

Abstract
The term ‘ornamental plant’ refers to all plants with ornamental value, which generally have beautiful flowers or special plant architectures. China is rich in ornamental plant resources and known as the “mother of gardens”. Genomics is the science of studying genomes and is useful for carrying out research on genome evolution, genomic variations, gene regulation, and important biological mechanisms based on detailed genome sequence information. Due to the diversity of ornamental plants and high sequencing costs, the progress of genome research on ornamental plants has been slow for a long time. With the emergence of new sequencing technologies and a reduction in costs since the whole-genome sequencing of the first ornamental plant (Prunus mume) was completed in 2012, whole-genome sequencing of more than 69 ornamental plants has been completed in <10 years. In this review, whole-genome sequencing and resequencing of ornamental plants will be discussed. We provide analysis with regard to basic data from whole-genome studies of important ornamental plants, the regulation of important ornamental traits, and application prospects.
Subject terms: Genome, Plant genetics, DNA sequencing
Introduction
Genomics is the science of studying genomes. It is used to summarize a branch of genetics involving genome mapping, sequencing, and whole-genome functional analysis. The whole genome is taken as the research object, with a focus on analyzing all of the genetic information in whole genomes of organisms. The main purpose of carrying out genomics research is to interpret the whole-genome sequence, including genomic variations and gene regulation, through mining and expression to gain a deeper understanding of biological mechanisms, formulate more effective breeding strategies, expand the mining breadth and depth of excellent alleles in germplasm resources, and increase the operability for improving complex traits and the efficiency of breeding new varieties.
Ornamental plants, a vital component of agriculture and horticulture, are of great significance for beautifying and improving humans’ living environment, cultivating human sentiment, and promoting structural adjustments in the agricultural industry. The first plant genome to be published was that of Arabidopsis thaliana in 20001. With the emergence of next-generation and high-throughput sequencing, sequencing technologies have continuously evolved, while their costs have continuously decreased, facilitating the whole-genome sequencing of many plants. According to incomplete statistics, whole-genome sequencing has been completed for ~400 plants2. With this progress, more abundant genetic data are provided for plant diversity studies, enabling breeders to perform comprehensive multidimensional research in the fields of genetics, genomics, and molecular breeding. This brings new development opportunities and driving forces for the breeding of more plants and thus leads to a new revolution of breeding technology. Since genome sequencing of the first ornamental plant (Prunus mume) was completed in 20123, whole-genome sequencing of more than 65 ornamental plants has been completed in <10 years. The whole-genome sequencing results from these ornamental plant species have built an enormous resource platform for molecular biology research in ornamental horticulture, which not only contributes to the understanding of genome structure and function in ornamental horticulture but also has substantial guiding significance for exploring the origin and evolution of ornamental plants, mapping and cloning the functional genes of important traits and accelerating the course of molecular breeding.
In this study, the research results from whole-genome sequencing and resequencing of ornamental plants are summarized. We provide a discussion with regard to basic data from whole-genome studies of important ornamental plants, the regulation of important ornamental traits, and application prospects.
Whole-genome sequences of ornamental plants
As of 30 October 2020, the whole-genome sequences and draft genome sequences of 69 ornamental plants have been published, including herbaceous plants, such as carnation (Dianthus caryophyllus), phalaenopsis (Phalaenopsis aphrodite), orchid (Apostasia odorata), sacred lotus (Nelumbo nucifera), chrysanthemum (Dendranthema morifolium) and Dionaea muscipula, and woody plants, such as mei (Prunus mume), Yoshino cherry (Prunus yedoensis), sweet osmanthus (Osmanthus fragrans), peony (Paeonia suffruticosa), and Chinese rose (Rosa chinensis) (Table 1). The number of sequenced genomes of ornamental plants completed each year significantly increased from 1 in 2012 to 17 in 2018. In particular, more than 10 species were sequenced for three consecutive years from 2016 to 2018 (Fig. 1a). China has independently completed or led genome sequencing for 32 ornamental plants, followed by Japan and the United States, which have also completed the genome sequencing of more than 10 species (Fig. 1b). Considering the sequencing material, except for the double-haploid material with relatively high homozygosity used for R. chinensis4,5, wild diploids or cultivars with relatively unclear genetic backgrounds and low heterozygosity were used for all of the other plants. Long-read sequencers in combination with optical maps6 are used to generate high-quality chromosome-level genome assemblies. For ornamental plants, the PacBio RS II system was first applied for the construction of the 1.27 Gb genome assembly of Dendrobium officinale7. Long-range scaffolding techniques such as high-throughput chromosome conformation capture (Hi-C) facilitate chromosome-scale assembly of contigs. In this respect, recently built genome assemblies of Rosa chinensis (515 Mb) have a contig N50 of 24 Mb, which is one of the most comprehensive plant genomes4. In consideration of the comprehensive utilization of Illumina HiSeq, Nanopore, PacBio, and Hi-C technologies, the contig N50 values of Gardenia jasminoides and Chimonanthus praecox can reach 44 and 65.35 Mb, respectively, which was unthinkable five years ago8,9. Generally, the sequencing technology that is predominantly used is next-generation sequencing on the Illumina platform (HiSeq 2000/2500/4000 and HiSeq X ten), coupled with third-generation sequencing (PacBio and Nanopore) and Hi-C technology. The assembled genome size of sequenced ornamental plants ranges from 237 Mb to 13.79 Gb with a scaffold N50 ranging from 13.8 Kb to 65.35 Mb (Fig. 2). We constructed phylogenetic trees for all species with a published genome, which belong to 21 orders and 35 families (Fig. 3). The representative species in Rosaceae, Orchidaceae, and Asteraceae for which high-quality sequencing has been completed were described and discussed.
Table 1.
List of current genome sequencing progress in ornamental plants
| Code | Date | Species | Estimated genome size | Chromosome number | Assembled genome size | Number of scaffolds | Scaffold N50 | Number of predicted genes | Sequencing platform | Objects/goals | Country of main contributor | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 27-Dec-12 | Prunus mume | 280 Mb | 2n = 16 | 237 Mb | 29,989 | 577.8 Kb | 31,390 | Illumina GA II | Early blooming, endodormancy, bacterial infection, biosynthesis of flower scent. | China | 3 |
| 2 | 10-May-13 | Nelumbo nucifera | 929 Mb | 2n = 16 | 804 Mb | 3,605 | 3.435 Mb | 26,685 | Illumina HiSeq 2000, Roche 454 | Aquatic lifestyle | China, USA | 21 |
| 3 | 17-Jun-13 | Nicotiana sylvestris | 2.64 Gb | 2n = 24 | 2.22 Gb | 253,984 | 79.7 Kb | 38,940 | Illumina HiSeq 2000 | Terpenoid metabolism, alkaloid metabolism, and heavy metal transport | Switzerland | 22 |
| 4 | 27-Aug-13 | Tarenaya hassleriana | 300 Mb | 2n = 20 | 290 Mb | 98 | 551.9 Kb | 28,917 | Illumina HiSeq 2000 | Floral developmental, self-incompatibility | China, Netherlands | 23 |
| 5 | 11-Oct-13 | Nelumbo nucifera | 879 Mb | 2n = 16 | 792 Mb | 3,031 | 986.5 Kb | 36,385 | Illumina HiSeq 2000 | Seed formation, embryonic development, seed dormancy, starch synthesis | China | 24 |
| 6 | 26-Nov-13 | Mimulus guttatus | – | 2n = 28 | 321.7 Mb | 2,216 | 1.1 Mb | 26,718 | Illumina HiSeq 2000 | Recombination activity | USA | 25 |
| 7 | 17-Dec-13 | Dianthus caryophyllus | 670 Mb | 2n = 30 | 622 Mb | 45,088 | 60.74 Kb | 43,266 | Illumina HiSeq 1000, GS FLXþ | Phenylpropanoid biosynthetic, betalain/chlorophyll and carotenoid synthesis, disease resistance, ethylene/carbohydrate metabolism, and cell wall modification during flower opening, floral scent | Japan | 26 |
| 8 | 28-Jul-14 | Amaranthus hypochondriacus | 466 Mb | 2n = 32 | 465.2 Mb | 4,897 | 35.09 Kb | 24,829 | Illumina HiSeq 2000 | Lysine biosynthetic pathway | India | 27 |
| 9 | 24-Nov-14 | Phalaenopsis equestris | 1.16 Gb | 2n = 38 | 1.09 Gb | 523 | 359.12 Kb | 29,431 | Illumina HiSeq 2000 | Crassulacean acid metabolism, MADS-box genes | China, Belgium | 13 |
| 10 | 23-Dec-14 | Dendrobium officinale | 1.27 Gb | 2n = 38 | 1.35 Gb | 33,364 | 76.49 Kb | 35,567 | Illumina HiSeq 2000, PacBio RS II | MADS-box genes, morphology of the flower, polysaccharides, alkaloids | China | 7 |
| 11 | 24-Jan-15 | Primula veris | 479.22 Mb | 2n = 22 | 301.8 Mb | 9,002 | 163.95 Kb | 19,507 | Illumina HiSeq 2000, PacBio RS II | Floral morphs | Switzerland, Norway | 28 |
| 12 | 11-Mar-15 | Catharanthus roseus | – | 2n = 16 | 523 Mb | 79,302 | 26.25 Kb | 33,829 | Illumina HiSeq 2000 | Monoterpene indole alkaloid pathway | UK, USA | 29 |
| 13 | 5-May-15 | Boea hygrometrica | 1.69 Gb | – | 1.55 Gb | 520,969 | 110.99 Kb | 49,374 | Illumina HiSeq 2000, Roche 454 | Desiccation tolerance | China | 30 |
| 14 | 26-Sep-15 | Lolium perenne | 1.99 Gb | 2n = 14 | 1.13 Gb | 48,415 | 70.1 Kb | 28,455 | Illumina HiSeq 2000 | Pollen allergens, self-incompatibility mechanism | Denmark | 31 |
| 15 | 30-Nov-15 | Trifolium pratense | 420 Mb | 2n = 14 | 309 Mb | 39,904 | 223 Kb | 40,868 | Illumina HiSeq 2000 | Forage nutrition traits | UK | 32 |
| 16 | 12-Jan-16 | Dendrobium catenatum | 1.11 Gb | 2n = 38 | 1.01 Gb | 723 | 391.46 Kb | 28,910 | Illumina HiSeq 2000 | Polysaccharide synthase, MADS-box genes | China, Belgium | 16 |
| 17 | 5-Feb-16 | Rosa roxburghii | 480.97 Mb | 2n = 14 | 409.36 Mb | 627,554 | 1.48 Kb | 22,721 | Illumina HiSeq 2500 | Ascorbate metabolism | China | 33 |
| 18 | 14-Mar-16 | Zoysia japonica | 340 Mb | 2n = 40 | 334.38 Mb | 11,786 | 2.37 Mb | 59,271 | Illumina HiSeq 2000, MiSeq | Comparative genome | Japan | 34 |
| 19 | 14-Mar-16 | Zoysia matrella | 423 Mb | 2n = 40 | 563.44 Mb | 13,609 | 108.90 Kb | 95,079 | Illumina HiSeq 2000, MiSeq | Comparative genome | Japan | 34 |
| 20 | 14-Mar-16 | Zoysia pacifica | 302 Mb | 2n = 40 | 397.01 Mb | 11,428 | 111.45 Kb | 65,252 | Illumina HiSeq 2000, MiSeq | Comparative genome | Japan | 34 |
| 21 | 12-May-16 | Phalaenopsis orchid | 3.45 Gb | 2n = 38 | 3.1 Gb | 149,151 | 100.94 Kb | 41,153 | Illumina HiSeq 2000 | Labellum organ development, flowering-time genes | China, Australia | 14 |
| 22 | 27-May-16 | Petunia axillaris | 1.4 Gb | 2n = 14 | 1.26 Gb | 83,639 | 1.24 Mb | 32,928 | Illumina HiSeq 2000, PacBio RS II | Floral color, pollination | USA, Switzerland | 35 |
| 23 | 27-May-16 | Petunia inflata | 1.4 Gb | 2n = 14 | 1.29 Gb | 136,283 | 884.43 Kb | 36,697 | Illumina HiSeq 2000, PacBio RS II | Floral color, pollination | USA, Switzerland | 35 |
| 24 | 13-Jul-16 | Drosera capensis | 293 Mb | – | 264 Mb | 13,142 | 82.65 Kb | – | Illumina HiSeq 2500 | 3D structures of cysteine protease | USA | 36 |
| 25 | 28-Jul-16 | Amaranthus hypochondriacus | 466 Mb | 2n = 32 | 377 Mb | 3,518 | 371 Kb | 23,059 | Illumina HiSeq 2500 | Systematic evolution | USA | 37 |
| 26 | 22-Aug-16 | Trifolium subterranum | 552.4 Mb | 2n = 16 | 471.8 Mb | 27,424 | 287.6 Kb | 42,706 | Illumina HiSeq 2000 | Evolutionary divergence | Japan | 38 |
| 27 | 8-Nov-16 | Ipomoea nil | 750 Mb | 2n = 30 | 734.8 Mb | 3,416 | 2.88 Mb | 42,783 | Illumina HiSeq2500, PacBio RS II | Dwarf trait | Japan | 39 |
| 28 | 21-Nov-16 | Ginkgo biloba | 10 Gb | 2n = 24 | 10.61 Gb | 6,459,773 | 1.36 Mb | 41,840 | Illumina HiSeq 2000/4000 | Multiple defense mechanisms, resistant genes | China | 40 |
| 29 | 21-Dec-16 | Hibiscus syriacus | 1.9 Gb | 2n = 80 | 1.75 Gb | 77,492 | 140 Kb | 87,603 | Illumina HiSeq 2000 | Flowering time, disease resistance | Korea | 41 |
| 30 | 26-Dec-16 | Fraxinus excelsior | 877.24 Mb | 2n = 22 | 867 Mb | 89,514 | 104 Kb | 38,852 | Illumina HiSeq 2000, MiSeq, Roche 454 | Disease resistance | UK | 42 |
| 31 | 5-May-17 | Rhodiola crenulata | 420.2 Mb | – | 344.5 Mb | 150,003 | 144.75 Kb | 31,517 | Illumina HiSeq 2000/4000 | Stress resistance, biosynthesis pathways of medicinal ingredients | China | 43 |
| 32 | 22-May-17 | Helianthus annuus | 3.6 Gb | 2n = 34 | 2.94 Gb | 12,318 | 524 Kb | 52,232 | PacBio RS II | Flowering time, oil production | France, Canada | 20 |
| 33 | 24-Jul-17 | Camptotheca acuminata | 516 Mb | 2n = 44 | 403.2 Mb | 1,394 | 1.75 Mb | 31,825 | Illumina HiSeq 2000 | Camptothecin biosynthesis | USA | 44 |
| 34 | 26-Aug-17 | Rhododendron delavayi | 697.94 Mb | 2n = 26 | 695.09 Mb | 313 | 637.83 Kb | 32,938 | Illumina HiSeq 2000 | Biosynthesis pathways of medicinal ingredients | China | 45 |
| 35 | 13-Sep-17 | Apostasia shenzhenica | 471 Mb | 2n = 68 | 349 Mb | 32 | 3.029 Mb | 21,841 | Illumina HiSeq 2000, PacBio | Flower development, seeds without endosperm, evolution of epiphytism | China, Belgium | 17 |
| 36 | 19-Sep-17 | Rosa multiflora | 711 Mb | 2n = 14 | 740 Mb | 83,189 | 90.8 Kb | 67,380 | Illumina HiSeq 2000, MiSeq | Flower color, flower scent, floral development | Japan | 12 |
| 37 | 7-Nov-17 | Carnegiea gigantea | 1.3 Gb | – | 980.3 Mb | 57,409 | 61.5 Kb | 28,292 | Illumina HiSeq 2000, MiSeq | Cactus phylogeny | USA | 46 |
| 38 | 30-Nov-17 | Handroanthus impetiginosus | 557 Mb | 2n = 40 | 503.7 Mb | 13,206 | 81.3 Kb | 31,688 | Illumina HiSeq 2000 | Biosynthetic pathway of specialized quinoids | Brazil | 47 |
| 39 | 1-Dec-17 | Kalanchoe fedtschenkoi | 260 Mb | 2n = 34 | 256 Mb | 1,324 | 2.45 Mb | 30,964 | MiSeq | Crassulacean acid metabolism | USA | 48 |
| 40 | 29-Dec-17 | Eschscholzia californica | 502 Mb | 2n = 12 | 489 Mb | 53,253 | 752.97 Kb | 41,612 | Illumina HiSeq 2500 | Benzylisoquinoline alkaloid biosynthesis | Japan | 49 |
| 41 | 25-Mar-18 | Nelumbo nucifera | – | 2n = 16 | 847.16 Mb | 14,630 | 1.48 Mb | 30,378 | BioNano | Chromosome fusions | China | 50 |
| 42 | 7-Apr-18 | Phalaenopsis aphrodite | 1.2 Gb | 2n = 38 | 1.03 Gb | 13,732 | 0.95 Mb | 28,902 | Illumina HiSeq 2000/2500 | Flower development | China | 15 |
| 43 | 30-Apr-18 | Rosa chinensis | 560 Mb | 2n = 14 | 515 Mb | 82 | 24 Mb | 36,377 | PacBio RS II, Hi-C | Recurrent blooming, flower scent, and flower color | France | 4 |
| 44 | 10-May-18 | Bombax ceiba | 809 Mb | – | 895 Mb | 125 | 2.06 Mb | 52,705 | Illumina HiSeq 2000, PacBio RS II | Evolutionary history | China | 51 |
| 45 | 11-Jun-18 | Rosa chinensis | 532.7 Mb | 2n = 14 | 512 Mb | 564 | 3.4 Mb | 39,669 | Illumina HiSeq 2500, PacBio RS-II | Rickle density, flower petals | France | 5 |
| 46 | 19-Jun-18 | Salvia splendens | 711 Mb | 2n = 44 | 808 Mb | 73 | 3.12 Mb | 54,008 | PacBio RS II | Flower color, bioactive secondary metabolites | China | 52 |
| 47 | 13-Jul-18 | Casuarina glauca | 314 Mb | 2n = 18 | 283 MB | 84 | 912.67 Kb | 26,282 | Illumina HiSeq 2000/4000 | Nitrogen-fixing root nodule symbiosis | Germany | 53 |
| 48 | 13-Jul-18 | Cercis canadensis | 301 Mb | – | 330 Mb | 193 | 421.03 Kb | 34,023 | Illumina HiSeq 2000/4000 | Nitrogen-fixing root nodule symbiosis | Germany | 53 |
| 49 | 13-Jul-18 | Mimosa pudica | 896 Mb | – | 557 Mb | 1,302 | 119.68 Kb | 33,108 | Illumina HiSeq 2000/4000 | Nitrogen-fixing root nodule symbiosis | Germany | 53 |
| 50 | 13-Jul-18 | Begonia fuchsioides | 935 Mb | – | 374 Mb | 569 | 154.27 Kb | 51,638 | Illumina HiSeq 2000/4000 | Nitrogen-fixing root nodule symbiosis | Germany | 53 |
| 51 | 4-Sep-18 | Prunus yedoensis | 257 Mb | 2n = 16 | 323.8 Mb | 519 | 199 Kb | 41,294 | HiSeq X Ten, PacBio RS II | S-locus genes | Korea | 10 |
| 52 | 29-Sep-18 | Lavandula angustifolia | 870 Mb | 2n = 50 | 688 Mb | 84,291 | 96.7 Kb | 62,141 | Illumina HiSeq 2000 | Pathways of isoprenoid metabolism | Canada | 54 |
| 53 | 17-Oct-18 | Chrysanthemum nankingense | 3.07 Gb | 2n = 18 | 2.53 Gb | 24,051 | 130.7 Kb | 56,870 | HiSeq2000, PacBio RS II | Flower trait, flavonoid biosynthesis | China | 18 |
| 54 | 14-Nov-18 | Casuarina equisetifolia | 300 Mb | – | 301 Mb | 366 | 1.06 Mb | 29,827 | Illumina HiSeq 2000, PacBio RS II | Secondary growth and DNA modification | China | 55 |
| 55 | 20-Nov-18 | Osmanthus fragrans | 733.5 Mb | 2n = 46 | 740.6 Mb | 145 | 1.59 Mb | 45,542 | HiSeq X ten, Hi-C | Flower scent | China | 56 |
| 56 | 17-Dec-18 | Liriodendron chinense | 1.8 Gb | 2n = 38 | 1.74 Gb | 3,711 | 3.53 Mb | 35,269 | Illumina HiSeq 2000, PacBio RS II, Bionano | Systematic evolution of angiosperms | China | 57 |
| 57 | 18-Dec-18 | Primula vulgaris | 474 Mb | 2n = 22 | 411.1 Mb | 67,491 | 294.8 Kb | 24,599 | Illumina HiSeq 2500 | Flower development | UK | 58 |
| 58 | 2-Jan-19 | Chrysanthemum seticuspe | 3.06 Gb | 2n = 18 | 2.72 Gb | 354,212 | 44.7 Kb | 71,057 | Illumina HiSeq 2000, MiSeq | Flowering time | Japan | 19 |
| 59 | 28-Jan-19 | Antirrhinum majus | 520 Mb | 2n = 16 | 510 Mb | 62 | 2.62 Mb | 37,714 | Illumina HiSeq 2000, PacBio RS II | Flower asymmetry, self-incompatibility | China | 59 |
| 60 | 14-Jun-19 | Sedum album | 305 Mb | 2n = 48 | 302 Mb | 6,038 | 93 Kb | 44,487 | PacBio RS II | Crassulacean acid metabolism | USA | 60 |
| 61 | 23-Jul-19 | Cerasus yedoensis | – | 2n = 16 | 350.1 Mb | 2,292 | 1.15 Mb | 48,280 | Illumina HiSeq 2000, MiSeq, HiSeq X | Dormancy- and flowering-associated genes | Japan | 11 |
| 62 | 23-Jul-19 | Cerasus yedoensis | – | 2n = 16 | 339.97 Mb | 2,279 | 800 Kb | 46,796 | Illumina HiSeq 2000, MiSeq, HiSeq X | Dormancy- and flowering-associated genes | Japan | 11 |
| 63 | 18-Nov-19 | Rhododendron williamsianum | – | 2n = 26 | 532 Mb | 11,985 | 218.8 Kb | 23,559 | Illumina HiSeq 2000, Hic | Evolutionary history | USA | 61 |
| 64 | 3-Dec-19 | Tanacetum cinerariifolium | 7.1 Gb | 2n = 18 | 7.08 Gb | 2,016,451 | 13.8 Kb | 60,080 | HiSeq X, HiSeq 4000 | Pyrethrin | Japan | 62 |
| 65 | 6-Dec-19 | Paeonia suffruticosa | 13.66–15.76 Gb | 2n = 10 | 13.79 Gb | 499,810 | 49.94 Kb | 35,687 | BGISEQ-500, PacBio RS II | MADS-box genes | China | 63 |
| 66 | 18-Dec-19 | Nymphaea colorata | 433 Mb | 2n = 28 | 409 Mb | 1,429 | 2.1 Mb | 31,580 | PacBio RS II, Hi-C | Flowering transition, flower development, floral scents, flower colors | China | 64 |
| 67 | 1-Apr-20 | Asparagus setaceus | 720 Mb | 2n = 20 | 710.15 Mb | 1,393 | 2.19 Mb | 28,410 | HiSeq X ten, Hi-C | Resistance R genes | China | 65 |
| 68 | 14-May-20 | Dionaea muscipula | 3.19 Gb | – | 1.5 Gb | 104,847 | 35 Kb | 21,135 | PacBio RS II | Carnivory genes | Japan, Germany | 66 |
| 69 | 10-Jun-20 | Chimonanthus salicifolius | 835.5 Mb | 2n = 22 | 820.1 Mb | 1,531 | 2.3 Mb | 36,651 | Illumina HiSeq 2000, PacBio RS II, Hi-C | Flower development, flavonoid biosynthesis | China | 67 |
| 70 | 18-Jun-20 | Gardenia jasminoides | 547.5 Mb | 2n = 22 | 535 Mb | 58,859 | 44 Mb | 35,967 | Illumina HiSeq 2000, Oxford Nanopore, Hi-C | Crocin and caffeine biosynthesis genes | China | 8 |
| 71 | 1-Aug-20 | Forsythia suspensa | 701.40 Mb | 2n = 28 | 737.47 Mb | 1,214 | 7.33 Mb | 33,062 | Illumina HiSeq 2500, Oxford Nanopore | Candidate genes associated with solar radiation, temperature, and water variables | China | 68 |
| 72 | 10-Aug-20 | Chimonanthus praecox | 778.71 Mb | 2n = 22 | 695.36 Mb | 1,623 | 65.35 Mb | 23,591 | Illumina HiSeq 2000, PacBio RS II, HiSeq X, Hi-C | Floral transition, floral organ specification, early blooming, strong cold resistance, terpene/benzenoid/phenylpropanoid biosynthesis | China | 9 |
| 73 | 1-Oct-20 | Cerasus serrulata | 256.65 Mb | 2n = 16 | 265.40 Mb | 304 | 31.12 Mb | 29,094 | Illumina X-ten, Nanopore, Hic-C | MADS-box, MYB, WRKY, and plant disease-resistance genes | China | 69 |
| 74 | 19-Oct-20 | Rhododendron simsii | 525 Mb | 2n = 26 | 528.6 Mb | 552 | 36.35 Mb | 34,170 | PacBio RS II, Hi-C | Metabolic pathways for anthocyanins and carotenoids | China | 70 |
Fig. 1. Statistics of ornamental plant species with sequenced genomes.
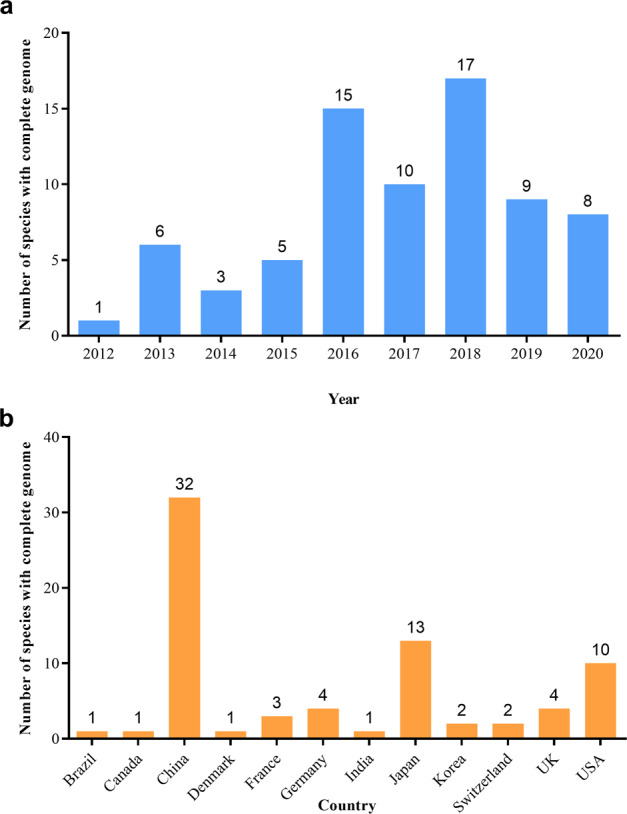
a Distribution of genome sequencing for ornamental plants completed from 2012 to 2020; b Distribution of genome sequencing for ornamental plants completed in different countries
Fig. 2. Summary of the representative ornamental plants with complete genome sequencing.

The x-axis represents the genome size of each plant, while the y-axis shows the scaffold N50 of the genome assembly. The sequencing platforms are indicated in different colors
Fig. 3. Phylogenetic relationships among ornamental plants with published sequenced genomes.

A maximum likelihood (ML) phylogenetic tree was built using low-copy orthologous sequences. All the published ornamental species belong to 21 orders and 35 families. The same background color was used for species in the same family
Rosaceae
Rosaceae contains more than 3300 species in 124 genera that are rich in economic and ornamental value and occupy an important position in gardens worldwide. The first flowering ornamental plant to be sequenced was Prunus mume (mei) from Rosaceae. In 2009, the National Engineering Research Center for Floriculture of Beijing Forestry University cooperated with the Beijing Genomics Institute (BGI) and other institutions to launch the mei genome project. First, a 237 Mb (84.6% of the estimated genome) genome of wild-type mei was assembled using the Illumina GA II. The scaffold N50 was 577.8 Kb, and 31,390 protein-coding genes were annotated. The genome data were published in Nature Communications in 2012, and this effort marked the first genome sequence map of a flowering crop worldwide3. Interestingly, equal to the status of mei in China, the “Yoshino cherry” tree (Prunus × yedoensis) is one of the most popular Prunus species in Japan, and its genome was sequenced by Korean researchers, revealing the parental origin and genomic delimitation of hybrid taxa using both Illumina and PacBio platforms in 201810. Soon afterwards, researchers from Japan also completed two similar genomes of Cerasus yedoensis, “Somei-Yoshino”, which were merged into a special genome11. At present, a large number of genome studies focusing on Prunus and Rosa in Rosaceae are underway.
Roses have high cultural and economic value as the most commonly cultivated ornamental and spice plants worldwide. The first ornamental Rosa to have its genome sequenced was Rosa multiflora, which was reported by Japanese scholars focusing on flower color, flower scent, and floral development traits12. Then, another well-known and long-awaited major study was published in Nature Genetics in May 2018. A team at the University of Lyon and Centre National de la Recherche Scientifique (CNRS) first revealed another parent of the modern rose, Rosa chinensis. The size of the Rosa genome is 560 Mb with a contig N50 of 24 Mb, which is one of the most comprehensive plant genomes4. Coincidentally, one month later, the same experimental material (a doubled haploid line from ‘Old Blush’) of Rosa chinensis was sequenced and republished in Nature Plants in June 2018. The high-quality genome was cross-verified, and ornamental and production traits of rose have been interpreted with the joint efforts of many research institutions from France, Belgium, Russia, etc.5.
Orchidaceae
As one of the most abundant families in the plant kingdom, Orchidaceae (orchid) plants are the flagship species of plant diversity protection, known as the “panda of the plant kingdom”. Orchids are divided into five subfamilies: Apostasioideae, Vanilloideae, Cypripedioideae, Orchidoideae, and Epidendroideae. Phalaenopsis and Dendrobium belong to Orchidoideae and Epidendroideae. Phalaenopsis plants are representative of Orchidaceae plants and have important ornamental value. Professor Zhongjian Liu of the National Orchid Conservation Center of China overcame technical problems resulting from high heterozygosity and completed the assembly of the whole-genome sequence of P. equestris with a scaffold N50 size of 359.1 Kb. As the first monocot flower for which genome-wide sequencing was completed, the genome of P. equestris was published as a cover paper in the journal Nature Genetics in November 201413. Phalaenopsis is an important potted flower with high economic value worldwide. A 3.1 Gb draft genome assembly of an important winter-flowering Phalaenopsis cultivar ‘KHM190’ was completed by researchers from China and Australia14. Another species of Phalaenopsis, P. aphrodite, also underwent high-quality genome sequencing with a scaffold N50 size of 19.7 Mb in April 201815. Scholars from China further analyzed the whole genomes of Dendrobium officinale and Dendrobium catenatuma, which were published in the journals Molecular Plant and Scientific Reports, respectively7,16. Apostasia shenzhenica is representative of one of two genera that form a sister lineage with the rest of the Orchidaceae; they have unique flower morphologies as well as diverse lifestyles and habitats. Professor Zhongjian Liu resequenced the high-quality genome of A. shenzhenica with a scaffold N50 size of 3.0 Mb. A 349 Mb genome was assembled and published in Nature in 201717. Vanilla fragrans is a plant of the vanilla family. Due to its unique fragrance that cannot be synthesized artificially, it is known as the “Perfume Queen”. In July 2014, the Fujian Agriculture & Forestry University and National Orchid Conservation Center of China (Shenzhen) officially launched the Vanilla shenzhenica genome project. As the first Orchidaceae vine plant to undergo complete sequencing, the genome of V. shenzhenica was ~800 Mb with a scaffold N50 size of 288 Kb, and its heterozygosity was ~1.14% (https://www.fafu.edu.cn/2015/0208/c132a18466/page.htm).
Asteraceae
There are ~24,000–35,000 species in Asteraceae; this family has very high plant diversity, accounting for ~10% of total angiosperms. Chrysanthemum, as a typical representative genus, is one of the most important ornamental crops in the world. The genome of Chrysanthemum morifolium is estimated to be more than 9 Gb (http://data.kew.org/cvalues/). Since the Chrysanthemum genus is large and complex, the genome of Chrysanthemum was not reported for a long time. In October 2018, the China Academy of Chinese Medical Sciences, Hubei University of Chinese Medicine cooperated with Nanjing Agricultural University and completed the sequencing of Chrysanthemum nankingense, a diploid species (2n = 18), which represents one of the progenitor genomes of domesticated chrysanthemums18. At around the same time, the de novo whole-genome assembly of Chrysanthemum seticuspe was announced by researchers from the Kazusa DNA Research Institute of Japan19. The 2.72 Gb of assembled sequences covered 89.0% of the 3.06 Gb C. seticuspe genome with 71,057 annotated genes19. Sunflower (Helianthus annuus L.), in the Asteraceae and the Helianthus genus, is a horticultural crop with important economic and ornamental value and a major research focus. In May 2017, a high-quality reference for the sunflower genome was published in the journal Nature by scientists from France and Canada20. The size of the sunflower genome was 2.94 Gb and covered 80% of the estimated genome; finally, 97% of annotated genes were anchored on a total of 17 pseudochromosomes.
Resequencing of ornamental plants
Whole-genome resequencing is a process of sequencing the genomes of different individuals of species with known genome sequences and analyzing the differences among individuals or populations. In recent years, to overcome the narrow genetic variation in current ornamental plant breeding programs, genome-scale investigations of wide germplasm panels and cultivated varieties have served to identify important genetic materials to study genomic variation dynamics during domestication and selective breeding71. For example, resequencing of multiple materials from different crop species based on genome-wide association study (GWAS) was facilitated to identify key genomic regions associated with plant domestication and selection/improvement72. Based on genome-wide resequencing technology, researchers can quickly screen resources, find a large number of genetic variations, and realize genetic evolution analysis and prediction of important candidate genes. Although great progress has been made in the de novo sequencing of ornamental plant genomes, only a few species of ornamental plants, such as sunflower, lotus, mei, rose, sakura, and Liriodendron chinense, have undergone genome resequencing (Table 2).
Table 2.
List of resequenced species of ornamental plants
| Code | Date | Species | Plant material | Average sequencing depth | Objects/goals | Reference |
|---|---|---|---|---|---|---|
| 1 | 26-Dec-16 | Fraxinus excelsior | 37 European diversity panel trees | 10.9× | Investigating genomic diversity | 42 |
| 2 | 1-Jun-17 | Helianthus annuus | 80 domesticated lines | 10–20× | Evolution of the cultivated sunflower | 20 |
| 3 | 1-Jun-17 | Helianthus annuus | 72 inbred lines | 9.3–19.5× | GWAS | 20 |
| 4 | 20-Oct-17 | Nelumbo nucifera | 19 individuals | 4× | Exploring genomic variation and evolution among different germplasms | 75 |
| 5 | 27-Apr-18 | Prunus mume | 333 cultivated landraces, 15 wild P. mume, and 3 close relatives of Prunus | 19.3× | Investigating the genetic architecture of floral traits and its domestication history | 74 |
| 6 | 30-Apr-18 | Rosa chinensis | 8 Rosa species, representing three of the four subgenera (Hulthemia: R. persica, Herperhodos: R. minutifolia and Rosa). | 36.5× | Genetic diversity within the Rosa genus | 5 |
| 7 | 11-Jun-18 | Rosa chinensis | 14 Rosa species, representing three sections (Synstylae, Chinenses, and Cinnamomeae) | 5–60× | Gaining insight into the makeup of the genomic relationship of modern roses | 4 |
| 8 | 4-Sep-18 | Prunus yedoensis | 9 accessions and 7 candidate parental species | 7.5–206.3× | Parental origin and genomic delimitation of hybrid taxa | 10 |
| 9 | 17-Dec-18 | Liriodendron chinense | 14 L. chinense individuals and six L. tulipifera individuals | 24.68–57.35× | Historical demographic fluctuations and present-day genetic diversity | 57 |
| 10 | 31-Dec-18 | Helianthus annuus | 287 cultivars, 17 Native American landraces, and 189 wild accessions | 1–25× | Genetic diversity and to quantify contributions from wild relatives | 73 |
Sunflower is not only an ornamental plant but also one of the four major oil crops in the world. In June 2017, genome sequencing of sunflower was completed, eighty domesticated lines (10–20× coverage) and 72 inbred lines (9.3–19.5× coverage) from 480 F1 hybrids were resequenced, and 35 genomic regions associated with flowering time were identified by GWAS20. Subsequently, to characterize genetic diversity in sunflower and to quantify contributions from wild relatives, scientists from the University of British Columbia sequenced 493 accessions, including cultivars, landraces, and wild relatives73. In all, 61,205 genes have been identified within the gene set of the sunflower pangenome, and a large number of candidate resistance genes and single nucleotide polymorphism (SNP) markers for downy mildew resistance were identified by GWAS, which may be of interest to other researchers and sunflower breeders73.
To reveal the evolutionary history of Prunus mume and the Prunus genus and the genetic mechanism of important ornamental characteristics of P. mume, 333 cultivated landraces, 15 wild P. mume, and three close relatives of Prunus (P. sibirica, P. davidiana, and P. salicina) were selected for genome-wide resequencing by Professor Qixiang Zhang from the National Engineering Research Center for Floriculture of China74. A total of 5.34 million high-quality SNPs were identified, and 24 important ornamental traits (such as petal color, stigma color, calyx color, bud color, stamina filament color, wood color, petal number, pistil character, bud aperture, and branching phenotype) of 333 cultivars of P. mume were analyzed by GWAS for the first time to confirm the hypothesis that P. mume exists due to introgression from P. sibirica and P. salicina74.
Three versions of the lotus genome have been published in five years21,24,50. To explore the genomic diversity and microevolution related to the rhizome growth pattern, especially the genomic markers of ecotype differentiation, researchers from the Wuhan Botanical Garden of the Chinese Academy of Sciences resequenced 19 individuals including rhizome lotus, seed lotus, flower lotus, wild lotus, Thai lotus and Nelumbo lutea75. Candidate genes associated with temperate and tropical lotus divergence always exhibited highly divergent expression patterns, which are valuable for the breeding and cultivation of lotus75.
Roses have high cultural and economic value because of their outstanding ornamental characteristics and essential oil composition. To analyze the genetic diversity and genetic regulation mechanism of important ornamental traits in roses, eight Rosa species representing three of the four subgenera (R. persica, R. minutifolia and Rosa) were resequenced, and the whole-genome sequence of a double-haploid rose line was completed5. At the same time, to gain insight into the makeup of modern roses, Raymond et al.4 resequenced representatives of three sections (“Synstylae”, “Chinenses” and “Cinnamomeae”) that participated in the domestication and breeding of the modern hybrid rose after the genome of homozygous Rosa chinensis ‘Old Blush’ was sequenced.
Sakura (Prunus yedoensis) is a woody ornamental plant with important cultural and economic value. To study the genomic relationship between P. yedoensis and its closely related species, nine P. yedoensis accessions and seven accessions of candidate parental species, including P. pendula, P. jamasakura and P. sargentii, were resequenced and compared to the assembled genome by researchers from Korea10. Resequencing data of six related taxa show that 41% of the genes were assigned to the parent state, suggesting that wild P. yedoensis is an F1 hybrid originating from a cross between P. pendula and P. jamasakura10.
Liriodendron chinense is an important woody ornamental plant known as a “woody tulip” in the UK and USA, as its flower shape is similar to that of the tulip. The high-quality genome of L. chinense was published in the journal Nature Plants in December 2018 in a project led by Professor Jisen Shi from Nanjing Forestry University57. To explore the historical demographic fluctuations and present-day genetic diversity between L. chinense and L. tulipifera, 14 L. chinense individuals and 6 L. tulipifera individuals were resequenced. Population analysis showed that Liriodendron can be divided into three subgroups: the Eastern China subgroup, Western China subgroup and North American subgroup. The species divergence time confirmed that the genetic diversity of L. chinense was much higher than that of L. tulipifera57.
Applications of whole-genome sequencing in ornamental plants
Gene annotation
Gene annotation is the process of attributing biological information to the completed sequence of a species using bioinformatics methods. It identifies gene fragments that do not encode proteins, recognizes elements on genes (gene prediction) and adds biological information to the elements for sequence repeat identification, noncoding RNA prediction, gene structure prediction, and gene function annotation. In this way, genes associated with ornamental horticultural traits such as flowering regulation, flower color, floral fragrance, plant type, dormancy, cold resistance, and disease resistance can be identified. The dormancy-associated MADS-box transcription factor (DAM) family, which is related to dormancy induction and release, is especially critical for ornamental plants76. Zhang et al.3 identified six DAM genes in the tandem array in the P. mume genome and confirmed that the distribution pattern was consistent with that from previous studies of the peach genome77. In Rosa, Raymond et al.4 identified new candidate genes potentially involved in recurrent blooming, such as TFL1, SPT, and DOG1.
Comparative genomics research
Based on genome mapping and sequencing technologies, comparative genomics research compares known genes and genome structures to understand the functions of associated genes, their expression mechanism, and the phylogenetic relationships of species. The acquisition of genomic information from multiple closely related species facilitates more comprehensive and in-depth research in comparative genomics. Moreover, it is crucial to perform in-depth comparative analysis of the collinear relationship between the genome sequences of two plants to analyze the origin and evolutionary relationship of plants and to explore important chromosome fragments or gene clusters that control major plant traits, which can provide essential reference information for the discovery and cloning of important genes. Zhang et al. constructed nine ancestral chromosomes of the Rosaceae family by comparing Rosaceae genomes. For the first time, these researchers revealed that ancestral chromosomes have evolved into eight existing chromosomes in P. mume via 11 fusions, seven existing chromosomes in strawberry (Fragaria ananassa) via 15 fusions and 17 existing chromosomes in apple (Malus domestica) via one whole-genome duplication event plus five fusions. These findings lay an important foundation for research to unravel the origin and evolution of Rosaceae3.
Resequencing
Whole-genome resequencing involves the sequencing of genomes in different individuals of species with known genome sequences and subsequent analysis of differences among individuals or populations. Whole-genome resequencing technology can be used to rapidly conduct resource screening, to find a large number of genetic variations and to implement genetic evolution analysis and candidate gene prediction for important traits. These results provide essential references for identifying valuable genetic resources and for horticultural crop breeding and are thus of significant research and industrial value. In P. mume, researchers investigated the genetic architecture of floral traits and plant domestication history by resequencing 348 P. mume accessions and three other Prunus species at an average sequencing depth of 19.3×. Highly admixed population structure and introgression from Prunus species were identified in mei accessions74. Huang et al.75 resequenced and analyzed the genomes of 19 lotus germplasms, provided a reliable and detailed understanding of the genome evolution of different lotus germplasms, and provided clues to key mutations responsible for rhizome enlargement.
GWAS
A GWAS is a genome-wide comparative analysis or correlation analysis using millions of SNPs in the genome as molecular genetic markers. It is a new strategy to find genetic variations that affect complex traits by comparison. With the development of genomics research and DNA microarray technology, a GWAS can provide an outlined overview of important traits simultaneously and is therefore suitable for the study of complex traits. At the genome-wide level, association studies between genes and traits are conducted with multiple centers, large samples, and repeated verifications. This method has been applied for the screening and identification of major genes for important economic traits in agriculture. In P. mume, through a GWAS, researchers have identified significant quantitative trait loci (QTLs) and genomic regions where several genes associated with petal color, stigma color, calyx color, bud color, stamina filament color, wood color, petal number, pistil character, bud aperture, and branching phenotype are located74. Taken together, the identification of genetic loci associated with floral and other traits provides more insight into the genetic mechanisms that underlie the domestication of P. mume and provides opportunities to design strategies for genomic selection to improve the performance of ornamental species. In sunflowers and roses, the key ornamental trait of flowering time was also identified by the GWAS method4,20.
Comparative analysis with transcriptome data
RNA sequencing is a newly emerging technology that uses next-generation sequencing for transcriptome analysis. It can comprehensively and rapidly acquire sequence information and expression information for almost all transcripts from specific cells or tissues in a particular state, including protein-coding mRNAs and various noncoding RNAs, as well as the expression abundance of different transcripts generated by alternative gene splicing. The transcriptome is an inevitable link that connects genetic information of the genome with the biological functions of the proteome. Currently, transcriptional regulation is the most well-studied and foremost regulatory method in organisms. Transcriptome studies are the foundation and starting point of gene function-structure studies and the first issue to address after the completion of whole-genome sequencing. Furthermore, transcriptome analysis provides large numbers of molecular markers, such as simple sequence repeats and SNPs. All of the sequence information, expression data, and molecular markers facilitate the localization of QTLs for key ornamental traits in ornamental plants through genetic mapping and contribute to the development of molecular markers in close linkage with excellent traits for use in the molecular marker-assisted breeding of flowers. Based on the genome sequence of P. mume, vital differences in gene expression between the bud stage and squaring stage were observed, and 7,813 DEGs were identified, which provided a special perspective on floral scent formation in P. mume78. The water lily genome revealed variable genomic signatures of ancient vascular cambium losses, and the expression profiles of floral ABCE genes, floral scent and color genes were screened from the DEGs in a comparative analysis of the transcriptome64.
Development of SNP microarrays
According to their position in genes, SNPs can occur in coding regions, noncoding regions, and gene spacer regions. They are DNA molecular markers that have the most abundant polymorphisms in the genome and are characterized by large numbers, a uniform distribution, and easy typing. SNPs can be used for the identification of genetic variation and genotyping of associated phenotypes. Using SNPs as molecular markers to construct genetic variation maps of the genome has become a vital part of the research for studying genome diversity, obtaining domesticated selection regions, and screening key genes of important traits. Based on the genome sequence and resequencing of P. mume, a total of 1,298,196 raw SNPs were located within coding regions of genes, 733,292 of which were nonsynonymous74. Furthermore, by combining transcriptome data, 76 SNPs within DEGs were identified that were associated with petal, stigma, calyx, and bud color74. In sacred lotus, wild and Thai lotus exhibited greater differentiation with a higher genomic diversity than cultivated lotus based on SNP sites in resequenced species75.
Exploiting genes associated with important ornamental traits
During the course of whole-genome sequencing, a very large number of genes, in the range of 19,507–87,603, are annotated for each flowering species (Table 1). Through further analysis, important genes associated with floral development, flower color formation, and stress resistance can be discovered. This is conducive to the breeding of unique, high-quality, and high-resistance varieties or types of a species and provides important references for improving ornamental and resistance qualities in other flowering species.
Candidate genes for controlling floral development
Flower blooming is a process that involves the formation of inflorescence meristems and flower meristem tissues through floral induction and a series of internal and external factors, followed by the generation of floral organ primordia and eventually the release of flora bud dormancy to form floral organs. The process of flowering is controlled by a complex regulatory network, with at least seven flowering regulation pathways found in A. thaliana79. The genes associated with floral development can be divided into two classes. One class consists of genes that control the formation of inflorescence meristems and determine the direction of newly formed floral primordia. These genes influence the flowering time of plants by controlling the formation of inflorescence meristems or flower meristems, and mutations in these genes can result in earlier or later flowering mutants. The other class consists of genes that determine the formation of floral organs, and mutations in these genes can result in homeoboxes79. In ornamental plants, the morphology and number of floral organs have undergone substantial variations, for example, double petals, multiple sepals, and multiple pistils and stamens, developing into independent flowers during the course of long-term artificial domestication and cultivation. These variations increase the ornamental value of ornamental plants while providing excellent materials for the study of floral organ development in plants. With genomic data analysis, as an important scientific issue, some key genes related to flowering transition and flower development have been analyzed, such as those in Tarenaya hassleriana23, Dendrobium officinale7, Primula veris28, Dendrobium catenatum16, Hibiscus syriacus41, Rosa4,5,12, Chrysanthemum18,19, and Nymphaea colorata64.
Candidate genes for controlling anthocyanin synthesis
Flower color is one of the most vital quality traits of ornamental plants. Anthocyanin is an essential pigment for coloring flowers, and its biosynthesis is catalyzed by a series of enzymes80. Various anthocyanins are formed due to differences in the substituent groups at varied positions on the basic skeleton, thus leading to different plant organ colors, such as red, purple, blue-purple, and blue. Anthocyanins are flavonoid secondary metabolites in plants and the most widely distributed water-soluble pigments in nature, playing a major role in the color formation and antioxidation in plant flowers and fruits. R2R3-MYB genes are involved in anthocyanin synthesis81. In P. mume, 96 R2R3-MYB genes were identified and divided into 35 subfamilies. Finally, the functions of PmMYB1 and PmMYBa1 were identified by overexpression in tobacco and significantly promoted the accumulation of anthocyanins in transgenic tobacco. The flower colors of PmMYB1-overexpressing transgenic plants were significantly deepened, and the anthocyanin contents in the corolla of transgenic plants were significantly higher than those of the control82. To understand the molecular basis of the blue color in water lily, delphinidin 3′-O was identified as the main blue anthocyanidin pigment, and some genes for an anthocyanidin synthase and a delphinidin-modification enzyme were screened by comparing the expression profiles between two N. colorata cultivars with white and blue petals64. Interestingly, after the butterfly pea UDP (uridine diphosphate)-glucose: anthocyanin 3′,5′-O-glucosyltransferase gene was introduced in chrysanthemums, blue flowers appeared83. In Rosa rugosa, two MYB transcription factors have been confirmed to affect flower color by regulating flavonoid biosynthesis in response to wounding and oxidation84. In Paeonia, a chalcone synthase (PhCHS) involved in flavonoid biosynthesis and two anthocyanin O-methyltransferase (AOMT) genes were consistent with anthocyanin accumulation in petals85,86.
Candidate genes for controlling floral scent biosynthesis
Floral scent, as one of the quality traits of ornamental plants, has great aesthetic, economic, and application value. The scent components present in petals primarily include secondary metabolites such as esters, alcohols, ketones, aldehydes, terpenes, and volatile phenols, mainly derived from terpene metabolism, phenylpropane metabolism, and the lipoxygenase pathway87. There are various types of scent components in different petals, thereby forming distinct scents among various flower species. In a study on the molecular mechanism responsible for the floral scent in P. mume, Zhang et al.3 first discovered that the benzylalcohol acetyltransferase (BEAT) gene can directly catalyze the formation of benzyl acetate, a crucial component of the floral scent in P. mume. Moreover, based on genomic data from P. mume and P. persica, 44 unique PmBEATs were found in P. mume, far more than the 16 in apple, 14 in strawberry, and four in grape. These PmBEAT genes originated from gene duplication events during the species evolution of P. mume, and retroduplication and tandem duplication were the two dominant duplication patterns. Overexpression of the PmBEAT36 or PmBEAT37 genes increased benzyl acetate production in the petal protoplasts of P. mume, and interference in the expression of these genes slightly decreased the benzyl acetate content88. Zhao et al.78 conducted a comparative transcriptome analysis of different developmental stages and tissues of flower genes associated with floral traits and preliminarily selected 12 new genes involved in floral scent formation in P. mume. Furthermore, five of the TFs (bHLH4, bHLH6, bZIP4, ERF1, and NAC1) from Phalaenopsis bellina have been proven to be involved in orchid floral monoterpenes89. In Plumeria rubra, PrCYP79D73 is involved in floral volatile organic compounds and other nitrogen-containing volatiles90.
Candidate genes for controlling plant architecture
Rich and diverse plant architectures are the result of long-term evolution, natural selection, and a complex regulatory process of interaction between genetics and the environment. Diverse plant architecture traits are not only conducive to the creation of rich and diverse horticultural landscapes but are also favorable for plant adaptation to complex environments and competition and the utilization of light and nutrients. Along with the completion of whole-genome sequencing for multiple ornamental plants of the genus Prunus, the results lay an important data foundation for studying the molecular genetic mechanisms of pendulous traits3,91. According to the eight scaffolds of the P. mume genome, Zhang et al. constructed a high-density genetic map using specific-length amplified fragment sequencing (SLAF) and mapped QTLs for major traits such as plant type, flower color, petals, and leaves in P. mume. They found 10 SLAF markers that were closely linked to the pendulous traits of P. mume. Using these markers, the pendulous traits were finely mapped to a 1.14 cM region on chromosome 7, and 36 candidate genes that might be associated with the pendulous traits of P. mume were predicted92. Breakthroughs were also achieved in the mining and labeling of genes for weeping and dwarf traits in peach (P. persica) by using genome and bulked segregant analyses93.
Candidate genes for controlling dormancy release
Flowers of the genus Prunus, such as P. mume and P. yedoensis, are early flowering types in spring. Zhang et al.3 explored the molecular mechanisms underpinning dormancy break and flowering in P. mume at low temperature. These researchers identified a total of six dormancy-associated MADS-box (DAM) genes with a tandem repeat distribution in the genome. The six DAM genes in P. mume are derived from a series of duplication events in the following order: PmDAM1, PmDAM3, PmDAM2, PmDAM5, PmDAM4, and PmDAM6. The molecular evolution pattern of DAM genes is unique to Prunus plants and is present in P. persica, but tandem genes have not been found in M. domestica or F. ananassa. This phenomenon could be related to the earlier flowering of Prunus plants, including P. persica, P. mume, apricot (Armeniaca vulgaris) and sweet cherry (Prunus avium), than of most other flowering species3. DAM genes are regulated by C-repeat-binding transcription factors (CBFs). A conserved CBF site was found 1000 bp upstream of the transcription start site of DAM4-DAM6 in P. persica and plum (Prunus salicina). The latest research results show that a sense-response relationship between PmCBFs and PmDAMs is exhibited in cold-induced dormancy and is jointly regulated by six PmCBFs and PmDAM4–694.
Candidate genes for controlling self-incompatibility
Self-incompatibility has always been an important research topic in the molecular genetic biology of flowers. According to different hereditary patterns of pollen incompatibility phenotypes, the regeneration disorder whereby plants reject self-pollen can be divided into sporophytic self-incompatibility and gametophytic self-incompatibility95. Various flowers of the Rosaceae family, including P. mume, P. yedoensis and P. persica, all exhibit gametophytic self-incompatibility, which is controlled by an S-locus with multiple alleles, including two linked genes: one is the S-RNase gene specifically expressed in pistil tissue, and the other is the S-haplotype-specific F-box gene specifically expressed in pollen96. In Tarenaya hassleriana, three syntenic regions containing most of the genes of the S-locus were found, and it was assumed that the single-copy ancestral region contained homologs of Pub8, ARK3, and B12023.
Candidate genes for controlling disease resistance
Disease resistance is an essential trait that attracts research attention across all flowering plants. Thus, the whole-genome analysis also focuses on the genes associated with disease resistance. The genes involved in plant disease resistance are mainly R genes, which encode proteins with extremely high structural similarities, such as leucine zippers, nucleotide-binding sites, transmembrane domains, leucine-rich repeats, and similar extracellular regions of drosophilid toll protein and mammalian toll and interleukin-1 receptor (TIR). Nucleotide-binding site leucine-rich repeat genes constitute the gene family with the widest distribution and largest number of plant R genes. In their encoded proteins, the nucleotide-binding site is present near the N-terminus, while the leucine-rich repeat exists near the C-terminus. The N-terminus of proteins encoded by different genes may also include one or more of the following two conserved structures: the coiled-coil motif and TIR motif. In the P. mume genome, 253 leucine-rich repeats receptor-like kinase (LRR-RLK) genes were identified, and most pathogenesis-related (PR) gene families were notably expanded and arranged in tandem, especially PR103. In Hibiscus syriacus, resistance (R) genes account for 0.53% of its total predicted genes, which is lower than that of other plants evaluated in genomic studies (0.63 to 1.35%)41. The Asparagus setaceus genome included 76 R genes with nucleotide-binding sites (NBSs), and the R genes belonged to five groups: TIR-NBS, CC-NBS-LRR, NBS-LRR, NBS, and CC-NBS. NBS-LRR was the largest group, including a total of 29 genes65.
Candidate genes for controlling abiotic stress resistance
Adverse conditions such as low temperature, humidity, heat, drought, and saline-alkali conditions severely inhibit the growth and development of ornamental plants. These conditions can cause changes in plant physiology, biochemistry, and morphology and even lead to death. Due to this issue, cultivation facilities for ornamental plants are cumbersome and cannot be widely promoted, which considerably affects their qualities and benefits. Low temperature is an important factor that constrains the normal growth, development, and geographical distribution of plants. Stress caused by low temperature can be divided into chilling stress (>0 °C) and freezing stress (<0 °C). Plants from the tropics and subtropics are more sensitive to cold; in contrast, plants from temperate regions have evolved complex mechanisms to resist and adapt to chilling (freezing) stress, protecting the plants from injury. Cold acclimation is a responsive protection mechanism for plant adaptation and resistance to low-temperature stress, and this process is regulated by a complex network97. In particular, the CBF pathway is considered the most important and well-studied pathway98. Based on the genome data for P. mume, 30 LEA genes were identified, and heterologous expression of PmLEA increased the cold resistance of Escherichia coli and tobacco (Nicotiana tabacum)99,100. Furthermore, a molecular regulation model of the PmDAM and PmCBF genes in response to dormancy and dormancy release of flower buds induced by low-temperature signals was proposed based on yeast two-hybrid and bimolecular fluorescence complementation experiments94.
Prospects for whole-genome sequencing data for ornamental plants
The Earth BioGenome Project (EBP) is a massive project in biology that aims to sequence, catalog, and characterize the genomes of all of Earth’s eukaryotic biodiversity over a period of 10 years. For plants, the core scientific problems are to improve crop yields and other agronomically important traits, biofuel production, gene editing, and conservation of endangered species101. The 10,000 Plant Genome Sequencing Project (10KP) initiated by the Beijing Genomics Institute in Shenzhen (BGI-Shenzhen) is a landmark effort to catalog plant genomic variation and represents a major step in understanding the tree of life102. A tentative plan of the 100 Flowers Genome Sequencing Project has been put forward by the National Engineering Research Center for Floriculture in China. Many ornamentals are marked by high ploidy levels and homologous polyploids (chrysanthemum and alfalfa) or extremely large genome sizes (lily and tulip), which limit the development and utilization of genome sequencing technology in ornamental plants. Along with the development of sequencing and bioinformatics analysis technologies and the continuous emergence of various new biological technologies, genomics research on ornamental plants has developed faster and better. Although genome sequencing and assembly of flowering plants face substantial difficulties, the quality of genome assembly results is relatively high in terms of the analytical results from 69 flower species that underwent genome sequencing, and four of them have been resequenced using updated sequencing technology5,11,37,50. As far as we know, there are at least a dozen ornamental plants undergoing the process of genome quality improvement. As more ornamental plant genomes are sequenced, further bioinformatics analysis could reveal crucial basic information on the origin of species and the genes that control flower traits. The development of genomics will surely address the knowledge gaps of traditional breeding methods. The ultimate goal is to obtain the optimal type of flower variety with fixed-point improvement and the aggregation of multiple elite traits by using the most effective and rapid method.
China has 30,000 species of higher (flowering) plants, and some ornamental flowering plants reached Europe quite early103. Chinese people love flowers and cultivate many kinds of brilliant flowers, such as mei, peony, chrysanthemum, rose, lily, lotus, and orchid. Due to the rapid development of genome sequencing technology worldwide, large quantities of whole-genome sequencing data are in urgent need of deep mining. A long-term strategic genomics research plan should be formulated that is not limited to cultivated species but considers thorough development of the sequencing of important wild relatives of ornamental species in China and promoting the mining, protection, and utilization of important genetic resources. It is essential to put an end to the dependence on the apparent phenotype, transform investigations into genotype-dependent research and shift from single-gene studies to GWAS. Efforts should be made to vigorously promote the application of genomics in gene cloning and molecular breeding in China and to improve the breeding capacity and level of horticultural crops.
Due to their complexity and particularity, plant genomes have always been an important focus of genomics. Before the second generation of high-throughput sequencing, sequencing costs were high, and the throughput was low. For species with highly repetitive sequences, it was too difficult or too expensive for researchers to obtain the whole-genome sequences of high repeat sequence species. Many species with important economic and ornamental value have not yet been submitted to complete genome sequencing. In short, due to the particularity and diversity of ornamental plants, there are challenges and opportunities in genome research of these species. Challenge: (1) Complex genome. The term complex genome refers to a kind of genome that cannot be directly analyzed by conventional sequencing and assembly methods. It usually refers to a genome containing a high proportion of repetitive sequences, high heterozygosity, extreme GC content, and difficulty in eliminating foreign DNA contamination. (2) Autopolyploidy. Autopolyploidy is common in ornamental plants. It is usually formed by doubling two or more sets of genomes, which is of great value in genetic breeding and agricultural production. Using conventional methods, it is easy to connect incorrect allele fragments together, resulting in the wrong connection of homologous chromosomes and a large number of chimeric assemblies; thus, assembly is still difficult. (3) Megagenome. Megagenome generally refers to species with genomes larger than 10 Gb. The sequencing and analysis of these species are very involved, especially for assembly analysis, which is a major challenge. Paris japonica is an unusual plant. Scientists have found that it has the world’s largest genome, with 150 Gb, which is 50 times more than that of humans. Although the genomes of some ornamental plants have been deemed complete, the assembly quality of some species is poor, and a small number of “holes” have not yet been completed due to technical limitations, although the interest of scientists in this regard is debatable. The latest research shows that the sequences that were once considered irrelevant, or “garbage”, in the genome have their own significance. These missing sequences play a very important role, and we now have the opportunity to mine them. Third-generation sequencing technology (PacBio and Nanopore) can make up for the holes in some genomic regions that are difficult to assemble due to sequencing errors, repeat regions, heterochromatin, genomic polymorphisms, and second-generation sequencing preferences. To solve the challenge of sequencing the genomes of ornamental plants, the following new technologies can be tried with third-generation sequencing technology. (1) Pangenome. The pangenome includes the core genome and the nonessential genome. Among them, the core genome refers to the genes that exist in all individuals; the nonessential genome refers to the genes that exist only in some individuals. (2) Hi-C. The advantages of Hi-C sequencing technology are as follows: on the one hand, there is no need to construct a large number of F1 populations, as only individuals are needed; on the other hand, the haplotype genome can be separated without parent purification, so this method is suitable for the assembly of a highly heterozygous genome that is not easy to purify.
With the development of sequencing technology, the concepts of difficult genome sequencing and assembly quality have also developed and changed. We cannot sequence everything for the sake of genome sequencing. The purpose of sequencing must be to reveal the key scientific problems of species. We should strengthen research related to transcriptomics, metabolomics, proteomics, degradomics, and phenomics. With more genomic data published, it has become a great challenge to analyze, store and share the massive amounts of genome sequencing data. A key problem is how to solve the time and cost problems faced by researchers to achieve the purpose of reducing repetitive research, improving the practicability of scientific research, mining research content, and improving the transparency of scientific research and data sharing with cross-research into other fields. Moreover, it is necessary to enhance bioinformatics education and apply bioinformatics in practice. With the continuous development of sequencing technology, we believe that the whole-genome sequencing of horticultural crops will enter a rapid development stage in the near future, leading to tremendous contributions to the world’s horticultural industry.
Acknowledgements
The research was supported by the National Natural Science Foundation of China (No. 31800595 and 31471906), the National Key Research and Development Program of China (2018YFD1000401), and the Special Fund for Beijing Common Construction Project.
Author contributions
T.Z. conceived and drafted the manuscript. T.Z., P.L., and L.L. analyzed the data. Q.Z. contributed to the conception of the study and finalized the manuscript. All authors read and approved the final manuscript.
Conflict of interest
The authors declare no competing interests.
References
- 1.Initiative AG. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 2.Fan, J. Plant Genomics. (Science Press, 2019).
- 3.Zhang, Q. et al. The genome of Prunus mume. Nat. Commun.10.1038/ncomms2290 (2012). [DOI] [PMC free article] [PubMed]
- 4.Raymond O, et al. The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 2018;50:772–777. doi: 10.1038/s41588-018-0110-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hibrand Saint-Oyant L, et al. A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat Plants. 2018;4:473–484. doi: 10.1038/s41477-018-0166-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwartz DC, et al. Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping. Science. 1993;262:110–114. doi: 10.1126/science.8211116. [DOI] [PubMed] [Google Scholar]
- 7.Yan L, et al. The genome of dendrobium officinale illuminates the biology of the important traditional Chinese orchid herb. Mol. Plant. 2015;8:922–934. doi: 10.1016/j.molp.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 8.Xu, Z. et al. Tandem gene duplications drive divergent evolution of caffeine and crocin biosynthetic pathways in plants. BMC Biol.10.1186/s12915-020-00795-3 (2020). [DOI] [PMC free article] [PubMed]
- 9.Shang, J. et al. The chromosome-level wintersweet (Chimonanthus praecox) genome provides insights into floral scent biosynthesis and flowering in winter. Genome Biol.21, 10.1186/s13059-020-02088-y (2020). [DOI] [PMC free article] [PubMed]
- 10.Baek, S. et al. Draft genome sequence of wild Prunus yedoensis reveals massive inter-specific hybridization between sympatric flowering cherries. Genome Biol.10.1186/s13059-018-1497-y (2018). [DOI] [PMC free article] [PubMed]
- 11.Shirasawa K, et al. Phased genome sequence of an interspecific hybrid flowering cherry, ‘Somei-Yoshino’ (Cerasus × yedoensis) DNA Res. 2019;26:379–389. doi: 10.1093/dnares/dsz016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakamura N, et al. Genome structure of Rosa multiflora, a wild ancestor of cultivated roses. DNA Res. 2018;25:113–121. doi: 10.1093/dnares/dsx042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai J, et al. The genome sequence of the orchid Phalaenopsis equestris. Nat. Genet. 2015;47:65–72. doi: 10.1038/ng.3149. [DOI] [PubMed] [Google Scholar]
- 14.Huang J, et al. The genome and transcriptome of Phalaenopsis yield insights into floral organ development and flowering regulation. PeerJ. 2016;4:e2017. doi: 10.7717/peerj.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chao Y, et al. Chromosome-level assembly, genetic and physical mapping of Phalaenopsis aphrodite genome provides new insights into species adaptation and resources for orchid breeding. Plant Biotechnol. J. 2018;16:2027–2041. doi: 10.1111/pbi.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang, G. et al. The Dendrobium catenatum Lindl. genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci. Rep.10.1038/srep19029 (2016). [DOI] [PMC free article] [PubMed]
- 17.Zhang G, et al. The Apostasia genome and the evolution of orchids. Nature. 2017;549:379–383. doi: 10.1038/nature23897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song C, et al. The Chrysanthemum nankingense genome provides insights into the evolution and diversification of chrysanthemum flowers and medicinal traits. Mol. Plant. 2018;11:1482–1491. doi: 10.1016/j.molp.2018.10.003. [DOI] [PubMed] [Google Scholar]
- 19.Hirakawa H, et al. De novo whole-genome assembly in Chrysanthemum seticuspe, a model species of Chrysanthemums, and its application to genetic and gene discovery analysis. DNA Res. 2019;26:195–203. doi: 10.1093/dnares/dsy048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Badouin H, et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature. 2017;546:148–152. doi: 10.1038/nature22380. [DOI] [PubMed] [Google Scholar]
- 21.Ming, R. et al. Genome of the long-living sacred lotus (Nelumbo nucifera Gaertn.). 10.1186/gb-2013-14-5-r41 (2013). [DOI] [PMC free article] [PubMed]
- 22.Sierro N, et al. Reference genomes and transcriptomes of Nicotiana sylvestris and Nicotiana tomentosiformis. Genome Biol. 2013;14:R60. doi: 10.1186/gb-2013-14-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng S, et al. The Tarenaya hassleriana genome provides insight into reproductive trait and genome evolution of crucifers. Plant Cell. 2013;25:2813–2830. doi: 10.1105/tpc.113.113480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, et al. The sacred lotus genome provides insights into the evolution of flowering plants. Plant J. 2013;76:557–567. doi: 10.1111/tpj.12313. [DOI] [PubMed] [Google Scholar]
- 25.Hellsten U, et al. Fine-scale variation in meiotic recombination in Mimulus inferred from population shotgun sequencing. Proc. Natl Acad. Sci. USA. 2013;110:19478–19482. doi: 10.1073/pnas.1319032110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yagi M, et al. Sequence analysis of the genome of carnation (Dianthus caryophyllus L.) DNA Res. 2014;21:231–241. doi: 10.1093/dnares/dst053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sunil M, et al. The draft genome and transcriptome of Amaranthus hypochondriacus: a C4 dicot producing high-lysine edible pseudo-cereal. DNA Res. 2014;21:585–602. doi: 10.1093/dnares/dsu021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nowak, M. D. et al. The draft genome of Primula veris yields insights into the molecular basis of heterostyly. Genome Biol.10.1186/s13059-014-0567-z (2015). [DOI] [PMC free article] [PubMed]
- 29.Kellner F, et al. Genome-guided investigation of plant natural product biosynthesis. Plant J. 2015;82:680–692. doi: 10.1111/tpj.12827. [DOI] [PubMed] [Google Scholar]
- 30.Xiao L, et al. The resurrection genome of Boea hygrometrica: A blueprint for survival of dehydration. Proc. Natl Acad. Sci. USA. 2015;112:5833–5837. doi: 10.1073/pnas.1505811112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Byrne SL, et al. A synteny-based draft genome sequence of the forage grass Lolium perenne. Plant J. 2015;84:816–826. doi: 10.1111/tpj.13037. [DOI] [PubMed] [Google Scholar]
- 32.De Vega, J. J. et al. Red clover (Trifolium pratense L.) draft genome provides a platform for trait improvement. Sci. Rep.10.1038/srep17394 (2015). [DOI] [PMC free article] [PubMed]
- 33.Lu M, An H, Li L. Genome survey sequencing for the characterization of the genetic background of Rosa roxburghii tratt and leaf ascorbate metabolism genes. PLoS ONE. 2016;11:e147530. doi: 10.1371/journal.pone.0147530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka H, et al. Sequencing and comparative analyses of the genomes of zoysiagrasses. DNA Res. 2016;23:171–180. doi: 10.1093/dnares/dsw006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bombarely, A. et al. Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nat. Plants10.1038/nplants.2016.74 (2016). [DOI] [PubMed]
- 36.Butts CT, Bierma JC, Martin RW. Novel proteases from the genome of the carnivorous plant Drosera capensis: Structural prediction and comparative analysis. Proteins. 2016;84:1517–1533. doi: 10.1002/prot.25095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clouse, J. W. et al. The Amaranth genome: Genome, transcriptome, and physical map assembly. Plant Genome10.3835/plantgenome2015.07.0062 (2016). [DOI] [PubMed]
- 38.Hirakawa, H. et al. Draft genome sequence of subterranean clover, a reference for genus Trifolium. Sci. Rep.10.1038/srep30358 (2016). [DOI] [PMC free article] [PubMed]
- 39.Hoshino, A. et al. Genome sequence and analysis of the Japanese morning glory Ipomoea nil. Nat. Commun.10.1038/ncomms13295 (2016). [DOI] [PMC free article] [PubMed]
- 40.Guan, R. et al. Draft genome of the living fossil Ginkgo biloba. Gigascience10.1186/s13742-016-0154-1 (2016). [DOI] [PMC free article] [PubMed]
- 41.Kim, Y. et al. Genome analysis of Hibiscus syriacus provides insights of polyploidization and indeterminate flowering in woody plants. DNA Res. 10.1093/dnares/dsw049 (2016). [DOI] [PMC free article] [PubMed]
- 42.Sollars ES, et al. Genome sequence and genetic diversity of European ash trees. Nature. 2017;541:212–216. doi: 10.1038/nature20786. [DOI] [PubMed] [Google Scholar]
- 43.Fu, Y. et al. Draft genome sequence of the Tibetan medicinal herb Rhodiola crenulata. Gigascience10.1093/gigascience/gix033 (2017). [DOI] [PMC free article] [PubMed]
- 44.Zhao D, et al. De novo genome assembly of Camptotheca acuminata, a natural source of the anti-cancer compound camptothecin. Gigascience. 2017;6:1–7. doi: 10.1093/gigascience/gix089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang, L. et al. The draft genome assembly of Rhododendron delavayi Franch. var. delavayi. Gigascience10.1093/gigascience/gix076 (2017). [DOI] [PMC free article] [PubMed]
- 46.Copetti D, et al. Extensive gene tree discordance and hemiplasy shaped the genomes of North American columnar cacti. Proc. Natl Acad. Sci. USA. 2017;114:12003–12008. doi: 10.1073/pnas.1706367114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silva-Junior, O. B., Grattapaglia, D., Novaes, E. & Collevatti, R. G. Genome assembly of the Pink Ipê (Handroanthus impetiginosus, Bignoniaceae), a highly valued, ecologically keystone Neotropical timber forest tree. Gigascience10.1093/gigascience/gix125 (2018). [DOI] [PMC free article] [PubMed]
- 48.Yang, X. et al. The Kalanchoë genome provides insights into convergent evolution and building blocks of crassulacean acid metabolism. Nat. Commun.10.1038/s41467-017-01491-7 (2017). [DOI] [PMC free article] [PubMed]
- 49.Hori K, et al. Mining of the uncharacterized cytochrome P450 genes involved in alkaloid biosynthesis in california poppy using a draft genome sequence. Plant Cell Physiol. 2018;59:222–233. doi: 10.1093/pcp/pcx210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gui S, et al. Improving Nelumbo nucifera genome assemblies using high‐resolution genetic maps and BioNano genome mapping reveals ancient chromosome rearrangements. Plant J. 2018;94:721–734. doi: 10.1111/tpj.13894. [DOI] [PubMed] [Google Scholar]
- 51.Gao, Y. et al. De novo genome assembly of the red silk cotton tree (Bombax ceiba). Gigascience10.1093/gigascience/giy051 (2018). [DOI] [PMC free article] [PubMed]
- 52.Dong, A. et al. High-quality assembly of the reference genome for scarlet sage, Salvia splendens, an economically important ornamental plant. Gigascience10.1093/gigascience/giy068 (2018). [DOI] [PMC free article] [PubMed]
- 53.Griesmann, M. et al. Phylogenomics reveals multiple losses of nitrogen-fixing root nodule symbiosis. Science361, 10.1126/science.aat1743 (2018). [DOI] [PubMed]
- 54.Malli RPN, Adal AM, Sarker LS, Liang P, Mahmoud SS. De novo sequencing of the Lavandula angustifolia genome reveals highly duplicated and optimized features for essential oil production. Planta. 2019;249:251–256. doi: 10.1007/s00425-018-3012-9. [DOI] [PubMed] [Google Scholar]
- 55.Ye G, et al. De novo genome assembly of the stress tolerant forest species Casuarina equisetifolia provides insight into secondary growth. Plant J. 2019;97:779–794. doi: 10.1111/tpj.14159. [DOI] [PubMed] [Google Scholar]
- 56.Yang, X. et al. The chromosome-level quality genome provides insights into the evolution of the biosynthesis genes for aroma compounds of Osmanthus fragrans. Hortic. Res.10.1038/s41438-018-0108-0 (2018). [DOI] [PMC free article] [PubMed]
- 57.Chen J, et al. Liriodendron genome sheds light on angiosperm phylogeny and species–pair differentiation. Nat. Plants. 2019;5:18–25. doi: 10.1038/s41477-018-0323-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cocker, J. M. et al. Primula vulgaris (primrose) genome assembly, annotation and gene expression, with comparative genomics on the heterostyly supergene. Sci. Rep.10.1038/s41598-018-36304-4 (2018). [DOI] [PMC free article] [PubMed]
- 59.Li M, et al. Genome structure and evolution of Antirrhinum majus L. Nat. Plants. 2019;5:174–183. doi: 10.1038/s41477-018-0349-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wai CM, et al. Time of day and network reprogramming during drought induced CAM photosynthesis in Sedum album. PLoS Genet. 2019;15:e1008209. doi: 10.1371/journal.pgen.1008209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soza VL, et al. The Rhododendron genome and chromosomal organization provide insight into shared whole-genome duplications across the heath family (Ericaceae) Genome Biol. Evol. 2019;11:3353–3371. doi: 10.1093/gbe/evz245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamashiro, T., Shiraishi, A., Satake, H. & Nakayama, K. Draft genome of Tanacetum cinerariifolium, the natural source of mosquito coil. Sci. Rep.10.1038/s41598-019-54815-6 (2019). [DOI] [PMC free article] [PubMed]
- 63.Lv S, et al. Draft genome of the famous ornamental plant Paeonia suffruticosa. Ecol. Evol. 2020;10:4518–4530. doi: 10.1002/ece3.5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang L, et al. The water lily genome and the early evolution of flowering plants. Nature. 2020;577:79–84. doi: 10.1038/s41586-019-1852-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li, S. et al. Chromosome-level genome assembly, annotation and evolutionary analysis of the ornamental plant Asparagus setaceus. Hortic. Res.10.1038/s41438-020-0271-y (2020). [DOI] [PMC free article] [PubMed]
- 66.Palfalvi G, et al. Genomes of the venus flytrap and close relatives unveil the roots of plant carnivory. Curr. Biol. 2020;30:2312–2320. doi: 10.1016/j.cub.2020.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lv Q, et al. The Chimonanthus salicifolius genome provides insight into magnoliid evolution and flavonoid biosynthesis. Plant J. 2020;103:1910–1923. doi: 10.1111/tpj.14874. [DOI] [PubMed] [Google Scholar]
- 68.Li, L., Cushman, S. A., He, Y. & Li, Y. Genome sequencing and population genomics modeling provide insights into the local adaptation of weeping forsythia. Hortic. Res.10.1038/s41438-020-00352-7 (2020). [DOI] [PMC free article] [PubMed]
- 69.Yi, X. et al. The genome of Chinese flowering cherry (Cerasus serrulata) provides new insights into Cerasus species. Hortic. Res.10.1038/s41438-020-00382-1 (2020). [DOI] [PMC free article] [PubMed]
- 70.Yang FS, et al. Chromosome-level genome assembly of a parent species of widely cultivated azaleas. Nat. Commun. 2020;11:5269. doi: 10.1038/s41467-020-18771-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou Z, et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015;33:408–414. doi: 10.1038/nbt.3096. [DOI] [PubMed] [Google Scholar]
- 72.Varshney RK, et al. Whole-genome resequencing of 292 pigeonpea accessions identifies genomic regions associated with domestication and agronomic traits. Nat. Genet. 2017;49:1082–1088. doi: 10.1038/ng.3872. [DOI] [PubMed] [Google Scholar]
- 73.Hübner S, et al. Sunflower pan-genome analysis shows that hybridization altered gene content and disease resistance. Nat. Plants. 2019;5:54–62. doi: 10.1038/s41477-018-0329-0. [DOI] [PubMed] [Google Scholar]
- 74.Zhang, Q. et al. The genetic architecture of floral traits in the woody plant Prunus mume. Nat. Commun.10.1038/s41467-018-04093-z (2018). [DOI] [PMC free article] [PubMed]
- 75.Huang L, et al. Whole genome re-sequencing reveals evolutionary patterns of sacred lotus (Nelumbo nucifera) J. Integr. Plant Biol. 2018;60:2–15. doi: 10.1111/jipb.12606. [DOI] [PubMed] [Google Scholar]
- 76.Sasaki R, et al. Functional and expressional analyses of PmDAM genes associated with endodormancy in Japanese apricot. Plant Physiol. 2011;157:485–497. doi: 10.1104/pp.111.181982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jimenez S, Lawtonrauh A, Reighard GL, Abbott AG, Bielenberg DG. Phylogenetic analysis and molecular evolution of the dormancy associated MADS-box genes from peach. BMC Plant Biol. 2009;9:81. doi: 10.1186/1471-2229-9-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao, K. et al. Comparative transcriptome reveals benzenoid biosynthesis regulation as inducer of floral scent in the woody plant Prunus mume. Front. Plant Sci.10.3389/fpls.2017.00319 (2017). [DOI] [PMC free article] [PubMed]
- 79.Fornara, F., De Montaigu, A. & Coupland, G. SnapShot: control of flowering in Arabidopsis. Cell10.1016/j.cell.2010.04.024 (2010). [DOI] [PubMed]
- 80.Tanaka Y, Ohmiya A. Seeing is believing: engineering anthocyanin and carotenoid biosynthetic pathways. Curr. Opin. Biotech. 2008;19:190–197. doi: 10.1016/j.copbio.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 81.Xu W, Dubos C, Lepiniec L. Transcriptional control of flavonoid biosynthesis by MYB–bHLH–WDR complexes. Trends Plant Sci. 2015;20:176–185. doi: 10.1016/j.tplants.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 82.Zhang Q, et al. Isolation and functional characterization of a R2R3-MYB regulator of Prunus mume anthocyanin biosynthetic pathway. Plant Cell Tiss. Org. 2017;131:417–429. doi: 10.1007/s11240-017-1294-4. [DOI] [PubMed] [Google Scholar]
- 83.Noda N, et al. Generation of blue chrysanthemums by anthocyanin B-ring hydroxylation and glucosylation and its coloration mechanism. Sci. Adv. 2017;3:e1602785. doi: 10.1126/sciadv.1602785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shen Y, et al. RrMYB5 and RrMYB10 regulated flavonoid biosynthesis plays a pivotal role in feedback loop responding to wounding and oxidation in Rosa rugosa. Plant Biotechnol. J. 2019;17:2078–2095. doi: 10.1111/pbi.13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Du H, et al. Methylation mediated by an anthocyanin, O -methyltransferase, is involved in purple flower coloration in. Paeonia. J. Exp. Bot. 2015;66:6563–6577. doi: 10.1093/jxb/erv365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gu Z, et al. Chalcone synthase is ubiquitinated and degraded via interactions with a RING-H2 protein in petals of Paeonia ‘He Xie’. J. Exp. Bot. 2019;70:4749–4762. doi: 10.1093/jxb/erz245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dudareva N, Pichersky E. Biochemical and molecular genetic aspects of floral scents. Plant Physiol. 2000;122:627–633. doi: 10.1104/pp.122.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bao F, et al. Expansion of PmBEAT genes in the Prunus mume genome induces characteristic floral scent production. Hortic. Res. 2019;6:24. doi: 10.1038/s41438-018-0104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chuang YC, Hung YC, Tsai WC, Chen WH, Chen HH. PbbHLH4 regulates floral monoterpene biosynthesis in Phalaenopsis orchids. J. Exp. Bot. 2018;69:4363–4377. doi: 10.1093/jxb/ery246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dhandapani S, Jin J, Sridhar V, Chua N, Jang I. CYP79D73 participates in biosynthesis of floral scent compound 2-Phenylethanol in Plumeria rubra. Plant Physiol. 2019;180:171–184. doi: 10.1104/pp.19.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Verde I, et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013;45:487–494. doi: 10.1038/ng.2586. [DOI] [PubMed] [Google Scholar]
- 92.Zhang J, et al. High-density genetic map construction and identification of a locus controlling weeping trait in an ornamental woody plant (Prunus mume Sieb. et Zucc) DNA Res. 2015;22:183–191. doi: 10.1093/dnares/dsv003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hollender CA, et al. Loss of a highly conserved sterile alpha motif domain gene (WEEP) results in pendulous branch growth in peach trees. Proc. Natl Acad. Sci. USA. 2018;115:201704515. doi: 10.1073/pnas.1704515115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhao K, et al. Crosstalk of PmCBFs and PmDAMs Based on the changes of phytohormones under seasonal cold stress in the stem of Prunus mume. Int. J. Mol. Sci. 2018;19:15. doi: 10.3390/ijms19020015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mcclure B, Franklintong VE. Gametophytic self-incompatibility: understanding the cellular mechanisms involved in “self” pollen tube inhibition. Planta. 2006;224:233–245. doi: 10.1007/s00425-006-0284-2. [DOI] [PubMed] [Google Scholar]
- 96.Donia A, Ghada B, Hend BT, Sana BM, Amel SH. Identification, evolutionary patterns and intragenic recombination of the gametophytic self incompatibility pollen gene (SFB) in tunisian Prunus species (Rosaceae) Plant Mol. Biol. Rep. 2016;34:339–352. doi: 10.1007/s11105-015-0922-6. [DOI] [Google Scholar]
- 97.Shi Y, Ding Y, Yang S. Molecular regulation of CBF signaling in cold acclimation. Trends Plant Sci. 2018;23:623–637. doi: 10.1016/j.tplants.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 98.Liu J, Shi Y, Yang S. Insights into the regulation of C‐repeat binding factors in plant cold signaling. J. Integr. Plant Biol. 2018;60:780–795. doi: 10.1111/jipb.12657. [DOI] [PubMed] [Google Scholar]
- 99.Bao F, et al. Overexpression of Prunus mume dehydrin genes in tobacco enhances tolerance to cold and drought. Front. Plant Sci. 2017;8:151. doi: 10.3389/fpls.2017.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Du D, et al. Genome-wide identification and analysis of late embryogenesis abundant (LEA) genes in Prunus mume. Mol. Biol. Rep. 2013;40:1937–1946. doi: 10.1007/s11033-012-2250-3. [DOI] [PubMed] [Google Scholar]
- 101.Lewin HA, et al. Earth BioGenome Project: sequencing life for the future of life. Proc. Natl Acad. Sci. USA. 2018;115:4325–4333. doi: 10.1073/pnas.1720115115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Twyford AD. The road to 10,000 plant genomes. Nat. Plants. 2018;4:312–313. doi: 10.1038/s41477-018-0165-2. [DOI] [PubMed] [Google Scholar]
- 103.Wilson, E. H. China, Mother of Gardens. (The Stratford Company, 1929).