

Abstract
Medulloblastoma (MB) is the most common primary malignant brain tumor of childhood and a significant contributor to pediatric morbidity and death. While metastatic dissemination is the predominant cause of morbidity and mortality for patients with this disease, most research efforts and clinical trials to date have focused on the primary tumor; this is due mostly to the paucity of metastatic tumor samples and lack of robust mouse models of MB dissemination. Most current insights into the molecular drivers of metastasis have been derived from comparative molecular studies of metastatic and non‐metastatic primary tumors. However, small studies on matched primary and metastatic tissues and recently developed mouse models of dissemination have begun to uncover the molecular biology of MB metastasis more directly. With respect to anatomical routes of dissemination, a hematogenous route for MB metastasis has recently been demonstrated, opening new avenues of investigation. The tumor micro‐environment of the primary and metastatic niches has also been increasingly scrutinized in recent years, and further investigation of these tumor compartments is likely to result in a better understanding of the molecular mediators of MB colonization and growth in metastatic compartments.
Keywords: extraneural, leptomeninges, medulloblastoma, metastasis, molecular subgroups, therapeutic targets, tumor microenvironment
Introduction
Medulloblastoma (MB) is the most common malignant brain tumor in children, arising in about 0.74 per 100 000 children per year in the United States 66. The current standard of care for children over the age of 3–5 years includes neurosurgical resection, craniospinal irradiation (CSI), and cisplatin‐based chemotherapy. In patients with average‐risk disease, the 5‐year survival rate is 85%, compared with 70% for children with a high‐risk designation 68, 89. Unfortunately the morbidity and mortality of therapy is high, and even those children who achieve remission and cure are left with lifelong sequalae including neurological, cognitive, and endocrinological impairment 59. In those cases where children do not survive, mortality almost always results from metastatic dissemination of the tumor from its primary site of origin in the posterior fossa to the leptomeninges. More rarely, MB spreads to extraneural sites such as bone, lung, liver, or lymph nodes 81. In 20–30% of affected children, metastasis will have occurred at time of first presentation, while in others this occurs as a metastatic recurrence 100. Integrative genomic studies have shown that this embryonal tumor comprises at least four molecular subgroups (SHH, WNT, Group/Grp 3, Group/Grp 4) with distinct clinical characteristics, including heterogenous recurrence patterns and propensity for metastatic dissemination 64, 65. SHH and Grp 4 patients have an intermediate prognosis and can show metastatic dissemination, especially Grp 4 tumors which comprise the majority of metastatic MB cases. WNT subgroup tumors are associated with an excellent prognosis, in contrast to Grp 3 tumors which are associated with poor outcomes and frequently show metastatic dissemination at first presentation or recurrence. 74, 75. More recent efforts to further resolve the molecular classification of MB have defined additional subtypes within the original four subgroups, permitting a more granular view of outcome and dissemination risk profiles 11, 87.
Because metastasis is the major cause of mortality in children with MB, there should be a special focus on the characterization of this phenomenon to identify novel therapeutic targets for prevention or amelioration of metastatic dissemination. Expression profiling of metastatic and non‐metastatic MB cases has informed much of the current understanding of the molecular drivers of tumor dissemination. Additional insights have been derived from comparative molecular analyses of matched primary and metastatic tissues, although these efforts have been limited by a paucity of such tissue samples 75. Various preclinical models including in vitro migration assays and in vivo murine models (transgenic and xenograft) have been used to uncover the molecular drivers of dissemination, the phylogenetic relationships between primary and metastatic tumor deposits, the anatomical routes of MB dissemination, and the biology of the primary and metastatic tumor microenvironments (TME) which facilitate dissemination and distant implantation. Current and emerging concepts in the field are reviewed and summarized here.
Patterns of relapse and metastasis by subgroup
The four molecular subgroups of MB listed in the WHO classification are SHH‐activated, WNT‐activated, Grp 3, and Grp 4 49. SHH tumors are characterized by aberrant activity of the Sonic hedgehog signaling pathway, which is frequently caused by mutations in pathway components, including PTCH, SMO, and GLI2. Tumors of this subgroup arise from the external granule layer of the cerebellum, frequently demonstrate desmoplastic/nodular histology, and represent the predominant subgroup in infants under age 3 and patients over age 16 63. Cohort studies have shown that metastases are rarely present at the time of first presentation and recurrences are more likely to occur locally (i.e. in the surgical cavity) than in the leptomeninges 75.
Tumors of the WNT subgroup arise from the lower rhombic lip and are driven by Wingless‐related integration site pathway activation 26 through frequent somatic mutations of beta‐catenin (CTNBB1) and monosomy 6. These usually show classic histology, tend to affect older children (ages 4–15) and are associated with an excellent prognosis, low rates of recurrence, and very low rates of metastatic dissemination 16. Grp 3 and Grp 4 tumors occupy the fourth ventricle, and are less clearly defined at the molecular level than SHH or WNT tumors. Grp 3 tumors harbor MYC amplification in 15–20%, are enriched for tumors with large cell/anaplastic (LCA) morphology, and predominantly affect infants and young children (never adults). Both Grp 3 and Grp 4 tumors can carry isochromosome 17q (i17q), though this finding is more frequent in Grp 4. Rates of metastatic dissemination at first presentation are high for Grp 3 MB which confers an especially poor prognosis, particularly in young infants treated with chemotherapy only 80. Recurrence patterns for both subgroups are similar, with a tendency towards dissemination in previously irradiated patients, with a particularly low latency period for Grp 3 tumors. Metastatic relapses of Grp 4 tumors tend to present with isolated deposits, whereas the metastatic relapses of Grp 3 tumors are typically multifocal or laminar 100. Importantly, evaluation of molecular subgroups in matched primary and metastatic tissues from multiple cohorts has confirmed that subgroup affiliation is unchanged between these compartments, further supporting the distinct biology of these subgroups 75.
Integrative data clustering methodologies such as similarity network fusion (SNF) analysis 97 have been applied to matched MB DNA methylation and gene expression profiles to further resolve the intra‐group heterogeneity of MB 85. This analysis has identified 12 unique subtypes, which show distinctive molecular and clinical characteristics, including four Shh‐activated (Shhα, Shhβ, Shhγ, Shhδ), two Wnt‐activated (Wntα, Wntβ), three Grp 3 (3α, 3β, 3γ) and three Grp 4 (4α, 4β, 4γ). Shhα tumors affect children aged 3–16 years, show various copy number alterations (resulting in enrichment of MYCN, and GLI2 amplifications), frequently carry TP53 mutations, and have the worst prognosis overall. In infants, Shh tumors show Shhβ and Shhγ affiliation, of which the Shhβ tumors are much more frequently metastatic and have a worse prognosis. SHHδ tumors are usually identified in adults, show frequent TERT promoter mutations, and have an overall better prognosis. Evaluation of the two Wnt subtypes shows that Wntα tumors mostly comprise pediatric patients and have ubiquitous monosomy 6, while Wntβ carries a diploid chromosome 6. Both subtypes however have a similar prognosis and equivalently low rates of metastasis. Within the Grp 3 subtypes, 3α tumors show frequent chromosome 8q loss (MYC locus is at 8q24), 3β tumors show higher frequency of GFI1 and GFI1B oncogene activation, OTX2 amplification, and loss of DDX31, and 3γ tumors show frequent 8q gain. While 3α and 3γ show similarly high rates of metastasis (as compared to 3β), the latter subtype shows a poorer prognosis overall, even when controlling for MYC amplification status. Finally, while 4α, 4β, and 4γ show differential molecular profiles, the rate of metastatic dissemination is homogenous between these subtypes.
Preclinical models
Various in vitro and in vivo experimental models of MB metastasis have been developed to characterize the biology of metastasis, and for preclinical testing of novel therapeutics. The in vitro assays which have been utilized primarily include assays of cell migration, such as scratch assays, radial migration assays, and Matrigel/Boyden chamber approaches 1, 10, 35, 36. In vivo xenograft and transgenic mouse models have been developed to permit more extensive molecular characterization of tumors in various compartments, and elucidation of the anatomical pathways of dissemination (Figure 1). A recurrent challenge with xenograft models of metastatic MB has been the mismatch between mouse survival times and the length of time required to achieve tumor cell dissemination and distant implantation. One solution to this problem, which has been utilized by some subgroups, is to inject xenografted cells subcutaneously in the flank, as this permits more effective surgical control of disease. Resection of primary tumor increases the survival period of the mouse model sufficiently so as to achieve dissemination and leptomeningeal/distant implantation22. A modified xenograft mouse model generated through tumor cell injection into the cisterna magna has also shown consistent leptomeningeal dissemination along the brain and spinal cord, and provides another in vivo model of metastasis 13. Transgenic models of metastatic MB have proven difficult to generate. One model utilized by our subgroup is an insertional mutagenesis mouse model developed using a Sleeping Beauty transposon system—consisting of a T2/Onc transposon and a Sleeping Beauty 11 (SB11) transposase encoded under the Math1 enhancer/promoter—on a Ptch +/− or Tp53mut background (Ptch1+/−/Math1‐SB11/T2Onc, Tp53mut/Math1‐SB11/T2Onc) 14, 15, 99. This random mutagenic approach permits the application of functional genomics approaches to interrogate the specific roles of individual genes in cell processes such as migration and metastasis. Gene sites which carry transposon insertions at rates higher than background are termed gene‐centric common insertion sites (gCISs), and these insertion events are assumed to be enriched due to survival benefits conferred by altered gene function. Comparison of the gCISs in matched primary and metastatic tumors permits inference of the functional contributions of individual gCIS to metastatic spread, and evaluation of the molecular and phylogenetic relationships of metastatic disease to primary tumor.
Figure 1.
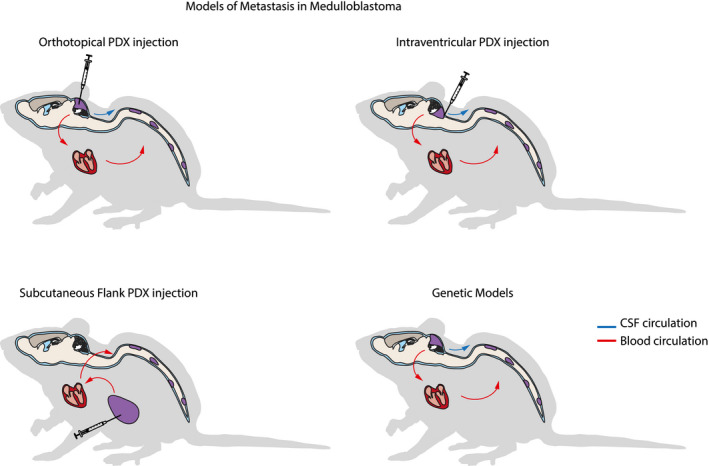
Models of metastasis in medulloblastoma. Mouse models of medulloblastoma dissemination and metastasis include traditional patient‐derived xenograft (PDX) models with injection of tumor cells orthotopically, into CSF spaces such as ventricles or cisterns, or into subcutaneous flank tissue. Mouse models created by injection into extra‐CNS sites such as the subcutaneous flank putatively achieve metastasis only through hematogenous dissemination, while metastasis in orthotopic or CSF injected PDX models may proceed hematogenously or through CSF. Genetic mouse models include those generated by germ line editing, allografting of edited mouse neural stem cells, or through mutagenic transposon approaches, and can putatively achieve dissemination through blood and/or CSF.
Molecular drivers of metastasis
Comparative molecular analysis of tumors with and without dissemination has identified activity of genes and signal transduction pathways enriched in metastatic populations, and which putatively drive metastasis in MB. One of the earliest such analyses identified enriched expression of RAS/MAPK pathway components in metastatic tumors, a finding which has been recapitulated in other studies 27, 51. Receptor tyrosine kinases (RTKs) upstream of RAS/MAPK such as PDGFR and ERBB2 (HER2/neu) have each been specifically implicated in MB pathogenesis and migration. Overexpression of ERBB2 in MB cell lines significantly increases in vitro invasion and cell migration compared to controls 32, and upregulates expression of downstream RAS/MAPK pathway genes (MAPK, MEK5) and of S100A4 (S100 calcium binding protein A4). S100A4 in particular has been implicated in regulation of motility, invasion, and tubulin polymerization, and is a known driver of tumor progression and metastasis in a number of epithelial and non‐epithelial malignancies 7. PDGFRA has been identified as a gCIS in both primary and metastatic compartments in Sleeping Beauty functional genomics screens, strongly suggesting a role for this gene in the maintenance of disease in both primary and metastatic compartments, and implicating it as a potential metastatic driver 99. Indeed, treatment of MB cell lines with anti‐PDGFRA antibody abrogates fibronectin‐dependent adhesion in in vitro assays, and results in the inhibition of downstream activating phosphorylation of pathway components including MAP2K1, MAP2K2, and MAPK1/3 51. HRAS over‐expression in SHH tumor cell lines results in increased cell migration in vitro, and significantly increases rates of pulmonary metastases when injected subcutaneously in mice, as compared to controls 101. The authors of that study further reported a case of matched human primary and metastatic MB tumor samples that demonstrated a higher degree of MAPK activation in metastatic tissue than primary tumor 101. Interestingly, immunostaining of tissue from a single primary tumor showed increased MAPK phosphorylation in the perivascular niche relative to tumor bulk, suggesting a role for RAS/MAPK signaling in tumor dissemination, especially pertinent in light of recent data showing that MB tumor cell dissemination may proceed through hematogenous routes 22, 101.
Like the RAS/MAPK pathway, the PI3K/AKT pathway is a powerful regulator of cellular proliferation and migration, with clearly demonstrated roles in tumor dissemination in other tumor types 12, 43. In MB, the pathway frequently shows aberrant overactivity 31, 76, and emerging data strongly imply a role in MB cell migration 28. In addition to RTK over‐expression, the pathway can also be activated in MB through loss of PTEN tumor suppressor, which is encoded on chromosome 10q, and also frequently deleted in MB 62, 77. Sleeping Beauty functional genomic screening has identified insertions in Pten, as well as Akt2 and Pik3r1 as being enriched in the metastatic compartment, further implicating the pathway as a regulator of metastatic dissemination 99. In vivo experiments using the RCAS/tv‐a system 76 have demonstrated that viral introduction of Shh and Akt genes to cerebella of Nestin‐TVA mice greatly increases the penetrance of tumor formation relative to Shh alone, and induces leptomeningeal metastasis in about 1/5 of mice 99. Interestingly, as with the RAS/MAPK pathway, the PI3K/AKT pathway can be upregulated in cells of the perivascular niche (in response to CSI), which could drive intravasation and hematogenous dissemination of malignant cells 29.
Various other pathways such as the HGF/cMET and NOTCH signaling pathways have also been shown to drive a pro‐migratory phenotype in MB. The HGF/cMET pathway is a known regulator of both cerebellar development and MB pathogenesis 33, 37, 44, 45, 93. Upregulated cMET expression has been identified in a subset of SHH and Grp 3 MB cases, and increased expression of the gene can drive pathway activity and dissemination. Activation of the HGF/cMET pathway can also be achieved through tumor suppressor loss by epigenetic and mutational silencing of SPINT2 (serine protease inhibitor kunitz‐type 2) 37. In vitro studies have demonstrated that the pathway has a pro‐migrational effect on MB cells, partly effected by increasing the expression and activity of a tissue factor (TF) which drives cellular motility through modulation of actin cytoskeleton 72, 73. In vitro pharmacological targeting of cMET inhibits the pathway, and impairs cell mobility in migration assays 41. The NOTCH signaling pathway has been implicated in dissemination of numerous cancers through roles in epithelial‐mesenchymal transition, escape from anoikis mediated cell death, and pro‐angiogenic functions. There is significant over‐representation of genes in the NOTCH signaling pathway in Grp 3 MB, and specific activating pathway mutations have been identified 18, 62. Early reports indicated significantly higher expression and activity of NOTCH2 compared to NOTCH1 in embryonal tumors such as MB, and implicated NOTCH2 specifically in cellular proliferation and tumor growth 18. More recently however, investigations with xenograft mouse models of metastatic Grp 3 MB showed higher expression of NOTCH1 and its transcriptionally active intracellular domain (NICD1) in spinal metastatic deposits than in primary tumors, with upregulated expression of NOTCH1 pathway genes. Consistent with earlier reports, NOTCH1 expression in primary MB tissue obtained from patients with metastatic disease was confirmed to be low, suggesting that the pathway is upregulated in a subclone with increased metastatic capacity. These pro‐metastatic effects may be mediated through activation of TWIST1 which in turn activates BMI1. BMI1, a member of Polycomb Repressive Complex I, is predictive of metastasis in various cancer types, associated with poor outcome in Grp 3 MB, and has been suggested as a potential therapeutic target in recurrent MB 3.
In addition to canonical signaling cascades, non‐coding RNAs such as microRNAs have gained attention as important drivers of tumor pathogenesis and dissemination 79. Recurrent amplification of the miR‐17/92 polycistron has been implicated as a driver of proliferation in SHH‐pathway dependent MB, whereas the highly conserved miR‐183~96~182 cluster encoded on chromosome 7q has been shown to be upregulated in non‐SHH‐MB 61, 94. The overexpression of this cluster (and miR‐182 in particular) has been very clearly associated with a pro‐metastatic phenotype in other cancers. In MB, the cluster has been shown to regulate the PI3K/AKT/mTOR signaling axis, suppress expression of pro‐apoptotic genes, and drive increased cell mobility in scratch assays in vitro 98. Intracranial injection of MB cell lines that overexpress miR‐182 generated tumors with large cell/anaplastic histology and significantly greater degrees of local cerebellar infiltration, as well as leptomeningeal metastatic dissemination 2.
Anatomic routes of metastatic dissemination
Historically, tumor dissemination to the leptomeninges was assumed to occur by passive spread through the cerebrospinal fluid (CSF), although this has never been empirically proven. Recently, dissemination has been shown to occur through the hematogenous compartment, which may inform efforts to identify inhibitors of metastatic dissemination22. The specific anatomical routes and protein mediators of tumor cell dissemination into the bloodstream—where they become circulating tumor cells (CTCs)—and exit to the leptomeninges, have not yet been elucidated, but if identified would present exciting and novel therapeutic targets. Expression analysis of Grp 3 primary and metastatic tumors has identified CCL2 as a differentially expressed gene between these compartments, and copy number gain of this protein in Grp 3 primary tumors predicts higher rates of metastatic dissemination
22. Forced expression, or knockdown, respectively, of CCL2 in rarely metastatic xenografted cell lines significantly increased and decreased metastatic rates, respectively, strongly suggesting a role in mediating metastatic dissemination22. Importantly, CCL2 and its cognate receptor CCR2 (together forming the CCL2/CCR2 signaling axis) have been shown to facilitate leukocyte trafficking out of the hematogenous compartment, and dysregulation of the axis has been implicated in driving the dissemination and progression of metastasis in a large number of other malignancies 48.
Like the CCL2/CCR2 axis, the pro‐metastatic role of the CXCR4/CXCL12 axis has also been demonstrated in numerous cancers and has received increasing attention as a possible driver of MB dissemination 67. CXCR4 expression by tumor cells facilitates homing of malignant cells to niches where stromal cells express the CXCL12 ligand (also known as SDF‐1, stromal cell‐derived factor 1) 8. In normal cerebellar development, CXCR4 appears to play important roles in the Shh‐induced proliferation and migration of granule cells 46, 50, 102. Overexpression of CXCR4 is common in MB, particularly in the SHH subgroup suggesting a role for metastatic dissemination of MB 82, 84, 86. Treatment of MB cell lines in vitro with CXCL12 has been shown to have proliferative and pro‐migratory effects. Interestingly, CXCL12 has known pro‐angiogenic functions and is expressed on tumor vessel walls, suggesting the possibility that this axis may be involved in the hematogenous dissemination of MB cells 34, 61, 65, 82. The cellular localization and activity of CXCR4 in MB has been shown to be mediated in part by protein phosphatase 2C delta (WIP1), through suppression of G protein coupled receptor 5 (GPCR5) 9. WIP1 is a known oncogene, with p53 inhibitory function, and encoded on 17q (17q22‐23). Interestingly, gene expression analysis has shown that metastatic MB has increased expression of WIP1, and that this is accompanied by higher expression levels of CXCR4 10.
Prior to introduction of routine chemotherapy for patients with MB, nearly 20% of all relapses included an extraneural component. Since the introduction of chemotherapy as standard of care for MB treatment, extraneural metastases have become exceedingly rare in children, though still seen in the adult population 20, 91. Overall, the most common extraneural site is bone, with lymph nodes and visceral organs relatively less affected. Recent advances in detection of CTCs has revealed that MB patients often have CTCs at diagnosis, suggesting there is an early propensity for intravasation of subclones of MB primary tumors 22. MB CTCs display positivity for CD56 (NCAM), which, coupled with CD45 exclusion (to exclude white blood cells) and morphological examination, allows the identification of such cells. CD56+ve MB cells are also found in the blood of patients that display overt extraneural disease at progression, indicating that the expression of the antigen is conserved at relapse and could be useful for tracking MB CTCs during the entire course of the disease 17, 23, 92. However, efforts to characterize the surfaceome of MB CTCs should be undertaken, because expression of CD56 is frequently focal in primary tumors. The ability of MB to intravasate is shared with neural stem cell progenitors, which are also able to leave the CNS to contribute to neurogenesis in tumors such as prostate cancer, a process which supports tumor development and progression in turn 56. In the context of a possible hematogenous route for MB leptomeningeal metastasis, it has become increasingly relevant to improve our understanding of the mechanism of MB cell intravasation, as this could represent the first step of both extraneural and leptomeningeal dissemination of MB 21, 22.
Tumor microenvironments in primary and metastatic compartments
Several cellular components of the brain parenchyma, such as neurons, astrocytes, and microglia establish extensive cross‐talk with MB tumor cells. Similarly, extensive interactions link MB tumor cells and non‐cellular components of the brain extracellular matrix, which also play a key role in regulating availability of nutrients and growth factors important for tumor cell proliferation and motility. The cellular components of primary MB TME have been investigated in small cohorts by immunohistochemistry to look at a few populations simultaneously, or more recently, in large transcriptomic datasets by bioinformatic approaches to highlight the relative abundance of cell‐type specific gene expression signatures 52, 54, 70, 71, 96. Immunohistochemistry‐based approaches return binary results about presence/absence of a given cell population, but results across studies from different institutions can be contradictory, and depend on the robustness of the biomarkers and antibodies used. Quantification of the different cellular components of the TME by immunohistochemistry is also complicated by the relatively low‐throughput nature of the technique but, on the contrary, immunohistochemistry provides spatial information about the location of TME cell populations, contributing to our understanding of spatial heterogeneity of MB. When analyzed through quantification of gene expression signatures, MB TME differs dramatically according to the subgroup affiliation of the tumor. SHH MBs are characterized by the highest levels of infiltration of macrophages and T cells compared to other subgroups, PD‐L1 is highest in a few WNT and SHH tumors, and Grp 3 tumors have the highest number of CD8+ T cells. Grp 3 and 4 tumors have the largest number of cytotoxic lymphocytes and also the largest number of endothelial cells 5. It is also important to highlight the limitations of such gene expression‐based approaches to profile the MB TME; when signature genes are expressed by both tumor cells and TME, confounding effects arise which have to be taken into account to robustly identify TME populations. Gene expression‐based approaches also do not allow evaluation of the spatial distribution of TME cell populations, which has important implications for generation of hypotheses about the immune‐suppressive status of the tumor 24. Taken together, recent findings from small immunohistochemistry‐based studies and larger bioinformatic studies converge on identifying SHH‐activated MBs as the most enriched in macrophages, which are more abundant in areas of active proliferation. The macrophage M1 vs. M2 polarization status and its correlation with progression is still under investigation 42, 53, 90.
While progress has been made in describing the TME in primary MB, there have been no advances in understanding the TME of leptomeningeal metastasis, the biggest hurdle being the lack of suitable samples for molecular profiling and a relevant model system in mice. Evidence on the importance of the TME in leptomeningeal metastasis comes from preclinical studies where macrophage‐targeted immunotherapies have been applied to Grp 3 orthotopic xenograft models 25. Gholamin and colleagues used the humanized anti‐CD47 antibody, Hu5F9‐G4, to block CD47‐SIRPα interaction which resulted in impaired local and metastatic growth of MB 25. CCL2 cytokine is a major chemoattractant for activated macrophages, and its overexpression in a small number of human Grp 3 primary tumors and metastases supports the relevance of macrophages in Grp 3 human leptomeningeal metastases 22.
MB tumor cells in the primary tumors also extensively interact with the unique components of the brain extracellular matrix. There are three recognized domains of the brain extracellular matrix: a neural interstitial matrix, a basal lamina, and perineuronal nets. The interstitial matrix is composed of loosely associated components such as hyaluronan, tenascin C and R, and a remarkably small amount of collagen and adhesive glycoproteins, such as laminin and fibronectin. In contrast, the basal lamina—which covers the endothelial cells of parenchymal blood vessels and the surface of the pia—is composed of a dense network of collagen, laminin and fibronectin (Figure 2). As a consequence, the ECM that comes in to play in primary MB—the interstitial matrix—is very different from the ECM that leptomeningeal metastases are in contact with, which is mostly composed of the basal lamina associated with the pia membrane. Primary MBs modify the ECM by active secretion of ECM proteins (collagen) and, at the same time, tumor cells are supported by the ECM which provides a reservoir of growth factors that increase invasion and migration properties 47, 83. In the leptomeningeal niche—the subarachnoid space—the concentrations of growth factors and nutrients is dramatically different from the primary tumor niche. Produced mostly by the choroid plexus in a process of active filtration and secretion, the CSF contains less glucose, amino acids (with the exception of glutamine), and lipoproteins than blood. How MB metastasis survive and then thrive in this rather different microenvironment is largely unknown, because early stages of metastasis in the leptomeninges are hard to access in humans and difficult to model in a pre‐clinical setting. Sampling and profiling of MB cells in the CSF and in blood might shed light on the intermediate stages of adaptation to the metastatic niche (i.e., anoikis). Further adaptation and selection might occur after extravasation or during CSF colonization, especially in the transition from micrometastasis to macrometastasis, in a manner similar to what has been shown for adult carcinomas with brain or leptomeningeal tropism 95. Breast cancer cell lines selected for a tropism toward the leptomeningeal space educate the subarachnoid microenvironment by secreting the complement protein C3, and this in turn permeabilizes the blood‐CSF‐barrier to better support leptomeningeal metastatic growth 6. To understand whether similar processes occur in MB metastasis and how relevant they are to explain the limited therapeutic response of MB metastasis to CSI and high‐dose chemotherapy will likely produce a better understanding on leptomeningeal metastasis vulnerabilities that could in turn be exploited to design more efficient and less toxic therapies for metastatic patients.
Figure 2.
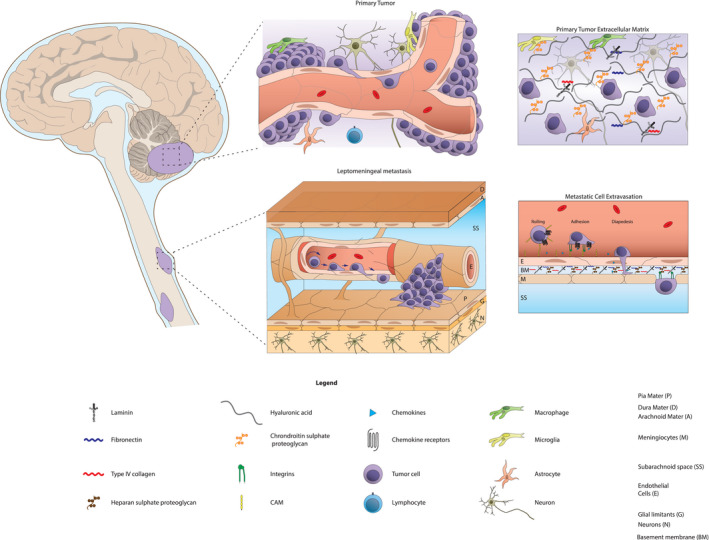
The primary and metastatic cell niche. Primary tumor cells reside in neural interstitial matrix composed of hyaluronan, collagen, adhesive glycoproteins, and cellular components such as microglia, macrophages, and parenchymal cells (i.e., neurons/astrocytes/oligodendrocytes). Extravasation of circulating tumor cells (CTCs) in response to cytokine secretion is mediated by cell adhesion molecules (CAMs) and integrins. The metastatic niche on the leptomeningeal (pial) surface is significantly different from neural interstitial matrix, and is composed of a dense network of collagen, laminin and fibronectin. Characterization of protein and molecular components which drive tumor cell adaptation to, and education of, the metastatic niche may uncover targets for inhibitors of leptomeningeal colonization and tumor growth.
Clinical implications and novel molecular targets
Current standard of care for all patients includes maximal safe surgical resection, followed by adjuvant CSI and chemotherapy. Historically, the choice of adjuvant therapy in North America has been based on binary stratification using a multivariate risk model which segregates patients into standard and high‐risk classes. The designation of standard or average risk is given to patients over the age of 3 years, who present without metastatic disease, have no histologic evidence of large cell anaplasia, and in whom near total gross section is achieved (residual tumor <1.5 cm2). Conversely, a high‐risk designation is assigned to those patients who have residual disease post‐resection (>1.5 cm2), show metastatic dissemination, or show large cell anaplastic histology; in these patients adjuvant therapy is intensified. CSI doses for standard risk patients are reduced to a total of 23.4 Gy, while high‐risk patients (including patients with metastatic dissemination) receive a standard dose of 36–39 Gy. In infants (i.e., children below 3 years of age), a protocol of high‐dose chemotherapy and autologous stem cell transplantation forms the standard of care in North America in order to avoid the neurocognitive toxicity of CSI at this developmental stage 58, 78.
The recognition that MB is a heterogenous disease entity with variable patterns of recurrence and outcome has prompted efforts to tailor both the intensity and delivery method of adjuvant therapy based on patient risk profile. The results of the SJYC07 trial have recently been published, these showed that treatment de‐escalation in patients with desmoplastic histology (predictive of good outcome) resulted in worsened progression‐free‐survival rates in this population, prompting premature trial termination 80. More recent trials designed since the development of molecular classification are now testing similar therapy de‐escalation strategies in patients with WNT‐activated MBs, including PNET5, ACNS1422, SJMB12, and NCT02212574 (which has also recently been terminated for reduced progression‐free survival rates). SJMB12 is additionally testing the utility of targeted SMO inhibition using vismodegib in skeletally mature patients with SHH‐activated MBs, and dose escalation of gemcitabine and premetrexed in Grp 3 and Grp 4 tumors. For patients with high‐risk disease or metastatic relapse, intrathecal delivery of highly active chemotherapy is a relatively safe, and potentially efficacious strategy for prevention or treatment of leptomeningeal metastatic growth. This delivery method is used routinely in some parts of the world, such as Europe where intrathecal administration of methotrexate is part of the standard of care for infants with MB. Early clinical trials with high risk/relapsed/disseminated MB patients have shown that intrathecal delivery of agents such as cytarabine (depocyte) or the radioimmunotherapeutic 131I‐3F8 is well‐tolerated and a promising strategy 39, 40, 55.
The identification of novel targets for inhibition of metastatic dissemination or metastatic disease has proven difficult because of a paucity of metastatic tissue samples (and therefore research data), and also by the inherent complexity of the biology underlying the process. In the clinical setting, treatment decisions are generally made based on evaluation of tissue obtained from primary, untreated tumor. However, previous whole genome sequencing efforts on matched primary and locally recurrent tumors has demonstrated that the highly mutagenic effects of anti‐tumor therapy induce profound genomic changes in tumor cells, even while molecular subgroup affiliation remains unchanged 57, 75. Specifically, recurrent tumors show dramatically increased somatic mutational burdens, and in one study SNVs and indels in these recurrent tumors were found to overlap their matched primaries by only 11.8% 57. These data suggest the possibility that additional molecular drivers of MB pathogenesis and migration may be acquired only after clonal generation and selection in response to adjuvant therapy, changes which would not be represented on genetic testing of a primary lesion.
The development of rational and personalized therapeutic strategies against MB dissemination is further complicated by the phylogenetic relationship of the metastatic tumor(s) to its matched primary lesion. While copy number analysis of matched human primary/metastatic samples, and sequencing of functional genomic models of MB dissemination clearly indicate a common cell‐of‐origin for all cases, the molecular features of metastatic disease suggest these are derived from (a) small subclone(s) of primary tumor. In the Sleeping Beauty mouse model of dissemination for example, gCIS overlap between primary and metastatic tumor was found to be just under 10%, and many gCISs highly clonal in metastatic deposits were present in only a very small subclone population of matched primary tumor 99. Even more concerning is the finding that molecular heterogeneity can exist between multiple metastatic deposits within a single patient; for example, copy number analysis of a single case with multiple metastatic deposits showed variable copy number alterations between deposits (e.g., deletion of chromosome 1p in one of three matched metastasis). These findings suggest the possibility that clonal evolution continues to occur to at least some degree after metastatic implantation on leptomeninges and/or distant sites, or that separate metastatic deposits could be seeded by molecularly unique subclones of the primary tumor 99. Together these data underline the difficulties which are likely to be encountered by initiatives to develop a single inhibitor of the metastatic compartment on the basis of tissue obtained from primary untreated tumor, or even from a single metastatic deposit.
Novel therapeutics for the treatment of MB and MB dissemination have been proposed and tested in preclinical MB models. Given the volume of data implicating them in MB maintenance and suggesting further roles in dissemination, the most obvious targets are members of the PI3K/AKT, RAS/MAPK, and HGF/cMET signal transduction pathways. One approach is the targeting of the upstream tyrosine kinase receptors such as ERBB2, PDGFRA, and cMET, given the relative availability of FDA‐approved therapies against these targets. For example, while ERBB2 expression levels have been deemed to be insufficiently high in MB for effective targeting by Trastuzumab (Herceptin), CAR T cells designed against this target have produced significant and sustained regression in xenograft models of MB (although impact on dissemination has not been tested in vitro or in vivo) 60. Targeting of PDGFR with imatinib mesylate (Gleevec) inhibits PDGF‐BB induced MB cell proliferation and migration in vivo, suggesting a role for this agent in abrogation of MB dissemination 1. Various small‐molecule inhibitors of cMET have also been evaluated in pre‐clinical models of MB, including a compound designated PHA665752 45, an assortment of flavonols (quercetin, kaempferol, and myricetin) 38, and the small‐molecule foretinib 41. In the case of foretinib, the drug was tested extensively in transgenic mouse models of Shh‐driven MB, and effectively caused tumor size reductions in both the primary and metastatic compartments 19.
Downstream of these RTKs, various other targets and therapies have been identified and tested, usually for inhibition of MB cell growth and proliferation, but occasionally for impacts on migration, or rarely, dissemination. These include therapeutics targeting histone deacetylases, DNA topoisomerase, and PI3K, among others 12, 28, 30, 31, 69, 88. Interestingly, PI3K inhibition has been shown to be cytotoxic to a radio‐resistant population of perivascular MB cancer stem cells in vitro, suggesting this as a possible strategy to prevent tumor cell intravasation 29. Finally, the emerging roles of CXCR4 and its ligand CXCL12 in MB proliferation and metastasis suggest a role for targeted inhibition of this axis as a therapeutic strategy both for treatment of primary tumor, and prevention of metastatic dissemination. Preclinical investigations with AMD 3100, a bicyclam small‐molecule inhibitor of CXCR4 and CXCL12 interaction, show that the drug effectively blocks the proliferative and pro‐migratory effects of CXCL12 exposure to MB cell lines in vitro, and inhibits downstream signaling through the PI3K/AKT and ERK pathways 4, 82.
Whether a single targeted therapeutic (or simple combination) can effectively inhibit the process of MB tumor cell dissemination and migration to leptomeningeal/extraneural sites, or effect sufficient cytotoxicity on metastatic tumor cell populations in human patients, remains to be seen. Well known challenges in the field of oncology including difficulties caused by tumor heterogeneity, clonal evolution, and pathway redundancy, are likely to present persistent challenges in this effort. The identification of indispensable gene targets required for bi‐compartmental tumor maintenance would be the most promising approach for therapies seeking to target primary, metastatic, and CTCs. The identification of these targets and development of therapeutics will require increased access to metastatic MB tissues and continued improvements in mouse models of MB dissemination. Further characterization of the TME in primary, leptomeningeal and extraneural compartments will also be important for the identification of novel targets/therapies against tumor niche elements. With progress on these fronts, emerging concepts in the laboratory are anticipated to translate into meaningful treatment advances for children with this debilitating disease.
References
- 1. Abouantoun TJ, MacDonald TJ (2009) Imatinib blocks migration and invasion of medulloblastoma cells by concurrently inhibiting activation of platelet‐derived growth factor receptor and transactivation of epidermal growth factor receptor. Mol Cancer Ther 8:1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bai AH, Milde T, Remke M, Rolli CG, Hielscher T, Cho Y‐J et al (2012) MicroRNA‐182 promotes leptomeningeal spread of non‐sonic hedgehog‐medulloblastoma. Acta Neuropathol 123:529–538. [DOI] [PubMed] [Google Scholar]
- 3. Bakhshinyan D, Venugopal C, Adile AA, Garg N, Manoranjan B, Hallett R et al (2019) BMI1 is a therapeutic target in recurrent medulloblastoma. Oncogene 38:1702–1716. [DOI] [PubMed] [Google Scholar]
- 4. Bertrand KC, Faria CC, Skowron P, Luck A, Garzia L, Wu X et al (2018) A functional genomics approach to identify pathways of drug resistance in medulloblastoma. Acta Neuropathol Commun 6:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bockmayr M, Mohme M, Klauschen F, Winkler B, Budczies J, Rutkowski S, Schüller U (2018) Subgroup‐specific immune and stromal microenvironment in medulloblastoma. Oncoimmunology 7:e1462430. 10.1080/2162402X.2018.1462430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boire A, Zou Y, Shieh J, Macalinao DG, Pentsova E, Massagué J (2017) Complement component 3 adapts the cerebrospinal fluid for leptomeningeal metastasis. Cell 168:1101–1113.e13. 10.1016/j.cell.2017.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boye K, Mælandsmo GM (2010) S100A4 and metastasis: a small actor playing many roles. Am J Pathol 176:528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burger JA, Kipps TJ (2006) CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood 107:1761–1767. [DOI] [PubMed] [Google Scholar]
- 9. Buss MC, Read TA, Schniederjan MJ, Gandhi K, Castellino RC (2012) HDM2 promotes WIP1‐mediated medulloblastoma growth. Neuro Oncol 14:440–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buss MC, Remke M, Lee J, Gandhi K, Schniederjan MJ, Kool M et al (2014) The WIP1 oncogene promotes progression and invasion of aggressive medulloblastoma variants. Oncogene 1126–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cavalli FM, Remke M, Rampasek L, Peacock J, Shih DJ, Luu B et al (2017) Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell 31:737–754.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen JS, Wang Q, Fu XH, Huang XH, Chen XL, Cao LQ et al (2009) Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: association with MMP‐9. Hepatol Res 39:177–186. [DOI] [PubMed] [Google Scholar]
- 13. Choi SA, Kwak PA, Kim S‐K, Park S‐H, Lee JY, Wang K‐C et al (2016) In vivo bioluminescence imaging for leptomeningeal dissemination of medulloblastoma in mouse models. BMC Cancer 16:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA (2005) Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature 436:221–226. [DOI] [PubMed] [Google Scholar]
- 15. Dupuy AJ, Clark K, Carlson CM, Fritz S, Davidson AE, Markley KM et al (2002) Mammalian germ‐line transgenesis by transposition. Proc Natl Acad Sci U S A 99:4495–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ellison DW, Onilude OE, Lindsey JC, Lusher ME, Weston CL, Taylor RE et al (2005) β‐Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children's Cancer Study Group Brain Tumour Committee. J Clin Oncol 23:7951–7957. [DOI] [PubMed] [Google Scholar]
- 17. Etzell JE, Keet C, McDonald W, Banerjee A (2006) Medulloblastoma simulating acute myeloid leukemia: case report with a review of ‘myeloid antigen’ expression in nonhematopoietic tissues and tumors. J Pediatr Hematol Oncol 28:703–710. [DOI] [PubMed] [Google Scholar]
- 18. Fan X, Mikolaenko I, Elhassan I, Ni X, Wang Y, Ball D et al (2004) Notch1 and Notch2 have opposite effects on embryonal brain tumor growth. Cancer Res 64:7787–7793. [DOI] [PubMed] [Google Scholar]
- 19. Faria CC, Golbourn BJ, Dubuc AM, Remke M, Diaz RJ, Agnihotri S et al (2015) Foretinib is effective therapy for metastatic sonic hedgehog medulloblastoma. Cancer Res 75:134–146. [DOI] [PubMed] [Google Scholar]
- 20. Friedrich C, von Bueren AO, von Hoff K, Kwiecien R, Pietsch T, Warmuth‐Metz M et al (2013) Treatment of adult nonmetastatic medulloblastoma patients according to the paediatric HIT 2000 protocol: a prospective observational multicentre study. Eur J Cancer 49:893–903. [DOI] [PubMed] [Google Scholar]
- 21. Fults DW, Taylor MD, Garzia L (2019) Leptomeningeal dissemination: a sinister pattern of medulloblastoma growth. J Neurosurg Pediatr 23:613–621. [DOI] [PubMed] [Google Scholar]
- 22. Garzia L, Kijima N, Morrissy AS, De Antonellis P, Guerreiro‐Stucklin A, Holgado BL et al (2018) A hematogenous route for medulloblastoma leptomeningeal metastases. Cell 172:1050–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gavini H, Arukala VD, Stewart J, Lee JH (2014) Medulloblastoma metastatic to the pancreas: a case report. Gastrointest Interv 3:116–119. 10.1016/j.gii.2014.09.007. [DOI] [Google Scholar]
- 24. Gholamin S, Mitra SS, Feroze AH, Liu J, Kahn SA, Zhang M et al (2017) Disrupting the CD47‐SIRPα anti‐phagocytic axis by a humanized anti‐CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci Transl Med 9:eaaf2968. [DOI] [PubMed] [Google Scholar]
- 25. Gholamin S, Mitra SS, Feroze AH, Liu J, Kahn SA, Zhang M et al (2017) Disrupting the CD47‐SIRPα anti‐phagocytic axis by a humanized anti‐CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci Transl Med 9:1–14. [DOI] [PubMed] [Google Scholar]
- 26. Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C et al (2010) Subtypes of medulloblastoma have distinct developmental origins. Nature 468:1095–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gilbertson RJ, Clifford SC (2003) PDGFRB is overexpressed in metastatic medulloblastoma. Nat Genet 35:197–198. [DOI] [PubMed] [Google Scholar]
- 28. Guerreiro AS, Fattet S, Fischer B, Shalaby T, Jackson SP, Schoenwaelder SM et al (2008) Targeting the PI3K p110α isoform inhibits medulloblastoma proliferation, chemoresistance, and migration. Clin Cancer Res 14:6761–6769. [DOI] [PubMed] [Google Scholar]
- 29. Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova‐Todorova K, Holland EC (2008) PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev 22:436–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hartmann W, Koch A, Brune H, Waha A, Schüller U, Dani I et al (2005) Insulin‐like growth factor II is involved in the proliferation control of medulloblastoma and its cerebellar precursor cells. Am J Pathol 166:1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hartmann W, Digon‐Söntgerath B, Koch A, Waha A, Endl E, Dani I et al (2006) Phosphatidylinositol 3′‐kinase/AKT signaling is activated in medulloblastoma cell proliferation and is associated with reduced expression of PTEN. Clin Cancer Res 12:3019–3027. [DOI] [PubMed] [Google Scholar]
- 32. Hernan R, Fasheh R, Calabrese C, Frank AJ, Maclean KH, Allard D et al (2003) ERBB2 up‐regulates S100A4 and several other prometastatic genes in medulloblastoma. Cancer Res 63:140‐148. [PubMed] [Google Scholar]
- 33. Ieraci A, Forni PE, Ponzetto C (2002) Viable hypomorphic signaling mutant of the met receptor reveals a role for hepatocyte growth factor in postnatal cerebellar development. Proc Natl Acad Sci U S A 99:15200–15205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ivins S, Chappell J, Vernay B, Suntharalingham J, Martineau A, Mohun TJ et al (2015) The CXCL12/CXCR4 axis plays a critical role in coronary artery development. Dev Cell 33:455–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jenkins NC, Kalra RR, Dubuc A, Sivakumar W, Pedone CA, Wu X et al (2014) Genetic drivers of metastatic dissemination in sonic hedgehog medulloblastoma. Acta Neuropathol Commun 2:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kahn SA, Wang X, Nitta RT, Gholamin S, Theruvath J, Hutter G et al (2018) Notch1 regulates the initiation of metastasis and self‐renewal of Group 3 medulloblastoma. Nat Commun 9:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kongkham PN, Northcott PA, Ra YS, Nakahara Y, Mainprize TG, Croul SE et al (2008) An epigenetic genome‐wide screen identifies SPINT2 as a novel tumor suppressor gene in pediatric medulloblastoma. Cancer Res 68:9945–9953. [DOI] [PubMed] [Google Scholar]
- 38. Kongkham PN, Onvani S, Smith CA, Rutka JT (2010) Inhibition of the MET receptor tyrosine kinase as a novel therapeutic strategy in medulloblastoma. Transl Oncol 3:336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kramer K, Humm JL, Souweidane MM, Zanzonico PB, Dunkel IJ, Gerald WL et al (2007) Phase I study of targeted radioimmunotherapy for leptomeningeal cancers using intra‐Ommaya 131‐I‐3F8. J Clin Oncol 25:5465–5470. [DOI] [PubMed] [Google Scholar]
- 40. Kramer K, Pandit‐Taskar N, Humm JL, Zanzonico PB, Haque S, Dunkel IJ et al (2018) A phase II study of radioimmunotherapy with intraventricular 131I–3F8 for medulloblastoma. Pediatr Blood Cancer 65:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Labbé D, Provençal M, Lamy S, Boivin D, Gingras D, Béliveau R (2009) The flavonols quercetin, kaempferol, and myricetin inhibit hepatocyte growth. J Nutr Biochem Mol Genet Mech 139:646–652. [DOI] [PubMed] [Google Scholar]
- 42. Lee C, Lee J, Choi SA, Kim S‐K, Wang K‐C, Park S‐H et al (2018) M1 macrophage recruitment correlates with worse outcome in SHH medulloblastomas. BMC Cancer 18:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee WJ, Chen WK, Wang CJ, Lin WL, Tseng TH (2008) Apigenin inhibits HGF‐promoted invasive growth and metastasis involving blocking PI3K/Akt pathway and β4 integrin function in MDA‐MB‐231 breast cancer cells. Toxicol Appl Pharmacol 226:178–191. [DOI] [PubMed] [Google Scholar]
- 44. Li Y, Guessous F, Johnson EB, Eberhart CG, Li XN, Shu Q et al (2008) Functional and molecular interactions between the HGF/c‐Met pathway and c‐Myc in large‐cell medulloblastoma. Lab Investig 88:98–111. [DOI] [PubMed] [Google Scholar]
- 45. Li Y, Lal B, Kwon S, Fan X, Saldanha U, Reznik TE et al (2005) The scatter factor/hepatocyte growth factor: c‐Met pathway in human embryonal central nervous system tumor malignancy. Cancer Res 65:9355–9362. [DOI] [PubMed] [Google Scholar]
- 46. Li M, Ransohoff RM (2008) Multiple roles of chemokine CXCL12 in the central nervous system: a migration from immunology to neurobiology. Prog Neurogibol 84:116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liang Y, Diehn M, Bollen AW, Israel MA, Gupta N (2008) Type I collagen is overexpressed in medulloblastoma as a component of tumor microenvironment. J Neurooncol 86:133–141. 10.1007/s11060-007-9457-5. [DOI] [PubMed] [Google Scholar]
- 48. Lim SY, Yuzhalin AE, Gordon‐weeks AN, Muschel RJ (2016) Targeting the CCL2‐CCR2 signaling axis in cancer metastasis. Oncotarget 7:28697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella‐Branger D, Cavenee WK et al (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131:803–820. [DOI] [PubMed] [Google Scholar]
- 50. Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T et al (1998) Impaired B‐lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4‐ and SDF‐1‐deficient mice. Proc Natl Acad Sci U S A 95:9448–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. MacDonald TJ, Brown KM, LaFleur B, Peterson K, Lawlor C, Chen Y et al (2001) Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat Genet 29:143–152. [DOI] [PubMed] [Google Scholar]
- 52. Majzner RG, Simon JS, Grosso JF, Martinez D, Pawel BR, Santi M et al (2017) Assessment of programmed death‐ligand 1 expression and tumor‐associated immune cells in pediatric cancer tissues. Cancer 123:3807–3815. 10.1002/cncr.30724. [DOI] [PubMed] [Google Scholar]
- 53. Margol AS, Robison NJ, Gnanachandran J, Hung LT, Kennedy RJ, Vali M et al (2015) Tumor‐associated macrophages in SHH subgroup of medulloblastomas. Clin Cancer Res 21:1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Margol AS, Robison NJ, Gnanachandran J, Hung LT, Kennedy RJ, Vali M et al (2015) Tumor‐associated macrophages in SHH subgroup of medulloblastomas. Clin Cancer Res 21:1457–1465. 10.1158/1078-0432.CCR-14-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mastronuzzi A, Del Bufalo F, Iacono A, Secco DE, Serra A, Colafati GS et al (2013) Intrathecal liposomal cytarabine and leptomeningeal medulloblastoma relapse: a valuable therapeutic option. Anticancer Res 33:3515–3518. [PubMed] [Google Scholar]
- 56. Mauffrey P, Tchitchek N, Barroca V, Bemelmans A, Firlej V, Allory Y et al (2019) Progenitors from the central nervous system drive neurogenesis in cancer. Nature 569:672–678. [DOI] [PubMed] [Google Scholar]
- 57. Morrissy AS, Garzia L, Shih DJH, Zuyderduyn S, Huang X, Skowron P et al (2016) Divergent clonal selection dominates medulloblastoma at recurrence. Nature 529:351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Moxon‐Emre I, Taylor MD, Bouffet E, Hardy K, Campen CJ, Malkin D et al (2016) Intellectual outcome in molecular subgroups of medulloblastoma. J Clin Oncol 34:4161–4170. [DOI] [PubMed] [Google Scholar]
- 59. Mulhern RK, Reddick WE, Palmer SL, Glass JO, Elkin TD, Kun LE et al (1999) Neurocognitive deficits in medulloblastoma survivors and white matter loss. Ann Neurol 46:834–841. [DOI] [PubMed] [Google Scholar]
- 60. Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T et al (2006) A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10:515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Northcott PA, Fernandez‐L A, Hagan JP, Ellison DW, Grajkowska W, Gillespie Y et al (2009) The miR‐17/92 polycistron is up‐regulated in sonic hedgehog‐driven medulloblastomas and induced by N‐myc in sonic hedgehog‐treated cerebellar neural precursors. Cancer Res 69:3249–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T et al (2017) The whole‐genome landscape of medulloblastoma subtypes. Nature 547:311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Northcott PA, Korshunov A, Pfister SM, Taylor MD (2012) The clinical implications of medulloblastoma subgroups. Nat Rev Neurol 8:340–351. [DOI] [PubMed] [Google Scholar]
- 64. Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S et al (2011) Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29:1408–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Northcott PA, Shih DJH, Peacock J, Garzia L, Sorana Morrissy A, Zichner T et al (2012) Subgroup‐specific structural variation across 1000 medulloblastoma genomes. Nature 488:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ostrom QT, de Blank PM, Kruchko C, Petersen CM, Liao P, Finlay J et al (2015) Alex’s lemonade stand foundation infant and childhood primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol 16(Suppl. 1):x1–x36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ozawa PM, Ariza CB, Ishibashi CM, Fujita TC, Banin‐Hirata BK, Oda JM, Watanabe MAE (2016) Role of CXCL12 and CXCR4 in normal cerebellar developmentand medulloblastoma.pdf. Int J Cancer 138:10–13. [DOI] [PubMed] [Google Scholar]
- 68. Packer RJ, Gajjar A, Vezina G, Rorke‐Adams L, Burger PC, Robertson PL et al (2006) Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average‐risk medulloblastoma. J Clin Oncol 24:4202–4208. [DOI] [PubMed] [Google Scholar]
- 69. Pei Y, Liu K‐W, Wang J, Garancher A, Tao R, Esparza LA et al (2016) Article HDAC and PI3K antagonists cooperate to inhibit growth of MYC‐ driven medulloblastoma article HDAC and PI3K antagonists cooperate to inhibit growth of MYC‐ driven medulloblastoma. Cancer Cell 29:311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pham CD, Flores C, Yang C, Pinheiro EM, Yearley JH, Sayour EJ et al (2016) Differential immune microenvironments and response to immune checkpoint blockade among molecular subtypes of murine medulloblastoma. Clin Cancer Res 22:582–595. 10.1158/1078-0432.CCR-15-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pham CD, Mitchell DA (2016) Know your neighbors: different tumor microenvironments have implications in immunotherapeutic targeting strategies across MB subgroups. Oncoimmunology 5:e1144002. 10.1080/2162402X.2016.1144002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Provencal M, Berger‐Thibault N, Labbé D, Veitch R, Boivin D, Rivard G‐É et al (2010) Tissue factor mediates the HGF/Met‐induced anti‐apoptotic pathway in DAOY medulloblastoma cells. J Neurooncol 97:365–372. [DOI] [PubMed] [Google Scholar]
- 73. Provençal M, Labbe D, Veitch R, Boivin D, Rivard G‐E, Sartelet H et al (2009) c‐Met activation in medulloblastoma induces tissue factor expression and activity: effects on cell migration. Carcinogenesis 30:1089–1096. [DOI] [PubMed] [Google Scholar]
- 74. Ramaswamy V, Northcott PA, Taylor MD (2011) FISH and chips: the recipe for improved prognostication and outcomes for children with medulloblastoma. Cancer Genet 204:577–588. [DOI] [PubMed] [Google Scholar]
- 75. Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJ et al (2013) Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol 14:1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rao G, Pedone CA, Valle LD, Reiss K, Holland EC, Fults DW. (2004) Sonic hedgehog and insulin‐like growth factor signaling synergize to induce medulloblastoma formation from nestin‐expressing neural progenitors in mice. Oncogene 23:6156–6162. [DOI] [PubMed] [Google Scholar]
- 77. Reardon DA, Michalkiewicz E, Boyett JM, Sublett JE, Entrekin RE, Ragsdale ST et al (1997) Extensive genomic abnormalities in childhood medulloblastoma by comparative genomic hybridization. Cancer Res 57:4042–4047. [PubMed] [Google Scholar]
- 78. Remke M, Ramaswamy V (2018) Infant medulloblastoma—learning new lessons from old strata. Nat Rev Clin Oncol 15:659–660. [DOI] [PubMed] [Google Scholar]
- 79. Kim J, Yao F, Xiao Z, Sun Y, Ma L (2018) MicroRNAs and metastasis: small RNAs play big roles. Cancer Metastasis Rev 37:5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Robinson GW, Rudneva VA, Buchhalter I, Billups CA, Waszak SM, Smith KS et al (2018) Risk‐adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol 19:768–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rochkind S, Blatt I, Sadeh M, Goldhammer Y (1991) Extracranial metastases of medulloblastoma in adults: literature review. J Neurol Neurosurg Psychiatry 54:80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rubin JB, Kung AL, Klein RS, Chan JA, Sun Y, Schmidt K et al (2003) A small‐molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci U S A 100:13513–13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Santhana Kumar K, Neve A, Guerreiro Stucklin AS, Kuzan‐Fischer CM, Rushing EJ, Taylor MD et al (2018) TGF‐β determines the pro‐migratory potential of bFGF signaling in medulloblastoma. Cell Rep 23:3798–3812.e8. 10.1016/j.celrep.2018.05.083. [DOI] [PubMed] [Google Scholar]
- 84. Schüller U, Koch A, Hartmann W, Garrè ML, Goodyer CG, Cama A et al (2005) Subtype‐specific expression and genetic alterations of the chemokine receptor gene CXCR4 in medulloblastomas. Int J Cancer 117:82–89. [DOI] [PubMed] [Google Scholar]
- 85. Schwalbe EC, Lindsey JC, Nakjang S, Crosier S, Smith AJ, Hicks D et al (2017) Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol 18:958–971. 10.1016/S1470-2045(17)30243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sengupta R, Dubuc A, Ward S, Yang L, Northcott P, Woerner BM et al (2012) CXCR4 activation defines a new subgroup of Sonic hedgehog‐driven medulloblastoma. Cancer Res 72:122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sharma T, Schwalbe EC, Williamson D, Sill M, Hovestadt V, Mynarek M et al (2019) Second‐generation molecular subgrouping of medulloblastoma: an international meta‐analysis of Group 3 and Group 4 subtypes. Acta Neuropathol 138:309–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Svalina MN, Kikuchi K, Abraham J, Lal S, Davare MA, Settelmeyer TP et al (2016) IGF1R as a key target in high risk, metastatic medulloblastoma. Sci Rep 6:27012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tarbell NJ, Friedman H, Polkinghorn WR, Yock T, Zhou T, Chen Z et al (2013) High‐risk medulloblastoma: a pediatric oncology group randomized trial of chemotherapy before or after radiation therapy (POG 9031). J Clin Oncol 31:2936–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Teo WY, Elghetany MT, Shen J, Man TK, Li X, Chintagumpala M e t al (2014) Therapeutic implications of CD1d expression and tumor‐infiltrating macrophages in pediatric medulloblastomas. J Neurooncol 120:293–301. [DOI] [PubMed] [Google Scholar]
- 91. Thomas PRM, Deutsch M, Kepner JL, Boyett JM, Krischer J, Aronin P et al (2000) Low‐stage medulloblastoma: final analysis of trial comparing standard‐dose with reduced‐dose neuraxis irradiation. J Clin Oncol 18:3004–3011. [DOI] [PubMed] [Google Scholar]
- 92. Tomita T, Das L, Radkowski MA (1990) Bone metastases of medulloblastoma in childhood; correlation with flow cytometric DNA analysis. J Neurooncol 8:113–120. [DOI] [PubMed] [Google Scholar]
- 93. Tong CY, Hui AB, Yin XL, Pang JC, Zhu XL, Poon WS, Ng H (2004) Detection of oncogene amplifications in medulloblastomas by comparative genomic hybridization and array‐based comparative genomic hybridization. J Neurosurg 100:187–193. [DOI] [PubMed] [Google Scholar]
- 94. Uziel T, Karginov FV, Xie S, Parker JS, Wang YD, Gajjar A et al (2009) The miR‐17~92 cluster collaborates with the Sonic Hedgehog pathway in medulloblastoma. Proc Natl Acad Sci U S A 106:2812–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH‐F, Lee DJ et al (2014) Serpins promote cancer cell survival and vascular Co‐option in brain metastasis. Cell 156:1002–1016. 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Vermeulen JF, Van Hecke W, Adriaansen EJM, Jansen MK, Bouma RG, Villacorta Hidalgo J et al (2018) Prognostic relevance of tumor‐infiltrating lymphocytes and immune checkpoints in pediatric medulloblastoma. Oncoimmunology 7:e1398877. 10.1080/2162402X.2017.1398877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wang B, Mezlini AM, Demir F, Fiume M, Tu Z, Brudno M et al (2014) Similarity network fusion for aggregating data types on a genomic scale. Nat Methods 11:333–337. [DOI] [PubMed] [Google Scholar]
- 98. Weeraratne SD, Amani V, Teider N, Pierre‐Francois J, Winter D, Kye MJ et al (2012) Pleiotropic effects of miR‐183~96~182 converge to regulate cell survival, proliferation and migration in medulloblastoma. Acta Neuropathol 123:539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wu X, Northcott PA, Dubuc A, Dupuy AJ, Shih DJH, Witt H et al (2012) Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 482:529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zapotocky M, Mata‐Mbemba D, Sumerauer D, Liby P, Lassaletta A, Zamecnik J et al (2018) Differential patterns of metastatic dissemination across medulloblastoma subgroups. J Neurosurg Pediatr 21:145–152. [DOI] [PubMed] [Google Scholar]
- 101. Zhao X, Ponomaryov T, Ornell KJ, Zhou P, Dabral SK, Pak E et al (2015) RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway‐dependent tumors. Cancer Res 75:3623–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zou YR, Kottman AH, Kuroda M, Taniuchi I, Littman DR (1998) Function of the chemokine receptor CXCR4 in heaematopolesis and in cerebellar development. Nature 393:595–599. [DOI] [PubMed] [Google Scholar]