

Abstract
Benign hepatosteatosis, affected by lipid uptake, de novo lipogenesis and fatty acid (FA) oxidation, progresses to non-alcoholic steatohepatitis (NASH) on stress and inflammation. A key macronutrient proposed to increase hepatosteatosis and NASH risk is fructose, whose excessive intake causes intestinal barrier deterioration and endotoxemia. However, how fructose triggers these alterations and their role in hepatosteatosis and NASH pathogenesis remain unknown. Here we show, using mice, that microbiota-derived Toll-like receptor (TLR) agonists promote hepatosteatosis without affecting fructose-1-phosphate (F1P) and cytosolic acetyl-CoA. Activation of mucosal-regenerative gp130 signaling, administration of the YAP-induced matricellular protein CCN1 or expression of the anti-microbial Reg3β peptide counteract fructose and endoplasmic reticulum stress-dependent barrier deterioration and subsequent endotoxemia. Endotoxin engages TLR4 to trigger TNF production by liver macrophages, thereby inducing lipogenic enzymes that convert F1P and acetyl-CoA to FA in both mouse and human hepatocytes.
Keywords: NASH, NAFLD, de novo lipogenesis, fructose, gut-liver axis, TNF
Non-alcoholic steatohepatitis (NASH), a severe manifestation of non-alcoholic fatty liver disease (NAFLD) which affects 30% of adult Americans1, is a leading cause of liver failure, cirrhosis and cancer2. NAFLD starts as simple steatosis with little liver damage or fibrosis, whose progression to NASH depends on multiple parallel hits acting in concert with hepatosteatosis. The latter can be due to increased lipid import, enhanced de novo lipogenesis (DNL), or impaired β oxidation3. Proposed hits include gut-derived inflammatory signals and endoplasmic reticulum (ER) stress3–7. Having established a role for ER stress and TNF signaling in NASH pathogenesis8,9, we sought to unravel the role of gut-derived inflammatory signals linked to fructose-induced barrier deterioration. Fructose, whose US consumption had increased several thousand-fold, is a key macronutrient that may contribute to NASH development10. Fructose causes barrier deterioration through a heretofore unknown mechanism entailing downregulation of tight junction proteins (TJP) in animal subjects and pediatric patients11–13. Fructose also triggers dysbiosis14 and its metabolism by gut bacteria generates short chain fatty acids (SCFA)15, proposed to improve barrier function16,17. Unlike glucose, which is generated by hydrolysis of starch and is metabolically mobilized by the highly regulated enzyme glucokinase, fructose is phosphorylated by constitutively active fructokinase (also called ketohexokinase/KHK)18, expressed in hepatocytes where it stimulates DNL and hepatic fat deposition10,19. DNL depends on two key enzymes, acetyl CoA carboxylase (ACC) and fatty acid synthase (FAS), whose expression is fructose induced20. Here we show that gut barrier deterioration and subsequent endotoxemia trigger hepatic induction of ACC1 and FAS and other SREBP1-activated lipogenic genes to upregulate DNL without an effect on KHK-dependent fructose-1 phosphate (F1P) and cytosolic acetyl-CoA accumulation. Fructose-elicited endotoxemia activates Toll-like receptor (TLR) signaling in liver macrophages, whose blockade via myeloid-specific MyD88 ablation prevents TNF, ACC1 and FAS induction and attenuates hepatosteatosis. Conversely, addition of TNF to cultured human hepatocytes enhances fructose conversion to lipids. Correspondingly, genetic and pharmacological manipulations that prevent ER stress-dependent fructose-induced barrier deterioration and endotoxemia block fructose-stimulated DNL, hepatosteatosis and the onset of steatohepatitis and cancer.
Results
Fructose stimulates DNL and induces steatosis, NASH and HCC
Transgenic (Tg) MUP-uPA mice expressing high amounts of urokinase plasminogen activator (uPA) from a liver-specific promoter demonstrate a causal role for hepatocyte ER stress in NASH pathogenesis8. Although MUP-uPA mice older than 8 weeks kept on normal chow diet (NCD) do not display obvious hepatic abnormalities, when given energy-rich high fat diet (HFD) they manifest classical NASH signs within 3-4 months and progress to hepatocellular carcinoma (HCC) by 9-10 months. Prevention of hepatocyte ER stress blocks NASH development8 by inhibiting IRE-1 dependent translation and subsequent activation of caspase-29. Induction of Caspase-2 mRNA requires TNF signaling, whose blockade also blocks NASH development8,9. How HFD induces TNF secretion by liver macrophages is unknown. We used the MUP-uPA model and parental BL6 mice to investigate whether fructose-induced DNL elicits NASH and subsequent HCC development. Both MUP-uPA and BL6 mice were given isocaloric low-fat and carbohydrate-rich diets containing either fructose or cornstarch (CS), a polymer that is readily hydrolyzed to glucose. Whereas high fructose diet (HFrD) contains 10% CS and 60% fructose, CS diet (CSD) derives 70% of its calories from CS. Although diet consumption was similar, HFrD, but not CSD, induced hepatosteatosis and increased liver triglycerides (TG) in both strains (Fig. 1a, b and Extended Data Fig. 1a–c). Steatohepatitis, ballooning degeneration and liver fibrosis were apparent in 6-month-old HFrD-fed MUP-uPA mice, but BL6 mice only showed benign steatosis. HFrD feeding did not cause adiposity and white adipose tissue (WAT) was somewhat smaller in HFrD-fed animals (Extended Data Fig. 1d, e). HFrD feeding induced colon shortening, indicating intestinal inflammation (Extended Data Fig. 1f) and caused more insulin resistance than CSD, as shown by glucose tolerance tests (GTT), while mildly decreasing energy expenditure in 6-month-old mice kept in metabolic cages, with similar weight gain (Extended Data Fig. 1g–j). Like insulin-resistance, non-fasting insulin was higher in HFrD-fed mice (Extended Data Fig. 1k). After 12 months, HFrD-fed MUP-uPA mice exhibited 3-fold more HCC nodules that were larger than those in CSD-fed mice, resulting in 10-fold higher tumor burden (Fig. 1c, d). HFrD also enhanced diethyl nitrosamine (DEN)-induced HCC in BL6 mice (Fig. 1e). Most tumors in HFrD-fed mice were poorly differentiated, steatotic HCCs (Extended Data Fig. 1l).
Figure 1. High-fructose diet induces hepatosteatosis and tumorigenesis.
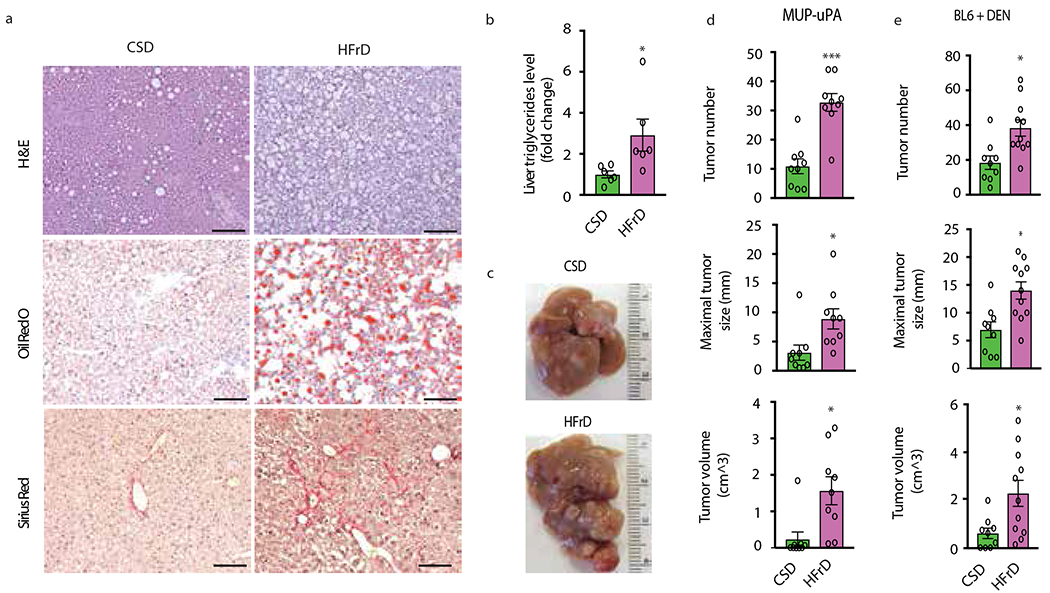
(a) MUP-uPA male mice were fed CSD or HFrD starting at 6 weeks of age. After 5 months, formalin-fixed, paraffin-embedded (FFPE) and frozen sections of non-tumor liver tissue were H&E or Oil Red O (ORO) stained to reveal general histology and lipid droplets (n = 6 males per group). Collagen deposition was visualized by Sirius Red staining of frozen sections. Scale bars, 100 μm. (b) Liver samples were analyzed for total triglyceride (TG) content (n=6 males per group), which is presented as fold- change relative to CSD-fed mice. (c) Representative liver morphology of CSD- and HFrD-fed MUP-uPA mice (n=9 and 11 males for the CSD and HFrD groups, respectively). (d) HCC development in CSD- or HFrD-fed MUP-uPA mice (n=9). (e) HCC in DEN- challenged BL6 mice fed CSD (n=9 males per group) or HFrD (n=11 males per group). Panels b, d and e show mean ± SEM determined by two-sided Student’s t test, *P < 0.05, ***P < 0.001.
The cytosolic pool of acetyl-CoA, needed to initiate DNL, was two-fold higher in HFrD- than CSD-fed MUP-uPA mice (Extended Data Fig. 2a). Prolonged HFrD consumption markedly upregulated hepatic mRNAs for SREBP1c (Srebf1), carbohydrate-responsive element-binding protein (ChREBP/Mixipl) and the SREBP1-regulated enzymes ACC1/Acaca and FAS/Fasn, while downregulating mRNAs coding for FA oxidizing enzymes (Fig. 2a and Extended Data Fig. 2b, c). Liver mRNAs for inflammatory cytokines and chemokines, including TNF, were elevated (Fig. 2b). Continuous HFrD feeding dramatically increased ACC1 and FAS amounts (Fig. 2c), and upregulated hepatic DNL, elevating C16:0 and C18:0 synthesis by 2-fold relative to CSD (Fig. 2d). Short-term (48-hour) HFrD feeding, however, did not elevate FA synthesis in liver or jejunum, although it strongly increased F1P amounts in both tissues (Extended Data Fig. 2d–f). Inflammatory cytokines, chemokines, and lipogenic mRNAs, other than Acaca, were not induced and ACC1 protein was barely elevated after a 48-hour exposure (Extended Data Fig. 2g).
Figure 2. HFrD induces inflammation, lipogenic gene expression and de novo lipogenesis.
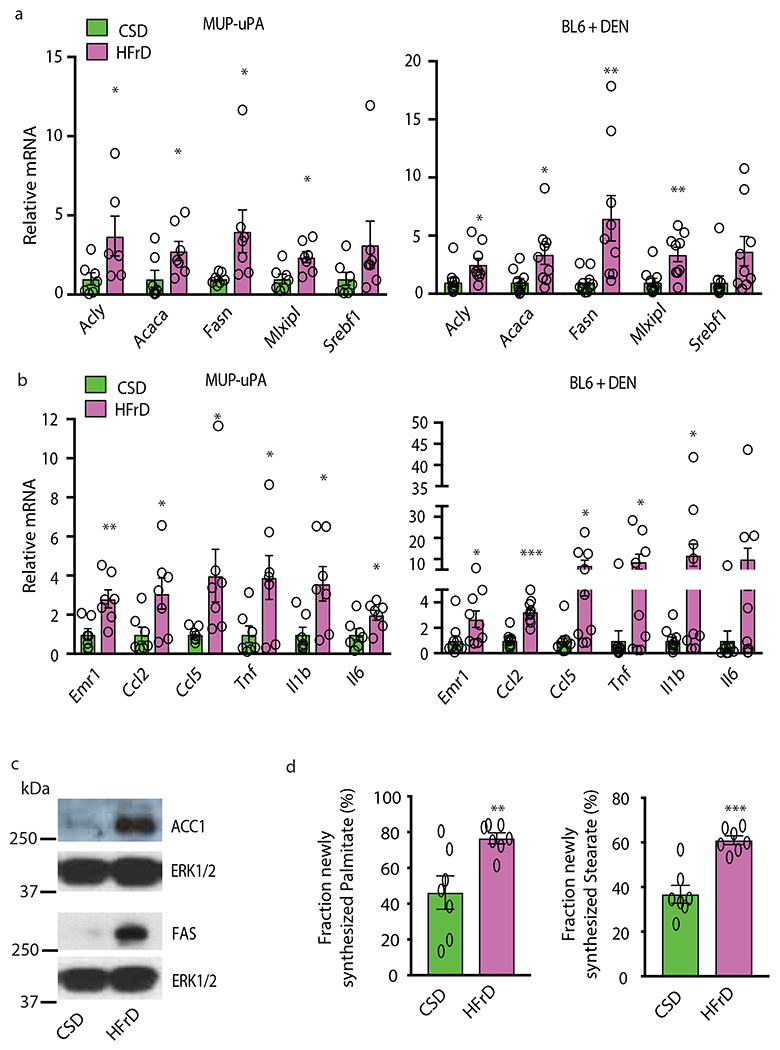
(a and b) Expression of lipogenic (a) and inflammatory (b) genes in MUP-uPA males (n=7 per group) and DEN-challenged BL6 (n=10 per CSD and n=9 per HFrD group) males fed CSD or HFrD for 5 months starting at 6 weeks. (c) Immunoblot (IB) analysis of ACC1 and FAS in livers of above MUP-uPA mice (representative of n=7). (d) Measurement of newly synthesized fatty acids in livers of above MUP-uPA mice using deuterated water (D2O) as a tracer (n =7 per group). Panels a, b and d show mean ± SEM determined by two-sided Student’s t test, *P < 0.05, **P < 0.01, ***P < 0.001. Benjamini-Hochberg FDR adjustment for P-value was performed on data presented in panels a and b.
Fructose-induced barrier deterioration and intestinal epithelial ER stress
Although short term HFrD feeding did not alter intestinal permeability, assessed by fecal albumin content and FITC-dextran translocation (Extended Data Fig. 2h), prolonged HFrD feeding caused intestinal barrier deterioration (Fig.3a). Fecal albumin elevation, however, was prevented by treatment with an antibiotics (Abx) cocktail21 that reduces fecal bacteria by more than 99.5%. Prolonged HFrD feeding reduced TJP mRNAs coding for Tjp2, occludin, and different claudins (including seal-forming claudin 3) in large and small intestine (Fig. 3b, c). Although some of these effects, especially in colon, were reversed by Abx treatment, most downregulated jejunal TJP mRNAs remained suppressed and occludin and claudin 1 were only partially restored (Extended Data Fig. 3a–c), Moreover, incubation of microbiota-free colonic organoids with 5-20 mM fructose decreased numerous TJP mRNAs (Fig. 3d). Il22 mRNA and some IL-22-induced antimicrobial protein (AMP) mRNAs (Lcn2, S100a9) were increased in colons of HFrD- fed mice (Extended Data Fig. 3d), presumably as an attempt at barrier restoration22, but Reg3b and Reg3g mRNAs were suppressed by HFrD-feeding. Abx treatment attenuated Il22, Lcn2, and S100a9 mRNA induction. Previous studies demonstrated that colonic inflammation upregulates S100 mRNAs but downregulates Reg genes23. Administration of a 30% wt:vol fructose solution, accounting for 32% of daily caloric intake in NCD-fed BL6 mice, also reduced TJP mRNAs, caused colonic shortening and increased FITC-dextran translocation (Extended Data Fig 3e, f). In contrast, a 30% sucrose drink did not reduce colon length or TJP mRNAs and barely increased ER stress markers (Extended Data Fig. 3g–i). These results are consistent with the weaker effect of sucrose vs. fructose on hepatic steatosis24
Figure 3. Fructose causes barrier deterioration, decreased tight junction protein expression and ER stress.
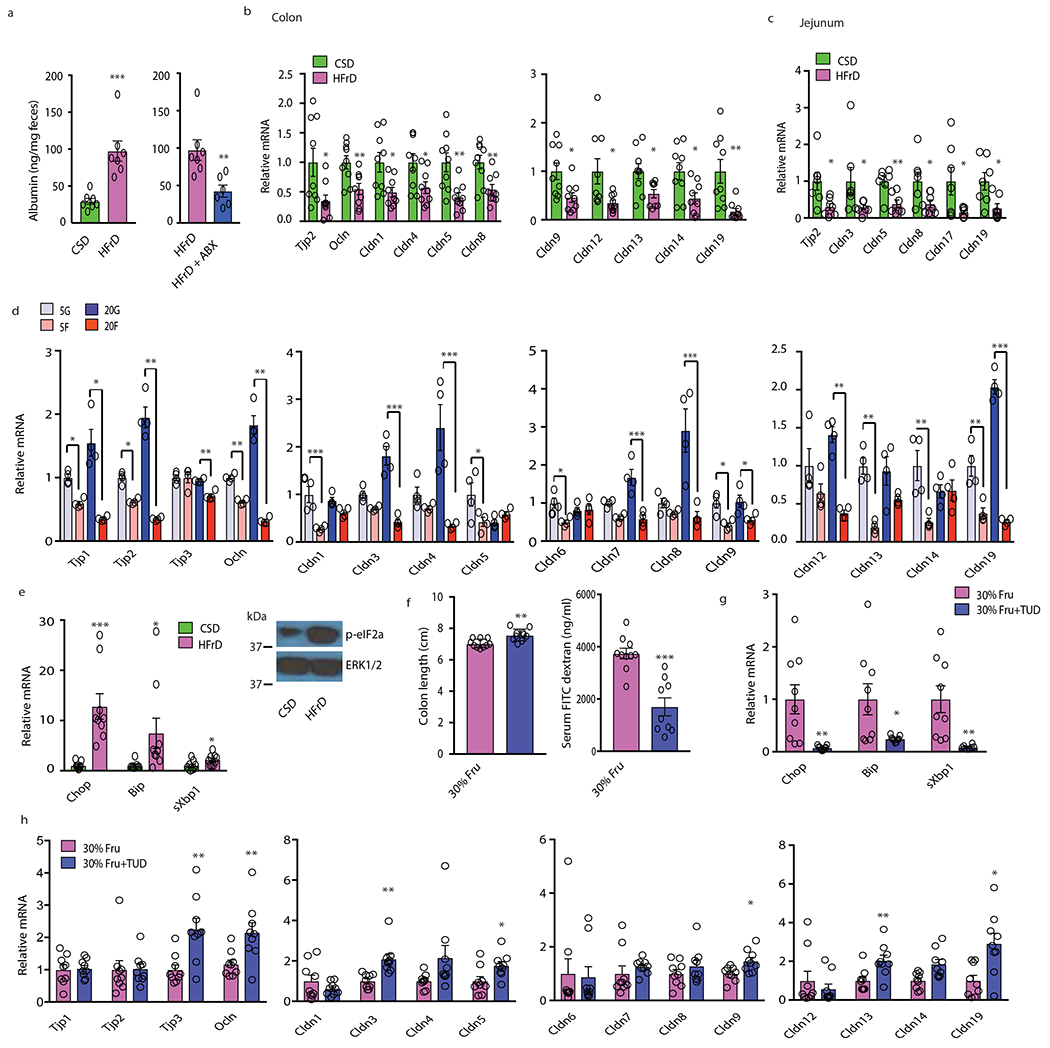
(a) Fecal albumin concentrations in MUP-uPA male mice kept on CSD (n=7), HFrD (n=7) or HFrD ± Abx (n=6). (b to c) Tight junction mRNAs in colonic (b) and jejunal (c) mucosa of CSD (b, n=9; c, n =7) or HFrD-fed (5 months; b, n = 9; c, n = 7) MUP-uPA males. (d) Tight junction mRNAs in BL6 enteroids incubated with 5 mM or 20 mM glucose (G) or fructose (F) (n=4 different mice). This experiment has been performed 2 times with similar results. (e) Expression of inflammatory and ER stress markers in colonic mucosa of above mice (n=9 each group) and a representative phospho-eIF2α blot of mucosal lysates (n=6 per group). (f) Colon length and serum FITC-dextran in male mice given 30% fructose drink and treated −/+ TUDCA (n=10 for -TUDCA and n=9 for +TUDCA group). (g) Mucosal ER stress marker mRNAs in above mice (n=9 per group). (h) Tight junction mRNAs in colonic mucosa of above mice (n=9 per group). Panels a-c, e-h show mean ± SEM determined by two-sided Student’s t test. In panel d mean ± SEM were determined by one-way ANOVA and Tuckey’s multiple - comparison test. *P < 0.05, **P < 0.01, ***P < 0.001. Benjamini-Hochberg FDR adjustment for P-value was performed on data presented in panels b-c, e and g-h.
Colon shortening is a sign of inflammation. To explore a plausible cause of mucosal inflammation, which can downregulate TJPs25, we posited that the fructose metabolite F1P interferes with N-glycosylation as reported in hereditary fructose intolerance26. Defective N-glycosylation triggers ER stress, a source of mucosal inflammation27, and impairs TJP transport and assembly28. Indeed, HFrD feeding strongly induced the ER stress markers/unfolded protein response (UPR) effectors Chop, Bip/Grp78, and spliced Xbp1 and increased eIF2α phosphorylation in intestinal mucosa along with elevated inflammatory markers (Fig. 3e). In vitro incubation of colonic organoids with fructose, but not glucose, also induced Chop and sXbp1 mRNAs and this was attenuated by glucose addition (Extended Data Fig. 3j), suggesting that interference with N-glycosylation may be involved. To explore the role of F1P in this response we incubated organoids with the KHK inhibitor 420640. This abrogated Chop and sXbp1 mRNA induction by fructose and diminished the decrease in most TJP mRNAs (Extended Data Fig. 3k, l). A strong protective effect was seen upon organoid incubation with the chemical chaperon tauroursodeoxycholic acid (TUDCA) (Extended Data Fig. 3m), which reduces liver ER stress and steatosis in MUP-uPA mice8. Importantly, TUDCA treatment of BL6 mice given a 30% fructose drink attenuated colon shortening, reduced FITC-dextran translocation, inhibited ER stress marker induction and prevented TJP mRNA downregulation (Fig. 3f–h).
HFrD induces liver inflammation and myeloid cell TLR signaling
Exploratory transcriptomic analysis independently validated by immunoblot (IB) and RT-PCR analyses revealed marked differences in innate immunity- and inflammation- related transcripts between tumor-free 6-month-old CSD- and HFrD-fed livers (Fig. 4 a and Extended Data Fig. 4a, b). HCC nodules, however, exhibited fewer diet-related transcriptomic changes, especially in HCC-related genes (Extended Data Fig. 4c,d), which were essentially identical to those upregulated in HFD-induced HCC29. HCC-specific genes were elevated in both CSD- and HFrD-associated tumors from 9-month-old mice relative to non-tumor liver, further demonstrating that HFrD mainly altered hepatic pathophysiology prior to tumor onset. Gene ontology enrichment analysis of the non-tumor transcriptome revealed large changes in genes involved in response to wounding, wound healing, cell adhesion, innate immunity, and responses to molecules of bacterial origins (Extended Data Fig. 4e). IB analysis confirmed elevated TLR2 and 4, MyD88, NLRP3 and STAT3 phosphorylation in HFrD-fed livers, changes that were abrogated by Abx treatment (Fig.4b).
Figure 4. HFrD-induced upregulation of liver TLR and cytokine signaling and role of myeloid MyD88.
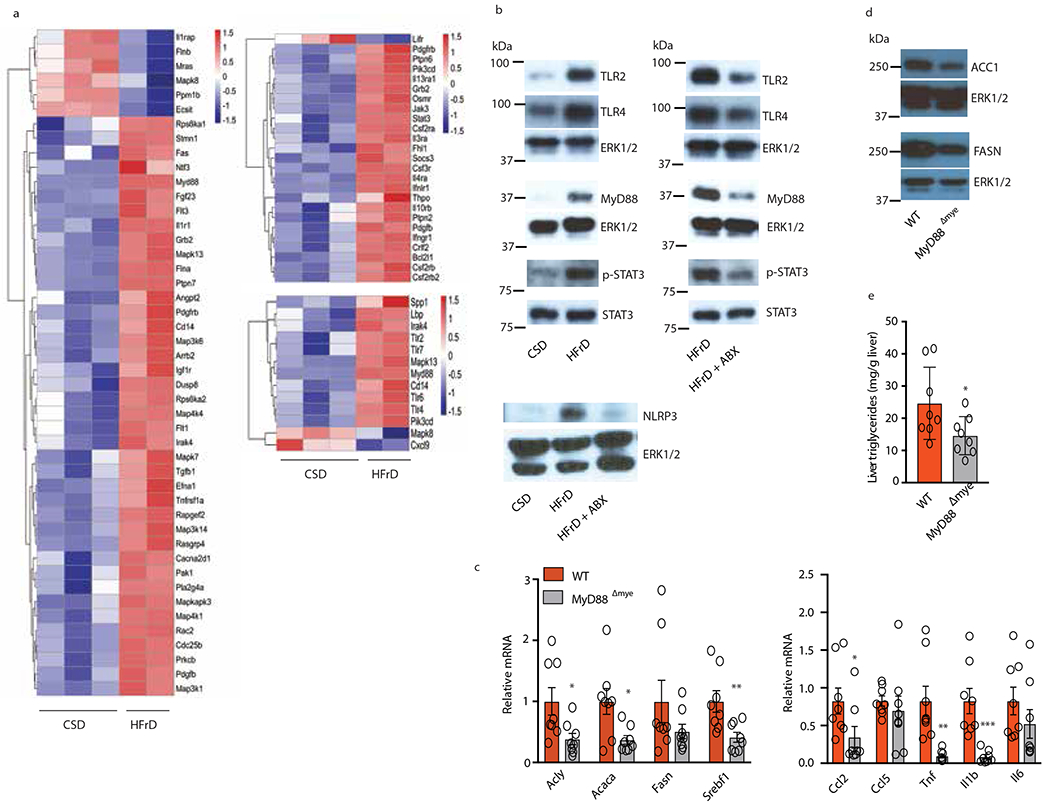
(a) Heatmaps depicting differential expression of genes related to inflammation wound healing and innate immunity in livers of CSD- or HFrD-fed (5 months) male MUP-uPA mice (CSD, n=3; HFrD, n=2). (b) IB analysis of TLRs, MyD88, phosphorylated and total STAT3 and NLRP3 in livers of 6-month-old MUP-uPA mice fed CSD or HFrD or HFrD ± Abx for 5 months (n=7 per group). (cand d) Expression of lipogenic and inflammatory genes (n=8 per group) and representative IB analysis of ACC1 and FAS (n=6) (d) in livers of male Myd88Δmye and W mice fed HFrD for 5 months. (e) Liver triglyceride content in above mice (n=8; mg/g tissue). Panels c and e show mean ± SEM determined by two-sided Student’s t test and Benjamini-Hochberg FDR adjustment for P-values *P < 0.05, **P < 0.01.
Increased TLR2, TLR4, MyD88 and NLRP3 expression most likely took place in liver-recruited macrophages. Accordingly, we ablated MyD88 in myeloid cells and examined the impact on HFrD-induced DNL controlling enzymes, inflammatory cytokines and hepatosteatosis. Although myeloid MyD88 ablation only inhibited Tnf, Il1β and Ccl2 mRNA induction with no effect on Ccl5 and Il6 mRNAs, it substantially decreased expression of DNL-related proteins and reduced hepatic TG accumulation in HFrD-fed mice (Fig. 4c–e).
Consistent with elevated TLR expression and the protective effect of macrophage MyD88 ablation, prolonged HFrD feeding increased circulating endotoxin in MUP-uPA and BL6 mice, whereas Abx administration inhibited endotoxemia and attenuated HFrD-stimulated liver tumorigenesis (Extended Data Fig. 5a, b). Abx treatment also blocked hepatosteatosis, TG accumulation and induction of DNL-related enzymes, inflammatory cytokines and chemokines (Extended Data Fig. 5c–g). Importantly, Abx treatment did not reduce liver F1P and had a marginal effect on cytosolic acetyl-CoA in HFrD-fed mice (Extended Data Fig. 5h, i), suggesting it does not affect fructolysis. Abx treatment, however, prevented HFrD-induced glucose intolerance and reduced non-fasting serum insulin without affecting body or WAT weight; it also reduced liver fibrosis and abrogated colon shortening (Extended Data Fig. 5j–n).
All HFrD-elicited phenotypic changes, including hepatosteatosis and elevated DNL-related enzymes, upregulation of innate immune proteins and inflammatory mediators and downregulation of TJP genes, were also observed in 6-month-old BL6 mice and were inhibited by Abx (Extended Data Fig. 6a–g). Thus, fructose-induced barrier deterioration and subsequent DNL and steatohepatitis are independent of genetic background or carcinogen-induced genetic alterations. Fructose drink (30%) administration also enhanced hepatosteatosis and upregulated liver mRNAs encoding inflammatory chemokines, cytokines and DNL enzymes in BL6 mice (Extended Data Fig. 7a, b). Fructose drink consumption, however, increased body and WAT weight, rendering dissociation of its liver-specific effects from those caused by increased caloric intake and peripheral obesity somewhat difficult (Extended Data Fig. 7c). To mimic human Western diet that includes both fat and fructose and reduce the caloric contribution of fructose from 32% in the above experiment to 20.9% an amount equivalent to that consumed by the 95th percentile of American adolescents30, we placed BL6 mice on HFD composed of 59% fat, 15% protein and 26% carbohydrates, and either regular water or a 30% fructose drink. Despite its reduced overall caloric contribution, fructose still increased body, WAT and liver weight, augmented hepatosteatosis and induced liver mRNAs encoding inflammatory chemokines, cytokines and DNL enzymes, while downregulating TJP mRNAs and increasing FITC-dextran translocation (Extended Data Fig. 7d–h).
Barrier restoration inhibits DNL and prevents steatohepatitis
Tight junction loss activates YAP and TAZ, closely related transcriptional activators that stimulate tissue repair31. Indeed, incubation of colonic organoids with fructose rapidly induced the YAP targets Ctgf and Ccne1 (Extended Data Fig. 8a). Mucosal Ctgf and Ccne1 mRNAs were also upregulated in HFrD-fed mice, but this chronic response was attenuated relative to the acute response manifested by fructose-incubated organoids (Extended Data Fig. 8b). Ccne1 encodes the matricellular protein CCN1, whose administration promotes barrier repair via integrin-mediated IL-6 incuction32, whereas engagement of IL-6 signal transducer (IL6ST/gp130) promotes mucosal repair via STAT3 and YAP33. Congruently, IEC-specific expression of constitutively active gp130 variant (gp130Act)33 conferred partial resistance to fructose-induced TJP mRNA downregulation in organoids (Extended Data Fig. 8c). IEC- specific gp130Act expression was even more effective in vivo, restoring Reg3b, Reg3g, Tjp mRNAs and claudin-1 and inhibiting HFrD-induced FITC-dextran translocation and endotoxemia (Extended Data Fig. 8d–h). Remarkably, MUP-uPA/gp130Act mice were resistant to fructose-induced hepatic tumorigenesis and failed to upregulate hepatic mRNAs encoding inflammatory and lipogenic proteins (Fig. 5a, b). Liver DNL rates were lower in HFrD-fed MUP-uPA/gp130Act mice than in MUP-uPA mice (Fig. 5c), but F1P accumulation in either liver or jejunum was not reduced (Extended Data Fig. 8i), indicating that YAP and STAT3 activation does not decrease fructose uptake. Administration of recombinant CCN1 was also protective, reversing fructose-induced colon-shortening and TJP mRNA downregulation and reducing fecal albumin (Extended Data Fig. 8j–l). Moreover, CCN1 treatment inhibited fructose-stimulated DNL, hepatosteatosis, steatohepatitis and liver FAS expression without affecting body weight (Extended Data Fig. 8m–p).
Figure 5. Activated gp130 suppresses fructose-induced hepatic tumorigenesis, DNL and lipogenic and inflammatory gene expression.
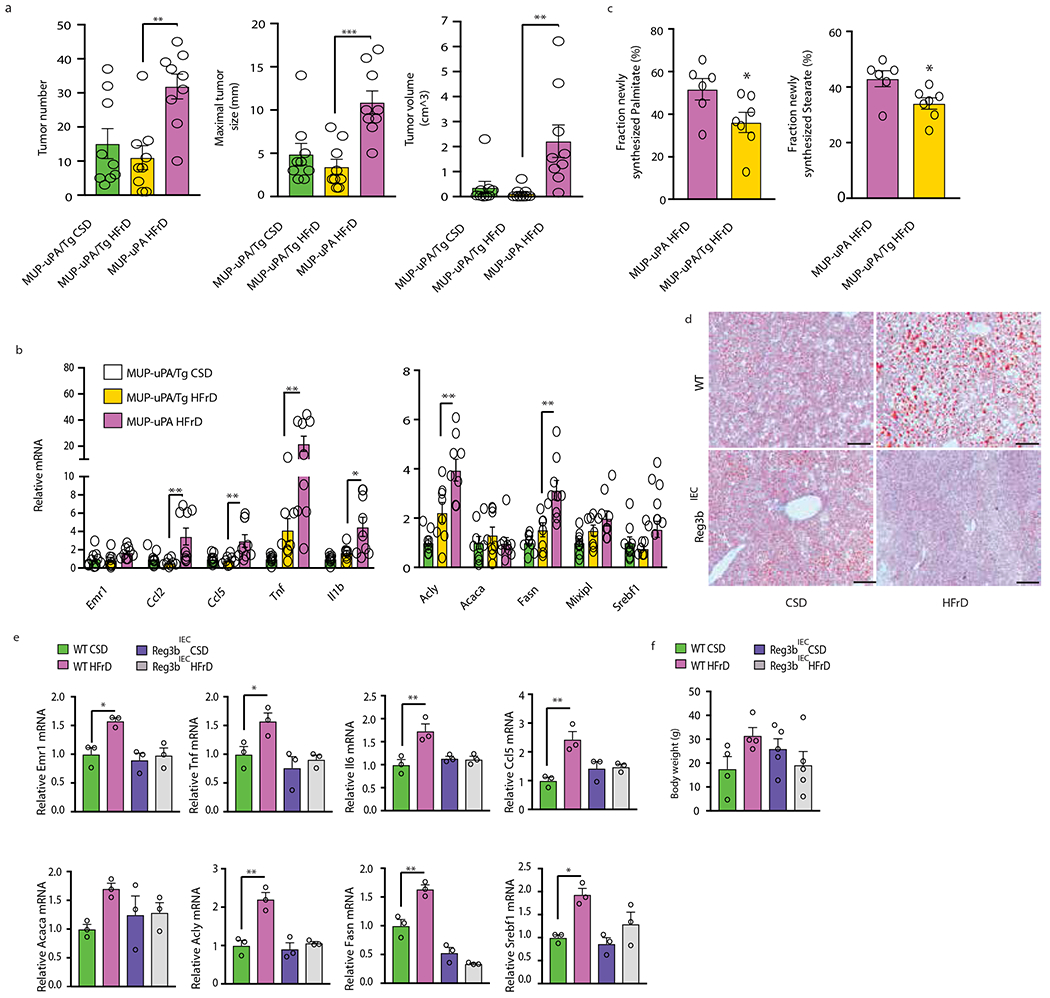
(a) HCC burden in CSD- or HFrD-fed MUP-uPA/gp130Act (MUP-uPA/Tg) and HFrD-fed MUP-uPA males (n=9 per group). (b) Liver lipogenic and inflammatory mRNAs in MUP-uPA/gp130Act males fed CSD or HFrD and MUP-uPA males fed HFrD (MUP-uPA/Tg CSD, n=9; MUP-uPA/Tg HFrD, n=8 and MUP-uPA HFrD n=9). (c) Liver fatty acid synthesis in MUP-uPA (n=6) and MUP-uPA/gp130Act (n=7) males administered D2O as a tracer. (d to f) Control FVB and mice overexpressing the antimicrobial protein Reg3β in intestinal epithelial cells were fed CSD or HFrD for 3 months. (d) Frozen liver sections were ORO stained to reveal lipid droplets (n=3 per group). Scale bars, 100 μm. (e) Relative mRNA amounts of liver lipogenic and inflammatory genes (n=3 per group) and (f) body weight (n=4 per group) in above mice. Panels a, b and e-f show mean ± SEM determined by one-way ANOVA and Tuckey’s multiple- comparison test. Panel c used two-sided Student’s t test. *P < 0.05.
Reg3β, whose expression is suppressed by HFrD, is an AMP that targets gram-negative bacteria and may neutralize endotoxin34. Accordingly, transgenic Reg3β expression from the IEC-specific Villin promoter prevented HFrD-induced hepatosteatosis (Fig. 5d). The Reg3b transgene also prevented fructose induction of liver mRNAs for inflammatory cytokines and lipogenic enzymes without affecting body weight (Fig. 5e, f). Reg3β IEC expression reduced HFrD-induced endotoxemia, but the increase in circulating endotoxin in HFrD-fed wt animals was of low statistical significance (Extended Data Fig. 8q). Conversely, low-dose LPS administration for 3 months caused TG accumulation and upregulation of DNL- and inflammation-related mRNAs in both CSD-fed MUP-uPA and HFrD-fed MUP- uPA/gp130Act livers (Extended Data Fig. 9a–f), supporting a role for circulating endotoxin in fatty liver disease.
Sequencing of fecal microbial 16S rRNA gene amplicons confirmed that prolonged HFrD consumption caused mild dysbiosis in MUP-uPA mice, characterized by decreased α diversity (Extended Data Fig. 9g). β diversity analysis by PERMANOVA based on unweighted UniFrac distances revealed significant (pseudo-F statistic = 2.88, P < 0.001) clustering of fecal samples according to diet (Extended Data Fig. 9h). Fecal 16S sequencing on CSD- and HFrD-fed MUP-uPA/gp130Act mice showed that prolonged HFrD feeding also caused dysbiosis in this mouse strain (Extended Data Fig. 9i, j).
TNF stimulates fructose-driven lipogenesis
We used primary hepatocyte cultures to examine whether TNF also stimulates fructose-driven steatosis in human liver. Primary hepatocytes from healthy donors were left unstimulated or treated with either LPS or TNF and placed in sugar-free culture medium or medium containing 5 mM glucose or fructose for 48 hours, after which lipid droplet accumulation was assessed. Whereas glucose or fructose alone or together with LPS barely affected lipid droplet formation, TNF treatment strongly enhanced lipid droplet buildup in both fructose and glucose incubated hepatocytes (Fig. 6a–c). TNF, but not LPS, induced ACACA, FASN and SREBF1 mRNAs.
Figure 6. TNF stimulates lipid droplet accumulation and lipogenic enzyme induction in human hepatocytes and schematic summary of the mechanism by which fructose intake stimulates hepatic DNL and steatosis.
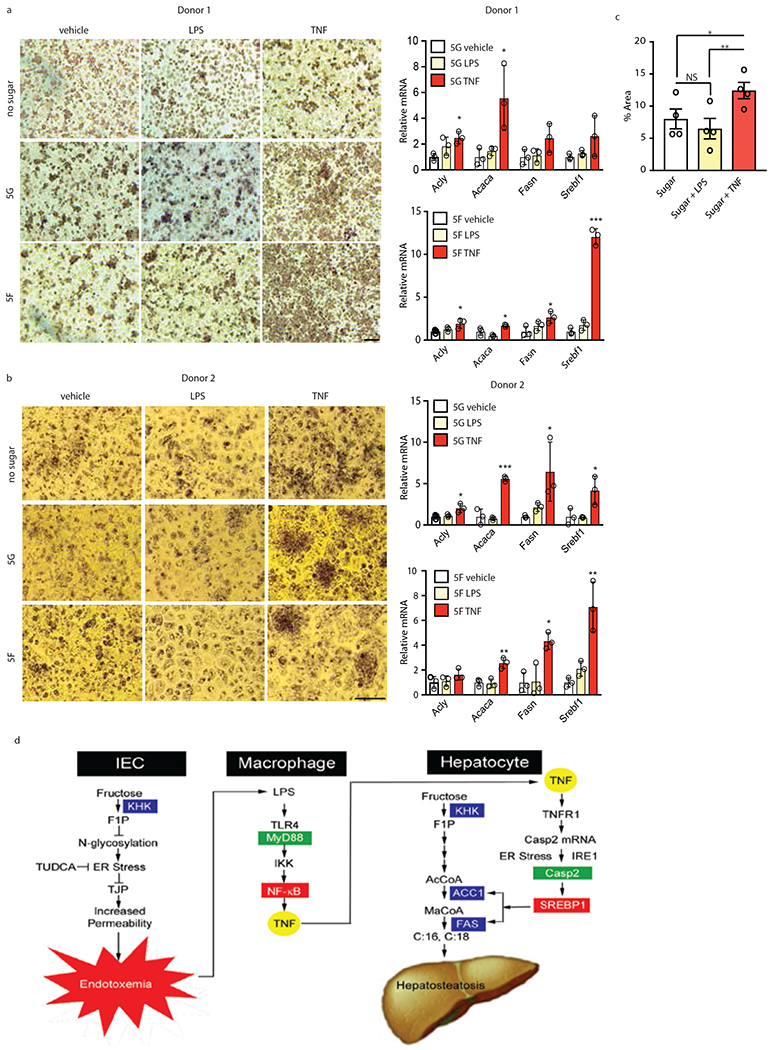
(a-c) Human hepatocytes isolated from two healthy donors were placed in sugar-free medium or in the presence of 5 mM fructose or glucose. LPS or TNF were added as indicated and 2 days later the cells were evaluated by ORO staining (a, b) for lipid accumulation, which was quantitated by image analysis (c; n=4 separate biological samples, 2 of which incubated with fructose and 2 incubated with glucose) and qRT-PCR for DNL-related mRNAs (a, b; n = 3 technical samples per each experiment), mean ± SEM by one-way ANOVA and Tukey’s multiple comparison test. (d) Schematic summary. In IEC dietary fructose is converted to F1P by KHK. F1P inhibits protein N-glycosylation triggering ER stress and intestinal inflammation to downregulate tight junction proteins (TJP) and trigger barrier deterioration and endotoxemia. Endotoxin (LPS) reaches the liver via the portal vein and engages TLR4 on macrophages to activate NF-κB via the adaptor protein MyD88 and induce TNF expression and secretion. TNF engages TNFR1 on hepatocytes to induce caspase-2 (Casp2) mRNA whose translation is stimulated by ER stress-activated IRE1. Casp2 activates SREBP1 to induce ACC1 and FAS, thereby priming the liver to convert KHK-generated F1P to FA (C:16/C:18), giving rise to hepatosteatosis.
Discussion
Excessive fructose intake causes dysbiosis and TJP downregulation, resulting in intestinal barrier deterioration and low-grade endotoxemia11–13. Fructose consumption stimulates hepatosteatosis and when combined with other risk factors can cause steatohepatitis and even HCC. It is well established that fructose is a more effective inducer of hepatic DNL than glucose19,20. However, the exact mechanism by which fructose preferentially drives DNL, other than its KHK-dependent conversion to acetyl-CoA via fructolysis, remains obscure. In particular, the metabolic effects of fructose-induced barrier deterioration and endotoxemia have not been investigated. We now show that fructose-induced endotoxemia, which can be prevented through IEC-restricted gp130 and Reg3β expression or CCN1 administration, activates MyD88-mediated inflammatory signaling in liver myeloid cells, thus triggering TNF induction (Fig. 6d). We previously showed that TNF signaling via TNFR1 together with ER stress-activated IRE1 stimulates hepatosteatosis through caspase-2 mediated SREBP1 activation9. Accordingly, myeloid cell MyD88 signaling is required for induction of the key lipogenic enzymes ACC1 and FAS in mouse liver and TNF treatment of sugar exposed human hepatocytes greatly enhances lipid droplet buildup, correlating with SREBF1, ACACA and FASN mRNA induction. Of note, fructose-induced endotoxemia does not affect fructose uptake, its conversion to F1P and subsequent acetyl-CoA generation via fructolysis. These new insights to fructose-induced hepatic lipid accumulation suggest that excessive fructose and other dietary factors that cause barrier deterioration contribute to the onset of steatohepatitis by stimulating net FA and TG accumulation in hepatocytes and decreasing mitochondrial β oxidation.
Our results explain why fructose rather than glucose leads to barrier deterioration. Fructose is converted by KHK to F1P, a toxic metabolite whose accumulation in aldolase B deficient individuals interferes with protein N-glycosylation26. Defective N-glycosylation interferes with protein transport and secretion, thereby triggering ER stress and inflammation35. We show that HFrD, but not CSD, consumption induces ER stress in fructose-metabolizing IEC and subsequent colonic inflammation. Fructose treatment of colonic organoids also triggers ER stress, preventable by KHK inhibition. ER stress also causes TJP downregulation27,28, increasing translocation of luminal microbial products to enhance the hepatic inflammatory response. Accordingly, the chemical chaperon TUDCA, activation of mucosal-regenerative gp130 signaling or administration of CCN1 prevent fructose-induced TJP downregulation and endotoxemia, thus blocking subsequent stimulation of hepatic DNL and steatohepatitis (Fig.6d). Future studies will tell whether CCN1 and other agents, such as aryl hydrocarbon receptor agonists36, that stimulate mucosal healing can reduce NAFLD and NASH incidence in humans. Another potential preventative/therapeutic approach that deserves further consideration is endotoxin neutralization. Although our mouse model experiments, employing high amounts of fructose, may not be immediately extended to humans most of whom rarely consume fructose without glucose, we suggest that barrier deterioration likely occurs only after continuous and excessive consumption of fructose and is probably influenced by other dietary and genetic factors or co-morbidities. Of note, diet composition determines the relative contribution of DNL to hepatosteatosis, such that DNL becomes important when fat intake is low. Nonetheless, barrier decline has been described in various experimental models and numerous liver diseases that entail hepatic fat accumulation and inflammation37,38. Thus, the finding that excessive fructose ingestion causes hepatosteatosis through a pathway that involves barrier deterioration, endotoxemia, activation of myeloid cell MyD88 signaling and TNF secretion, and induction of lipogenic enzymes in hepatocytes (Fig. 6d) may be of relevance to several common liver diseases and metabolic disorders.
Methods
Mouse Studies
MUP-uPA (generated by Dr. Eric Sandgren at School of Veterinary Medicine, University of Wisconsin-Madison) and gp130Act mice originating from the Karin lab8,33 were crossed to generate MUP-uPA/gp130Act mice, also in the Karin lab. Briefly, gp130Act cDNA was PCR amplified PCR and subcloned into a plasmid containing the 12.4-kb Villin promoter (A gift from Dr. D.L. Gumucio, University of Michigan). The 15.7-kb expression cassette was excised by PmeI digestion, purified, and injected into fertilized C57BL/6 oocytes to obtain founder mice, two of which transmitted the gp130Act transgene. Villin-Cre mice [Tg(Vil1-cre)997Gum] were obtained from The Jackson Laboratory (stock 021504). All mice were on the C57BL/6N background, due to backcrossing to C57BL/6N mice (strain code 027 from Charles River Laboratories), and were group-housed (3-4 per cage) in filter-topped cages on autoclaved food and water at UCSD according to NIH guidelines. Beddings were interchanged between the different strains to minimize microbiome alterations. All experiments, involving these mice were performed in accordance with UCSD and NIH guidelines and regulations. Reg3bIEC transgenic mice in which the Villin promoter drives Reg3b in intestinal epithelial cells (IEC) were generated at the University of Frankfurt by Polygene. Briefly, Flag-tagged mouse Reg3b cDNA was cloned into the 12.4 Villin promoter described above and the construct injected into the pro-nucleus. The resulting transgenic mice were propagated in the FVB background. In these experiments both male and female mice 6-10 weeks of age were used. All other experiments (at UCSD) were conducted on male mice, whose age is specified in the Figure Legends and Method Details section. All mice were put on the designated diets at 6 weeks of age and kept on the specified diets for the durations indicated in Figure Legends. Whenever possible we used littermates but as some experiments were conducted with a large number of mice, several different litters had to be used. To induce HCC in BL6 mice, DEN (25 mg/kg) was injected intraperitoneally (i.p.) into 14-day-old males. Mice were randomly placed on one of two isocaloric diets purchased from Research Diets Inc.: (a) control corn starch diet (CSD) in which 70% of calories are from corn starch, 10% from fat, and 20% from protein, totaling 4.1 kcal/g, or (b) high-fructose diet (HFrD) in which 60 % of calories are from fructose, 10% from corn starch, 10% from fat, and 20% from protein; 4.1 kcal/g. Both diets contain 4.7 gm % fiber. Mice were treated with antibiotics as described21,39. Mice were also given NCD or HFD as previously8 and allowed free access to either 30% fructose or sucrose in water solution or regular drinking water. Mice were given daily 250 mg/kg TUDCA or vehicle (PBS) i.p. injections for the last 10 weeks of fructose drink exposure. All experiments were performed using UCSD IACUC-approved protocols in accordance with UCSD and NIH guidelines and regulations. The Reg3bIEC experiments were performed at the University of Frankfurt in accordance with German law and regulations.
Human Hepatocyte Cultures
Donor livers rejected for transplantation were obtained via LifeSharing OPO as a part of Dr.T. Kisseleva’s research program. Donors, who authorized that their organs/tissues may be used for research, with no history of alcohol abuse, low BMI (18-22), no liver fibrosis, no diabetes, minimal steatosis, and normal levels of ALT and AST were qualified as normal in this study conducted under IRB 171883XX (approved on 11/9/17 by UCSD Human Research Protections Program, under the title Unused liver from deceased donors: role of myofibroblasts in liver fibrosis). For hepatocyte isolation the livers were placed on top of an ice pan covered by a plastic bag and then a sterile field. Catheters were inserted into the major portal and/or hepatic vessels and the tissue was perfused with cold organ preservation solution or EMEM (Corning Cellgro cat # 15-010-CM) + 25 mM HEPES (Corning Cellgro cat# 25-060-CI) to determine which vessels provide the most uniform tissue perfusion. This step also helps remove any remaining blood. The catheters were then secured into the vessels either by sutures or surgical grade glue. All remaining major vessels on the cut surfaces were closed with sutures or surgical grade glue. The liver tissue was then placed in a sterile plastic bag and connected to a peristaltic pump with flowrate dependent on the number of catheters and size of tissue.
The bag containing the tissue was placed in a water bath at 38.5°C and the tissue was perfused with HBSS (without calcium and magnesium or phenol red) (Hyclone cat # SH30588.02) supplemented with 1.0 mM EGTA without recirculation for 10-20 min. The EGTA chelates calcium which leads to the separation of cell junctions and helps remove any residual blood. Finally, the liver specimen was perfused with EMEM containing 0.1 mg/ml of collagenase (VitaCyte cat# 001-2030) and 0.02 mg/ml of protease (VitaCyte cat# 003-1000) which is recirculated as long as needed to complete the digestion (15-30 min total digestion time). Perfusion was stopped when the liver tissue began to show fissures and separation from the liver capsule. The liver tissue was then removed from the plastic bag and placed in a sterile plastic beaker that contains warmed (37°C) DMEM supplemented with 5% FBS, 1% Sodium pyruvate, 1% antibiotic. The specimen was then gently dissociated with sterile scissors to release hepatocytes. The cell suspension was filtered through sterile nylon mesh covered funnels to remove cellular debris and clumps of undigested tissue. The above steps were repeated as many times as needed to obtain the maximum numbers of cells. Hepatocytes were isolated from other cell types in the suspension by low speed centrifugation at 80x g for 5 min at 18°C. The supernatant was decanted and saved (NPC fraction).The hepatocyte pellets were gently resuspended in a Percoll (Sigma # GE17-0891-09) gradient and centrifuged at 100x g for 10 minutes at 18°C.The supernatant was removed by aspiration and the pellets were saved and resuspended in warmed (37°C) DMEM and the centrifugation step was repeated. The supernatant was removed and the hepatocytes were resuspended in 4°C Hypothermosol FRS (BioLIfe Solutions # 101104) to make the Hepatocyte Stock. An aliquot of the Hepatocyte Stock (150 μl) was removed and placed in a 1.5ml Eppendorf tube, mixed with an equal volume of trypan blue and assessed by dye exclusion to determine cell viability and number using a hemocytometer. Cell viability was expressed as a percentage of the total cells counted (# of live cells/[# of live cells + # of dead cells] x 100 = % viable). Usually there were 515X106 total viable cells in the Hepatocyte Stock. An aliquot of 6X106 viable hepatocytes was removed from the hepatocyte stock and diluted with 5 volumes of ice cold EMEM. The aliquot was centrifuged at 100x g and 18°C for 5 min. The supernatant was decanted and the pellet was resuspended in 12 ml of Hepatocyte Plating Medium**. The hepatocytes were then seeded on a collagen coated 24 well plate and allowed to attach in a 37°C, 5% CO2 incubator for 2-12 hours and then the medium is changed to serum free Hepatocyte Maintenance Medium. The remaining Hepatocyte Stock was allowed to settle at 4°C for 1-2 hours. The excess liquid was carefully removed from the bottle with care not to disturb the hepatocytes at the bottom. The cell suspension was then brought to a final concentration of 8 × 106 hepatocytes/ml using a solution of Cryostor CS10 (BioLife Solutions # 210102) +DMSO+Fructose giving a final concentration of 10% DMSO + 100 mM Fructose. The cell suspension was placed at 1.5ml/vial, into 2ml freezing vials and frozen in control rate freezer till used.
*Hepatocyte Plating Medium: Williams’ E medium from Life Technologies (Cat. # A12176-01) containing 10mM HEPES, 2mM L-Glutamine, 10%FBS, 1X10-7M Dexamethasone, 1X10-7M Insulin and 1% Antibiotic/Antimycotic solution. **Hepatocyte Maintenance Medium = Hepatocyte Plating Medium without FBS
Histology
Freshly collected liver tissues from MUP-uPA, MUP-uPA;gp130Act, Reg3bIEC and C57BL/6 mice were fixed in 10% neutral-buffered formalin or 4% paraformaldehyde, embedded in paraffin, sectioned and stained with hematoxylin and eosin (H&E) and Sirius Red. For frozen-block preparation, tissues were embedded in Tissue-Tek OCT compound and stained with Oil Red O and H&E. For Oil Red O and Sirius Red analysis, multiple images were examined for each section. Representative images were captured on an upright light/fluorescent microscope (Zeiss) equipped with an AxioCam camera.
Lipidomics
Triacylglycerol (TAG) lipid species were measured as described40 with minor modifications. Briefly, freshly isolated liver samples were homogenized in one volume of phosphate buffered saline (PBS) (pH 7.4), and sonicated. Next, a BCA protein assay was performed and 25 μg of each liver sample aliquoted and utilized for lipid extraction. An internal standard mixture and CHCl3/methanol (2:1) mixture were added to each sample before being vortexed, mixed, sonicated, and centrifuged. Lipid-containing supernatants were removed and dried before being resuspended in H2O-saturated butanol and sonicated again. Methanol with 10 mM ammonium were added and the resuspended samples were centrifuged and the supernatants were transferred to individual glass vials. Lipidomic determination was performed by liquid chromatography electrospray ionization tandem mass spectrometry (LC-MS) combined with a triple quadrupole mass spectrometer. Data were analyzed using Multiquant software v1.2 and TAG normalized to the total phosphatidylcholine (PC) levels of each sample.
Indirect calorimetry
Mice were placed into Comprehensive Lab Animal Monitoring System (CLAMS; Columbus Instruments) metabolic cages to adapt to their surroundings for 48 hours before study. Data from 3 light/dark cycles were used in the analysis. Rates of O2 consumption (VO2; ml/kg/hour) and CO2 production (VCO2) were continuously acquired.
Immunoblotting and antibodies
IB analysis was performed on tissue or cell lysates that were SDS-PAGE separated and transferred to nitrocellulose membranes. Blots were incubated with 5% (wt:vol) nonfat dry milk in PBS with 0.05% (wt:vol) Tween 20 (Millipore Sigma) (PBST) at room temperature for 1 hour to block nonspecific binding, and overnight at 4° C with primary antibodies in 3% BSA (wt:vol) in PBST and finally with HRP-conjugated secondary antibody in blocking buffer. Blots were developed using the Western Lightning Plus-ECL enhanced chemiluminescence detection kit (PerkinElmer). Immunoblotting was performed with antibodies against TLR2 (Santa Cruz, sc-21760), TLR4 (Santa Cruz, sc-293072), MyD88 (Santa Cruz, sc-74532), phospho-STAT3 (Cell Signaling, CS9145), STAT3 (Cell Signaling, CS12640), NLRP3 (LSBio, LS- C334192), FAS (Cell Signaling, #3180), ACC (Cell Signaling, #3662), ERK1/2 (Cell Signaling, #9102), Occludin (Santa Cruz, sc-271842) and Claudin 1 (Santa Cruz, sc- 166338). All primary antibodies were diluted 1:1000. Secondary antibodies were diluted 1:5000.
Glucose tolerance test
Mice were transferred to clean cages with no food and fasted for approximately 12 hours with access to drinking water. A 20% glucose solution was i.p. injected (2 g/kg mouse body mass) and small drops of blood were obtained from tail cuts and assessed for baseline glucose levels using a One-touch Ultra 2 (Lifescan, Johnson & Johnson) glucometer at 30, 60, 90 and 120 minutes.
Intestinal permeability
Fecal albumin measurements were performed on dried fecal pellets using the Mouse Albumin ELISA Quantitation Set from Bethyl Laboratories according to manufacturer’s instructions. An in vivo permeability assay to assess barrier function was performed using the FITC-labeled dextran method. Food and water were withdrawn overnight, and mice were gavaged with 45 mg/100 g body weight of FITC-labeled dextran (FD-4; Sigma-Aldrich). Serum was collected 5 hours after FD-4 gavage, and fluorescence intensity of each sample was measured (excitation, 492 nm; emission, 525 nm). Serum or plasma LPS concentrations were determined with the Mouse Lipopolysaccharides (LPS) ELISA Kit obtained from Cusabio following manufacturer’s instructions.
Cytosolic acetyl-CoA measurement
Frozen liver pieces were weighed, thawed, cut into smaller pieces and homogenized using a pestle in 1.5 ml microfuge tubes on ice. The cytoplasmic fraction was separated from the mitochondria and deproteinized using Mitochondria/Cytosol Fractionation Kit (Bio Vision, # K256) and Deproteinizing Sample Preparation Kit (Bio Vision, # K808), respectively. Acetyl-CoA concentrations were determined using PicoProbe™ Acetyl-CoA Assay Kit (Bio Vision, #K317).
Intestinal organoid culture
Intestinal crypts were isolated from MUP-uPA and gp130Act mouse colons and were cultured using standard procedure41. Two days after passaging, when the crypts started budding, the medium was changed to DMEM (Gibco, 11966-025) supplemented with 5 mM or 20 mM glucose or fructose or a combination of both sugars (5 mM glucose and 20 mM fructose) and 10% FBS. After changing the medium, the organoids were cultured for 24 hours.
RNA-seq analysis
RNA was extracted from snap-frozen liver using the NucleoSpin® RNA kit (Macherey-Nagel) according to manufacturer’s instructions. Library preparation for RNA-seq was performed using TruSeq Stranded mRNA Library Prep Kit (Illumina) following manufacturer’s guidelines and best practices. For quality control of the libraries, the samples were inspected with a Bioanalyzer chip (DNA 1000, Agilent). Sequencing was performed for paired-end samples on a HiSeq 2500 v4.0 system. The resulting fastq files were quality controlled using FastQC, and adapters were trimmed using TrimGalore! v0.4.0 (both developed at The Babraham Institute) for paired-end sequenced samples. The trimmed fastq files were aligned to the mouse reference genome (Mus_musculus.GRCm38.83) using STAR aligner (v2.5.1) 42. RSEM (v1.3.0)43 was used to estimate gene expression levels. Downstream analysis for differential gene expression of the raw count table was performed using DESeq244 in R. Heatmaps were drawn using the R package pheatmap (Pretty Heatmaps; R package version 1.0.10; https://CRAN.R-project.org/package=pheatmap) clustering columns and rows and the values for each row being centered and scaled. Mapping of gene expression levels to corresponding KEGG pathways was performed using the R packages gage45 and path view46.
Quantitative real-time PCR analysis
RNA samples were prepared using RNeasy kit (Qiagen). RNA was reverse transcribed using a Super Script VILO cDNA synthesis kit (Thermo Scientific) and qPCR was performed using SYBR green (Biorad) based real-time PCR on a Biorad CFX96 machine. Relative mRNA expression was calculated from the comparative threshold cycle (Ct) values relative to ribosomal protein S18 mRNA. PCR primers were designed using online primer tools Primer3Plus and purchased from Integrated DNA Technologies. Primer sequences are provided in table S1.
Microbiome analysis
Samples were processed following the Earth Microbiome Project47 DNA extraction and 16S rRNA gene amplicon sequencing protocol, updated for the Kingfisher liquid-handling robot48. In brief, the 96-well MoBio Powersoil DNA kit was used to extract DNA from fecal samples and barcoded 515F-806R primers targeting the V4 region of the 16S rRNA were used for 16S amplification. Sequencing was performed at UCSD Institute for Genomic Medicine (IGM) on an Illumina MiSeq. 16S sequencing data were uploaded to Qiita (https://qiita.ucsd.edu/) for QC, demultiplexing and processing with Deblur49. The resulting deblurred feature table was downloaded for analysis with QIIME 2 (https://qiime2.org) v 2017.1050. The feature table was rarified to 8150 sequences per sample and the rarified table used for alpha diversity and beta diversity analysis. Taxonomic assignment was performed with QIIME 2 using a naive bayes sklearn classifier trained on the 515F/806R region of Greengenes 13_8 99% OTUs.
D2O administration and measurement of de novo lipogenesis
MUP-uPA and MUP-uPA/gp130Act mice were fed ad libitum CSD or HFrD for either 12 weeks or 48 hours from 6-8 weeks of age. One day before termination, mice were i.p. injected with 0.028 ml/g body weight 0.9% NaCl D2O, and drinking water was replaced with 6% D2O-enriched water. 19-20 hours later, plasma and tissue samples were immediately snap-frozen in liquid nitrogen.
The 2H labeling of water from samples or standards was determined by deuterium acetone exchange. 5 μl of sample or standard were reacted with 4 μl of 10 M NaOH and 4 μl of a 5% (vol:vol) solution of acetone in acetonitrile for 24 hours. Acetone was extracted by addition of 600 μl chloroform and 0.5 g Na2SO4 followed by vigorous mixing. 80 μl of the chloroform was then transferred to a glass GC–MS vial. Acetone was measured using an Agilent DB-35MS column (30 m x 0.25 mm i.d. x 0.25 μm, Agilent J&W Scientific) installed mass spectrometer (MS) with the following temperature program: 60° C initial, increase by 20° C/min to 100° C, increase by 50° C/min to 220° C, and hold for 1 minute. The split ratio was 40:1 with a helium flow of 1 ml/min. Acetone eluted at approximately 1.5 minutes. The mass spectrometer was operated in the electron impact mode (70 eV). The mass ions 58, 59, and 60 were integrated and the percent M1 and M2 (m/z 59 and 60) calculated. Known standards were used to generate a standard curve and plasma percent enrichment was determined from this. All samples were analyzed in triplicate. In vivo calculation of the fraction of newly synthesized fatty acids (FNS) was based on the method described in51 where FNS is described by the following equation:1). where ME is the average number of deuterium atoms incorporated per molecule (ME = 1 × m1 + 2 × m2 + 3 × m3…), p is the deuterium enrichment in water and n is the maximum number of hydrogen atoms from water incorporated per molecule. n was determined using the equation:2). , as described in51, where q is the fraction of hydrogen atoms and p + q = 1.
GC–MS analysis of fatty acids and polar metabolites
For tissue and plasma, total fatty acids were extracted from tissues and plasma using a Folch-based methanol/chloroform/saline extraction at a ratio of 1:2:1 with inclusion of 10 nmol/mg [2H31]palmitate as a lipid internal standard as described previously52. Briefly, 250 μl methanol, 500 μl chloroform, 250 μl saline and isotope internal standards were added to ~20 mg weighed tissue in a 2 mL Eppendorf. This was pulverized in a Retsch mill with 3 ball bearings for 5 minutes followed by removal of ball bearings and centrifugation at 21,000 g for 10 minutes. The lower chloroform phase was dried and then derivatized to form fatty acid methyl esters (FAMEs) via addition of 500 μl 2% H2SO4 in MeOH and incubation at 50° C for 2 hours. FAMEs were extracted via addition of 100 μl saturated salt solution and 2 successive additions of 500 μl hexane and these were analyzed using a Select FAME column (100 m x 0.25 mm i.d.) installed in an Agilent 7890A GC interfaced with an Agilent 5975C MS using the following temperature program: 80° C initial, increase by 20° C/min to 170° C, increase by 1° C/min to 204° C, then 20° C/min to 250° C and hold for 10 minutes. The percent isotopologue distribution of each fatty acid was determined and corrected for natural abundance.
F1P analysis in liver and intestine
Optima LC/MS grade water was sourced from Fisher Scientific (Part number: W6-4). Pure HPLC ethyl alcohol was purchased from Sigma-Aldrich (Part number: 459828-2L). HPLC grade acetonitrile, methanol, and isopropyl alcohol were purchased from Honeywell (Part numbers: LC015-4, LC230-4, and LC323-4). Pre-weighed frozen tissue was transferred to a pre-chilled 2 mL homogenization tube containing 300 mg of 1 mm zirconium disruption beads. To each sample, a −20° C mixture of methanol:water (1:1) was added to achieve a normalized concentration of 50 μg of tissue per 1 μL of solvent. The samples were then homogenized using a BeadRuptor Elite homogenizer at a speed of 8 m/s for 3 cycles involving 10 seconds of homogenization followed by 10 seconds of dwell. The samples were then immediately placed on dry ice and the homogenate was transferred to clean 1.5 mL microfuge tubes and allowed to deproteinize for 30 minutes at −20° C. Samples were then centrifuged at a speed of 14,000 rpm at 4° C for 10 minutes. 50 μL of each sample’s supernatant was then transferred to an amber autosampler vial containing a Wheaton 300 μL glass insert. Samples were stored in an Agilent Infinity 1290 UHPLC Autosampler maintained at 4° C until 20 μL of each sample was injected for targeted analysis by LC-MS/MS performed on an Agilent Infinity 1290 UHPLC equipped with a Shodex HILICpak VT-50 2D Column (100 Å, 5 μm particle size, 2.0 × 150 mm, P/N SH-F7630400) maintained at 60° C. Elution was performed with an isocratic elution of mobile phase containing 80:20 water:acetonitrile with 25 mM ammonium formate at a constant flow rate of 0.250 mL/min over the course of 14 minutes. Data acquisition was performed on an Agilent 6495 Triple Quadrupole Mass Spectrometer equipped with an Agilent Jet Stream electrospray ionization source operating in negative mode under the following source settings: Gas Temp 200° C; Gas Flow 11 l/min; Nebulizer 35 psi; Sheath Gas Temp 400° C; Sheath gas flow 11 l/min; Capillary voltage 3000 V; Nozzle Voltage 1500 V; High Pressure RF 50 V; Low Pressure RF 40 V. Fructose-1- phosphate was monitored using a single MRM with the settings described in Table S2.
In vivo treatment with LPS
Six-week-old MUP-uPA and MUP-uPA/gp130Act mice were fed CSD or HFrD diet and i.p. injected daily with LPS (E. coli, Sigma Chemical, St. Louis, MO; 0.25 mg/kg) or vehicle (PBS). The mice were sacrificed after twelve weeks of treatment and the tissues were collected for further analysis.
In vivo treatment with CCN1
Recombinant CCN1 was expressed in E. coli and purified as described53. Six-week-old MUP-uPA mice were fed HFrD for twelve weeks and i.p. injected with vehicle or 2 ug CCN1 in 200 μl of PBS every other day for the last four weeks of the experiment. The mice were sacrificed at 18 weeks of age and the tissues were collected for further analysis.
Quantification and Statistical Analysis
Data are presented either as means ± SEM or medians of continuous values and were analyzed by an unpaired two-sided Students’ t-test or Mann–Whitney test for comparison of two groups, respectively, followed by Benjamini-Hochberg FDR adjustment for P-values. To compare multiple groups we used one-way ANOVA and Tukey’s multiple comparison test or two-way ANOVA and Sidak’ multiple comparison test. Statistical significance is defined as a P-value greater than 0.05. Significance testing for differentially abundant bacteria was performed with the QIIME2 implementation of ANCOM54.
Extended Data
Extended Data Figure 1. HFrD stimulates HCC development without affecting body weight or white adipose tissue.
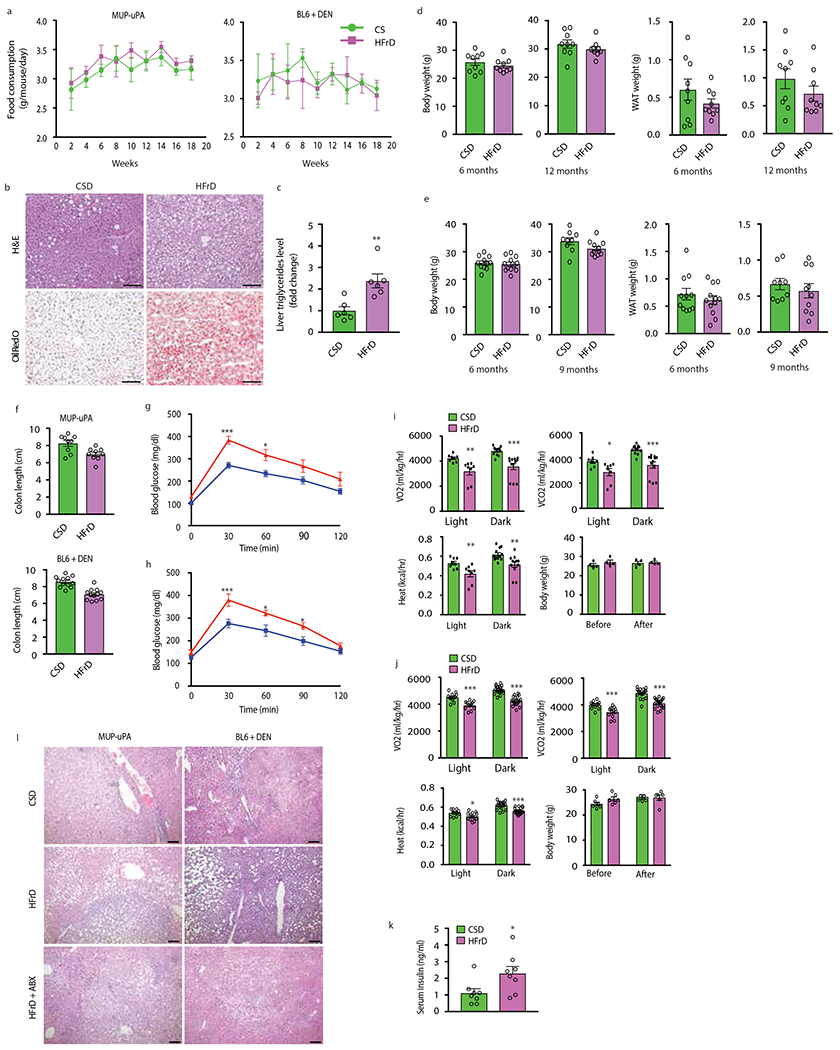
(a) Food consumption by male MUP- uPA and BL6 mice given CSD (MUP-uPA n=4 cages/9 males; BL6 n=5 cages/11 males) or HFrD (MUP-uPA n=4 cages/12 males; BL6 n=5 cages/12 males). (b) Formalin-fixed, paraffin-embedded (FFPE) and frozen liver tissue sections of DEN- challenged BL6 males were stained with H&E or ORO (n=6 per group). Representative images are shown. Scale bars, 100 μm. (c) Total triglycerides in livers of DEN-challenged BL6 males (n=6 per group) are presented as fold-change relative to CSD-fed mice. (d and e) Body and WAT weight of MUP-uPA males (n=9 per group) (d) and DEN- challenged BL6 males (n=11 CSD at 6 months and n=9 CSD at 9 months; n=12 HFrD at 6 months and n=9 HFrD at 12 months) (e) mice given CSD or HFrD were measured at the indicated time points. (f) Colon length in indicated male mice (MUP-uPA n=9, BL6 n=12, per group). (g and h) MUP-uPA males (n=15 per group) (g) or DEN-challenged BL6 males (n = 14 per group) (h) fed HFrD (▴) or CSD (▪) for 5 months were subjected to glucose tolerance tests (GTT). (i and j) CSD- or HFrD- fed MUP-uPA males (n=4 per group) (i) or DEN-challenged BL6 males (n=6 per group) (j) were placed in metabolic cages for two-and-a-half 24-hour cycles composed of two 12-hour light periods and three dark periods. VO2, VCO2, heat production and body weights were measured. (k) Non-fasting serum insulin was measured by ELISA in indicated males (n=8 per group). (l) Liver tumor histology in MUP-uPA and DEN-challenged BL6 males given either CSD, HFrD, or HFrD + Abx cocktail (MUP-uPA n=7, BL6 n=12 per group). FFPE tumor sections were stained with hematoxylin and eosin (H&E). Representative images are shown. Scale bars, 100 μm. Unpaired two-sided Student’s t test was used in panels c to f and k, whereas two-way ANOVA and Sidak’s multiple comparison test were used in panels g-j. Mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
Extended Data Figure 2. Fructose effects on DNL and lipogenic enzymes.
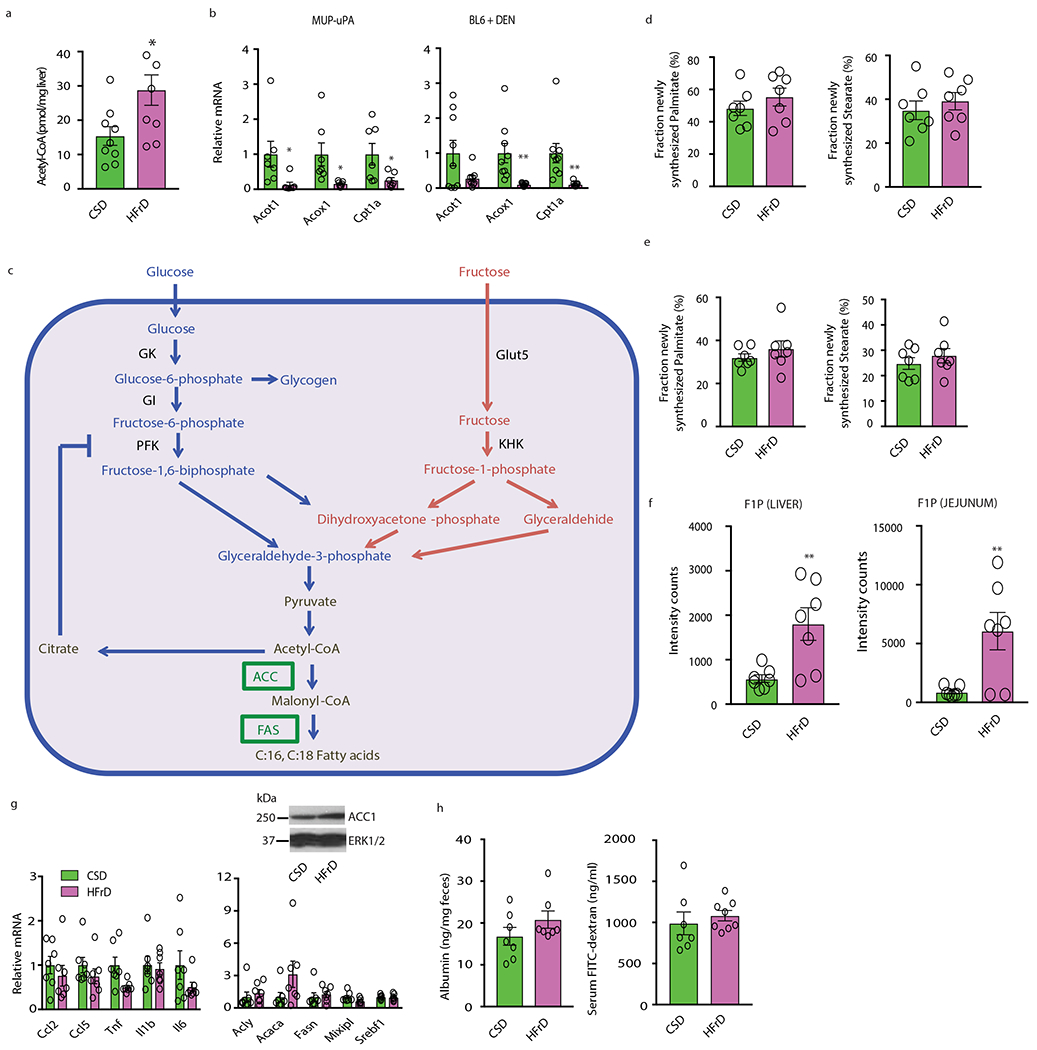
(a) Cytosolic acetyl-CoA concentrations in 6-month-old CSD and HFrD fed MUP - uPA males (n=9 CSD group, n=7 HFrD group). (b) Expression of genes encoding FA-oxidizing enzymes in CSD- or HFrD- fed (5 months) MUP-uPA (n=7 per group) and DEN-challenged BL6 male mice (n=9 per group). (c) A metabolic chart comparing pathways through which glucose and fructose are converted to acetyl-CoA in the cytoplasm. GK-glucokinase; KHK-ketohexokinase; GI-glucose isomerase; PFK-phosphofructokinase. Cytoplasmic acetyl-CoA is converted to malonyl-CoA by acetyl-CoA carboxylase (ACC) and malonyl-CoA serves as the building block for synthesis of C16:0 (palmitate) and C18:0 (stearate) by fatty acid synthase (FAS). Expression of ACC1 and FAS is strongly upregulated by prolonged HFrD feeding. (d and e) Measurement of newly synthesized FA in livers (d) and jejunum (e) of MUP-uPA males using D2O as a tracer after a short-term feeding period (48 hours; n=7 per group). (f) Amounts of F1P in liver and jejunum expressed in arbitrary units (48 hours feeding; n=7 per group). (g and h) Expression of inflammatory and lipogenic genes and ACC1 protein in livers (g) and fecal albumin and serum FITC-dextran concentrations (h) in CSD- or HFrD-fed (48 hours) males (n=7 per group). Unpaired two-sided Student’s t test was used in panels a, b and d-h. Mean ± SEM, *P < 0.05, **P < 0.01. Benjamini-Hochberg FDR adjustment for P-values was performed on data in panels b and g.
Extended Data Figure 3. HFrD feeding downregulates TJP mRNAs and induces ER stress, colonic inflammation and barrier deterioration.
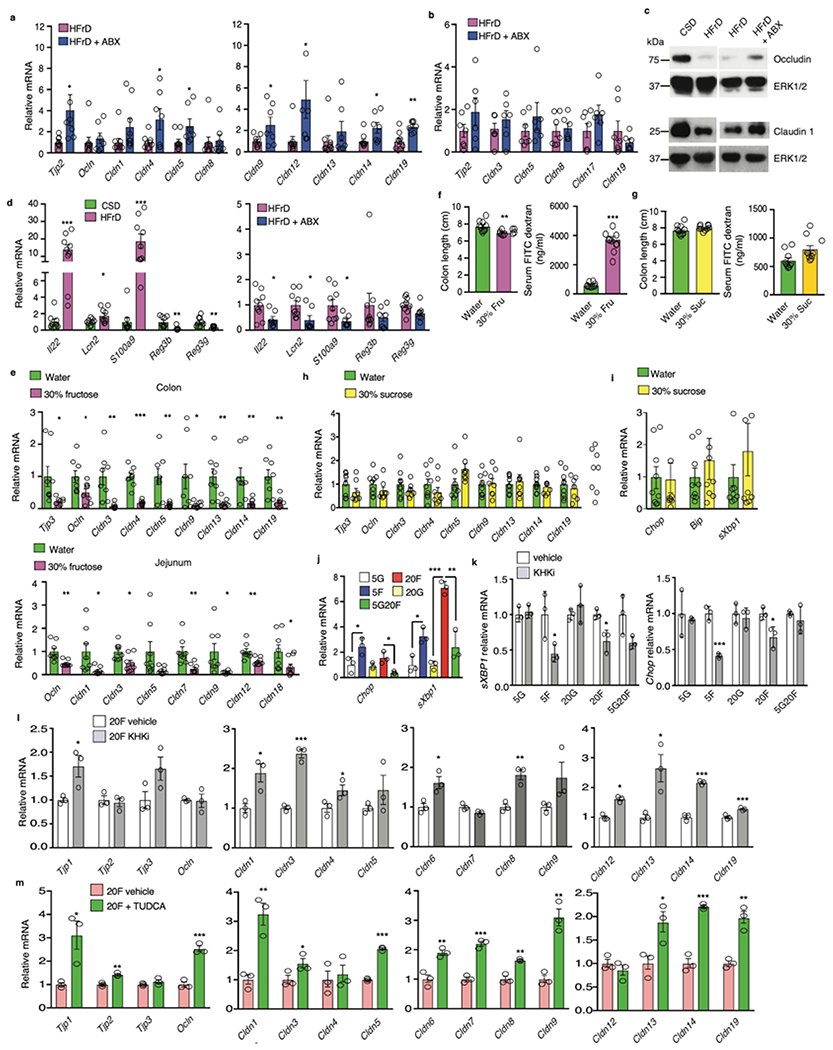
(a and b) Expression of TJP genes in colon (a) and jejunum (b) of MUP- uPA males fed HFrD with (a, n=6 or 7; b, n=6) or without (a, n=9; b, n=7) concomitant Abx treatment. (c) IB analysis of indicated TJPs in colons of above mice (n=9 no Abx, n=7 plus Abx). Representative blots are shown. (d) Expression of Il22 mRNA and IL-22 regulated genes in colons of CSD-, HFrD-, or HFrD + Abx-fed MUP- uPA male mice (n=9 no Abx, n=7 plus Abx). (e and f) BL6 males (e, n=8 per group; f, n=10 per group) were given 30% fructose in drinking water and fed NCD for 5 (e) or 3 (f) months and mRNA amounts of TJP genes in colon and jejunum (e), colon length and FITC-dextran serum levels were measured (f). (g to h) BL6 males were given regular water with or without 30% sucrose and fed NCD for 3 months. Colon length and FITC-dextran serum levels (n=10 per group) (g), colon TJP mRNAs (h) and ER stress marker mRNAs (i) (h and i, n=9 regular water group and n=8 30% sucrose group) were measured. (j) Chop and sXbp1 mRNA amounts in fructose and glucose treated organoids (isolated from 3 different male mice). (j-m) Expression of indicated mRNAs in BL6 enteroids (isolated from 3 different male mice) maintained in media containing the indicated glucose and/or fructose concentrations (mM) and treated with KHK inhibitor (k and l), TUDCA (m) or vehicle. Unpaired two-sided Student’s t test was used in all panels, other than c, to determine Mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001. Benjamini-Hochberg FDR adjustment for P-values was performed on data in panels a, b, d, e and h-m. Experiments on enteroids were repeated twice with similar results.
Extended Data Figure 4. Fructose-induced alterations in the liver transcriptome.
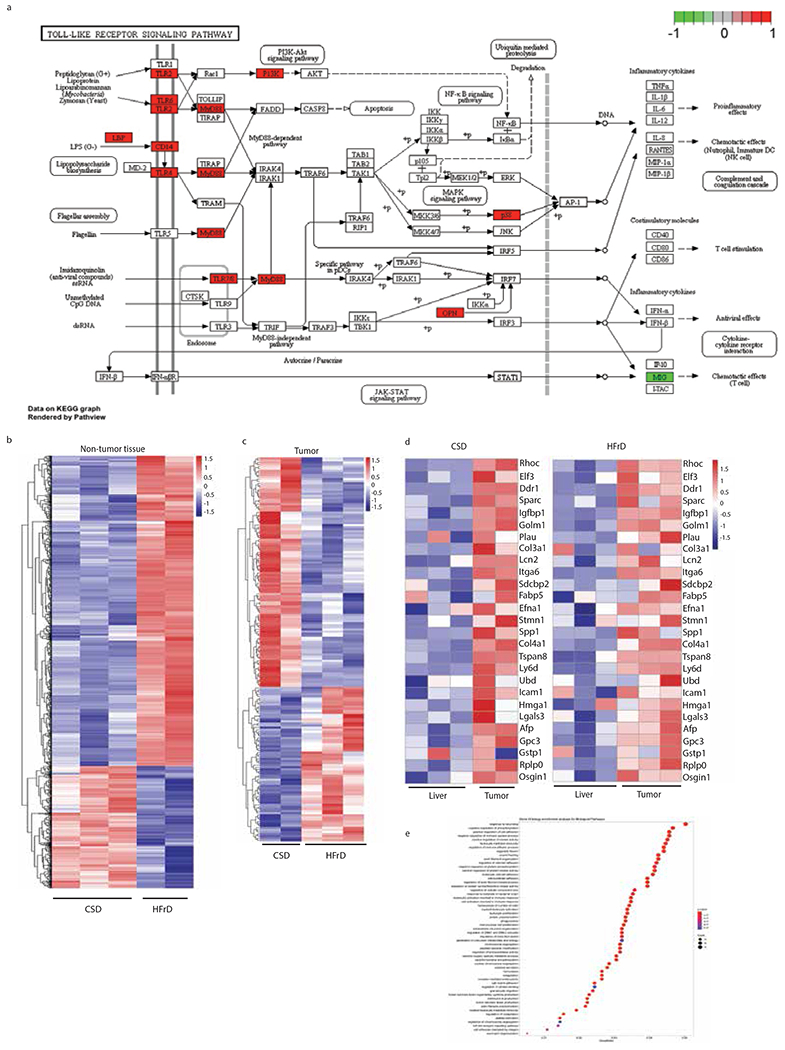
(a) KEGG pathway map of innate immune signaling with HFrD-induced genes indicated in red. (b) Heatmap depicting the entire transcriptome of non-tumor liver tissue from DEN-treated BL6 males kept on CSD (n=3) or HFrD (n=2) for 5 months and analyzed immediately thereafter. (c) Heatmap depicting the entire transcriptome of tumor-bearing liver tissue from DEN-treated BL6 males kept on CSD (n=2) or HFrD (n=3) until 9 months old. (d) Heatmap depicting expression of HCC-related genes in non-tumor and tumor liver tissues from mice described in (b). The color code represents relative mRNA abundance where red shows overexpression and blue shows under expression. (e) Most significant Gene Ontology enrichment terms for Biological Pathways for the genes shown in heatmap (b) comparing DEN-treated BL6 mice fed a CSD or HFrD at 6 months of age. The bigger the dot, the more genes fall in the respective category. The color code represents the adjusted P-value (all are significant and below a cut-off level of 0.01) determined using Benjamini-Hochberg FDR adjustment.
Extended Data Figure 5. Fructose-induced endotoxemia, hepatosteatosis, HCC, liver fibrosis, glucose intolerance and colonic inflammation are inhibited by antibiotics.
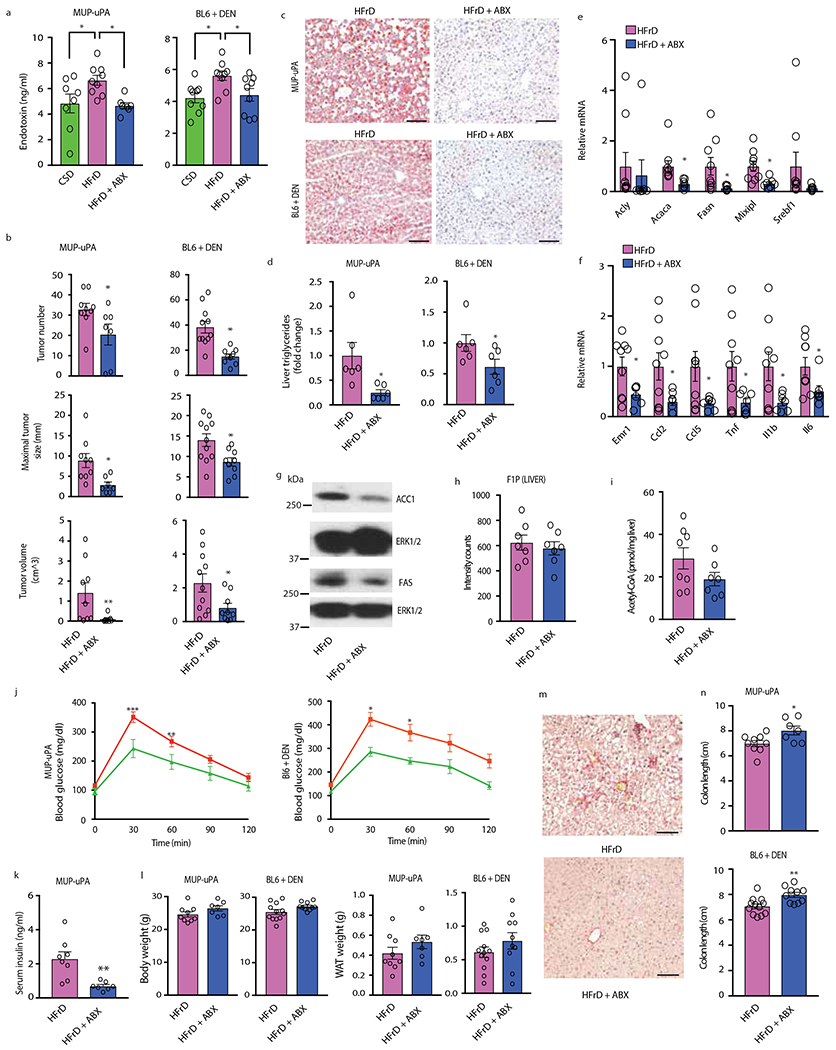
(a) CSD and HFrD-fed MUP-uPA (n=8 CSD, n=9 HFrD, n=7 HFrD+Abx) and DEN- challenged BL6 (n=9 per group) male mice were treated or not with Abx cocktail for 5 months, after which serum endotoxin concentrations were measured. (b) HCC burden in MUP-uPA (n=9 HFrD, n=7 HFrD+Abx) and DEN-challenged BL6 (n=11 HFrD, n=9 HFrD+Abx) males given HFrD alone or HFrD plus Abx cocktail. (c) Hepatosteatosis in MUP-uPA and DEN-challenged BL6 males given HFrD ± Abx for 5 months determined by ORO staining of liver sections (n=6). Scale bars, 100 μm. (d) Liver triglycerides in above mice (n=6 per group). (e-g) Expression of lipogenic (e) and inflammatory (f) genes (n=9 HFrD, n=7 HFrD+Abx) and IB analysis of liver ACC1 and FAS (g). (h, i) Liver F1P abundance (MUP-uPA; n=7 samples from different males per group) (h) and cytosolic acetyl-CoA (n=8 HFrD, n=7 HFrD+Abx samples from different males per group; the HFrD bar is same as in Extended Data Figure 2a). (j, k) glucose tolerance (MUP-uPA, n=10 serum samples from different males per group, BL6, n=7 serum samples from different males per group) (j) and non-fasting serum insulin (n=8 HFrD, n = 7 HFrD+Abx serum samples from different males per group) (k). (l) Total body and white adipose tissue weights in indicated males (MUP-UPA, n = 9 HFrD, n = 7 HFrD+Abx; BL6, n = 12 HFrD, n = 10 HFrD+Abx) (m) Liver fibrosis in HFrD-fed MUP-uPA males was examined by Sirius Red staining (n=7), representative images. (n) Colon length in indicated males (MUP-uPA, n=9 HFrD, n=7 HFrD+Abx; BL6 n=12 HFrD, n=10 HFrD+Abx ; HFrD bars are same as in E.D. Fig. 1f). Scale bars, 100 μm. Unpaired two-sided Student’s t test was used in panels b, d-f, h, I, k, l and n to determine mean ± SEM, whereas panel a was analyzed by one-way ANOVA and Tukey’s multiple comparison test and panel j was analyzed using two-way ANOVA and Sidak’s multiple comparison test. *P < 0.05, **P < 0.01, ***P < 0.001. Benjamini-Hochberg FDR adjustment for P-values was performed on data in panels e and f.
Extended Data Figure 6. Antibiotics attenuate fructose-induced hepatosteatosis, barrier deterioration and other alterations in BL6 mice.
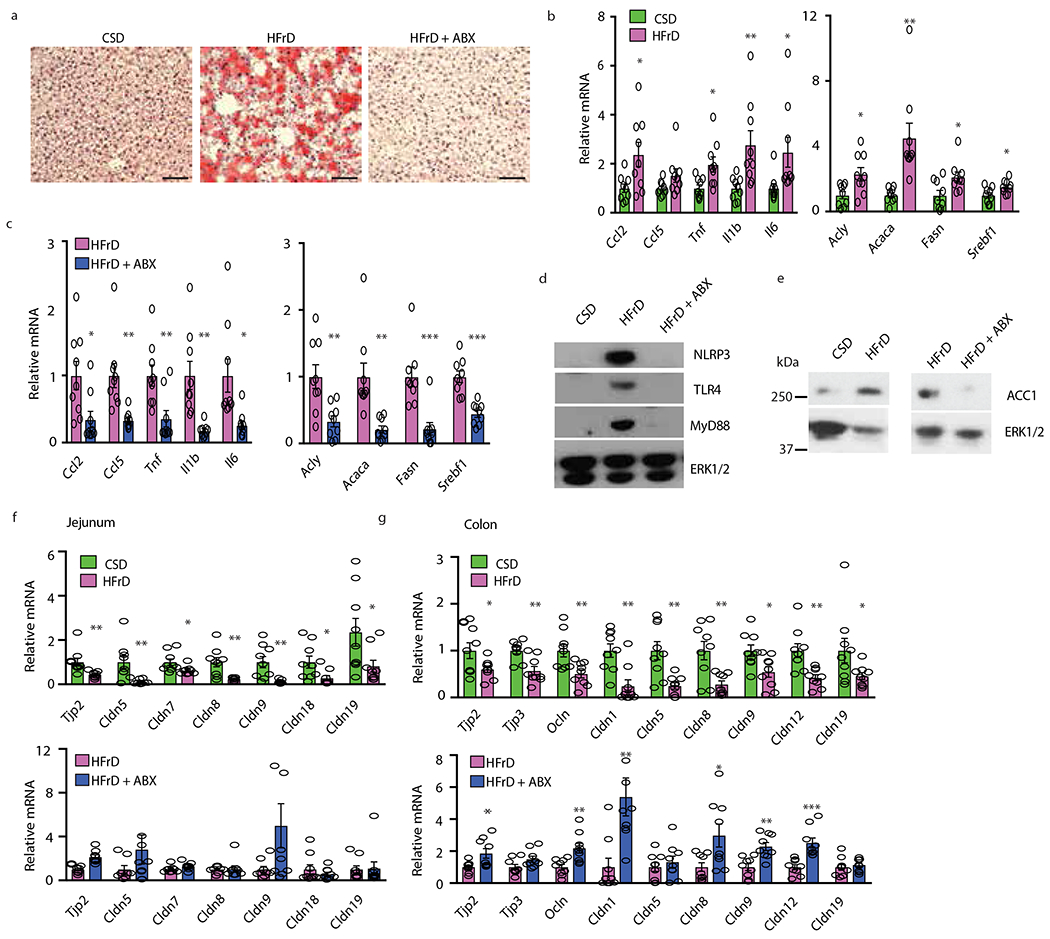
(a to g) Male BL6 mice were fed CSD or HFrD ± Abx. (a) After five months, liver tissue sections were stained with ORO (n=6 samples from different mice per group). Representative images are shown. Scale bars, 100 μm. (b and c) Inflammatory and DNL-related mRNAs in liver were determined by Q-RT- PCR (n=9 per group). (d and e) Inflammatory and lipogenic proteins in liver lysates were IB analyzed (n=7 protein lysates per group). Representative blots are shown. (f and g) Q-RT-PCR analysis of TJPmRNAs in jejunum (f) and colon (g) (n=9 per group). Two-sided Student’s t test was used to determine mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001. Benjamini-Hochberg FDR adjustment for P-values was performed on data in panels b,c,f and g.
Extended Data Figure 7. Fructose drink enhances hepatosteatosis, expression of liver mRNAs encoding inflammatory cytokines and DNL-related enzymes and barrier deterioration.
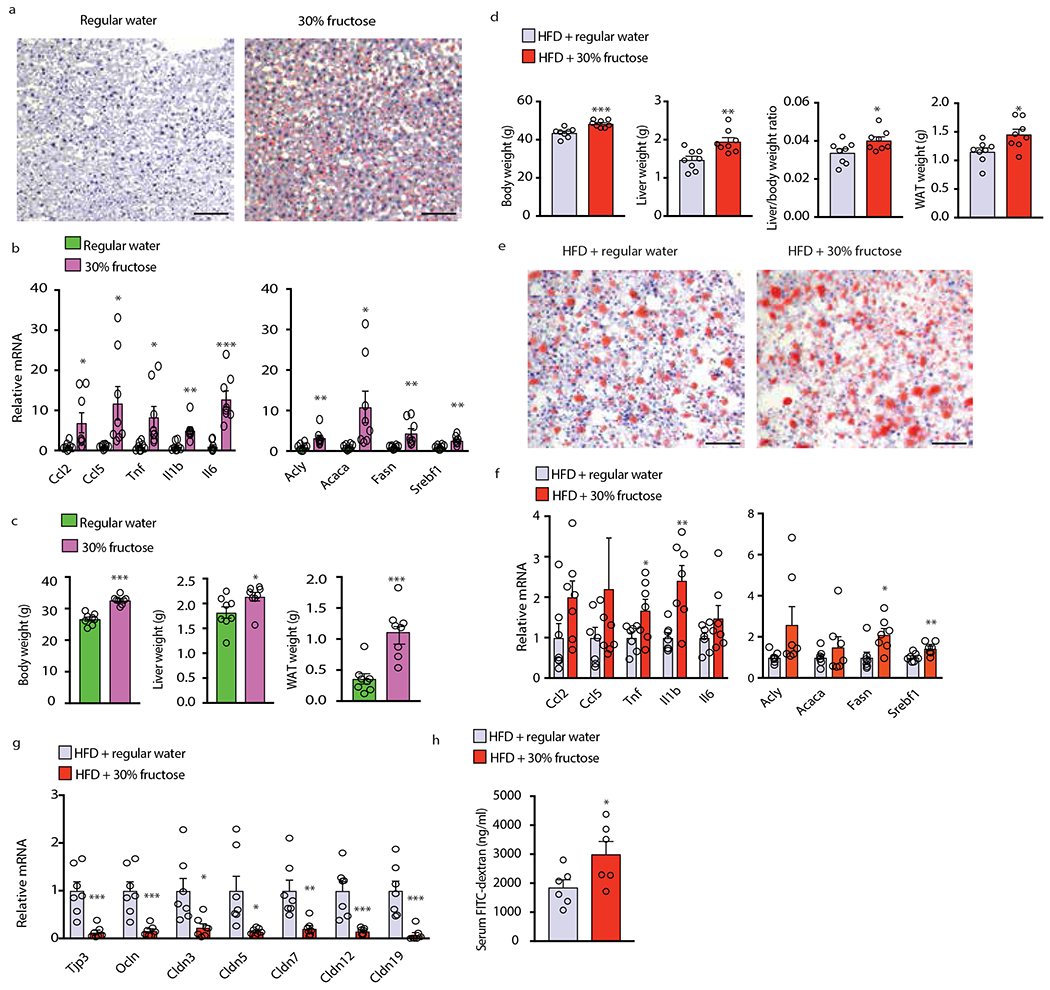
(a-c) BL6 males were placed on 30% fructose in drinking water or regular water and NCD for 5 months (n=8 per group). (a) Liver sections were ORO stained and representative images are shown (n=6 sections per group). (b) Liver inflammatory and DNL-related mRNAs were PCR analyzed (n=8 samples per group). (c) Total body, liver and white adipose tissue weights were measured (n=8). (d - h) BL6 males were placed on 30% fructose drink or regular water and HFD for 3 months. Body, liver and WAT weight and liver/body weight ratio were determined (n=8 per group) (d). Liver sections were analyzed by ORO staining (n=6 sections per group). Representative images are shown (e). Liver mRNAs encoding cytokines and DNL-related proteins were measured using Q-RT-PCR (n=7 per group) (f). Expression of TJP mRNAs in colon (n=7 per group) (g) and FITC-dextran in serum (n=6 per group) (h) were also measured. Scale bars, 100 μm. Two-sided Student’s t test was used to determine mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001. Benjamini-Hochberg FDR adjustment for P-values was performed on data in panels b,f and g.
Extended Data Figure 8. Fructose-induced upregulation of YAP target genes and attenuation of TJP mRNA downregulation, barrier deterioration, hepatosteatosis and induction of DNL-related proteins in MUP-uPA/gp130Act and CCN1-treated mice.
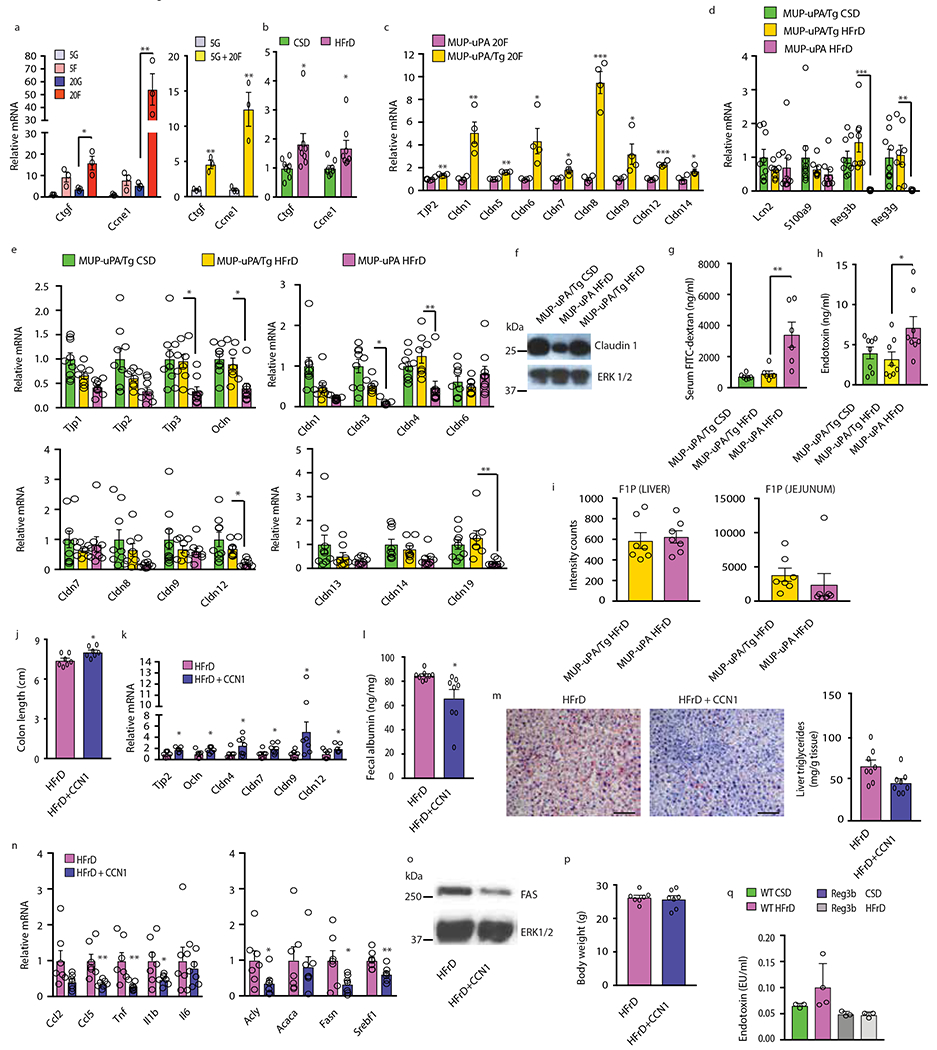
(a, b) Expression of indicated mRNAs in BL6 enteroids maintained in media containing the indicated glucose and fructose concentrations (mM) (n=3, enteroids from three male mice; two repeats with similar results). (a) and colonic mucosa of MUP-uPA males (n=8 per group) (b) was measured by Q-RT-PCR. (c) Enteroids from MUP-uPA and MUP- uPA/gp130Act mice (n=4, enteroids from four mice per group) were incubated with 20 mM fructose and mRNAs were measured by Q-RT-PCR. Two repeats with similar results. (d and e) Anti-microbial (d) and TJP (e) mRNAs in colon tissues of MUP-uPA/gp130Act (MUP-uPA/Tg) mice fed CSD (n=9 per group) or HFrD (n=8 per group) compared to MUP-uPA mice fed HFrD for five months (n=9 per group). Asterisks indicate significant changes between MUP-uPA and MUP-uPA/Tg mice fed HFrD. (f) IB of claudin-1 in colons of MUP-uPA/gp130Act and MUP-uPA mice kept on CSD or HFrD. A representative blot is shown. (g) FITC-dextran serum concentrations in indicated mice (n=6 serum samples per group). (h) Serum endotoxin concentrations in indicated mice (n=8 serum samples per group). (i) F1P abundance in liver and jejunum of indicated mice (arbitrary units; n=7 samples per group). (j to p) 6-week-old MUP-uPA males were fed HFrD for 3 months and i.p. injected 2 μg CCN1 or vehicle (PBS) every other day for the last 4 weeks of HFrD feeding. Length (j) and TJP mRNA expression (k) were determined on excised colons (n=7 per group). Fecal albumin was measured by ELISA (n=8 serum samples per group) (l). Liver ORO staining (n=6 tissue samples) and triglyceride concentrations (n=8 samples per group) (m), inflammatory and lipogenic gene expression (n=7 per group) (n) and FAS protein amounts (n=7 per group), representative blots (o). (p) Body weight at the end of the CCN1 treatment course (n=7 males per group). (q) Serum endotoxin in indicated mice (n=3 per group). Scale bars, 100 μm. In panels a, d, e, g, h and q one-way ANOVA with Tukey’s multiple comparison test was used to determine mean ± SEM. Two-sided Student’s t test was used to determine mean ± SEM in panels b, c, i-n, and p. *P < 0.05, **P < 0.01, ***P < 0.001. Benjamini-Hochberg FDR adjustment for P-values was performed on data in panels c, k and n.
Extended Data Figure 9. Low-dose LPS treatment stimulates hepatosteatosis and increases lipogenic and inflammatory gene expression and effect of fructose on microbiome diversity.
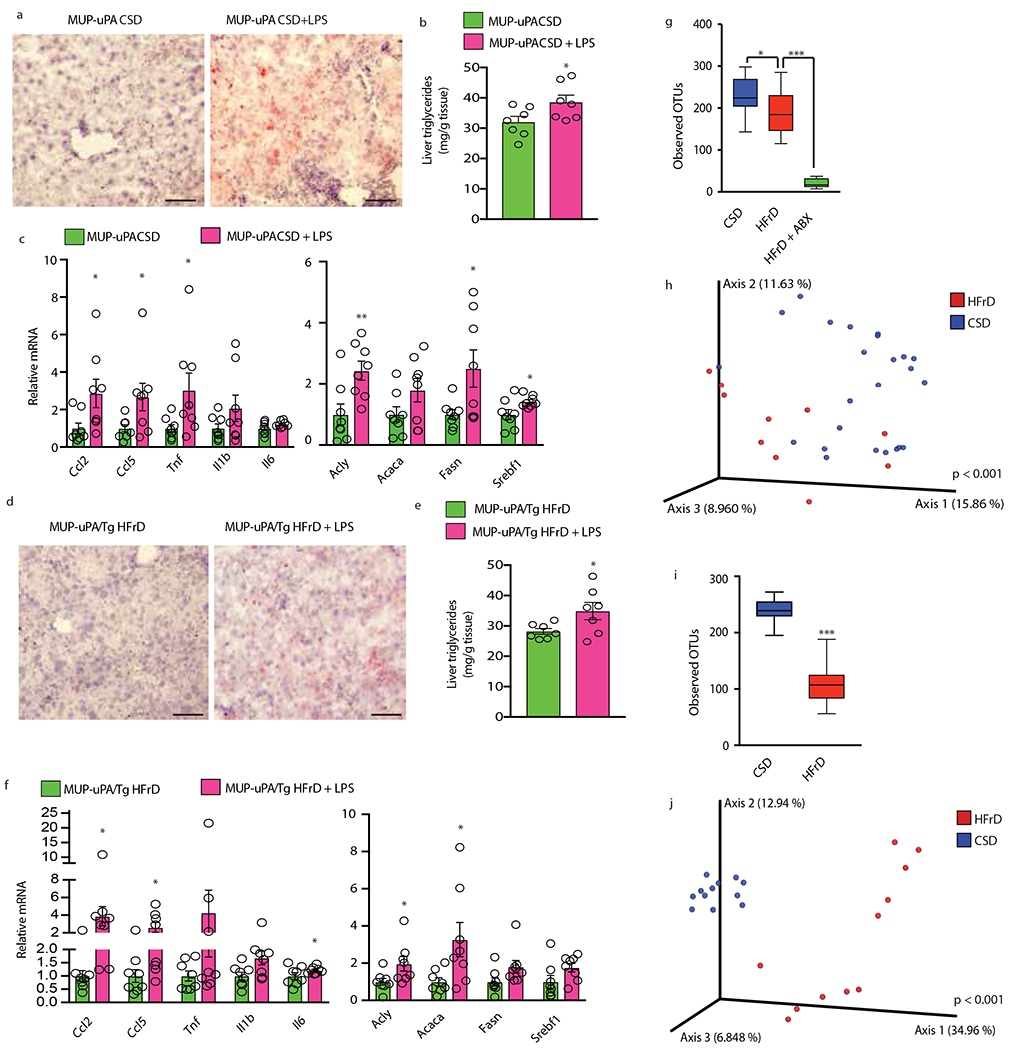
(a to c) Six-week-old MUP-uPA males were fed CSD and given daily i.p. injections of LPS (0.25 mg/kg) or vehicle (PBS). (a) ORO staining of liver sections (n = 7 per group; scale bars-100 μm), representative images are shown. (b) Liver triglycerides in above mice (n=7 per group). (c) Q-RT-PCR analysis of lipogenic and inflammatory mRNAs (n=8 per group). (d to f) Six-week-old MUP-uPA/Tg males were fed HFrD and received daily i.p. injections of LPS or PBS. (d) ORO staining of liver sections (n=7 per group; scale bars-100 μm), representative images are shown. (e) Liver triglycerides (n = 7 per group). (f) Relative mRNA amounts of lipogenic and inflammatory genes (n = 8 per group). (g and i) Relative abundance of significantly differing taxa. Significance was determined by ANCOM. Box-plots of observed operational taxonomic units (OTUs) α-diversity indices comparing stool samples of MUP-uPA (g) and MUP-uPA/Tg (i) mice treated as indicated. (h and j) Principal coordinate analysis of microbiome data using unweighted UniFrac distances; difference between CSD and HFrD was significant in both MUP-uPA (h) (pseudo-F statistic = 2.88, P < 0.001, n = 10 and 24) and MUP-uPA/Tg (j) (pseudo-F statistic = 9.19, P < 0.001, n = 11 and 12) mice as determined by PERMANOVA. Two-sided Student’s t test was used to determine mean ± SEM in panels b, c, e and f. *P < 0.05, **P < 0.01, ***P < 0.001. Benjamini-Hochberg FDR adjustment for P-values was performed on data in panels c and f.
Supplementary Material
Acknowledgments
We thank Dr. Manuela Raffatellu for advice and discussion and Vivian Sheen, Winnie Gong, Jean Yung and Kevin Lam for technical support. Research was supported by grants from the NIH (P42ES010337, DK120714, R01CA198103, R01AI043477, R01CA211794, R01CA234128 and to M.K.; R03CA223717 to J.T.; T32AI007469 and K22AI139444 to R.MN..; R01CA192642, R01CA218254 to M.T.D.-M.; R01DK108743, R01CA207177 and R01CA211794 to J.M.; U01AA027681 to S.S. and M.K.; and R01CA188652 to C.M.M.), JSPS KAKENHI (JP15K21775) and “Kibou Projects” Startup Support for Young Researchers in Immunology (to K.T.), and the Australian NHMRC (APP112227), to M.A.F. and M.K. who holds the Ben and Wanda Hildyard Chair for Mitochondrial and Metabolic Diseases; work in F.R.G. laboratory was supported by institutional funds from the Georg-Speyer-Haus and by the LOEWE Center Frankfurt Cancer Institute (FCI) funded by the Hessen State Ministry for Higher Education, Research and the Arts [III L 5 - 519/03/03.001 - (0015)], NIH K01DK116917 and P30DK063491 pilot award to J.D.W.; and S10OD020025, R01ES027595, and P42ES010337 to M.J. M.A.F is a Senior Principal Research Fellow of the NHMRC Australia (APP 1116936).
Footnotes
Competing Interests Statement
M.K. holds a US patent on the use of MUP-uPA mice to study NASH and HCC (10,034,462 B2) and had received research support from Jansen Pharmaceuticals. All other authors declare no competing interests.
Data Availability
RNA-Seq data are deposited in NCBI Gene Expression Omnibus (GEO) database under accession number GSE119080. Microbiome sequencing data are deposited in EMBL-EBI under accession number ERP110352.
References
- 1.Spengler EK & Loomba R Recommendations for Diagnosis, Referral for Liver Biopsy, and Treatment of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Mayo Clin Proc 90, 1233–1246 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stickel F & Hellerbrand C Non-alcoholic fatty liver disease as a risk factor for hepatocellular carcinoma: mechanisms and implications. Gut 59, 1303–7 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Tilg H & Moschen AR Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 52, 1836–1846 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Fukui H Increased Intestinal Permeability and Decreased Barrier Function: Does It Really Influence the Risk of Inflammation? Inflamm. Intest. Dis 1, 135–145 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lebeaupin C et al. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol 69, 927–947 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Puri P et al. Activation and Dysregulation of the Unfolded Protein Response in Nonalcoholic Fatty Liver Disease. Gastroenterology 134, 568–576 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Rahman K et al. Loss of Junctional Adhesion Molecule A Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology 151, 733–746.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakagawa H et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26, 331–343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim JY et al. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 175, 133–145.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vos MB & Lavine JE Dietary fructose in nonalcoholic fatty liver disease. Hepatology 57, 2525–2531 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Jin R et al. Fructose induced endotoxemia in pediatric nonalcoholic Fatty liver disease. Int J Hepatol 2014, 560620 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kavanagh K et al. Dietary fructose induces endotoxemia and hepatic injury in calorically controlled primates. Am J Clin Nutr 98, 349–357 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spruss A, Kanuri G, Stahl C, Bischoff SC & Bergheim I Metformin protects against the development of fructose-induced steatosis in mice: role of the intestinal barrier function. Lab. Investig 92, 1020–1032 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Lambertz J, Weiskirchen S, Landert S & Weiskirchen R Fructose: A Dietary Sugar in Crosstalk with Microbiota Contributing to the Development and Progression of Non-Alcoholic Liver Disease. Front Immunol 8, 1159 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oh J-H et al. Dietary Fructose and Microbiota-Derived Short-Chain Fatty Acids Promote Bacteriophage Production in the Gut Symbiont Lactobacillus reuteri. Cell Host Microbe 25, 273–284.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Chang PV, Hao L, Offermanns S & Medzhitov R The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. U. S. A 111, 2247–52 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelly CJ et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 17, 662–71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geidl-Flueck B & Gerber PA Insights into the Hexose Liver Metabolism-Glucose versus Fructose. Nutrients 9, 1026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Softic S, Cohen DE & Kahn CR Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig Dis Sci 61, 1282–1293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Softic S et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Invest 127, 4059–4074 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grivennikov SI et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491, 254–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karin M & Clevers H Reparative inflammation takes charge of tissue regeneration. Nature 529, 307–315 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barnett MPG et al. Changes in colon gene expression associated with increased colon inflammation in interleukin-10 gene-deficient mice inoculated with Enterococcus species. BMC Immunol. 11, 39 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siddiqui RA et al. Comparative study of the modulation of fructose/sucrose-induced hepatic steatosis by mixed lipid formulations varying in unsaturated fatty acid content. Nutr. Metab. (Lond) 12, 41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Landy J et al. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J. Gastroenterol 22, 3117–3126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaeken J, Pirard M, Adamowicz M, Pronicka E & van Schaftingen E Inhibition of phosphomannose isomerase by fructose 1-phosphate: an explanation for defective N-glycosylation in hereditary fructose intolerance. Pediatr. Res 40, 764–6 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Kaser A & Blumberg RS Endoplasmic reticulum stress and intestinal inflammation. Mucosal Immunol. 3, 11–6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balda MS & Matter K Tight junctions. J. Cell Sci 111 (Pt 5), 541–7 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Shalapour S et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 551, 340–345 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vos MB, Kimmons JE, Gillespie C, Welsh J & Blanck HM Dietary fructose consumption among US children and adults: the Third National Health and Nutrition Examination Survey. Medscape J. Med 10, 160 (2008). [PMC free article] [PubMed] [Google Scholar]
- 31.Low BC et al. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett 588, 2663–2670 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Choi JS, Kim K-H & Lau LF The matricellular protein CCN1 promotes mucosal healing in murine colitis through IL-6. Mucosal Immunol. 8, 1285–1296 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taniguchi K et al. A gp130–Src–YAP module links inflammation to epithelial regeneration. Nature 519, 57–62 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miki T, Holst O & Hardt W-D The bactericidal activity of the C-type lectin RegIIIβ against Gram-negative bacteria involves binding to lipid A. J. Biol. Chem 287, 34844–55 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schröder M & Kaufman RJ ER stress and the unfolded protein response. Mutation Research - Fundamental and Molecular Mechanisms of Mutagenesis vol. 569, 29–63 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Metidji A et al. The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity 49, 353–362.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fukui H Increased Intestinal Permeability and Decreased Barrier Function: Does It Really Influence the Risk of Inflammation? Inflamm. Intest. Dis 1, 135–145 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wiest R, Albillos A, Trauner M, Bajaj JS & Jalan R Targeting the gut-liver axis in liver disease. J. Hepatol 67, 1084–1103 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Ammirante M, Luo J-L, Grivennikov S, Nedospasov S & Karin M B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 464, 302–305 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu S et al. Polysome profiling in liver identifies dynamic regulation of endoplasmic reticulum translatome by obesity and fasting. PLoS Genet 8, e1002902 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sato T et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–72 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo W, Friedman MS, Shedden K, Hankenson KD & Woolf PJ GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo W & Brouwer C Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 29, 1830–1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson LR et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marotz C et al. DNA extraction for streamlined metagenomics of diverse environmental samples. Biotechniques 62, 290–293 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Amir A et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuczynski J et al. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Bioinforma Chapter 10, Unit 10.7. (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee WN et al. In vivo measurement of fatty acids and cholesterol synthesis using D2O and mass isotopomer analysis. Am. J. Physiol. Metab 266, E699–E708 (1994). [DOI] [PubMed] [Google Scholar]
- 52.Wallace M et al. Enzyme promiscuity drives branched-chain fatty acid synthesis in adipose tissues. Nat. Chem. Biol 14, 1021–1031 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kireeva ML, MO FE, Yang GP & Lau LF Cyr61, a product of a growth factor-inducible immediate-early gene, promotes cell proliferation, migration, and adhesion. Mol. Cell. Biol 16, 1326–1334 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mandal S et al. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb. Ecol. Health Dis 26, 27663 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.