

Abstract
Cumulative data suggest the involvement of Fyn tyrosine kinase in Alzheimer's disease (AD). Previously, our group has shown increased immunoreactivities of the FynT isoform in AD neocortex (with no change in the alternatively spliced FynB isoform) which associated with neurofibrillary degeneration and reactive astrogliosis. Since both the aforementioned neuropathological features are also variably found in Lewy Body dementias (LBD), we investigated potential perturbations of Fyn expression in the post‐mortem neocortex of patients with AD, as well as those diagnosed as having one of the two main subgroups of LBD: Parkinson's disease dementia (PDD) and dementia with Lewy bodies (DLB). We found selective upregulation of FynT expression in AD, PDD, and DLB which also correlated with cognitive impairment. Furthermore, increased FynT expression correlated with hallmark neuropathological lesions, soluble β‐amyloid, and phosphorylated tau, as well as markers of microglia and astrocyte activation. In line with the human post‐mortem studies, cortical FynT expression in aged mice transgenic for human P301S tau was upregulated and further correlated with accumulation of aggregated phosphorylated tau as well as with microglial and astrocytic markers. Our findings provide further evidence for the involvement of FynT in neurodegenerative dementias, likely via effects on tauopathy and neuroinflammation.
Keywords: alternative splicing, Alzheimer's disease, Fyn kinase, glial activation, Lewy body dementia, tauopathy
Splice variant‐specific upregulation of FynT kinase correlated with clinical and neuropathological markers of Alzheimer's Disease (AD) and Lewy body Dementias (LBD). Our results suggest that FynT should be further evaluated as a novel therapeutic target in AD and LBD.

Abbreviations
- AD
Alzheimer's disease
- ANOVA
analysis of variance
- AP
amyloid plaques
- BA
Brodmann area
- BA21
mid temporal gyrus
- BA9
dorsolateral and medial prefrontal cortex
- CDK5
cyclin‐dependent kinase 5
- DLB
dementia with Lewy bodies
- FDR
false discovery rate
- FynB
Fyn kinase isoform B
- FynT
Fyn kinase isoform T
- GO
gene ontology
- GSK3
glycogen synthase kinase 3
- HTA
Human Transcriptome Array
- HTRF
Homogenous Time‐Resolved Fluorescence
- LB
Lewy bodies
- LBD
Lewy body dementias
- MMSE
Mini‐Mental State Examination
- NFT
neurofibrillary tangles
- PDD
Parkinson's disease dementia
- PMI
post‐mortem interval
- RIN
RNA Integrity Number
- RT‐PCR
reverse transcription polymerase chain reaction
- sAβ42
soluble fraction of β‐amyloid1‐42 peptide
1. INTRODUCTION
Alzheimer's disease (AD) and Lewy body dementias (LBD) are respectively the number one and two causes of neurodegenerative dementia in the elderly, and contribute to significant morbidity for both sufferers and their caregivers, as well as to global health‐care burden (1, 2, 3). LBD consists of two clinical subtypes: Parkinson's disease dementia (PDD) and dementia with Lewy bodies (DLB), where differentiation is mainly based on the “one‐year rule” in which patients presenting with dementia before, or within a year of, Parkinsonism symptoms are diagnosed as DLB, whereas dementia occurring >1 year after Parkinsonism is considered to be PDD (4). While PDD and DLB share similar neuropathological features, especially the presence of cortical aggregated α‐synuclein‐containing Lewy bodies (LB), significant differences in clinical presentation as well as neurochemical perturbations have been reported between them (5, 6, 7, 8, 9, 10), giving rise to ongoing debate on whether PDD and DLB are separate entities or part of the same spectrum of LBD (11, 12, 13, 14). Interestingly, in addition to LB, both PDD and DLB manifest variable burdens of hallmark AD lesions, namely intercellular amyloid plaques (AP) consisting mainly of insoluble, aggregated β‐amyloid peptides (Aβ); and intracellular neurofibrillary tangles (NFT) formed from aggregation of hyperphosphorylated tau proteins into paired helical filaments (15, 16, 17, 18). Furthermore, the presence of chronic neuroinflammation and glial activation, which are intimately linked to both AP and NFT (19, 20), are also salient features of AD and LBD (21, 22). Therefore, studies focused on the pathophysiological mechanisms and effects of AP and NFT formation (23) should consider the applicability and implications of their findings to LBD.
Given the diverse biological functions (ranging from regulating brain function to modulating T‐cell signaling) attributed to Fyn, a member of the Src family tyrosine kinases (24), it is perhaps unsurprising that Fyn kinase has been implicated in AD pathophysiology (25). As Fyn is alternatively spliced into two major isoforms: brain‐predominant FynB and immune cells‐predominant FynT, we have measured FynB versus FynT immunoreactivities in post‐mortem AD neocortex, and found selective increases in FynT which also associated with neurofibrillary degeneration and reactive astrogliosis (26). These findings are in line with in vitro data showing that prolonged inflammatory response and activation in astrocytes are mediated by pro‐inflammatory cytokine‐induced FynT (27). However, the status of Fyn isoform expression as well as potential associations with neuropathological features and neuroinflammatory markers in LBD is currently unknown. In this study, we identified differential splicing of Fyn using a human transcriptome microarray approach, and further confirmed our findings with real‐time RT‐PCR of Fyn isoform expression in neocortex of AD and LBD patients together with aged non‐demented subjects, and correlated the measures with Aβ and phospho‐Tau concentrations, neuroinflammatory markers, neuropathological and clinical scores. To further study possible links between Fyn perturbations and NFT formation, we also monitored longitudinal changes of Fyn isoform expression in association with tau pathology and neuroinflammation in a transgenic mouse model of human tauopathy which harbors the human P301S tau mutation (28).
2. METHODS
2.1. Patients, clinical and neuropathological assessments
Post‐mortem brain tissues from the frontal (Brodmann Area, BA9, dorsolateral/medial prefrontal cortex) and temporal (BA21: middle temporal gyrus) regions of subjects with AD, DLB, PDD, as well as elderly controls (CTRL) without neurological or psychiatric diseases were obtained from the University Hospital Stavanger, Newcastle Brain Tissue Resource, the London Neurodegenerative Diseases Brain Bank and the Thomas Willis Brain Collections at Oxford University, the UK sites being part of the Brains for Dementia Research network (29). Subjects with dementia were part of longitudinal studies and were followed up annually with clinical assessments including the Mini‐Mental State Examination (MMSE) (30) to measure cognitive decline until death, at which time informed consent was obtained from next‐of‐kin before removal of brains. Consensus criteria used for clinical diagnoses of AD, DLB, and PDD with neuropathologic confirmation have been previously described in detail (31). In addition, semiquantitative scoring (0 = None, 1 = Sparse, 2 = Moderate, and 3 = Abundant) of AP, NFT/neuropil threads, and LB/Lewy neurites as visualized by immunostaining with 4G8, AT8, and α‐synuclein antibodies, respectively, were performed by neuropathologists blinded to clinical diagnosis as previously described (18). For the neurochemical and gene expression studies, 1 cm3 frozen brain chunks were obtained from BA9 and BA21 of the hemisphere contralateral to the one used for neuropathologic studies, then, stored at −80°C before further processing for homogenate preparation and RNA isolation.
2.2. Brain tissues from transgenic mice
The P301S tau transgenic mouse line PS19 (B6 N.Cg‐Tg (Prnp‐MAPT*P301S)PS19Vle/J) were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and bred with wild‐type (WT) control, C57BL/6 NJ to generate hemizygotes and WT littermate controls. The strain harbors the 1N4R isoform of human MAPT gene with the P301S mutation, driven by mouse prion protein promoter which expressed the human P301S tau at levels fivefold higher than endogenous mouse tau (28). Brains from both genotypes were harvested directly after CO2 euthanasia before 6 months (mo) old (WT: n = 13, 8 M/5F, 4.9 ± 0.2 mo; P301S: n = 13, 6 M/7F, 4.7 ± 0.2 mo) and after 6 months old (WT: n = 18, 10 M/8F, 8.6 ± 0.4 mo; P301S: n = 18, 9 M/9F, 8.9 ± 0.4 mo). After removing cerebellum, olfactory bulb, and meninges, the cortex were separated into left and right hemispheres for RNA isolation and brain homogenate preparation, respectively. All experimental procedures conducted in the study were approved by the SingHealth Institutional Animal Care and Use Committee and carried out in accordance with the approved guidelines and regulations.
2.3. Brain tissue processing
Unless otherwise specified, all chemicals and reagents were purchased from Sigma Aldrich (St Louis, MO, USA) and of analytical grade. Frozen human brain chunks from BA9 and BA21 were thawed on ice, dissected free of meninges and white matter, then, homogenized (21) with an Ultra‐Turrax® T‐25 homogenizer (IKA‐Werke, Staufen, Germany) in ice‐cold buffer (50 mM Tris‐HCl, 120 mM NaCl, 5 mM KCl, and pH 7.4) with cOmplete™ protease inhibitor cocktail and PhosSTOP™ phosphatase inhibitor tablets (Roche Life Science, Penzberg, Germany). Brain homogenates from mice were prepared as above without the prior dissection step. For both human and mice brains, representative portions were kept in TRIzol® reagent (Invitrogen Inc., Carlsbad, CA, USA) at −80°C for subsequent RNA extraction.
2.4. Enzyme‐linked immunosorbent assays (ELISA) of pS396 Tau and soluble Aβ42 in human neocortex
Aliquots of brain homogenates was treated with 5 M Guanidine for detection of tau with serine phosphorylation at position 396 (pS396 Tau) by ELISA according to manufacturer's instructions (KHB7031, Thermo Fisher Scientific, Waltham, MA, USA). Separate homogenate aliquots were agitated using a sonicator for 30 mins at 4°C before centrifugation at 6000 g, 30 min at 4°C, with the supernatant used for detection of soluble Aβ1‐42 peptides (sAβ42) by ELISA according to manufacturer's instructions (KHB3441, Thermo Fisher Scientific). Concentrations values in pg/mL were normalized to protein content of each sample (determined using Pierce Coomassie Plus Reagent, Thermo Fisher Scientific) and presented as pg/mg protein.
2.5. RNA isolation, reverse transcription polymerase chain reaction (RT‐PCR), and capillary electrophoresis
Brain tissue kept frozen in TRIzol® reagent (Invitrogen Inc., Carlsbad, CA, USA) were thawed and processed for RNA extraction following manufacturer's instructions. A Fragment Analyzer™ system (Advanced Analytical Technologies Inc, Ankeny, IA, USA) was used for evaluating RNA quality, with RIN <3.0 excluded from further analysis. 2 μg of RNA was reverse‐transcribed to cDNA using High‐Capacity cDNA RT kit (Applied Biosystems, Foster City, CA, USA) in accordance to manufacturer's protocol. Semiquantitative measurements of gene expression were performed on the 7500 Fast Real‐time PCR system (Applied Biosystems) or CFX96 TM Real‐time PCR system (BioRad) using GoTaq® qPCR Master Mix (Promega, Madison, WI, USA). The primer sequences used in this study are listed in Table S1. All real‐time RT‐PCR assays were performed in duplicate. Standard curves of each gene were generated independently by 10x serial dilution of template DNA. The relative signal intensity of each sample was calculated according to the corresponding standard curve. Normalization was performed in each sample by dividing the relative signal intensity of gene of interest to geometric mean of β‐actin, GAPDH, and 18S rRNA. To determine FynT to FynB ratios using capillary electrophoresis, a pair of common primers spanning the alternative spliced exon of Fyn were used as described previously (26). Peak areas of FynT (219 bp DNA amplicon) and FynB (228 bp DNA amplicon) in each electropherogram were translated into expression level and used to determine ratios of FynT to FynB expression.
2.6. Human Transcriptome Array (HTA) data and Gene Ontology (GO) term enrichment analyses
A subset of samples (9 CTRL, 9 AD, 12 DLB, and 12 PDD) from BA9 were processed for high‐throughput transcriptome profiling using the GeneChip® HTA 2.0 array (Affymetrix, Santa Clara, CA, USA). The data set has been deposited with the Gene Expression Omnibus (GEO) (Access #GSE150696). Using the Gene View module of Partek Genomics Suite® 7.0 software (Partek Inc, St Louis, MO, USA), we were able to allocate differentially expressed probesets to specific transcript variants of Fyn. Expressed probesets which were significantly correlated with the FynT‐specific probesets (PSR06025242.hg.1) at false discovery rate (FDR)(32) cutoff of 5% were retrieved. Genes with ≥10 representative probesets that consistently correlated with FynT were subject to Gene Ontology (GO) term enrichment analyses using DAVID Bioinformatics Resources 6.7 (http://david.abcc.ncifcrf.gov/) (33, 34), with Benjamini's enrichment p values adjustment for multiple testing using FDR of 5%. For this study, we focused on GO categories of Biological Processing and Cellular components, after removing general GO terms with enrichment factor <2.
2.7. Homogenous Time‐Resolved Fluorescence (HTRF) assays of mouse brain homogenates
Cisbio® HTRF assays (PerkinElmer Inc. Waltham, MA, USA), a technology combining standard fluorescence resonance energy transfer (FRET) with time‐resolved measurement of fluorescence, were used for measuring total human tau protein (MAP‐Tau Kit), phosphorylated tau (Phospho‐Ser202/Thr205 human TAU kit), and aggregated tau (Tau aggregation kit) according to manufacturer's instructions. A portion of the right hemisphere was homogenized in Cisbio lysis buffer with blocking reagent using 5 mm stainless steel beads in a TissueLyser LT (Qiagen, Venlo, the Netherlands) tissue disruptor (two rounds of 50 Hz, 2 min). After centrifugation at 21,000 g for 20 min at 4°C, supernatants were measured for protein concentration using the Bradford protein assay kit (BioRad, Hercules, CA, USA). Equal amounts of brain homogenate were then diluted and incubated with respective antibodies for 24 h at room temperature. Samples were excited at 340 nm and emission values collected at 620 nm (Donor) and 665 nm (Acceptor) using EnSpire® multimode plate reader (Perkin Elmer). For data analysis, HTRF ratio was calculated for each individual well by the ratio of the two emission values (Signal 665 nm/Signal 620 nm) and multiplied by 10,000 to ease data processing. Data were presented as Delta F% which reflected the signal to background of the assay, and calculated as [HTRF ratio (sample) ‐ HTRF ratio (negative control)]/HTRF ratio (negative control) x 100.
2.8. Statistical analyses
Pairwise differences among groups were analyzed by Kruskal–Wallis analysis of variance (ANOVA) with post hoc Dunn's test or data transformed to log2 base value and analyzed using one‐way ANOVA with Bonferroni's post hoc tests. Two‐way ANOVA with Bonferroni's post hoc tests were applied to transgenic mice study with genotype and age as independent variables. Pearson's or Spearman's correlations was used to for parametric or nonparametric variables, respectively. For all analyses, two‐tailed p values of <0.05 were considered to be statistically significant. Statistical analyses and plotting of graphs were performed using the PRISM Version 5 software (GraphPad, San Diego, CA, USA).
3. RESULTS
3.1. Demographic and disease variables of the study cohort
Table 1 shows the maximum available numbers of aged controls (CTRL) as well as a community‐based cohort of PDD, DLB, and AD patients. Groups were not significantly different in age at death and post‐mortem interval (Kruskal–Wallis ANOVA p > 0.05). Within the dementia subgroups (AD, PDD, and DLB), no significant difference in dementia severity (indicated by MMSE decline per year) was observed (Kruskal–Wallis ANOVA p > 0.05). Braak staging (35) for extent of AD pathological changes showed all but one of the aged CTRL in Braak stage ≤II, with one in III–IV (another four CTRL did not have Braak staging data). In contrast, 10 out of 13 available AD cases were Braak V/VI, with the rest in Braak III/IV. PDD and DLB showed variable AD pathology ranging from Braak ≤II to V/VI, with DLB showing higher numbers of those with more advanced stages compared to PDD. Furthermore, ELISA measures of sAβ42 and pS396Tau, used respectively as markers of amyloid and neurofibrillary tangle burden, were also raised in the dementia subgroups, with the highest levels in AD, and the lowest in PDD (Table 1). These neuropathological and biochemical features are in line with previous clinical and post‐mortem observations of AD and Lewy body dementias, especially with regards to PDD harboring the lowest AD pathological burden among the dementia subgroups (17, 18).
TABLE 1.
Demographic and disease variables of a cohort of patients with neuropathologically confirmed AD and LBD
| CTRL | AD | DLB | PDD | |
|---|---|---|---|---|
| (n = 16) | (n = 13) | (n = 39) | (n = 27) | |
| Demographics a | ||||
| Gender (male/female) | 9 M/7F | 5 M/8F | 26 M/13F | 15 M/12F |
| Age at death (years) | 82.0 ± 1.7 | 86.1 ± 1.9 | 80.9 ± 1.1 | 79.3 ± 1.2 |
| Postmortem interval (hours) | 45.7 ± 5.9 | 36.7 ± 7.1 | 43.7 ± 4.9 | 33.5 ± 3.0 |
| MMSE before death | – | 9 ± 2.1 (n = 13) | 14 ± 1.5 (n = 32) | 12 ± 1.6 (n = 25) |
| Braak staging | ||||
| 0–II | 11 (91.7%) | 0 (0%) | 6 (15.4%) | 18 (66.7%) |
| III–IV | 1 (8.3%) | 3 (23%) | 20 (51.3%) | 7 (26%) |
| V–VI | 0 (0%) | 10 (77%) | 13 (33.3%) | 2 (7.3%) |
| ELISA assay | ||||
| Soluble Aβ42 (pg/mg) (BA9) | 5.6 ± 1.5 (n = 15) | 33.2 ± 2.2* (n = 13) | 29.6 ± 2.4* (n = 37) | 10.9 ± 1.8**, *** (n = 26) |
| Soluble Aβ42 (pg/mg) (BA21) | 6.2 ± 1.5 (n = 15) | 27.5 ± 2.4* (n = 13) | 25.2 ± 1.9* (n = 39) | 10.3 ± 1.8**, *** (n = 26) |
| pS396Tau (pg/mg) (BA9) | 5.4 ± 2.7 (n = 16) | 168.4 ± 33.3* (n = 13) | 47.6 ± 13.8** (n = 39) | 2.2 ± 0.6**, *** (n = 27) |
| pS396Tau (pg/mg) (BA21) | 25.1 ± 5.6 (n = 15) | 524.8 ± 92.9* (n = 13) | 215.1 ± 48** (n = 38) | 61.9 ± 25.6** (n = 26) |
Data are presented as mean ± S.E.M.
Maximum available n. Not all samples were available for all assessments. Available n listed in individual assessments.
Significantly different compared to CTRL (Kruskal–Wallis ANOVA with post hoc Dunn's test, p < 0.05).
Significantly different compared to AD (Kruskal–Wallis AONVA with post hoc Dunn's test, p < 0.05).
Significantly different compared to DLB (Kruskal–Wallis ANOVA with post hoc Dunn's test, p < 0.05).
3.2. Selective upregulation of FynT isoform expression in neocortex of AD and LBD
We have previously reported specific increases of FynT immunoreactivities which contrasted with unchanged brain‐predominant FynB in AD (26). Here, using a high‐throughput gene profiling (HTA) platform, we found similar FynT upregulation in a separate cohort of AD and further extended this observation to DLB and PDD. Figure 1 shows Gene View plot where probeset PSR06025242.hg.1 (indicated by blue asterisk) corresponding to sequence for FynT‐specific exon was significantly upregulated in AD (~3 fold), DLB (~2.5 fold), and PDD (~1.8 fold) prefrontal cortex compared with CTRL. In contrast, probesets corresponding to common exons and FynB‐specific exons (yellow boxes in FynB transcript, Figure 1) showed no significant induction in the dementia subgroups. To confirm these findings, we determined the proportional amounts of FynB and FynT in two brain regions (BA9 and BA21) of aged CTRL and dementia subgroups using common primers spanning the alternative spliced exon of Fyn for RT‐PCR followed by capillary electrophoresis. The representative electropherograms in Figure 2A indicated that the FynT peaks were about 10 times lower than FynB peaks in CTRL, while elevations of FynT peaks relative to FynB were detected in the dementia subgroups. Indeed, AD, DLB, and PDD all had significantly higher FynT to FynB ratios compared to CTRL in both BA9 and BA21 (Figure 2B), with the ratios showing a high degree of correlation between the two brain regions (Pearson's r = 0.79, p < 0.001).
FIGURE 1.
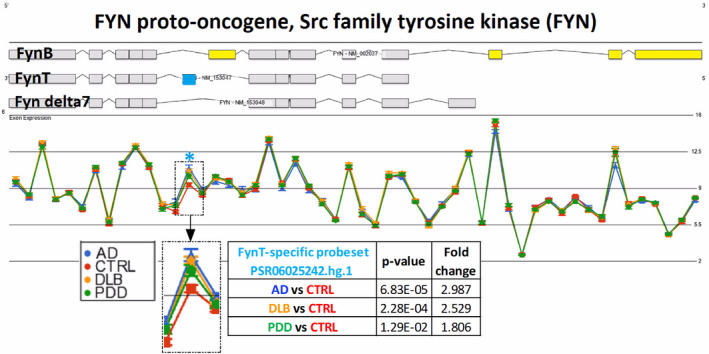
Specific upregulation of FynT isoform expression in prefrontal cortex of AD and LBD. Gene View plot demonstrates differential alternative splicing of Fyn in BA9 samples of CTRL (n = 9, red), AD (n = 9, blue), DLB (n = 12, amber), and PDD (n = 12, green), from data derived from Affymetrix HTA array. Light blue asterisk indicated the PSR06025242.hg.1 probeset, with a magnified view shown in the inset, corresponding to the FynT‐specific exon highlighted in blue. FynB‐specific exons are highlighted in yellow. The Y‐axis is in Log2 scale intensity value. The p values shown are of Alt‐splicing ANOVAs using the Partek Genomic Suite® software
FIGURE 2.
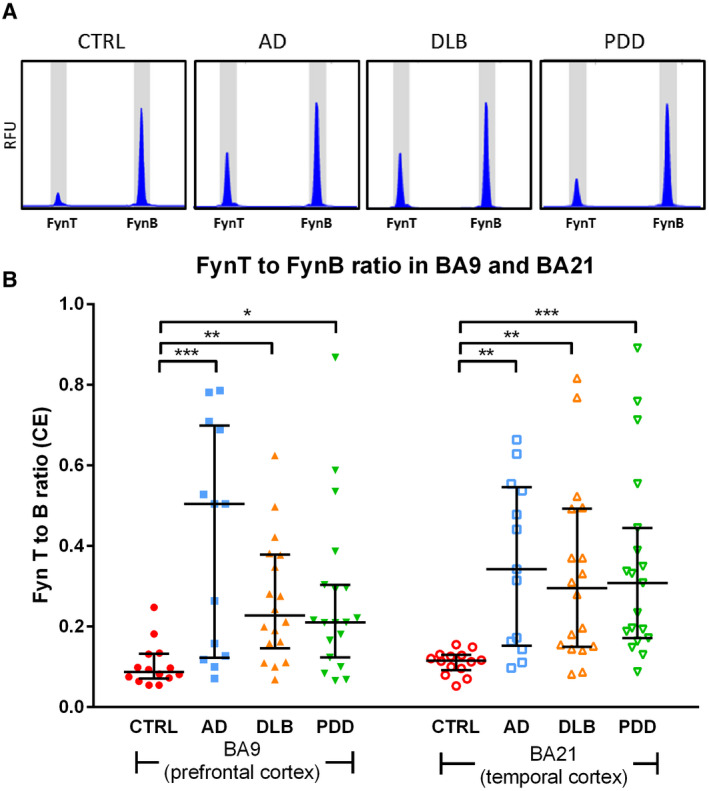
Increased FynT to FynB ratios in prefrontal and temporal cortex of AD and LBD. (A) Representative electropherograms of the proportional levels of FynB and FynT in each sample as determined by RT‐PCR using common primers spanning the alternatively spliced exon of Fyn followed by capillary electrophoresis. Relative fluorescence units (RFU) of FynT peak (219 bp DNA amplicon) and FynB peak (128 bp DNA amplicon) are shown for each diagnostic group. (B) Peak areas of FynT and FynB in each electropherogram were converted to gene transcript levels and used to determine ratio of FynT to FynB expression, depicted as dot plots of CTRL (n = 14, red), AD (n = 13, blue), DLB (n = 18, amber), and PDD (n = 19, green) values with horizontal lines in each group indicating median and interquartile range. *p < 0.05, **p < 0.01, ***p < 0.001, significantly different from CTRL by Kruskal–Wallis ANOVA with post hoc Dunn's tests
3.3. FynT upregulation correlated with cognitive impairment and neuropathological features in AD and LBD
In order to investigate potential associations between Fyn perturbations and clinical or neuropathologic features, FynT and FynB expression levels were determined by real‐time RT‐PCR and correlated with pre‐death MMSE, neuropathologic scores as well as sAβ42 and pS396 Tau concentrations. Consistently, FynT was observed to be upregulated in the dementia subgroups, reaching statistical significance for AD and DLB (with a trend toward increase for PDD) in BA9, and for all three dementia subgroups in BA21 (Figure 3A left panel). We also confirmed that FynB expression were not significantly altered in the dementia subgroups (Figure 3A right panel).
FIGURE 3.
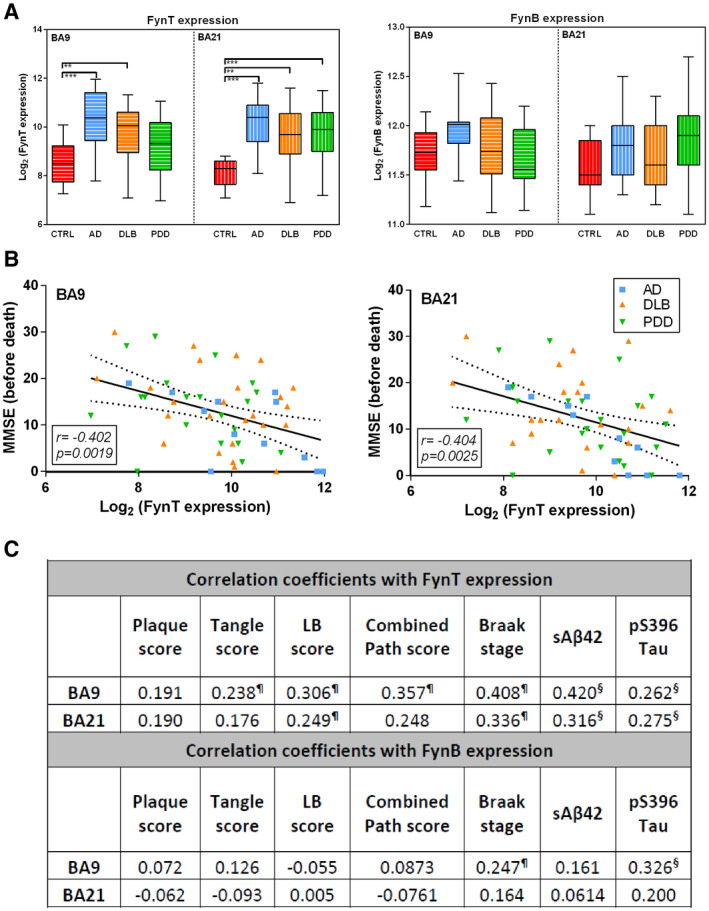
FynT upregulation in AD and LBD correlates with cognitive impairment and neuropathological features. (A) FynT and FynB expression were determined by real‐time RT‐PCR in BA9 and BA21, derived from CTRL (n = 13–15, red), AD (n = 11–12, blue), DLB (n = 25–31, amber), and PDD (n = 14–21, green). Data were Log2 transformed and depicted as box plots, with the horizontal intersecting lines and borders indicating median and interquartile range for each group, while whiskers show the highest and lowest values not considered outliers. (B) Scatter plots of Log2‐transforemd FynT expression against pre‐death cognition (MMSE) scores in BA9 (left) and BA21 (right) in the combined dementia cohort. Solid lines indicated linear regressed best‐fit curves while dashed lines indicated their respective 95% confidence intervals, with insets showing Pearson's correlation coefficients (r) and their respective p values. (C) Table of correlation coefficients for FynT or FynB expression with neuropathologic scores, Braak stage and biochemical measures of pS396Tau and soluble Aβ42 (in pg/mg protein) were analyzed using samples derived from CTRL, AD, and LBD. **p < 0.01, ***p < 0.001, significant differences compared to CTRL by one‐way ANOVA with post hoc Bonferroni's tests. ¶Pearson's (r) and §Spearman's (ρ) correlation coefficients with p < 0.05
Figure 3B shows that FynT expression in both BA9 and BA21 negatively correlated with pre‐death MMSE scores in the combined dementia cohort, suggesting that FynT upregulation may be associated with more severe cognitive impairment. Furthermore, Figure 3C lists the correlation coefficients between Fyn expression and (i) semiquantitative scores of amyloid plaques, neurofibrillary tangles (NFT), Lewy bodies (LB), and combined neuropathologic scores (which we have previously shown to be variably increased in LBD (18)); (ii) Braak staging and (iii) concentrations of soluble Aβ (sAβ42) and phosphorylated Tau (pS396 Tau). Interestingly, FynT expression was widely correlated with neuropathological scores (NFT, LB, and combined scores), Braak stage, sAβ42 and pS396 Tau measures, especially in BA9, while FynB did not correlate with neuropathologic scores and biochemical measures except for Braak stage and pS396 Tau in BA9 (Figure 3C). Taken together, our data suggest that FynT isoform‐specific upregulation in the neocortex may be associated with clinical severity and neuropathological burden in AD and LBD.
3.4. FynT upregulation correlated with markers of neuroinflammation in AD and LBD
We have previously found that FynT induction was associated with astrocyte activation under neuroinflammatory conditions (27). In PD, microglial activation was also reported to be dependent on Fyn kinase activity (36). Here, we investigated potential associations between Fyn expression and neuroinflammatory responses using glial fibrillary acidic protein (GFAP) and CD11b expression to indicate astrogliosis and microglial activation, respectively (37, 38). Figure 4A shows significant increases of GFAP expression in BA9 for AD, and in BA21 for DLB, while expression remained unchanged for PDD in both regions (left panel). For microglia marker CD11b, expression was significantly increased in BA9 for AD, and in BA21 for all dementia subgroups (right panel). Figure 4B indicates that FynT expression correlated with both GFAP and CD11b in both brain regions in the combined cohort. To study potential sources of Fyn isoform expression in brain, we measured FynT to FynB ratios, as well as FynT, FynB, GFAP, and CD11b expression in isolated rat primary astrocyte, microglia, and neurons. Among the three cell types, we observed the highest expression of FynT in microglia, followed by astrocytes, with neurons showing the lowest expression (see Figure S1). Taken together, our results suggest that FynT induction has a role in astrogliosis and microglial activation in the neocortex of AD and LBD.
FIGURE 4.
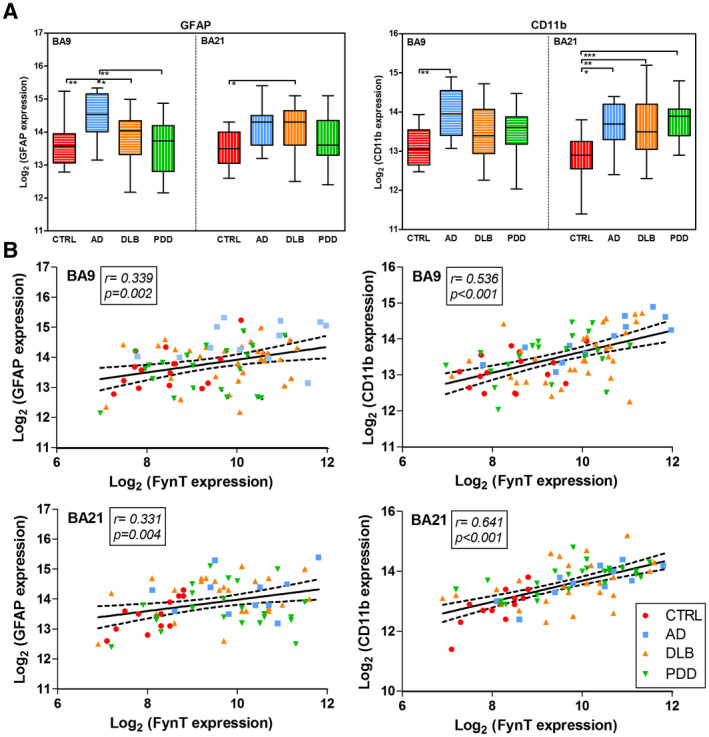
Association of FynT expression with neuroinflammatory markers in CTRL, AD, and LBD. (A) Gene expression of GFAP astrocytic marker (left) and CD11b microglial marker (right) were determined by real‐time RT‐PCR in BA9 and BA21, derived from CTRL (n = 13–15, red), AD (n = 11–12, blue), DLB (n = 25–31, amber), and PDD (n = 14–21, green). Data were Log2 transformed and depicted as box plots, with the horizontal intersecting lines and borders indicating median and interquartile range for each group, while whiskers show the highest and lowest values not considered outliers. (B) Scatter plots of Log2‐transforemd FynT expression against GFAP (left) and CD11b (right) in BA9 (top) and BA21 (bottom) of the combined cohort. Solid lines indicated linear regressed best‐fit curves while dashed lines indicated their respective 95% confidence intervals, with insets showing Pearson's correlation coefficients (r) and their respective p values. *p < 0.05, **p < 0.01, ***p < 0.001, significant pairwise differences between diagnostic groups by one‐way ANOVA with post hoc Bonferroni's tests
3.5. Synaptic dysfunction is a potential consequence of FynT upregulation in AD and LBD
To investigate the potential pathophysiological impact of FynT induction in neurodegenerative dementias, we utilized the high‐throughput transcriptome profiling data on BA9 of a subset of the cohort (9 CTRL, 9 AD, 12 DLB, and 12 PDD). The subset was selected based on RNA quality (RIN > 4) and matched demographic factors (gender, age, and PMI), and not on neuropathological scores or neurochemical variables. Stringent criteria (see details in methodology) were set to identify candidate genes which were correlated either positively or negatively with FynT. In summary, 515 FynT‐positively correlated genes and 1437 FynT‐negatively correlated genes were uncovered and independently analyzed by DAVID (34) to gain an overview of the functional profiles of associated genes in order to understand the underlying processes. Table 2 summarizes the top five GO (gene ontogeny) terms found in each category (the full list of GO terms and corresponding genes can be found in Table S2). In general, genes positively correlated with FynT were mainly associated with cell adhesion, whereas the negatively correlated genes were associated with neuronal function and components found in dendrites and synapses (Table 2). These results suggest that synaptic dysfunction and synaptopathology may be associated with FynT upregulation in AD and LBD.
TABLE 2.
Top five* GO Terms under BP and CC categories for FynT positively and negatively correlated genes derived from CTRL, AD, and LBD transcriptome profiling data
| GO term ID (categories) | Description (FynT positively correlated genes) | Count a | Fold enrichment b | Benjamini c |
|---|---|---|---|---|
| GO:0005913 (CC) | Cell–cell adherens junction | 34 | 4.07 | 2.64E‐09 |
| GO:0005925 (CC) | focal adhesion | 37 | 3.66 | 4.69E‐09 |
| GO:0030027 (CC) | lamellipodium | 18 | 4.35 | 5.62E‐05 |
| GO:0030175 (CC) | filopodium | 12 | 6.54 | 9.64E‐05 |
| GO:0001726 (CC) | ruffle | 13 | 5.59 | 1.53E‐04 |
| GO:0098609 (BP)* | Cell–cell adhesion | 24 | 3.23 | 4.00E‐03 |
| GO:0016569 (BP)* | covalent chromatin modification | 14 | 4.51 | 1.52E‐02 |
| GO term ID (categories) | Description (FynT negatively correlated genes) | Count a | Fold enrichment b | Benjamini c |
|---|---|---|---|---|
| GO:0043209 (CC) | myelin sheath | 54 | 4.75 | 3.64E‐20 |
| GO:0030054 (CC) | cell junction | 91 | 2.65 | 1.62E‐15 |
| GO:0014069 (CC) | postsynaptic density | 50 | 3.64 | 2.66E‐13 |
| GO:0008021 (CC) | synaptic vesicle | 33 | 4.80 | 5.41E‐12 |
| GO:0045211 (CC) | postsynaptic membrane | 49 | 3.11 | 2.28E‐10 |
| GO:0016192 (BP) | vesicle‐mediated transport | 40 | 3.41 | 2.46E‐08 |
| GO:0034220 (BP) | ion transmembrane transport | 48 | 2.96 | 2.57E‐08 |
| GO:0006886 (BP) | intracellular protein transport | 52 | 2.85 | 4.34E‐08 |
| GO:0000165 (BP) | MAPK cascade | 54 | 2.67 | 6.03E‐08 |
| GO:0016241 (BP) | regulation of macroautophagy | 20 | 5.88 | 1.06E‐07 |
Gene ontology (GO) category: biological process (BP) and cellular component (CC).
Count represents the total number of genes correlated with FynT in the respective GO term.
Fold enrichment is used to measure the magnitude of enrichment.
p values adjusted for multiple testing using FDR < 0.05.
For FynT positively correlated genes, only two BP terms were significant at FDR < 0.05.
3.6. Selective upregulation of FynT is associated with tauopathy and neuroinflammation in aged P301S mice
Finally, the potential pathophysiological links between FynT induction, tau pathology, and neuroinflammation were studied in a P301S Tau transgenic mouse model of tauopathy at two age groups: <6 mo when there is minimal brain pathology; and ≥6 mo, when the mice are known to manifest neurofibrillary tangles and neuroinflammation during disease progression. HTRF assays confirmed that P301S Tau mice expressed high levels of human tau in the brain when compared with wild‐type (WT) littermate controls at both age groups (Figure 5A, left panel). However, the younger P301S mice (<6 mo) showed minimal phosphorylated tau and aggregation, similar to WT (Figure 5A, middle and right panels). In contrast, older mice (≥ 6 mo) showed significantly increased levels of phosphorylated, aggregated tau (Figure 5A, middle and right panels). Similar to observations of human post‐mortem brains, we found significantly increased FynT expression as well as higher FynT to FynB ratio in older, but not younger, P301S mice (Figure 5B left and right panels), while FynB expression was unchanged in both age groups (Figure 5B middle panel). Furthermore, markers of neuroinflammatory responses as indicated by GFAP and CD11b expression were significantly increased only in older P301S mice (Figure 5C). Finally, Figure 5D lists the correlation coefficients between expression of Fyn isoforms, GFAP and CD11b as well as HTRF measurements of phosphorylated and aggregated Tau, and showed that both FynT and FynT:FynB correlated with the neuroinflammatory markers as well as pathologic Tau markers. Therefore, our findings suggest that selective FynT upregulation likely occurs downstream of tauopathy and is associated with disease progression, including neuroinflammation, in P301S mice.
FIGURE 5.
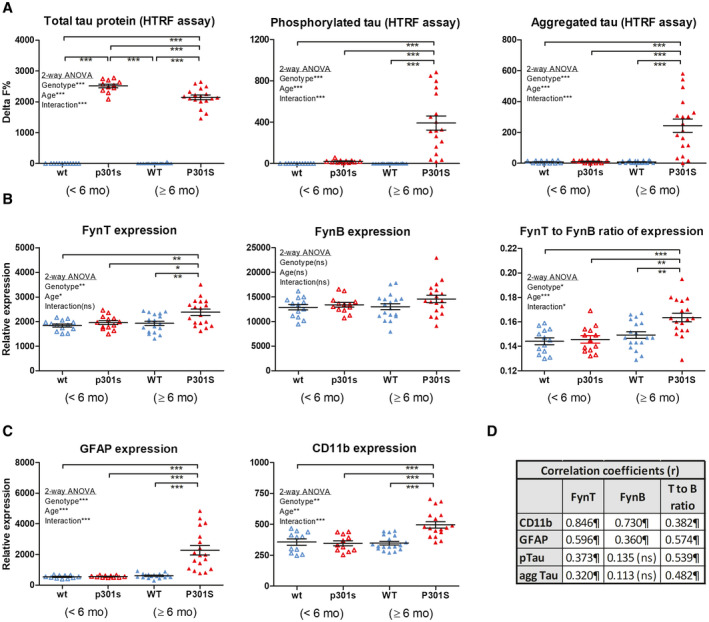
Selective upregulation of FynT in aged transgenic P301S tau mice correlates with tauopathy and neuroinflammation. (A) Determination of phosphorylated tau (pS202/T205 tau), aggregated tau and total tau protein by HTRF in the brain homogenates of P301S Tau transgenic mice and wild‐type littermate controls at age <6 mo (before onset of pathology, labels in small letters: p301 s, wt) and ≥6 mo (with disease progression, labels in capital letters: P301S, WT). RT‐PCR derived values for relative expressions of (B) FynT, FynB (together with FynT to FynB ratios) as well as (C) GFAP and CD11b in the samples as described above. Horizontal lines on the dot plots in each group indicate median and interquartile range. (D) Table of correlation coefficients for FynT, FynB, FynT:FynB with phosphorylated tau, aggregated tau, and neuroinflammation markers. *p < 0.05, **p < 0.01, ***p < 0.001, significant pairwise differences by two‐way ANOVA with post hoc Bonferroni's tests (Significance of the Genotype, Age, and Interaction factors are given in the respective insets of each graph). ¶Pearson's (r) correlation coefficients with p < 0.05
4. DISCUSSION
4.1. FynT kinase as a potential mediator of tauopathy
While processes associated with amyloid precursor protein mismetabolism leading to accumulation of insoluble Aβ plays a major role in AD pathogenesis (39), it is the hyperphosphorylation of tau and ensuing formation of neurofibrillary tangles (NFT) which correlated more closely with disease progression, and may indeed mediate the clinical severity of AD (40, 41). As NFT (together with amyloid plaques) are a common feature of AD, PDD, and DLB, these neurodegenerative dementias can be defined as tauopathies. In this paper, we reported further evidence for a selective upregulation of the FynT isoform in AD neocortex, and extended these observations to PDD and DLB. As basal FynT expression was around one tenth of FynB expression in the normal brain (see Figure 2), we speculate that the inability of some previous studies to detect Fyn induction in AD (42, 43) was likely because of the use of non‐isoform‐specific measurements, since the amount of FynT increase would likely be masked by the unchanged, predominant FynB isoform. Importantly, we showed here that FynT upregulation was associated with astrogliosis, microglial activation, neuropathologic burden, and dementia severity, therefore, suggesting, as others have (25), that Fyn may mediate the pathophysiological link between amyloid and tau. Indeed, Fyn can regulate Aβ‐induced excitotoxicity by interacting with dendritic tau, the latter targeting Fyn postsynaptically to glutamate NMDA receptors where excitotoxic signals are transmitted (44). Aβ‐induced tau accumulation at somatodendritic compartments was also found to be Fyn‐dependent (45). In another study, binding of Aβ oligomers to postsynaptic prion proteins was shown to trigger Fyn‐mediated signaling cascades leading to tauopathy and synaptotoxicity (46, 47). Interestingly, more recent studies suggest that Fyn can also be a key regulator of tau‐associated pathology in the absence of Aβ (48, 49, 50). Conversely, tau seem to directly regulate Fyn targeting to (44); and nanodomain organization (51) within, neuronal dendrites. The complex interactions between Fyn and tau have been further demonstrated by tau's induction of Fyn autophosphorylation and kinase activity (52), contrasting with Fyn‐mediated direct (on tyrosine18) (53) and indirect (via CDK5 and GSK3) (54, 55) phosphorylation of tau. In summary, Fyn seems to be able to signal to tau and regulate tau‐associated pathology in both Aβ‐dependent and ‐independent pathways. Our own observations of FynT upregulation and correlations with tauopathy in AD and LBD, as well as FynT changes in P301S mice which do not manifest Aβ burden, suggest that Fyn interacts with tau in these conditions in an isoform‐specific manner, and that at least some of the pathogenic mechanisms mentioned above may be mediated by altered regulation of alternative splicing which favors FynT.
4.2. Potential mechanisms of FynT‐associated neuronal dysfunction and neuroinflammation in AD and LBD
Several studies have demonstrated that Fyn kinase can modulate synaptic plasticity and learning (56, 57, 58, 59). In this study, we speculated that the detrimental consequences of FynT upregulation during the dementia progression could be associated with synaptic dysfunction, based on the consistent negative correlations of FynT with genes coding for functional components of neurons and synapses (Table 2), as well as with pre‐death cognitive scores (Figure 3B). Recently, mutant P301L tau has been shown to promote aberrant Fyn nanoclustering in dendritic spines which likely contributes to synaptic dysfunction (51). In this study, we used a similar mutant P301S tau mice to demonstrate that age‐dependent increment of phosphorylated and aggregated tau was associated with FynT upregulation (Figure 5). Interestingly, while perturbations of neuronal networks and long‐term potentiation have been reported in young (2 month old) mice (60), our data suggest that these processes are unlikely to involve FynT changes, since FynT upregulation was not apparent until 6 months or older (Figure 5B). Instead, FynT might mediate the previously reported contributions of tau pathology to the development of chronic inflammatory responses during reactive astrogliosis (61). We have reported increased FynT immunoreactivity in hypertrophic cytoplasm of reactive astrocytes in the AD brain (27), which is corroborated here by the finding of significant correlations between FynT and GFAP expression (Figures 4B and 5D).
Additionally, Fyn induction in post‐mortem PD brains has been reported to be associated with neuroinflammatory responses resulting from microglial activation (62). Interestingly, the present in vitro data indicate the highest expression of FynT in microglia, followed by astrocyte, while neurons had the lowest expression (see Figure S1). As the correlation coefficient for FynT and CD11b expression was higher than those between FynT and GFAP or pS396Tau in the post‐mortem study, our data suggest FynT upregulation in the AD and LBD brain could due in large part to activation of predominantly FynT‐expressing microglia, which may in turn contribute to chronic neuroinflammation‐associated neuronal damage (63). However, given the significant (albeit weaker) correlations between FynT and GFAP, as well as previously reported localization of FynT in reactive astrocytes (26), we postulate that astrogliosis underlie at least part of the observed FynT upregulation. Here, it is worth noting that FynT likely plays a critical role in astrocyte activation, as we have previously shown that neuroinflammatory responses to tumor necrosis factor is abolished in astrocytes expressing a kinase‐dead mutant of FynT (27). Therefore, when considered in the context of previous studies, our current in vitro and post‐mortem data suggest that in AD and LBD, FynT upregulation likely occurs in response to tau phosphorylation and NFT formation, and mediates astrocyte and/or microglial activation, leading in turn to neuroinflammation‐associated synaptic dysfunction and neuronal damage.
4.3. Isoform‐specific role of FynT as therapeutic target for neurodegenerative dementias
Fyn kinase has already been proposed as a potential therapeutic target for AD (64). In this regard, a previous study showed that inhibiting Fyn in an AD mouse model restored synaptic density, limited tau aggregation, reduced microglial activation, and subsequently reversed memory deficits without attenuating Aβ load (58). Recent findings consistently demonstrated that both pharmacological Fyn inhibition and depletion of Fyn (via gene knockout) in tauopathy models resulted in behavioral improvements, recovery of synaptic loss, together with reduced tau phosphorylation and gliosis, suggesting that Fyn is an essential pathogenic factor in tauopathies (48, 49, 50). This has expanded the application of Fyn as a therapeutic target for tauopathies in addition to AD. However, it is worth noting that AZDO530, a Src family kinase inhibitor which showed promise in animal studies, demonstrated no statistically significant improvement on neuroimaging outcome in AD patients in a year‐long Phase IIa clinical trial (although a trend toward less shrinkage of the hippocampus and entorhinal cortex was observed in the treated group)(65). One insight that may be gleaned from our study is the need to refine the target to Fyn, and specifically the FynT isoform, for potentially improved efficacy as well as reduced off‐target adverse effects, since the major brain variant FynB has important physiological functions and should not be targeted. Furthermore, when a FynT‐specific compound does become available, our data suggest extending assessments of their clinical utility to LBD in addition to AD.
4.4. Study limitations
There are several limitations apparent in this study which are related to the limited number of available samples. Therefore, in interpreting findings where changes are found in one, but not both of the brain regions examined (e.g., significant correlation between FynT and neurofibrillary tangle (NFT) scores in BA9 prefrontal cortex but not BA21 temporal cortex, see Figure 3C), it is unclear whether the observed regional differences has a genuine pathophysiological basis (for, e.g., more active disease process in BA9 versus widespread cell loss in BA21(66)) or was because of higher data variability in BA21 resulting in nonsignificance of the association. On the contrary, FynT expression seemed to be increased in both regions in LBD, but did not reach statistical significance in BA9 for PDD (Figure 3A). While higher AP and NFT loads are detected in DLB compared with PDD (12, 67), and NFT (which we have shown here to be strongly correlated with FynT) is known to originate from the entorhinal, hippocampal, and neocortical regions of the temporal lobe followed by progression into the prefrontal and other neocortical areas (68), we cannot at present confirm whether these regional differences are DLB‐ or PDD‐specific because of associations with pathological burden, necessitating follow‐up studies on larger cohorts looking at multiple brain regions.
Second, the current study has not studied Fyn associations with LBD‐specific lesions (α‐synuclein‐containing Lewy bodies, LB) in detail. While the focus of our study has been on AD pathological features which are present in both AD and LBD, FynT may also be linked to LB, as suggested by Figure 3C. However, it is at present unclear whether, or how, FynT may be related to α‐synuclein aggregation and related pathophysiology per se, as assays of soluble α‐synuclein revealed unchanged baseline cortical concentrations across controls and dementia subgroups in agreement with previous studies (69, 70) which did not correlate with Fyn or other neuropathological measures (data not shown). These negative findings suggest that other potentially pathogenic species of α‐synuclein, for example, Ser129‐phosphorylated α‐synuclein (71) need to be measured in follow‐up studies of potential Fyn involvement in LBD‐specific pathology.
Finally, the current study employed a genomics approach with the use of microarrays and RT‐PCR to study Fyn expression and associations with a wide range of gene expression changes. There is therefore a lack of histological and cellular localization data, which is mitigated somewhat by (i) the use of primary cultures to provide in vitro validation of the likely sources of FynT, namely, astrocytes and microglia (Figure S1); and (ii) our previous study which showed the localization of FynT in activated astrocytes in AD (26). However, follow‐up studies are needed to confirm the findings for microglia and also extend them to LBD.
5. CONCLUSIONS
Using microarray platforms, we report FynT isoform‐specific upregulation in the neocortex of AD, DLB, and PDD. The observed induction of FynT was closely associated with tauopathy and neuroinflammation, and may mediate their deleterious effects on synaptic and cognitive functions. In corroboration, age‐dependent FynT upregulation was detected in P301S mutant tau transgenic mice, which was significantly correlated with phosphorylated and aggregated tau, as well as with markers of microglial and astrocytic activation. Our findings point to FynT as the pathogenic Fyn isoform for AD and LBD, and propose a refinement of therapeutic approach from nonspecific inhibition of Src family kinases to a more focused search for compounds which specifically target FynT as potential treatment for AD‐related chronic neuroinflammation.
AUTHOR CONTRIBUTIONS
MGKT and MKPL conceived the study and designed the project; CYBL, JHL, FTWL, and CL performed the experiments; CB and PTF provided post‐mortem and clinical data; CYBL, JHL, and MGKT analyzed the data; MGKT and MKPL wrote the first draft. All authors have read and approved the manuscript.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
CONSENT FOR PUBLICATION
All authors gave consent for publication.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
For the post‐mortem study, ethics approval for the collection and study of brain tissues received Institutional Review Board approval in both the United Kingdom (08/H1010/4) and Singapore institutions (NUS 12‐062E), and informed consent was obtained from participants’ next‐of‐kin prior to removal of brain. The mice studies were approved by the SingHealth Group Institutional Animal Care and Use Committee (2017/SHS/1280).
Supporting information
Clara Y. B. Low and Jasinda H. Lee contributed equally to this work.
Funding information
This work was supported by research grants (NMRC/OFIRG/0028/2016 and NMRC/CSA/CSA‐SI/007/2016) by the National Medical Research Council, Singapore. JHL is a recipient of a NUSMed Post‐Doctoral Fellowship (NUHSRO/2017/075/PDF/05) from Yong Loo Lin School of Medicine, National University of Singapore. Post‐mortem tissue for this study was provided by the Newcastle Brain Tissue Resource, which is funded in part by a grant from the UK Medical Research Council (grant number G0400074) and by Brains for Dementia research, a joint venture between Alzheimer's Society and Alzheimer's Research UK
Contributor Information
Mitchell K. P. Lai, Email: mitchell.lai@dementia-research.org.
Michelle G. K. Tan, Email: michelle.tan.g.k@sgh.com.sg.
DATA AVAILABILITY STATEMENT
The human transcriptome data set has been deposited in Gene Expression Omnibus (GEO) data repository (Access #GSE150696). Other data that support the findings of this study are available on reasonable request from the corresponding authors.
REFERENCES
- 1. Alves LCS, Monteiro DQ, Bento SR, Hayashi VD, Pelegrini LNC, Vale FAC. Burnout syndrome in informal caregivers of older adults with dementia: a systematic review. Dement Neuropsychol. 2019;13(4):415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hurd MD, Martorell P, Delavande A, Mullen KJ, Langa KM. Monetary costs of dementia in the United States. N Engl J Med. 2013;368(14):1326–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zweig YR, Galvin JE. Lewy body dementia: the impact on patients and caregivers. Alzheimers Res Ther. 2014;6(2):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863–72. [DOI] [PubMed] [Google Scholar]
- 5. Alghamdi A, Vallortigara J, Howlett DR, Broadstock M, Hortobagyi T, Ballard C, et al. Reduction of RPT6/S8 (a proteasome component) and proteasome activity in the cortex is associated with cognitive impairment in lewy body dementia. J Alzheimers Dis. 2017;57(2):373–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ballard CG, Aarsland D, McKeith I, O'Brien J, Gray A, Cormack F, et al. Fluctuations in attention: PD dementia vs DLB with parkinsonism. Neurology. 2002;59(11):1714–20. [DOI] [PubMed] [Google Scholar]
- 7. Francis PT, Perry EK. Cholinergic and other neurotransmitter mechanisms in Parkinson's disease, Parkinson's disease dementia, and dementia with Lewy bodies. MovDisord. 2007;22(Suppl 17):S351–S357. [DOI] [PubMed] [Google Scholar]
- 8. Mohamed NE, Howlett DR, Ma L, Francis PT, Aarsland D, Ballard CG, et al. Decreased immunoreactivities of neocortical AMPA receptor subunits correlate with motor disability in Lewy body dementias. J Neural Transm (Vienna). 2014;121(1):71–8. [DOI] [PubMed] [Google Scholar]
- 9. Whitfield DR, Vallortigara J, Alghamdi A, Howlett D, Hortobagyi T, Johnson M, et al. Assessment of ZnT3 and PSD95 protein levels in Lewy body dementias and Alzheimer's disease: association with cognitive impairment. Neurobiol Aging. 2014;35(12):2836–44. [DOI] [PubMed] [Google Scholar]
- 10. Xing H, Lim YA, Chong JR, Lee JH, Aarsland D, Ballard CG, et al. Increased phosphorylation of collapsin response mediator protein‐2 at Thr514 correlates with b‐amyloid burden and synaptic deficits in Lewy body dementias. Mol Brain. 2016;9(1):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aarsland D, Ballard CG, Halliday G. Are Parkinson's disease with dementia and dementia with Lewy bodies the same entity? J Geriatr Psychiatry Neurol. 2004;17(3):137–45. [DOI] [PubMed] [Google Scholar]
- 12. Jellinger KA, Korczyn AD. Are dementia with Lewy bodies and Parkinson's disease dementia the same disease? BMC Med. 2018;16(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lippa CF, Duda JE, Grossman M, Hurtig HI, Aarsland D, Boeve BF, et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology. 2007;68(11):812–9. [DOI] [PubMed] [Google Scholar]
- 14. Tsuboi Y, Dickson DW. Dementia with Lewy bodies and Parkinson's disease with dementia: are they different? Parkinsonism Relat Disord. 2005;11(Suppl 1):S47–51. [DOI] [PubMed] [Google Scholar]
- 15. Edison P, Rowe CC, Rinne JO, Ng S, Ahmed I, Kemppainen N, et al. Amyloid load in Parkinson's disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry. 2008;79(12):1331–8. [DOI] [PubMed] [Google Scholar]
- 16. Gomperts SN, Locascio JJ, Makaretz SJ, Schultz A, Caso C, Vasdev N, et al. Tau positron emission tomographic imaging in the Lewy body diseases. JAMA Neurol. 2016;73(11):1334–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gomperts SN, Locascio JJ, Marquie M, Santarlasci AL, Rentz DM, Maye J, et al. Brain amyloid and cognition in Lewy body diseases. Mov Disord. 2012;27(8):965–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Howlett DR, Whitfield D, Johnson M, Attems J, O'Brien JT, Aarsland D, et al. Regional multiple pathology scores are associated with cognitive decline in Lewy body Dementias. Brain Pathol. 2015;25(4):401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Laurent C, Buee L, Blum D. Tau and neuroinflammation: What impact for Alzheimer's disease and tauopathies? Biomed J. 2018;41(1):21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Minter MR, Taylor JM, Crack PJ. The contribution of neuroinflammation to amyloid toxicity in Alzheimer's disease. J Neurochem. 2016;136(3):457–74. [DOI] [PubMed] [Google Scholar]
- 21. Calsolaro V, Edison P. Neuroinflammation in Alzheimer's disease: Current evidence and future directions. Alzheimers Dement. 2016;12(6):719–32. [DOI] [PubMed] [Google Scholar]
- 22. Surendranathan A, Rowe JB, O'Brien JT. Neuroinflammation in Lewy body dementia. Parkinsonism Relat Disord. 2015;21(12):1398–406. [DOI] [PubMed] [Google Scholar]
- 23. Sharma P, Srivastava P, Seth A, Tripathi PN, Banerjee AG, Shrivastava SK. Comprehensive review of mechanisms of pathogenesis involved in Alzheimer's disease and potential therapeutic strategies. Prog Neurogibol. 2019;174:53–89. [DOI] [PubMed] [Google Scholar]
- 24. Resh MD. Fyn, a Src family tyrosine kinase. Int J Biochem Cell Biol. 1998;30(11):1159–62. [DOI] [PubMed] [Google Scholar]
- 25. Haass C, Mandelkow E. Fyn‐tau‐amyloid: a toxic triad. Cell. 2010;142(3):356–8. [DOI] [PubMed] [Google Scholar]
- 26. Lee C, Low CY, Francis PT, Attems J, Wong PT, Lai MK, et al. An isoform‐specific role of FynT tyrosine kinase in Alzheimer's disease. J Neurochem. 2016;136(3):637–50. [DOI] [PubMed] [Google Scholar]
- 27. Lee C, Low CY, Wong SY, Lai MK, Tan MG. Selective induction of alternatively spliced FynT isoform by TNF facilitates persistent inflammatory responses in astrocytes. Sci Rep. 2017;7:43651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337–51. [DOI] [PubMed] [Google Scholar]
- 29. Francis PT, Costello H, Hayes GM. Brains for dementia research: evolution in a longitudinal brain donation cohort to maximize current and future value. J Alzheimers Dis. 2018;66(4):1635–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–98. [DOI] [PubMed] [Google Scholar]
- 31. Chai YL, Chong JR, Weng J, Howlett D, Halsey A, Lee JH, et al. Lysosomal cathepsin D is upregulated in Alzheimer's disease neocortex and may be a marker for neurofibrillary degeneration. Brain Pathol. 2019;29(1):63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 2008;57(1):289–300. [Google Scholar]
- 33. Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. [DOI] [PubMed] [Google Scholar]
- 35. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82(4):239–59. [DOI] [PubMed] [Google Scholar]
- 36. Panicker N, Saminathan H, Jin H, Neal M, Harischandra DS, Gordon R, et al. Fyn kinase regulates microglial neuroinflammatory responses in cell culture and animal models of Parkinson's disease. J Neurosci. 2015;35(27):10058–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eng LF, Ghirnikar RS. GFAP and astrogliosis. Brain Pathol. 1994;4(3):229–37. [DOI] [PubMed] [Google Scholar]
- 38. Ling EA, Wong WC. The origin and nature of ramified and amoeboid microglia: a historical review and current concepts. Glia. 1993;7(1):9–18. [DOI] [PubMed] [Google Scholar]
- 39. Reitz C. Alzheimer's disease and the amyloid cascade hypothesis: a critical review. Int J Alzheimers Dis. 2012;2012:369808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004;61(3):378–84. [DOI] [PubMed] [Google Scholar]
- 41. Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Larson M, Sherman MA, Amar F, Nuvolone M, Schneider JA, Bennett DA, et al. The complex PrP(c)‐Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer's disease. J Neurosci. 2012;32(47):16857–71. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43. Lau DH, Hogseth M, Phillips EC, O'Neill MJ, Pooler AM, Noble W, et al. Critical residues involved in tau binding to fyn: implications for tau phosphorylation in Alzheimer's disease. Acta Neuropathol Commun. 2016;4(1):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid‐b toxicity in Alzheimer's disease mouse models. Cell. 2010;142(3):387–97. [DOI] [PubMed] [Google Scholar]
- 45. Li C, Gotz J. Somatodendritic accumulation of Tau in Alzheimer's disease is promoted by Fyn‐mediated local protein translation. EMBO J. 2017;36(21):3120–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, et al. Alzheimer amyloid‐beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci. 2012;15(9):1227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang H, Ren CH, Gunawardana CG, Schmitt‐Ulms G. Overcoming barriers and thresholds ‐ signaling of oligomeric Abeta through the prion protein to Fyn. Mol Neurodegener. 2013;8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Briner A, Gotz J, Polanco JC. Fyn kinase controls tau aggregation in vivo. Cell Rep. 2020;32(7):108045. [DOI] [PubMed] [Google Scholar]
- 49. Liu G, Fiock KL, Levites Y, Golde TE, Hefti MM, Lee G. Fyn depletion ameliorates tau(P301L)‐induced neuropathology. Acta Neuropathol Commun. 2020;8(1):108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tang SJ, Fesharaki‐Zadeh A, Takahashi H, Nies SH, Smith LM, Luo A, et al. Fyn kinase inhibition reduces protein aggregation, increases synapse density and improves memory in transgenic and traumatic Tauopathy. Acta Neuropathol Commun. 2020;8(1):96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Padmanabhan P, Martinez‐Marmol R, Xia D, Gotz J, Meunier FA. Frontotemporal dementia mutant Tau promotes aberrant Fyn nanoclustering in hippocampal dendritic spines. Elife. 2019;8:e45040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sharma VM, Litersky JM, Bhaskar K, Lee G. Tau impacts on growth‐factor‐stimulated actin remodeling. J Cell Sci. 2007;120(Pt 5):748–57. [DOI] [PubMed] [Google Scholar]
- 53. Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, et al. Phosphorylation of tau by fyn: implications for Alzheimer's disease. J Neurosci. 2004;24(9):2304–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lesort M, Jope RS, Johnson GV. Insulin transiently increases tau phosphorylation: involvement of glycogen synthase kinase‐3beta and Fyn tyrosine kinase. J Neurochem. 1999;72(2):576–84. [DOI] [PubMed] [Google Scholar]
- 55. Sasaki Y, Cheng C, Uchida Y, Nakajima O, Ohshima T, Yagi T, et al. Fyn and Cdk5 mediate semaphorin‐3A signaling, which is involved in regulation of dendrite orientation in cerebral cortex. Neuron. 2002;35(5):907–20. [DOI] [PubMed] [Google Scholar]
- 56. Chin J, Palop JJ, Puolivali J, Massaro C, Bien‐Ly N, Gerstein H, et al. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2005;25(42):9694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chin J, Palop JJ, Yu GQ, Kojima N, Masliah E, Mucke L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J Neurosci. 2004;24(19):4692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kaufman AC, Salazar SV, Haas LT, Yang J, Kostylev MA, Jeng AT, et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol. 2015;77(6):953–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Trepanier CH, Jackson MF, MacDonald JF. Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS J. 2012;279(1):12–9. [DOI] [PubMed] [Google Scholar]
- 60. Przybyla M, van Eersel J, van Hummel A, van der Hoven J, Sabale M, Harasta A, et al. Onset of hippocampal network aberration and memory deficits in P301S tau mice are associated with an early gene signature. Brain. 2020;143(6):1889–904. [DOI] [PubMed] [Google Scholar]
- 61. Metcalfe MJ, Figueiredo‐Pereira ME. Relationship between tau pathology and neuroinflammation in Alzheimer's disease. Mt Sinai J Med. 2010;77(1):50–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Panicker N, Sarkar S, Harischandra DS, Neal M, Kam TI, Jin H, et al. Fyn kinase regulates misfolded alpha‐synuclein uptake and NLRP3 inflammasome activation in microglia. J Exp Med. 2019;216(6):1411–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer's disease. J Cell Biol. 2018;217(2):459–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nygaard HB. Targeting fyn kinase in Alzheimer's disease. Biol Psychiat. 2018;83(4):369–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. van Dyck CH, Nygaard HB, Chen K, Donohue MC, Raman R, Rissman RA, et al. Effect of AZD0530 on cerebral metabolic decline in Alzheimer disease: a randomized clinical trial. JAMA Neurol. 2019;76(10):1219–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kirvell SL, Esiri M, Francis PT. Down‐regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer's disease. J Neurochem. 2006;98(3):939–50. [DOI] [PubMed] [Google Scholar]
- 67. Hepp DH, Vergoossen DL, Huisman E, Lemstra AW, Berendse HW, Rozemuller AJ, et al. Distribution and load of amyloid‐beta pathology in parkinson disease and dementia with Lewy bodies. J Neuropathol Exp Neurol. 2016;75(10):936–45. [DOI] [PubMed] [Google Scholar]
- 68. Braak H, Del Tredici K. Spreading of tau pathology in sporadic Alzheimer's disease along cortico‐cortical top‐down connections. Cereb Cortex. 2018;28(9):3372–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cantuti‐Castelvetri I, Klucken J, Ingelsson M, Ramasamy K, McLean PJ, Frosch MP, et al. Alpha‐synuclein and chaperones in dementia with Lewy bodies. J Neuropathol Exp Neurol. 2005;64(12):1058–66. [DOI] [PubMed] [Google Scholar]
- 70. Klucken J, Ingelsson M, Shin Y, Irizarry MC, Hedley‐Whyte ET, Frosch M, et al. Clinical and biochemical correlates of insoluble a‐synuclein in dementia with Lewy bodies. Acta Neuropathol. 2006;111(2):101–8. [DOI] [PubMed] [Google Scholar]
- 71. Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. a‐Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4(2):160–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The human transcriptome data set has been deposited in Gene Expression Omnibus (GEO) data repository (Access #GSE150696). Other data that support the findings of this study are available on reasonable request from the corresponding authors.