

SUMMARY
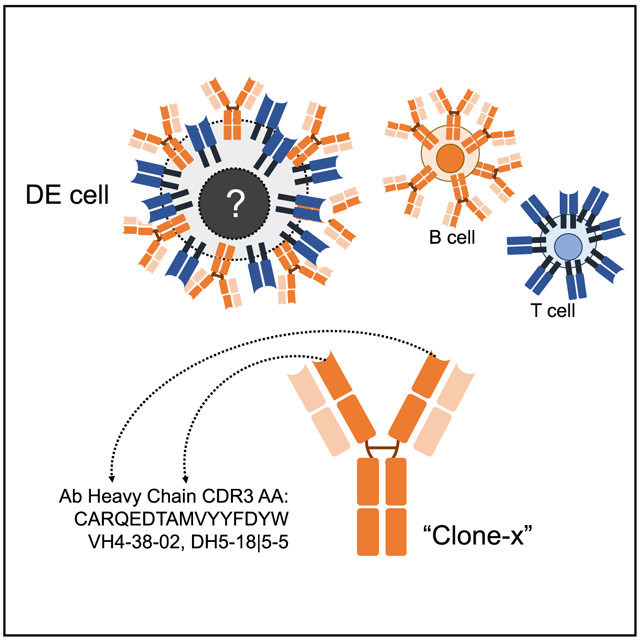
Ahmed and colleagues recently described a novel hybrid lymphocyte expressing both a B and T cell receptor, termed double expresser (DE) cells. DE cells in blood of T1D subjects were present at increased numbers and enriched for a public B cell clonotype. Here, we attempted to reproduce these findings. While we could identify DE cells by flow cytometry, we found no association between DE cell frequency and T1D status. We were unable to identify the reported public B cell clone, or any similar clone, in bulk B cells or sorted DE cells from T1D subjects or controls. We also did not observe increased usage of the public clone VH or DH genes in B cells or in sorted DE cells. Taken together, our findings suggest that DE cells and their alleged public clonotype are not enriched in T1D. This Matters Arising paper is in response to Ahmed et al. (2019), published in Cell. See also the response by Ahmed et al. (2021), published in this issue.
Graphical Abstract
In Brief
There does not appear to be increased abundance of dual-expresser TCR+/BCR+ lymphocytes in type I diabetes patients, nor a public clonotype, challenging the results of a previous study.
INTRODUCTION
Type 1 diabetes (T1D) is characterized by the destruction of insulin-producing β cells within the pancreatic islets of Langerhans (Opie, 1901). Genetic studies alongside extensive histological analyses support the concept of an autoimmune-mediated pathogenesis (Eisenbarth, 1986; Gepts, 1965). T cells and B cells are both present within the insulitic lesions and have been implicated in disease progression (Herold et al., 2013). T cells are thought to act as an effector population producing inflammatory cytokines and/or directly killing pancreatic β cells, whereas B cells appear to serve as producers of prognostic autoantibodies to β cell antigens in addition to their role as antigen-presenting cells (Atkinson et al., 2015). A number of lymphocyte-targeting agents have demonstrated clinical efficacy in preserving endogenous C-peptide in new onset T1D (e.g., anti-CD3, LFA3-Ig, and anti-thymocyte globulin), with anti-CD3 recently showing the ability to impart a 2-year delay of disease onset in high-risk subjects (Herold et al., 2019). Moreover, agents that impact B cells directly via a targeting antibody (anti-CD20 [Pescovitz et al., 2009]) or indirectly via costimulatory blockade (Lenschow et al., 1992) have also demonstrated some capacity for preserving β cell function. But so far these and other therapies have not resulted in durable prevention or reversal of the disease. The triggering event(s) and immunopathologic mechanisms of T1D in humans still remain outstanding questions.
Recently, Ahmed and colleagues (Ahmed et al., 2019), described the presence and mechanistic association of a novel B:T cell hybrid with T1D. They identified a population of CD5+ cells expressing both a T cell receptor (TCR) and a B cell receptor (BCR) that they termed “double expresser” or DE cells. They documented increased frequencies of these DE cells in the blood of individuals with T1D, suggesting a link to disease pathogenesis. They also observed that a public B cell clone with a CDR3 (complementarity determining region) amino acid (AA) sequence of “CARQEDTAMVYYFDYW” was highly enriched in DE cells sorted from patients with T1D, but not in DE cells from healthy controls. Peptides derived from the BCR of this public clone could bind and stimulate T cells in the context of the T1D-associated HLA class II molecule, DQ8. Finally, they also suggested that secreted forms of the public clone antibody directly stimulated insulin-specific CD4+ T cells. These findings not only challenge traditional paradigms of lymphocyte development and function but also have important diagnostic and therapeutic potential as DE cells could provide a novel means of generating and responding to autoantigens.
DE cells blur the distinction between T and B cells as separate lymphocyte lineages. The conventional paradigm is that B and T cell lineage commitment signals tend to be conveyed early in the developmental program, well before the expression of a fully functional antigen receptor (Allman et al., 1999). A combination of molecules and processes, including transcription factor expression, epigenetic changes, signaling receptors, homing molecules, and V(D)J recombination accompany the sequential and progressive narrowing of cell fate that ultimately specifies commitment to the B cell orT cell lineage (Rothenberg, 2011). Transdifferentiation of mature B cells to T cells or vice versa appears to be largely prohibited under physiologic conditions, based in part on positive and negative regulators of lineage commitment (Nutt et al., 1999; Maillard et al., 2005). However, there is increasing evidence that commitment to the T or B cell lineage can be modified. Manipulation of Oct4, Sox2, Klf4, c-Myc, C/EBPα, and/or Pax5 can be used to reprogram mature B cells to give rise to mouse embryos (Hanna et al., 2008; Bueno et al., 2016). In addition, cell culture conditions have been described in which mature B cells can be converted into T cells and vice versa (Cobaleda et al., 2007; Ikawa et al., 2016). There are also rare “mixed lineage” leukemias with features of both B cells and T cells (Mi et al., 2018). These examples beg the question of whether B:T cell hybrids are transient intermediates, a bona fide lineage, or rare “exceptions” to the rule. The description of a B:T cell hybrid by Ahmed and colleagues provides welcome insights into these fundamental paradigms: if verified, the dual receptor-expressing (BCR+TCR+) DE cells would represent a novel cell type and evidence for a physiologic state of developmental plasticity.
Another novel aspect of the claims made by Ahmed and colleagues is that DE cells in individuals with T1D are enriched for a public clone that may be linked to disease pathogenesis. The existence of public or shared clones suggests that common antigens or selective pressures generate similar immune responses in different people. Sharing of immunoglobulins has been described in diseases such as chronic lymphocytic leukemia (Stamatopoulos et al., 2007) and systemic lupus erythematosus (Isenberg et al., 1993; Pugh-Bernard et al., 2001) and specific antibody heavy/light-chain pairs are associated with binding to a hapten (Cumano and Rajewsky, 1986). A public clone is an even more stringent claim than stereotypy or skewed VH or VH/VL gene usage as public clones are typically identical or highly similar at the amino acid level.
Given that the provocative findings reported by Ahmed and colleagues have potential importance as both a diagnostic measure and a novel mechanism for T1D pathogenesis, we attempted to replicate and expand on their findings by analyzing three independent cohorts of T1D subjects and controls, including studies of sorted DE cells. We leveraged two unique data and cell resources for the analysis of T1D immunopathogenesis in humans: (1) the Human Pancreas Analysis Program (HPAP), an NIH-funded resource for performing deep phenotyping of tissues from human organ donors designed to understand the anatomic, physiologic, metabolic, and immune abnormalities contributing to β cell loss in T1D (Kaestner et al., 2019); (2) the University of Florida Diabetes Institute (UFDI) T1D biorepository comprised of cross-sectional peripheral blood mononuclear cell (PBMC) samples from controls, T1D subjects, and individuals at varying levels of risk based on genetics, first-degree relative status, and autoantibody status (Perry et al., 2018). In addition to the analysis of previously available datasets from HPAP and the UFDI, we performed a robust, statistically powered, and blinded analysis at both sites independently, using the same methodology described by Ahmed and colleagues to identify the DE subset. We also present a large-scale analysis of antibody heavy-chain VH region rearrangements in 40 T1D and 52 control individuals to identify the public clone identified by Ahmed and colleagues in samples stratified by disease, tissue, and HLA DQ8 status. We also analyzed VH rearrangements in sorted DE cells of T1D and control individuals. Finally, we discuss potential reasons why our findings differ from those of Ahmed and colleagues.
RESULTS
Lymphocytes coexpressing BCR and TCR are not enriched in T1D
In their published work (Ahmed et al., 2019), Ahmed and colleagues described double expresser (DE) cells as TCRαβ+IgD+ that they recovered from the circulating CD5+CD19+ B cell subpopulation. Within PBMCs, they reported highly significant enrichment of DE cells in T1D compared to healthy controls (HC), with frequencies in the range of ~1%-2% of CD5+CD19+ cells and 0.1%-0.7% of CD5−CD19+ cells (Figure 1A of Ahmed et al. [2019]). We sought to reproduce their findings.
First, we re-analyzed previously generated flow cytometry data on whole blood samples that were stained using a modified human immunophenotyping panel (HIPC [Maecker et al., 2012]) on UFDI biorepository subjects (T1D n = 232, first-degree relatives [FDR] n = 320, second-degree relatives [SDR] n = 24, 32 islet autoantibody (AAb+) at-risk individuals [RSK] n = 23, and healthy controls n = 246). T1D individuals were shown by Ahmed and colleagues to have increased proportions of DE cells in both CD19+CD5+ and CD19+CD5− populations. While TCRαβ and CD5 were not included in the HIPC panels, we postulated that we could detect DE cells using CD3 as a surrogate for TCRαβ. Using this method, DE events (CD3+CD19+) were detected in similar frequencies to those reported by Ahmed and colleagues in HC, but the difference between HC and individuals with T1D was 0.04 percentage points (Figure 1A). When the distributions of DE events of HC and T1D subjects were overlaid, there was substantial overlap, unlike the clear separation in DE cell frequencies observed by Ahmed and colleagues (Figure 1B). To model the level of similarity between the distributions, we performed a non-parametric bootstrap analysis yielding a 95% confidence interval of (−0.0165, 0.0887) for the difference in the mean DE percentage between control and T1D individuals. Since this interval included zero and the effect size was very modest, these data do not convincingly support a biologically meaningful difference in the DE fraction between subjects with T1D and controls. However, because these data were acquired from whole blood and included a limited number of relevant lineage markers, we were concerned that our analysis strategy was not optimized for rare-event detection of DE cells (see STAR Methods).
Figure 1. IgD+CD19+ cells expressing CD3 are not expanded in T1D patients, their relatives, or seropositive at-risk individuals.

(A) Dot plot from the UFDI biorepository showing the % of CD19+ cells defined as IgD+CD3+ in whole blood of healthy controls (HC), second-degree relatives (SDR), first-degree relatives with 1 or fewer AAb (FDR), 32 AAb+ at-risk individuals (RSK), and type 1 diabetes patients (T1D). Lines indicate mean. Kruskal-Wallis with Dunn’s post-test, ***p < 0.001.
(B) Distribution of DE cells in T1D versus HC, showing substantial overlap. The 95% confidence interval (−0.0165, −0.0887) for the difference in means of a bootstrap analysis included zero.
(C) HPAP dataset of tissues obtained from organ donors. DE cells were defined as CD3+ events out of the total CD19+ population. PBMC, peripheral blood mononuclear cells; Mes LN, mesenteric lymph node; SMA LN, superior mesenteric artery lymph node; PLN pancreatic lymph node from head, body, or tail of the pancreas. ND, non-diabetic; Aab+, auto-antibody-positive; T1D, type 1 diabetic. Bars indicate mean and SD. Two-way ANOVA with Sidak’s correction, ns, non-significant (p > 0.05).
Therefore, we also accessed available flow cytometry data generated by the HPAP consortium (Kaestner et al., 2019; https://hpap.pmacs.upenn.edu) to determine whether DE cells could be detected in PBMCs with a more stringent gating scheme and a live-dead gate. The HPAP analysis also included diabetes-relevant organs, particularly the pancreatic lymph node (PLN), wherein we expected to find DE cells at higher frequencies in T1D and potentially also in autoantibody-positive (AAb+) individuals, who are at elevated risk of developing T1D compared to HC (Sosenko et al., 2013). We re-analyzed data from a broad immune lineage panel (23 markers) to detect DE cells. Because CD5, IgD, and TCRαβ were not included in the panel, we again measured the frequency of DE cells as the fraction of CD19+ cells that co-expressed CD3 in the blood, spleen, mesenteric lymph node (MLN), superior mesenteric artery lymph node (SMA LN), and PLN (segregated by head, body, and tail of the pancreas) of normal donors (ND), T1D, and AAb+donors from the HPAP cohort (Table S1). The tissue samples had comparable and high cell viability (Figure S1A). We found no significant differences in the frequencies of CD3+CD19+ DE events between the groups across all tissues analyzed (Figure 1C). Notably, CD3+CD19+ DE events were not enriched in the PLN of T1D or AAb+ individuals.
Because neither of the preceding analyses used the methodology of Ahmed and colleagues to enumerate the DE cells, we next designed a statistically powered replication experiment using an exactly matching flow cytometry panel (CD19-BV421, CD5-APC, IgD-PE, and TCRαβ-AF488) to the one used by Ahmed and colleagues (disclosed by personal communication with Dr. Hamad) with the addition of a viability dye to distinguish dead cells. Two aliquots of PBMCs per participant (11 HC and 17 T1D, Table S1) were run independently at two sites, UPenn and UFDI. The viability of the cells after thawing was high and comparable between HC and T1D at both sites (Figures S1B and S1C). Investigators at both sites were blinded to donor disease status for the duration of the processing and flow cytometry data analysis. Gating of DE cells included singlet event gating and viability staining (Figures S1D and S1E). Isotype staining and fluorescence minus one controls were used to identify positive events (Figure S1F). In contrast to Ahmed and colleagues, we observed no enrichment of DE cells (IgD+TCRαβ+) within the CD5+CD19+ or CD5−CD19+ population in T1D. Results from both the UPenn (Figures 2A and 2B) and the UFDI cohorts (Figures 2C and 2D) showed a similarly low frequency of DE cells within CD5+CD19+ B cells of T1D and HC, each comparable to the level observed in HC by Ahmed and colleagues.
Figure 2. Two-site replication experiment to assess DE cells in PBMCs.

(A and C) DE events were identified as IgD+TCRαβ+, as in Ahmed et al. (2019), independently at UPenn and UFDI
(B and D) Frequencies of DE events in CD5+CD19+, CD5−CD19+, and CD5+CD19− populations. Two-way ANOVA with Sidak’s correction performed with DE populations matched by subject, ns, non-significant (p > 0.05).
(E and F) Spearman correlations between the frequencies of parent populations (E) or DE events (F) measured by UPenn and UFDI in the PBMCs from the same donors.
To cross-validate the results obtained by UPenn and UFDI, we performed correlations on the frequencies of DE cells and the parent populations (CD5+CD19+, CD5−CD19+, and CD5+CD19−). We observed strong positive correlations between the frequencies of the parent populations obtained at UPenn and UFDI (Spearman’s ρ > 0.75, Figure 2E), indicating the validity of the independent experiments performed on the same PBMCs at the two sites and acquired using different flow cytometers. The correlations between the frequencies of DE cells were weaker (Spearman’s ρ < 0.52) (Figure 2F), likely due to the variability in measuring very low numbers of DE events. For example, in the UFDI dataset using the staining panel of Ahmed and colleagues, DE cells (CD19+CD5+TCRαβ+IgD+) were rare events, comprising 0.013% ± 0.01% (mean ± SD) of live singlet lymphocytes.
Non-specific binding of fluorescently labeled antibodies is a common source of technical artifacts in flow cytometry leading to the apparent presence of rare events with unique, often biology-defying, phenotypes. To further qualify DE events and exclude possible non-specific antibody binding, we stained an aliquot of the same T1D and HC PBMCs with a 22-marker flow cytometry panel (performed at UPenn), including the same markers used by Ahmed and colleagues in addition to lineage markers for all major peripheral blood leukocyte subsets. After size and live/dead exclusion, we identified IgD+TCRαβ+ DE cells and then performed a “clean-up” by gating on events positive for CD45 (pan-leukocyte marker) and excluding events positive for CD15 (neutrophils), CD14 (monocytes), CD16 (monocytes/neutrophils/natural killer [NK] cells), CD56 (NK cells), and CD34 (stem cells) (Figure 3A). The remaining DE events were then expressed as a frequency of their original parent population. As with the lower-dimensional panels, no significant difference was observed in the frequency of DE events between T1D patients and HC (Figure 3B). The frequency of DE cells in each of the parental populations on the 22-color panel was approximately 60% of that observed for the 4-color panel (Figure 3C). Furthermore, DE cells exhibited inconsistent expression for several of the T and B cell markers in the 22-color panel (Figure S2). Only about 50% of the DE cells in both T1D and HC samples co-expressed CD3, an essential component of the TCR complex, suggesting that these cells are not bona fide T cells (Figure 3D). Similar results were obtained at UFDI with an 11-color panel (data not shown). Additionally, the frequency of DE cells measured within subjects between alternative panels (4- versus 22-color markers at UPenn and 4- versus 11-color markers at UFDI) did not show a strong correlation (Figures S3A and S3C), suggesting that detection of DE events is subject to high variability. On the other hand, the parent populations showed a strong correlation, confirming a high degree of reproducibility in the detection of those cell populations using different combinations of reagents (Figures S3B and S3D). Lastly, to exclude that the use of cryopreserved as opposed to fresh samples could have led to our observed low DE cell frequency in T1D, we performed the 4-color panel on PBMCs from four subjects (two HC and two T1D), before and after cryopreservation for 12 weeks. We observed negligible effects of a freeze/thaw cycle on the frequency of CD5+CD19+ DE cells, which were under 0.3% for both HC and T1D (Figures S3E and S3F). Together, these data do not replicate the enrichment of DE cells in the peripheral blood in T1D reported by Ahmed and colleagues and call into question the validity and reproducibility of DE cells as an important feature of T1D.
Figure 3. Exclusion of cells expressing other immune lineage markers reduces the frequency of DE events.

(A) “Clean-up” gating strategy to include CD45+ events and exclude events positive for CD14, CD15, CD16, CD56, and CD34.
(B) Frequency of DE events in the PBMCs of HC and T1D after “clean-up.” Two-way ANOVA with Sidak’s correction, ns, non-significant (p > 0.05).
(C) Comparison of DE events obtained with the minimal 4-color marker panel and the expanded 22-color marker panel. Two-way ANOVA with Sidak’s correction, *p < 0.05, **p < 0.01, ns, non-significant.
(D) Proportion of CD3+ DE cells in patients with T1D versus controls. Unpaired t test, ns, not significant.
The clone-x CDR3 sequence, “CARQEDTAMVYYFDYW,” is not found in bulk cells or in “DE cells” sorted from individuals with T1D or controls
We next set out to identify the public B cell clone (i.e., clone-x) described by Ahmed and colleagues. Clone-x refers to B cells that share a specific antibody heavy-chain and light-chain pair. Here, we are focusing on the heavy-chain rearrangement of the clone-x antibody, which consists of VH4-38-02 (previously called V04b), DH (DH5-18 and/or DH5-5, which are identical in sequence and hereafter referred to as DH5-18∣5-5 [Lefranc et al., 2015]) and JH4. The third complementarity determining region (CDR3) of clone-x, which spans the VH, DH, and JH gene segments and the junctions between them, consists of the following AA sequence: “CARQEDTAMVYYFDYW.” Embedded within this CDR3 sequence is a core epitope consisting of the sequence “DTAMVYYFD” (hereafter referred to as the “core motif”) that Ahmed and colleagues identified as a peptide that can bind strongly to the T1D risk-associated class II HLA, DQ8.
To find clone-x, we analyzed antibody heavy-chain gene rearrangements from bulk populations of B cells in the peripheral blood and spleen from two independent cohorts of T1D subjects and controls (Table S2). Our method for amplifying and sequencing antibody heavy-chain rearrangements (Meng et al., 2017) was agnostic to the VH gene and generated longer reads for more reliable VH gene identification than the assay used by Ahmed and colleagues (Zhang et al., 2015). We chose to include spleen samples in the analysis because we already had existing data from the HPAP cohort and because the spleen is a rich source of B cells, including memory cells, which might be enriched for pathogenic clones. We analyzed 92 different individuals from the HPAP and the UFDI cohorts, amassing a total dataset with over 1.6 million clones and 514 sequencing libraries (Figure 4A).
Figure 4. Analysis of shared CDR3 sequences and clones.

(A) Overview of study subjects and IgH sequencing data. Numbers of subjects with peripheral blood (PBL) or spleen (SPL) samples, numbers of sequencing libraries, clones (same VH, same JH, same CDR3 length and 85% amino acid similarity in CDR3), numbers of valid reads, and numbers of times that sequences matching the clone-x CDR3 amino acid (AA) sequence of “CARQEDTAMVYYFDYW” are found.
(B) Levenshtein distance (in AA) of all rearrangements from the clone-x CDR3 string (sequences within 4 AA are listed in Table S3).
(C) Number of public CDR3 sequences (sequences shared by two or more subjects) per subject (each dot is a subject). The median frequency of CDR3 sequences per subject is not significantly different between individuals with T1D (orange) and controls (blue), p = 0.49, Mann-Whitney.
(D) Correlation plot with total clone count (subject level) versus number of public CDR3 sequences (CDR3 AA sequences shared by two or more subjects). r2 is 0.77 for controls and 0.88 for T1D.
(E) Total number of identical CDR3 AA sequences shared versus the number of subjects that share them.
(F) Correlation between shared CDR3 number and the DE cell fraction. r2 is 0.31 for controls and 0.09 for T1D.
We were unable to find a single instance of the clone-x CDR3 AA sequence in any sample from any subject in our dataset. We further searched for sequences that were similar to the clone-x CDR3, differing by replacement, addition, or deletion of amino acids from the clone-x CDR3 (Levenshtein, 1966). When we analyzed the resulting CDR3 AA edit distances from bulk sequenced B cells from blood and spleen samples (Figure 4B), the distributions between T1D and non-T1D subjects overlapped, implying that patients with T1D did not have antibody repertoires that were enriched for CDR3 sequences that were similar to clone-x. There were no clones within a single AA of the clone-x CDR3 sequence, and only three clones, each occurring in only one individual, were within 2 AA of clone-x. Clones within 4 AA of the clone-x CDR3 AA mostly used DH5-18∣5-5, without evidence of skewing toward the clone-x VH gene, VH4-38-02 (Table S3). We also performed this edit distance analysis on the same samples with the clone-x core motif sequence and did not observe enrichment for the core motif in T1D or DQ8+ individuals (Figures S4A and S4B).
We searched for evidence of other public CDR3 sequences in T1D versus control subjects in the bulk sequencing libraries from peripheral blood and spleen samples. The average number of shared CDR3 sequences per subject was not increased in individuals with T1D compared to controls (Figure 4C). Instead, the major driver of the number of shared sequences was the number of clones in the sequencing library of each subject (Figure 4D). Furthermore, the number of public CDR3 sequences shared by 2, 3, 4, 5, and 6 or more subjects was similar in T1D and controls (Figure 4E). Finally, the number of public CDR3 sequences did not correlate with the DE fraction in either T1D or controls (Figure 4F).
As antibody sequences can undergo somatic hypermutation, we also considered the possibility that searching for sequences with identical CDR3 AA sequences was too restrictive a definition of public antibodies. We therefore also searched for clonally related sequences between different subjects in our dataset (defining clones as sharing 85% AA identity in the CDR3 and having the same VH, JH, and CDR3 length). Here too, we noted that the numbers of shared clones (Figure S4C) and that the numbers of subjects who shared them (Figure S4D) did not differ between T1D and controls. The number of shared clones in each subject also did not correlate with the DE cell fraction in subjects with or without T1D (Figure S4E).
The clone-x antibody consists of sequences that closely match the corresponding germline VH, DH, and JH gene segments (i.e., they are templated and unmutated). Therefore, we wondered whether the antibody repertoire in T1D might be skewed toward the VH or DH genes themselves. We also considered the possibility that clones harboring these VH, DH genes or the clone-x core motif (“DTAMVYYFD”) might be larger or more numerous in individuals who have T1D, are DQ8+, or exhibit a higher proportion of DE cells. Out of the 92 subjects with IgH sequencing data on bulk PBMC or spleen samples, DQ8 typing information was available in 90, with the DE cell fraction measured by flow cytometry in 57 (Figure 5). For VH gene usage, we combined all of the sequencing libraries for each subject and weighted the clones based upon their copy-number fractions (a proxy for clone size). VH gene usage between T1D individuals and controls was similar overall, with subtle differences, consistent with earlier reports (Figure S5; Seay et al., 2016; Thomas, 1993). VH4-38-02 was deleted from several individuals, consistent with previous findings (Figure S6; Gidoni et al., 2019). When it was present, the frequency of VH4-38-02 usage did not differ significantly between T1D and controls, nor was it enriched in DQ8+ individuals (Figure 5B). We also did not observe a significant difference in DH5-18∣5-5 usage in T1D and controls or between groups that were stratified by DQ8 status (Figure 5C). We also did not observe a statistically significant correlation between the frequency of DE cells and the copy-number fraction of VH4-38-02 (Figure 5D) or DH5-18∣5-5 rearrangements (Figure 5E). We also did not observe enrichment for clones that harbored the clone-x core motif in T1D or in DQ8+ individuals (Figure 5F).
Figure 5. Clone-x VH, DH, and core motif usage by disease and DQ8 status.

(A) Number of subjects (N) who were DQ8 positive and negative, stratified by disease status.
(B) Total copy-number fraction of VH4-38-02 clones in each donor versus DQ8; donors (n = 27) with no IGHV4-38-2 clones are excluded (see Figure S4 and STAR Methods).
(C) Total copy-number fraction of IgD5-18∣5-5 clones (excluding minor rearrangements that were tied with other DHs) versus DQ8; donors with no IGHD5-18∣5-5 clones are excluded.
(D–F) (D) VH4-38-02 copy fraction versus DE cell (percentage of B cell gate). (E) DH5-18∣5-5 copy fraction versus DE cell (percentage of B cell gate). (F) Fraction of clones (weighted by copies) containing the clone-x core motif CDR3 AA sequence (“DTAMVYYFD”) in HC versus T1D and in DQ8+ versus DQ8− individuals. Each dot is a subject. Comparisons in (B), (C), and (F) were made on median values by rank sum test (Mann-Whitney). DE calculations used in (D) and (E) combine PBMC and spleen samples from UFDI and HPAP (DE data shown in Figures 1 and 2) and used different flow panels for DE quantification, ns, not significant with a one-sided cut-off p < 0.01.
Based upon our analysis of bulk IgH sequence data on blood and spleen samples, we conclude that clone-x and clones with similar heavy-chain sequences are infrequent in the global repertoire of individuals with or without T1D. However, it remained possible that clone-x could be more highly represented in the rare DE cell subset and was insufficiently sampled. We therefore also searched for the clone-x CDR3 in sorted DE cells from spleens of three organ donors: HPAP015 and HPAP032 (both T1D) were DQ8 positive, and HPAP017 (control) was DQ8 negative (sorting strategy in Figure S7A). We recovered >2,700 clones from the BCR/TCR double positive gate (“DE cells”) and >30,000 clones from CD5+ B cells (Figure 6A). As with the repertoire analysis on bulk PBMCs and spleen samples, we found no instance of the clone-x CDR3 within the sorted DE cells from these donors. The CDR3 AA edit distance distribution of sequences with similarity to the clone-x sequence was the same, irrespective of disease or B cell subset (Figure 6B; Table S3). Unlike the findings of Ahmed and colleagues, the IgH sequences in both CD5+ B cells and DE cells were diverse, with no clearly dominant clonal expansions (Figures 6C, 6D, and S7). VH4-38-02 and DH5-18∣5-5 rearrangements were very infrequent in both CD5+ B cells and DE cells (Figure 6E). Among VH4-38-02 rearrangements, there was no appreciable skewing toward DH5-18∣5-5 usage (Figure 6F). In fact, none of the 32 VH4-38-02 rearrangements recovered from DE cells used DH5-18∣5-5. With respect to the clone-x core motif, only one clone in the entire sorted cell dataset contained the motif and was present in the non-DE cell subset (CD5+TCRαβ− B cells) in HPAP015 at a frequency of 0.000066. Also in contrast to the findings reported by Ahmed and colleagues, we observed that DE cells and CD5+ TCRαβ− B cells included some clones with somatic hypermutations (Figure S8).
Figure 6. Antibody VH rearrangement data on sorted DE cells from T1D versus control subjects.

(A) Overview of study subjects and sorted B cell subsets from organ donor spleen (SPL). CD5+ B cells are CD5+, CD19+, TCRαβ− lymphocytes. DE, double expresser cells are TCRαβ+, CD19+, CD5+ lymphocytes. Libraries, clones, and reads (copies) are as defined in Figure 4.
(B) Levenshtein distance distribution of sorted cells, by population and disease status.
(C and D) (C) Copy-number percent of top 20 clones in the DE and (D) CD5+ B cell sorts. Orange wedges in pie charts show percentage of total library copies taken up by the top 20 clones. None of the clones in the top 20 copy number rearrangements in HPAP-032 had VH4-38-02 rearrangements (data on the other two donors are in Figure S7).
(E and F) (E) Fractions of clones with VH4-38-02 rearrangements from sorted DE and CD5+ B cells; (F) DH gene usage for VH4-38-02 rearrangements in sorted subsets. DH gene usage is shown, with each unique rearrangement only counted once. DH gene usage is given by family, except for DH5-18∣5-5, where sequences with this specific DH gene assignment are highlighted in orange. “Other DH5” indicates a DH5 gene other than DH5-18∣5-5. TIES indicate DH genes with more than one DH family assignment. UNKNOWN indicates no DH gene assignment. For (B), (E), and (F), the data for HPAP-015 and HPAP-032 were combined for T1D and HPAP-017 was the control. Each symbol is an individual. ns, not significant by Mann-Whitney rank sum test, α = 0.01.
DISCUSSION
Identifying predictive biomarkers of T1D susceptibility and targetable mechanisms underlying T1D pathogenesis is of paramount importance in the effort to reduce T1D risk and develop effective therapies. The findings of Ahmed and colleagues provided an exciting and novel avenue for T1D and for immunological research. We therefore sought to confirm two of their central findings that, in our opinion, represent the lynch pin of their study, namely: (1) the association of a novel BCR/TCR dual-positive cell subset (the DE cell) with T1D disease status, and (2) the presence of a public B cell clone (clone-x) identified in DE cells of T1D individuals. If either of these points could not be confirmed, the remaining findings of the Ahmed study would remove their immediate interest as a biomarker or potential therapeutic target in T1D. We did not try to confirm the existence of the DE subset, nor the mechanistic association between the clone-x CDR3-derived core motif peptide and T cell stimulation, for the naive T cell repertoire of most individuals is capable of responding to many MHC-binding neo-epitopes in vitro (Wooldridge et al., 2012; Caserta et al., 2008; Jedema et al., 2011). As we have presented, we found no association between the DE cell phenotype and T1D and no evidence that clone-x or highly similar antibody sequences are enriched in T1D, DQ8+, or in cells harboring the DE phenotype.
Why were Ahmed and colleagues able to find DE cells at increased frequency in T1D and a public B cell clone enriched in DE cells in patients with T1D, yet we failed to do so? We are concerned that technical artifacts during data generation and analysis may have led to their results. Regarding the immunophenotyping data, many of the DE cells may be doublets or other forms of noise for the following reasons. (1) Their analysis scheme to identify DE cells focused on larger events within the lymphocyte gate (see their Figure S1A). If one compares a conventional small lymphocyte gate to a large gate (Figure 7A), the large gate enriches for DE cells, which may include doublets (Figure 7B). (2) There is discordance between the frequency of DE cells identified by the 4-color panel that they use, compared to our more stringent 22-color panel with multiple lineage markers (Figure 3C). (3) The flow plot in Figure 1A from Ahmed and colleagues shows off-scale events on the top and right axes that may contribute toward spuriously elevated DE cell frequencies. (4) The expression of other markers on DE cells, including CD3, CD45, and CD34 (Figures 3D and S2), is heterogeneous and inconsistent, suggesting the absence of a distinct DE cell lineage. This marker heterogeneity carries through in the single-cell RNA sequencing (scRNA-seq) data (see Figure 1 in Ahmed et al.), with some cells expressing monocyte markers. These heterogeneous scRNA-seq phenotypes could also be due to ambient RNA contamination from dead or dying cells (Lun et al., 2019). There are several potential explanations for sources of noise in rare-event detection by flow cytometry including doublets (Burel et al., 2020), trogocytosis, mRNA transfer by exosomes (Valadi et al., 2007), or binding of the BCR on the DE cell to the anti-TCR antibody.
Figure 7. Technical considerations.

(A) Gating on lymphocytes on the basis of forward versus side scatter area using a small versus less restrictive (large) lymphocyte gate.
(B) Differences in DE cell frequencies with small versus large lymphocyte gate. Two-way ANOVA with Sidak’s correction, ****p < 0.0001.
(C) All top-copy sequences corresponding to clone-x and a second dominant clonotype are identical in T1D1, T1D2, and T1D3. Nucleotide sequence alignments of top-copy clones found in three donors in the Ahmed paper. Dashes indicate identity between sequences and carets indicate identity in the CDR3 nucleotide sequence (given in capitals).
(D) Nucleic acid sequence alignments of top-copy clone (clone-x) with different VHs and identical CDR3 nt sequence (consistent with hybrid PCR products). To the left of each sequence is the sample name followed by an underscore and then the most similar VH gene assignment (e.g., 1-18 = VH1-18). The clone-x CDR3 amino acid (AA) sequence is shown above the corresponding nucleotides.
Regardless of the DE cell and its questionable validity as a bona fide cell type, the DE cell frequency reported by Ahmed and colleagues clearly separated T1D subjects from controls, whereas we found no such difference. The discrepant outcome in the Ahmed study might have arisen due to batch effects, for example if there was uncontrolled variation in cell processing, instrument usage, analysis procedure, or operators during the analysis of one cohort versus the other. In our experiments, we used samples from T1D subjects and controls subjected to identical processing conditions. We also controlled for operator differences, instruments, and methodology by performing a blinded analysis of aliquots of the same randomly selected T1D and control samples at two geographically separate sites (UPenn and UFDI). We used a 4-color analysis panel identical to that employed by Ahmed and colleagues, as well as a more robust 22-color panel to quantify the DE cell population.
The other major finding reported by Ahmed and colleagues was the presence of a public clone (clone-x) dominantly expressed in DE cells from persons with T1D. In fact, they observed two shared clones for the three T1D individuals from whom they sorted CD19+CD5−TCRαβ− “Bcon” cells and CD19+CD5+TCRαβ+ DE cells. We could not find clones with identical CDR3 sequences to either clone in bulk sequencing libraries from peripheral blood and spleen in 92 individuals, including 40 subjects with T1D. We also did not observe enrichment of rearrangements similar to the clone-x heavy chain in sorted DE cells compared to CD5+ B cells in individuals with or without T1D.
Public clones arise from different cell progenitors, typically resulting from different TCR or antibody gene rearrangements. By virtue of amino acid code degeneracy, many different independently generated nucleotide sequences in public clones can encode the same CDR3 AA sequences (Quigley et al., 2010; Venturi et al., 2008). Curiously, the two top-copy-number public clone nucleotide sequences reported by Ahmed and colleagues (Figure 7C, immunoSEQ 10.21417/RA042019), one of which is clone-x (with the CDR3 AA sequence of “CARQEDTAMVYYFDYW”), are identical at the nucleotide level in all sequencing libraries from all three T1D subjects. This raises the specter of contamination or mixing of samples.
The authors comment on independent sequences related to clone-x with different VH genes but shared CDR3 sequence, offering this as added proof that they have bona fide public clones. However, these sequences (Figure 7D) exhibit a sharp boundary between dissimilar VH genes and still have identical CDR3 nucleotide sequences, a finding more consistent with hybrid PCR products. While Ahmed and colleagues claim that clone-x is public, their own searches of 37 million unique IgH sequences (DeWitt et al., 2016) and insulin-binding B cell clonotypes from the nPOD archive (Seay et al., 2016), did not reveal the clone-x CDR3. Rather, their searches mirror our findings that the clone-x CDR3 AA sequence is absent from all 23 million sequences generated in this study.
The authors provide additional support for their public clone in the form of a sequence-specific PCR (SSP) assay to amplify sequences related to clone-x in subjects with and without T1D. However, the fact that not all of the subjects amplify with the SSP assay could be due to deletions of the gene encoding VH4-38-02 in some people but not in others (Figure S6; Gidoni et al., 2019). We did not find an increased frequency of VH4-38-02 or DH5-18∣5-5 rearrangements (or their combination) in individuals with T1D or in sorted DE cells. We also could not confirm their finding that DE cells had unmutated antibodies. In our analysis, at least some DE cells harbored somatically mutated IgH. It is possible that our analysis of DE cells, which used spleen samples, detected more memory B cells with somatic mutations than the analysis of Ahmed and colleagues, which was performed on DE cells sorted from the peripheral blood.
One notable feature of the DE cells described by Ahmed and colleagues is CD5 expression. CD5 marks a subset of B1 B cells in mice (so-called B1a cells [Baumgarth, 2011]) as well as ~75% of circulating B1-like cells in humans (Griffin et al., 2011). B1 B cells often harbor polyreactive receptors likely useful for binding to commonly encountered pathogens and cleaning up cellular debris (Hayakawa et al., 1983). Many B1 B cells are created in fetal life at a time when terminal deoxynucleotidyl transferase is not expressed at high levels, leading to shorter N-additions (Feeney, 1990). The paucity of non-templated nucleotides in clone-x could reflect the intrinsic “B1-ness” of the CD5+ cells being studied by Ahmed and colleagues. Whether B1-like B cells are enriched for public clones is unresolved.
Finally, Ahmed and colleagues show that peptides derived from the hypervariable sequences within the antibody heavy-chain CDR3 sequence of clone-x can stimulate T cells in a DQ8-restricted manner. We did not find an association between disease or DQ8 status and the usage of the rearrangements in clone-x, namely VH4-38-02 and DFH5-18∣5-5 or the core motif. Furthermore, the frequency of VH4-38-02 and DFH5-18∣5-5 usage did not correlate with the DE cell fraction, and there were too few rearrangements containing the core motif for further analysis. In previous studies of insulin-binding B cells (Thomas, 1993) and PLNs from T1D compared to controls (Seay et al., 2016), there was increased usage of VH3 family members but not increased usage of VH4-38-02.
Taken together, our results call into question the reported association of the DE cell phenotype with T1D and the existence of a public B cell clone within the DE population. While these efforts raise concerns over the validity of DE cells as a biomarker or mechanism of T1D pathogenesis, our data provide an opportunity for further discovery. The data can be used to determine whether potential immune cell or immune repertoire motifs are associated with T1D, to compare against libraries of antibodies with known auto-specificities (Seay et al., 2016) as well as for sample normalization, motif discovery, and disease-specific immune subset analysis (Olson et al., 2019; Miho et al., 2019). Another interesting and under-explored aspect of our data is the analysis of the autoantibody-positive control subjects, who have broken tolerance to a subset of T1D-associated autoantigens (Battaglia et al., 2020). These data, along with detailed information on the study subjects, provide a valuable resource for investigators who are studying T1D and autoimmunity.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Michael R. Betts (betts@pennmedicine.upenn.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Immune repertoire profiling data have been shared in an AIRR-compliant fashion (Rubelt et al., 2017); the data have been uploaded onto GenBank/SRA (SRA: PRJNA604535). Flow cytometric data for the 2-site blinded study are available at ImmPort (https://www.immport.org) under study accession number SDY1625.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human samples obtained by the UFDI
Informed consent for participants termed the University of Florida Diabetes Institute (UFDI) biorepository cohort was obtained from subjects enrolled from outpatient clinics of the University of Florida, Gainesville, Florida; Nemours Children’s Hospital, Orlando, Florida; and Emory University, Atlanta, Georgia, under Institutional Review Board (IRB)- approval at each facility (IRB #201400709). Organ Procurement Organizations (OPO) partnering with nPOD to provide research resources are listed at https://www.jdrfnpod.org/for-partners/npod-partners/. Coded nPOD human donor spleen samples were obtained and processed by nPOD core laboratory under a University of Florida Institutional Review Board-approved protocol (IRB201600029). Peripheral blood was collected in sodium heparin for processing PBMCs, EDTA for DNA isolation and whole blood immunophenotyping, and clotting tubes for serum collection. Serum samples were stored at −20°C. Genomic DNA was purified using QiaAmp DNA Blood spin columns in conjunction with QiaCube automated processors (QIAGEN) and was stored at −20°C. Splenocytes were obtained after mechanical dissociation, collagenase digestion, ACK buffer treatment to remove erythrocytes, and filtration, followed by cryopreservation using standard procedures. Autoantibodies were measured from serum as previously described (Perry et al., 2018). Subjects were genotyped using a custom UFDIchip Axiom array with the Precision Medicine Research Array as the base. UFDIchips were processed on an Affymetrix Gene Titan instrument with external sample handling on a BioMek FX dual arm robotic workstation. Genetic data underwent standard quality control procedures at the SNP, sample, and plate levels using AxiomTM Analysis Suite 3.0 (ThermoFisher Scientific) set to the default stringency thresholds as recommended. HLA was then imputed to 4-digits using AxiomTM HLA Analysis software (ThermoFisher Scientific).
Human samples obtained by the HPAP
Details on the samples obtained through the HPAP are available online at https://hpap.pmacs.upenn.edu and described (Kaestner, et al., 2019). Whole organ samples are obtained via the local organ procurement organization or via University of Florida/nPOD. The consent for procurement of research pancreata was obtained by staff of the organ procurement organization. All human samples were de-identified and IRB exempt. Donors were evaluated for autoantibody status (GAD, IA-2, IAA and ZnT8), C-peptide levels and undergo clinical chart review. Clinical data including diagnosis of T1D, HbA1c, BMI, age, gender and HLA type are recorded in the HPAP database. Additional data from histology, immunophenotyping, repertoire profiling, metabolic and transcriptomic studies are generated on all HPAP donors, providing a shared and publicly available comprehensive resource.
METHOD DETAILS
Design of two-site blinded experiment to assess DE cell frequency
Previously defined T1D and age/sex matched T1D- controls were randomly selected from the UFDI biorepository. Sample numbers per group were set to be equivalently powered to Ahmad and colleagues. Paired aliquots of cryopreserved PBMC were selected to include DQ8 subjects, and to be as age- and sex-matched as possible (Table S1). Samples were de-identified by UFDI convention and 1 aliquot from each donor was shipped in liquid nitrogen vapor phase to UPenn. Subject status was unblinded following completion of all flow cytometry and analyses.
Cell isolation from human specimens
PBMCs were isolated by density gradient centrifugation using Ficoll-Paque (GE Healthcare). Pancreatic and mesenteric LN obtained from brain-dead organ donors were cleaned of surrounding fat, cut into small pieces (~2mm x 2mm) and placed in R10 media (RPMI + 10% FBS + 1% penicillin/streptomycin + 2mM L-glutamine) supplemented with 10U/mL DNase I (Roche). Cells were extracted from the tissue with the blunt end of a 5mL syringe plunger and filtered through a 70 μm strainer. Approximately 30 g of spleen tissue was placed in R10 supplemented with 10U/mL DNase I and X collagenase D (source) and cut into 4mm x 4mm pieces. The tissue was then mechanically dissociated using a GentleMACS™ (Miltenyi Biotech) and incubated at 37°C for 15 min. After digestion cell suspension was filtered through a 100 μm strainer and erythrocytes were lysed by ACK buffer (ThermoFisher Scientific). Spleen mononuclear cells were isolated by density gradient centrifugation using Ficoll-Paque. Viable cell suspensions from peripheral blood, LN and spleen were cryopreserved in FBS + 10% DMSO and stored at −150°C until thawing.
Flow cytometry on whole blood (UFDI samples, Figure 1A)
Whole blood was stained fresh at 200 μl/test with antibody cocktail, followed by simultaneous red cell lysis and fixation (Fix/Lyse, ThermoFisher) per manufacturer protocol. Cells were washed in 2% FBS in PBS and stored at 4oC until acquisition on a BD LSR Fortessa flow cytometer. The B cell antibody cocktail was emulated from the “B cells” antibody panel suggested by the Human Immunophenotyping Consortium (Maecker et al., 2012) and consisted of PacBlue-CD3, FITC-IgD, PerCP/Cy5,5-CD19, PE-CD24, PE/Cy7-CD27, APC-CD38, and APC/H7-CD20. Data were analyzed with FlowJo v9 (FlowJo LLC). Singlet events were gated by FSC-H/FSC-W as well as SSC- H/SSC-W, followed by a FSC-A/SSC-A lymphocyte gate. Total B cells were gated as CD19+ lymphocytes, and DE cells were gated as CD3+IgD+ within total B cells, where CD3 was used as surrogate for TCRαβ.
Flow cytometry on cryopreserved cells (HPAP samples, Figures 1C and 2 and 3)
Cryopreserved cells were thawed by gentle shaking in a 37°C water bath, washed with R10 medium and assessed for viability. Up to 3x106 viable cells were used per sample. Cells were first stained with a fixable amine reactive viability dye (ThermoFisher) together with TruStain Fc’ receptor blocker (BioLegend) for 10 min at room temperature. Cells were subsequently surface stained with a cocktail of fluorochrome- conjugated antibodies at predetermined optimal dilutions for 20 min at room temperature (see Key Resources Table for antibody clones and conjugates). Cells were then washed with FACS buffer (PBS + 2% FCS + 0.05% sodium azide), fixed with 1% formaldehyde and stored at 4°C until acquisition. Data were acquired on a flow cytometer (BD LSR II, BD FACSymphony or Cytek Aurora [405nm, 488nm, 640nm]) and analyzed with FlowJo v9 and v10 (FlowJo LLC).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BUV395 anti-human CD11c (B-ly6) | BD Biosciences | Cat#563788;RRID: AB_2744274 |
| BUV496 anti-human CD8 (RPA-T8) | BD Biosciences | Cat# 564804; RRID: AB_2744460 |
| BUV563 anti-human CD45RA (HI100) | BD Biosciences | Cat# 565702; RRID: AB_2744407 |
| BUV661 anti-human CD38 (HIT2) | BD Biosciences | Cat# 565069; RRID: AB_2744377 |
| BUV737 anti-human CD25 (2A3) | BD Biosciences | Cat# 564385; RRID: AB_2744342 |
| BUV805 anti-human CD3 (UCHT1) | BD Biosciences | Cat# 565515; RRID: AB_2739277 |
| BV421 anti-human CD5 (L17F12) | BioLegend | Cat# 364030; RRID: AB_2734408 |
| APC anti-human CD5 (L17F12) | BioLegend | Cat# 364016; RRID: AB_2565726 |
| BV480 anti-human CD14 (MϕP9) | BD Biosciences | Cat# 566190; RRID: AB_2739585 |
| BV605 anti-human HLA-DR (G46-6) | BD Biosciences | Cat# 562845; RRID: AB_2744478 |
| BV650 anti-human CD27 (O323) | BioLegend | Cat# 302828; RRID: AB_2562096 |
| BV711 anti-human CD16 (3G8) | BioLegend | Cat# 302044; RRID: AB_2563802 |
| BV785 anti-human IgM (MHM-88) | BioLegend | Cat# 314544; RRID: AB_2800832 |
| FITC anti-human CD15 (HI98) | BD Biosciences | Cat# 555401; RRID: AB_395801 |
| BB790 anti-human CD4 (SK3) | BD Biosciences | Cat# 624296; RRID: N/A |
| PE anti-human CD19 (HIB19) | BioLegend | Cat# 302208; RRID: AB_314238 |
| BV421 anti-human CD19 (HIB19) | BD Biosciences | Cat# 562440; RRID: AB_11153299 |
| PE-CF594 anti-human IgD (IA6-2) | BD Biosciences | Cat# 562540; RRID: AB_11153129 |
| PE anti-human IgD (IA6-2) | BioLegend | Cat# 348204; RRID: AB_10553900 |
| PE-Cy5 anti-human CD56 (B159) | BD Biosciences | Cat# 555517; RRID: AB_395907 |
| PE-Cy5.5 anti-human CD34 (581) | Thermo Fisher | Cat# CD34-581-18; RRID: AB_2536504 |
| PE-Cy7 anti-human CD21 (Bu32) | BioLegend | Cat# 354912; RRID: AB_2561577 |
| APC anti-human TCRαβ (IP26) | BioLegend | Cat# 306718; RRID: AB_10612569 |
| AF488 anti-human TCRαβ (IP26) | BioLegend | Cat# 306712; RRID: AB_528967 |
| AF700 anti-human CD45 (HI30) | BD Biosciences | Cat# 560566; RRID: AB_1645452 |
| APC-Cy7 anti-human CCR7 (G043H7) | BioLegend | Cat# 353212; RRID: AB_10916390 |
| PacBlue anti-human CD3e (UCHT1) | BioLegend | Cat# 300417; RRID: AB_493094 |
| AF488 anti-human IgD (IA6-2) | BioLegend | Cat# 348216; RRID: AB_11150595 |
| PerCP/Cy5.5 anti-human CD19 (HIB19) | BioLegend | Cat# 302230; RRID: AB_2073119 |
| PE anti-human CD24 (ML5) | BioLegend | Cat# 311106; RRID: AB_314855 |
| PE/Cy7 anti-human CD27 (O323) | BioLegend | Cat# 302838; RRID: AB_2561919 |
| APC anti-human CD38 (HB7) | BioLegend | Cat# 356606; RRID: AB_2561902 |
| APC/H7 anti-human CD20 (2H7) | BD Biosciences | Cat# 560853; RRID: AB_10561681 |
| Biological Samples | ||
| Human peripheral blood mononuclear cells | This paper (Table S1) | N/A |
| Human blood and lymphoid tissues from organ donors | This paper (Table S1) | N/A |
| Critical Commercial Assays | ||
| Live/Dead Fixable Aqua Dead Cell Stain Kit | Thermo Fisher | Cat# L34957 |
| Deposited Data | ||
| Immune repertoire profiling data | Luning Prak Lab (UPenn) | SRA: PRJNA604535 |
| Flow cytometry data | This paper (2-site replication expt.) | ImmPort study accession SDY1625 |
| Oligonucleotides | ||
| Primers for amplification (FR1 + JH cocktail) and sequencing of IgH rearrangements (Methods are provided in the STAR Methods, primer sequences are listed in the supplemental data of the Meng et al. paper). | Meng et al., 2017 | N/A |
| Software and Algorithms | ||
| FlowJo | FlowJo | https://www.flowjo.com/solutions/flowjo |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImmuneDB (analysis and visualization of IgH rearrangement data) | Rosenfeld et al., 2018b | https://immunedb.readthedocs.io/en/latest/ |
| R: The R Project for Statistical Computing | The R Foundation | https://www.r-project.org |
Cell sorting for antibody heavy chain rearrangement sequencing
Single cell suspensions from 3 HPAP spleen samples (two T1D and one autoantibody positive, donors HPAP015, HPAP032 and HPAP017, respectively) were stained as described above with the exception of the last formaldehyde fixation step. CD19+CD5+IgD+TCRαβ+, CD19+CD5+IgD−TCRαβ+ DE cells and CD19+CD5+TCRαβ− B cells were sorted based on the sorting strategy published by Ahmed and colleagues (see Figure S7, this paper). Cells were sorted into cell lysis solution (QIAGEN, Valencia, CA) for genomic DNA extraction.
Antibody heavy chain rearrangement sequencing
Genomic DNA was isolated from sorted or bulk cell populations using the QIAGEN Gentra Puregene Kit (QIAGEN, Cat. No. 158388). Antibody heavy chain variable region rearrangements were amplified using framework region (FR)1 and JH primers as described in (Meng et al., 2017) and (Rosenfeld et al., 2018a) with 100 ng of input DNA per library (replicate) and up to 6 replicates per donor. Sequencing libraries were prepared using 2 × 300 bp paired-end kits (Illumina MiSeq Reagent Kit v3, 600-cycle, Illumina Inc., San Diego, Cat. No. MS-102-3003).
Antibody heavy chain sequence data analysis
Sequencing data were quality controlled with pRESTO (Vander Heiden et al., 2014), annotated with IgBLAST, (Ye et al., 2013) and imported into ImmuneDB v0.29.6 (Rosenfeld et al., 2018b) for further processing and data visualization. To group related sequences together into clones, ImmuneDB hierarchically clusters sequences with the same VH gene, same JH gene, same CDR3 length, and 85% identity at the amino acid level within the CDR3 sequence (Meng et al., 2017). Clones with consensus CDR3 sequences within 2 nucleotides (Bolotin et al., 2015) of each other were further collapsed to reduce the impact of incorrect gene calls. A subject was considered to have VH4- 38-02 if there existed at least one clone in that subject that had both a VH-gene call of VH4-38-02 (including ties) and an average level of mutation from the closest germline of less than 6%. For the edit distance analysis relative to clone-x, the Levenshtein distance (Levenshtein, 1966) was calculated from CARQEDTAMVYYFDYW to each clone’s consensus CDR3 amino acid sequence. No copy number cutoff or filtering was applied to the clones in our dataset in order to find potentially rare clones that were similar to clone-x.
HLA genotyping
High resolution HLA typing of class I and class II alleles on all HPAP donors was performed by next generation sequencing in the Clinical Immunology Laboratory at the Hospital of the University of Pennsylvania. For UFDI subjects, class I and class II HLA was imputed to 4-digits using AxiomTM HLA Analysis software on a custom SNP array with a Precision Medicine Research Array base (ThermoFisher).
QUANTIFICATION AND STATISTICAL ANALYSIS
Immunophenotyping data
The percent of DE cells for all groups in the UFDI biorepository violated the assumption of normality, thus the data were analyzed using the nonparametric Kruskal-Wallis test followed by Dunn’s post-test (Prism version 7.0). P value < 0.05 was set as the level of significance for all comparisons. A 95% confidence interval for the mean difference in DE percent between T1D and Controls was calculated using a non-parametric bootstrap procedure (Efron and Tibshirani, 1986). We repeatedly calculated the mean difference of DE % in 100,000 bootstrap samples. The mean difference values at the 2.5th and 97.5th percentile of all values obtained were used as the boundaries of the 95% confidence interval. Calculations were performed in R v3.6.1 with set.seed(99) used for reproducibility.
Levenshtein distance
The Levenshtein distance was calculated from the clone-x CDR3 AA, “CARQEDTAMVYYFDYW,” to every other clone’s consensus amino-acid CDR3 sequence. The distance indicates the number of insertions, deletions, and substitutions required to change the x-clonotype CDR3 AA sequence into the consensus CDR3 sequence of each clone (Levenshtein, 1966). The fraction of total clones represents the number of clones at a given Levenshtein distance divided by the total number of clones in the dataset. A similar calculation was performed for edit distances from the core motif AA sequence of “DTAMVYYFD.”
Public CDR3s in controls and individuals with diabetes
The number of subjects sharing identical CDR3 AA sequences were tabulated for both controls and individuals with T1D. The distributions of these frequencies were compared using a Mann-Whitney test yielding p values which were not significant at α = 0.01.
Correlation between number of clones and public clones
The number of total clones was calculated for each subject and plotted versus the number of clones in that subject that share an exact CDR3 amino-acid sequence with at least one other subject, regardless of disease status. The coefficient of determination, or r2, was calculated independently for individuals with T1D and controls.
Comparing VH4-38-02 and DH5-18∣5-5 usage between T1D/Control and DQ8 ±
The total number of clones with a VH-gene call of VH4-38-02 or DH-gene call DH5-18∣5-5 was independently calculated. Within each subject, the sum of total sequence copies within the clones with that gene call was divided by the total number of copies across all the clones. The donors were then stratified by DQ8 and disease status. Mann-Whitney tests between the different populations yielded no significant β values at α = 0.01.
ADDITIONAL RESOURCES
This study has used flow cytometric and immune repertoire sequence data generated by the HPAP at the University of Pennsylvania and flow cytometry data generated at the University of Florida. Clinical information and the datasets used on HPAP subjects can be accessed at https://hpap.pmacs.upenn.edu. Please refer to Table S1 (immunophenotyping subjects) and Table S2 (immune repertoire subjects) in the electronic supplement for more details.
Supplementary Material
Highlights.
DE cells not increased in blood or lymphoid tissue of T1D subjects compared to controls
Failure to replicate results of Ahmed et al. with identical FACS assay
“Public” clone-x BCR sequence not found in blood or spleen of T1D and controls
CDR3 AA sequences similar to clone-x are not enriched in B cells or sorted DE cells
ACKNOWLEDGMENTS
Funding was provided by NIH grants UC4 DK112217 (to A.N., K.H.K., and M.R.B.), P30-CA016520 (to E.T.L.P.), and P01 AI42288 (to T.M.B. and M.A.A.) and the Human Islet Research Network (HIRN). This research was performed with the support of the Network for Pancreatic Organ donors with Diabetes (nPOD; RRID: SCR_014641), a collaborative type 1 diabetes research project sponsored by JDRF (nPOD: 5-SRA-2018-557-Q-R), and The Leona M. & Harry B. Helmsley Charitable Trust (grant 2018PG-T1D053). We thank S. Chamala and M. Kamoun for HLA immunogenetics expertise and I. Kusmartseva, E. Walters, and K. McGrail for help with sample shipping, management and metadata. We further thank D. Farber, M. Shlomchik, M. Kamoun, and M. Cancro for helpful discussions.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.11.035.
DECLARATION OF INTERESTS
The authors declare no competing interests.
WEB RESOURCES
HPAP consortium, https://hpap.pmacs.upenn.edu
REFERENCES
- Ahmed R, Omidian Z, Giwa A, Cornwell B, Majety N, Bell DR, Lee S, Zhang H, Michels A, Desiderio S, et al. (2019). A Public BCR Present in a Unique Dual- Receptor-Expressing Lymphocyte from Type 1 Diabetes Patients Encodes a Potent T Cell Autoantigen. Cell 177, 1583–1599.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed R, Omidian Z, Giwa A, Donner T, Jie C, and Hamad ARA (2021). A reply to “TCR+/BCR+ dual-expressing cells and their associated public BCR clonotype are not enriched in type 1 diabetes”. Cell 184, 840–843, this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allman D, Li J, and Hardy RR (1999). Commitment to the B lymphoid lineage occurs before DH-JH recombination. J. Exp. Med 189, 735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson MA, von Herrath M, Powers AC, and Clare-Salzler M (2015). Current concepts on the pathogenesis of type 1 diabetes–considerations for attempts to prevent and reverse the disease. Diabetes Care 38, 979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia M, Ahmed S, Anderson MS, Atkinson MA, Becker D, Bingley PJ, Bosi E, Brusko TM, DiMeglio LA, Evans-Molina C, et al. (2020). Introducing the Endotype Concept to Address the Challenge of Disease Heterogeneity in Type 1 Diabetes. Diabetes Care 43, 5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgarth N (2011). The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat. Rev. Immunol 11, 34–46. [DOI] [PubMed] [Google Scholar]
- Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, and Chudakov DM (2015). MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods 12, 380–381. [DOI] [PubMed] [Google Scholar]
- Bueno C, Sardina JL, Di Stefano B, Romero-Moya D, Muñoz-López A, Ariza L, Chillón MC, Balanzategui A, Castaño J, Herreros A, et al. (2016). Reprogramming human B cells into induced pluripotent stem cells and its enhancement by C/EBPα. Leukemia 30, 674–682. [DOI] [PubMed] [Google Scholar]
- Burel JG, Pomaznoy M, Lindestam Arlehamn CS, Seumois G, Vijayanand P, Sette A, and Peters B (2020). The Challenge of Distinguishing Cell-Cell Complexes from Singlet Cells in Non-Imaging Flow Cytometry and Single-Cell Sorting. Cytometry A 97, 1127–1135. 10.1002/cyto.a.24027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caserta S, Alessi P, Guarnerio J, Basso V, and Mondino A (2008). Synthetic CD4+ T cell-targeted antigen-presenting cells elicit protective antitumor responses. Cancer Res. 68, 3010–3018. [DOI] [PubMed] [Google Scholar]
- Cobaleda C, Jochum W, and Busslinger M (2007). Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature 449, 473–477. [DOI] [PubMed] [Google Scholar]
- Cumano A, and Rajewsky K (1986). Clonal recruitment and somatic mutation in the generation of immunological memory to the hapten NP. EMBO J. 5, 2459–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt WS, Lindau P, Snyder TM, Sherwood AM, Vignali M, Carlson CS, Greenberg PD, Duerkopp N, Emerson RO, and Robins HS (2016). A Public Database of Memory and Naive B-Cell Receptor Sequences. PLoS ONE 11, e0160853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B, and Tibshirani R (1986). Bootstrap methods for standard errors, confidence intervals, and other measures of statistical accuracy. Stat Sci 1, 54–77. [Google Scholar]
- Eisenbarth GS (1986). Type I diabetes mellitus. A chronic autoimmune disease. N. Engl. J. Med 314, 1360–1368. [DOI] [PubMed] [Google Scholar]
- Feeney AJ (1990). Lack of N regions in fetal and neonatal mouse immunoglobulin V-D-J junctional sequences. J. Exp. Med 172, 1377–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gepts W (1965). Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 14, 619–633. [DOI] [PubMed] [Google Scholar]
- Gidoni M, Snir O, Peres A, Polak P, Lindeman I, Mikocziova I, Sarna VK, Lundin KEA, Clouser C, Vigneault F, et al. (2019). Mosaic deletion patterns of the human antibody heavy chain gene locus shown by Bayesian haplotyping. Nat. Commun 10, 628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin DO, Holodick NE, and Rothstein TL (2011). Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70−. J. Exp. Med 208, 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Markoulaki S, Schorderet P, Carey BW, Beard C, Wernig M, Creyghton MP, Steine EJ, Cassady JP, Foreman R, et al. (2008). Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell 133, 250–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Hardy RR, Parks DR, and Herzenberg LA (1983). The “Ly-1 B” cell subpopulation in normal immunodefective, and autoimmune mice. J. Exp. Med 157, 202–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold KC, Vignali DA, Cooke A, and Bluestone JA (2013). Type 1 diabetes: translating mechanistic observations into effective clinical outcomes. Nat. Rev. Immunol 13, 243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, Gitelman SE, Gottlieb PA, Krischer JP, Linsley PS, et al. ; Type 1 Diabetes TrialNet Study Group (2019). An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med 381, 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa T, Masuda K, Endo TA, Endo M, Isono K, Koseki Y, Nakagawa R, Kometani K, Takano J, Agata Y, et al. (2016). Conversion of T cells to B cells by inactivation of polycomb-mediated epigenetic suppression of the B-lineage program. Genes Dev. 30, 2475–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenberg D, Spellerberg M, Williams W, Griffiths M, and Stevenson F (1993). Identification of the 9G4 idiotope in systemic lupus erythematosus. Br. J. Rheumatol 32, 876–882. [DOI] [PubMed] [Google Scholar]
- Jedema I, van de Meent M, Pots J, Kester MG, van der Beek MT, and Falkenburg JH (2011). Successful generation of primary virus-specific and anti-tumor T-cell responses from the naive donor T-cell repertoire is determined by the balance between antigen-specific precursor T cells and regulatory T cells. Haematologica 96, 1204–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaestner KH, Powers AC, Naji A, and Atkinson MA; HPAP Consortium (2019). NIH Initiative to Improve Understanding of the Pancreas, Islet, and Autoimmunity in Type 1 Diabetes: The Human Pancreas Analysis Program (HPAP). Diabetes 68, 1394–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc MP, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, Carillon E, Duvergey H, Houles A, Paysan-Lafosse T, et al. (2015). IMGT®, the international ImMunoGeneTics information system® 25 years on. Nucleic Acids Res. 43, D413–D422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenschow DJ, Zeng Y, Thistlethwaite JR, Montag A, Brady W, Gibson MG, Linsley PS, and Bluestone JA (1992). Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4lg. Science 257, 789–792. [DOI] [PubMed] [Google Scholar]
- Levenshtein V (1966). Binary codes capable of correcting deletions, insertions and reversals. Sov. Phys. Dokl 10, 707–710. [Google Scholar]
- Lun ATL, Riesenfeld S, Andrews T, Dao TP, Gomes T, and Marioni JC; participants in the 1st Human Cell Atlas Jamboree (2019). EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 20, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maecker HT, McCoy JP, and Nussenblatt R (2012). Standardizing immunophenotyping for the Human Immunology Project. Nat. Rev. Immunol 12, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillard I, Fang T, and Pear WS (2005). Regulation of lymphoid development, differentiation, and function by the Notch pathway. Annu. Rev. Immunol 23, 945–974. [DOI] [PubMed] [Google Scholar]
- Meng W, Zhang B, Schwartz GW, Rosenfeld AM, Ren D, Thome JJC, Carpenter DJ, Matsuoka N, Lerner H, Friedman AL, et al. (2017). An atlas of B-cell clonal distribution in the human body. Nat. Biotechnol 35, 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi X, Griffin G, Lee W, Patel S, Ohgami R, Ok CY, Wang S, Geyer JT, Xiao W, Roshal M, et al. (2018). Genomic and clinical characterization of B/T mixed phenotype acute leukemia reveals recurrent features and T-ALL like mutations. Am. J. Hematol 93, 1358–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miho E, Roškar R, Greiff V, and Reddy ST (2019). Large-scale network analysis reveals the sequence space architecture of antibody repertoires. Nat. Commun 10, 1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt SL, Heavey B, Rolink AG, and Busslinger M (1999). Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature 401, 556–562. [DOI] [PubMed] [Google Scholar]
- Olson BJ, Moghimi P, Schramm CA, Obraztsova A, Ralph D, Vander Heiden JA, Shugay M, Shepherd AJ, Lees W, and Matsen FA 4th. (2019). sumrep: A Summary Statistic Framework for Immune Receptor Repertoire Comparison and Model Validation. Front. Immunol 10, 2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opie EL (1901). On the Relation of Chronic Interstitial Pancreatitis to the Islands of Langerhans and to Diabetes Melutus. J. Exp. Med 5, 397–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry DJ, Wasserfall CH, Oram RA, Williams MD, Posgai A, Muir AB, Haller MJ, Schatz DA, Wallet MA, Mathews CE, et al. (2018). Application of a Genetic Risk Score to Racially Diverse Type 1 Diabetes Populations Demonstrates the Need for Diversity in Risk-Modeling. Sci. Rep 8, 4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, Gottlieb PA, Marks JB, McGee PF, Moran AM, et al. ; Type 1 Diabetes TrialNet Anti-CD20 Study Group (2009). Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N. Engl. J. Med 361, 2143–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh-Bernard AE, Silverman GJ, Cappione AJ, Villano ME, Ryan DH, Insel RA, and Sanz I (2001). Regulation of inherently autoreactive VH4-34 B cells in the maintenance of human B cell tolerance. J. Clin. Invest 108, 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley MF, Greenaway HY, Venturi V, Lindsay R, Quinn KM, Seder RA, Douek DC, Davenport MP, and Price DA (2010). Convergent recombination shapes the clonotypic landscape of the naive T-cell repertoire. Proc. Natl. Acad. Sci. USA 107, 19414–19419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld AM, Meng W, Chen DY, Zhang B, Granot T, Farber DL, Hershberg U, and Luning Prak ET (2018a). Computational Evaluation of B-Cell Clone Sizes in Bulk Populations. Front. Immunol 9, 1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld AM, Meng W, Luning Prak ET, and Hershberg U (2018b). ImmuneDB, a Novel Tool for the Analysis, Storage, and Dissemination of Immune Repertoire Sequencing Data. Front. Immunol 9, 2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg EV (2011). T cell lineage commitment: identity and renunciation. J. Immunol 186, 6649–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubelt F, Busse CE, Bukhari SAC, Bürckert JP, Mariotti-Ferrandiz E, Cowell LG, Watson CT, Marthandan N, Faison WJ, Hershberg U, et al. ; AIRR Community (2017). Adaptive Immune Receptor Repertoire Community recommendations for sharing immune-repertoire sequencing data. Nat. Immunol 18, 1274–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seay HR, Yusko E, Rothweiler SJ, Zhang L, Posgai AL, Campbell-Thompson M, Vignali M, Emerson RO, Kaddis JS, Ko D, et al. (2016). Tissue distribution and clonal diversity of the T and B cell repertoire in type 1 diabetes. JCI Insight 1, e88242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosenko JM, Skyler JS, Palmer JP, Krischer JP, Yu L, Mahon J, Beam CA, Boulware DC, Rafkin L, Schatz D, and Eisenbarth G; Type 1 Diabetes TrialNet Study Group; Diabetes Prevention Trial-Type 1 Study Group (2013). The prediction of type 1 diabetes by multiple autoantibody levels and their incorporation into an autoantibody risk score in relatives of type 1 diabetic patients. Diabetes Care 36, 2615–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatopoulos K, Belessi C, Moreno C, Boudjograh M, Guida G, Smilevska T, Belhoul L, Stella S, Stavroyianni N, Crespo M, et al. (2007). Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: Pathogenetic implications and clinical correlations. Blood 109, 259–270. [DOI] [PubMed] [Google Scholar]
- Thomas JW (1993). V region diversity in human anti-insulin antibodies. Preferential use of a VHIII gene subset. J. Immunol 150, 1375–1382. [PubMed] [Google Scholar]
- Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, and Lötvall JO (2007). Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol 9, 654–659. [DOI] [PubMed] [Google Scholar]
- Vander Heiden JA, Yaari G, Uduman M, Stern JN, O’Connor KC, Hafler DA, Vigneault F, and Kleinstein SH (2014). pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics 30, 1930–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturi V, Price DA, Douek DC, and Davenport MP (2008). The molecular basis for public T-cell responses? Nat. Rev. Immunol 8, 231–238. [DOI] [PubMed] [Google Scholar]
- Wooldridge L, Ekeruche-Makinde J, van den Berg HA, Skowera A, Miles JJ, Tan MP, Dolton G, Clement M, Llewellyn-Lacey S, Price DA, et al. (2012). A single autoimmune T cell receptor recognizes more than a million different peptides. J. Biol. Chem 287, 1168–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Ma N, Madden TL, and Ostell JM (2013). IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 41, W34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Meng W, Prak ET, and Hershberg U (2015). Discrimination of germline V genes at different sequencing lengths and mutational burdens: A new tool for identifying and evaluating the reliability of V gene assignment. J. Immunol. Methods 427, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Immune repertoire profiling data have been shared in an AIRR-compliant fashion (Rubelt et al., 2017); the data have been uploaded onto GenBank/SRA (SRA: PRJNA604535). Flow cytometric data for the 2-site blinded study are available at ImmPort (https://www.immport.org) under study accession number SDY1625.