

Abstract
Model‐informed drug development (MIDD) has a long and rich history in infectious diseases. This review describes foundational principles of translational anti‐infective pharmacology, including choice of appropriate measures of exposure and pharmacodynamic (PD) measures, patient subpopulations, and drug‐drug interactions. Examples are presented for state‐of‐the‐art, empiric, mechanistic, interdisciplinary, and real‐world evidence MIDD applications in the development of antibacterials (review of minimum inhibitory concentration‐based models, mechanism‐based pharmacokinetic/PD (PK/PD) models, PK/PD models of resistance, and immune response), antifungals, antivirals, drugs for the treatment of global health infectious diseases, and medical countermeasures. The degree of adoption of MIDD practices across the infectious diseases field is also summarized. The future application of MIDD in infectious diseases will progress along two planes; “depth” and “breadth” of MIDD methods. “MIDD depth” refers to deeper incorporation of the specific pathogen biology and intrinsic and acquired‐resistance mechanisms; host factors, such as immunologic response and infection site, to enable deeper interrogation of pharmacological impact on pathogen clearance; clinical outcome and emergence of resistance from a pathogen; and patient and population perspective. In particular, improved early assessment of the emergence of resistance potential will become a greater focus in MIDD, as this is poorly mitigated by current development approaches. “MIDD breadth” refers to greater adoption of model‐centered approaches to anti‐infective development. Specifically, this means how various MIDD approaches and translational tools can be integrated or connected in a systematic way that supports decision making by key stakeholders (sponsors, regulators, and payers) across the entire development pathway.
Model‐informed drug development (MIDD) has been defined as a “quantitative framework for prediction and extrapolation centered on knowledge and inference generated from integrated models of compound‐, mechanism‐, and disease‐level data aimed at improving the quality, efficiency, and cost‐effectiveness of decision making.” 1 We apply a non‐reductionist perspective of this definition. This means that model‐based methods and competencies to improve decision making in drug development are best sought from multiple disciplines, via diverse methods, and in an integrated manner. The full potential of MIDD requires recognition and adoption by stakeholders across the drug development value chain.
MIDD has been extensively used in infectious diseases, significantly assisted by the fact that pathogens can be meaningfully evaluated outside the patient. The types of MIDD approaches applied are inextricably linked to the types of translational tools used in infectious diseases research and development. In bacteriology, using the minimum inhibitory concentration (MIC), with its many flaws, might be regarded as the first example of exposure‐response and translational pharmacokinetics/pharmacodynamics (PKs/PDs), traced back to early efforts examining the antibacterial activity of penicillium by Alexander Fleming in 1944. 2
From the early 1980s, using an ever‐expanding array of preclinical infection approaches and clinical outcomes, regression models linking dose, PKs, and MIC to outcome have been increasingly applied during the development of antibacterial drugs. In vitro systems range from MIC testing to static concentration kill curve experiments to dynamic hollow‐fiber approaches. In vitro systems to establish PKs/PDs for infectious diseases outside of bacteriology have had more complex evolution aligned with the greater complexity of laboratory methods required for detection and quantification. For example, in virology, there are additional specific cell culture or reverse transcription‐polymerase chain reaction (RT‐PCR) systems required (e.g., flavivirus, influenza, and severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2)), and in parasitology, specific methods for staging a pathogen life‐cycle (e.g., Plasmodium (P.) falciparumand P. vivax) and quantification via microscopy or RT‐PCR are also necessary. In vivo models provide complementary information to in vitro systems when they can be reliably established. An extensive array of animal infection models has been used across the infectious disease spectra to explore different intervention modes (e.g., prophylaxis and treatment) and different end points (quantitative changes in pathogen load and signs of infection ranging from change in activity, weight, and temperature to moribund/mortality end points), including approaches to “humanize” such systems to better support translation (e.g., humanizing PK and immunosuppression). Controlled human infection models (CHIMs), where healthy volunteers are infected with a pathogen, represent an important version of the search for translational models, and are an important part of the translational armamentarium for diseases, such as malaria and influenza and infections with respiratory syncytial virus (RSV).
Empiric rubrics based on PK/PD target patterns (e.g., time‐free concentrations exceed MIC, time above MIC (T > MIC), and free area under the curve (fAUC)/MIC ratio), (sub)population PK/PD modeling, regression modeling, and Monte Carlo simulation for probability of target attainment, have all been applied for the development and breakpoint determination of antibacterials and antifungals. Assumptions on the transferability of empiric PK/PD MIDD (Figure 1 ) concepts across infectious diseases has likely both hastened progress in some areas and delayed progress in others. For example, correlating PK/PD indices with patient outcomes 3 from bacteriology has also been a consistent predictor of outcomes with antivirals in HIV minimum concentration/50% inhibitory concentration (C min/IC50), yet has not been particularly informative for influenza. 4 We speculate that extensive adoption of empiric PK/PD MIDD for antibacterials and over‐reliance on the MIC, primarily due to the ease of determining MIC and applying in dose decisions and the lack of clinical data to strongly validate mechanistic approaches, has delayed (relative to other therapeutic areas) adoption of mechanistic MIDD methods (Figure 1 ) such as quantitative systems pharmacology (QSP) approaches, which may define more effective genotypic‐targeted treatment paradigms for antibacterials informed by deeper mechanistic understanding. QSP approaches offer potential for increased precision over MIC measures and account for important factors, such as inoculum effect, development of resistance overtime, context of biofilm, and immune function. More emphasis on mechanism‐based PKs/PDs has become an important part of antimalarial drug development. In contrast to QSP, physiologically‐based PK (PBPK) modeling is a mechanistic MIDD approach that is broadly adopted in infectious diseases. 5 , 6 PBPK is routinely used to support drug development decisions, including lead candidate selection, dosing in pediatrics and other populations, hazard assessment in breast feeding and pregnancy, and the clinical context of potential drug‐drug interactions (DDIs). It is also increasingly used to translate anti‐infective PK/PD relationships to unbound drug exposures at the sites of infection, which is widely accepted as the most relevant biophase for translational pharmacology efforts in the infectious diseases arena.
Figure 1.
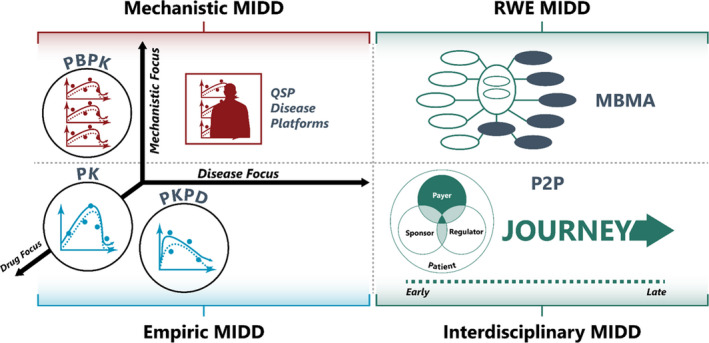
Schematic overview of MIDD approaches that have been applied to the development of anti‐infectives. MBMA, model‐based meta‐analysis; MIDD, model‐informed drug development; P2P, pharmacology to the payer; PBPK, physiologically‐based pharmacokinetics; PD, pharmacodynamics; PK, pharmacokinetics; QSP, quantitative systems pharmacology; RWE, real‐world evidence.
Uniquely, therapeutics targeting infectious diseases have potential for benefit and harm to be amplified beyond the individual patient. When effective, anti‐infectives can reduce infectiousness and spread of a disease in a community. However, their use can also result in the emergence and propagation of microbial resistance. Extending interdisciplinary MIDD (Figure 1 ) approaches to include stochastic or agent‐based epidemiological models has allowed new anti‐infective treatment strategies to be evaluated from a public health perspective and has applications, including stewardship, disease eradication campaigns, or management of outbreaks and pandemics. 7 , 8 , 9 Extending interdisciplinary MIDD approaches to health economic models enables earlier engagement with potential payers to collaborate on approaches to increase the commercial viability of anti‐infectives.
The world’s response to identifying therapeutic interventions for coronavirus disease 2019 (COVID‐19) has highlighted real‐world evidence (RWE) of clinical trial inefficiencies, economic damage, and lives lost. 10 Although there were heroic efforts by researchers and volunteers to rapidly develop a vaccine and design and conduct studies, there are calls for the clinical and quantitative pharmacology professions to do more 11 and drive broader adoption of MIDD to improve decision making across the drug development ecosystem. 11 , 12 , 13 , 14 , 15 Model‐based meta‐analysis (MBMA) approaches have been applied to explore emerging COVID‐19 clinical trials data for signal detection and may offer potential as an RWE MIDD tool (Figure 1 ).
This review showcases state‐of‐the‐art, empiric, mechanistic, interdisciplinary, and RWE MIDD applications in the development of antibacterials, antivirals, antifungals, drugs for the treatment of global health infectious diseases, and medical countermeasures. We also seek to describe the degree of adoption of MIDD practices across the field and future applications. This review is constrained to MIDD activities of lead candidates through to regulatory and payer approval and does not include MIDD as it pertains to drug discovery or precision dosing, such as therapeutic drug monitoring, application of virtual twin approaches using PBPK, 16 or dual individualization principles where PKs, pathogen sensitivity, and PKs/PDs are integrated to guide dosing decisions for individual patients. 17
GENERAL CONSIDERATIONS FOR MIDD IN INFECTIOUS DISEASES
Although infectious diseases are extremely diverse, there are foundational principles in translational pharmacology that repeat frequently in the application of MIDD principles to developing anti‐infectives. A nonexhaustive list of these factors is shown in Figure 2 . Important features to highlight include: (i) establishing the PK/PD relationship as close to the pathogen as possible at the site of infection, including the consideration of distribution and unbound exposures; (ii) the impact of comorbid conditions or other patient characteristics impacting PKs; (iii) how the pathogen end point is quantified, such as direct culture, 50% tissue culture infective dose, RT‐PCR, and counts of specific stage of pathogen life‐cycle; (iv) acknowledgement that the infecting pathogen is a heterogeneous population; and (v) the impact of the immune system on individual susceptibility and disease course, such as with acute respiratory distress syndrome and sepsis.
Figure 2.
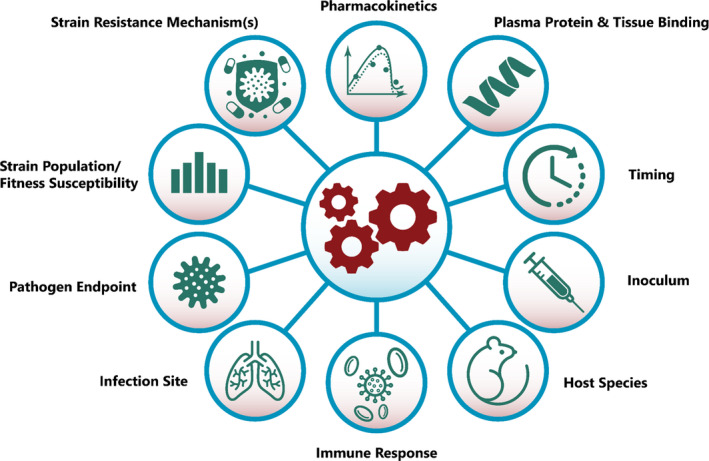
General considerations important for translational pharmacology in infectious diseases.
It is also important to highlight that although specific applications of MIDD and general level of adoption of approaches may differ in certain infectious diseases, similar MIDD approaches are almost uniformly and routinely applied to inform dosing of subpopulations (pediatric, pregnant, elderly, renally or hepatically impaired, or critically ill patients) as well as informing DDIs (see sections below). As such, the omission of specific case examples within each infectious disease does not imply lack of adoption of such MIDD approaches. MIDD adoption across infectious disease areas is summarized in Table 1 .
Table 1.
Relative importance and adoption of MIDD approaches across infectious diseases
| MIDD intensive translational models | Empiric MIDD | Mechanistic MIDD | RWE MIDD | Interdisciplinary MIDD | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| In vitro | In vivo | CHIM | Ph2 | Individual PK/PD/TD | Population PK/PD/TD | PTA MCS | PBPK | QSP | MBMA | Combined pharmacology epidemiology and HEOR | |
| Antibacterials | ++ | ++ | − | ++ | ++ | ++ | ++ | ++ | − | − | − |
| Antifungal | ++ | ++ | − | ++ | ++ | ++ | ++ | ++ | − | − | − |
| Global health | |||||||||||
| Tuberculosis | + | ++ | − | ++ | ++ | ++ | ++ | ++ | − | − | − |
| Malaria | + | + | ++ | ++ | ++ | ++ | + | ++ | +/− | − | − |
| Onchocerciasis | − | − | − | ++ | ++ | ++ | + | ++ | +/− | − | + |
| Antivirals | |||||||||||
| HIV | ++ | ++ | − | ++ | ++ | ++ | ++ | ++ | − | − | − |
| Hep C | ++ | ++ | − | ++ | ++ | ++ | + | ++ | − | − | − |
| RSV | + | ++ | ++ | ++ | ++ | ++ | + | ++ | − | − | − |
| Medical countermeasures | |||||||||||
| Smallpox | − | ++ | |||||||||
| Anthrax | + | ++ | − | − | ++ | ++ | ++ | ++ | − | − | − |
| Influenza | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | − | − | ++ |
| COVID | + | + | − | + | ++ | ++ | ++ | ++ | +/− | ++ | + |
The rankings represent a consensus view based on authors’ experience in application of MIDD approaches across the field of infectious diseases whereby ++, important to mainstream development; +, some use, but not broadly adopted or consider critical; +/−, starting to be used; −, limited/no experience.
CHIM, controlled human infection model; COVID, coronavirus disease; HEOR, health economics and outcomes research; Hep C, hepatitis C; MBMA, model‐based meta‐analyses; MCS, Monte Carlo simulation; MIDD, model‐informed drug development; PBPK, physiologically‐based PK; PD, pharmacodynamics; Ph 2, phase II; PK, pharmacokinetics; PTA, probability of target attainment; QSP, quantitative systems pharmacology; RSV, respiratory syncytial virus; RWE, real‐world evidence; TD, toxicodynamics.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Choice of appropriate exposure and pharmacodynamic measures
It was realized more than 50 years ago that protein binding has a big impact on anti‐infective activity and that only the free concentration, inferred from the unbound fraction in serum or plasma, is pharmacologically active. 18 Ignoring protein binding in dose estimation of daptomycin led to early termination of unsuccessful clinical trials. 19 Hence, it became clear that only unbound concentrations of anti‐infective agents should be considered in any dose estimation. 20 Because protein binding is frequently linear and concentration independent, this can often be achieved by simply multiplying the plasma or serum concentration with a constant fraction unbound. 21 There are exceptions, including where there is significant variability within the patient population or where nonlinearities in protein binding require concentration‐dependent adjustments. 22 In addition, there have been reports of rare exceptions where antibiotic activity was enhanced in the presence of proteins. 23 However, it is routine practice today to convert measured plasma or serum concentrations to their unbound values and report them as free peak concentrations (fCmax) or fAUC. 24
Furthermore, the site of infection can be extravascular, so it is important to determine the unbound concentration at the infection site. 25 , 26 Differences in tissue distribution are not adequately assessed with plasma concentrations only. 27 Drug concentrations in the epithelial lining fluid (ELF) can be estimated using bronchoalveolar lavage, and microdialysis has emerged in recent years as an experimental method to directly measure free drug concentrations in various tissues. 28 , 29 , 30 , 31 Microdialysis has been used extensively to measure unbound drug concentrations in large interstitial spaces, such as in subcutaneous tissues or muscle, as well as in specialized infection sites, such as lung cavities in patients with tuberculosis and in diabetic foot infections, 32 , 33 , 34 , 35 , 36 , 37 and direct measurement of infected biological fluids, such as cereberospinal fluid, peritoneal fluid, and ELF in pneumonia. A special warning should be given to the frequently applied collection of biopsies to measure “tissue concentrations.” The average concentrations usually do not reflect the actual free drug concentration at the infection site but represent an average number that should not be related to PD activity, and for xenobiotics that accumulate intracellularly homogenization destroys tissue architecture resulting in overestimation of unbound interstitial concentrations where many pathogens reside. This is a source of misinterpretation, particularly when anti‐infective drugs accumulate intracellularly in lysosomes at very high concentrations. 38 , 39 , 40
Analogous to using average target‐site concentrations, summary measures, such as fAUC or fCmax, can also distort the direct relationship between the concentration and drug action, as well as any time dependencies. This can have impact, particularly when defining optimal dosage for targets with a fast turnover, such as microbial pathogens. It is, therefore, of importance to consider if and how the use of a summary PK measurement is likely to influence the choice of dosing regimen when extrapolations to untested dosing regimens are made. In such situations, PK/PD models that enable characterization of the time course of PK and PD are preferred. PBPK methods that capture mathematically complex anatomic, physiological, physical, and chemical descriptions of the phenomena involved in drug absorption, distribution, metabolism, and excretion are also increasingly applied to estimate the full time course of drug concentrations at the infection site, such as tuberculous granuloma, 41 as well as within organelles or other sites in addition to or in lieu of microdialysis methods, and can be linked to PDs. 42
As described in further detail below, the fact that pathogens have a diverse range of in vitro sensitivity, as shown with MIC, 50% MIC (MIC50), and 90% MIC (MIC90) or IC50 and 90% IC (IC90), is a foundational observation in microbiology, parasitology, and virology. For decades, PK exposure measures have also been compared with in vitro and in vivo susceptibility measures in describing critical PK/PD indices (AUC/MIC, T > MIC, and Cmin/IC50) 43 , 44 and thresholds that are correlated with microbiological and clinical outcome and serve as breakpoints that guide approval and clinical use of a wide range of antimicrobials (an example with lefamulin is shown in Figure 3 ). 45 , 46 , 47 Even within the antimicrobial area, the observation that PK/PD indices and thresholds differ by drug class, 43 within drug class by infection site, 48 or by pathogen (gram positive vs gram negative), 49 highlights the increasing need for sophistication when applying PK/PD indices. 50 Consequently, for MIDD, there has been increasing focus on specific drug‐pathogen pharmacology and increasing use of QSP methods, 51 whereby more detailed PK/PD models are established that can explicitly describe both the time course of the concentrations of a specific drug and the time course of PD of a specific pathogen, thereby removing the limitations associated with using average PK measures.
Figure 3.
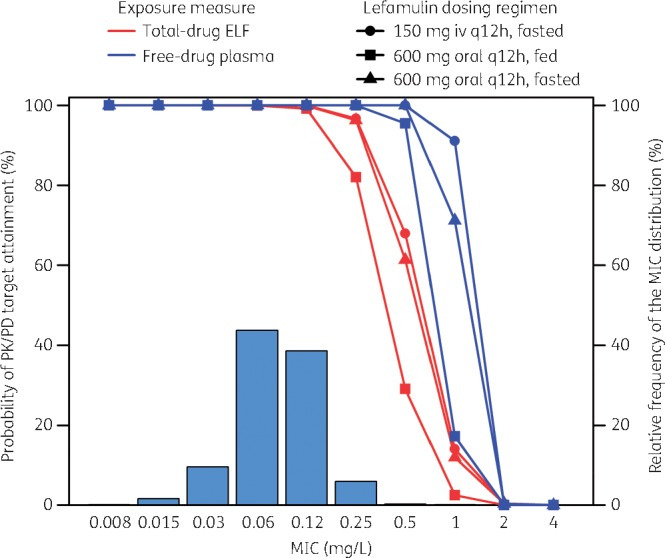
Percentage probabilities of PK/PD target attainment by MIC for lefamulin intravenous and oral dosing regimens based on the evaluation of the total‐drug ELF and free‐drug plasma AUC/MIC ratio targets associated with a 1 log10 CFU reduction from baseline for Streptococcus pneumoniae (S. pneumoniae), overlaid on the MIC distribution for S. pneumoniae. AUC, area under the curve; CFU, colony‐forming units; ELF, epithelial lining fluid; MIC, minimum inhibitory concentration; PD, pharmacodynamic; PK, pharmacokinetic. Reprinted with permission from Bhavnani et al. 47
Patient subpopulations
Specific patient characteristics can determine the need for dose adjustments in individual patient scenarios. 52 Although the systemic and infection‐site PK of many anti‐infectives can change in various patient groups (e.g., critically ill, burn, trauma, and patients wth obesity), changes in immune system function and severity of infection can also affect parameter estimates and these can be accounted for by MIDD. 53 Where necessary, infection site‐specific anti‐infective doses may be needed; for example, in the treatment of meningitis/ventriculitis (e.g., meropenem and ceftriaxone) 54 and ventilator‐associated pneumonia (e.g., ceftolozane and tazobactam). 55 Understanding the breadth of patient scenarios that will be encountered is of fundamental importance to provide sufficient data and guidance to enable clinicians to optimally use a drug after its licensed indication. The spectra of MIDD methods that can be applied to support established dosing in various subpopulations is extensive. 53
In pediatrics, for example, dosing recommendations may be derived from modeling and simulation based on a combination of PK modeling of adult data, knowledge of disease impact on PKs, and incorporation of covariates that influence pediatric PKs, such as body weight, age, or ontogeny of drug‐metabolizing enzymes and transporters. 56 , 57 Dosing for pediatric patients is determined based on full extrapolation of efficacy by identifying a dose that delivers exposures that are comparable to those in adults at the efficacious dose. This approach assumes that the PK/PD relationship is the same for adults and pediatrics. Population PK methods incorporating allometric scaling or PBPK approaches can be used to estimate the drug exposure in the pediatric population when data are sparse or not available. Having data from the patient population of interest is greatly preferred over estimations based on prior knowledge from other drugs or allometry alone. 58 Other patient factors incorporated into such MIDD methods to establish dosing include:
Weight—Obese and underweight patients are likely to also have altered drug clearance and volume of distribution
Changes in renal and/or hepatic function—Renally cleared drugs and/or metabolites, acute or chronic kidney diseases, and the acute emergence of augmented renal clearance can change drug clearance
Genetics—May affect metabolic clearance of some drugs where there is genetic variation of enzymes or transporters 59
Disease—Acute and chronic diseases can affect drug absorption and clearance
Extracorporeal treatments—Renal replacement therapies like hemodialysis; heart‐lung support devices, including extracorporeal membrane oxygenation; or other treatments, including plasma exchange, can all affect drug clearance and, in some cases, may cause drug adsorption to materials of the device 60 , 61
Infection site—Some infection pathologies may require higher doses because of impaired site penetration (e.g., meningitis). 54
Drug‐drug interactions
DDIs are of great clinical importance for anti‐infectives, as these drugs are often prescribed in patients with multiple comorbidities who are taking multiple drugs. Population PK methods with specific concomitant medications as covariates impacting clearance are common MIDD approaches applied to guide dosing in the setting of DDIs. PBPK is particularly useful in addressing the clinical relevance of potential DDIs that arise via induction or inhibition of several cytochrome P450 enzymes. For example, Siccardi and colleagues 62 developed a PBPK model to determine the dose adjustment that was required for artemether to overcome the DDI with efavirenz in HIV‐infected patients being treated for malaria. Similarly, a PBPK model was used to assess whether HIV‐positive women receiving efavirenz‐based antiretroviral therapy (ART) and levonorgestrel via contraceptive implants could attain high enough exposure of levonorgestrel to avoid unintended pregnancies. 63
ANTIBACTERIALS
Review of MIC‐based models
The MIC is defined as the lowest concentration of an antimicrobial agent that inhibits the visible in vitro growth of microorganisms. The MIC test determines the antimicrobial activity of a test agent against a specific bacterium after an incubation time of 16–20 hours. This measure of potency has been used as a PD target that is then related to the respective PK properties of the anti‐infective agent of interest. Three different PK/PD‐indices comparing these properties are most common: free T > MIC (fT > MIC), the ratio of fAUC over 24 hours at steady‐state and MIC (fAUC24/MIC), and the ratio of fCmax and MIC (fCmax/MIC). 24 These indices are easy to calculate and have become very popular. New compounds under development are classified into one of these three groups based on dose fractionation studies in animals. 43 A large number of different doses and dosing intervals are administered to infected animals, such as mice with thigh infections. The response observed at 24 hours after dosing is then compared with the respective exposure parameters, and the PK/PD index that shows the best correlation is selected to classify the compound. Beta‐lactam antibiotics usually fall into the fT > MIC class (also called time dependent), whereas quinolones and macrolides belong to the fAUC24/MIC class (also called concentration dependent). These terms are not very accurate because, obviously, the effect of all drugs depend on both the time course as well as the magnitude of their exposure. Few drugs seem to belong to the third class (fCmax/MIC), although there have been studies to show that may be the case for some aminoglycosides. 64 Once the PK/PD index type is chosen, the next step is to identify the target value that best correlates with clinical efficacy. Some of the recommended index values are consistent with the drug’s in vitro potency (quinolones have an fAUC24/MIC target of 25 for gram‐positive pathogens, which is equivalent to an average concentration of MIC), whereas others are not. Vancomycin is a prominent exception to in vitro‐in vivo correlations given it has an AUC/MIC target of 400, which is equivalent to 16 × MIC. Although there appears little reason why the required in vivo exposure has to be so much higher than that proposed from in vitro studies, various factors related to methods for MIC measurement (E‐test vs. broth microdilution method) and pathophysiological changes associated with infection appear to have greater effects for vancomycin than other drugs.
MIC‐based PK/PD indices have major limitations. One example is ciprofloxacin, which is a typical “AUC‐driven” drug, so AUC/MIC should be predictive of outcome. Figure 4 shows the results of two treatment schedules of ciprofloxacin for B. anthracis, with the same total daily dose given once or twice daily. 65 The fAUC24 is the same for both treatment schedules and, therefore, the fAUC24/MIC is also the same. However, both treatment schedules result in completely different outcomes; twice‐daily dosing worked very well without resistance development and once‐daily dosing failed. Hence, fAUC24/MIC was not predictive for outcome for this quinolone, likely related to the short half‐life in mice. 50 Similarly, it has been suggested that meropenem is a “time‐driven” drug, where fT > MIC is predictive. However, although the PK/PD indices provide an exposure parameter for dose calculations, they do not inform dosing frequency (usually determined empirically based on PKs) or treatment duration, because the PK/PD magnitude is based on response following 24 hours postdosing.
Figure 4.
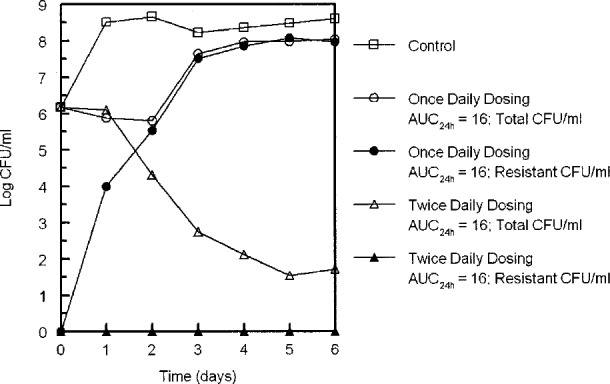
Effect of dose schedule on efficacy of ciprofloxacin against B. anthracis. A ciprofloxacin exposure of an AUC24h of mg h/liter (AUC24h/MIC 256) was given as two equal doses at 12‐hour intervals or as a single dose at 24‐hour intervals. AUC24h, area under the curve over 24 hours; CFU, colony‐forming units; MIC, minimum inhibitory concentration. Reprinted with permission from Deziel et al. 65
Review of mechanism‐based PK/PD models
To overcome the shortcomings of MIC‐based models, mechanism‐based models have been developed over the years that depend not only on a single bacteria measurement but follow the effect of the drug over time. The effect over time can be determined in in vitro and in vivo experiments. In vitro the microorganisms of interest are incubated at a starting inoculum, usually measured in colony‐forming units/mL, and then with the anti‐infective agent. A large number of these in vitro systems have been proposed. 66 They can be classified as those where the drug concentration is constant over the duration of the experiment (static models) or where the drug concentration is altered to mimic the expected in vivo concentration profile (dynamic models). 67 , 68 , 69 The advantage of this approach over simple MIC measurements is that it provides detailed information about the magnitude and time course of anti‐infective activity, which also requires a more sophisticated data analysis that will attempt to capture the observed kill curve profiles in mathematical models to allow for translation. The number of parameters used for this approach are larger than the single MIC value and, at a minimum, usually include a control growth rate constant and a killing rate, often expressed as a maximum kill rate and a potency value such as the half‐maximal effect concentration (EC50). Müller and colleagues compared both approaches and suggested that, for a given drug/bug combination, there is a quantitative link between MIC and EC50. 25 However, because MIC is defined solely by the absence of visible growth at a certain time point, it is not dependent on the maximum kill rate and, therefore, provides less detail. Different combinations of growth and kill rates can result in similar MIC values. Moreover, emergence of resistance during the incubation period is not accounted for. Over the years, the mathematical models to describe the results of these in vitro studies have matured and become more detailed and sophisticated. 70 Some of these models are shown in Figure 5 . This approach has clear advantages over the simpler MIC‐based PK/PD indices. For example, it has been shown that the choice of the best PK/PD index changes with changing PK parameters of the drug, 50 and the magnitude of the PK/PD index is sensitive to the experimental design, the MIC, and the PKs. Therefore, it may be preferable to perform simulations for dose selection based on an integrated PK/PD model rather than using a fixed PK/PD index target. 71 , 72 These integrated PK/PD models are much more powerful than simple MIC‐based PK/PD indices. 73 , 74 They also can be used for dose identification and optimization of anti‐infective drug combinations. 75 , 76 , 77 , 78 Another advantage of mechanism‐based PK/PD models are that they can be built on data from static experimental models and the resource‐demanding dynamic experiments can be saved for verification or minor adjustments given their predictive capacity. 79 It is anticipated that PK/PD models describing the time courses of bacterial growth and killing will be increasingly used in drug development.
Figure 5.
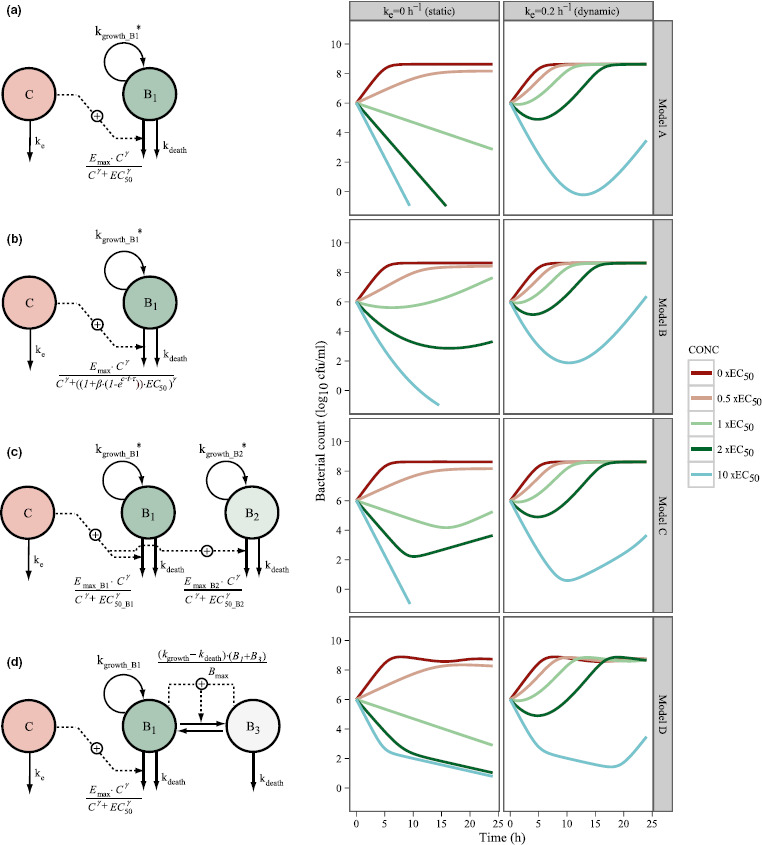
Schematic illustrations of four different PK/PD model structures (left panel) with typical predicted bacterial time‐kill curves following a static (middle panel) or dynamic (right panel) drug exposure. *The logistic growth was applied according to k growth (1 − (B 1 + B 2)/B max). C, drug compartment; B 1, compartment with drug‐sensitive bacteria; B 2, compartment with less drug‐sensitive bacteria; B 3, compartment with nongrowing, drug‐insensitive bacteria; B max, maximal bacterial count in the system; EC50, drug concentration producing 50% of E max; E max, maximum achievable effect; g, sigmoidicity factor; k e, first order drug elimination rate constant; k growth, rate constants for multiplication of bacteria; k death, rate constant for natural death of bacteria; PD, pharmacodynamic; PK, pharmacokinetic. Reprinted with permission from Nielsen and Friberg. 70
PK/PD models characterizing antibiotic resistance
Regrowth is most often observed in experimental systems (i.e., despite the constant antibiotic concentration during the experimental time), the bacterial number increases after an initial decline. This could be because: (i) the bacteria population in the starting inocula has a heterogeneous susceptibility distribution and the resistant bacteria are selected during the experiment, (ii) mutations occur during the experimental time, or (iii) there is a phenotypic switch during antibiotic pressure leading to the bacteria becoming less susceptible (i.e., the bacteria are adapting and become more tolerant to the antibiotic). In any situation, it is important to investigate the presence of different populations and resistance in these experiments, and to incorporate this into the PK/PD model. There are multiple variants of semimechanistic models that describe regrowth; most suggest the existence of subpopulations at start of the experiment, 80 mainly for aminoglycosides 71 and polymyxins, 72 where adaptive resistance has been experimentally demonstrated to be of relevance. It is less plausible that stochastic mutations, leading to reduced susceptibility, will also gain sufficient fitness to contribute to regrowth during the relatively short duration of time‐kill experiments. The clinical relevance of the regrowth observed in experimental systems remains unclear and could be viewed as a necessary part of a PK/PD model to accurately quantify the actual PK/PD relationship in the absence of resistance development.
Another phenomenon that is frequently apparent in experimental systems is an inoculum effect and a reduced log killing as the number of bacteria approaches a maximum count. In PK/PD models, these characteristics have been described by transition of bacteria to a resting and insusceptible state as the bacterial density increases in the system 81 or an effect of signal molecules that become activated as the bacterial number increases. 82 A limitation of logistic growth, which could be viewed to be a more empirical description of the asymptotic growth, is that additional functions are needed to describe the inoculum effect because the same log kill will be predicted regardless of the bacterial density. As more functions and parameters are needed to describe the same data in a model, there is a risk that the parameters become less precise, and the predictive capacity may not be better than a more parsimonious model structure.
Models of immune responses
In patients treated with antibiotics, the immune system also contributes to bacterial killing. However, the quantitative impact of neutrophils, a key component of the innate immune system, has rarely been studied or considered in the development of PK/PD models of antibiotics. The main reason for this is likely that neutrophils are less viable and functioning in the in vitro time‐kill systems. In the in vivo models used to define PK/PD indices, the mice are typically made neutropenic prior to the infection and, therefore, it could be perceived that both the in vitro and in vivo systems are reflecting a worst‐case situation (i.e., an immunocompromised patient). The immune system will, however, play an important role in the presence of antibiotic drug resistance, as it does not distinguish between a resistant bacterium and a susceptible one, and can, therefore, contribute to eradicate the residual bacteria. Based on in vitro and in vivo data, neutrophil‐mediated phagocytosis has only recently been quantitatively described over a wider range of conditions, 83 and interactions between immune system and antibiotics have rarely been quantified. 84
The bacterial load in patients is, for practical reasons, difficult to measure quantitatively for most bacterial infections. There may, however, be an opportunity through PK/PD modeling to make better use of the immune system response to understand the severity of the disease. 83 In addition to the dynamics of immune cells, the release of infection biomarkers and cytokines may guide the efficiency of an anti‐infective therapy. So far, such biomarkers are not considered in the development of antibiotics but, based on models describing the complex interplay between such variables in advanced animal infection models, 85 the information could be efficiently translated and linked to clinical situations to optimize therapy.
ANTIFUNGALS
For antifungals, the primary MIDD approaches applied to date have involved mainstream empiric PK/PD methods to establish dosing rationale, including the use of PK/PD indices, application of population PK models, and the use of Monte Carlo simulation for target attainment of PK/PD break points, and are canvassed below. Subpopulation and DDI dosing, although not featured below, are supported by PBPK methods, whereas QSP and other MIDD methods described are uncommon in the antifungal area.
The majority of the approaches applied for antibacterial MIDD have translated well for antifungals. 43 First, there are reproducible and regulatory approved methodologies that provide a measure of in vitro potency or MIC. 86 Second, multiple in vitro and small‐animal infection models have assessed the impact of antifungal concentration. Third, reliable organism burden end points exist for most fungal pathogens, including quantitative culture, quantitative PCR, other fungal biomarkers, and animal survival. In most instances, exposure‐response relationships utilizing these measures have been congruent. Most importantly, clinical antifungal PK/PD datasets have validated preclinical model end points for two of the most prevalent invasive fungal genera, Candida and less so for Aspergillus spp. A number of preclinical models have addressed other important fungal pathogens and await clinical data to explore their translational relevance.
Common PK/PD studies in the model include dose fractionation, time kill, and multi‐organism escalating drug exposure. 87 The exposure‐response analysis for each of these study designs helps to inform important aspects about a drug’s activity, including a PK/PD index that drives efficacy (e.g., AUC/MIC, Cmax/MIC, or T > MIC), the presence and extent of post‐antifungal effect, and, finally, the PK/PD target exposure linked to various therapeutic effects, such as net stasis, 1‐log kill, or half‐maximal effective dose (ED50), respectively. For both triazoles and echinocandins, dose‐fractionation and time‐kill studies have demonstrated that AUC/MIC is the PK/PD driver associated with efficacy and that there are moderate to prolonged post‐antifungal effects. 88 , 89 , 90 , 91 , 92 , 93 , 94 , 95 Conversely, for flucytosine and polyenes, T > MIC and Cmax/MIC have been the PK/PD‐linked drivers of efficacy. 96 , 97 Recent investigations with novel mode of action antifungals have also demonstrated the utility of these models. For the Gwt1 inhibitor, fosmanogepix, AUC/MIC has been the clearly linked dosing parameter, whereas for the orotomide antifungals, time above threshold is the driving index. 98 , 99 An additional finding of note is the congruence of the PK/PD driver within drug class. For example, the driver is consistently AUC/MIC within the triazole class. Furthermore, the PK/PD driver for a single drug will remain the same between organisms. For example, isavuconazole has the same PK/PD driver (AUC/MIC) for C. albicans, C. glabrata, and A. fumigatus. 93 , 100 Given the standardization and experience over time with these models, they are easily adaptable to new or emerging Candida pathogens, for example, C. auris, and to new antifungals. 98 , 99 , 101 , 102
Incorporation of multi‐isolate studies are designed to estimate the PK/PD target exposure (for the previously defined PK/PD driver) associated with a therapeutic effect (e.g., net stasis, 1‐log kill, or ED50). This is a critical element to capture inherent variability between organisms, variability in the model, and variability in clinical medicine to be able to robustly estimate PK/PD target exposures. There is extensive experience in preclinical PK/PD studies for IC. In studies examining the PK/PD relationship of triazoles, multiple laboratories have demonstrated that the target PK/PD exposure‐linked ED50 against C. albicans is a free drug AUC/MIC of ~ 25–50. 89 , 90 , 91 , 93 The triazole PK/PD target for other Candida species has not been studied as thoroughly, but limited data suggest that the PK/PD targets may be incrementally lower compared with C. albicans. 93 Echinocandins have also undergone robust preclinical PK/PD study, including multiple Candida species. Free drug AUC/MIC targets associated with net stasis against C. albicans were 10–20, but numerically 2‐fold to 3‐fold lower for other species (e.g., C. glabrata and C. parapsilosis). 88 , 92 , 94 , 103 , 104 In experimental models of invasive pulmonary aspergillosis, PK/PD target exposures for ED50 or net stasis for voriconazole, posaconazole, and isavuconazole were noted at free drug AUC/MIC exposures of 1.7–11. 100 , 105 , 106 A novel ex vivo lung epithelial cell model has also been explored for determination of the antifungal PK/PD target using a measure of Aspergillus cell wall. 107
Translating PK/PD targets to clinical medicine has been approached in one of three ways. First, if the PK/PD target exposure and the pathogen MIC distribution is known, the PK exposure can be determined, and, therefore, what dose may be necessary to optimize chances of clinical success. If the PK/PD target exposure is known and there are preclinical PK data for a defined dosing regimen, appropriate MIC thresholds (i.e., breakpoints) can be set where one would expect success or failure. Finally, if there are both a defined dosing regimen with known PK and pathogen MIC distribution, it is possible to determine the probability that clinical PK/PD target exposures, incorporating both PK variability and variability in MIC of the infecting pathogen, would meet or exceed the preclinical targets observed in the animal model. This last method is commonly done via Monte Carlo simulation with the output of probability of target attainment. 108
Clinical validation of PK/PD targets is a final and necessary step to harness the full translational potential of these studies. In the case of invasive candidiasis, there are multiple clinical datasets that have validated the predictive effect for patient outcome if meeting or exceeding the PK/PD targets originally identified in the animal model. 109 , 110 , 111 , 112 , 113 For example, PK/PD analysis of micafungin trials of invasive candidiasis identified an AUC/MIC target that was identical to the murine PK/PD model AUC/MIC exposure needed to achieve a net stasis end point. 92 , 114 In the absence of robust clinical data, the preclinical model results have been utilized as the predominant evidence for disease state approval. Specifically, PK/PD data from the rabbit Candida meningitis model were a predominant reason for approval of the echinocandin, micafungin, for neonatal meningoencephalitis. 115 This decision was based in large part on the perceived near impossibility of a clinical trial in this disease state.
Clinical PK/PD datasets for triazoles and invasive pulmonary aspergillosis that include organism data (e.g., MIC), dosing regimen, PK data, and outcome are more limited but do exist for voriconazole. 116 , 117 For both candidiasis and aspergillosis, the preclinical PK/PD results and congruent clinical datasets have been utilized in the development of susceptibility breakpoints for the Clinical Laboratory Standards Institute (CLSI) and the European Medicines Agency (EMA). Most recently, the results from preclinical investigations in murine invasive candidiasis and aspergillosis have been incorporated into dose selection decisions for clinical trials with novel agents. 98 , 99 , 118
In addition to established models for Candida and Aspergillus genera, there are a number of groups exploring PK/PD models for other clinically important species, including Cryptococcus and fungi from the zygomycetes group. 119 , 120 , 121 Further preclinical and clinical validation studies are needed for these infections, which are associated with exorbitant morbidity and mortality.
ANTIVIRALS
Hepatitis C
The development of drugs for the treatment of hepatitis C virus (HCV) infection has elucidated a number of important learnings for viral diseases focused on a particular site of infection, in this case the liver. Small‐molecule drugs, which have become a cornerstone of HCV treatment, include protease inhibitors, non‐nucleoside polymerase inhibitors, and nucleos(t)ide analogue inhibitors. The linkage of viral kinetic models and PK models to characterize antiviral PK/PD has been used extensively to optimize treatment regimens for HCV. 122
The PD predictors of clinical potency for both protease inhibitors and non‐nucleoside polymerase inhibitors were reported by Reddy and colleagues using a PK/viral kinetic model. 123 For both drug classes, increasing clinical potency was associated with increasing Cmin concentrations as a function of the in vitro protein‐shifted EC50. Interestingly, given the dependence of HCV protease inhibitors on the hepatic uptake transporters, organic anion transporting polypeptide (OATP) 1B1/1B3, for liver penetration, additional consideration of the liver‐to‐plasma ratio significantly enhanced the prediction of antiviral activity (Figure 6 ). Similar results further verified the importance of the liver‐to‐plasma ratio for HCV protease inhibitors. 124
Figure 6.
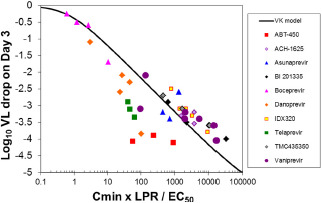
Predicted (curve) and observed (symbols) VL decrease from baseline on day three of PI monotherapy (log10 IU/mL) as a function of C min × LPR/EC50. ABT‐450 and two of the four narlaprevir doses were coadministered with ritonavir. C min, minimum concentration; EC50, drug concentration producing 50% of E max; E max, maximum achievable effect; LPR, liver to plasma ratio; PI, protease inhibitor; VL, viral load. Reprinted with permission from Reddy et al. 123
Although these PK/PD markers are based on early antiviral potency, they may not predict the ultimate clinical outcome of HCV therapy, which is a sustained virological response 12 weeks following the cessation of treatment. Both protease inhibitors and non‐nucleoside polymerase inhibitors also have a low barrier to resistance, making monotherapy insufficient for successful treatment. Therefore, combination therapy, including a drug that has a high barrier to resistance, is necessary to ultimately lead to successful clinical outcomes. 125 Modeling efforts have characterized the relationship between pre‐existing resistant mutations and HCV viral kinetics, providing further insights into treatment optimization. 124 , 126 , 127
The dependence of HCV protease inhibitors on hepatic uptake transporters to achieve adequate liver concentrations has had important implications for OATP‐related DDIs. Many HCV protease inhibitors, including simeprevir 128 and asunaprevir, 129 demonstrate significant increases in plasma concentrations as a result of co‐administration with OATP1B1/1B3 inhibitors. For example, asunaprevir plasma exposures increased 15‐fold to 21‐fold when co‐administered with an OATP inhibitor. 129 The clinical consequence of such an interaction is less drug taken up into hepatocytes, which may compromise clinical efficacy. In such circumstances, dose reductions due to the increase in plasma concentrations are not advised, as such reductions would be expected to further reduce hepatocyte concentrations.
The third class of drugs used in the treatment of HCV include nucleos(t)ide analogues. These agents are metabolized intracellularly to the active triphosphate form. The PD of these drugs are, therefore, driven by intracellular concentrations, making the utility of measuring plasma concentrations questionable. For certain nucleosides, when the intracellular half‐life is faster than the plasma half‐life, plasma concentrations may serve as a relevant matrix to drive the PD effect, although this is rare. In most cases, an appreciation for intracellular hepatocyte PK is required, which will determine the frequency of administration and ability to achieve therapeutic exposures. 130 A number of investigators have applied modeling approaches to understand the mechanisms of action of nucleos(t)ide analogues 131 and to optimize both dosing and treatment duration of nucleos(t)ides containing interferon‐free combination regimens. 132
Respiratory syncytial virus
Similar to the focus of HCV treatment on the liver as the site of action, the focus of RSV treatment is on the lung as the site of action. Mechanistic models have been leveraged to identify and achieve therapeutic plasma concentrations, which is particularly important given that the patient population includes children 2 years of age and below. In this population, model‐based approaches have been utilized to improve the ability to dose infected infants at therapeutic levels, minimizing the number of children who may be exposed to subtherapeutic doses. Definition of therapeutic concentrations may come from in vitro studies, animal studies, and/or human challenge studies, which provides a therapeutic target to achieve in children. Allometric scaling from adults can then be used to predict doses that will provide the anticipated therapeutic exposures in RSV‐infected infants. An example of this approach includes a dose‐finding study of the nucleoside analogue, lumicitabine (ALS‐8176), conducted in healthy adult volunteers infected with RSV. 133 The results of this study were utilized to construct a mathematical model linking intracellular triphosphate concentrations in lung epithelial cells to an RSV viral kinetic model (Figure 7 ). 134 This model developed in adults allowed for the allometric scaling of lumicitabine triphosphate intracellular lung concentrations to infants to design initial dosing regimens to be tested in infected patients who were anticipated to be within the therapeutic range. This example highlights the particular utility of model‐based approaches for pediatric development, in particular where the initial testing of a novel drug in an infant population can be challenging.
Figure 7.
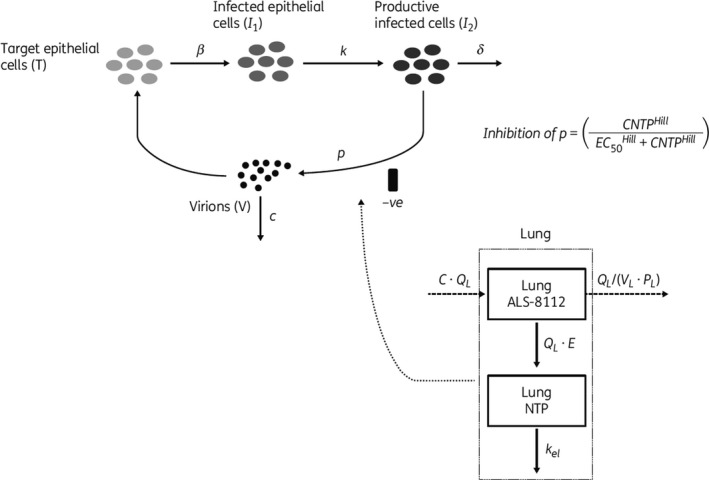
A structural representation of the model describing the kinetics of respiratory syncytial virus, and the antiviral inhibition effect of ALS‐8112 (lumicitabine)/NTP in lung tissue. β, infectivity rate constant; c, clearance of free virus; CNTPHill, predicted NTP concentration in the lung; EC50 Hill, concentration producing half maximal inhibition; δ, mortality rate constant; κ, latency “eclipse” period between the infection time of non‐productive epithelial cells and those actively producing virons; k el, elimination rate constant; NTP, 5′‐nucleoside triphosphate; p, ALS‐8112 (lumicitabine) inhibition of virus production; P L, lung to blood partition coefficient; Q L, epithelial lung blood perfusion. Reprinted with permission from Patel et al. 134
Human immunodeficiency virus
HIV is a chronic viral infection with a validated surrogate marker (plasma HIV‐ribonucleic acid [RNA], also known as viral load) used for predicting efficacy of antiretroviral agents that is highly dynamic. Reductions in viral load are highly predictive of clinical benefit and used as appropriate end points for clinical studies. The treatment goal for ART therapy is to suppress and maintain undetectable viral load. In addition to clinical benefit, viral suppression effectively removes the risk of sexually transmitting HIV to uninfected partners. As such, viral load is the primary monitoring parameter of effectiveness of ART.
A basic concept for maintenance of virologic control is the principle of inhibitory quotient (IQ), or an indicator of the ratio of antiviral exposure, usually adjusted for protein binding, to the susceptibility of the virus. There are various iterations of this concept depending upon which measures of exposure and susceptibility are used: IQ, genotypic IQ ratio (trough concentration/#genotypic mutations from genotype), phenotypic IQ ratio (trough concentration/IC50 from phenotype), and normalized IQ (PIQ from an individual/reference IQ). 135 IQ values in plasma and/or the intracellular milieu can be used to assess the dose‐response of HIV therapy, such that high IQ values indicate potential to inhibit HIV and minimize the development of resistance. 136 Typically, the IQ is derived from early phase I studies of individual antiretroviral agents as monotherapy in patients with HIV for a limited duration (e.g., 10 days of monotherapy). From this early assessment, target concentrations based on achievable IQ values can be used to inform dose selection decisions for chronic dosing in combination with other antiretroviral agents. Such short‐term phase I studies generally offer the only opportunity to evaluate IQ for individual antiretroviral agents, given that subsequent studies are usually conducted with combination regimens. As such, the relationship between exposure and antiviral outcomes in longer‐term studies needs to be considered within the context various other factors, such as the study design, the target population, the other drugs in the combination, and the pre‐existence/emergence of resistance mutations.
Given the clinical significance of nonadherence with antiretroviral treatment, relatively high IQ values are targeted in the development of ART agents to provide regimen “forgiveness,” such that missed or late doses should have limited impact. In the context of long‐acting parenteral administration, oral bridging regimens are needed to provide an oral alternative for interruptions (either planned or unplanned) with parenteral dosing using similar PK/IQ targets. PK modeling has been used to inform oral dosing regimens and strategies for switching between them and injectable formulations. As an example, Rossenu and colleagues used a population PK model of rilpivirine to inform strategies for managing dose interruptions in long‐acting intramuscular (i.m.) injection. 137 Modeling and simulation was performed to maintain rilpivirine trough concentration at or above target values based on the fifth percentile of observed values at week 4 after the initial rilpivirine long‐acting 900‐mg dose in the ATLAS/FLAIR studies. The analysis addressed questions such as monthly vs. every 4 weeks dosing, impact of delays in i.m. dosing, and bridging with oral rilpivirine to cover missed i.m. dosing. In addition, population PK for both plasma and intracellular PK of islatravir was used to select a phase III dose for both treatment‐naïve and highly treatment‐experienced patients from a flat dose‐response curve. Simulations of IQ values were evaluated to identify a regimen that would predict 100% of patients having full IQ activity (IQ > 5) by day 8 and > 90% of patients with a rare variant with reduced susceptibility having full IQ activity at steady‐state. 138 Similar analyses have been conducted for cabotegravir to model various dosing delays and predict trough concentrations against the fifth percentile trough concentration in phase III studies. Simulations of 1 to 2 months of oral bridging regimens were conducted to support dosing recommendations for oral bridging and to inform recommendations for delayed dosing. 139
One particular utility of exposure‐response modeling includes the potential to evaluate various tissues and reservoirs that have clinical importance for transmission and/or latency, such as vaginal and rectal tissue, breast milk, semen, genital tract fluid, lymphoid tissue, and intra‐cellular milieu. Because HIV transmission occurs primarily through mucosal fluids and early infection occurs within mucosal tissues, PK within these compartments is particularly important for pre‐exposure prophylaxis. For certain ART agents, intracellular PK of active metabolite is more critical compared with plasma PK, often indicating a more prolonged duration of activity, such as tenofovir alafenamide, with a very short plasma half‐life of ~ 0.5 hours, but tenofovir disoproxil fumarate‐diphosphate has terminal half‐life of up to 150 hours. 140 Likewise, islatravir is phosphorylated to an intracellular metabolite with a half‐life that is longer relative to parent drug in plasma (50–60 hours for parent vs. 130–210 hours for islatravir‐triphosphate). 141
As described in the DDI section, PBPK has been applied in the HIV setting to evaluate potential DDIs and optimize dosing. Some further examples include the use of PBPK to evaluate the interaction potential of cabotegravir both as a victim of uridine diphosphate glucuronosyltransferase‐based interactions 142 and as a perpetrator of OAT‐based interactions, 143 and its use to simulate the effect of rifampin in the PK of cabotegravir and rilpivirine long‐acting injectable using PK data from oral regimens. 144 Further uses of PBPK have included the optimization of fostemsavir dosing in pediatrics. Recently published data utilized a PBPK model to inform fostemsavir doses (600 mg twice daily (adult dose) for patients ≥ 35 kg and 400 mg twice daily for patients 20–35 kg, to maintain a balance in safety and efficacy and optimal PK sampling strategy for pediatric patients) 145 and characterization of the efficacy of broadly neutralizing antibodies for the treatment of HIV. 146
GLOBAL HEALTH
Onchocerciasis
Onchocerciasis or “river blindness” is a neglected tropical disease caused by the parasitic filarial worm Onchocerca volvulus and transmitted by black flies in riverine areas of Africa and South America. 147 Following an infective bite by the black fly, filaria, both macrofilaria and microfilaria ultimately develop in humans. This can cause intense dermal pruritus and, in some instances, transient or permanent visual impairment. For the last 30 years, the macrocyclic lactone, ivermectin, has been the mainstay of treatment of onchocerciasis. Ivermectin as a single dose produced a tolerable, rapid, and sustained reduction in microfilaria that permitted annual or semi‐annual mass drug administration, 147 , 148 most likely due to embryostasis in female macrofilaria. 149 A related macrocyclic lactone, moxidectin, was recently approved by the US Food and Drug Administration for onchocerciasis. 150 Elegant pharmacometric analyses of dose/exposure response demonstrated that the 8‐mg dose provided a superior sustained reduction in microfilaria at 12 months relative to lower doses and provided a more sustained reduction of microfilaria relative to ivermectin. 150 , 151 , 152
A model‐based meta‐analysis of the dynamic time‐related effects of ivermectin on microfilaria and macrofilaria has also been published. 153 The mathematical model developed from aggregated response‐time data describes the dynamics of macrofilarial fecundity and quantitative microfiladermia determined via skin snips. Application of the published model allows investigators and epidemiologists to explore various scenarios of the PD impact of degrees of macrofilaricidal activity, microfilaricidal activity, or some combination of the two on the probability of sustained reduction of parasites and probability of parasite elimination as a function of mass drug administration programs. Incorporation of PK/PD relationships of novel macrofilaricides and/or microfilaricides could facilitate clinical trial design and end point determinations and also the development of new drug combinations.
A stochastic, individual‐based model analogue of the EPIONCHO deterministic transmission model used for planning onchocerciasis control was able to capture skin microfilaria dynamics in response to treatment with ivermectin and moxidectin from the phase III clinical trial in Figure 8 . 154 , 155 The model was used to predict possible effects of treatment strategies on the control and, ultimately, elimination of onchocerciasis. 154 , 156 Even though the clinical program for moxidectin had only evaluated single‐dose regimens, the EPIONCHO model was combined with PK modeling to support retreatment recommendations for moxidectin during its regulatory approval process.
Figure 8.

Skin microfilaria dynamics for ivermectin (red) and moxidectin (blue) matching phase III clinical trial baseline endemicity. EPIONCHO model inclusion/exclusion criteria were used. Solid lines represent arithmetic mean microfilarial load and dashed lines the standard error calculated from the EPIONCHO‐IBM model trial cohort. Solid markers show the arithmetic mean microfilarial load data from the phase III trial with standard error bars. PIII, phase III. Reprinted with permission from Milton et al. 154
A full PBPK model for ivermectin that allows prediction of plasma and tissue drug concentrations was developed as part of a program of work to facilitate drug development for parasitic disease, including onchocerciasis. The model was able to capture observed ivermectin exposures in plasma, adipose tissue, and skin to within 1.3‐fold of the observed data. 157 In order for mass drug administration to be used safely, especially in situations of (potential) pregnancy and lactation, the ability to simulate possible exposures in the fetus and infant could be helpful. PBPK models that have been verified for maternal‐fetal transfer of drug can assist in the risk‐benefit assessments. PBPK models that have been verified for lactation can also help predict potential neonatal/infant exposures to drugs and weighed against findings in toxicology studies. The ultimate goal of MIDD and repurposing is the elimination of Onchocerca volvulus through test‐and‐treat and mass drug administration. PK/PD and epidemiologic modeling can help reduce empiricism to more efficiently achieve the elimination goal.
Tuberculosis
Accelerating the decline of the tuberculosis (TB) epidemic and improving outcomes for patients requires a pan‐TB regimen for treatment of both drug sensitive and drug resistant forms of TB with significant emphasis on treatment duration shortening. 158 The path toward the successful selection and design of new treatment‐shortening regimens is further complicated by the complexity of the TB lung lesion itself. The granulomatous lesions are highly dynamic and shaped by both host immune response elements and the pathogen itself 159 ; treatment regimen components that penetrate the granulomatous lesions and have an effect on a metabolically altered bacillus in the granulomas are emerging as a critical consideration. Experimental tools that support the development of TB drugs (in vitro hollow‐fiber system 160 , 161 ; acute, chronic, and relapsing mouse models; the Kramnik mouse model; and marmoset and rabbit models 162 , 163 ) provide important qualitative information on the agents’ potential for microbiologic activity and capacity for sterilization but generate limited PK/PD data. 164 , 165
In the absence of quantitative PK/PD translational data, regimens move into late‐phase trials with important uncertainties regarding the treatment‐shortening potential of a regimen and the potential to produce cure at rates equal to or better than that of the standard‐of‐care regimen. Advancements have been made toward the development of a universal preclinical to clinical mechanistic in silico PK/PD model for assessing TB drug combinations. These models have the potential to inform selection of the human‐equivalent dose and dosing schedule of a candidate drug used in a combination to then determine the likelihood of achieving treatment durations of 1 to 3 months with efficacy in both drug sensitive and drug resistant patients. Significant recent progress has been realized in the development of such systems pharmacology models (see below) to enable comparative efficacy evaluation and intended treatment‐shortening potential of novel regimens based on preclinical data and optimized translational simulations. 166 , 167 , 168 , 169 , 170 These model systems seek to integrate: (i) the quantification of the bacterial growth dynamics in the absence of treatment, (ii) the impact of immune system response in the absence and presence of treatment, (iii) the contribution of each drug (concentration–response relationship) to the observed total efficacy of drug combinations, and (iv) the interplay between disease pathology and drug response, including description of tissue penetration.
Recently, a significant advancement was made toward the aspirational goal of achieving a shorter treatment duration and serves as an exemplary integration of model‐informed PK/PD best practices in TB regimen selection and design. A clinical trial of rifapentine‐containing TB treatment‐shortening regimens (Study 31) investigated a 4‐month regimen that achieved noninferiority compared with the 6‐month standard‐of‐care regimen. MIDD played a pivotal role in the success of this trial. 170 , 171 , 172 , 173 First, a direct comparison in the chronic mouse model showed that the standard rifapentine dose (10 mg/kg) was superior to the standard rifampicin dose (10 mg/kg) in the mouse; however, this finding did not translate well to the earlier phase IIB study (Study 29 (https://clinicaltrials.gov/ct2/show/NCT00694629)), which evaluated rifapentine substitution for rifampin at this dose. Therefore, the choice of an optimal rifapentine dose was based on clinical PK/PD work from a separate dose‐ranging study. The design of Study 29 initially relied on weight‐based dosing, but after further investigation, a quantitative model‐based approach that considered drug exposure and response was applied to select a single flat dose that would be optimal for most of patients. However, phase IIB data has shown that for patients at higher risk for delayed culture conversion and consequently treatment failure (i.e., those with large cavities demonstrated on chest radiograph), a single drug substitution, even with a high dose of 1,200 mg, might not be sufficient. The study team ultimately selected the 1,200 mg rifapentine flat dose rather than the weight‐based dose, which led to variability in exposures in patients. Furthermore, it was later confirmed in a rabbit lesion model that the reason for suboptimal efficacy of rifapentine in patients with large cavitations was a result of poor lesion penetration 163 and further supported the inclusion of moxifloxacin in Study 31 due to its previously established favorable lesion penetration properties. For this reason, the integration of lesion penetration data and PK/PD modeling is integral to regimen selection and dose optimization in future TB studies.
The penetration of TB treatments into tissues, specifically into the lung lesions observed with the disease, has been investigated using PBPK models. A PBPK model was developed to predict tissue exposures of rifapentine, particularly in the lung, for different dosage regimens and to identify doses that could potentially cause efficacy or safety issues. 174 A more complex lung model consisting of a number of compartments, including pulmonary capillary blood, lung tissue, ELF, and alveolar air, was developed to investigate the lung exposures of a range of anti‐TB drugs. 175 Simulations of plasma and pulmonary concentrations of various drugs, including rifampin, isoniazid, and ethambutol, demonstrated reasonable recovery of observed exposures of each of the drugs in ELF, thus, providing confidence in a framework for predicting lung PKs of novel anti‐TB drugs. 175
Three large multicenter randomized phase III trials of 4‐month fluoroquinolone‐containing regimens (REMoxTB, Rifaquin, and OFLOTUB) failed individually to achieve noninferiority compared with the standard 6‐month control regimen. A pooled MBMA integrating the standardized patient‐level data from these phase III clinical trials concluded that these experimental regimens achieved 80% or better outcomes after 4 months, suggesting that addition of a fluoroquinolone could result in a shorter regimen; however, other adjustments and/or substitutions to the regimen would likely be required to achieve the regimen‐shortening goal. Additionally, this work suggested that a significant proportion of global TB burden may be eligible for shorter treatment courses if major determinants of unfavorable outcome are identified. The MBMA analyses helped identify the hard‐to‐treat patient phenotypes as potential keys to the development of novel, adaptive TB clinical trial designs to achieve the primary goal of durable cure for all patients. 176 Both Study 31 learnings and MBMA analysis speak toward the need to design regimens for the hardest‐to‐treat patients for whom high bacillary load and presence of cavitary lung lesions present the greatest challenge for treatment further exacerbated by the presence of malnutrition, comorbidities, and sex. Therefore, it is essential to ensure that features such as drug penetration and granulomatous lesion‐centric efficacy are integrated in the translational approaches for regimen selection.
Malaria
Malaria is a deadly but preventable and treatable disease caused by a single‐celled organism, the protozoan Plasmodium parasite. There are five different species of this parasite that can infect humans (Plasmodium (P.) falciparum, P. vivax, P. ovale, P. malariae, and P. knowlesi), each with a slightly nuanced pathogenesis. The multiple stages of the parasite’s life cycle, differences in pathology among the five species, and variable presence of drug resistance necessitate combination treatment for malaria. After an infection is confirmed via microscopy or a rapid diagnostic test, treatment depends on the species of malaria, severity of infection, regional resistance profiles, and patient‐specific factors, such as age, weight, pregnancy, breastfeeding, and/or immune status, and concomitant infections or known metabolic deficiencies. 177 Maintaining therapeutic concentrations of drug for a sufficient duration is necessary to eliminate the parasite from the human host. This attainment is dependent on selection and administration of the right combination, dose, and frequency of administration. A thorough understanding of PK/PD can efficiently facilitate optimal treatment regimen selection.
Some antimalarials have been introduced to vulnerable populations (i.e., children and pregnant women) at suboptimal doses. 178 Mathematical modeling of exposure‐response data has enabled revised treatment recommendations, including the use of weight bands for young children to avoid underdosing, 179 implementing loading doses to more rapidly achieve therapeutic concentrations, 180 supporting dose adjustments in children living in malaria‐endemic regions, 181 investigating drug exposures in malnourished populations, 182 and optimizing doses due to altered PKs in pregnant women. 183
Modeling and simulation have been increasingly used to support the discovery and development of new anti‐malarial drugs, 184 and in vitro assays have been optimized to be able to measure physicochemical properties, permeability, binding, and metabolic intrinsic clearance data for highly lipophilic compounds needed to meet ambitious single dosing and subpopulation target product profile requirements. 185 These renewed in vitro datasets populate PBPK models to simulate a range of scenarios, including early evaluation of feasibility of dose projections in pregnant women and children. A number of PBPK models that have been verified for maternal‐fetal transfer of drug are available and may assist in risk‐benefit assessments of drug treatment. 186 , 187 , 188 Furthermore, PBPK models that have been verified for lactation can also help predict potential neonatal/infant exposures to drugs and be weighed against findings in toxicology studies.
Other approaches and applications of MIDD for malaria therapeutics have been thoroughly discussed in the literature. 184 Allometric modeling approaches of preclinical data (e.g., from NOD/SCID/γcnull mouse studies) and population PK modeling data from first‐in‐human studies can be leveraged for the design of malaria CHIM studies. Malaria CHIM studies have been used as proof‐of‐concept studies for the development of malaria therapeutics, for the first and only malaria vaccine (RTS, S), and, more recently, for prophylactic malaria monoclonal antibodies. 189
The designs of malaria CHIM studies vary based on intervention. In CHIM studies testing malaria therapeutics, healthy volunteers are infected with malaria either via mosquito bite, injection of lyophilized/reconstituted sporozoites, or direct venous infusion of malaria‐infected erythrocytes. Drug concentrations sampled, frequently postdose, facilitate PK parameter estimates. Response is characterized by parasitemia detected serially by quantitative PCR over time. As such, infected volunteers typically remain asymptomatic or oligosymptomatic (few or minor symptoms) and data obtained postdose are rich for characterizing PDs (e.g., time to recrudescence or parasite reduction ratio). Real‐time population PK/PD modeling on a cohort‐by‐cohort basis can inform dose selection of subsequent cohorts to investigate doses that will be most informative of the exposure response. 190 The strain of parasite administered in malaria CHIM studies is most often 3D7 or NF54, both P. falciparum strains that are well‐characterized and drug sensitive. More recently, researchers have developed CHIM studies using an artemisinin‐resistant strain of malaria to evaluate the efficacy of therapeutics against drug‐resistant malaria. 191
The combination of data generated from malaria CHIM studies and mathematical modeling allows for a much more efficient development pathway to understand drug combinations. The current development paradigm for combination therapy includes two separate monotherapy, single‐ascending‐dose, malaria CHIM studies, a combination malaria CHIM study, and, typically, two to four phase II dose‐response studies. After the efficacy and safety characteristics of monotherapy are understood in these phase II trials, one recommended method for establishing optimal doses in combination has been factorial design. Factorial design studies are conducted in endemic areas with patients infected with malaria and include varying dose levels for each of the combination drugs that are factorially paired into combination study arms. This approach is fraught with high costs, difficulty recruiting patients, especially in areas of seasonal transmission, and the presence of confounding factors. Phase II studies produce critical information required to demonstrate the contribution of each drug, establish efficacy of the combination, and optimize the combination dose levels. Innovative alternative phase II designs, specific to malaria, can leverage data from malaria CHIM studies with modeling and simulation techniques to better understand dose/exposure response and enable efficient and optimal dose or doses of the combination for phase II dose‐response and phase III confirmatory clinical trials. 190 Adaptive and variable sample sizes could potentially provide opportunity for subsets of populations to be studied on an as‐needed basis without the confounding factors present in a field study, thereby increasing safety for patients and de‐risking antimalarial clinical trial design. Although field studies will always be necessary to demonstrate that the dose‐response relationship is similar between naturally infected patients living in endemic areas and subjects in a malaria CHIM study with no prior exposure, the use of modeling and simulation could potentially decrease the number and size of these confirmatory studies.
Finally, the harmonization of techniques and procedures for malaria CHIM studies for the development of therapeutics, similar to the World Health Organization’s recommendations for controlled human infection studies for vaccine development, would enable the industrialization of reproducible, consistent malaria CHIM studies. 192 The need to understand similarities and differences between malaria CHIM and field data is important for extrapolation and is currently being investigated; nonetheless, the malaria CHIM studies allow estimates of dose response in humans for both monotherapy and combination treatment to be efficiently estimated prior to field trials.
MEDICAL COUNTERMEASURES
Medical countermeasures (MCMs) include drugs and biologics (vaccines and antibodies) to be used in the event of a potential public health emergency occurring from terrorist attack with a biological, chemical, or radiological/nuclear material, or a naturally occurring emerging infectious disease. Efficacy must be demonstrated against the intended indication prior to approval of MCM products. Preferably, a dose range should be tested in at least two animal models, as recommended by the Animal Rule, 193 to characterize response and identify the fully efficacious doses. Clinical efficacy studies are neither ethical nor feasible to conduct, and the Animal Rule introduces the potential for drug approval based on efficacy in well‐controlled studies in animals, alongside appropriate human safety and PK information. 193 , 194 The natural history of pathophysiology in the animal models for efficacy and correlation to human disease needs to be demonstrated, as many infectious diseases can cause different infection patterns in animals than in humans. 195 Animal models with the ability to demonstrate a response to the countermeasure are key to allowing PK/PD relationships derived from the animal model to be extrapolated to humans. 196 , 197
MIDD has been applied to support approval of MCMs. Application of logistic regression analysis demonstrated a relationship between survival and bacteremia in the efficacy studies of obiltoxaximab against anthrax, allowing for the assignment of the variability observed in survival between studies to differences in bacteria load at the start of treatment. A Weibull cure rate model was applied to combined efficacy data from rabbits and monkeys to define the fully efficacious doses and determine impact of bacteremia on response. Higher bacteremia was associated with a decrease in survival, resulting in higher efficacious doses. The survival model was subsequently applied to simulate response of various dose levels, which could not be experimentally tested, and identify the dose above which there was no efficacy benefit, which was subsequently used to determine the human dose selection. 198 Dose‐response modeling was also applied to tecovirimat efficacy against smallpox. Efficacy data combined from multiple studies in the monkeypox model, which vary in dose, size, and treatment initiation day, was described using a nonparametric Kaplan–Meier analysis. A receiver operating characteristic analysis was used to identify cutoff points for dose and exposures that best correlated with survival. Analysis identified a dose of 3 mg/kg and associated exposures as the minimum efficacious dose. 199
To support identification of a clinical dose, PK is characterized in healthy and infected animals. PK modeling of nonclinical data allows determination of whether disease affects PK and the magnitude of the effect, which can be applied to humans because determination of PKs in patients may not be feasible. In addition, population PK analyses provide the flexibility to simulate PK profiles for the entire disease course and estimate PK parameters when PK sampling is limited, which is often the case in animal models, and serves as input for exposure‐response modeling. Efficacy is extrapolated to humans based on exposures and achievement of the target efficacious exposures derived from the animal model and dose/exposure‐response relationships as described above. Modeling and simulation approaches allow for effective dosing regimens in humans to be identified, which are expected to deliver exposures in humans that exceed the efficacious exposures in the animal models and produce a benefit similar to that observed in the animal models. 193 , 200
Animal models can facilitate rapid responses to emerging infection diseases by providing means of demonstrating proof of pharmacological plausibility for a new or approved therapy, deriving potential efficacious exposures, and providing initial data to support development decisions. More importantly, modeling and simulation approaches and translational medicine are critical for translating information to support clinical trials. Application of MIDD in response to emerging infectious diseases allows for the development of smarter trials and identification of treatment and optimal doses in cases where there is an urgency to get medicines to patients. MIDD was applied to develop a model‐informed analytical framework to test the impact of modulating specific steps in the SARS‐CoV‐2 viral dynamic model and to determine the most viable drugs and drug combinations for the treatment of COVID‐19. Simulations indicate that early treatment and drugs that promote infected cell death and reduction of virions per infections have the potential to provide the best response. 201 The use of viral kinetic models to select initial therapeutic combinations for COVID‐19 has been proposed as a starting point for a novel adaptive‐platform clinical trial that could deconvolute initial efficacious combinations into a parsimonious treatment combination for SARS‐CoV2. 202 Such a potential model‐informed combination repurposing strategy for COVID‐19 has been summarized in Figure 9 , and the novel approach has general applicability for rapid combination repurposing for emerging infectious diseases.
Figure 9.
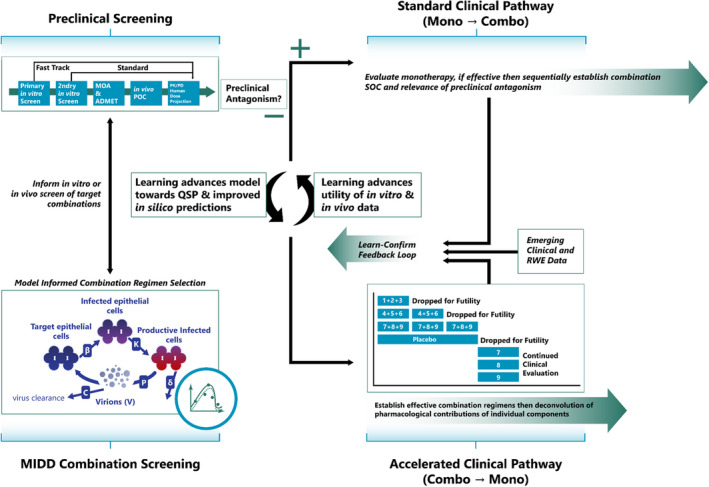
Model‐informed combination repurposing strategy for COVID‐19: candidate antiviral combinations are proposed through guidance from a QSP model (bottom left scheme). The model depicted in the figure is a simple viral cell cycle model adapted from Dodds et al. 201 The candidate combination regimens are then evaluated in preclinical screens for potential antagonism (upper left scheme). Preclinical evidence of antagonism triggers standard clinical evaluation of monotherapy first before combination assessment (upper right scheme). Absence of preclinical evidence of antagonism triggers an accelerated clinical evaluation of combinations using an adaptive deconvolution approach described by Rayner et al. 202 (bottom right scheme). In all circumstances, emerging clinical and RWE data is fed back to improve both preclinical screening methodologies and QSP models. Model‐based methods are implicitly applied throughout the model informed combination repurposing strategy including evaluation of emerging preclinical data, PK/PD, disease model and exposure‐response assessments from all clinical trials, and MBMA of emerging RWE clinical data. COVID‐19, coronavirus disease 2019; MBMA, model‐based meta‐analyses; MIDD, model‐informed drug development; MOA, mechanism of action; PK/PD, pharmacokinetic/pharmacodynamic; POC, proof‐of‐concept; QSP, quantitative systems pharmacology; RWE, real‐world evidence; SOC, standard of care; Mono, monotherapy; Combo, combination therapy; β, viral cell cycle rate constants of β, target cell infection; κ, conversion to productive cells; δ, productive cell death; p, virion release; c, virion clearance; 1, 2, 3… to 9 indicate individual therapeutics 1, 2, 3.. to 9; +, “in combination” (for example, 1 + 2 + 3 means triple combination of therapeutics 1, 2, and 3).
Application of MIDD is key for advancing the process of evaluating new COVID‐19 therapies and combinations and supports go/no‐go decisions for most promising candidates. 11 , 203 Given the volume of clinical trials initiated during COVID‐19, there is a particular need to be able to gather emerging RWE, including potential variability in treatment regimens, and use meta‐analysis methods to support signal detection of potential efficacious regimens. MBMAs are an extension of network meta‐analyses and incorporate pharmacological models, where possible, to support comparison of different dose regimens of a given therapeutic and regimen combinations in situations where they may not have been directly evaluated. 204 It is envisaged that MBMA will be an RWE MIDD method that will provide insights into emerging treatment options for COVID‐19 with the recent establishment of a precompetitive outcomes database. 205
In situations like pandemics, MIDD can support decisions on treatment selection and initiation to prevent spread and investigate economic impact. In a model developed for oseltamivir, 8 a framework integrating PK/PD from CHIM, epidemiology, and health economics models was developed to evaluate different pandemic scenarios and inform decisions on the best treatment to minimize spread and increase health economic impact (Figure 10 ). 4 , 7 The results of the modeling demonstrated that oseltamivir treatment in a flu pandemic could reduce the median number of individuals infected and prevent death, resulting in a decrease in health care use and cost savings. Such a modeling framework could be applied to optimize COVID‐19 treatment decisions in order to minimize spread and improve cost benefits.
Figure 10.
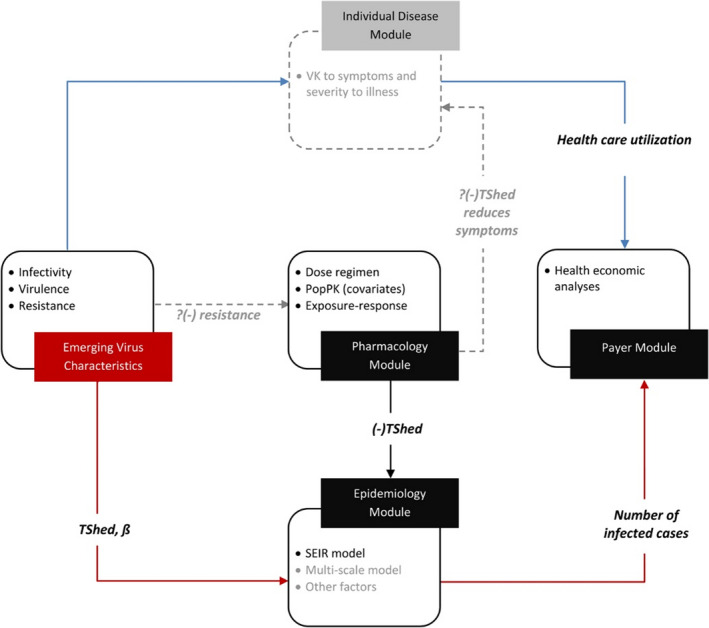
Integrated MIDD system, including multidisciplinary “modules” applied to optimizing oseltamivir use for pandemic planning. The solid lines indicate that adequate data exists to be able to create semi‐mechanistic links to each of adjacent “modules.” The dotted lines and light grey describe where significant unknowns remain and are not mature enough to have been incorporated into the current framework. ß, the rate of infectivity; MIDD, model‐informed drug development; PopPK, population pharmacokinetic; SEIR, susceptible‐exposed‐infected‐recovered; TShed, viral shedding; VK, viral kinetics. Reprinted with permission from Wu et al. 7
PROSPECTUS AND FUTURE
Anti‐infectives have a long history in leading the charge for PK/PD innovation and advancing MIDD methods in accelerating medicines to patients. This review demonstrates that the application of empiric PK/PD methods is entrenched in anti‐infective drug development; the adoption of PBPK for DDI and special populations across the field is extensive and, in specific infectious diseases, translational systems, such as the hollow‐fiber model for TB and CHIM for malaria, are central to MIDD pathways. The interdisciplinary linkage of models of pharmacology to epidemiology and health economics represent more recent MIDD innovations from the infectious diseases field that have assisted in shifting the regulatory and commercial value conversation of anti‐infectives from the individual patient journey to one including a public health perspective.
MIDD is broadly adopted across many regulatory guidances. The inclusion of MIDD‐related performance goals in the latest reauthorized Prescription Drug User Fee Act (PDUFA VI) reflects the acceptance and commitment surrounding the use of quantitative approaches to advance effective drug development and regulatory evaluation. 206 Population PK, exposure‐response, and MIC‐based PK/PD are standard approaches where there is an expectation of evaluation. PBPK modeling is generally accepted for the assessment of DDIs by cytochrome P450 inhibition mechanisms, whereas QSP is less often applied with fewer examples of QSP being leveraged to guide regulatory decisions (about 10 in 2018). 207 Most examples involving the use of QSP focus on key decisions in drug discovery and development, such as hypothesis generation, compound prioritization, translational biomarker development, and nonclinical and clinical trial design.
Over the last 2 decades, however, the pace of adoption of novel MIDD approaches, like QSP and MBMA, have been slower in the anti‐infectives arena vs. high‐growth areas, such as immunology and particularly immuno‐oncology. There are two key reasons for this. First, macroeconomic factors, including reduction of investment in infectious diseases research and development, seeing MIDD talent, resources, and innovation pivot to more commercially viable areas. Second, the entrenched application of pragmatic microbiological methods, like MIC for antibacterials in clinical practice and in‐turn microbiological breakpoint determination that dictates use, also create a challenge for new practice paradigms, such as genotypic‐informed treatment selection (analogous to genotypic mutations from genotype ratio in HIV), that may arise through application of QSP innovations.
We predict the future application of MIDD in infectious diseases will progress along two planes; “depth” and “breadth” of MIDD methods. MIDD depth refers to deeper incorporation of the specific pathogen biology and intrinsic‐resistance and acquired‐resistance mechanisms; host factors, such as immunologic response and infection site, to enable deeper interrogation of pharmacological impact on pathogen clearance, clinical outcome, and emergence of resistance from a pathogen, patient, and population perspective. Improved early assessment of emergence of resistance potential, in particular, will become a greater focus in MIDD approaches, as it represents a key risk that is poorly mitigated by current development approaches.
MIDD breadth refers to greater adoption of a model‐centered approach to anti‐infective development, and, specifically, how various MIDD approaches and translational tools can be integrated or connected together in a systematic way that supports decision making by key stakeholders (sponsor, regulator, and payer) across the entire development pathway. Specifically, MIDD breadth includes interconnectedness of MIDD models like PBPK and QSP to predict pharmacological response at infection site; aligning MIDD translational tools, including in vitro systems like hollow‐fiber, animal models, and CHIM, to inform and validate such model frameworks, incorporating emerging RWE clinical and microbiologic data to support model development, and extending epidemiological and health economic models to explore population and economic impact of forecasted outcomes, like probability of emergence of resistance. We also anticipate the application of deep learning and artificial intelligence approaches to drive efficiencies in and insights from such model frameworks and informatively influence MIDD practices in infectious diseases. 208
We forecast that adoption of MIDD depth and breadth of the sophisticated approaches described above will be driven by innovative developers in the field, fueled primarily by the demonstration of increasing effectiveness in internal decision making and supporting interactions with regulators and payers on aspects such as dose justification and economic impact supporting pricing discussions. Such innovative practices will be more easily adopted in emerging infectious diseases areas or global health where there are less established clinical practices. In contrast, in the setting of antimicrobials and antifungals, MIC and breakpoint‐guided/informed decisions will continue to be a clinical practice paradigm that must also be catered to in an MIDD program until the clinical impact of alternative approaches becomes accepted. These issues highlight the tight interdependency and competing forces between clinical practice and drug development innovation. We forecast that as drug development innovations permeate established clinical practice areas in infectious diseases, we will gradually see increased adoption of advanced model‐informed therapeutic(s) selection and precision dosing approaches informed by detailed host and pathogen characteristics, including increasing genotypically guided approaches analogous to tumor genotyping in immuno‐oncology.
In conclusion, this review outlines the current state of the art of MIDD for anti‐infectives and future considerations. This paper is dedicated to our co‐author, friend, colleague, and mentor, Professor Hartmut Derendorf, who passed away unexpectedly as we were concluding the writing of this review. We believe it is appropriate that he has the final word that captures the spirit of the future evolution of MIDD of anti‐infectives not being constrained to current dogma, “MIC is poison for the mind—we need to be much smarter in the way we approach dosing of anti‐infectives.”
FUNDING
No funding was received for this work.
CONFLICT OF INTEREST
C.R.R., P.F.S., T.M.P., V.S., M.J.S., D.W., and K.R.Y. are employees of Certara, a consulting firm in integrated drug development, and have directly consulted with a variety of not‐for‐profit global health organizations, biotechnology and pharmaceutical companies, and governments with an interest in infectious diseases. J.A.R. has been on consultancy/advisory boards for Pfizer, Sandoz, Wolters Kluwer, Merck, QPEX, Discuva, Accelerate Diagnostics, Bayer, and Biomerieux; has received speaking fees from Merck, Biomerieux, Pfizer, and Cipla; and has received industry grants from Merck, The Medicines Company, Cardeas Pharma, Biomerieux, QPEX, and Pfizer. D.A. has received consultation fees and grant support from Amplyx, Cidara, Fedora, Merck, Roche, Workhart, Matrina, sFunga, and Astellas. All other authors declared no competing interests for this work.
Acknowledgments
The authors would like to thank Professor Hartmut Derendorf for his contributions to the field of MIDD for anti‐infectives by generating knowledge to guide the development of anti‐infectives and by training professionals to consider the application of MIDD. Professor Derendorf’s love for the field knew no barriers, as he collaborated with colleagues, pharmaceutical companies, and universities across the globe. Professor Derendorf will be greatly missed. His contributions will always be remembered by his publications and by the colleagues he trained. We also gratefully acknowledge the help of Dr. Holly Capasso‐Harris with referencing, manuscript and figure preparation, and submission of this paper.
References
- 1. Marshall, S.F. et al. Good practices in model‐informed drug discovery and development: practice, application, and documentation. CPT Pharmacometrics Syst. Pharmacol. 5, 93–122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wheat, P.F. History and development of antimicrobial susceptibility testing methodology. J. Antimicrob. Chemother. 48(suppl 1), 1–4 (2001). [DOI] [PubMed] [Google Scholar]
- 3. McAleenan, A. et al. Methodological features of clinical pharmacokinetic‐pharmacodynamic studies of antibacterials and antifungals: a systematic review. J. Antimicrob. Chemother. 75, 1374–1389 (2020). [DOI] [PubMed] [Google Scholar]
- 4. Rayner, C.R. et al. Pharmacokinetic‐pharmacodynamic determinants of oseltamivir efficacy using data from phase 2 inoculation studies. Antimicrob. Agents Chemother. 57, 3478–3487 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jones, H. & Rowland‐Yeo, K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics Syst. Pharmacol. 2, e63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jamei, M. Where do PBPK models stand in pharmacometrics and systems pharmacology? CPT Pharmacometrics Syst. Pharmacol. 9, 75–76 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu, D.B.C. et al. Cost‐utility analysis of antiviral use under pandemic influenza using a novel approach ‐ linking pharmacology, epidemiology and heath economics. Epidemiol. Infect. 146, 496–507 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kamal, M.A. et al. Interdisciplinary pharmacometrics linking oseltamivir pharmacology, influenza epidemiology and health economics to inform antiviral use in pandemics. Br. J. Clin. Pharmacol. 83, 1580–1594 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fidler, M. , Murillo, M. , Rayner, C. , Kamal, M.A. & Smith, P. Modeling the spread of pandemic influenza in the United States: impact of antiviral interventions, pharmacology, and resistance . American Conference on Pharmacometrics, October 2014; Las Vegas, Nevada.
- 10. Thorlund, K. , Dron, L. , Park, J. , Hsu, G. , Forrest, J.I. & Mills, E.J. A real‐time dashboard of clinical trials for COVID‐19. Lancet Digit. Health 2, e286–e287 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hartman, D. , Kern, S. , Brown, F. , Minton, S.K. & Rayner, C.R. Time to step up: a call to action for the clinical and quantitative pharmacology community to accelerate therapeutics for COVID‐19. Clin. Transl. Sci. 13, 646–648 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rayner, C.R. , Smith, P.F. , Hershberger, K. & Wesche, D. Optimizing COVID‐19 candidate therapeutics: thinking without borders. Clin. Transl. Sci. 13, 830–834 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baker, E.H. , Gnjidic, D. , Kirkpatrick, C.M.J. , Pirmohamed, M. , Wright, D.F.B. & Zecharia, A.Y. A call for the appropriate application of clinical pharmacological principles in the search for safe and efficacious COVID‐19 (SARS‐COV‐2) treatments. Br. J. Clin. Pharmacol. https://doi.org/ 10.1111/bcp.14416. [e‐pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 14. Krishnaswami, S. et al. MID3: mission impossible or model‐informed drug discovery and development? Point‐counterpoint discussions on key challenges. Clin. Pharmacol. Ther. 107, 762–772 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van der Graaf, P.H. & Giacomini, K.M. COVID‐19: a defining moment for clinical pharmacology? Clin. Pharmacol. Ther. 108, 11–15 (2020). [DOI] [PubMed] [Google Scholar]
- 16. Polasek, T.M. & Rostami‐Hodjegan, A. Virtual twins: understanding the data required for model‐informed precision dosing. Clin. Pharmacol. Ther. 107, 742–745 (2020). [DOI] [PubMed] [Google Scholar]
- 17. Tillotson, G. PK‐PD compass, a novel computerized decision support system. Lancet Infect. Dis. 17, 908 (2017). [Google Scholar]
- 18. Kunin, C.M. , Craig, W.A. , Kornguth, M. & Monson, R. Influence of binding on the pharmacologic activity of antibiotics. Ann. N. Y. Acad. Sci. 226, 214–224 (1973). [DOI] [PubMed] [Google Scholar]
- 19. Lee, B.L. , Sachdeva, M. & Chambers, H.F. Effect of protein binding of daptomycin on MIC and antibacterial activity. Antimicrob. Agents Chemother. 35, 2505–2508 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zeitlinger, M.A. et al. Protein binding: do we ever learn? Antimicrob. Agents Chemother. 55, 3067–3074 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gonzalez, D. , Schmidt, S. & Derendorf, H. Importance of relating efficacy measures to unbound drug concentrations for anti‐infective agents. Clin. Microbiol. Rev. 26, 274–288 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deitchman, A.N. , Singh, R.S.P. & Derendorf, H. Nonlinear protein binding: not what you think. J. Pharm. Sci. 107, 1754–1760 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ghobrial, O. , Derendorf, H. & Hillman, J.D. Human serum binding and its effect on the pharmacodynamics of the lantibiotic MU1140. Eur. J. Pharm. Sci. 41, 658–664 (2010). [DOI] [PubMed] [Google Scholar]
- 24. Mouton, J.W. , Dudley, M.N. , Cars, O. , Derendorf, H. & Drusano, G.L. Standardization of pharmacokinetic/pharmacodynamic (PK/PD) terminology for anti‐infective drugs: an update. J. Antimicrob. Chemother. 55, 601–607 (2005). [DOI] [PubMed] [Google Scholar]
- 25. Müller, M. , Delapena, A. & Derendorf, H. Issues in pharmacokinetics and pharmacodynamics of anti‐infective agents: distribution in tissue. Antimicrob. Agents Chemother. 48, 1441–1453 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mouton, J.W. , Theuretzbacher, U. , Craig, W.A. , Tulkens, P.M. , Derendorf, H. & Cars, O. Tissue concentrations: do we ever learn? J. Antimicrob. Chemother. 61, 235–237 (2008). [DOI] [PubMed] [Google Scholar]
- 27. Estes, K.S. & Derendorf, H. Comparison of the pharmacokinetic properties of vancomycin, linezolid, tigecyclin, and daptomycin. Eur. J. Med. Res. 15, 533–543 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brunner, M. , Derendorf, H. & Müller, M. Microdialysis for in vivo pharmacokinetic/pharmacodynamic characterization of anti‐infective drugs. Curr. Opin. Pharmacol. 5, 495–499 (2005). [DOI] [PubMed] [Google Scholar]
- 29. de la Peña, A. , Liu, P. & Derendorf, H. Microdialysis in peripheral tissues. Adv. Drug Deliv. Rev. 45, 189–216 (2000). [DOI] [PubMed] [Google Scholar]
- 30. Deitchman, A.N. , Heinrichs, M.T. , Khaowroongrueng, V. , Jadhav, S.B. & Derendorf, H. Utility of microdialysis in infectious disease drug development and dose optimization. AAPS J. 19, 334–342 (2017). [DOI] [PubMed] [Google Scholar]
- 31. Li, Y. , Peris, J. , Zhong, L. & Derendorf, H. Microdialysis as a tool in local pharmacodynamics. AAPS J. 8, E222–E235 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kempker, R.R. et al. Cavitary penetration of levofloxacin among patients with multidrug‐resistant tuberculosis. Antimicrob. Agents Chemother. 59, 3149–3155 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kempker, R.R. et al. A comparison of linezolid lung tissue concentrations among patients with drug‐resistant tuberculosis. Eur. Respir. J. 51, 1702166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kempker, R.R. et al. Lung tissue concentrations of pyrazinamide among patients with drug‐resistant pulmonary tuberculosis. Antimicrob. Agents Chemother. 61, e00226‐00217 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ray, A. , Malin, D. , Nicolau, D.P. & Wiskirchen, D.E. Antibiotic tissue penetration in diabetic foot infections: a review of the microdialysis literature and needs for future research. J. Am. Podiatr. Med. Assoc. 105, 520–531 (2015). [DOI] [PubMed] [Google Scholar]
- 36. So, W. , Kuti, J.L. , Shepard, A. , Nugent, J. & Nicolau, D.P. Tissue penetration and exposure of cefepime in patients with diabetic foot infections. Int. J. Antimicrob. Agents 47, 247–248 (2016). [DOI] [PubMed] [Google Scholar]
- 37. Wiskirchen, D.E. , Shepard, A. , Kuti, J.L. & Nicolau, D.P. Determination of tissue penetration and pharmacokinetics of linezolid in patients with diabetic foot infections using in vivo microdialysis. Antimicrob. Agents Chemother. 55, 4170–4175 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Derendorf, H. Excessive lysosomal ion‐trapping of hydroxychloroquine and azithromycin. Int. J. Antimicrob. Agents 55, 106007 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zheng, S. , Matzneller, P. , Zeitlinger, M. & Schmidt, S. Development of a population pharmacokinetic model characterizing the tissue distribution of azithromycin in healthy subjects. Antimicrob. Agents Chemother. 58, 6675–6684 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matzneller, P. et al. Blood, tissue, and intracellular concentrations of azithromycin during and after end of therapy. Antimicrob. Agents Chemother. 57, 1736–1742 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pienaar, E. et al. Comparing efficacies of moxifloxacin, levofloxacin and gatifloxacin in tuberculosis granulomas using a multi‐scale systems pharmacology approach. PLoS Comput. Biol. 13, e1005650 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rowland Yeo, K. et al. Impact of disease on plasma and lung exposure of chloroquine, hydroxychloroquine and azithromycin: application of PBPK modeling. Clin. Pharmacol. Ther. 108, 976–984 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Craig, W.A. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin. Infect. Dis. 26, 1–10; quiz 11–12 (1998). [DOI] [PubMed] [Google Scholar]
- 44. Mouton, J.W. et al. Conserving antibiotics for the future: new ways to use old and new drugs from a pharmacokinetic and pharmacodynamic perspective. Drug Resist. Updat. 14, 107–117 (2011). [DOI] [PubMed] [Google Scholar]
- 45. Mouton, J.W. et al. The role of pharmacokinetics/pharmacodynamics in setting clinical MIC breakpoints: the EUCAST approach. Clin. Microbiol. Infect. 18, E37–E45 (2012). [DOI] [PubMed] [Google Scholar]
- 46. Asín, E. , Isla, A. , Canut, A. & Rodriguez Gascon, A. Comparison of antimicrobial pharmacokinetic/pharmacodynamic breakpoints with EUCAST and CLSI clinical breakpoints for gram‐positive bacteria. Int. J. Antimicrob. Agents 40, 313–322 (2012). [DOI] [PubMed] [Google Scholar]
- 47. Bhavnani, S.M. et al. Pharmacokinetic/pharmacodynamic target attainment analyses to support intravenous and oral lefamulin dose selection for the treatment of patients with community‐acquired bacterial pneumonia. J. Antimicrob. Chemother. 74, iii35–iii41 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rayner, C.R. , Forrest, A. , Meagher, A.K. , Birmingham, M.C. & Schentag, J.J. Clinical pharmacodynamics of linezolid in seriously ill patients treated in a compassionate use programme. Clin. Pharmacokinet. 42, 1411–1423 (2003). [DOI] [PubMed] [Google Scholar]
- 49. Ambrose, P.G. , Bhavnani, S.M. & Owens, R.C. Jr Clinical pharmacodynamics of quinolones. Infect. Dis. Clin. North Am. 17, 529–543 (2003). [DOI] [PubMed] [Google Scholar]
- 50. Kristoffersson, A.N. et al. Simulation‐based evaluation of PK/PD indices for meropenem across patient groups and experimental designs. Pharm. Res. 33, 1115–1125 (2016). [DOI] [PubMed] [Google Scholar]
- 51. Eckhardt, M. , Hultquist, J.F. , Kaake, R.M. , Huttenhain, R. & Krogan, N.J. A systems approach to infectious disease. Nat. Rev. Genet. 21, 339–354 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roberts, J.A. et al. Individualised antibiotic dosing for patients who are critically ill: challenges and potential solutions. Lancet Infect. Dis. 14, 498–509 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tängden, T. et al. The role of infection models and PK/PD modelling for optimising care of critically ill patients with severe infections. Intensive Care Med. 43, 1021–1032 (2017). [DOI] [PubMed] [Google Scholar]
- 54. Kumta, N. , Roberts, J.A. , Lipman, J. & Cotta, M.O. Antibiotic distribution into cerebrospinal fluid: can dosing safely account for drug and disease factors in the treatment of ventriculostomy‐associated infections? Clin. Pharmacokinet. 57, 439–454 (2018). [DOI] [PubMed] [Google Scholar]
- 55. Sulaiman, H. , Abdul‐Aziz, M.H. & Roberts, J.A. Pharmacokinetic/pharmacodynamics‐optimized antimicrobial therapy in patients with hospital‐acquired pneumonia/ventilator‐associated pneumonia. Semin. Respir. Crit. Care Med. 38, 271–286 (2017). [DOI] [PubMed] [Google Scholar]
- 56. Kamal, M.A. et al. The posology of oseltamivir in infants with influenza infection using a population pharmacokinetic approach. Clin. Pharmacol. Ther. 96, 380–389 (2014). [DOI] [PubMed] [Google Scholar]
- 57. Larson, K.B. et al. Ceftolozane‐tazobactam population pharmacokinetics and dose selection for further clinical evaluation in pediatric patients with complicated urinary tract or complicated intra‐abdominal infections. Antimicrob. Agents Chemother. 63, e02578‐18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dorofaeff, T. , Bandini, R.M. , Lipman, J. , Ballot, D.E. , Roberts, J.A. & Parker, S.L. Uncertainty in antibiotic dosing in critically ill neonate and pediatric patients: can microsampling provide the answers? Clin. Ther. 38, 1961–1975 (2016). [DOI] [PubMed] [Google Scholar]
- 59. Gumbo, T. Integrating pharmacokinetics, pharmacodynamics and pharmacogenomics to predict outcomes in antibacterial therapy. Curr. Opin. Drug Discov. Devel. 11, 32–42 (2008). [PubMed] [Google Scholar]
- 60. Cheng, V. , Abdul‐Aziz, M.H. , Roberts, J.A. & Shekar, K. Optimising drug dosing in patients receiving extracorporeal membrane oxygenation. J. Thorac. Dis. 10, S629–S641 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Roberts, J.A. et al. The effect of renal replacement therapy and antibiotic dose on antibiotic concentrations in critically ill patients: data from the multinational SMARRT study. Clin. Infect. Dis. https://doi.org/ 10.1093/cid/ciaa224. [e‐pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 62. Siccardi, M. et al. Use of a physiologically‐based pharmacokinetic model to simulate artemether dose adjustment for overcoming the drug‐drug interaction with efavirenz. Silico Pharmacol. 1, 4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Roberts, O. et al. Physiologically based pharmacokinetic modelling prediction of the effects of dose adjustment in drug‐drug interactions between levonorgestrel contraceptive implants and efavirenz‐based ART. J. Antimicrob. Chemother. 73, 1004–1012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Burkhardt, O. , Lehmann, C. , Madabushi, R. , Kumar, V. , Derendorf, H. & Welte, T. Once‐daily tobramycin in cystic fibrosis: better for clinical outcome than thrice‐daily tobramycin but more resistance development? J. Antimicrob. Chemother. 58, 822–829 (2006). [DOI] [PubMed] [Google Scholar]
- 65. Deziel, M.R. et al. Effective antimicrobial regimens for use in humans for therapy of bacillus anthracis infections and postexposure prophylaxis. Antimicrob. Agents Chemother. 49, 5099–5106 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gloede, J. , Scheerans, C. , Derendorf, H. & Kloft, C. In vitro pharmacodynamic models to determine the effect of antibacterial drugs. J. Antimicrob. Chemother. 65, 186–201 (2010). [DOI] [PubMed] [Google Scholar]
- 67. McSharry, J.J. , Deziel, M.R. , Zager, K. , Weng, Q. & Drusano, G.L. Pharmacodynamics of cidofovir for vaccinia virus infection in an in vitro hollow‐fiber infection model system. Antimicrob. Agents Chemother. 53, 129–135 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Drusano, G.L. et al. Pharmacodynamics of abacavir in an in vitro hollow‐fiber model system. Antimicrob. Agents Chemother. 46, 464–470 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bilello, J.A. , Bauer, G. , Dudley, M.N. , Cole, G.A. & Drusano, G.L. Effect of 2',3'‐didehydro‐3'‐deoxythymidine in an in vitro hollow‐fiber pharmacodynamic model system correlates with results of dose‐ranging clinical studies. Antimicrob. Agents Chemother. 38, 1386–1391 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nielsen, E.I. & Friberg, L.E. Pharmacokinetic‐pharmacodynamic modeling of antibacterial drugs. Pharmacol. Rev. 65, 1053–1090 (2013). [DOI] [PubMed] [Google Scholar]
- 71. Mohamed, A.F. , Nielsen, E.I. , Cars, O. & Friberg, L.E. Pharmacokinetic‐pharmacodynamic model for gentamicin and its adaptive resistance with predictions of dosing schedules in newborn infants. Antimicrob. Agents Chemother. 56, 179–188 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mohamed, A.F. , Cars, O. & Friberg, L.E. A pharmacokinetic/pharmacodynamic model developed for the effect of colistin on Pseudomonas aeruginosa in vitro with evaluation of population pharmacokinetic variability on simulated bacterial killing. J. Antimicrob. Chemother. 69, 1350–1361 (2014). [DOI] [PubMed] [Google Scholar]
- 73. Sy, S.K. , Zhuang, L. & Derendorf, H. Pharmacokinetics and pharmacodynamics in antibiotic dose optimization. Expert Opin. Drug Metab. Toxicol. 12, 93–114 (2016). [DOI] [PubMed] [Google Scholar]
- 74. Sy, S.K.B. & Derendorf, H. Experimental design and modelling approach to evaluate efficacy of beta‐lactam/beta‐lactamase inhibitor combinations. Clin. Microbiol. Infect. 24, 707–715 (2018). [DOI] [PubMed] [Google Scholar]
- 75. Sy, S.K.B. et al. A mathematical model‐based analysis of the time‐kill kinetics of ceftazidime/avibactam against Pseudomonas aeruginosa. J. Antimicrob. Chemother. 73, 1295–1304 (2018). [DOI] [PubMed] [Google Scholar]
- 76. Sy, S.K.B. , Zhuang, L. , Sy, S. & Derendorf, H. Clinical pharmacokinetics and pharmacodynamics of ceftazidime‐avibactam combination: a model‐informed strategy for its clinical development. Clin. Pharmacokinet. 58, 545–564 (2019). [DOI] [PubMed] [Google Scholar]
- 77. Sy, S.K.B. , Zhuang, L. , Xia, H. , Schuck, V.J. , Nichols, W.W. & Derendorf, H. A model‐based analysis of pharmacokinetic‐pharmacodynamic (PK/PD) indices of avibactam against Pseudomonas aeruginosa. Clin. Microbiol. Infect. 25, 904.e9–904.e16 (2019). [DOI] [PubMed] [Google Scholar]
- 78. Brill, M.J.E. , Kristoffersson, A.N. , Zhao, C. , Nielsen, E.I. & Friberg, L.E. Semi‐mechanistic pharmacokinetic‐pharmacodynamic modelling of antibiotic drug combinations. Clin. Microbiol. Infect. 24, 697–706 (2018). [DOI] [PubMed] [Google Scholar]
- 79. Nielsen, E.I. , Cars, O. & Friberg, L.E. Predicting in vitro antibacterial efficacy across experimental designs with a semimechanistic pharmacokinetic‐pharmacodynamic model. Antimicrob. Agents Chemother. 55, 1571–1579 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Khan, D.D. et al. A mechanism‐based pharmacokinetic/pharmacodynamic model allows prediction of antibiotic killing from MIC values for WT and mutants. J. Antimicrob. Chemother. 70, 3051–3060 (2015). [DOI] [PubMed] [Google Scholar]
- 81. Nielsen, E.I. , Viberg, A. , Lowdin, E. , Cars, O. , Karlsson, M.O. & Sandstrom, M. Semimechanistic pharmacokinetic/pharmacodynamic model for assessment of activity of antibacterial agents from time‐kill curve experiments. Antimicrob. Agents Chemother. 51, 128–136 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bulitta, J.B. , Ly, N.S. , Yang, J.C. , Forrest, A. , Jusko, W.J. & Tsuji, B.T. Development and qualification of a pharmacodynamic model for the pronounced inoculum effect of ceftazidime against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53, 46–56 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Thorsted, A. Pharmacometrics to characterize innate immune response and antibacterial treatments (dissertation). Uppsala University, Uppsala, Sweden: (2020). [Google Scholar]
- 84. Drusano, G.L. et al. Interaction of drug‐ and granulocyte‐mediated killing of Pseudomonas aeruginosa in a murine pneumonia model. J. Infect. Dis. 210, 1319–1324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Thorsted, A. et al. A non‐linear mixed effect model for innate immune response: in vivo kinetics of endotoxin and its induction of the cytokines tumor necrosis factor alpha and interleukin‐6. PLoS One 14, e0211981 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, CLSI Standard m27 (Clinical and Laboratory Standards Institute (CLSI), Wayne, PA, 2017) [Google Scholar]
- 87. Lepak, A.J. & Andes, D.R. Antifungal pharmacokinetics and pharmacodynamics. Cold Spring Harb. Perspect. Med. 5, a019653 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Andes, D. et al. In vivo pharmacodynamic characterization of anidulafungin in a neutropenic murine candidiasis model. Antimicrob. Agents Chemother. 52, 539–550 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Andes, D. et al. Pharmacodynamics of a new triazole, posaconazole, in a murine model of disseminated candidiasis. Antimicrob. Agents Chemother. 48, 137–142 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Andes, D. , Marchillo, K. , Stamstad, T. & Conklin, R. In vivo pharmacokinetics and pharmacodynamics of a new triazole, voriconazole, in a murine candidiasis model. Antimicrob. Agents Chemother. 47, 3165–3169 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Andes, D. & van Ogtrop, M. Characterization and quantitation of the pharmacodynamics of fluconazole in a neutropenic murine disseminated candidiasis infection model. Antimicrob. Agents Chemother. 43, 2116–2120 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Andes, D.R. , Diekema, D.J. , Pfaller, M.A. , Marchillo, K. & Bohrmueller, J. In vivo pharmacodynamic target investigation for micafungin against Candida albicans and C. Glabrata in a neutropenic murine candidiasis model. Antimicrob. Agents Chemother. 52, 3497–3503 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lepak, A.J. , Marchillo, K. , VanHecker, J. , Diekema, D. & Andes, D.R. Isavuconazole pharmacodynamic target determination for Candida species in an in vivo murine disseminated candidiasis model. Antimicrob. Agents Chemother. 57, 5642–5648 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Louie, A. , Deziel, M. , Liu, W. , Drusano, M.F. , Gumbo, T. & Drusano, G.L. Pharmacodynamics of caspofungin in a murine model of systemic candidiasis: importance of persistence of caspofungin in tissues to understanding drug activity. Antimicrob. Agents Chemother. 49, 5058–5068 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Warn, P.A. , Sharp, A. , Parmar, A. , Majithiya, J. , Denning, D.W. & Hope, W.W. Pharmacokinetics and pharmacodynamics of a novel triazole, isavuconazole: mathematical modeling, importance of tissue concentrations, and impact of immune status on antifungal effect. Antimicrob. Agents Chemother. 53, 3453–3461 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Andes, D. & van Ogtrop, M. In vivo characterization of the pharmacodynamics of flucytosine in a neutropenic murine disseminated candidiasis model. Antimicrob. Agents Chemother. 44, 938–942 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Andes, D. , Stamsted, T. & Conklin, R. Pharmacodynamics of amphotericin B in a neutropenic‐mouse disseminated‐candidiasis model. Antimicrob. Agents Chemother. 45, 922–926 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhao, M. et al. In vivo pharmacokinetics and pharmacodynamics of APX001 against Candida spp. In a neutropenic disseminated candidiasis mouse model. Antimicrob. Agents Chemother. 62, e02542‐02517 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hope, W.W. et al. Pharmacodynamics of the orotomides against Aspergillus fumigatus: new opportunities for treatment of multidrug‐resistant fungal disease. MBio 8, e01157‐01117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lepak, A.J. , Marchillo, K. , Vanhecker, J. & Andes, D.R. Isavuconazole (BAL4815) pharmacodynamic target determination in an in vivo murine model of invasive pulmonary aspergillosis against wild‐type and cyp51 mutant isolates of Aspergillus fumigatus. Antimicrob. Agents Chemother. 57, 6284–6289 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lepak, A.J. , Zhao, M. & Andes, D.R. Pharmacodynamic evaluation of rezafungin (CD101) against Candida auris in the neutropenic mouse invasive candidiasis model. Antimicrob. Agents Chemother. 62, e01572‐01518 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lepak, A.J. , Zhao, M. , Berkow, E.L. , Lockhart, S.R. & Andes, D.R. Pharmacodynamic optimization for treatment of invasive Candida auris infection. Antimicrob. Agents Chemother. 61, e00791‐00717 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Groll, A.H. et al. Pharmacokinetic and pharmacodynamic modeling of anidulafungin (LY303366): reappraisal of its efficacy in neutropenic animal models of opportunistic mycoses using optimal plasma sampling. Antimicrob. Agents Chemother. 45, 2845–2855 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Andes, D. , Diekema, D.J. , Pfaller, M.A. , Bohrmuller, J. , Marchillo, K. & Lepak, A. In vivo comparison of the pharmacodynamic targets for echinocandin drugs against Candida species. Antimicrob. Agents Chemother. 54, 2497–2506 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Howard, S.J. et al. Pharmacokinetics and pharmacodynamics of posaconazole for invasive pulmonary aspergillosis: clinical implications for antifungal therapy. J. Infect. Dis. 203, 1324–1332 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lepak, A.J. , Marchillo, K. , Vanhecker, J. & Andes, D.R. Posaconazole pharmacodynamic target determination against wild‐type and Cyp51 mutant isolates of Aspergillus fumigatus in an in vivo model of invasive pulmonary aspergillosis. Antimicrob. Agents Chemother. 57, 579–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Al‐Nakeeb, Z. et al. Pharmacodynamics of itraconazole against Aspergillus fumigatus in an in vitro model of the human alveolus: perspectives on the treatment of triazole‐resistant infection and utility of airway administration. Antimicrob. Agents Chemother. 56, 4146–4153 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Trang, M. , Dudley, M.N. & Bhavnani, S.M. Use of Monte Carlo simulation and considerations for PK‐PD targets to support antibacterial dose selection. Curr. Opin. Pharmacol. 36, 107–113 (2017). [DOI] [PubMed] [Google Scholar]
- 109. Clancy, C.J. , Yu, V.L. , Morris, A.J. , Snydman, D.R. & Nguyen, M.H. Fluconazole MIC and the fluconazole dose/MIC ratio correlate with therapeutic response among patients with candidemia. Antimicrob. Agents Chemother. 49, 3171–3177 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Rex, J.H. et al. Development of interpretive breakpoints for antifungal susceptibility testing: conceptual framework and analysis of in vitro‐in vivo correlation data for fluconazole, itraconazole, and Candida infections. Subcommittee on antifungal susceptibility testing of the national committee for clinical laboratory standards. Clin. Infect. Dis. 24, 235–247 (1997). [DOI] [PubMed] [Google Scholar]
- 111. Baddley, J.W. , Patel, M. , Bhavnani, S.M. , Moser, S.A. & Andes, D.R. Association of fluconazole pharmacodynamics with mortality in patients with candidemia. Antimicrob. Agents Chemother. 52, 3022–3028 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Pai, M.P. , Turpin, R.S. & Garey, K.W. Association of fluconazole area under the concentration‐time curve/MIC and dose/MIC ratios with mortality in nonneutropenic patients with candidemia. Antimicrob. Agents Chemother. 51, 35–39 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Rodriguez‐Tudela, J.L. et al. Correlation of the MIC and dose/MIC ratio of fluconazole to the therapeutic response of patients with mucosal candidiasis and candidemia. Antimicrob. Agents Chemother. 51, 3599–3604 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Andes, D. et al. Use of pharmacokinetic‐pharmacodynamic analyses to optimize therapy with the systemic antifungal micafungin for invasive candidiasis or candidemia. Antimicrob. Agents Chemother. 55, 2113–2121 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Hope, W.W. et al. The pharmacokinetics and pharmacodynamics of micafungin in experimental hematogenous Candida meningoencephalitis: implications for echinocandin therapy in neonates. J. Infect. Dis. 197, 163–171 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Troke, P.F. , Hockey, H.P. & Hope, W.W. Observational study of the clinical efficacy of voriconazole and its relationship to plasma concentrations in patients. Antimicrob. Agents Chemother. 55, 4782–4788 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Smith, J. et al. Voriconazole therapeutic drug monitoring. Antimicrob. Agents Chemother. 50, 1570–1572 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Zhao, M. et al. APX001 pharmacokinetic/pharmacodynamic target determination against Aspergillus fumigatus in an in vivo model of invasive pulmonary aspergillosis. Antimicrob. Agents Chemother. 63, e02372‐02318 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sudan, A. et al. Pharmacokinetics and pharmacodynamics of fluconazole for cryptococcal meningoencephalitis: implications for antifungal therapy and in vitro susceptibility breakpoints. Antimicrob. Agents Chemother. 57, 2793–2800 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Gebremariam, T. et al. Fosmanogepix (APX001) is effective in the treatment of pulmonary murine mucormycosis due to Rhizopus arrhizus. Antimicrob. Agents Chemother. 64, e00178‐00120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kovanda, L.L. et al. Pharmacodynamics of isavuconazole in a rabbit model of cryptococcal meningoencephalitis. Antimicrob. Agents Chemother. 63, e00546‐00519 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Chatterjee, A. , Smith, P.F. & Perelson, A.S. Hepatitis C viral kinetics: the past, present, and future. Clin. Liver Dis. 17, 13–26 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Reddy, M.B. et al. Pharmacokinetic/pharmacodynamic predictors of clinical potency for hepatitis C virus nonnucleoside polymerase and protease inhibitors. Antimicrob. Agents Chemother. 56, 3144–3156 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Duan, J. et al. The liver partition coefficient‐corrected inhibitory quotient and the pharmacokinetic‐pharmacodynamic relationship of directly acting anti‐hepatitis C virus agents in humans. Antimicrob. Agents Chemother. 56, 5381–5386 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Canini, L. , Guedj, J. , Chatterjee, A. , Lemenuel‐Diot, A. , Smith, P.F. & Perelson, A.S. Modelling the interaction between danoprevir and mericitabine in the treatment of chronic HCV infection. Antivir. Ther. 21, 297–306 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Adiwijaya, B.S. et al. A multi‐variant, viral dynamic model of genotype 1 HCV to assess the in vivo evolution of protease‐inhibitor resistant variants. PLoS Comput. Biol. 6, e1000745 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Rong, L. , Ribeiro, R.M. & Perelson, A.S. Modeling quasispecies and drug resistance in hepatitis C patients treated with a protease inhibitor. Bull. Math. Biol. 74, 1789–1817 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ouwerkerk‐Mahadevan, S. , Snoeys, J. , Peeters, M. , Beumont‐Mauviel, M. & Simion, A. Drug‐drug interactions with the NS3/4a protease inhibitor simeprevir. Clin. Pharmacokinet. 55, 197–208 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Eley, T. et al. Organic anion transporting polypeptide‐mediated transport of, and inhibition by, asunaprevir, an inhibitor of hepatitis C virus NS3 protease. Clin. Pharmacol. Ther. 97, 159–166 (2015). [DOI] [PubMed] [Google Scholar]
- 130. Wang, T. et al. Species differences in liver accumulation and metabolism of nucleotide prodrug sofosbuvir. Drug Metab. Pharmacokinet. 35, 334–340 (2020). [DOI] [PubMed] [Google Scholar]
- 131. Guedj, J. et al. Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half‐life. Proc. Natl. Acad. Sci. USA 110, 3991–3996 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Dahari, H. et al. HCV kinetic and modeling analyses indicate similar time to cure among sofosbuvir combination regimens with daclatasvir, simeprevir or ledipasvir. J. Hepatol. 64, 1232–1239 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. DeVincenzo, J.P. et al. Activity of oral ALS‐008176 in a respiratory syncytial virus challenge study. N. Engl. J. Med. 373, 2048–2058 (2015). [DOI] [PubMed] [Google Scholar]
- 134. Patel, K. et al. Respiratory syncytial virus‐A dynamics and the effects of lumicitabine, a nucleoside viral replication inhibitor, in experimentally infected humans. J. Antimicrob. Chemother. 74, 442–452 (2019). [DOI] [PubMed] [Google Scholar]
- 135. la Porte, C. Inhibitory quotient in HIV pharmacology. Curr. Opin. HIV AIDS 3, 283–287 (2008). [DOI] [PubMed] [Google Scholar]
- 136. Guidance for industry: antiviral product development‐conducting and submitting virology studies to the agency. Rockville, MD, USA: United States Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) <https://www.fda.gov/media/71223/download> (2006). Accessed December 14, 2020.
- 137. Rossenu, S. , Neyens, M. , VanSolingen‐Ristea, R. , Baugh, B. & Crauwels, H. Population pharmacokinetic (PopPK) modeling and simulation of monthly intramuscular (IM) long‐acting rilpivirine (RPV LA) to inform strategies following dosing interruption in HIV‐1 infected subjects (abstract 6572). International Aids Conference July 6–10, 2020; Virtual <https://www.natap.org/2020/IAC/IAC_104.htm> Accessed December 14, 2020.
- 138. Rudd, D.J. et al. Modeling‐supported islatravir dose selection for phase 3. Conference on Retroviruses and Opportunistic Infections March, 8–11, 2020; Boston, MA, USA <https://www.natap.org/2020/CROI/croi_52.htm> Accessed December 14, 2020.
- 139. Han, K. et al. Population pharmacokinetic (PPK) modeling and simulation of long‐acting (la) cabotegravir (cab) to inform strategies following dosing interruptions in HIV‐1 infected subjects (abstract 1532). Open Forum Infect. Dis. 6(Suppl 2), S558 (2019). [Google Scholar]
- 140. Ruane, P.J. et al. Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of tenofovir alafenamide as 10‐day monotherapy in HIV‐1‐positive adults. J. Acquir. Immune Defic. Syndr. 63, 449–455 (2013). [DOI] [PubMed] [Google Scholar]
- 141. Markowitz, M. & Grobler, J.A. Islatravir for the treatment and prevention of infection with the human immunodeficiency virus type 1. Curr. Opin. HIV AIDS 15, 27–32 (2020). [DOI] [PubMed] [Google Scholar]
- 142. Taskar, K. et al. Utilization of physiologically based pharmacokinetic modelling (PBPK) to predict the effect of UGT enzyme inhibition and induction on the systemic exposure of cabotegravir (abstract). 20th International Workshop on Clinical Pharmacology of HIV, Hepatitis, & Other Antiviral Drugs May 14‐16, 2019; Noordwijk, The Netherlands <https://www.natap.org/2019/Pharm/Pharm_34.htm> Accessed December 14, 2020.
- 143. Taskar, K.S. et al. Prediction of renal OAT1 and OAT3 inhibition by cabotegravir using PBPK modelling (abstract 470). Conference on Retroviruses and Opportunistic Infections March 4–7, 2019; Seattle, WA, USA <https://www.croiconference.org/abstract/prediction‐renal‐oat1‐and‐oat3‐inhibition‐cabotegravir‐using‐pbpk‐modelling/>. Accessed December 14, 2020.
- 144. Rajoli, R.K.R. et al. Predicting drug‐drug interactions between rifampicin and long‐acting cabotegravir and rilpivirine using physiologically based pharmacokinetic modeling. J. Infect. Dis. 219, 1735–1742 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Thakkar, N. et al. Model‐based approach of dose selection and optimal PK sampling of fostemsavir for pediatric patients with multidrug resistant HIV‐1 infection (abstract peb0291). International AIDS Conference July 6‐10, 2020; San Francisco, CA, USA <https://www.natap.org/2020/IAC/IAC_106.htm> Accessed December 14, 2020.
- 146. Baker, M. , Patel, P. , Piscitelli, J. & van Lunzen, J. PK/PD modelling of bnabs for HIV treatments identifying knowledge gaps (abstract PE4/6). European AIDS Conference November 6–9, 2019; Basel, Switzerland <https://d201nm4szfwn7c.cloudfront.net/5f95dbd7‐245e‐4e65‐9f36‐1a99e28e5bba/a0b884c5‐e33a‐45b5‐a8a4‐f364d3c81db0/a0b884c5‐e33a‐45b5‐a8a4‐f364d3c81db0_viewable_rendition__v.pdf?medcommid=REF–ALL‐002437>. Accessed December 14, 2020.
- 147. Onchocerciasis fact sheet. Geneva, Switzerland: World Health Organization <https://www.who.int/news‐room/fact‐sheets/detail/onchocerciasis> (2019). Accessed December 14, 2020.
- 148. Approval package for mectizan (ivermectin). Rockville, MD, USA: United States Food and Drug Administration Center for Drug Evaluation and Research <https://www.accessdata.fda.gov/drugsatfda_docs/nda/96/050742ap.pdf> (1996). Accessed November 25, 2020.
- 149. Stolk, W.A. , Walker, M. , Coffeng, L.E. , Basáñez, M.G. & de Vlas, S.J. Required duration of mass ivermectin treatment for onchocerciasis elimination in Africa: a comparative modelling analysis. Parasit. Vectors 8, 552 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Drug approval package: moxidectin. United States Department of Health and Human Services Food and Drug Administration <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210867Orig1s000TOC.cfm> (2018). Accessed November 25, 2020.
- 151. Opoku, N.O. et al. Single dose moxidectin versus ivermectin for Onchocerca volvulus infection in Ghana, Liberia, and the Democratic Republic of the Congo: a randomised, controlled, double‐blind phase 3 trial. Lancet 392, 1207–1216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Jamsen, K. et al. Determining the optimal dose of moxidectin for onchocerciasis via pharmacokinetic‐pharmacodynamic (PK‐PD) modelling of data from healthy volunteers with onchocerciasis (abstract A762). American Society of Tropical Medicine and Hygiene November 7, 2017; Washington, DC.
- 153. Basáñez, M.G. , Pion, S.D. , Boakes, E. , Filipe, J.A. , Churcher, T.S. & Boussinesq, M. Effect of single‐dose ivermectin on Onchocerca volvulus: a systematic review and meta‐analysis. Lancet Infect. Dis. 8, 310–322 (2008). [DOI] [PubMed] [Google Scholar]
- 154. Milton, P. et al. The potential for six‐monthly mass administration of moxidectin to accelerate onchocerciasis elimination (abstract LB‐5102). American Society of Tropical Medicine and Hygiene November 5–9, 2017; Baltimore, MD.
- 155. Turner, H.C. et al. The potential impact of moxidectin on onchocerciasis elimination in Africa: an economic evaluation based on the phase II clinical trial data. Parasit. Vectors 8, 167 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Milton, P. et al. Modelling alternative strategies for onchocerciasis elinimation: The case for moxidectin. Am. J. Trop. Med. Hyg. 97(Suppl 5), 559–560 (2017). [Google Scholar]
- 157. Bray, M. , Rayner, C. , Noel, F. , Jans, D. & Wagstaff, K. Ivermectin and COVID‐19: a report in antiviral research, widespread interest, an FDA warning, two letters to the editor and the authors' responses. Antiviral Res. 178, 104805 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Target regimen profiles for TB treatment: candidates: rifampicin‐susceptible, rifampicin‐resistant and pan‐TB treatment regimens. Geneva, Switzerland: World Health Organization <https://apps.who.int/iris/bitstream/handle/10665/250044/9789241511339‐eng.pdf?sequence=1> (2016). Accessed December 14, 2020.
- 159. Pagan, A.J. & Ramakrishnan, L. Immunity and immunopathology in the tuberculous granuloma. Cold Spring Harb. Perspect. Med. 5, a018499 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Gumbo, T. , Lenaerts, A.J. , Hanna, D. , Romero, K. & Nuermberger, E. Nonclinical models for antituberculosis drug development: a landscape analysis. J. Infect. Dis. 211(Suppl 3), S83–95 (2015). [DOI] [PubMed] [Google Scholar]
- 161. Draft qualification opinion: in vitro hollow fiber system model of tuberculosis (HFS‐TB), ema/chmp/sawp/381716/2014. London, UK: European Medicines Agency Committee for Medicinal Products for Human Use (CHMP) <https://www.ema.europa.eu/en/documents/regulatory‐procedural‐guideline/draft‐qualification‐opinion‐vitro‐hollow‐fibre‐system‐model‐tuberculosis‐hfs‐tb_en.pdf> (2014). Accessed December 14, 2020.
- 162. Via, L.E. et al. A sterilizing tuberculosis treatment regimen is associated with faster clearance of bacteria in cavitary lesions in marmosets. Antimicrob. Agents Chemother. 59, 4181–4189 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Rifat, D. et al. Pharmacokinetics of rifapentine and rifampin in a rabbit model of tuberculosis and correlation with clinical trial data. Sci. Transl. Med. 10, eaai7786 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Nuermberger, E. & Hanna, D. Assessing the landscape of tools and approaches for novel tuberculosis regimen development. J. Infect. Dis. 211(Suppl 3), S81–S82 (2015). [DOI] [PubMed] [Google Scholar]
- 165. Nuermberger, E. , Sizemore, C. , Romero, K. & Hanna, D. Toward an evidence‐based nonclinical road map for evaluating the efficacy of new tuberculosis (TB) drug regimens: proceedings of a critical path to TB drug regimens‐National Institute of Allergy and Infectious Diseases In Vivo Pharmacology Workshop for TB Drug Development. Antimicrob. Agents Chemother. 60, 1177–1182 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Fors, J. , Strydom, N. , Fox, W.S. , Keizer, R.J. & Savic, R.M. Mathematical model and tool to explore shorter multi‐drug therapy options for active pulmonary tuberculosis. PLoS Comput. Biol. 16, e1008107 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Zhang, N. et al. Mechanistic modeling of mycobacterium tuberculosis infection in murine models for drug and vaccine efficacy studies. Antimicrob. Agents Chemother. 64, e01727–19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Blanc, L. et al. High‐resolution mapping of fluoroquinolones in TB rabbit lesions reveals specific distribution in immune cell types. Elife 7, e41115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Strydom, N. et al. Tuberculosis drugs' distribution and emergence of resistance in patient's lung lesions: a mechanistic model and tool for regimen and dose optimization. PLoS Med. 16, e1002773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Ernest, J.P. et al. Development of new tuberculosis drugs: translation to regimen composition for drug‐sensitive and multidrug‐resistant tuberculosis. Annu. Rev. Pharmacol. Toxicol. https://doi.org/ 10.1146/annurev-pharmtox-030920-011143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Rosenthal, I.M. et al. Dose‐ranging comparison of rifampin and rifapentine in two pathologically distinct murine models of tuberculosis. Antimicrob. Agents Chemother. 56, 4331–4340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Dorman, S.E. et al. Substitution of rifapentine for rifampin during intensive phase treatment of pulmonary tuberculosis: Study 29 of the tuberculosis trials consortium. J. Infect. Dis. 206, 1030–1040 (2012). [DOI] [PubMed] [Google Scholar]
- 173. Bartelink, I.H. et al. New paradigm for translational modeling to predict long‐term tuberculosis treatment response. Clin. Transl. Sci. 10, 366–379 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Zurlinden, T.J. , Eppers, G.J. & Reisfeld, B. Physiologically based pharmacokinetic model of rifapentine and 25‐desacetyl rifapentine disposition in humans. Antimicrob. Agents Chemother. 60, 4860–4868 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Gaohua, L. et al. Development of a multicompartment permeability‐limited lung PBPK model and its application in predicting pulmonary pharmacokinetics of antituberculosis drugs. CPT Pharmacometrics Syst. Pharmacol. 4, 605–613 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Imperial, M.Z. et al. A patient‐level pooled analysis of treatment‐shortening regimens for drug‐susceptible pulmonary tuberculosis. Nat. Med. 24, 1708–1715 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Guidelines for the Treatment of Malaria (Global Malaria Programme, World Health Organization, Geneva, Switzerland, 2015). [Google Scholar]
- 178. Kloprogge, F. et al. Artemether‐lumefantrine dosing for malaria treatment in young children and pregnant women: a pharmacokinetic‐pharmacodynamic meta‐analysis. PLoS Med. 15, e1002579 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Hendriksen, I.C. et al. Population pharmacokinetics of intramuscular artesunate in African children with severe malaria: implications for a practical dosing regimen. Clin. Pharmacol. Ther. 93, 443–450 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Hendriksen, I.C. et al. Population pharmacokinetic and pharmacodynamic properties of intramuscular quinine in Tanzanian children with severe falciparum malaria. Antimicrob. Agents Chemother. 57, 775–783 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Byakika‐Kibwika, P. , Ssenyonga, R. , Lamorde, M. , Blessborn, D. & Tarning, J. Piperaquine concentration and malaria treatment outcomes in Ugandan children treated for severe malaria with intravenous artesunate or quinine plus dihydroartemisinin‐piperaquine. BMC Infect. Dis. 19, 1025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Chotsiri, P. et al. Severe acute malnutrition results in lower lumefantrine exposure in children treated with artemether‐lumefantrine for uncomplicated malaria. Clin. Pharmacol. Ther. 106, 1299–1309 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Kloprogge, F. et al. Population pharmacokinetics of lumefantrine in pregnant and nonpregnant women with uncomplicated Plasmodium falciparum malaria in Uganda. CPT Pharmacometrics Syst. Pharmacol. 2, e83 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Andrews, K.A. et al. Model‐informed drug development for malaria therapeutics. Annu. Rev. Pharmacol. Toxicol. 58, 567–582 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Charman, S.A. et al. An in vitro toolbox to accelerate anti‐malarial drug discovery and development. Malar. J. 19, 1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Ke, A.B. , Greupink, R. & Abduljalil, K. Drug dosing in pregnant women: challenges and opportunities in using physiologically based pharmacokinetic modeling and simulations. CPT Pharmacometrics Syst. Pharmacol. 7, 103–110 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Zhang, Z. & Unadkat, J.D. Development of a novel maternal‐fetal physiologically based pharmacokinetic model II: verification of the model for passive placental permeability drugs. Drug Metab. Dispos. 45, 939–946 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Mian, P. et al. Integration of placental transfer in a fetal‐maternal physiologically based pharmacokinetic model to characterize acetaminophen exposure and metabolic clearance in the fetus. Clin. Pharmacokinet. 59, 911–925 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Vrc 612: a phase 1, dose escalation, open‐label clinical trial with experimental controlled human malaria infections (CHMI) to evaluate the safety and protective efficacy of an anti‐ malaria human monoclonal antibody, VRC‐MALMAB0100‐00‐AB (CIS43LS), in healthy, malaria‐naive adults. National Institutes of Health Clinical Center <https://clinicaltrials.gov/ct2/show/NCT04206332> (2020). Accessed November 8, 2020.
- 190. Andrews, K.A. , McCarthy, J. , Wesche, D. , Gobeau, N. , Grasela, T. & Mohrle, J. Retrospective PK/PD analysis offers improvements in design efficiency of malaria volunteer infection studies for antimalarial drug development. Clin. Transl. Sci. https://doi.org/ 10.1111/cts.12934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Watts, R.E. et al. Safety and parasite clearance of artemisinin‐resistant Plasmodium falciparum infection: a pilot and a randomised volunteer infection study in Australia. PLoS Medicine 17, e1003203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Laurens, M.B. et al. A consultation on the optimization of controlled human malaria infection by mosquito bite for evaluation of candidate malaria vaccines. Vaccine 30, 5302–5304 (2012). [DOI] [PubMed] [Google Scholar]
- 193. Animal rule information. United States Food and Drug Administration <https://www.fda.gov/emergency‐preparedness‐and‐response/mcm‐regulatory‐science/animal‐rule‐information> (2019). Accessed December 14, 2020.
- 194. Roberts, R. & McCune, S.K. Animal studies in the development of medical countermeasures. Clin. Pharmacol. Ther. 83, 918–920 (2008). [DOI] [PubMed] [Google Scholar]
- 195. Kurilla, M. Animal models facilitate rapid responses to emerging infectious diseases. ILAR J. 52S, 501–506 (2011). [Google Scholar]
- 196. Perry, M.R. , Warren, R. , Merchlinsky, M. , Houchens, C. & Rogers, J.V. Rabbitpox in New Zealand white rabbits: a therapeutic model for evaluation of poxvirus medical countermeasures under the FDA animal rule. Front. Cell Infect. Microbiol. 8, 356 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Phipps, A.J. , Premanandan, C. , Barnewall, R.E. & Lairmore, M.D. Rabbit and nonhuman primate models of toxin‐targeting human anthrax vaccines. Microbiol. Mol. Biol. Rev. 68, 617–629 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Obiltoxaximab clinical pharmacology review. Rockville, MD, USA: United States Food and Drug Administration Center for Drug Evaluation and Research <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/125509Orig1s000ClinPharmR.pdf> (2015). Accessed December 14, 2020.
- 199. Leeds, J.M. et al. Pharmacokinetic and pharmacodynamic modeling to determine the dose of ST‐246 to protect against smallpox in humans. Antimicrob. Agents Chemother. 57, 1136–1143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Bergman, K.L. , Krudys, K. , Seo, S.K. & Florian, J. Modeling and simulation in dose determination for biodefense products approved under the FDA animal rule. J. Pharmacokinet. Pharmacodyn. 44, 153–160 (2017). [DOI] [PubMed] [Google Scholar]
- 201. Dodds, M.G. , Krishna, R. , Goncalves, A. & Rayner, C.R. Model‐informed drug repurposing: viral kinetic modelling to prioritize rational drug combinations for COVID‐19. Br. J. Clin. Pharmacol. https://doi.org/ 10.1111/bcp.14486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Rayner, C.R. et al. Accelerating clinical evaluation of repurposed combination therapies for COVID‐19. Am. J. Trop. Med. Hyg. 103, 1364–1366 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Addressing the COVID‐19 challenge with computer modeling & simulation. The Science Advisory Board <https://www.scienceboard.net/index.aspx?sec=sup&sub=can&pag=dis&ItemID=1052> (2020). Accessed November 25, 2020.
- 204. Mandema, J.W. , Gibbs, M. , Boyd, R.A. , Wada, D.R. & Pfister, M. Model‐based meta‐analysis for comparative efficacy and safety: application in drug development and beyond. Clin. Pharmacol. Ther. 90, 766–769 (2011). [DOI] [PubMed] [Google Scholar]
- 205. Outcomes database. COVID‐19 Pharmacology Resource Center <https://www.covidpharmacology.com/outcomes‐database/> (2020).
- 206. Madabushi, R. , Wang, Y. & Zineh, I. A holistic and integrative approach for advancing model‐informed drug development. CPT Pharmacometrics Syst. Pharmacol. 8, 9–11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Zineh, I. Quantitative systems pharmacology: a regulatory perspective on translation. CPT Pharmacometrics Syst. Pharmacol. 8, 336–339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Kannan, S. , Subbaram, K. , Ali, S. & Kannan, H. The role of artificial intelligence and machine learning techniques: race for COVID‐19 vaccine. Arch. Clin. Infect. Dis. 15, e103232 (2020). [Google Scholar]