

Abstract
Background
Avapritinib, a novel inhibitor of KIT/PDGFRA, is approved in the U.S. for the treatment of adults with PDGFRA exon 18‐mutant unresectable or metastatic gastrointestinal stromal tumors (U/M GISTs). We assessed the safety of avapritinib and provide evidence‐based guidance on management of avapritinib‐associated adverse events (AEs), including cognitive effects and intracranial bleeding.
Materials and Methods
We performed a post hoc analysis of data from a two‐part, single‐arm dose escalation/expansion phase I study (NAVIGATOR; NCT02508532) in patients with U/M GISTs treated with oral avapritinib 30–600 mg once daily. The primary endpoints were safety and tolerability; the impact of dose modification (interruption and/or reduction) on progression‐free survival (PFS) was a secondary endpoint. Efficacy analyses were limited to patients who started avapritinib at 300 mg (approved dose).
Results
Of 250 patients enrolled in the study, 74.0% presented with KIT mutation and 24.8% presented with PDGFRA exon 18‐mutation; 66.8% started avapritinib at 300 mg. The most common treatment‐related AEs (any grade) were nausea (59.2%), fatigue (50.0%), periorbital edema (42.0%), anemia (39.2%), diarrhea (36.0%), vomiting (36.0%), and increased lacrimation (30.8%). No treatment‐related deaths occurred. Among 167 patients starting on 300 mg avapritinib, all‐cause cognitive effects rate (grade 1–2) was 37.0% in all patients and 52.0% in patients ≥65 years. Cognitive effects improved to a lower grade more quickly with dose modification (1.3–3.1 weeks) than without (4.9–7.6 weeks). Median PFS was 11.4 months with dose modification and 7.2 months without.
Conclusion
Tolerability‐guided dose modification of avapritinib is an effective strategy for managing AEs in patients with GISTs.
Implications for Practice
Early recognition of adverse events and tailored dose modification appear to be effective approaches for managing treatment‐related adverse events and maintaining patients on avapritinib. Dose reduction does not appear to result in reduced efficacy. Patients' cognitive function should be assessed at baseline and monitored carefully throughout treatment with avapritinib for the onset of cognitive adverse events. Dose interruption is recommended at the first sign of any cognitive effect, including grade 1 events.
Keywords: Avapritinib, Gastrointestinal stromal tumor, Cognitive effects, PDGFRA, KIT
Short abstract
This report provides a comprehensive assessment of the safety and tolerability of an avapritinib once‐daily regimen and evidence‐based guidance on the management of patients with adverse events, including cognitive effects and intracranial bleeding, and any potential effects of dose interruption and/or reduction on efficacy of this agent.
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common sarcoma subtype of the gastrointestinal tract [1, 2]. The estimated GIST incidence rate is 5–20 cases per million, and the prevalence is 10 times greater than the incidence [3]. More than 85% of GISTs are characterized by oncogenic activating mutations in either the KIT or platelet‐derived growth factor receptor alpha (PDGFRA) gene [2, 4].
Treatment guidelines recommend sequential treatment with the tyrosine kinase inhibitors (TKIs) imatinib, sunitinib, regorafenib, and ripretinib [5, 6, 7, 8]. These agents are approved for the treatment of unresectable or metastatic (U/M) GISTs in the first‐, second‐, third‐, and fourth‐line settings, respectively [9, 10, 11, 12]. Treatment guidelines also recommend mutational testing prior to initiation of therapy, because the presence/absence and types of KIT and PDGFRA mutations have been shown to affect the clinical response to multitargeted TKIs. For example, patients with KIT exon 9 mutant GISTs may benefit from high‐dose imatinib therapy [7, 13, 14, 15, 16] or from sunitinib [17]. Despite this evidence, in the U.S., one study demonstrated that only 27.0% of patients with GISTs undergo mutational testing of the tumor [18]. Patients with PDGFRA exon 18 D842V‐mutant GISTs have a poor prognosis; prior to the approval of avapritinib, standard‐of‐care treatment in the advanced GIST setting achieved median progression‐free survival (PFS) of only 3–5 months and overall survival (OS) of approximately 15 months [19, 20, 21].
Avapritinib (formerly BLU‐285; Blueprint Medicines Corporation, Cambridge, MA) is approved in the U.S. for the treatment of adults with U/M GISTs with a PDGFRA exon 18 mutation, including PDGFRA D842V mutations, regardless of line of therapy [22]. Avapritinib is a potent and selective inhibitor of KIT and PDGFRA, with high potency for the KIT exon 17 mutation D816V and PDGFRA D842V‐mutant kinases, which are associated with resistance to imatinib, sunitinib, and regorafenib [23]. Owing to the selectivity of avapritinib, the potential for off‐target effects is reduced compared with other multitarget TKIs [23], which are associated with off‐target effects [24, 25]. A dose‐escalation/expansion phase I study (NAVIGATOR; NCT02508532) in patients with GISTs identified oral, once‐daily avapritinib 300 mg as the recommended phase II dose. This phase I study was the basis for the Food and Drug Administration approval of avapritinib in adults with U/M GISTs harboring a PDGFRA exon 18 mutation, including D842V mutations. It demonstrated an objective response rate of 86% in patients with PDGFRA exon 18‐mutant GISTs, and 22% in patients who received avapritinib in the fourth‐line or higher treatment setting (including one partial response pending confirmation), regardless of mutation status [26]. Findings from the NAVIGATOR study have recently been reported [27]. The most common adverse events (AEs; all grades) related to 300 mg avapritinib in ≥35% of patients were nausea, fatigue, and anemia [26]. Supportive care and flexible dosing strategies (including dose interruptions and/or reductions), which are common strategies for managing AEs associated with multitargeted TKIs [28, 29, 30, 31, 32], were used to manage the AEs seen with avapritinib in the NAVIGATOR phase I study.
This report provides a comprehensive assessment of the safety and tolerability of an avapritinib once‐daily regimen and evidence‐based guidance on the management of patients with AEs, including cognitive effects and intracranial bleeding (ICB), and any potential effects of dose interruption and/or reduction on efficacy of this agent.
Materials and Methods
Study Design and Patients
A post hoc analysis was performed on data from the first‐in‐human, two‐part, single‐arm, multicenter, dose escalation/expansion phase I study (NAVIGATOR; NCT02508532); the study design of this trial has been reported previously [27]. Briefly, the study was designed to evaluate the safety, tolerability, pharmacokinetics, pharmacodynamics, and antitumor activity of avapritinib in adults (≥18 years) with unresectable GISTs. The study included a dose escalation (Part 1) and a dose expansion phase (Part 2). Eligible patients had histologically or cytologically confirmed unresectable KIT‐mutant GISTs that progressed following treatment with ≥2 TKIs, or PDGFRA exon 18‐mutant GISTs, regardless of prior therapy.
Oral avapritinib 30–600 mg was administered once daily in 28‐day cycles until unacceptable toxicity, nonadherence, withdrawal, physician decision to discontinue treatment, progressive disease, or death. In Part 1, treatment interruption was required for patients who experienced a dose‐limiting toxicity. After resolution of the event, patients could resume therapy at the previous dose level tested during dose escalation. In Part 2, treatment interruption was required for any grade ≥3 treatment‐related AEs (TRAEs). If the event resolved to grade ≤2, patients could resume avapritinib at a lower dose (100 mg less than the initial dose; supplemental online Table 1).
The study was approved by the institutional review board or independent ethics committee at each participating site and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. All study participants provided written informed consent. Reporting of AEs and serious AEs was performed according to institutional, national, and international law. Serious AEs were defined as meeting any of the following criteria: death, life‐threatening, causes or prolongs in‐patient hospitalization, persistent or significant incapacity, congenital anomaly in offspring of patient, or other important events based on medical judgment.
Outcomes
The primary objective of this post hoc analysis was to investigate the safety and tolerability of avapritinib in adult patients with U/M GISTs and to provide evidence‐based guidance on the management of patients with AEs. Safety was assessed at all study visits from the first dose of study treatment until 30 days after the last dose. The AE terms were coded according to the Medical Dictionary for Regulatory Activities version 18.1 (MedDRA v18.1). AE severity was graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. Medical history was taken into consideration in grading of cognitive effects. The full description of symptoms for cognitive effects and ICB is detailed in supplemental online Table 2. The umbrella term of cognitive effects included the Preferred Terms of memory impairment, cognitive disorder, confusional state, and encephalopathy. The terms to include in the cognitive effect group were determined based on medical review of reported AEs (frequency, severity, seriousness, relatedness, and medical concept) of Preferred Terms within the system organ classes (SOC) of Nervous System Disorders and Psychiatric Disorders. In the Nervous System Disorders SOC, the terms consistent with cognitive effects were memory impairment, cognitive disorder, and encephalopathy. Among the Psychiatric Disorders SOC, the AE of confusional state did not appear to be attributed to an exacerbation of pre‐existing conditions or medications other than avapritinib and thus was included. The terms to include in the ICB groups were determined based on Preferred Terms within the SOC of Injury Poisoning and Procedural Complications. The ICB group included cerebral hemorrhage, subdural hematoma, and intracranial hemorrhage.
The secondary objective of this post hoc analysis was to assess the impact of dose modification (interruption and/or reduction) on PFS in patients receiving avapritinib. The guidelines applied for dose modification are detailed in supplemental online Table 1. Efficacy was evaluated by magnetic resonance imaging or computed tomography; images were assessed by a central radiology laboratory (Virtual Scopics, Rochester, NY) using modified RECIST version 1.1 for patients with GISTs [11].
Statistical Analysis
Safety analyses included patients with U/M GISTs who were started on avapritinib 300 mg or 400 mg; efficacy analyses were limited to patients who were started on avapritinib 300 mg, which is the recommended phase II dose. Most data were summarized using descriptive statistics. The correlations between cognitive effects and race, gender, baseline Eastern Cooperative Oncology Group (ECOG) performance status, number of prior TKIs, total duration of prior TKI use, and cumulative dose of avapritinib were determined by multivariable logistic regression. Cumulative avapritinib dose was analyzed per 1,000‐mg increase. Time to improvement of grade ≥2 cognitive effects and impact of dose modification on the occurrence of cognitive effects were assessed using the Kaplan‐Meier method. The cutoff date for these analyses was April 2, 2019.
Results
Patients
A total of 250 patients with GISTs were enrolled in the study between October 12, 2015, and March 9, 2019. The median age was 61 years, 155 (61.6%) of the patients were male, 181 (72.4%) were white, 185 (74.0%) presented with a tumor harboring a KIT mutation, 62 (24.8%) presented with a tumor harboring a PDGFRA exon 18‐mutation, and 139 (55.6%) had received ≥3 prior lines of multitargeted TKIs. A starting dose of avapritinib 300 mg was assigned to 167 (66.8%) patients, and 50 (20.0%) patients were assigned a starting dose of 400 mg (Table 1).
Table 1.
Baseline demographics and characteristics
| Demographics/characteristic | Patients (n = 250) |
|---|---|
| Median age, years (range) | 61 (25–90) |
| Male, n (%) | 154 (61.6) |
| Race, n (%) | |
| White | 181 (72.4) |
| Asian | 22 (8.8) |
| Black or African American | 12 (4.8) |
| Unknown | 27 (10.8) |
| Other a | 8 (3.2) |
| ECOG performance status, n (%) | |
| 0 | 101 (40.4) |
| 1 | 141 (56.4) |
| 2 | 8 (3.2) |
| Metastatic disease, n (%) | 242 (96.8) |
| GIST mutational subtype, n (%) | |
| KIT | 185 (74.0) |
| PDGFRA exon 18 | 62 (24.8) |
| PDGFRA D842V | 56 (22.4) |
| PDGFRA non‐D842V | 6 (2.4) |
| Largest target lesion size, n (%) | |
| ≤10 cm | 194 (77.6) |
| >10 cm | 52 (20.8) |
| Unknown | 4 (1.6) |
| Prior lines of multitargeted TKIs, n (%) | |
| <4 | 111 (44.4) |
| ≥4 | 139 (55.6) |
| Starting avapritinib dose, n (%) | |
| <300 mg | 30 (12.0) |
| 300 mg | 167 (66.8) |
| 400 mg | 50 (20.0) |
| 600 mg | 3 (1.2) |
Includes individuals self‐identified as American Indian, Alaska Native, Native Hawaiian, other Pacific Islander, or other.
Abbreviations: ECOG, Eastern Cooperative Oncology Group; GIST, gastrointestinal stromal tumor; KIT, KIT proto‐oncogene receptor tyrosine kinase; PDGFRA, platelet‐derived growth factor receptor alpha; TKI, tyrosine kinase inhibitor.
Safety
In the safety population, 248/250 (99.2%) patients experienced AEs regardless of causality (all‐cause); the most common were nausea (160/250, 64.0%), fatigue (147/250, 58.8%), and anemia (128/250, 51.2%). The overall incidence of all‐cause AEs (any grade) was similar between patients who started on avapritinib 300 mg (166/167, 99.4%) and 400 mg (49/50, 98.0%; Table 2). All‐cause grade ≥3 AEs were reported in 121/167 (72.5%) patients who started on avapritinib 300 mg and 42/50 (84.0%) patients who started on 400 mg. In the safety population, all‐cause grade ≥3 AEs were reported in 188/250 (75.2%) patients, most commonly anemia (76/250, 30.4%), fatigue (19/250, 7.6%), abdominal pain (14/250, 5.6%), disease progression (14/250, 5.6%), and hypophosphatemia (14/250, 5.6%). The overall incidence of all‐cause AEs in elderly patients (≥65 years) who started on avapritinib 300 mg appeared to be slightly higher compared with younger patients (<65 years; supplemental online Table 3). Elderly patients were also more susceptible to all‐cause grade ≥3 AEs compared with younger patients, with a rate of 84.6% (55/65) versus 64.7% (66/102), respectively (p = .006). The most common all‐cause grade ≥3 AE in the older population was anemia (27/65, 41.5%).
Table 2.
Summary of all‐cause AEs in patients initiated with avapritinib 300 mg or 400 mg
| Avapritinib 300 mg (n = 167) | Avapritinib 400 mg (n = 50) | |||
|---|---|---|---|---|
| AEs | Any grade | Grade ≥3 | Any grade | Grade ≥3 |
| Patients with AEs, n (%) | 166 (99.4) | 121 (72.5) | 49 (98.0) | 42 (84.0) |
| Nausea | 103 (61.7) | 2 (1.2) | 38 (76.0) | 3 (6.0) |
| Fatigue | 88 (52.7) | 10 (6.0) | 35 (70.0) | 17 (34.0) |
| Anemia | 87 (52.1) | 51 (30.5) | 26 (52.0) | 17 (34.0) |
| Diarrhea | 70 (41.9) | 8 (4.8) | 20 (40.0) | 3 (6.0) |
| Periorbital edema | 64 (38.3) | 2 (1.2) | 26 (52.0) | 0 (0) |
| Decreased appetite | 61 (36.5) | 4 (2.4) | 21 (42.0) | 3 (6.0) |
| Vomiting | 58 (34.7) | 4 (2.4) | 27 (54.0) | 1 (2.0) |
| Increased lacrimation | 51 (30.5) | 0 (0) | 21 (42.0) | 0 (0) |
| Peripheral edema | 45 (26.9) | 1 (<1.0) | 18 (36.0) | 1 (2.0) |
| Memory impairment | 45 (26.9) | 1 (<1.0) | 20 (40.0) | 1 (2.0) |
| Abdominal pain | 41 (24.6) | 11 (6.6) | 10 (20.0) | 1 (2.0) |
| Constipation | 38 (22.8) | 3 (1.8) | 13 (26.0) | 0 (0) |
| Face edema | 38 (22.8) | 0 (0) | 14 (28.0) | 1 (2.0) |
| Blood bilirubin increased | 37 (22.2) | 8 (4.8) | 10 (20.0) | 1 (2.0) |
| Hair color changes | 33 (19.8) | 0 (0) | 14 (28.0) | 1 (2.0) |
| Hypokalemia | 30 (18.0) | 7 (4.2) | 7 (14.0) | 2 (4.0) |
| Dysgeusia | 30 (18.0) | 0 (0) | 5 (10.0) | 0 (0) |
| Dizziness | 27 (16.2) | 1 (<1.0) | 21 (42.0) | 0 (0) |
| Dyspnea | 27 (16.2) | 4 (2.4) | 12 (24.0) | 2 (4.0) |
| Headache | 27 (16.2) | 1 (<1.0) | 10 (20.0) | 0 (0) |
| Dyspepsia | 27 (16.2) | 0 (0) | 7 (14.0) | 0 (0) |
| Weight decreased | 27 (16.2) | 2 (1.2) | 6 (12.0) | 0 (0) |
| Hypophosphatemia | 24 (14.4) | 8 (4.8) | 8 (16.0) | 3 (6.0) |
| Pyrexia | 22 (13.2) | 0 (0) | 10 (20.0) | 1 (2.0) |
| AST increased | 21 (12.6) | 0 (0) | 12 (24.0) | 1 (2.0) |
| Alopecia | 19 (11.4) | — | 10 (20.0) | — |
| Rash | 18 (10.8) | 1 (<1.0) | 10 (20.0) | 0 (0) |
| Cough | 14 (8.4) | 0 (0) | 9 (18.0) | 0 (0) |
| Upper respiratory tract infection | 13 (7.8) | 0 (0) | 8 (16.0) | 0 (0) |
| Feeling cold | 8 (4.8) | 0 (0) | 9 (18.0) | 0 (0) |
| Cognitive effects | 67 (40.1) | 5 (3.0) | 25 (50.0) | 4 (8.0) |
| Memory impairment | 45 (26.9) | 1 (<1.0) | 20 (40.0) | 1 (2.0) |
| Cognitive disorder | 21 (12.6) | 1 (<1.0) | 3 (6.0) | 1 (2.0) |
| Confusional state | 11 (6.6) | 2 (1.2) | 5 (10.0) | 2 (4.0) |
| Encephalopathy | 1 (<1.0) | 1 (<1.0) | 2 (4.0) | 1 (2.0) |
| Intracranial bleeding | 4 (2.4) | 2 (1.2) | 0 (0) | 0 (0) |
| Intracranial hemorrhage | 3 (1.8) | 2 (1.2) | 0 (0) | 0 (0) |
| Subdural hematoma | 1 (<1.0) | 0 (0) | 0 (0) | 0 (0) |
| Cerebral hemorrhage | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Serious AEs, n (%) | 93 (55.7) | — | 29 (58.0) | — |
| AE leading to dose interruption, n (%) | 110 (65.9) | — | 34 (68.0) | — |
| AE leading to dose reduction, n (%) | 75 (44.9) | — | 33 (66.0) | — |
| AE leading to treatment discontinuation, n (%) | 28 (16.8) | — | 9 (18.0) | — |
| Treatment‐related AE leading to discontinuation, n (%) | 16 (9.6) | — | 6 (12.0) | — |
The cutoff date for these analyses was April 2, 2019.
Preferred Terms for any‐grade AEs reported in ≥15% of patients in either dose group.
Abbreviations: AE, adverse event; AST, aspartate aminotransferase.
TRAEs were reported in 245/250 (98.0%) patients; those occurring in ≥30% of patients included nausea (148/250, 59.2%), fatigue (125/250, 50.0%), periorbital edema (105/250, 42.0%), anemia (98/250, 39.2%), diarrhea (90/250, 36.0%), vomiting (83/250, 33.2%), and increased lacrimation (77/250, 30.8%). TRAEs leading to discontinuation of avapritinib occurred in 28/250 (11.2%) patients; the most common were cognitive effects (10/245, 4.1%), ICB (3/245, 1.2%), fatigue (2/245, <1%), and vomiting (2/245, <1%). Discontinuation due to TRAEs occurred in 16/167 (9.6%) patients who started on avapritinib 300 mg and 6/50 (12.0%) patients who started on avapritinib 400 mg. There were no treatment‐related deaths in the study.
Dose interruptions due to all‐cause AEs occurred in 169/250 (67.6%) patients in the safety population, and the rate was similar in patients who started on avapritinib 300 mg (110/167, 65.9%) and 400 mg (34/50, 68.0%). Dose reductions occurred in 81/250 (32.4%) patients in the safety population. Among patients who experienced a dose reduction, the rate of onset of all‐cause AEs was markedly lower after dose reduction compared with the rate of onset prior to dose reduction (Fig. 1). Among patients with PDGFRA exon 18‐mutant GISTs, dose interruptions occurred in 53/62 (85.5%) and dose reductions occurred in 40/62 (64.5%) patients. Among patients receiving avapritinib in the fourth‐ or higher‐line treatment setting regardless of mutation type, dose interruptions occurred in 89/139 (64.0%) and dose reductions occurred in 69/139 (49.6%) patients. The difference in the rate of dose modification between patients with PDGFRA exon 18‐mutant GISTs and patients receiving avapritinib in the fourth‐ or higher‐line treatment may be related to the longer treatment duration observed in the patients with PDGFRA exon 18‐mutant GISTs.
Figure 1.
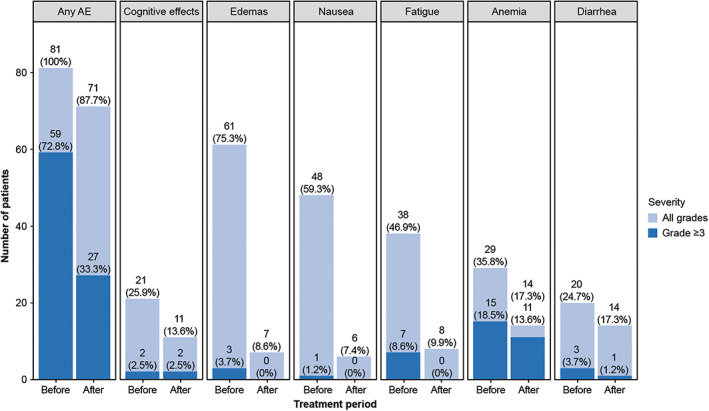
Onset of all‐cause AEs before and after dose reduction in the safety population. Worst‐grade period is presented for the onset of any AE, cognitive effects, edemas, nausea, fatigue, anemia, and diarrhea in patients who experienced dose reduction (n = 81).Abbreviation: AE, adverse event.
In the dose‐escalation part of the study, preliminary antitumor activity was observed with all tested doses (30–600 mg) of avapritinib [27]; however, the time‐to‐response was of shorter duration with the higher doses [33]. Based on the safety data, 400 mg was determined as the maximum tolerated dose for avapritinib. Owing to the similar preliminary antitumor activity observed with avapritinib 300 mg and 400 mg and subsequent safety findings, a dose of 300 mg was determined as the recommended dose for administration of avapritinib [22]. The recommended dose of avapritinib 300 mg is considered to maximize efficacy, with dose modifications being considered to maximize the risk‐benefit for each individual patient.
Cognitive Effects
Cognitive effects were reported overall in 104/250 (41.6%) patients treated with any dose of avapritinib in the safety population. The rate of cognitive effects was lower with 300 mg (67/167, 40.1%) compared with 400 mg avapritinib (25/50, 50.0%). Memory impairment was the most common cognitive effect, with higher prevalence in the 400‐mg group (20/50, 40.0%) compared with the 300‐mg group (45/167, 26.9%; Table 2). Cognitive effects occurred more frequently in elderly patients (≥65 years) than in younger patients (38/65 [58.5%] vs. 40/102 [39.2%]; p = .018; supplemental online Table 3). Cognitive effects were not associated with cumulative dose, and there was no difference in the incidence of cognitive effects by race, gender, baseline ECOG performance status, number of prior TKIs, or total duration of prior TKI use (supplemental online Table 4).
Among the 67 patients who started treatment with 300 mg avapritinib and experienced cognitive effects, 47/67 (70.1%) experienced grade 1 cognitive effects that did not affect activities of daily living (supplemental online Table 5). Grade 2 cognitive effects that interfere with activities of daily living were reported in 15/67 (22.4%) patients. Grade 3 cognitive effects occurred in 5/67 (7.5%) patients, significantly limiting the ability of a patient to perform routine activities. Among the five patients with grade 3 cognitive effects, confusional state occurred in two (3.0%) patients, and memory impairment, cognitive disorder, and encephalopathy occurred in one (1.5%) patient each. No patient who started on avapritinib 300 mg experienced life‐threatening (grade 4) or fatal (grade 5) cognitive effects.
Dose Modifications in Patients with Cognitive Effects
To better evaluate the impact of dose modification (interruption and/or reduction) on cognitive effects, data from a total of 20 patients who started on 300 mg and experienced grade ≥2 cognitive effects were analyzed. With dose modification, the median time for improvement of at least one grade from grade ≥2 was 7.6 weeks (95% confidence interval [CI], 2.2 to not evaluable; Fig. 2A), as was median time for improvement to grade 1 or resolution (95% CI, 2.0–8.3; Fig. 2B).
Figure 2.
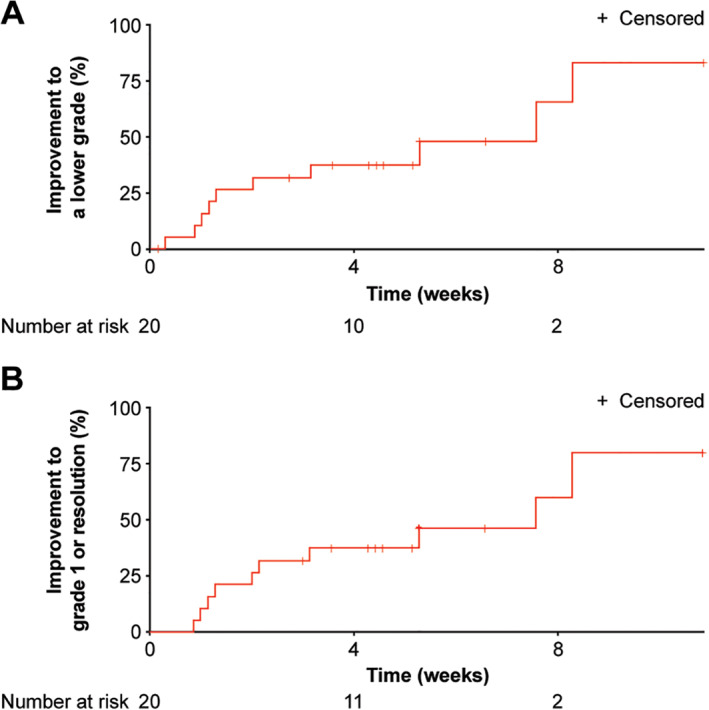
Time to improvement of grade ≥2 cognitive effects in patients who started treatment with avapritinib 300 mg. Kaplan‐Meier estimated time (weeks) to improvement of cognitive effects from grade ≥2 to (A) a lower grade or (B) grade 1 or resolution.
In these 20 patients, a total of 30 cognitive effects events of grade ≥2 were reported. Dose modifications occurred in response to 14 of those events (47.0%; Table 3). Among the 30 grade ≥2 cognitive effects events reported, the dose of avapritinib was reduced in 1 (3.3%), interrupted in 10 (33.3%), and both interrupted and subsequently reduced in 3 (10.0%); the dose was not modified in response to 16 of these events (53.3%; Table 3). About half (16/30, 53.3%) of grade ≥2 cognitive effects events showed improvement to a lower grade in a median time of 2 weeks (range: 0.3–8.3; Table 3). Either dose interruption or dose interruption with subsequent reduction resulted in improvement of cognitive effects (to a lower grade, or to grade 1 or symptom resolution) in a median time of 1.3 weeks or 3.1 weeks, respectively. Without dose modification, the median time for symptom improvement to a lower grade was 4.9 weeks, and the median time for symptom improvement to grade 1 or resolution was 7.6 weeks (Table 3). Time for resolution could not be determined for patients who had unresolved ongoing AEs or who were lost to follow‐up. The median dose intensity of avapritinib in the 20 patients who started on 300 mg and experienced grade ≥2 cognitive effects was 233 mg (mean dose intensity 228 mg ± 68 mg). In comparison, among all 167 patients who started on avapritinib 300 mg, dose interruption due to AEs occurred in 110/167 (65.9%) patients and dose reduction in 75/167 (44.9%; Table 2). The median dose intensity of avapritinib in these 167 patients was 255 mg (mean dose intensity 244 mg ± 62 mg).
Table 3.
Outcome of grade ≥2 cognitive effect eventsa by dose action taken in patients receiving a starting dose of avapritinib 300 mg
| Outcome | Dose reduction (n = 1) | Dose interruption (n = 10) | Dose reduction and interruption (n = 3) | Any dose modification (n = 14) | No dose modification (n = 16) | Total (n = 30) |
|---|---|---|---|---|---|---|
| Event improved to grade 1 or resolved, n (%) | 0 (0) | 9 (90.0) | 3 (100) | 12 (85.7) | 3 (18.8) | 15 (50.0) |
| Median time, weeks (range) | — | 1.3 (0.3–5.3) | 3.1 (1.1–4) | 1.6 (0.3–5.3) | 7.6 (2.1–8.3) | 2 (0.3–8.3) |
| Event improved to a lower grade, n (%) | 0 (0) | 9 (90.0) | 3 (100.0) | 12 (85.7) | 4 (25.0) | 16 (53.3) |
| Median time, weeks (range) | — | 1.3 (0.3–5.3) | 3.1 (1.1–4) | 1.6 (0.3–5.3) | 4.9 (1–8.3) | 1.9 (0.3–8.3) |
| Event unchanged, n (%) | 1 (100) | 1 (10.0) | 0 (0) | 2 (14.3) | 10 (62.5) | 12 (40.0) |
| Median time, weeks (range) | 6.6 (6.6–6.6) | 4.3 (4.3–4.3) | — | 5.4 (4.3–6.6) | 4.5 (0.1–10.9) | 4.5 (0.1–10.9) |
| Events worsened, n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (12.5) | 2 (6.7) |
| Median time, weeks (range) | — | — | — | — | 0.9 (0.3–1.6) | 0.9 (0.3–1.6) |
Approximately one quarter of grade 1 events resolved, and the majority were ongoing regardless of what action (or lack thereof) was taken. These analyses only considered dose modifications directly related to cognitive events; modifications may have been made for reasons other than cognitive effects.
Abbreviation: —, not applicable.
Intracranial bleeding
ICB occurred in 4/167 (2.4%) patients who started on 300 mg avapritinib. One patient had grade 1 subdural hematoma, and three patients had intracranial hemorrhage (grades 1, 3, and 4). The patient with grade 1 subdural hematoma presented with multiple traumatic events that were assessed as nonserious grade 1 or 2 events, on the same day, suggesting a head trauma. The patient with grade 1 intracranial hemorrhage was asymptomatic and was diagnosed during routine imaging; this patient had a history of hypertension at the time of the event. Both patients continued avapritinib. The patient with grade 3 intracranial hemorrhage started on avapritinib <2 months prior to the event, and the event led to treatment discontinuation. The grade 4 intracranial hemorrhage was considered serious and led to permanent discontinuation of avapritinib. Brain imaging studies of these patients showed nonsignificant minimal microangiopathy at baseline, and defined microangiopathy at the time of the event. A fifth patient experienced grade 3 cerebral hemorrhage. This patient had received an initial avapritinib dose of 90 mg and had been using avapritinib 200 mg for approximately 8 months prior to the event. The patient had a history of hypertension and was receiving aspirin at the time of the event. In addition, brain imaging showed cavernous angiomas in this patient. The event was considered a serious AE and resulted in discontinuation of avapritinib. All five ICB events resolved or resolved with sequelae (seizures), and no patient died because of ICB.
Impact of Dose Modification on PFS
To assess the impact of dose modification on clinical outcomes, we examined the duration of PFS in the efficacy‐evaluable population (n = 97) of patients who started on avapritinib 300 mg. Patients who experienced dose reduction had a longer PFS compared with those who had no reduction of dose. The median PFS was 11.4 (95% CI, 8.1–20.3) months with dose reduction and 7.2 (95% CI, 5.5–24.0) months without dose reduction. With dose reduction, PFS rates (interval definition = min/max interquartile range 95% CI) at 3, 6, and 12 months were 96.8% (92.5%–100%), 76.6% (65.8%–87.4%), and 48.6% (35.2%–61.9%), respectively. In contrast, the rates of PFS without dose reduction at 3, 6, and 12 months were 85.0% (72.9%–97.1%), 56.4% (39.2%–73.7%), and 38.5% (20.9%–56.1%), respectively (Fig. 3).
Figure 3.

Effect of dose reduction on progression‐free survival in patients who started treatment with avapritinib 300 mg. Kaplan‐Meier estimate of progression‐free survival in the safety population (n = 97) with or without dose reduction.
Discussion
In this post hoc analysis of the registration‐enabling phase I NAVIGATOR study, avapritinib was generally well tolerated at the recommended dose of 300 mg once daily. The most common AEs in this study were nausea, fatigue, and anemia, which are consistent with on‐target inhibition of KIT and PDGFRA, and the safety profile of oral kinase inhibitors in patients with GISTs [6, 10, 34, 35]. Reports of thyroid abnormalities and hand‐foot syndrome/reactions, a common side effect of some multitargeted TKIs, were infrequent in this study [10, 11]. Cognitive effects (memory impairment, cognitive disorder, confusional state, and encephalopathy) were mostly grade 1 or 2 and manageable with dose modification (interruption and/or reduction). Cognitive effects (any grade) occurred in 41.6% of patients who were exposed to avapritinib. The rate of cognitive effects was lower in patients who started on avapritinib 300 mg (40.1%) compared with patients who started on 400 mg (50.0%). Among patients who experienced these events in the 300‐mg avapritinib group, most (70.1%) had grade 1 cognitive effects, whereas no patients experienced grade 4 or 5 cognitive effects. Improvement of grade ≥2 cognitive effects to a lower grade or symptom resolution were achieved faster with dose modification compared with maintaining the dose. Importantly, dose modification did not appear to result in reduced median PFS. These data suggest that early dose adjustment may be a beneficial approach for managing grade ≥2 cognitive effects in patients treated with avapritinib. We recommend continued monitoring of patients who experience cognitive effects even after the AEs improve to grade 1.
As seen with other oral TKIs, there may be differences in intrapatient drug exposures at a given dose of avapritinib. In this post hoc analysis, the rate of cognitive effects was associated with the cumulative exposure to avapritinib and the rate of AEs, including cognitive effects, and was higher in patients who started on 400 mg compared with 300 mg. An analysis of pharmacokinetic data from this phase I study and a phase III study (VOYAGER; NCT03465722) with avapritinib in patients with GISTs suggested a trend for higher occurrence of grade 3 or 4 AEs in patients who presented with higher concentrations of avapritinib, regardless of their actual prescribed dose, given differing interpatient variability [26]. Therefore, dose adjustments based on individual patient tolerability and benefit/risk assessments are needed to optimize maintenance of patients on therapy. In this post hoc analysis, dose reduction did not appear to negatively impact PFS in the safety population.
Early AE recognition and tailored dose modification appears to be an effective approach for managing TRAEs and maintaining patients on avapritinib. The use of a lower starting dose may help to minimize AE occurrence. For patients with PDGFRA exon 18‐mutant GISTs who respond well to treatment, maintaining treatment at a lower dose may warrant further investigation. It is important to educate patients and their caregivers on the potential for AEs, including cognitive effects, prior to initiating avapritinib.
Evaluation of cognitive status at the first visit and consistency in care providers can help in early detection of cognitive effects and foster early interventions, including dose modifications that can expedite improvement in cognitive AEs. Cognitive tests can be used to quantify changes in cognitive effects; these tests can be short (<5 minutes) and adapted to each patient. Encouraging family members, friends, and/or caregivers to attend visits may help with identification of cognitive effects not reported or not recognized by the patient, or subtle changes in the patient's cognitive state, for example, the patient starting to ask the same question repeatedly. Because cognitive effects may develop early, close monitoring of patients after initiating treatment is important. Dose interruption is recommended at the first sign of any cognitive effect, including grade 1 events (supplemental online Table 1). In elderly patients (≥65 years) with PDGFRA exon 18‐mutant GISTs, starting treatment at a lower avapritinib dose can be considered. If the patient responds well to treatment, the lower dose could be maintained [36, 37]. Patients who experience cognitive events can benefit from close monitoring even after the AEs have improved to grade 1.
There is a need to raise awareness among oncologists of early signs of cognitive effects, and the potential benefits of dose modification to improve symptoms. Awareness of cognitive changes specific to avapritinib may help the oncologist rule out other etiologies. In light of the long half‐life of avapritinib (~25 hours) [38], dose interruption for 1–2 weeks rather than dose reduction may be preferred for AE management. We recommend dose interruption with or without subsequent reduction at the first sign of any cognitive impairment, including grade 1 events. Avapritinib should be withheld until the event improves or resolves. Treatment may then be resumed at 300 mg or reduced to 200 mg; if needed, avapritinib can be further reduced to 100 mg. However, in patients who experience grade 3 or 4 cognitive effects, avapritinib should be interrupted for a minimum of 14 days and discontinuation should be considered. If resuming treatment is in the best medical interest of the patient owing to the underlying GIST, dosing may be resumed with a dose reduction of 100 mg when the cognitive effect has improved to grade ≤1, or when it has improved to grade 2 in patients who require therapy more urgently.
In this phase I study, ICB occurred in four patients (2.4%) treated with a starting dose of avapritinib 300 mg. Although not all ICBs were considered treatment related, such events could be related to KIT or PDGFRA inhibition, as subdural hematomas have been reported in patients treated with imatinib [39, 40, 41]. In patients with ICB, avapritinib should be interrupted until resolution. In patients who experience grade 1 or 2 ICB, treatment can be resumed at a reduced dose if the benefit/risk assessment favors continued therapy. However, treatment should be permanently discontinued in patients who develop grade ≥3 ICB and in those in whom the event recurs (irrespective of severity) following the resumption of treatment.
One of the risk factors for ICB is severe thrombocytopenia (platelet count <50,000 cells/μL) associated with mast cell infiltration of bone marrow, observed in trials of avapritinib in patients with advanced systemic mastocytosis [42]. However, grade 3 thrombocytopenia has not been observed in patients with GISTs who are treated with avapritinib. Grade 1 or 2 thrombocytopenia occurred in 7/167 (4.2%) patients who started on 300 mg avapritinib, and none of those patients experienced intracranial hemorrhage. Signs and/or symptoms associated with ICB, such as sudden severe headache, somnolence, weakness, or difficulty speaking, should be assessed, and subsequent brain imaging should be considered based on presentation of emerging symptoms. Other AEs, such as anemia, neutropenia, nausea, diarrhea, constipation, edema, and fatigue, should be managed using standard guidelines for AE management.
This study had several limitations. With only the four most prevalent Preferred Terms included under the grouping “cognitive effects,” there is potential for cognitive effects to have been underreported or under‐recognized at study entry during patient screening. Other Preferred Terms, such as “headache” and “dizziness,” were purposefully omitted from these analyses because they were regarded as common and nonspecific. In addition, preferred CTCAE terms can be broadly interpreted, leading to a lack of specificity for the cognitive domains affected, and thus may not have comprehensively collected all the cognitive AEs observed during the trial. The analysis of dose modification impact on cognitive effects was limited by small patient numbers. Because some patients left the study before they had the opportunity for dose modification, or had ongoing cognitive AEs that were not resolved, the time to cognitive AE resolution was only available for a subgroup of patients. Moreover, only dose modifications directly related to cognitive effects were considered. As the dose of avapritinib could have been interrupted and/or reduced for reasons other than cognitive effects, the reported results may be reflective of a specific subpopulation of patients.
Conclusion
This post hoc analysis supports tolerability‐guided dose adjustment of avapritinib as an effective way to manage TRAEs without compromising efficacy. Patients should be assessed for cognitive impairment at baseline and monitored carefully throughout treatment with avapritinib for the onset of cognitive AEs. Action should be taken at the earliest sign of any cognitive effect, regardless of severity.
Author Contributions
Conception/design: Robin L. Jones, Patrick Schöffski, Teresa Zhou, Sebastian Bauer.
Provision of study material or patients: Cissimol P. Joseph, Yoon‐Koo Kang, Richard F. Riedel, Eric D. Tetzlaff, Tracy Havnaer
Collection and/or assembly of data: Cissimol P. Joseph, Robin L. Jones, Yoon‐Koo Kang, Patrick Schöffski, César Serrano, Jonathan Trent, Eric D. Tetzlaff, Teresa Zhou, Sebastian Bauer, Maria Roche, Tracy Havnaer
Data analysis and interpretation: Sarah N. Abaricia, Michelle A. Angelis, Kathleen Polson, Robin L. Jones, Yoon‐Koo Kang, Richard F. Riedel, Patrick Schöffski, César Serrano, Jonathan Trent, Tuan Dong Si, Teresa Zhou, Ashely Doyle, Sebastian Bauer, Maria Roche
Manuscript writing: Cissimol P. Joseph, Sarah N. Abaricia, Michelle A. Angelis, Kathleen Polson, Robin L. Jones, Yoon‐Koo Kang, Richard F. Riedel, Patrick Schöffski, César Serrano, Jonathan Trent, Eric D. Tetzlaff, Tuan Dong Si, Teresa Zhou, Ashley Doyle, Sebastian Bauer, Maria Roche, Tracy Havnaer
Manuscript approval: Cissimol P. Joseph, Sarah N. Abaricia, Michelle A. Angelis, Kathleen Polson, Robin L. Jones, Yoon‐Koo Kang, Richard F. Riedel, Patrick Schöffski, César Serrano, Jonathan Trent, Eric D. Tetzlaff, Tuan Dong Si, Teresa Zhou, Ashley Doyle, Sebastian Bauer, Maria Roche, Tracy Havnaer
Disclosures
Cissimol P. Joseph: Deciphera (C/A); Kathleen Polson: Deciphera (C/A); Robin L. Jones: Blueprint Medicines Corporation (C/A); Richard F. Riedel: Limbguard, LLC (OI [spouse]), AADi, AROG, Daiichi‐Sankyo, Epizyme, GlaxoSmithKline, Karyopharm, Ignyta, Immune Design, Eli Lilly & Co., NanoCarrier, Novartis, Oncternal, Philogen, Plexxikon, Roche, SpringWorks, Threshold, Tracon (RF [institutional]), Bayer, Blueprint Medicines Corporation, EISAI, EMD Serono, Janssen, Eli Lilly & Co., Ignyta, NanoCarrier, SpringWorks (C/A); Patrick Schöffski: Blueprint Medicines Corporation, Genmab (RF [institutional]), Deciphera, Ellipses (C/A), Exelixis (Other: travel support), Plexxikon, Transgene (Other: independent data monitoring service); César Serrano: Blueprint Medicines Corporation, Deciphera (SAB), Blueprint Medicines Corporation, Bayer (H), Deciphera, Pfizer (RF), Pfizer, Bayer, Eli Lilly & Co., Novartis, Pharmamar (Other: travel expenses); Jonathan Trent: Blueprint Medicines Corporation, Deciphera, Daiichi‐Sankyo, Epizyme, Agios (C/A); Eric D. Tetzlaff: Blueprint Medicines Corporation (C/A, H), Association of PAs in Oncology (RF); Tuan Dong Si: Blueprint Medicines Corporation (E, OI); Ashley Doyle: Blueprint Medicines Corporation (E, OI); Sebastian Bauer: Blueprint Medicines Corporation, Incyte, Novartis, (RF), Blueprint Medicines Corporation, ADC Therapeutics, Eli Lilly & Co., Novartis, Daichii‐Sankyo, Plexxikon, Deciphera, Exelixis, Janssen‐Cilag (SAB), Novartis, Pfizer, Bayer, Eli Lilly & Co., Pharmamar, GlaxoSmithKline (Other: CME‐related honoraria); Maria Roche: Blueprint Medicines Corporation (E). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board; (CRH) CME‐related honoraria; (RTE) Reimbursed travel expenses; (TS) Travel support; (IDMS) Independent data monitoring services
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Table S1 Protocol‐specified dose‐modification guidelines
Table S2. Presentation of Common Terminology Criteria for Adverse Events (CTCAE) [1] cognitive effects adverse events
Table S3. All‐cause AEs by age among patients starting with avapritinib 300 mg
Table S4. Correlation between patient characteristics and cognitive effects. Univariable logistic regression of cognitive events in patients who started on avapritinib 300 mg
Table S5. Summary of cognitive effects in patients starting with avapritinib 300 mg
Acknowledgments
We thank the patients and their families, as well as the global network of investigators, research nurses, study coordinators, and operations staff. Medical writing support was provided by Miriam Cohen, Ph.D., and editorial support by Michelle Seddon, both of Paragon, Knutsford, U.K., supported by Blueprint Medicines Corporation. Blueprint Medicines Corporation follows all current policies established by the International Committee of Medical Journal Editors and Good Publication Practice guidelines (Link). The sponsor was involved in the study design and collection, analysis, and interpretation of data, as well as data checking of information provided in the manuscript. However, ultimate responsibility for opinions, conclusions, and data interpretation lies with the authors. The NAVIGATOR study (NCT02508532) was funded by Blueprint Medicines Corporation, Cambridge, Massachusetts. Dr Teresa Zhou is currently affiliated with Kiniksa Pharmaceuticals Corporation, Lexington, Massachusetts, USA.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Demetri GD, von Mehren M, Antonescu CR et al. NCCN Task Force report: Update on the management of patients with gastrointestinal stromal tumors. J Natl Compr Canc Netw 2010;8(suppl 2):S1–41; quiz S42–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ducimetiere F, Lurkin A, Ranchere‐Vince D et al. Incidence of sarcoma histotypes and molecular subtypes in a prospective epidemiological study with central pathology review and molecular testing. PLoS One 2011;6:e20294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Soreide K, Sandvik OM, Soreide JA et al. Global epidemiology of gastrointestinal stromal tumours (GIST): A systematic review of population‐based cohort studies. Cancer Epidemiol 2016;40:39–46. [DOI] [PubMed] [Google Scholar]
- 4. Nilsson B, Bumming P, Meis‐Kindblom JM et al. Gastrointestinal stromal tumors: The incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era–A population‐based study in western Sweden. Cancer 2005;103:821–829. [DOI] [PubMed] [Google Scholar]
- 5. Casali PG, Abecassis N, Aro HT et al. Gastrointestinal stromal tumours: ESMO‐EURACAN clinical practice guidelines for diagnosis, treatment and follow‐up. Ann Oncol 2018;29:iv68–iv78. [DOI] [PubMed] [Google Scholar]
- 6. Rammohan A, Sathyanesan J, Rajendran K et al. A gist of gastrointestinal stromal tumors: A review. World J Gastrointest Oncol 2013;5:102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. National Comprehensive Cancer Network . NCCN clinical practice guidelines in oncology: Soft tissue sarcoma (version 6.2019). Available at https://www.nccn.org/professionals/physician_gls/pdf/sarcoma.pdf. Accessed March 5, 2020.
- 8. Smith BD, Kaufman MD, Lu WP et al. Ripretinib (DCC‐2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug‐resistant KIT and PDGFRA variants. Cancer Cell 2019;35:738–751.e739. [DOI] [PubMed]
- 9. Demetri GD, von Mehren M, Blanke CD et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347:472–480. [DOI] [PubMed] [Google Scholar]
- 10. Demetri GD, van Oosterom AT, Garrett CR et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006;368:1329–1338. [DOI] [PubMed] [Google Scholar]
- 11. Demetri GD, Reichardt P, Kang Y‐K et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet 2013;381:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deciphera Pharmaceuticals LLC. Qinlock (ripretinib). Prescribing information. 2020. Available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213973s000lbl.pdf. Accessed June 10, 2020. [Google Scholar]
- 13. Maleddu A, Pantaleo MA, Nannini M et al. The role of mutational analysis of KIT and PDGFRA in gastrointestinal stromal tumors in a clinical setting. J Transl Med 2011;9:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schoffski P, Wozniak A, Schoffski O et al. Overcoming cost implications of mutational analysis in patients with gastrointestinal stromal tumors: A pragmatic approach. Oncol Res Treat 2016;39:811–816. [DOI] [PubMed] [Google Scholar]
- 15. Serrano C, Mariño‐Enríquez A, Tao DL et al. Complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib‐resistant gastrointestinal stromal tumours. Br J Cancer 2019;120:612–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Y, Call J. Mutational testing in gastrointestinal stromal tumor. Curr Cancer Drug Targets 2019;19:688–697. [DOI] [PubMed] [Google Scholar]
- 17. Hsu CC, Wu CE, Chen JS et al. Imatinib escalation or sunitinib treatment after first‐line imatinib in metastatic gastrointestinal stromal tumor patients. Anticancer Res 2014;34:5029–5036. [PubMed] [Google Scholar]
- 18. Florindez J, Trent J. Low frequency of mutation testing in the United States: An analysis of 3866 GIST patients. Am J Clin Oncol 2020;43:270–278. [DOI] [PubMed] [Google Scholar]
- 19. Dematteo RP, Heinrich MC, El‐Rifai WM et al. Clinical management of gastrointestinal stromal tumors: Before and after STI‐571. Hum Pathol 2002;33:466–477. [DOI] [PubMed] [Google Scholar]
- 20. Yoo C, Ryu MH, Jo J et al. Efficacy of imatinib in patients with platelet‐derived growth factor receptor alpha‐mutated gastrointestinal stromal tumors. Cancer Res Treat 2016;48:546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cassier PA, Fumagalli E, Rutkowski P et al. Outcome of patients with platelet‐derived growth factor receptor alpha‐mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin Cancer Res 2012;18:4458–4464. [DOI] [PubMed] [Google Scholar]
- 22. Blueprint Medicines Corporation . Ayvakit (avapritinib). Prescribing information. 2020. Available at https://www.blueprintmedicines.com/uspi/AYVAKIT.pdf. Accessed March 6, 2020.
- 23. Evans EK, Gardino AK, Kim JL et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci Transl Med 2017;9:eaao1690. [DOI] [PubMed] [Google Scholar]
- 24. Giampieri R, Prete MD, Prochilo T et al. Off‐target effects and clinical outcome in metastatic colorectal cancer patients receiving regorafenib: The TRIBUTE analysis. Sci Rep 2017;7:45703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bhullar KS, Lagaron NO, McGowan EM et al. Kinase‐targeted cancer therapies: Progress, challenges and future directions. Mol Cancer 2018;17:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Joseph CP, Abaricia SN, Angelis MA et al. Avapritinib for the treatment of GIST: Analysis of efficacy, safety, and patient management strategies at the recommended phase 2 dose. Presented at: Connective Tissue Oncology Society Annual Meeting; 2019; Tokyo, Japan. [Google Scholar]
- 27. Heinrich MC, Jones RL, von Mehren M et al. Avapritinib in advanced PDGFRA D842V‐mutant gastrointestinal stromal tumour (NAVIGATOR): A multicentre, open‐label, phase 1 trial. Lancet Oncol 2020;21:935–946. [DOI] [PubMed] [Google Scholar]
- 28. Seiler S, Kosse J, Loibl S et al. Adverse event management of oral mucositis in patients with breast cancer. Breast Care (Basel) 2014;9:232–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moreau P, Dimopoulos MA, Richardson PG et al. Adverse event management in patients with relapsed and refractory multiple myeloma taking pomalidomide plus low‐dose dexamethasone: A pooled analysis. Eur J Haematol 2017;99:199–206. [DOI] [PubMed] [Google Scholar]
- 30. Ikeda M, Kobayashi M, Tahara M et al. Optimal management of patients with hepatocellular carcinoma treated with lenvatinib. Expert Opin Drug Saf 2018;17:1095–1105. [DOI] [PubMed] [Google Scholar]
- 31. McShane TM, Wolfe TA, Ryan JC. Updates on managing advanced breast cancer with palbociclib combination therapy. Ther Adv Med Oncol 2018;10:1758835918793849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Si X, Zhang L, Wang H et al. Management of anlotinib‐related adverse events in patients with advanced non‐small cell lung cancer: Experiences in ALTER‐0303. Thorac Cancer 2019;10:551–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jones RL, Serrano C, von Mehren M et al. Avapritinib in unresectable or metastatic PDGFRA D842V‐mutant gastrointestinal stromal tumours: long‐term efficacy and safety data from the NAVIGATOR phase 1 trial. Eur J Cancer 2020; In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dematteo RP, Ballman KV, Antonescu CR et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: A randomised, double‐blind, placebo‐controlled trial. Lancet 2009;373:1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shitara K, Doi T, Dvorkin M et al. Trifluridine/tipiracil versus placebo in patients with heavily pretreated metastatic gastric cancer (TAGS): A randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol 2018;19:1437–1448. [DOI] [PubMed] [Google Scholar]
- 36. Chamberlain F, Farag S, Williams‐Sharkey C et al. Toxicity management of regorafenib in patients with gastro‐intestinal stromal tumour (GIST) in a tertiary cancer centre. Clin Sarcoma Res 2020;10:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nakamura M, Yamada T, Ishiyama S et al. Regorafenib dose escalation therapy for patients with refractory metastatic colorectal cancer (RECC Study). J Clin Oncol 2020;38(suppl 4):116a. [DOI] [PubMed] [Google Scholar]
- 38. Gilreath JA, Tchertanov L, Deininger MW. Novel approaches to treating advanced systemic mastocytosis. Clin Pharmacol 2019;11:77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Feki J, Marrekchi G, Boudawara T et al. Subdural hematoma during therapy of gastro‐intestinal stromal tumor (GIST) with imatinib mesylate. Gulf J Oncolog 2015;1:92–95. [PubMed] [Google Scholar]
- 40. Kim MS, Lee DH, Lee YR et al. A case of subdural hematoma in patient with chronic myeloid leukemia treated with high‐dose imatinib mesylate. Korean J Hematol 2010;45:73–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Theodotou CB, Shah AH, Ivan ME et al. Subdural hematoma in a patient taking imatinib for GIST: A case report and discussion of risk with other chemotherapeutics. Anticancer Drugs 2016;27:259–263. [DOI] [PubMed] [Google Scholar]
- 42. Gotlib J, Radia DH, George TI et al. Avapritinib induces responses in patients with advanced systemic mastocytosis (AdvSM), regardless of prior midostaurin therapy. Presented at: 25th European Hematology Association Congress; 2020; Virtual Format.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Table S1 Protocol‐specified dose‐modification guidelines
Table S2. Presentation of Common Terminology Criteria for Adverse Events (CTCAE) [1] cognitive effects adverse events
Table S3. All‐cause AEs by age among patients starting with avapritinib 300 mg
Table S4. Correlation between patient characteristics and cognitive effects. Univariable logistic regression of cognitive events in patients who started on avapritinib 300 mg
Table S5. Summary of cognitive effects in patients starting with avapritinib 300 mg