | RT | 96 | 61 | |||
| 8 | H2SO4 | CH2Cl2 | 0 | 72 | 5 | 4 | 11 | |
| 9 | H2SO4 | Tol | RT | 96 | NR | NR | NR | NR |
| 10 | ascorbic acid | CH2Cl2 | 0 | 24 | NR | NR | NR | NR |
| 11 | ascorbic acid | Tol | RT | 96 | NR | NR | NR | NR |
| 12 | citric acid | EtOH | RT | 96 | NR | NR | NR | NR |
| 13 | HOAc | CH2Cl2 | 0 | 24 | NR | NR | NR | NR |
| 14 | HOAc | Tol | RT | 96 | NR | NR | NR | NR |
| 15 | H3PO4 | Tol | –10 to 50 | 48 | NR | NR | NR | NR |
RT, room temperature; NR, no reaction.
Determined via HPLC and 1H NMR analysis.
pTSA 10% catalytic amount.
Some Δ4(8)-iso-THC formation was detected in three cases (Table 1, entries 5 and 7; Table 2, entry 8), and this compound was isolated and characterized.
The experimental results indicate that toluene is the most suitable solvent for the conversion of CBD into THC isomers. This solvent particularly affects the selectivity of the isomers according to the other experimental conditions (the reaction temperature and nature of the acid cyclization promoter). Increasing the temperature reduces the selectivity of the activation of the double bond and favors the formation of the corresponding most stable isomers. The Lewis acids BF3·OEt2, In(OTf)3, and TMSOTf have a proven effect and affected the major formation of Δ8-THC and the product mixtures. As for protic acids, pTSA promotes the reaction to selectively afford Δ9- and Δ8-THCs, depending on the reaction time. CSA emerges as an interesting cyclization inducer, giving Δ9-THC in good yields through readily accessible reaction conditions.
With these encouraging screening results, the influence of CSA was more thoroughly investigated; therefore, the solvent, temperature, and time were considered variables. Using toluene as the solvent at room temperature gave a 61% yield of Δ9-THC accompanied by unreacted CBD (Table 2, entry 7). A longer reaction time led to isomerization of Δ9-THC and to decomposition of compounds (Table 3, entry 3). Increasing the temperature led to the formation of a mixture that was enriched with the Δ8 isomer over time (Table 3, entries 4–8). Increasing the temperature further drastically reduced the CBD conversion time and isomerization, obtaining Δ9-THC as a kinetic reaction product (Table 3, entries 9 and 10). In CH2Cl2, the reaction was faster and less selective toward Δ9-THC formation (Table 3, entries 11–13). n-Hexane and MTBE (t-butyl methyl ether) induced a high degree of CBD conversion without a marked preferential selectivity for the formation of a THC isomer even at a short reaction time (Table 3, entries 14 and 15). In all experiments, iso-THC isomers were not detected except when the reaction was performed in MTBE (Table 3, entry 15).
Table 3. Reaction Conditions of the Acid-Catalyzed Cyclization of CBD Using CSAa.
| reaction mixture composition (%)b |
||||||
|---|---|---|---|---|---|---|
| entry | solvent | T (°C) | time (h) | Δ9-THC | Δ8-THC | Δ8-iso-THC |
| 1 | Tol | RT | 48 | |||
| 2 | Tol | RT | 96 | 61 | ||
| 3 | Tol | RT | 120 | 20 | 28 | |
| 4 | Tol | 30 | 96 | 53 | 20 | |
| 5 | Tol | 40 | 24 | 48 | 19 | |
| 6 | Tol | 40 | 48 | 45 | 52 | |
| 7 | Tol | 40 | 72 | 28 | 72 | |
| 8 | Tol | 40 | 96 | 13 | 87 | |
| 9 | Tol | 50 | 3 | 37 | 10 | |
| 10 | Tol | 50 | 4 | 62 | 19 | |
| 11 | CH2Cl2 | RT | 24 | 33 | 5 | |
| 12 | CH2Cl2 | RT | 48 | 64 | 36 | |
| 13 | CH2Cl2 | 30 | 24 | 48 | 52 | |
| 14 | n-Hex | 30 | 96 | 31 | 41 | |
| 15 | MTBE | 30 | 96 | 54 | 26 | 9 |
| 16 | cyclohexane | 30 | 96 | NR | NR | NR |
RT, room temperature; NR, no reaction.
Determined via HPLC and 1H NMR analysis.
Toluene showed the best selectivity; however, the long reaction time seemed to be a drawback. CH2Cl2 appeared to be promising in this respect, but continuous monitoring of the reaction was required to avoid the prevalence of isomerization.
Δ9-THC, Δ8-THC, Δ8-iso-THC, and Δ4(8)-iso-THC were fully characterized using NMR data, and the complete 1H and 13C NMR assignments (Table 4–6) have been determined on the basis of 1D and 2D NMR spectra (1H and 13C NMR, correlation spectroscopy (COSY), heteronuclear single quantum coherence (HSQC), and heteronuclear multiple bond correlation (HMBC)). The data were compared with those available in the literature.15,16
Table 4. NMR Spectroscopy Data (400 MHz, Methanol-d4) of CBD.

Chemical shifts (in ppm) were determined with reference to TMS.
Spectra recorded at 101 MHz.
Spectra recorded at 400 MHz.
Chemical shifts bearing the same symbol overlap.
Table 6. NMR Spectroscopy Data (300 MHz, Acetone-d6) of Δ8-iso-THC and (400 MHz, CDCl3) of Δ4(8)-iso-THC.
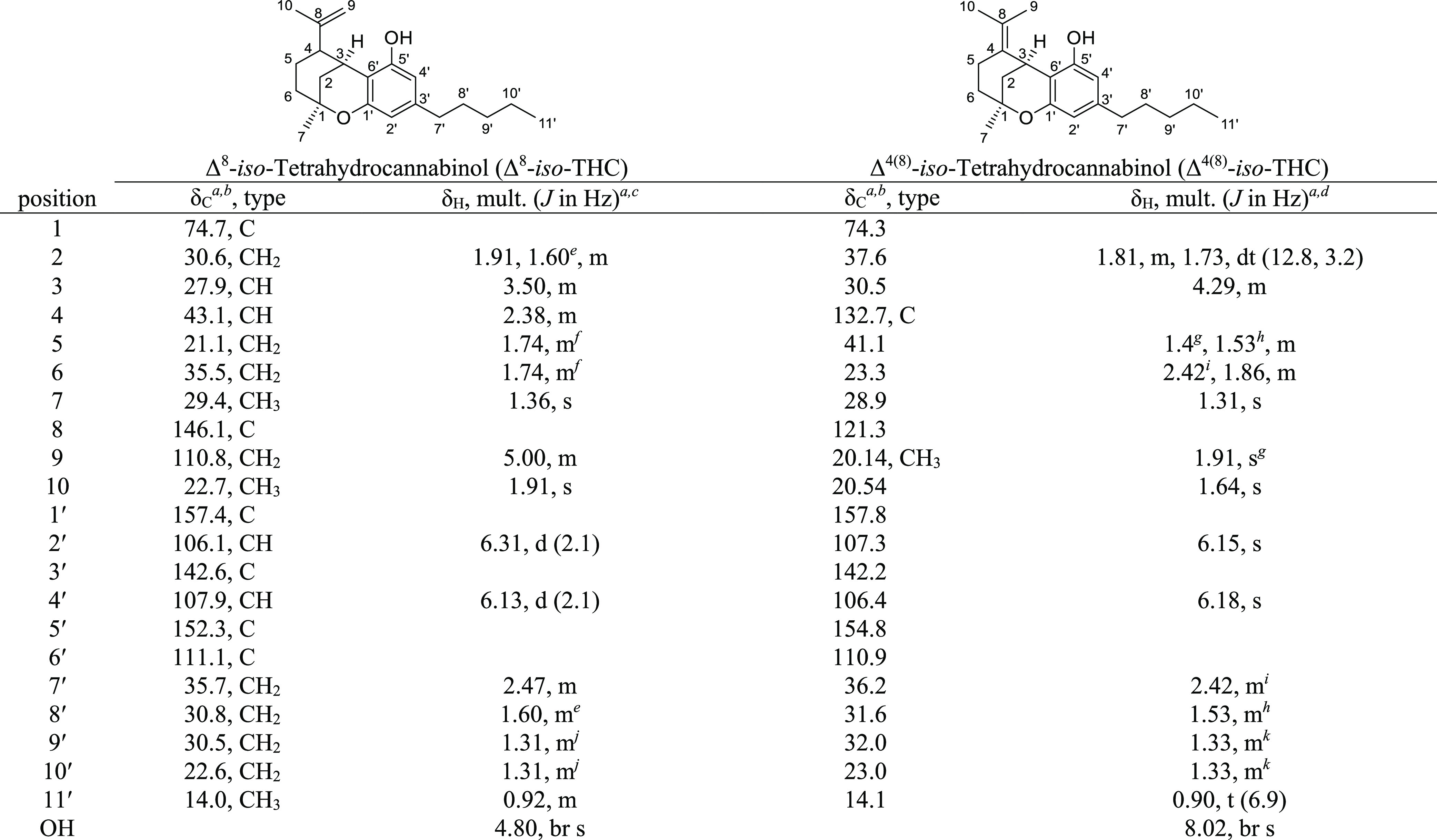
Chemical shifts (in ppm) were determined with reference to TMS.
Spectra recorded at 101 MHz.
Spectra recorded at 300 MHz.
Spectra recorded at 400 MHz.
Chemical shifts bearing the same symbol overlap.
Diagnostic and distinguishable NMR peaks permit identification of compounds derived from intramolecular cyclization within the crude reaction mixture and also allow the composition percentage to be determined from integration ratios (Figure 2). In CDCl3, Δ9-THC is characterized by the presence of signals at 6.34 ppm (H-10), 3.23 ppm (H-10a), 2.22–2.16 ppm (H-8), and 4.88 ppm (OH). The 1H NMR data of Δ8-THC show two signals for H-10 (3.21 and 2.19–2.15 ppm), while the signal due to H-10a is present at 2.71 ppm. The olefinic (H-8) and the hydroxy proton appear at 5.45 and 4.63 ppm, respectively. For Δ8-iso-THC, the corresponding characteristic signals are the doublet at 4.98 ppm (H-9) and the two signals at 3.49 and 2.37 ppm that match the protons H-3 and H-4, respectively. The spectrum of Δ4(8)-iso-THC shows a characteristic signal at 4.29 ppm (H-3).
Figure 2.

1H NMR spectrum in CDCl3 of a mixture of CBD, Δ9-THC, Δ8-THC, and Δ8-iso-THC.
On the basis of the literature data,17 an HPLC method was used to follow the cyclization of CBD. The reactions monitored via HPLC provided a composition of the reaction mixture comparable with that of the 1H NMR data analysis.
The analysis was performed on an ASCENTIS RP-C18 column (5 μm × 4.6 × 150 mm). The pressure was set at 101 bar, and the temperature was maintained at 40 °C with a constant flow rate of 0.95 mL/min. UV spectra were recorded at 228.8 nm using a gradient elution method. The mobile phase consisted of a mixture of A (0.1% v/v HCOOH in H2O) and B (0.1% v/v HCOOH in MeCN). The gradient elution program was adapted to a 30 min duration to obtain RRT 1.00 for CBD and RRT 1.28 for Δ9-THC. After 30 min, the column was purged with 100% B in 7 min; subsequently, the system was washed under these conditions for 3 min and restored to the initial conditions. The retention times were CBD, 23.63 min; Δ4(8)-iso-THC, 29.62 min; Δ9-THC, 29.92 min; Δ8-THC, 30.77 min; and Δ8-iso-THC, 30.77 min (Figure 3).
Figure 3.
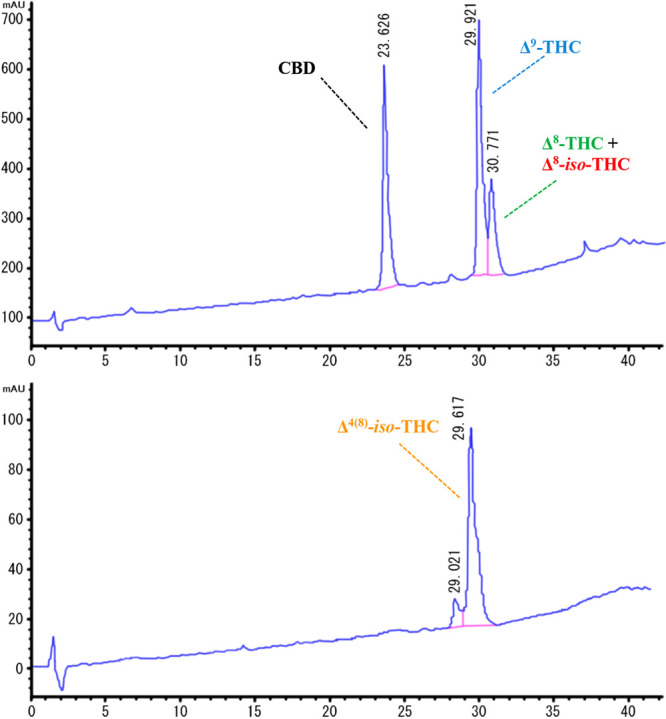
Representative chromatogram of the standard cannabinoid mixture.
The method allowed an excellent separation of CBD from the THC isomers; in particular, it was possible to recognize Δ9-THC from Δ8-THC and Δ8-iso-THC; however, the peaks were quite close. The drawback was that it remained difficult to obtain a better resolution between the peaks of Δ9-THC/Δ4(8)-iso-THC, and Δ8-THC/Δ8-iso-THC, which have similar retention times. For this reason, the HPLC results were always compared with those obtained from the 1H NMR data.
In conclusion, all THC isomers were fully characterized via 1H and 13C NMR spectroscopy. An analytical method was optimized to monitor the course of the reactions. In particular, it was found that CSA in toluene at room temperature (RT) for 96 h and pTSA in toluene for 48 h at RT were the best conditions for the selective formation of Δ9-THC. TMSOTf in CH2Cl2 at −10 °C for 6 h, In(OTf)3 in toluene at 0 °C for 24 h, pTSA in CH2Cl2 at RT for 36 h, and CSA in toluene at 40 °C for 96 h selectively afforded Δ8-THC in high yields. The use of BF3·OEt2 in toluene led to the formation of the iso-THC isomer, depending on the reaction temperature. At −10 °C, a separable mixture of Δ9-THC and Δ8-iso-THC was obtained, whereas a temperature increase to 0 °C shifted the result toward the corresponding most stable isomers, Δ8-THC and Δ4(8)-iso-THC. CBD is a challenging substrate that permits the chemical reactivity of natural alkenes and phenols to be addressed and exploited.
Experimental Section
General Experimental Procedures
Unless otherwise stated, reagents and solvents were purchased from Sigma-Aldrich (Milan, Italy), Fluorochem (Hadfield, United Kingdom), or TCI (Zwijndrecht, Belgium) and used without further purification. All reactions were carried out in oven-dried glassware and dry solvents under a nitrogen atmosphere and were monitored by TLC on silica gel (Merck precoated 60F254 plates), with detection by UV light (254 nm) or by cerium molybdate stain (Hanessian’s stain). Analytical HPLC was performed on an ASCENTIS RP-C18 column (5 μm × 4.6 × 150 mm). The pressure was set at about 101 bar, and the temperature was maintained at 40 °C, with a constant flow rate of 0.95 mL/min. UV spectra were recorded at 228 nm using a gradient elution method. The mobile phase consisted of a mixture of A (0.1% v/v HCOOH in H2O) and B (0.1% v/v HCOOH in MeOH). The gradient was programmed linearly from 60% B to 90% B in 30 min. Flash column chromatography (FCC) was performed using silica gel (240–400 mesh, Merck) as a stationary phase. 1H NMR spectra were recorded on a Bruker Avance Spectrometer 300 or 400 MHz, and chemical shifts are reported relative to residual CDCl3, methanol-d4, or acetone-d6. 13C NMR spectra were recorded on the same instrument (101 MHz), and chemical shifts are reported relative to residual CDCl3, methanol-d4, or acetone-d6. All 1D and 2D NMR spectra were acquired using the standard pulse sequences available with Bruker Topspin 1.3. Chemical shifts (δ) for proton and carbon resonances are quoted in parts per million (ppm) relative to TMS, used as an internal standard. Data for 1H NMR are reported as follows: chemical shift (δ/ppm), multiplicity, and coupling constants (Hz). Multiplicities are reported as follows: s = singlet, d = doublet, t = triplet, m = multiplet, and br s= broad singlet. Data for 13C NMR are reported in terms of chemical shifts (δ/ppm). MS spectra were recorded using the Electrospray Ionization (ESI) technique on a Waters Micromass quadrupole time-of-flight micro-mass spectrometer, and HR-ESI mass spectra were recorded on a FT-ICR APEXII instrument (Bruker Daltonics). EI mass spectra were recorded at an ionizing voltage of 6 kEv on a VG 70–70 EQ. Optical rotation values were measured on a Jasco P-1030 polarimeter at 20 °C, using a sodium D line wavelength λ = 589 nm.
General Procedure Using Lewis or Protic Acids
All the reactions were performed under a nitrogen atmosphere in different anhydrous solvents and at different temperatures. To a CBD stirred solution at the specified temperature (more details follow below) was slowly added the corresponding Lewis or protic acids, and the mixture was stirred. The reaction was quenched with a saturated aqueous NaHCO3 solution, stirred for 30 min, and washed with a saturated aqueous NaHCO3 solution and with brine. The organic phase was dried over Na2SO4, filtered, and evaporated under reduced pressure. All the reactions were monitored by TLC (CH2Cl2/n-hexane 1:3) developed by cerium molybdate stain, and the crudes were analyzed by 1H NMR spectroscopy in CDCl3 and HPLC to determine composition. All the residues were purified by FCC on silica gel (CH2Cl2/n-hexane 1:3), providing four possible THC isomers.
CBD
[α]D20 −113 (c 1, EtOH); [HPLC ASCENTIS C18; RT CBD = 23.63 min]; 1H and 13C NMR data see Table 4; HRMS (ESI) m/z [M + Na]+ 337.2137 (calcd. for C21H30O2Na, 337.213).
Δ9-THC
[α]D20 −159 (c 1, CHCl3); [HPLC ASCENTIS C18; RT Δ9-THC = 29.92 min]; 1H and 13C NMR data see Table 5; HRMS (ESI) m/z [M + Na]+ 337.2132 (calcd for C21H30O2Na, 337.2138).
Table 5. NMR Spectroscopy Data (400 MHz, CDCl3) of Δ9-THC and (300 MHz, CDCl3) of Δ8-THC.
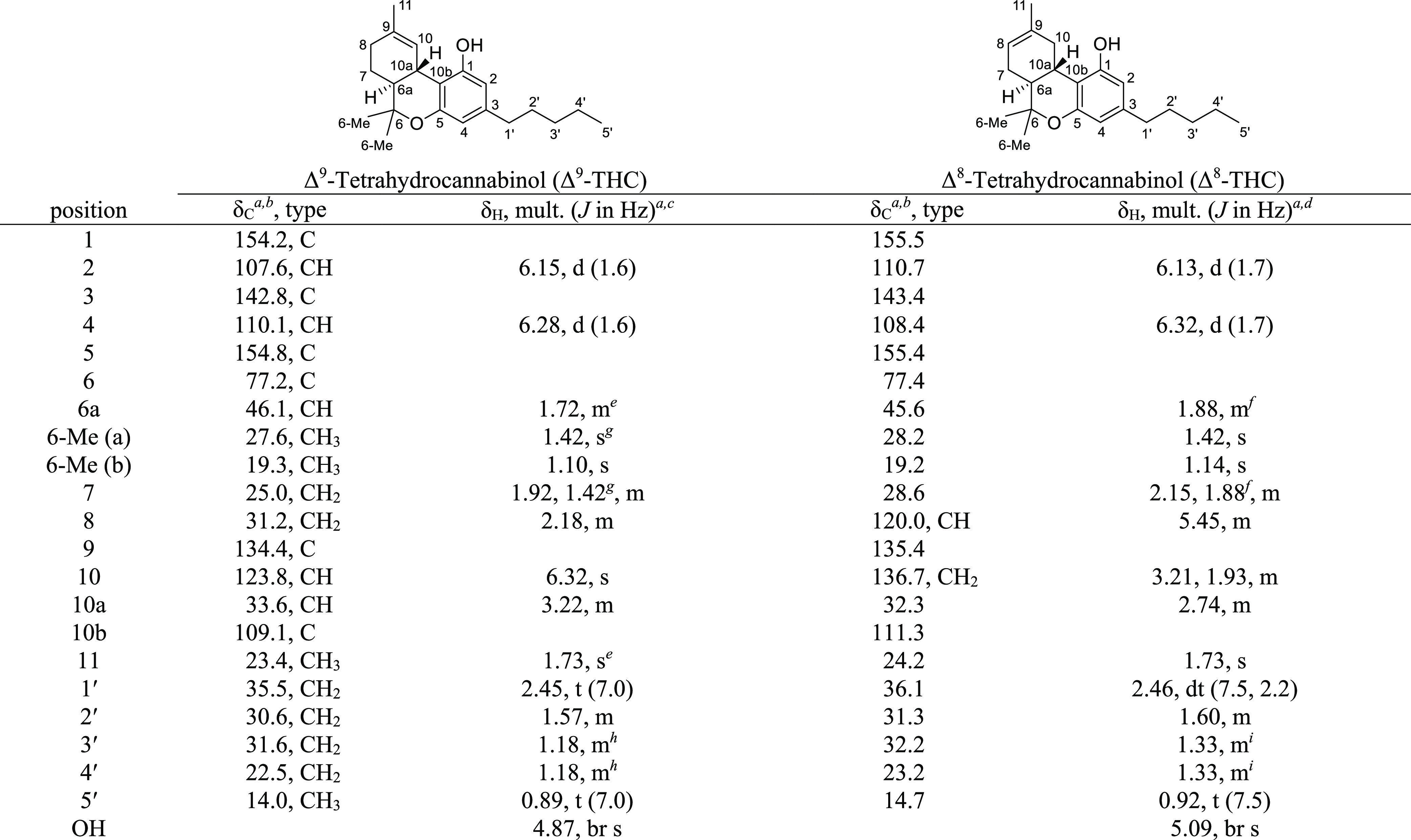
Chemical shifts (in ppm) were determined with reference to TMS.
Spectra recorded at 101 MHz.
Spectra recorded at 400 MHz.
Spectra recorded at 300 MHz.
Chemical shifts bearing the same symbol overlap.
Δ8-THC
[α]D20 −238 (c 1, CHCl3); [HPLC ASCENTIS C18; RT Δ8-THC = 30.77 min]; 1H and 13C NMR data see Table 5; HRMS (ESI) m/z [M + Na]+ 337.2136 (calcd. for C21H30O2Na, 337.2138).
Δ8-iso-THC
[α]D20 −249 (c 1, CHCl3); [HPLC ASCENTIS C18; RT Δ8-iso-THC = 30.77 min]; 1H and 13C NMR data see Table 6; HRMS (ESI) m/z [M + Na]+ 337.2141 (calcd. for C21H30O2Na, 337.2138).
Δ4(8)-iso-THC
[α]D20 −236 (c 1, CHCl3); [HPLC ASCENTIS C18; RT Δ4(8)-iso-THC = 29.62 min]; 1H and 13C NMR data see Table 6; HRMS (ESI) m/z [M + Na]+ 337.2133 (calcd. for C21H30O2Na, 337.2138).
BF3·OEt2-Catalyzed Reactions (Table 1)
Reactions were performed as specified in the general procedure for Lewis acids.
Conditions (Table 1, entry 1): CBD (315 mg, 1 mmol); solvent, anhydrous CH2Cl2 (5 mL); T = −10 °C; BF3·OEt2 (151 μL, 1.2 mmol); reaction time, 4 h. Yields: Δ9-THC, 138 mg (44%); Δ8-THC, 4 mg (1%); Δ8-iso-THC, 11 mg (3%).
Conditions (Table 1, entry 2): CBD (315 mg, 1 mmol); solvent, anhydrous CH2Cl2 (5 mL); T = 0 °C; BF3·OEt2 (151 μL, 1.2 mmol); reaction time, 6 h. Yields: Δ9-THC, 5 mg (2%); Δ8-THC, 164 mg (52%).
Conditions (Table 1, entry 3): CBD (315 mg, 1 mmol); solvent, anhydrous CH2Cl2 (5 mL); T = −78 to −30 °C; BF3·OEt2 (151 μL, 1.2 mmol); reaction time, 48 h. Yields: Δ9-THC, 32 mg (10%); Δ8-THC, 35 mg (11%); Δ8-iso-THC, 16 mg (5%).
Conditions (Table 1, entry 4): CBD (156 mg, 0.5 mmol); solvent, anhydrous toluene (2.5 mL); T = −10 °C; BF3·OEt2 (76 μL, 0.6 mmol); reaction time, 3 h. Yields: Δ9-THC, 64 mg (41%); Δ8-THC, 3 mg (2%); Δ8-iso-THC, 45 mg (29%).
Conditions (Table 1, entry 5): CBD (316 mg, 1 mmol); solvent, anhydrous toluene (5 mL); T = 0 °C; BF3·OEt2 (151 μL, 1.2 mmol); reaction time, 6 h. Yields: Δ8-THC, 115 mg (36%); Δ4(8)-iso-THC, 83 mg (26%).
Conditions (Table 1, entry 7): CBD (315 mg, 1 mmol); solvent, anhydrous MeCN (5 mL); T = −10 °C; BF3·OEt2 (151 μL, 1.2 mmol); reaction time, 6 h. Yields: Δ8-THC, 16 mg (5%); Δ8-iso-THC, 95 mg (30%); Δ4(8)-iso-THC, 17 mg (5%).
TMSOTf-Catalyzed Reactions (Table 1)
Reactions were performed as specified in the general procedure for Lewis acids.
Conditions (Table 1, entry 8): CBD (315 mg, 1 mmol); solvent, anhydrous CH2Cl2 (5 mL); T = −10 °C; TMSOTf (217 μL, 1.2 mmol); reaction time, 6 h. Yields: Δ8-THC, 293 mg (93%).
Conditions (Table 1, entry 9): CBD (80 mg, 0.25 mmol); solvent, anhydrous toluene (1.25 mL); T = −10 °C; TMSOTf (91 μL, 0.5 mmol); reaction time, 6 h. Yields: Δ9-THC, 10 mg (12%); Δ8-THC, 61 mg (75%).
In(OTf)3-Catalyzed Reactions (Table 1)
Reactions were performed as specified in the general procedure for Lewis acids.
Conditions (Table 1, entry 10): CBD (317 mg, 1 mmol); solvent, anhydrous CH2Cl2 (5 mL); T = −10 °C; In(OTf)3 (675 mg, 1.2 mmol); reaction time, 6 h. Yields: Δ9-THC, 165 mg (52%); Δ8-THC, 18 mg (6%); Δ8-iso-THC, 12 mg (4%).
Conditions (Table 1, entry 11): CBD (317 mg, 1 mmol); solvent, anhydrous CH2Cl2 (5 mL); T = 0 °C to RT; In(OTf)3 (58 mg, 0.1 mmol); reaction time, 48 h. Yields: Δ8-THC, 228 mg (72%).
Conditions (Table 1, entry 13): CBD (156 mg, 0.5 mmol); solvent, anhydrous toluene (2.5 mL); T = 0 °C; In(OTf)3 (563 mg, 1 mmol); reaction time, 24 h. Yields: Δ8-THC, 153 mg (98%).
TiCl4-Catalyzed Reaction (Table 1)
The reaction was performed as specified in the general procedure for Lewis acids.
Conditions (Table 1, entry 15): CBD (315 mg, 1 mmol); solvent, anhydrous CH2Cl2 (5 mL); T = −10 °C; TiCl4 (167 μL, 1.2 mmol); reaction time, 6 h. Yields: CBD, 38 mg (12%); Δ9-THC, 108 mg (34%); Δ8-THC, 27 mg (9%).
HCl-Catalyzed Reaction (Table 2)
Reaction was performed as specified in the general procedure for protic acids.
Conditions (Table 2, entry 1): CBD (156 mg, 0.5 mmol); solvent, H2O (1.6 mL); T = RT; HCl 37% (1.6 mL); reaction time, 72 h. Yields: Δ8-THC, 89 mg (57%).
pTSA·H2O-Catalyzed Reactions (Table 2)
Reactions were performed as specified in the general procedure for protic acids.
Conditions (Table 2, entry 2): CBD (154 mg, 0.5 mmol); solvent, anhydrous CH2Cl2 (2.5 mL); T = RT; pTSA·H2O (189 mg, 1 mmol); reaction time, 36 h. Yields: Δ8-THC, 145 mg (94%).
Conditions (Table 2, entry 3): CBD (155 mg, 0.5 mmol); solvent, n-hexane (2.5 mL); T = RT; pTSA·H2O (190 mg, 1 mmol); reaction time, 36 h. Yields: Δ9-THC, 20 mg (13%); Δ8-THC, 102 mg (66%); Δ8-iso-THC, 20 mg (13%).
Conditions (Table 2, entry 5): CBD (318 mg, 1 mmol); solvent, anhydrous toluene (5 mL); T = RT; pTSA·H2O (386 mg, 2 mmol); reaction time, 48 h. Yields: Δ9-THC, 262 mg (82%); Δ8-THC, 34 mg (11%).
Conditions (Table 2, entry 6): CBD (79 mg, 0.25 mmol); solvent, anhydrous toluene (1.25 mL); T = RT; pTSA·H2O (6 mg, 0.025 mmol); reaction time, 96 h. Yields: Δ9-THC, 7 mg (9%); Δ8-THC, 70 mg (89%).
CSA-Catalyzed Reaction (Table 2)
The reaction was performed as specified in the general procedure for protic acids.
Conditions (Table 2, entry 7): CBD (79 mg, 0.25 mmol); solvent, anhydrous toluene (1.25 mL); T = RT; CSA (117 mg, 0.5 mmol); reaction time, 96 h. Yields: CBD, 28 mg (36%); Δ9-THC, 48 mg (61%).
H2SO4-Catalyzed Reactions (Table 2)
Reactions were performed as specified in the general procedure for protic acids.
Conditions (Table 2, entry 8): CBD (315 mg, 1 mmol); solvent, anhydrous CH2Cl2 (5 mL); T = 0 °C; H2SO4, 98% (54 μL, 1 mmol); reaction time, 72 h. Yields: Δ8-THC, 16 mg (5%); Δ8-iso-THC, 13 mg (4%); Δ4(8)-iso-THC, 37 mg (11%).
General Procedure for CSA Screening Reactions (Table 3)
The procedure is the same as described in the general procedure for Lewis acids, but the reactions were quenched by diluting with EtOAc and were monitored by TLC (n-hexane/EtOAc 7:3, eluted 2 times) developed by cerium molybdate stain. Crudes were analyzed by 1H NMR spectroscopy in CDCl3 and HPLC to determine the composition. Conditions: CBD (1 equiv); CSA (2 equiv); solvent, toluene (0.2 M) or as specified in Table 3; T as specified in Table 3.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jnatprod.0c00436.
NMR spectra of compounds (PDF)
Author Present Address
‡ For F.F.: Dipartimento di Scienza e Alta Tecnologia, Università degli Studi dell’Insubria, Como 22100, Italy.
Author Present Address
§ For M.L.: LINNEA SA, Riazzino (TI) 6595, Switzerland.
Author Contributions
† These authors contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Adams R.; Baker B. R.; Wearn R. B. J. Am. Chem. Soc. 1940, 62 (8), 2204–2207. 10.1021/ja01865a083. [DOI] [Google Scholar]
- Jacob A.; Todd A. R. Cannabidiol and Cannabol, Constituents of Cannabis indica Resin. Nature 1940, 145 (3670), 350–350. 10.1038/145350a0. [DOI] [Google Scholar]
- Hanus L. O.; Meyer S. M.; Munoz E.; Taglialatela-Scafati O.; Appendino G. Phytocannabinoids: a unified critical inventory. Nat. Prod. Rep. 2016, 33, 1357–1392. 10.1039/c6np00074f. [DOI] [PubMed] [Google Scholar]
- Izzo A. A.; Borrelli F.; Capasso R.; Di Marzo V.; Mechoulam R. Trends Pharmacol. Sci. 2009, 30, 515–527. 10.1016/j.tips.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Pertwee R. G. Br. J. Pharmacol. 2008, 153 (2), 199–215. 10.1038/sj.bjp.0707442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casajuana Koguel C.; Lopez-Pelayo H.; Balcells-Olivero M. M.; Colom J.; Gual A. Constituyentes psicoactivos del cannabis y sus implicaciones clínicas: una revisión sistemática. Adicciones 2018, 30 (2), 140–151. 10.20882/adicciones.858. [DOI] [PubMed] [Google Scholar]
- Ibeas Bih C.; Chen T.; Nunn A. V. W.; Bazelot M.; Dallas M.; Whalley B. J. Neurotherapeutics 2015, 12 (4), 699–730. 10.1007/s13311-015-0377-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laun A. S.; Shrader S. H.; Brown K. J.; Song Z.-H. Acta Pharmacol. Sin. 2019, 40 (3), 300–308. 10.1038/s41401-018-0031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaoni Y.; Mechoulam R. Tetrahedron 1966, 22 (4), 1481–1488. 10.1016/S0040-4020(01)99446-3. [DOI] [Google Scholar]
- Mechoulam R.; Hanus L.ır Cannabidiol: an overview of some chemical and pharmacological aspects. Part I: chemical aspects. Chem. Phys. Lipids 2002, 121 (1–2), 35–43. 10.1016/S0009-3084(02)00144-5. [DOI] [PubMed] [Google Scholar]
- Gaoni Y.; Mechoulam R. J. Am. Chem. Soc. 1966, 88 (23), 5673–5675. 10.1021/ja00975a071. [DOI] [Google Scholar]
- Gaoni Y.; Mechoulam R. Isolation and structure of .DELTA.+- tetrahydrocannabinol and other neutral cannabinoids from hashish. J. Am. Chem. Soc. 1971, 93 (1), 217–224. 10.1021/ja00730a036. [DOI] [PubMed] [Google Scholar]
- Nikas S. P.; Grzybowska J.; Papahatjis D. P.; Charalambous A.; Banijamali A. R.; Chari R.; Fan P.; Kourouli T.; Lin S.; Nitowski A. J.; Marciniak G.; Guo Y.; Li X.; Wang C. L. J.; Makriyannis A. The role of halogen substitution in classical cannabinoids: A CB1 pharmacophore model. AAPS J. 2004, 6 (4), 23. 10.1208/aapsj060430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis P.; Mortreux A.; Petit F.; Buono G.; Peiffer G. Ligand-assisted catalysis: new active and selective nickel modified homogeneous catalysts for linear dimerization of butadiene. J. Org. Chem. 1984, 49 (26), 5274–5276. 10.1021/jo00200a060. [DOI] [Google Scholar]
- Choi Y. H.; Hazekamp A.; Peltenburg-Looman A. M. G.; Frédérich M.; Erkelens C.; Lefeber A. W. M.; Verpoorte R. Phytochem. Anal. 2004, 15 (6), 345–354. 10.1002/pca.787. [DOI] [PubMed] [Google Scholar]
- de A. Leite J.; de Oliveira M. V.L.; Conti R.; de S. Borges W.; Rosa T. R.; Filgueiras P. R.; Lacerda V.; Romao W.; Neto A. C. Extraction and isolation of cannabinoids from marijuana seizures and characterization by 1H NMR allied to chemometric tools. Sci. Justice 2018, 58 (5), 355–365. 10.1016/j.scijus.2018.06.005. [DOI] [PubMed] [Google Scholar]
- Gul W.; Gul S. W.; Radwan M. M.; Wanas A. S.; Mehmedic Z.; Khan I. I.; Sharaf M. H. M.; ElSohly M. A. J. AOAC Int. 2015, 98 (6), 1523–1528. 10.5740/jaoacint.15-095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.