

Abstract
Background:
Increased circulating myeloid-derived suppressor cells (MDSCs) are independently associated with poor long-term clinical outcomes in sepsis. Studies implicate subsets of MDSCs having unique roles in lymphocyte suppression; however, characterization of these cells after sepsis remains incomplete. We performed a pilot study to determine the transcriptomic landscape in MDSC subsets in sepsis using single-cell RNAseq (scRNA-seq).
Methods:
A mixture of whole blood myeloid-enriched and Ficoll-enriched PBMC’s from two late septic patients on post-sepsis day 21 and two control subjects underwent Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-seq).
Results:
We successfully identified the three MDSC subset clusters – granulocytic (G-), monocytic (M-), and early (E-) MDSCs. Sepsis was associated with a greater relative expansion of G-MDSCs versus M-MDSCs at 21 days as compared to control subjects. Genomic analysis between septic patients and control subjects revealed cell-specific and common differential expression of genes in both G-MDSC and M-MDSC subsets. Many of the common genes have previously been associated with MDSC proliferation and immunosuppressive function. Interestingly, there was no differential expression of several genes demonstrated in the literature to be vital to immunosuppression in cancer-induced MDSC.
Conclusion:
This pilot study successfully demonstrated that MDSCs maintain a transcriptomic profile that is immunosuppressive in late sepsis. Interestingly, the landscape in chronic critical illness is partially dependent on the original septic insult. Preliminary data would also indicate immunosuppressive MDSCs from late sepsis patients appear to have a somewhat unique transcriptome from cancer and/or other inflammatory diseases.
Keywords: transcriptome, CITE-seq, myeloid derived suppressor cells, MDSC, Sepsis, chronic critical illness
Introduction
Advancements in detection and treatment of surgical sepsis have led to decreases in acute mortality and increases in two clinical trajectories: rapidly recovery and chronic critical illness (CCI) (1). CCI is defined as prolonged stays in the ICU with unresolved organ dysfunction (2). More than 40% of surgical sepsis patients develop CCI and have poor one-year outcomes, defined as poor one-year quality of life and increased mortality after sepsis, as compared to those that rapidly recover (1). Additionally, CCI patients use an increased amount of hospital resources and are responsible for a larger financial burden as compared to other sepsis patients (3).
The mechanisms of CCI after sepsis remain undefined (4, 5). Our laboratory has determined that CCI can manifest itself as a persistent inflammation, immunosuppression and catabolism syndrome (PICS) (6–10). We have demonstrated that part of the PICS pathobiology is induced by a host ‘myelodyscrasia,’ which includes increased bone marrow and extramedullary proliferation of myeloid-derived suppressor cells (MDSCs) (2, 3). MDSCs are a heterogeneous group of immature myeloid cells described in many diseases and physiologic states including cancer, transplantation, pregnancy, obesity, autoimmunity and sepsis (10–12). In acute sepsis, MDSCs are thought to suppress acute inflammatory responses and resolve inflammation (13–15). However, continued MDSC expansion and infiltration, as seen in CCI, contributes to host immunosuppression (13, 14, 16). Dmitry Gabrilovich, one of the first individuals to identify MDSCs in cancer patients, has defined this MDSC differentiation, accumulation, and persistence in the host as ‘pathologic myeloid cell activation’ (17).
Our laboratory and others have demonstrated that circulating MDSCs are increased after sepsis are independently associated with poor long-term clinical outcomes (18, 19). Previous studies have revealed an expansion of multiple subsets of MDSCs, each thought to have unique roles in inflammation and immune suppression [reviewed in (17)]. However, differences in the numbers and expression patterns of different MDSC subsets after sepsis remain controversial. Therefore, we sought to determine the phenotype and transcriptome of circulating MDSCs in CCI patients 21 days after sepsis, when these cells are known to be immunosuppressive (20), using single cell RNA sequencing (scRNA-seq).
Materials and Methods
Study Design, Patient Enrollment and Classification
The study was registered with clinicaltrials.gov (NCT02276417) and conducted by the Sepsis and Critical Illness Research Center at the University of Florida. All patients eligible for inclusion in the study were enrolled within 12 hours of sepsis protocol onset on a delayed waiver of consent, which was approved by our Institutional Review Board. If written informed consent could not be obtained from the patient or their legally assigned representative within 96 hours of study enrollment, the patient was removed from the study and all collected biologic samples and clinical data were destroyed. Screening for sepsis was carried out using the Modified Early Warning Signs-Sepsis Recognition System (MEWS-SRS), which quantifies derangements in vital signs, white blood cell count, and mental status. All patients with sepsis were managed using a standardized, evidence-based protocol that emphasizes early goal-directed fluid resuscitation as well as other time-appropriate interventions such as administration of broad-spectrum antibiotics. Empiric antibiotics were chosen based on current hospital antibiograms in conjunction with the suspected source of infection. Antimicrobial therapy was then narrowed based on culture and sensitivity data.
Inclusion criteria consisted of the following: (a) admission to the surgical or trauma ICU; (b) age ≥18 years; (c) clinical diagnosis of sepsis or septic shock as defined by the 2016 SCCM/ESICM International Sepsis Definitions Conference (21) with this being the patient’s first septic episode; and, (d) entrance into our sepsis clinical management protocol as previously described (22). Exclusion criteria consisted of: (a) refractory shock (i.e. patients expected to die within the first 24 hours); (b) an inability to achieve source control (i.e. irreversible disease states such as unresectable dead bowel); (c) pre-sepsis expected lifespan <3 months; (d) patient/family not committed to aggressive management; (e) severe CHF (NYHA Class IV); (f) Child-Pugh Class C liver disease or pre-liver transplant; (g) known HIV with CD4+ count <200 cells/mm3; (h) organ transplant recipient or use of chronic corticosteroids or immunosuppressive agents; (i) pregnancy; (j) institutionalized patients; (k) chemotherapy or radiotherapy within 30 days; (l) severe traumatic brain injury (i.e. evidence of neurological injury on CT scan and a GCS <8); (m) spinal cord injury resulting in permanent sensory and/or motor deficits; or, (n) inability to obtain informed consent.
Patients were diagnosed with sepsis or septic shock using the Sepsis-3 definitions (21). CCI was defined as an ICU length of stay greater than or equal to 14 days with evidence of persistent organ dysfunction, measured using components of the Sequential Organ Failure Assessment (SOFA) score (i.e. cardiovascular SOFA ≥ 1, or score in any other organ system ≥ 2). Patients with an ICU LOS less than 14 days would also qualify for CCI if they were discharged to another hospital, a long-term acute care facility, or to hospice and demonstrated continuing evidence of organ dysfunction at the time of discharge. Those patients experiencing death within 14 days of sepsis onset were excluded from the analyses.
Human Blood Collection and Sample Preparation
Ethylenediaminetetraacetic acid-anticoagulated human whole blood samples were collected by venipuncture from two patients on day 21 post meeting Sepsis-3 criteria (21) and two control subjects. Samples were stored on ice and processed within six hours after blood draw. Each sample was divided to undergo two separate enrichment processes. Peripheral blood mononuclear cells (PBMC) from the half of each human whole blood sample were collected using Ficoll-Paque™ PLUS (GE Healthcare) and density gradient centrifugation. Myeloid cells were collected from the other half of each whole blood sample using RosetteSep™ HLA Myeloid Cell Enrichment Kit (Stemcell). A 1:3 mixture of enriched PBMCs to myeloid cells was created for the two CCI sepsis patients and two healthy control subjects to undergo further analysis. Although single-cell technology allows for detection of smaller cell populations, the original samples were enriched to ensure our ability to adequately analyze and compare the small target population of MDSCs (especially in healthy controls, which have very limited numbers/percentages of MDSCs), while also allowing us to characterize other important immune cells present in late sepsis (e.g. lymphocytes).
scRNA-seq/CITE-seq and Library Construction
Gene expression libraries were prepared from 5,000 cells using the Chromium Single Cell 5’ Bead and Library Kit v1 (10x Genomics). Libraries were sequenced on an Illumina HiSeq™ instrument at a target read depth of 50,000 reads per cell. After quality control to separate high quality cells from empty cells and doublets, the average number of genes detected per cell was 592, and an average of 1421 total Unique Molecular Identifier (UMI) counts per cell.
Myeloid cells were labeled with oligo-tagged antibodies to CD33, CD11b, CD14, CD15, CD66b, Lox1 and HLA-DR. Labeled cells (5,000) were encapsulated for droplet-based CITE-seq utilizing the 10x Genomics Chromium Controller™ platform. Granulocytic (G-), Monocytic (M-), and Early (E-) MDSCs were identified as preciously described by Bronte et al (23)): G-MDSCs (Lin− CD33+ CD11b+ CD14− and CD15+ or CD66b+); M-MDSCs (Lin− HLADRlow/− CD33+ CD11b+ CD14+ CD15− CD66b−); and, E-MDSCs (Lin− HLADRlow/− CD33+ CD11b+ CD14− CD15− CD66b−). Complementary DNA (cDNA) libraries were constructed to assess gene expression (RNA) and surface phenotype (protein) for MDSC subpopulations simultaneously. We also performed CITE-seq analysis on T lymphocytes (CD4+ T, CD8+ T and activated T cells), activated macrophages, monocytes, dendritic plasmacytoid cells, and platelets from the same PBMC fraction, using additional oligo-tagged antibodies to CD3, CD127, CD183, CD4, CD196, and CD25.
Processing of Sequencing Reads and Generation of Gene-Barcode Matrices
Raw sequencing reads were processed using Cell Ranger v3.0.0 to create a raw (unfiltered) gene-barcode matrix. Briefly, Cell Ranger mkfastq was used to make fastq files from bcl files. Next, Cell Ranger count was used for aligning sequencing reads to the hg19 reference genome (refdata-cellranger-hg19–3.0.0), obtained from (https://support.10xgenomics.com/single-cell-gene-expression/software/release-notes/build) using STAR (33). For confidently mapped reads, UMI sequences were collapsed and the number of UMI reads per gene were stored in the raw gene-barcode matrix (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/algorithms/overview). The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Filtering of Barcodes/Quality Control
We distinguished true cells from background droplets using the emptyDrops method implemented in the DropletUtils Bioconductor R package (24). By testing each cell versus an ambient RNA distribution, barcodes with a false discovery rate adjusted p-value < 0.01 were retained for further consideration. We performed a second quality control step to identify cells with low RNA content, possible doublets, or dead/damaged cells, where we filtered cells based on the total number of UMIs per cell, the number of genes expressed, and the percentage of mitochondrial reads per cell. We used the scatter R package to identify outlier identify cells in any of these metrics, where outlier was defined as three median absolute deviations (MADs) from the median (25). The remaining number of cells in each sample were 1,471 and 869 for the two healthy controls and 1,209 and 3,935 in the two sepsis subjects. The scRNA-seq data were normalized using the NormalizeData function in the Seurat R package v 3.1.5 (21) in which the total counts for each cell were scaled to have 10000 total counts. The antibody counts were normalized using the same function with the centered log ratio transformation method.
Dataset Integration and Dimensionality Reduction
The datasets were integrated as detailed in McCarthy et al (25). Briefly, canonical correlation analysis (CCA) was performed to identify shared sources of variation across the datasets, and mutual nearest neighbors in the CCA space were identified to produce anchors between datasets. Briefly, highly variable genes accounting for the majority of the heterogeneity within each sample were identified with the FindVariableFeatures function in the Seurat R package v 3.1.5 (26), which fits a local polynomial regression (loess) model to the mean-variance relationship and selects the top 2000 genes with greatest standardized deviation from the fitted model. Using these features, anchors between the datasets which correspond to similar cells across datasets were identified using the FindIntegrationAnchors function, and this was used as input into the IntegrateData function to generate an integrated dataset. For dimensionality reduction, expression values for each gene in the integrated dataset were scaled to have a mean of zero and standard deviation of one using the ScaleData function. Principal component analysis (PCA) was run on this matrix using the RunPCA function in Seurat. For visualization Uniform Manifold Approximation and Projection (UMAP), a common dimensionality reduction method in scRNA-seq (26), plots were created based on the top 30 principal components using the RunUMAP function in Seurat.
Clustering and Cluster Differential Expression Analysis
The Seurat package also includes functions to identify clusters of cells based on their similarity. Cells were clustered into groups of similar transcriptomic profiles based on the first 30 principal components of the integrated dataset. A k-nearest neighbors (KNN) graph used was constructed based on the first 30 principle components to create a shared nearest neighbors graph based on the Jaccard index between each cell and its 20 nearest neighbors, as implemented in the FindNeighbors function in Seurat. Clusters were then identified by partitioning this graph using the Louvain community detection algorithm with a resolution of 0.5, as implemented in the FindClusters function in Seurat. Marker genes for each cluster were identified by comparing each individual cluster with the remaining pooled clusters for each sample using the Wilcoxon rank sum test implemented in the Seurat R package. Differentially expressed genes across conditions (sepsis versus healthy) and pooling cells across subjects was performed for each cell cluster using the Wilcoxon rank sum test. P-values were adjusted for multiple testing using the Bonferroni method. Enrichment analysis of gene sets was also performed using the R package clusterProfiler (27). All genes with adjusted P-value for differential expression < 0.01 were included for the enrichment analysis. For KEGG, the enrichment P-value cutoff was set to pvalueCutoff = 0.05 and for GO we set qvalueCutoff = 0.01. For gene expression heatmaps, hierarchical clustering with method=“ward.D2” and based on the Euclidean distance matrix was used for the rows (genes) as well as the columns (cells). Cells were clustered within the highlighted groups separately for clear visualization. For calculating the distance matrix, counts were natural logged after a pseudocount of one was added. All analyses were performed using R version 3.6.3.
Annotation of Cell Clusters
Cell clusters were manually annotated based on their expression of known marker genes and leveraging both the RNA expression counts and antibody derived tag counts. Details of this analysis are provided in supplemental methods, Supplemental Digital Content (SDC) 1.
Results
Identification of Cell Types
We used scRNA-seq to identify cells in a 3:1 myeloid:PBMC mixture from two sepsis patients, post-sepsis day 21 with CCI, and two healthy control subjects. Sepsis patient 1 (Sep1) was enrolled due to bacterial sepsis (as defined by Sepsis-3 guidelines), while sepsis patient 2 (Sep2) was enrolled for fungemia-causing septic shock (21). Patient characteristics are detailed in Supplemental Digital Content 2. We leveraged the CITE-seq data to better annotate the cell clusters and have described this in detail in the Methods along with relevant marker expression plots noted in Figure 1A, which shows mRNA and corresponding epitope Antibody Derived Tag (ADT) signals for selected CITE-seq markers. We identified the three MDSC subset clusters as classified by Gabrilovich and colleagues (23): Granulocytic (G-CD33+CD11b+HLA-DRlow/−CD14−), Monocytic (M-;CD33+CD11b+HLA-DRlow/−CD14+), and, Early (E;CD33+CD11b+ HLA-DRlow/−CD14−CD15−) (Figure 1). Absolute cell counts of MDSC subsets for each sepsis patient and healthy subject from a target cell read of 5000 cells can be found in SDC2. Sepsis-induced CCI was associated with a greater relative number of G-MDSCs versus M-MDSCs when compared to control patients (64% versus 26% G-MDSCs, 33% versus 74% M-MDSCs, and 3% and 0 % E-MDSCs of total MDSCs). CCI after fungemic septic shock had similar expansion of G-MDSCs when compared to bacteremic sepsis (67%% vs 60% G-MDSCs of total MDSC count, respectively). E-MDSCs were only detectable in the bacterial sepsis patient (Sep1); thus, differential expression with control subjects could not be ascertained (Figure 1).
Figure 1.
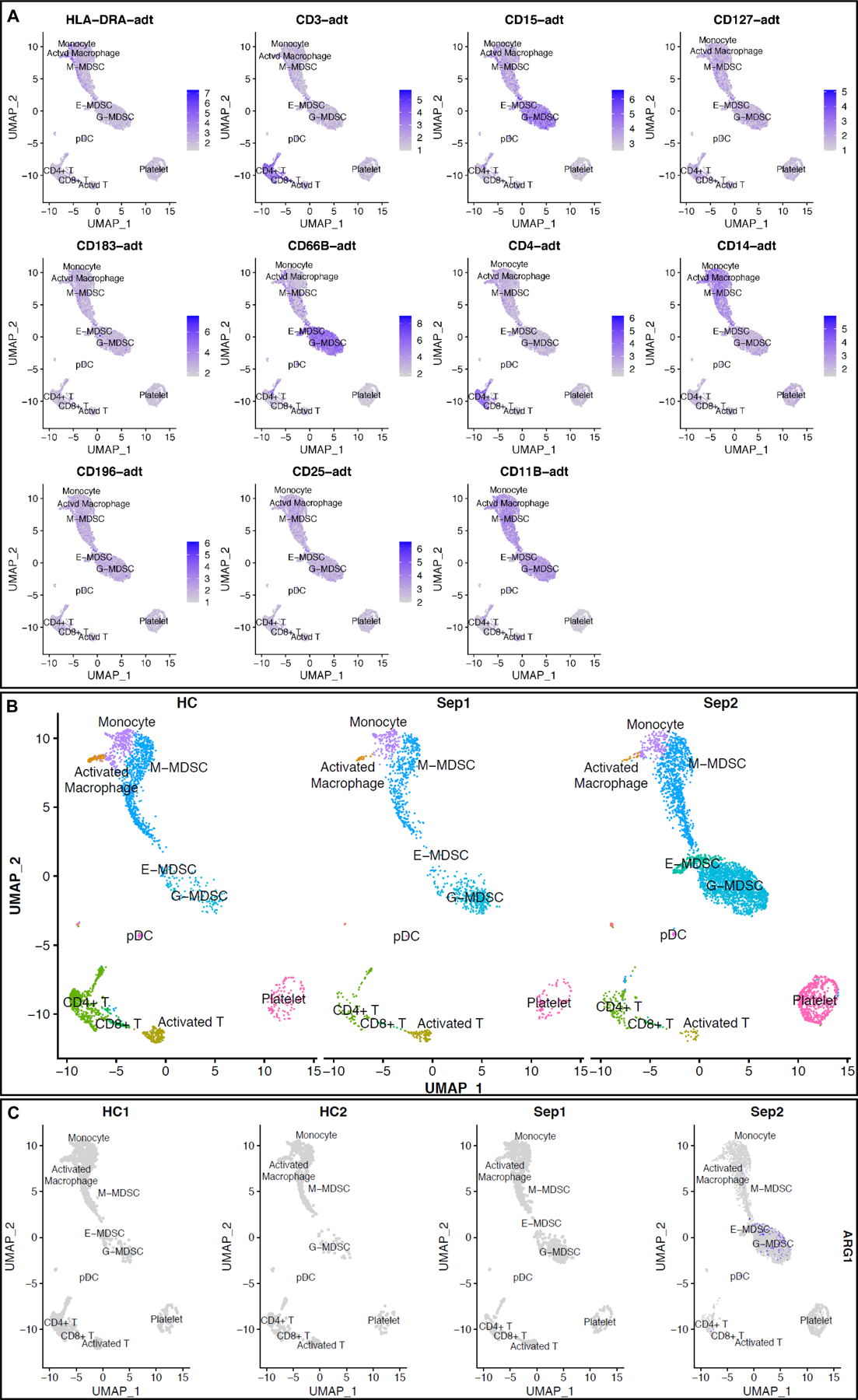
scRNA-seq analysis at 21 days post-sepsis versus healthy control. Cells depicted are from all subjects in the study, in each corresponding group (sepsis n=2, healthy n=2). Using Seurat’s method of integrating data across conditions/batches, the integration allows for joint clustering and to identify shared (or possibly unshared) cell clusters. Cells are visualized on uniform manifold approximation and projection (UMAP) plots colored by cell types. (A) mRNA and corresponding epitope ADT signal projections over UMAP representation for select CITE-seq markers. (B) UMAP representation of cell clusters identified in healthy patients (n=2340 cells) versus sepsis 1 (bacteremia, sepsis; n= 1544 cells) and sepsis 2 (fungemia, septic shock; n= 5587 cells) showing three distinct MDSC subsets. (C) UMAP representation of ARG1 expression noted in only a few sepsis MDSCs in Sepsis 2. Importantly, ARG1 does not have significant differential expression based on our analysis. (E-MDSC = early myeloid derived suppressor cell, G-MDSC = granulocytic myeloid derived suppressor cell, M-MDSC = monocytic myeloid derived suppressor cell, plasmacytoid = plasmacytoid dendritic cells).
Differential Gene Expression
We utilized scRNA-seq to determine differential expressed genes for each cell cluster of interest. In order to reveal potential novel targets for identification of the difference cell types, we also performed an analysis for marker genes, genes that are highly differentially expressed in a given cell type compared to all other cell types (Data File, SDC 3, which shows significant markers for each given cell cluster). Gene expression analysis of each sepsis patient versus the heathy control subjects was conducted separately as sepsis and septic shock as potentially having distinct pathologies (21, 28–31).
Bacteremic Sepsis Patient (Sep1)
Comparison of Sep1 (sepsis due to bacteremia) and healthy controls revealed differential expression (FDR-adjusted p-value < 0.01) of 16 genes in G-MDSCs and 67 genes in M-MDSCs, with only 3 genes in common (Figure 2, SDC 4).
Figure 2.

Volcano plots for each cell cluster of the top up and down regulated genes between healthy subjects (H) versus sepsis patient 1 (S1) at day 21. Each dot represents a gene statistically enriched or reduced within the cell cluster. The volcano plot compares natural log fold-change (healthy mean divided by sepsis mean; x-axis) with adjusted p-values (y-axis). Significance of differential gene expression was determined with adjusted p-value (p adj.) < 0.01.
Septic Shock Patient Due to Fungemia (Sep 2)
Comparison of Sep2 (septic shock due to fungemia) and healthy controls revealed differential expression (FDR-adjusted p-value < 0.01) of 82 genes in G-MDSCs and 331 genes in M-MDSCs, with only 55 genes in common (Figure 3, SDC 5). Between the Sep1 and Sep2 data sets, there were 9 common genes between the G-MDSCs and 40 common genes between the M-MDSCs.
Figure 3.
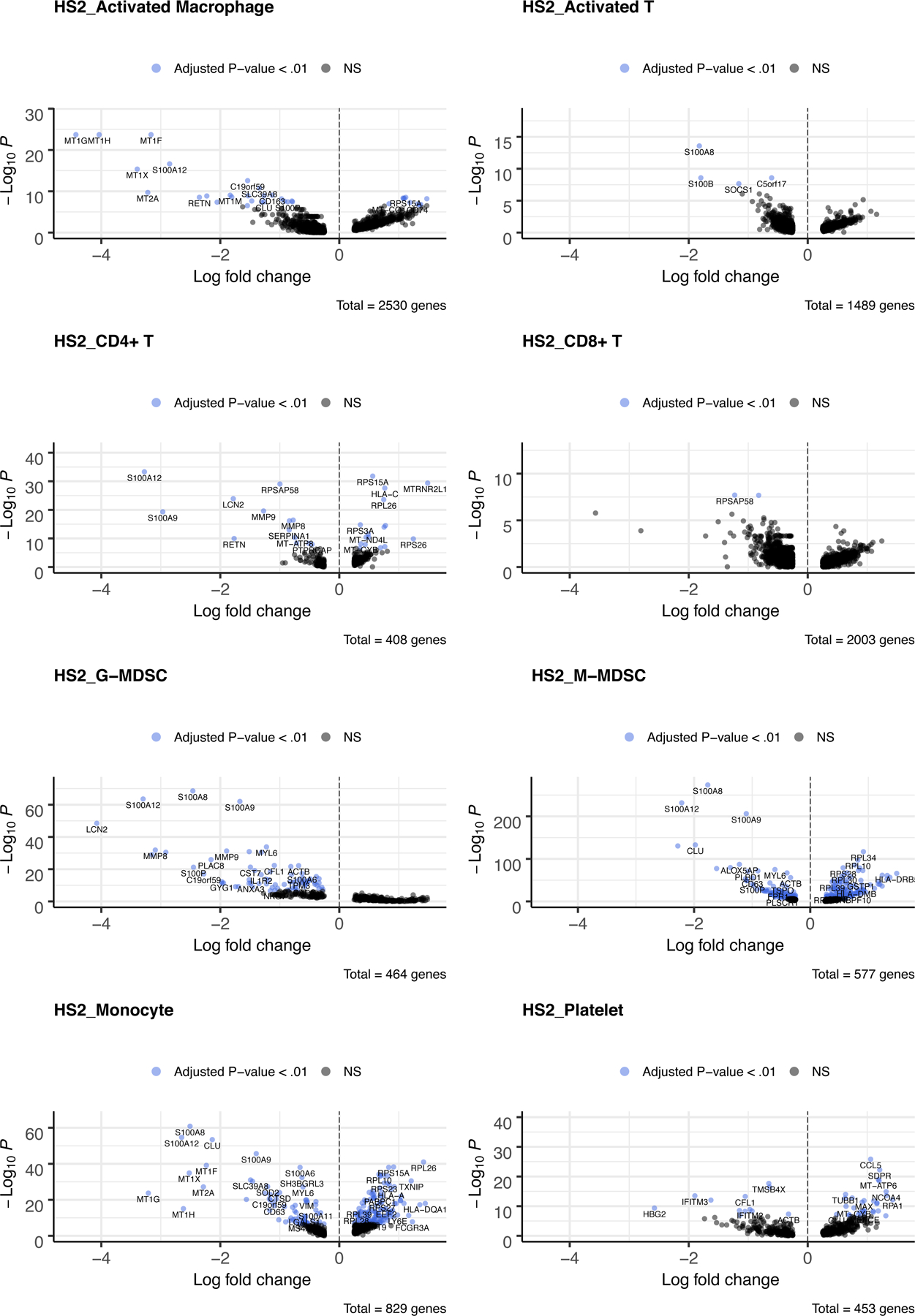
Volcano plots for each cell cluster of the top up and down regulated genes between healthy subjects (H) versus sepsis patient 2 (S2) at day 21. Each dot represents a gene statistically enriched or reduced within the cell cluster. The volcano plot compares natural log fold-change (healthy mean divided by sepsis mean; x-axis) with adjusted p-values (y-axis). Significance of differential gene expression was determined with adjusted p-value (p adj.) < 0.01.
Further Sepsis CITE-seq Analysis
Differential expression of some genes important to MDSC function were specific to cell type and initial insult, with LCN2 and MMP8/9 only upregulated in G-MDSCs and with LGALS2 only upregulated in M-MDSCs in the CCI patient after septic shock. However, many genes identified as common between the sepsis patient datasets and MDSC subsets were previously reported to be associated with MDSC proliferative and immunosuppressive functions, such as CYBB/NOX2 (↑), S100A8/9 (↑) and RETN (↑), which were (20). Overall, it appears that the Sep1 was more transcriptomically similar to controls than Sep2 at post-sepsis day 21.
Comparing Sepsis Due to Bacteremia vs Fungemic Septic Shock (Sep1 vs Sep2)
We also performed a gene expression analysis between the CCI patients to further determine the differences between the MDSC subsets at post-sepsis day 21 based on initial septic insult and severity (SDC 6). Interestingly, gene expression analysis between the CCI patients revealed differential expression (FDR-adjusted p-value < 0.01) of 223 genes in G-MDSCs and 211 genes in M-MDSCs, with 72 genes in common. However, many of these genes differed only modestly in expression thus we applied a further cutoff value of log fold-change (FC) > │1.0│ to select significant genes. Gene analysis with the more stringent criteria (FDR-adjusted p-value < 0.01 and log FC > │1.0│) revealed differential expression of 83 genes in G-MDSCs (includes S100A8, S100A12, MMP8/9 RETN, CYBB) and 22 genes in M-MDSCs, with 10 genes in common between the subsets including RETN and S100A8. LCN2, MMPP8/9, RETN, S100A8/9 were among the 40 genes in the G-MDSC overexpressed in Sep2 compared to Sep1. In the M-MDSC subset, 7 genes were overexpressed in Sep2 compared to Sep1.
Interestingly, there was no differential expression of many MDSC genes vital to cancer immunosuppression, such as ARG1, CD274, COX2, PGE2 and NOS2 (17) (Table 1). Of note, the evidence of differential expression of ARG1 was not considered significant by analysis, although we do observe that ARG1 expression was detected in a small percentage of MDSCs from sepsis patients, but undetected in MDSCs from both healthy subjects (Figure 1C, SDC 6). For NOS2, however, the lack of significant differential expression was due to the absence of detectable expression (expression level/UMI counts of zero).
Table 1.
List of genes previously reported to important for MDSC function but not differentially expressed (not significant per analysis or no expression levels detected) in the G-MDSCs or M-MDSCs of either 21 day post sepsis patient.
|
ARG1, CCL3, CCL4, CCL5, CCR2, CCR3, CD124, CD274, CD38, CD39, CD3G, CD8, Cebpb, COX2, ENTPD1, G-CSF, IDO, IFNy, IL10, Il-1b, IL4, IL6, IL8, miR126a, miR223, NFKB1, NLRP3, NOS1, NOS2, PTGS2, RIPK3, ROS, SIRT1, STAT1, STAT3, STAT4, STAT5, TACE, TNF, VEGFA. |
We utilized Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and gene ontology (GO) categories significant for further overall and pathway analysis of our genomic data. The use of these software allows greater biological insight into the functional processes activated or inhibited in late sepsis MDSCs (Data File, SDC 7, which shows significant identified KEGG and GO pathways for septic G-MDSCs and M-MDSCs in analysis of each sepsis patient versus healthy controls). Clustering and enrichment analysis identified KEGG pathways significant for IL-17 signaling and leukocyte migration in late sepsis G-MDSCs for both sepsis patients when compared to controls; while it identified involvement in antigen processing and leukocyte migration only in Sep2. KEGG enrichment showed significant overlap in many pathways including Th17 and hematopoietic cell lineage pathways in M-MDSCs for both sepsis patients; however, some immune pathways such as Th1 and Th2 cell differentiation were only identified in Sep2. GO analysis of the differentially expressed genes illustrated overlap in late sepsis G-MDSC transcriptome involvement of biological processes related to immunity, such as neutrophil activation and leukocyte migration, with processes such as immune cell differentiation only seen in Sep2. GO analysis of the M-MDSC transcriptome identified significant involvement in categories of neutrophil function and antigen processing/presentation in both sepsis patients; however, only the Sep2 M-MDSC transcriptome was identified to be involved in immune cell migration, leukocyte activation and proliferation, and cytokine production. The late sepsis G-MDSC transcriptome for both Sep1 and Sep2 is implicated in biological processes mainly related to immune processes, while the late sepsis M-MDSC transcriptome is mainly implicated in biological processes related to energy and protein metabolism in Sep1 and immune processes in Sep2.
Discussion
Studies have demonstrated that MDSCs play an important role in the persistent low-grade inflammation and immunosuppression displayed in post-sepsis CCI patients (18, 19, 32). We have previously demonstrated that bulk-isolated MDSCs from surgical sepsis survivors after 14–21 days become potently immunosuppressive; not only do they block T-cell proliferation, but they suppress T-lymphocyte cytokine production (20). The data presented here cannot reveal whether these immunosuppressive properties are attributable to either G-MDSCs and/or M-MDSCs. What this pilot data does illustrate is that transcriptionally, MDSCs from sepsis survivors potentially differ from phenotypically similar cells reported in cancer and autoimmunity (17).
Arginase-1 (ARG1) and programmed death-ligand 1 (PD-L1) have been suggested to have an important role in the immunosuppressive function of MDSCs in cancer. In fact, many studies aim to target arginase-1 as an MDSC suppressive by-product (33). Preclinical mouse models demonstrated that phosphodiesterase-5 inhibitors downregulate iNOS and ARG1 activities, inhibiting MDSC immunosuppressive function and leading to activation of antitumor host immunity and prolonged survival in mice (34–36). In one clinical trial, patients with renal carcinoma that received tyrosine kinase inhibitors, blocking VEGF and c-KIT signaling pathways, had decreased levels of circulating MDSCs, reduced STAT3 activation and ARG1 expression, and displayed elevated activity and proliferation of CD8+ cells (37). Surprisingly, our pilot data did reveal that ARG1 and CD274 (PD-L1) expression are not significantly increased in these MDSC subsets at day 21, and therefore, potentially are not essential to immunosuppressive mechanisms of MDSCs in late sepsis (Figure 4). Our post-transcriptomic analysis did reveal the differential expression of genes in many immune related pathways. Genes and their translated proteins, such as S100A8/9, implicated in the sustained proliferation and functional activation of MDSCs (38), may be an important immunosuppressive mechanisms by MDSCs in late surgical sepsis regardless of initial insult.
Figure 4.
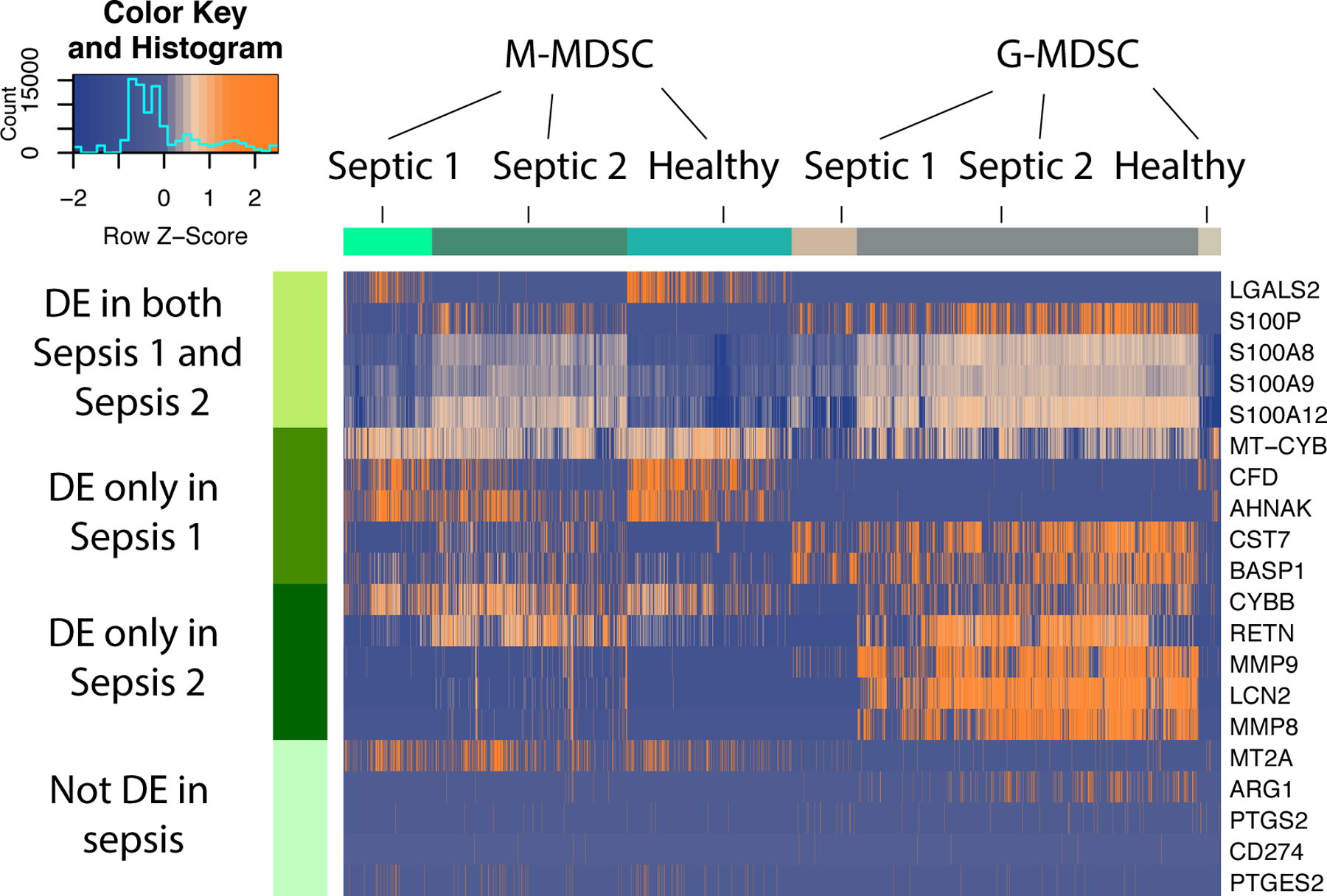
Heatmap illustrating expression patterns of MDSC subsets at 21 days post-sepsis versus healthy control subjects. Rows represent the specific genes of interest differentially expressed in both sepsis patients; only sepsis patient 1; only sepsis patient 2; and genes not differentially expressed in this study, but previously determined to be important to MDSC function in cancer and autoimmunity. Number of columns represent number of cells analyzed in each group. DE = differentially expressed genes in sepsis vs healthy controls. Colors represent mean normalized relative expression with blue representing reduced expression and orange, increased expression. Associated Z-scores are provided in the insert.
Interpretation is limited by the study of MDSCs at a single point in time, late in the course of sepsis when patients are experiencing CCI and a predominant immunosuppressive phenotype. In a previous study, we found a significant upregulation of ARG1 in septic MDSCs at day 14 compared to healthy controls (20) which is not upregulated in this this study of MDSCs at day 21. This could suggest that septic MDSCs may lose their ARG1 expression over time, which is consistent with the known plasticity of MDSCs (39). In fact, it is important to note that the septic MDSC transcriptome may resemble cancer or inflammatory diseases at earlier time points. Therefore, it is important that these studies be repeated in septic patients at earlier time points as well as in patients who are rapidly recovering to discern the plasticity of MDSCs in sepsis. Additionally, although the G-MDSC subset is expanded in both septic patients, the analysis of expression patterns could be skewed towards one patient. Furthermore in human patients, the proportion of the different subsets of MDSCs are also noted to expand differently depending on the microbial origin of sepsis (40–42). Therefore, further analysis with a larger sample size to confidently determine the unique transcriptomic signatures of sepsis is also warranted.
Despite the limitations, this pilot study suggests that MDSC subsets are universally expanded in surgical sepsis survivors who develop CCI. Although MDSCs in sepsis may be phenotypically similar to populations from cancer and other chronic diseases, they may have diverse and unique transcriptomic expression patterns. Importantly, these cells may not only differ based on traditional patient defining characteristics, such as sex and age, but factors such as the source of infection, the type of microbe that is infecting the host and in the severity of the sepsis. All of these listed factors may be important to the surgical sepsis endotype, and thus the post-septic immune response (43, 44). Immunomodulation of MDSCs may be a potential future method of improving the outcomes of humans; however, this treatment may require precision/personal medicine to be employed, as the therapy may be disease and host specific in order to have the desired effects (45–49).
Supplementary Material
Acknowledgments
Funding: This work was supported, in part, by the following National Institutes of Health grants: R01 GM-113945 (PAE), R01 GM-040586 and R01 GM-104481 (LLM), and P50 GM-111152 (LLM, PAE), awarded by the National Institute of General Medical Sciences (NIGMS). In addition, this work was supported, in part, by a postgraduate training grant T32 GM-008721 (DBD) in burns, trauma, and perioperative injury by NIGMS.
Footnotes
Note: This work was accepted for an oral presentation at the Shock Society’s 43rd Annual Conference on Shock (Toronto, Canada) before cancellation due to the COVID-19 pandemic.
Declarations: The authors declare that they have no relevant conflicts of interests.
References
- 1.Brakenridge SC, Efron PA, Cox MC, Stortz JA, Hawkins RB, Ghita G, Gardner A, Mohr AM, Anton SD, Moldawer LL, et al. : Current Epidemiology of Surgical Sepsis: Discordance Between Inpatient Mortality and 1-year Outcomes. Ann Surg 270(3):502–510, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horiguchi H, Loftus TJ, Hawkins RB, Raymond SL, Stortz JA, Hollen MK, Weiss BP, Miller ES, Bihorac A, Larson SD, et al. : Innate Immunity in the Persistent Inflammation, Immunosuppression, and Catabolism Syndrome and Its Implications for Therapy. Front Immunol 9:595, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hawkins RB, Raymond SL, Stortz JA, Horiguchi H, Brakenridge SC, Gardner A, Efron PA, Bihorac A, Segal M, Moore FA, et al. : Chronic Critical Illness and the Persistent Inflammation, Immunosuppression, and Catabolism Syndrome. Front Immunol 9:1511, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Efron PA, Mohr AM, Moore FA, Moldawer LL: The future of murine sepsis and trauma research models. Journal of leukocyte biology, 2015. [DOI] [PMC free article] [PubMed]
- 5.Stortz JA, Raymond SL, Mira JC, Moldawer LL, Mohr AM, Efron PA: Murine Models of Sepsis and Trauma: Can We Bridge the Gap? ILAR J 58(1):90–105, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, Moldawer LL, Moore FA: Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg 72(6):1491–1501, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodwin AJ, Rice DA, Simpson KN, Ford DW: Frequency, cost, and risk factors of readmissions among severe sepsis survivors. Critical care medicine 43(4):738–746, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yende S, Austin S, Rhodes A, Finfer S, Opal S, Thompson T, Bozza FA, LaRosa SP, Ranieri VM, Angus DC: Long-Term Quality of Life Among Survivors of Severe Sepsis: Analyses of Two International Trials. Crit Care Med 44(8):1461–1467, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stortz JA, Mira JC, Raymond SL, Loftus TJ, Ozrazgat Baslanti T, Wang Z, Ghita GL, Leeuwenburgh C, Segal M, Bihorac A, et al. : Benchmarking clinical outcomes and the immunocatabolic phenotype of chronic critical illness after sepsis in surgical intensive care unit patients. Journal of Trauma and Acute Care Surgery:in press., 2017. [DOI] [PMC free article] [PubMed]
- 10.Mira JC, Gentile LF, Mathias BJ, Efron PA, Brakenridge SC, Mohr AM, Moore FA, Moldawer LL: Sepsis Pathophysiology, Chronic Critical Illness, and Persistent Inflammation-Immunosuppression and Catabolism Syndrome. Critical care medicine 45(2):253–262, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathias B, Delmas AL, Ozrazgat-Baslanti T, Vanzant EL, Szpila BE, Mohr AM, Moore FA, Brakenridge SC, Brumback BA, Moldawer LL, et al. : Human Myeloid-derived Suppressor Cells are Associated With Chronic Immune Suppression After Severe Sepsis/Septic Shock. Annals of surgery 265(4):827–834, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pawelec G, Verschoor CP, Ostrand-Rosenberg S: Myeloid-Derived Suppressor Cells: Not Only in Tumor Immunity. Front Immunol 10:1099, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cuenca AG, Cuenca AL, Winfield RD, Joiner DN, Gentile L, Delano MJ, Kelly-Scumpia KM, Scumpia PO, Matheny MK, Scarpace PJ, et al. : Novel role for tumor-induced expansion of myeloid-derived cells in cancer cachexia. J Immunol 192(12):6111–6119, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moreno C, Scumpia PO, Laface DM, Heyworth PG, Efron PA, Moldawer LL: A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Molecular medicine (Cambridge, Mass: 17(3–4):281–292, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuenca AG, Moldawer LL: Myeloid-derived suppressor cells in sepsis: friend or foe? Intensive care medicine 38(6):928–930, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hotchkiss RS, Monneret G, Payen D: Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 13(12):862–874, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veglia F, Perego M, Gabrilovich D: Myeloid-derived suppressor cells coming of age. Nat Immunol 19(2):108–119, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mathias B, Delmas AL, Ozrazgat-Baslanti T, Vanzant EL, Szpila BE, Mohr AM, Moore FA, Brakenridge SC, Brumback BA, Moldawer LL, et al. : Human Myeloid-derived Suppressor Cells are Associated With Chronic Immune Suppression After Severe Sepsis/Septic Shock. Ann Surg 265(4):827–834, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uhel F, Azzaoui I, Gregoire M, Pangault C, Dulong J, Tadie JM, Gacouin A, Camus C, Cynober L, Fest T, et al. : Early Expansion of Circulating Granulocytic Myeloid-derived Suppressor Cells Predicts Development of Nosocomial Infections in Patients with Sepsis. Am J Respir Crit Care Med 196(3):315–327, 2017. [DOI] [PubMed] [Google Scholar]
- 20.Hollen MK, Stortz JA, Darden D, Dirain ML, Nacionales DC, Hawkins RB, Cox MC, Lopez MC, Rincon JC, Ungaro R, et al. : Myeloid-derived suppressor cell function and epigenetic expression evolves over time after surgical sepsis. Crit Care 23(1):355, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. : The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315(8):801–810, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loftus TJ, Mira JC, Ozrazgat-Baslanti T, Ghita GL, Wang Z, Stortz JA, Brumback BA, Bihorac A, Segal MS, Anton SD, et al. : Sepsis and Critical Illness Research Center investigators: protocols and standard operating procedures for a prospective cohort study of sepsis in critically ill surgical patients. BMJ Open 7(7):e015136, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, et al. : Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 7:12150, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lun ATL, Riesenfeld S, Andrews T, Dao TP, Gomes T, participants in the 1st Human Cell Atlas J, Marioni JC: EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol 20(1):63, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCarthy DJ, Campbell KR, Lun AT, Wills QF: Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33(8):1179–1186, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R: Comprehensive Integration of Single-Cell Data. Cell 177(7):1888–1902 e1821, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu G, Wang LG, Han Y, He QY: clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16(5):284–287, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL: Sepsis and septic shock. Nat Rev Dis Primers 2:16045, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shankar-Hari M, Phillips GS, Levy ML, Seymour CW, Liu VX, Deutschman CS, Angus DC, Rubenfeld GD, Singer M, Sepsis Definitions Task F: Developing a New Definition and Assessing New Clinical Criteria for Septic Shock: For the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315(8):775–787, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salomao R, Brunialti MK, Gomes NE, Mendes ME, Diaz RS, Komninakis S, Machado FR, da Silva ID, Rigato O: Toll-like receptor pathway signaling is differently regulated in neutrophils and peripheral mononuclear cells of patients with sepsis, severe sepsis, and septic shock. Crit Care Med 37(1):132–139, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Cannon JG, Tompkins RG, Gelfand JA, Michie HR, Stanford GG, van der Meer JW, Endres S, Lonnemann G, Corsetti J, Chernow B, et al. : Circulating interleukin-1 and tumor necrosis factor in septic shock and experimental endotoxin fever. J Infect Dis 161(1):79–84, 1990. [DOI] [PubMed] [Google Scholar]
- 32.Schrijver IT, Theroude C, Roger T: Myeloid-Derived Suppressor Cells in Sepsis. Front Immunol 10:327, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maples KR, Mason RP: Free radical metabolite of uric acid. J Biol Chem 263(4):1709–1712, 1988. [PubMed] [Google Scholar]
- 34.Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, Dolcetti L, Bronte V, Borrello I: Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med 203(12):2691–2702, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyer C, Sevko A, Ramacher M, Bazhin AV, Falk CS, Osen W, Borrello I, Kato M, Schadendorf D, Baniyash M, et al. : Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc Natl Acad Sci U S A 108(41):17111–17116, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin S, Wang J, Wang L, Wen J, Guo Y, Qiao W, Zhou J, Xu G, Zhi F: Phosphodiesterase-5 inhibition suppresses colonic inflammation-induced tumorigenesis via blocking the recruitment of MDSC. Am J Cancer Res 7(1):41–52, 2017. [PMC free article] [PubMed] [Google Scholar]
- 37.Chen H-M, Ma G, Gildener-Leapman N, Eisenstein S, Coakley BA, Ozao J, Mandeli J, Divino C, Schwartz M, Sung M, et al. : Myeloid-Derived Suppressor Cells as an Immune Parameter in Patients with Concurrent Sunitinib and Stereotactic Body Radiotherapy. Clinical Cancer Research 21(18):4073–4085, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G: Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol 181(7):4666–4675, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ben-Meir K, Twaik N, Baniyash M: Plasticity and biological diversity of myeloid derived suppressor cells. Curr Opin Immunol 51:154–161, 2018. [DOI] [PubMed] [Google Scholar]
- 40.Janols H, Bergenfelz C, Allaoui R, Larsson AM, Ryden L, Bjornsson S, Janciauskiene S, Wullt M, Bredberg A, Leandersson K: A high frequency of MDSCs in sepsis patients, with the granulocytic subtype dominating in gram-positive cases. J Leukoc Biol 96(5):685–693, 2014. [DOI] [PubMed] [Google Scholar]
- 41.Patel JJ, Rosenthal MD, McClave SA, Martindale RG: Tempering the Clinical Effects of Early Myeloid-derived Suppressor Cell Expansion in Severe Sepsis and Septic Shock. Am J Respir Crit Care Med 197(5):677–678, 2018. [DOI] [PubMed] [Google Scholar]
- 42.Stoll H, Ost M, Singh A, Mehling R, Neri D, Schafer I, Velic A, Macek B, Kretschmer D, Weidenmaier C, et al. : Staphylococcal Enterotoxins Dose-Dependently Modulate the Generation of Myeloid-Derived Suppressor Cells. Front Cell Infect Microbiol 8:321, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cox MC, Brakenridge SC, Stortz JA, Hawkins RB, Darden DB, Ghita GL, Mohr AM, Moldawer LL, Efron PA, Moore FA: Abdominal sepsis patients have a high incidence of chronic critical illness with dismal long-term outcomes. Am J Surg, 2020. [DOI] [PMC free article] [PubMed]
- 44.Stortz JA, Mira JC, Raymond SL, Loftus TJ, Ozrazgat-Baslanti T, Wang Z, Ghita GL, Leeuwenburgh C, Segal MS, Bihorac A, et al. : Benchmarking clinical outcomes and the immunocatabolic phenotype of chronic critical illness after sepsis in surgical intensive care unit patients. J Trauma Acute Care Surg 84(2):342–349, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Efron PA, Mohr AM, Bihorac A, Horiguchi H, Hollen MK, Segal MS, Baker HV, Leeuwenburgh C, Moldawer LL, Moore FA, et al. : Persistent inflammation, immunosuppression, and catabolism and the development of chronic critical illness after surgery. Surgery 164(2):178–184, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dorhoi A, Glaria E, Garcia-Tellez T, Nieuwenhuizen NE, Zelinskyy G, Favier B, Singh A, Ehrchen J, Gujer C, Munz C, et al. : MDSCs in infectious diseases: regulation, roles, and readjustment. Cancer Immunol Immunother 68(4):673–685, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kyvelidou C, Sotiriou D, Zerva I, Athanassakis I: Protection Against Lipopolysaccharide-Induced Immunosuppression by IgG and IgM. Shock (Augusta, Ga: 49(4):474–482, 2018. [DOI] [PubMed] [Google Scholar]
- 48.Seo EH, Song GY, Kwak BO, Oh CS, Lee SH, Kim SH: Effects of Glycyrrhizin on the Differentiation of Myeloid Cells of the Heart and Lungs in Lipopolysaccharide-Induced Septic Mice. Shock (Augusta, Ga: 48(3):371–376, 2017. [DOI] [PubMed] [Google Scholar]
- 49.Martire-Greco D, Rodriguez-Rodrigues N, Castillo LA, Vecchione MB, de Campos-Nebel M, Cordoba Moreno M, Meiss R, Vermeulen M, Landoni VI, Fernandez GC: Novel Use of All-Trans-Retinoic Acid in A Model of Lipopolysaccharide-Immunosuppression to Decrease the Generation of Myeloid-Derived Suppressor Cells by Reducing the Proliferation of CD34+ Precursor Cells. Shock (Augusta, Ga: 48(1):94–103, 2017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.