

Abstract

Nitrogen heterocycles (azacycles) are common structural motifs in numerous pharmaceuticals, agrochemicals, and natural products. Many powerful methods have been developed and continue to be advanced for the selective installation and modification of nitrogen heterocycles through C–H functionalization and C–C cleavage approaches, revealing new strategies for the synthesis of targets containing these structural entities. Here, we report the first total syntheses of the lycodine-type Lycopodium alkaloids casuarinine H, lycoplatyrine B, lycoplatyrine A, and lycopladine F as well as the total synthesis of 8,15-dihydrohuperzine A through bioinspired late-stage diversification of a readily accessible common precursor, N-desmethyl-β-obscurine. Key steps in the syntheses include oxidative C–C bond cleavage of a piperidine ring in the core structure of the obscurine intermediate and site-selective C–H borylation of a pyridine nucleus to enable cross-coupling reactions.
Introduction
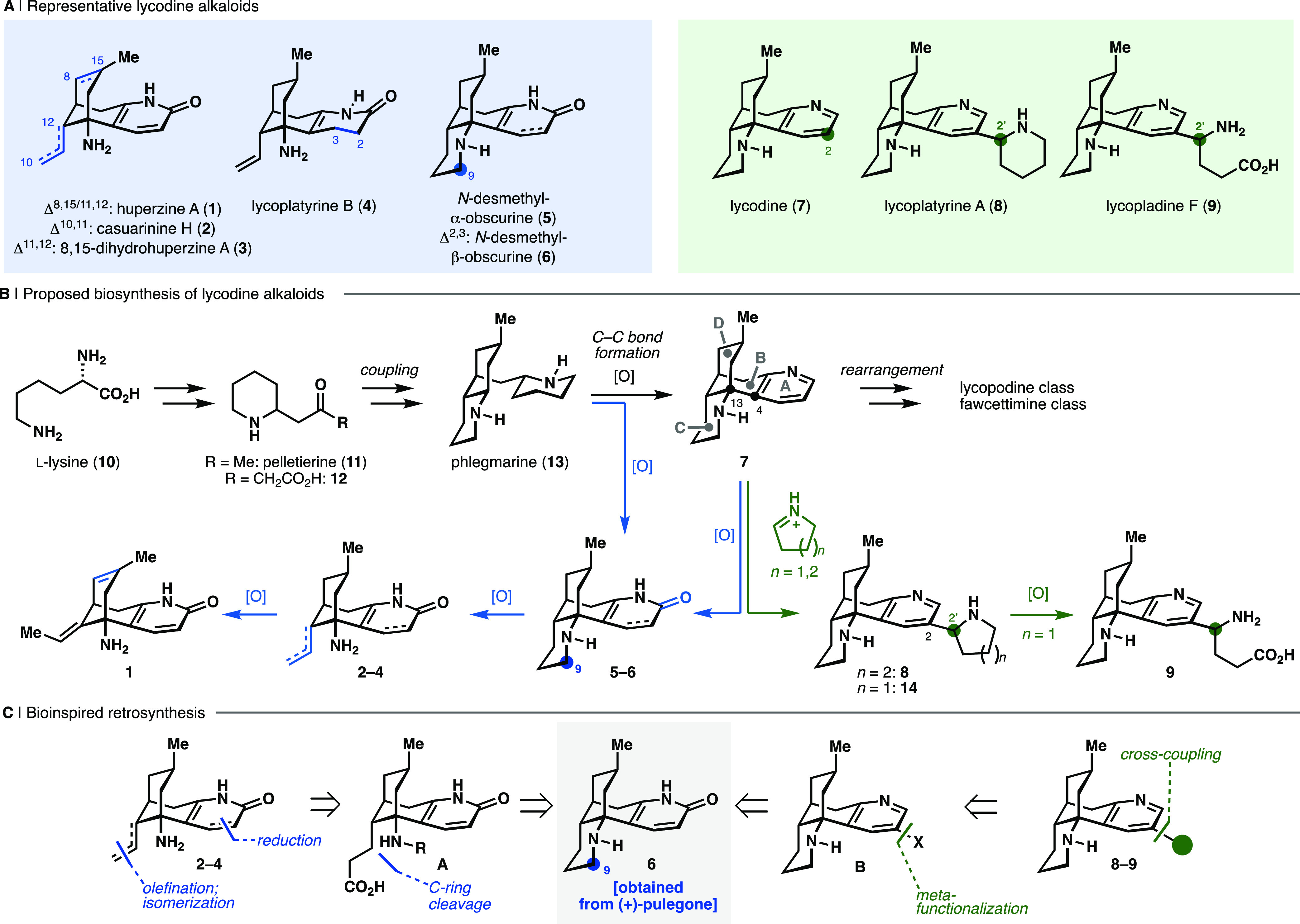
The Lycopodium alkaloids are a diverse group of natural products found in plants of the widely distributed Lycopodium genus, commonly known as clubmosses.1,2 Since the isolation of the first of these alkaloids, lycopodine, in 1881,3 a wealth of biosynthetically related alkaloids have also been isolated and characterized. These natural products are organized into four main classes (lycodine, lycopodine, fawcettimine, and a miscellaneous class) on the basis of their distinct carbon backbones, which arise as a consequence of C–C bond formation and rearrangement events during their putative biosyntheses.2 Many Lycopodium alkaloids possess intriguing and complex molecular architectures, and also display promising bioactivity profiles. The archetypical lycodine alkaloid huperzine A (1, Figure 1a), for example, is a potent and selective acetylcholinesterase (AChE) inhibitor and also demonstrates noncholinergic neuroprotective effects.4−6 This bioactivity is of interest for the symptomatic treatment of Alzheimer’s disease and other neurodegenerative disorders.2,4−6 The combination of interesting structural features and noteworthy bioactivity continue to drive synthetic studies toward Lycopodium alkaloids and their analogues.7−12
Figure 1.
Bioinspired plans for the synthesis of lycodine alkaloids.
Synthetic strategies that enable late-stage structural modification and diversification of a common advanced intermediate can provide versatility that facilitates efficient access to a range of products that might otherwise each require significant synthetic investment. A rapidly growing catalog of C–H bond functionalization technologies has powerfully expanded the processes available for such structural alterations, typically elaborating around the periphery of a molecule.13 Alternatively, C–C bond cleavage and functionalization strategies represent a key complementary approach which can be applied to remodel not only the periphery but also the core carbon skeleton of organic compounds.14 Although C–C cleavage tactics typically result in a decrease in molecular complexity—in contrast to Corey’s retrosynthetic paradigm15—they can lead to the identification of new retrosynthetic disconnections. In turn, such methods could enable rapid access to a diverse range of natural products or bioactive agents from a single compound, which, albeit more structurally complex, is easily obtained through chemical synthesis, biosynthesis, or synthetic biology.
The ubiquity of nitrogen heterocycles in pharmaceuticals,16 agrochemicals, and alkaloids17 render them attractive structural motifs for diversification to efficiently access underexplored chemical space.18 Therefore, a variety of methods for both the introduction and selective functionalization of azacycles continue to be reported.19−21 Inspired by these contributions, we envisioned nitrogen heterocycles as versatile synthetic handles that would enable the expedient preparation of a collection of lycodine-type alkaloids (2–4, 8, 9, Figure 1a) from a common, readily prepared, precursor through a series of programmed oxidation and C–C bond cleavage events in analogy to their biosynthesis.2,22
Although the complete biosynthetic pathways to the Lycopodium alkaloids remain to be fully elucidated,23 biochemical studies have suggested that these compounds derive from phlegmarine (13), which arises from the coupling of pelletierine (11) and 4-(2-piperidyl) acetoacetate (12), both of which originate from l-lysine (10, Figure 1b).2
Subsequent closure of ring B through bond formation between C13 and C4 furnishes the characteristic [3.3.1]-bicyclic scaffold of the lycodine class. A series of oxidative modifications, which include oxidation of the A-ring to the corresponding pyridone (e.g., in N-desmethyl-β-obscurine, 6) or pyridine (e.g., in lycodine, 7), C-ring cleavage, and excision of C9 further diversifies the parent scaffold, yielding a range of alkaloids including 1–6 (Figure 1b, blue arrows).
On the basis of these presumed biosynthetic events, we envisioned a retrosynthesis (Figure 1c) in which 8,15-dihydrohuperzine A (3)24 could arise from casuarinine H (2)25 through olefin isomerization, whereas lycoplatyrine B (4)26 could be accessed from 2 through semireduction of the pyridone. Casuarinine H (2) was traced back to functionalized tricyclic intermediate A through decarboxyolefination. In turn, A could be formed from the readily accessible key precursor N-desmethyl-β-obscurine (6) through oxidative functionalization and cleavage of the C9–N bond.
Another small set of structurally unique lycodine alkaloids bearing substitution at the C2 position of the pyridine A-ring (e.g., lycoplatyrine A,268, and lycopladine F,279) is proposed to arise biosynthetically through electrophilic substitution on lycodine (7) or the corresponding dihydropyridine by a Δ1-piperidinium or Δ1-pyrrolinium cation (or the corresponding imines; Figure 1b, green arrows).26,27 Subsequent oxidative cleavage of the pyrrolidine ring in 14 is suggested to provide lycopladine F (9), analogous to the oxidative ring cleavage pathway that leads to metabolic products of nicotine.28 Overall, we envisioned lycoplatyrine A (8) and lycopladine F (9) could be accessed through cross-coupling of appropriate C(sp3) nucleophiles with a functionalized lycodine analog (B), which again would be prepared from the key obscurine scaffold 6. The required deoxygenation of precursor 6 and site-selective functionalization at C2 would rely upon precedent demonstrated by our laboratories in the total synthesis of the dimeric lycodine alkaloids complanadine A and B.29,30
Results and Discussion
Preparation of the Key Diversifiable Precursor
Our investigations commenced with the development of a robust synthesis of N-desmethyl-β-obscurine (6), the late-stage common intermediate for the synthesis of all of the alkaloids described here. A convergent route featuring a diastereoselective formal (3 + 3)-cycloaddition to form the three contiguous stereocenters and two C–C bonds in ring B of 6(31) was adapted from literature protocols by Schuster,32 Caine,33 Dake,34 and Jung35 as well as our own previous studies.29
The coupling partner that would lead to ring A, dihydropyridone 17, was prepared from β-ketoester 15 through a Michael addition into acrylonitrile followed by decarboxylation to give nitrile 16. Subsequent nitrile hydration and cyclization in vacuo delivered 17 in 18% overall yield (Scheme 1a).29,31
Scheme 1. Synthesis of the Bicyclo[3.3.1]nonane Core in N-Desmethyl-α-obscurine through Formal (3 + 3)-Cycloaddition.
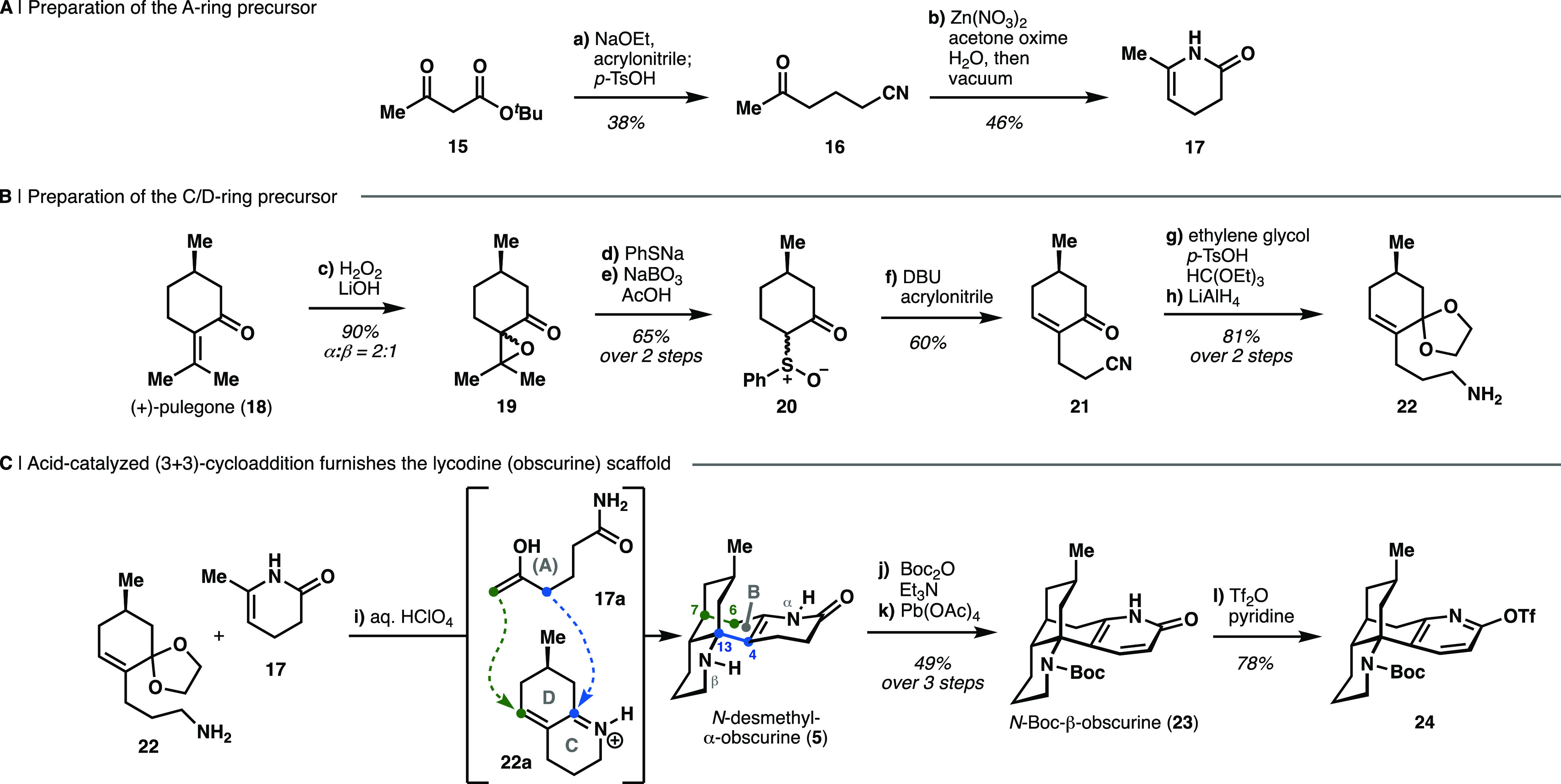
Reagents and conditions: (a) NaOEt, EtOH, 21 °C, then acrylonitrile, 0 to 21 °C, then TsOH, 145 °C (38%, > 13 g scale); (b) Zn(NO3)2·6H2O, acetone oxime, H2O, 90 °C, then vacuum, 120 °C (46%, > 2 g scale); (c) aq. H2O2, LiOH·H2O, MeOH, H2O, 21 °C (90%, 30 g scale); (d) PhSH, Na, THF, 21 °C, then 19, 85 °C; (e) NaBO3·H2O, AcOH, 40 °C (65%, 2 steps, > 17 g scale); (f) DBU, iPrOH, 0 °C, then acrylonitrile, 0 to 40 °C (60%, > 7 g scale); (g) ethylene glycol, p-TsOH, HC(OEt)3, 75 °C (97%); (h) LiAlH4, Et2O, 0 °C (84%, > 2 g scale); (i) aq. HClO4, 1,4-dioxane, 105 °C; (j) Boc2O, Et3N, THF, 60 °C (54%, 2 steps); (k) Pb(OAc)4, CHCl3, 21 °C (90%); (l) Tf2O, pyridine, CH2Cl2, −78 to 21 °C (78%).
The C/D ring cycloaddition partner 22 was prepared from (+)-pulegone (18) in six steps and 28% overall yield (Scheme 1b).32,33 The sequence was initiated by Weitz–Scheffer-type epoxidation of the exocyclic olefin group of 18, which provided a 1:2 mixture of epoxide isomers (19).38,39 Subsequent nucleophilic opening of the epoxide with sodium thiophenolate and concomitant retro-aldol reaction delivered the phenylthioether,33 which was selectively oxidized to sulfoxide 20 with sodium perborate.34 α-Alkylation of 20 with acrylonitrile, followed by thermal syn-elimination of phenylsulfenic acid gave enone 21,35,36 which was protected as the ethylene glycol ketal and reduced with LiAlH4 to deliver primary amine 22.32 The two building blocks (17 and 22) were ultimately coupled upon heating with perchloric acid (Scheme 1c). Under these conditions, oxygen-sensitive α,β-unsaturated iminium ion 22a and the open-chain enolamide 17a are presumably formed in situ and undergo the desired formal cycloaddition to furnish N-desmethyl-α-obscurine (5).29,31,37 Boc-protection of the piperidine nitrogen in 5 and dehydrogenation of the dihydropyridone ring using lead(IV) acetate provided N-Boc-β-obscurine (23) in 49% yield over three steps.
As an alternative to the oxidation of Boc-protected 5 using stoichiometric lead(IV) acetate, we investigated a photocatalytic dehydrogenation protocol.40,41 Our preliminary results demonstrated that N-Boc-5 was readily oxidized to 23 (57% yield) in the presence of an iridium(III) photoredox catalyst (Ir[dF(CF3)ppy]2(dtbbpy)PF6) with potassium persulfate as the terminal oxidant upon irradiation with blue light (λ = 450 nm) under anoxic conditions. In the absence of light or the photoredox catalyst, only traces of product (6%) were formed in the best case, whereas under aerobic conditions complete decomposition of the substrate was noted (see Section S3.1 in the Supporting Information, SI). Despite attempts to optimize this reaction, we were unable to obtain yields comparable with those achieved with lead(IV) acetate (90%). Therefore, the latter conditions were employed for the preparation of large quantities of material. Finally, pyridone O-triflation of 23 delivered fully protected β-obscurine scaffold 24 in 78% yield.29
Synthesis of (−)-Casuarinine H, (−)-8,15-Dihydrohuperzine A, and (+)-Lycoplatyrine B
Our envisioned route toward the lycodine alkaloids casuarinine H (2), 8,15-dihydrohuperzine A (3), and lycoplatyrine B (4) required the identification of suitable conditions to effect the bioinspired oxidative cleavage of the C9–N bond in protected tetracycle 24 or a related obscurine congener. To this end, we pursued several conditions for C–N cleavage and functionalization that included biocatalytic and transition metal-mediated approaches.
Biocatalytic methods were explored as a means to achieve a protecting group-free oxidation of the C9–N bond, reminiscent of the proposed biosynthetic tailoring process. Although the requisite biosynthetic enzymes have not been identified, we posited that other established biocatalysts capable of oxidizing C-heteroatom bonds could accept the bicyclo[3.3.1]nonane scaffold of 5 as a substrate while retaining site-selectivity. A screening set composed of 14 commercial and in-house heterologously expressed copper-42,43 and flavin-dependent oxidases,44−46 a pyrroloquinoline (PQQ) dependent dehydrogenase,47 a horseradish peroxidase (HRP),48 and a laccase/TEMPO redox mediator system49 was assembled. However, overview screenings under representative conditions did not identify any oxidation activity with unprotected substrate 5 (see SI Section S3.2 for details).
We therefore sought to examine other established chemical conditions for the oxidation of carbamate-protected saturated nitrogen heterocycles. While methods employing iron50 and copper51 redox mediators in combination with peroxides failed to generate the anticipated enamine or enamide products, we observed that substoichiometric quantities of RuO2 with sodium periodate as stoichiometric oxidant in a mixture of tBuOH and water resulted in piperidine oxidation to yield 25 (Scheme 2a).52,53 Although oxidation under these conditions by the presumed in situ generated RuO4 catalyst was expected to give the corresponding amino acid (i.e., following hydrolysis of an intermediate C-ring iminium ion and oxidation of the resulting aldehyde), cyclic imide 25 was obtained in 86% yield. Additional experiments demonstrated that the electronically deactivating triflyl group on the pyridone oxygen was critical to the success of the piperidine oxidation–oxidation of derivatives of 25 bearing methyl, benzyl, or SEM groups instead of the triflyl moiety proved unsuccessful under identical conditions.
Scheme 2. Synthesis of C-Ring Cleaved Lycodine Alkaloids from Protected β-Obscurine.

Reagents and conditions: (a) RuO2·H2O, NaIO4, H2O, 21 °C, then 24, tBuOH, 60 °C (86%); (b) aq. 1 M LiOH, THF, 30 °C; (c) MeI, Ag2CO3, CHCl3, 75 °C (86%, 2 steps); (d) aq. 1 M LiOH, THF, 65 °C (97%); (e) PdBr2, DPE-Phos, Piv2O, Et3N, DMPU, 130 °C (65%); (f) TMSI, CHCl3, 65 °C (89%); (g) Sm, aq. 3 M HCl, 0 to 21 °C (84%); (h) Pd(dba)2, P(tBu)3, iPrCOCl, toluene, 90 °C (81%); (i) TMSI, CHCl3, 65 °C (41%).
We envisioned that hydrolysis of imide 25 followed by decarboxyolefination of the resulting carboxylic acid could offer an attractive strategy to excise C9 and install the required unsaturation at C10–C11. Treatment of 25 with aqueous LiOH at the elevated temperatures required for imide hydrolysis resulted in undesired concomitant triflate cleavage. Therefore, a methyl ether was introduced in place of the triflate prior to imide hydrolysis to yield carboxylic acid 26. Unfortunately, subjecting 26 to classic Kochi oxidative decarboxylation conditions54 failed to deliver alkene 27. Additionally, an attempted Hunsdiecker-type decarboxyhalogenation55 resulted in the C-ring contracted pyrrolidine 28 (Scheme 2b), presumably the result of an SN2 displacement of the intermediate alkyl halide. While more recently developed decarboxyolefination conditions using metallo-organo-56 or organo-photocatalysts57 in combination with cobalt-based dehydrogenation catalysts furnished olefin 27 in 50% yield, a competing protodemetalation pathway leading to ethyl derivative 29 hindered further optimization of this process. Alternatively, desired terminal olefin 27 was obtained in higher yield (65%) through a Pd(0)-catalyzed decarbonylative elimination of an in situ-generated mixed anhydride of 26.58 Deprotection of 27 using TMSI12 completed the first total synthesis of the neuroprotective compound (−)-casuarinine H (2, Scheme 2c).25 Semireduction of the pyridone moiety in 2 with samarium metal in aqueous HCl59 cleanly yielded (+)-lycoplatyrine B (4)26 in 84% yield, also constituting the first total synthesis of this Lycopodium alkaloid. Furthermore, treatment of terminal olefin 27 with an in situ-generated palladium hydride catalyst effected isomerization to the internal (E)-alkene in 81% yield.60 A subsequent TMSI-mediated deprotection delivered (−)-8,15-dihydrohuperzine A (3).24,61
The spectroscopic data for synthetic (−)-casuarinine H (2), (+)-lycoplatyrine B (4), and (−)-8,15-dihydrohuperzine A (3) were in full agreement with those reported upon isolation of these natural products from the producing organisms.24−26 Taking advantage of this late-stage diversification approach, the target alkaloids 2–4 were prepared in 16 to 17 steps (longest linear sequence, LLS) and 1.7–4.5% overall yield from (+)-pulegone.
Synthesis of Lycoplatyrine A and Lycopladine F
For the synthesis of Lycopodium alkaloids bearing substituents at C2 (i.e., 8–9), we envisioned a cross-coupling approach in which the key β-obscurine intermediate 24 would be elaborated to a selectively C2-functionalized lycodine derivative to serve as a common coupling partner. Accordingly, protected β-obscurine 24 was deoxygenated in the presence of a palladium catalyst and ammonium formate as reductant to deliver N-Boc lycodine (30). Subsequent iridium-catalyzed meta-selective C–H borylation29,62 of the pyridine A-ring and bromodeborylation63 furnished 2-bromolycodine (31) (Scheme 3a).
Scheme 3. Couplings of a Site-Selectively Functionalized Lycodine Congener in the Syntheses of C2-Substituted Alkaloids.
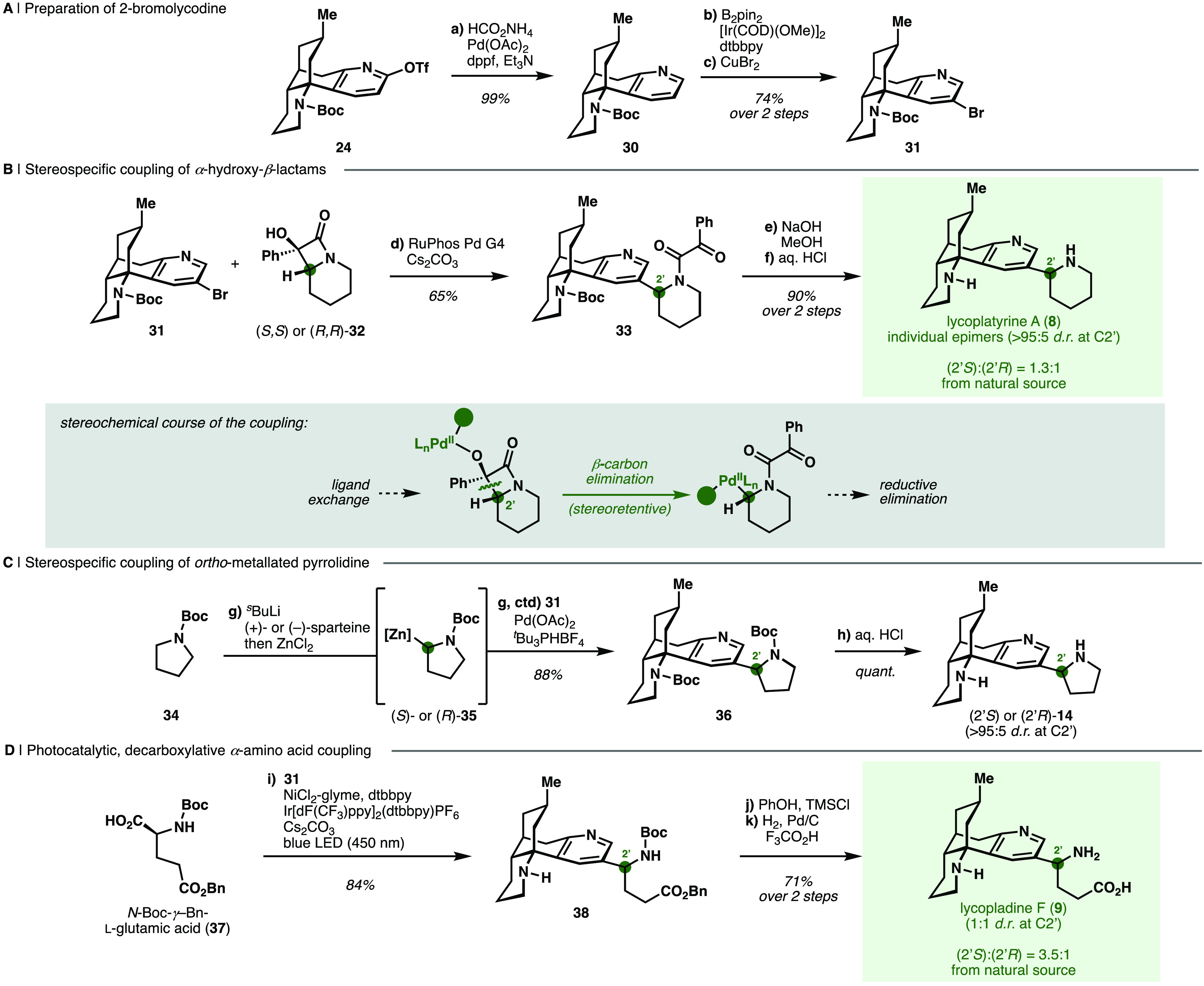
Reagents and Conditions: (a) HCO2NH4, Pd(OAc)2, dppf, Et3N, DMF, 60 °C (99%); (b) B2 pin2, [Ir(COD)(OMe)]2, dtbbpy, THF, 80 °C; (c) CuBr2, MeOH, H2O, 80 °C (74%, 2 steps); (d) RuPhos Pd G4, Cs2CO3, toluene, 70 °C [(2′S)-33: 65%, (2′R)-33: 65%, 33 as epimeric mixture at C2′ with rac-32: 72%]; (e) NaOH, MeOH, 1,4-dioxane, 70 °C (f) aq. 6 M HCl, 70 °C [(2′S)-8: 90%, (2′R)-8: 68%; 2 steps]; (g) sBuLi, (+)- or (−)-sparteine, MTBE, −78 °C, then ZnCl2, THF, −78 to 21 °C, then 31, Pd(OAc)2, tBu3PHBF4, MTBE, 60 °C [(2′S)-36: 55%, (2′R)-36: 88%]; (h) aq. 6 M HCl, 21 °C [(2′S)-14: quantitative, (2′R)-14: 70%]; (i) 31, NiCl2-glyme, dtbbpy, Ir[dF(CF3)ppy]2(dtbbpy)PF6, Cs2CO3, DMF, 450 nm LED, 21 °C (84%); (j) PhOH, TMSCl, CH2Cl2, 21 °C; (k) 500 psi H2, Pd/C, CF3CO2H, MeOH, 21 °C (71%, 2 steps).
Lycoplatyrine A (8) features a C2 piperidine substituent as an epimeric mixture of undetermined absolute configuration,26 which we anticipated could be installed through the coupling of 31 with an α-functionalized piperidine derivative (Scheme 3b). We specifically envisioned the application of a method recently disclosed by our laboratory in which α-hydroxy-β-lactams such as 32 serve as surrogates for α-metalated N-heterocycles in a palladium-catalyzed coupling with aryl halides.20 This method was particularly attractive due to the mild and stereospecific nature of the cross-coupling, although the use of pyridyl bromides had not been previously demonstrated. As proposed, the coupling of 31 with racemic lactam 32, prepared from the corresponding piperidine-derived 2-oxophenylacetamide through a Norrish-Yang reaction,20 delivered 33 as a mixture of epimers at C2′. Cleavage of the 2-oxophenylacetamide and Boc-protecting groups under sequential basic and acidic conditions yielded lycoplatyrine A (8) as a 1:1.5 mixture of the anticipated C2′ epimers.
According to the previously proposed mechanism for this coupling, the hydroxy group of the lactam coordinates to the palladium center before irreversible C–C bond cleavage (β-carbon elimination) driven by the release of ring strain in 32 delivers a C2′-palladated species in a stereoretentive manner (Scheme 3b, gray box).20,64 We therefore anticipated that the use of enantiomerically pure lactams (2′S)- and (2′R)-32 would enable the stereospecific piperidinylation of the lycodine scaffold at C2, and thus allow the assignment of absolute configurations at C2′ in naturally occurring alkaloid 8.
To obtain α-hydroxy-β-lactam 32 in enantioenriched form, we first investigated enzymatic resolution methods. Despite extensive reaction engineering, selectivity for an enzymatic hydrolytic kinetic resolution65,66 of acetylated tertiary alcohol 32 with pig liver esterase (PLE) and lipase A from C. antarctica (CalA) was poor and therefore not synthetically useful (E ≤ 7) (see SI Section S3.5). Alternatively, enantiomerically resolved lactams (2′S)- and (2′R)-32 were obtained from preparative chiral supercritical fluid chromatography (SFC).20 Coupling of lactams (2′S)- and (2′R)-32 with lycodine bromide 31 gave single epimers of 33 in 65% yield, which were deprotected to provide single epimers of lycoplatyrine A (8) in 4.7% overall yield over 16 steps from (+)-pulegone (LLS). Comparison of the spectral data of single epimers of synthetic 8 with data for naturally derived 8 revealed a slight excess of the (2′S)-8 epimer in material isolated from natural sources (d.r. 1.3:1).26 The cross-coupling product obtained using racemic 32 was also enriched in the same epimer (d.r. 1.5:1, vide supra), suggesting that the chiral lycodine scaffold exerts a low level of enantiodiscrimination and enantiotopic face discrimination in both the synthetic and natural coupling processes.
Indeed, our success in preparing single epimers of lycoplatyrine A (8) rested on the stereospecific coupling of α-hydroxy-β-lactams as surrogates for α-metalated piperidines, which otherwise typically suffer from low yields and poor stereoselectivities in the metalation step.67,68 Although an analogous β-lactam-based cross-coupling for five-membered nitrogen heterocycles is precluded due to the inaccessibility of the five-membered analogues of 32 with established photochemical methods,69 α-metalated pyrrolidines are excellent stereoselective coupling partners. These reagents set the stage for the preparation of the pyrrolidine analog of lycoplatyrine A (“pyrrolo-lycoplatyrine A”, 14), which is hypothesized to be an intermediate in the biosynthesis of other lycodine-derived congeners including lycopladine F (9).27 For the synthesis of N-Boc pyrrolo-lycoplatyrine A (36), we turned to a method by Campos and co-workers70 for the stereoselective α-arylation of N-Boc-pyrrolidine (34) (Scheme 3c). Enantioselective ortho-lithiation of 34 in the presence of either (+)- or (−)-sparteine,71 transmetalation to form the corresponding organozinc species (35), and subsequent palladium-catalyzed coupling to lycodine bromide (31) delivered single C2′-epimers of the desired product (36) in high yield (88%). Subsequent deprotection provided each of the two C2′-epimers of pyrrolo-lycoplatyrine A (14) in 15 steps from (+)-pulegone (7% overall, LLS).
We sought to similarly access lycopladine F (9) via a direct coupling approach where the necessary amino acid moiety is appended at C2 of lycodine bromide (31, Scheme 3d). To this end, iridium-catalyzed photoredox conditions effected activation of bis-protected glutamic acid 37 through single-electron oxidation of the cesium carboxylate, followed by decarboxylative C–C bond scission and nickel-catalyzed C(sp3)–C(sp2) coupling with aryl bromide 31 to deliver protected lycopladine F (38) in 84% yield.72 A low nickel loading (1 mol %) was necessary to attenuate consumption of bromide 31 in a nonproductive protodehalogenation pathway and achieve good yields of 38. Removal of both Boc protecting groups followed by hydrogenolytic cleavage of the benzyl ester in the presence of trifluoroacetic acid yielded lycopladine F (9) in 71% yield as a 1:1 mixture of epimers (4.8% yield over 16 steps LLS). The analytical data obtained for the synthetic material matched those reported for the natural material, which was isolated from Lycopodium complanatum as a 3.5:1 mixture of (2′S):(2′R)-epimers.27 We expect access to pyrrolo-lycoplatyrine A (14) and lycopladine F (9) to set the stage for studies into the biosynthesis of the latter natural product.27
Conclusions
In summary, we have developed the first total syntheses of lycodine alkaloids casuarinine H (2), lycoplatyrine B (4), lycoplatyrine A (8), and lycopladine F (9) and a total synthesis of 8,15-dihydrohuperzine A (3) employing the readily accessible tetracycle N-desmethyl-β-obscurine (6) as a common intermediate. A series of bioinspired modifications of the piperidine C-ring in 6, including oxidative ring cleavage, C–C bond scission with carbon atom excision, and olefin isomerization delivered tricyclic congeners 2–4. Conversion of the pyridone A-ring in 6 to the corresponding pyridine (7) and site-selective C–H functionalization to ultimately afford bromopyridine 31 enabled direct cross-couplings with saturated azacycles or an amino acid to complete the syntheses of C2-derivatized lycodine alkaloids lycoplatyrine A (8) and lycopladine F (9). The general late-stage peripheral derivatization and C–C functionalization strategies outlined herein may provide a basis for synthetic access to an even wider range of Lycopodium alkaloids. Our synthetic studies toward these compounds should also set the stage for a broader, more systematic assessment of their biosynthesis and bioactivity.25,26,61 Biological activities exerted by these natural products may include a range of neuroprotective effects such as those observed for huperzine A,4,5 for example the attenuation of both glutamate-induced neurotoxicity and free radical-mediated oxidative stress.
Acknowledgments
This research was funded in part by the Austrian Science Fund (FWF) [J 4227-B21]. S.E.P. acknowledges funding by the Austrian FWF through an Erwin Schrödinger fellowship. J.N. and S.C. acknowledge funding through a PROMOS-scholarship by the DAAD (Deutscher Akademischer Austauschdienst). Financial support for this research was provided to R.S. by the National Institutes of General Medical Sciences (NIGMS R35 GM130345A). Prof. Nick Turner (University of Manchester), Prof. Marco Fraaje (University of Groningen), Prof. Kurt Faber (University of Graz), and Prof. Wolfgang Kroutil (University of Graz) are thanked for the donation of plasmids for various oxidase enzymes. We are grateful to Prof. John F. Hartwig (UC Berkeley) and Prof. Jon Rittle (UC Berkeley) for generously sharing their equipment for the preparation of biocatalysts. We are grateful to Frederick M. Tomlin and James N. Newton (UC Berkeley) for early contributions on the RuO4 oxidation chemistry. We thank Dr. Charles Yeung and colleagues at Merck Research Laboratories for the chiral SFC separation of the enantiomers of 32. We thank Dr. Hasan Celik and UC Berkeley’s NMR facility in the College of Chemistry (CoC-NMR) for spectroscopic assistance. Instruments in CoC-NMR are supported in part by NIH S10OD024998. We are also grateful to Dr. Miao Zhang (DOE Catalysis Facility, UC Berkeley) for support with the acquisition of HRMS and IR data.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c00457.
Experimental procedures, additional experimental results, and compound characterization (PDF)
Author Present Address
† Institute of Chemistry, University of Graz, Heinrichstrasse 28, A-8010 Graz, Austria, Europe; orcid.org/0000–0001–9665–1710.
Author Present Address
‡ Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1–5/10, 8093 Zürich, Switzerland, Europe.
Author Present Address
§ Max-Planck Institute for Medical Research, Jahnstrasse 29, 69120 Heidelberg, Germany, Europe.
Author Present Address
∥ Institute of Technical Chemistry, Leibniz University Hannover, Callinstrasse 5, 30167 Hannover, Germany, Europe.
Author Contributions
⊥ These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Siengalewicz P.; Mulzer J.; Rinner U.. Chapter 1: Lycopodium Alkaloids—Synthetic Highlights and Recent Developments. In The Alkaloids: Chemistry and Biology; Knölker H.-J., Ed.; Academic Press: Cambridge, MA, 2013; Vol. 72, pp 1–151. [DOI] [PubMed] [Google Scholar]
- Ma X. Q.; Gang D. R. The Lycopodium alkaloids. Nat. Prod. Rep. 2004, 21, 752–772. 10.1039/b409720n. [DOI] [PubMed] [Google Scholar]
- Bödeker K. Lycopodin, das erste Alkaloïd der Gefässkryptogamen. Liebigs Ann. 1881, 208, 363–367. 10.1002/jlac.18812080308. [DOI] [Google Scholar]
- Herzon S. B.; Tun M. K. M. The pharmacology and therapeutic potential of (−)-huperzine A. J. Exp. Pharmacol. 2012, 4, 113–123. 10.2147/JEP.S27084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z. M.; Ke Y. Huperzine A: Is it an Effective Disease-Modifying Drug for Alzheimer’s Disease?. Front. Aging Neurosci. 2014, 6, 216. 10.3389/fnagi.2014.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozikowski A. P.; Tuckmantel W. Chemistry, pharmacology, and clinical efficacy of the Chinese nootropic agent huperzine A. Acc. Chem. Res. 1999, 32, 641–650. 10.1021/ar9800892. [DOI] [Google Scholar]
- Takayama H. Total Syntheses of Lycopodium and Monoterpenoid Indole Alkaloids Based on Biosynthesis-Inspired Strategies. Chem. Pharm. Bull. 2020, 68, 103–116. 10.1248/cpb.c19-00872. [DOI] [PubMed] [Google Scholar]
- Kaneko H.; Takahashi S.; Kogure N.; Kitajima M.; Takayama H. Asymmetric Total Synthesis of Fawcettimine-Type Lycopodium Alkaloid, Lycopoclavamine-A. J. Org. Chem. 2019, 84, 5645–5654. 10.1021/acs.joc.9b00586. [DOI] [PubMed] [Google Scholar]
- Vertorano M. C.; Johnson K. L.; He P.; Wei Z.; Wang Z. A transannular approach toward lycopodine synthesis. J. Antibiot. 2019, 72, 494–497. 10.1038/s41429-019-0155-2. [DOI] [PubMed] [Google Scholar]
- Burtea A.; DeForest J.; Li X. T.; Rychnovsky S. D. Total Synthesis of (−)-Himeradine A. Angew. Chem., Int. Ed. 2019, 58, 16193–16197. 10.1002/anie.201910129. [DOI] [PubMed] [Google Scholar]
- Shao H.; Fang K.; Wang Y.-P.; Zhang X.-M.; Ding T.-M.; Zhang S.-Y.; Chen Z.-M.; Tu Y.-Q. Total Synthesis of Fawcettimine-Type Alkaloid, Lycojaponicumin A. Org. Lett. 2020, 22, 3775–3779. 10.1021/acs.orglett.0c00961. [DOI] [PubMed] [Google Scholar]
- White J. D.; Li Y.; Kim J.; Terinek M. Cyclobutane synthesis and fragmentation. A cascade route to the Lycopodium alkaloid (−)-huperzine A. J. Org. Chem. 2015, 80, 11806–11817. 10.1021/acs.joc.5b01619. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F. Catalyst-Controlled Site-Selective Bond Activation. Acc. Chem. Res. 2017, 50, 549–555. 10.1021/acs.accounts.6b00546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Perea M. A.; Sarpong R. Transition Metal-Mediated Cleavage of C−C Single Bonds: Making the Cut in Total Synthesis. Angew. Chem., Int. Ed. 2020, 59, 18898–18919. 10.1002/anie.201915657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey E. J.; Cheng X.-M.. The Logic of Chemical Synthesis; Wiley: New York, 1995; pp 2–3. [Google Scholar]
- Vitaku E.; Smith D. T.; Njardarson J. T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles Among US FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- O’Hagan D. Pyrrole, pyrrolidine, pyridine, piperidine and tropane alkaloids. Nat. Prod. Rep. 2000, 17, 435–446. 10.1039/a707613d. [DOI] [PubMed] [Google Scholar]
- Campos K. R.; Coleman P. J.; Alvarez J. C.; Dreher S. D.; Garbaccio R. M.; Terrett N. K.; Tillyer R. D.; Truppo M. D.; Parmee E. R. The importance of synthetic chemistry in the pharmaceutical industry. Science 2019, 363, eaat0805 10.1126/science.aat0805. [DOI] [PubMed] [Google Scholar]
- Antermite D.; Bull J. A. Transition Metal-Catalyzed Directed C(sp3)−H Functionalization of Saturated Heterocycles. Synthesis 2019, 51, 3171–3204. 10.1055/s-0037-1611822. [DOI] [Google Scholar]
- Roque J. B.; Kuroda Y.; Jurczyk J.; Xu L. P.; Ham J. S.; Gottemann L. T.; Roberts C. A.; Adpressa D.; Sauri J.; Joyce L. A.; Musaev D. G.; Yeung C. S.; Sarpong R. C−C Cleavage Approach to C−H Functionalization of Saturated Aza-Cycles. ACS Catal. 2020, 10, 2929–2941. 10.1021/acscatal.9b04551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.; Paul A.; Abboud K. A.; Seidel D. Rapid functionalization of multiple C−H bonds in unprotected alicyclic amines. Nat. Chem. 2020, 12, 545–550. 10.1038/s41557-020-0438-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.; You W.; Wu S.; Fan Z.; Xu B.; Zhu M.; Li X.; Xiao Y. Global transcriptome analysis of Huperzia serrata and identification of critical genes involved in the biosynthesis of huperzine A. BMC Genomics 2017, 18, 245. 10.1186/s12864-017-3615-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Zhang Z.-K.; Jiang F.-F.; Qi B.-W.; Ding N.; Hnin S. Y. Y.; Liu X.; Li J.; Wang X.-H.; Tu P.-F.; Abe I.; Morita H.; Shi S.-P. Deciphering the Biosynthetic Mechanism of Pelletierine in Lycopodium Alkaloid Biosynthesis. Org. Lett. 2020, 22, 8725–8729. 10.1021/acs.orglett.0c03339. [DOI] [PubMed] [Google Scholar]
- Thorroad S.; Worawittayanont P.; Khunnawutmanotham N.; Chimnoi N.; Jumruksa A.; Ruchirawat S.; Thasana N. Three new Lycopodium alkaloids from Huperzia carinata and Huperzia squarrosa. Tetrahedron 2014, 70, 8017–8022. 10.1016/j.tet.2014.08.042. [DOI] [Google Scholar]
- Tang Y.; Fu Y.; Xiong J.; Li M.; Ma G.-L.; Yang G.-X.; Wei B.-G.; Zhao Y.; Zhang H.-Y.; Hu J.-F. Casuarinines A−J, Lycodine-Type Alkaloids from Lycopodiastrum casuarinoides. J. Nat. Prod. 2013, 76, 1475–1484. 10.1021/np4003355. [DOI] [PubMed] [Google Scholar]
- Yeap J. S.-Y.; Lim K.-H.; Yong K.-T.; Lim S.-H.; Kam T.-S.; Low Y.-Y. Lycopodium Alkaloids: Lycoplatyrine A, an Unusual Lycodine-Piperidine Adduct from Lycopodium platyrhizoma and the Absolute Configurations of Lycoplanine D and Lycogladine H. J. Nat. Prod. 2019, 82, 324–329. 10.1021/acs.jnatprod.8b00754. [DOI] [PubMed] [Google Scholar]
- Ishiuchi K.; Kubota T.; Hayashi S.; Shibata T.; Kobayashi J. Lycopladines F and G, new C16N2-type alkaloids with an additional C4N unit from Lycopodium complanatum. Tetrahedron Lett. 2009, 50, 4221–4224. 10.1016/j.tetlet.2009.04.139. [DOI] [Google Scholar]
- McKennis H.; Turnbull L. B.; Bowman E. R. Metabolism of Nicotine to (+)-γ-(3-Pyridyl)-γ-methylaminobutyric Acid. J. Am. Chem. Soc. 1958, 80, 6597–6600. 10.1021/ja01557a036. [DOI] [Google Scholar]
- Fischer D. F.; Sarpong R. Total Synthesis of (+)-Complanadine A Using an Iridium-Catalyzed Pyridine C−H Functionalization. J. Am. Chem. Soc. 2010, 132, 5926–5927. 10.1021/ja101893b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton J. N.; Fischer D. F.; Sarpong R. Synthetic Studies on Pseudo-Dimeric Lycopodium Alkaloids: Total Synthesis of Complanadine B. Angew. Chem., Int. Ed. 2013, 52, 1726–1730. 10.1002/anie.201208571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann D.; Naumann A. Synthese des Lycopodium-alkaloids rac-α-Obscurin durch 1,3-Anellierung eines Enimins. Liebigs Ann. Chem. 1983, 1983, 220–225. 10.1002/jlac.198319830206. [DOI] [Google Scholar]
- Schuster E.; Jas G.; Schumann D. An Improved Synthesis of 2,3,4,6,7,8-Hexahydroquinolines. Org. Prep. Proced. Int. 1992, 24, 670–672. 10.1080/00304949209356242. [DOI] [Google Scholar]
- Caine D.; Procter K.; Cassell R. A. A Facile Synthesis of (−)-(R)-5-Methyl-2-cyclohexen-1-one and Related 2-Substituted Enones from (+)-Pulegone. J. Org. Chem. 1984, 49, 2647–2648. 10.1021/jo00188a032. [DOI] [Google Scholar]
- Kozak J. A.; Dake G. R. Total Synthesis of (+)-Fawcettidine. Angew. Chem., Int. Ed. 2008, 47, 4221–4223. 10.1002/anie.200800522. [DOI] [PubMed] [Google Scholar]
- Jung M. E.; Chang J. J. Enantiospecific Formal Total Synthesis of (+)-Fawcettimine. Org. Lett. 2010, 12, 2962–2965. 10.1021/ol1009762. [DOI] [PubMed] [Google Scholar]
- For a different preparation of 21 via radical addition into acrylonitrile see:; Liu K.-M.; Chau C.-M.; Sha C.-K. Intermolecular radical addition reactions of α-iodo cycloalkenones and a synthetic study of the enantiopure fawcettimine. Chem. Commun. 2008, 1, 91–93. 10.1039/B714078A. [DOI] [PubMed] [Google Scholar]
- A similar cyclization via a hydroxylated derivative of 22a was used in the synthesis of a huperzine-type alkaloid:; Wu B.; Bai D. The First Total Synthesis of (±)-Huperzine B. J. Org. Chem. 1997, 62, 5978–5981. 10.1021/jo970248f. [DOI] [Google Scholar]
- Katsuhara J. Absolute Configuration of Pulegone Oxide and Piperitenone Dioxide. J. Org. Chem. 1967, 32, 797–799. 10.1021/jo01278a062. [DOI] [Google Scholar]
- Elgendy E. M.; Khayyat S. A. Oxidation Studies on Some Natural Monoterpenes: Citral, Pulegone, and Camphene. Russ. J. Org. Chem. 2008, 44, 814–822. 10.1134/S1070428008060067. [DOI] [Google Scholar]
- West J. G.; Huang D.; Sorensen E. J. Acceptorless dehydrogenation of small molecules through cooperative base metal catalysis. Nat. Commun. 2015, 6, 10093. 10.1038/ncomms10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadhu P. S.; Ravinder M.; Kumar P. A.; Rao V. J. Photochemical dehydrogenation of 3,4-dihydro-2-pyridones. Photochem. Photobiol. Sci. 2009, 8, 513–515. 10.1039/b813921k. [DOI] [PubMed] [Google Scholar]
- Sun L. H.; Bulter T.; Alcalde M.; Petrounia I. P.; Arnold F. H. Modification of Galactose Oxidase to Introduce Glucose 6-Oxidase Activity. ChemBioChem 2002, 3, 781–783. . [DOI] [PubMed] [Google Scholar]
- Vilim J.; Knaus T.; Mutti F. G. Catalytic Promiscuity of Galactose Oxidase: A Mild Synthesis of Nitriles from Alcohols, Air, and Ammonia. Angew. Chem., Int. Ed. 2018, 57, 14240–14244. 10.1002/anie.201809411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghislieri D.; Green A. P.; Pontini M.; Willies S. C.; Rowles I.; Frank A.; Grogan G.; Turner N. J. Engineering an Enantioselective Amine Oxidase for the Synthesis of Pharmaceutical Building Blocks and Alkaloid Natural Products. J. Am. Chem. Soc. 2013, 135, 10863–10869. 10.1021/ja4051235. [DOI] [PubMed] [Google Scholar]
- Gandomkar S.; Jost E.; Loidolt D.; Swoboda A.; Pickl M.; Elaily W.; Daniel B.; Fraaije M. W.; Macheroux P.; Kroutil W. Biocatalytic Enantioselective Oxidation of Sec-Allylic Alcohols with Flavin-Dependent Oxidases. Adv. Synth. Catal. 2019, 361, 5264–5271. 10.1002/adsc.201900921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrittwieser J. H.; Groenendaal B.; Resch V.; Ghislieri D.; Wallner S.; Fischereder E. M.; Fuchs E.; Grischek B.; Sattler J. H.; Macheroux P.; Turner N. J.; Kroutil W. Deracemization by Simultaneous Bio-Oxidative Kinetic Resolution and Stereoinversion. Angew. Chem., Int. Ed. 2014, 53, 3731–3734. 10.1002/anie.201400027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K.; Umezawa K.; Varnai A.; Eijsink V. G. H.; Igarashi K.; Yoshida M.; Nakamura N. Fungal PQQ-dependent dehydrogenases and their potential in biocatalysis. Curr. Opin. Chem. Biol. 2019, 49, 113–121. 10.1016/j.cbpa.2018.12.001. [DOI] [PubMed] [Google Scholar]
- Thiel D.; Doknic D.; Deska J. Enzymatic aerobic ring rearrangement of optically active furylcarbinols. Nat. Commun. 2014, 5, 5278. 10.1038/ncomms6278. [DOI] [PubMed] [Google Scholar]
- Diaz-Rodriguez A.; Martinez-Montero L.; Lavandera I.; Gotor V.; Gotor-Fernandez V. Laccase/2,2,6,6-Tetramethylpiperidinoxyl Radical (TEMPO): An Efficient Catalytic System for Selective Oxidations of Primary Hydroxy and Amino Groups in Aqueous and Biphasic Media. Adv. Synth. Catal. 2014, 356, 2321–2329. 10.1002/adsc.201400260. [DOI] [Google Scholar]
- Takasu N.; Oisaki K.; Kanai M. Iron-Catalyzed Oxidative C(3)−H Functionalization of Amines. Org. Lett. 2013, 15, 1918–1921. 10.1021/ol400568u. [DOI] [PubMed] [Google Scholar]
- Chen C.; Kattanguru P.; Tomashenko O. A.; Karpowicz R.; Siemiaszko G.; Bhattacharya A.; Calasans V.; Six Y. Synthesis of functionalised azepanes and piperidines from bicyclic halogenated aminocyclopropane derivatives. Org. Biomol. Chem. 2017, 15, 5364–5372. 10.1039/C7OB01238A. [DOI] [PubMed] [Google Scholar]
- Kaname M.; Yoshifuji S.; Sashida H. Ruthenium tetroxide oxidation of cyclic N-acylamines by a single layer method: formation of ω-amino acids. Tetrahedron Lett. 2008, 49, 2786–2788. 10.1016/j.tetlet.2008.02.127. [DOI] [Google Scholar]
- Nishikawa K.; Noguchi T.; Kikuchi S.; Maruyama T.; Araki Y.; Yotsu-Yamashita M.; Morimoto Y. Tetrodotoxin Framework Construction from Linear Substrates Utilizing a Hg(OTf)2-Catalyzed Cycloisomerization Reaction: Synthesis of the Unnatural Analogue 11-nor-6,7,8-Trideoxytetrodotoxin. Org. Lett. 2021, 23, 1703. 10.1021/acs.orglett.1c00125. [DOI] [PubMed] [Google Scholar]
- Bacha J. D.; Kochi J. K. Alkenes from acids by oxidative decarboxylation. Tetrahedron 1968, 24, 2215–2226. 10.1016/0040-4020(68)88124-4. [DOI] [Google Scholar]
- Kulbitski K.; Nisnevich G.; Gandelman M. Metal-Free Efficient, General and Facile Iododecarboxylation Method with Biodegradable Co-Products. Adv. Synth. Catal. 2011, 353, 1438–1442. 10.1002/adsc.201100145. [DOI] [Google Scholar]
- Sun X.; Chen J.; Ritter T. Catalytic dehydrogenative decarboxyolefination of carboxylic acids. Nat. Chem. 2018, 10, 1229–1233. 10.1038/s41557-018-0142-4. [DOI] [PubMed] [Google Scholar]
- Nguyen V. T.; Nguyen V. D.; Haug G. C.; Dang H. T.; Jin S.; Li Z.; Flores-Hansen C.; Benavides B. S.; Arman H. D.; Larionov O. V. Alkene Synthesis by Photocatalytic Chemoenzymatically Compatible Dehydrodecarboxylation of Carboxylic Acids and Biomass. ACS Catal. 2019, 9, 9485–9498. 10.1021/acscatal.9b02951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goossen L. J.; Rodriguez N. A mild and efficient protocol for the conversion of carboxylic acids to olefins by a catalytic decarbonylative elimination reaction. Chem. Commun. 2004, 6, 724–725. 10.1039/B316613A. [DOI] [PubMed] [Google Scholar]
- Ding R.; Fu J.-G.; Xu G.-Q.; Sun B.-F.; Lin G.-Q. Divergent total synthesis of the Lycopodium alkaloids huperzine A, huperzine B, and huperzine U. J. Org. Chem. 2014, 79, 240–250. 10.1021/jo402419h. [DOI] [PubMed] [Google Scholar]
- Gauthier D.; Lindhardt A. T.; Olsen E. P. K.; Overgaard J.; Skrydstrup T. In Situ Generated Bulky Palladium Hydride Complexes as Catalysts for Efficient Isomerization of Olefins. Selective Transformation of Terminal Alkenes to 2-Alkenes. J. Am. Chem. Soc. 2010, 132, 7998–8009. 10.1021/ja9108424. [DOI] [PubMed] [Google Scholar]
- Kozikowski A. P.; Miller C. P.; Yamada F.; Pang Y. P.; Miller J. H.; McKinney M.; Ball R. G. Delineating the Pharmacophoric Elements of Huperzine A: Importance of the Unsaturated Three-Carbon Bridge to Its AChE Inhibitory Activity. J. Med. Chem. 1991, 34, 3399–3402. 10.1021/jm00116a010. [DOI] [PubMed] [Google Scholar]
- Ishiyama T.; Takagi J.; Ishida K.; Miyaura N.; Anastasi N. R.; Hartwig J. F. Mild Iridium-Catalyzed Borylation of Arenes. High Turnover Numbers, Room Temperature Reactions, and Isolation of a Potential Intermediate. J. Am. Chem. Soc. 2002, 124, 390–391. 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]
- Bromodeborylation of a Cbz-protected lycodine derivative was previously demonstrated:; Zhao L.; Tsukano C.; Kwon E.; Shirakawa H.; Kaneko S.; Takemoto Y.; Hirama M. Competent Route to Unsymmetric Dimer Architectures: Total Syntheses of (−)-Lycodine and (−)-Complanadines A and B, and Evaluation of Their Neurite Outgrowth Activities. Chem. - Eur. J. 2017, 23, 802–812. 10.1002/chem.201604647. [DOI] [PubMed] [Google Scholar]
- Xu L.-P.; Roque J. B.; Sarpong R.; Musaev D. G. Reactivity and Selectivity Controlling Factors in the Pd/Dialkylbiarylphosphine-Catalyzed C−C Cleavage/Cross-Coupling of an N-Fused Bicyclo α-Hydroxy-β-Lactam. J. Am. Chem. Soc. 2020, 142, 21140–21152. 10.1021/jacs.0c10220. [DOI] [PubMed] [Google Scholar]
- Muller M. Enzymatic Synthesis of Tertiary Alcohols. ChemBioEng Rev. 2014, 1, 14–26. 10.1002/cben.201300005. [DOI] [Google Scholar]
- Hari Krishna S.; Persson M.; Bornscheuer U. T Enantioselective transesterification of a tertiary alcohol by lipase A from Candida antarctica. Tetrahedron: Asymmetry 2002, 13, 2693–2696. 10.1016/S0957-4166(02)00739-5. [DOI] [Google Scholar]
- Bailey W. F.; Beak P.; Kerrick S. T.; Ma S.; Wiberg K. B. An Experimental and Computational Investigation of the Enantioselective Deprotonation of Boc-piperidine. J. Am. Chem. Soc. 2002, 124, 1889–1896. 10.1021/ja012169y. [DOI] [PubMed] [Google Scholar]
- Coldham I.; O’Brien P.; Patel J. J.; Raimbault S.; Sanderson A. J.; Stead D.; Whittaker D. T. E. Asymmetric deprotonation of N-Boc-piperidines. Tetrahedron: Asymmetry 2007, 18, 2113–2119. 10.1016/j.tetasy.2007.09.001. [DOI] [Google Scholar]
- Under the established photochemical conditions for generation of piperidine-derived α-hydroxy-β-lactam 32 (see ref (20)), the corresponding pyrrolidine-derived species is not formed. Alternatively, irradiation of the same pyrrolidine-derived phenyl keto amide substrate in solution with blue LEDs results in the formation of the corresponding pyrrolidine-fused 4-oxazolidinone (N,O-acetal) product.
- Campos K. R.; Klapars A.; Waldman J. H.; Dormer P. G.; Chen C. Y. Enantioselective, Palladium-Catalyzed α-Arylation of N-Boc-pyrrolidine. J. Am. Chem. Soc. 2006, 128, 3538–3539. 10.1021/ja0605265. [DOI] [PubMed] [Google Scholar]
- Nikolic N.; Beak P. (R)-(+)-2-(Diphenylhydroxymethyl) Pyrrolidine. Org. Synth. 1997, 74, 23–29. 10.15227/orgsyn.074.0023. [DOI] [Google Scholar]
- Zuo Z.; Ahneman D. T.; Chu L.; Terrett J. A.; Doyle A. G.; MacMillan D. W. C. Merging photoredox with nickel catalysis: Coupling of α-carboxyl sp3-carbons with aryl halides. Science 2014, 345, 437–440. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.