

An individual’s risk of coronary artery disease (CAD) is determined by the interplay of physiologic, lifestyle, and genetic factors. Whereas measurements of low-density lipoprotein cholesterol (LDL-C) form the basis of CAD prevention strategies, the extent to which CAD risk varies with different lipid levels in individuals with varying genetic risk remains unknown. We used the polygenic risk score (PRS) to quantify the genetic risk of CAD and explore its interplay with LDL-C.
We analyzed 408 186 White British individuals from the prospective UK Biobank study.1 UK Biobank received ethical approval from the North West Multi-Center Research Ethics Committee (11/NW/03820). This dataset was further split to produce independent validation and testing datasets using the UK Biobank first release (genotyping batch from 11 to 22) and second release (genotyping batch from 23 to 95), respectively. CAD outcome was based on self-reported diagnoses and Hospital Episodes Statistics as described in Inouye et al.2 Data analyzed in this article are available for researchers interested in replicating our results through application to the UK Biobank.
We first developed a novel CAD PRS using a new method that implements a stacked clumping and thresholding (SCT) algorithm.3 SCT is able to learn new single nucleotide polymorphism (SNP) weights using genomewide association study summary statistics and a validation dataset, as opposed to other methods that can only identify optimal hyperparameters. As input, we used the same genomewide association study summary statistics used by Khera and colleagues4 and selected a subset of SNPs with MAF >0.01 and P≤0.1, leading to a training dataset of 894 239 SNPs. The method generated ≈123 200 vectors of PRS by applying clumping and thresholding over a 4-dimensional grid of parameters (squared correlation, P value, clumping window size, and imputation quality). In a subsequent stacking phase, SCT learned an optimal linear combination of all computed PRS using a penalized regression and the validation dataset comprising 7912 CAD cases and 121 941 controls. The resulting SCT PRS included 300 238 SNPs.
For comparison, we computed 2 previously published PRS by Khera et al4 and Inouye et al2 and built 2 additional PRS by combining the SNPs from SCT with the Khera et al4 PRS (SCT-K: 6 695 156 SNPs) and with the Inouye et al2 PRS (SCT-I: 1 926 521 SNPs), retaining the effect size of the SCT PRS for matching SNPs. The SCT-I PRS panel is available from the corresponding author for noncommercial use.
We assessed the predictive performance of the 5 PRS in the testing dataset of 15 433 CAD cases and 262 900 controls. The SCT-I PRS had the highest predictive performance, with an area under the receiver operating characteristic curve (AUC, 0.645 [95% CI, 0.637–0.653]) that was significantly higher than the other 4: Khera et al4 (P=9.94×10−7; AUC, 0.634 [95% CI, 0.626–0.642]), Inouye et al2 (P=2.26×10−7; AUC, 0.636 [95% CI, 0.629–0.644]), SCT (P=2.27×10−4; AUC, 0.640 [95% CI, 0.632–0.648]), and SCT-K (P=0.0046; AUC, 0.641 [95% CI, 0.633–0.649]). Differences between AUC pairs were tested through a nonparametric 2-sided test using the roc.test function (with the following parameters: method = DeLong and alternative = 2-sided) in the pROC R package.
We applied SCT-I to a target dataset of 126 499 controls and 2215 incident CAD cases, selected from the testing dataset by excluding individuals on the basis of the following criteria: previous history of CAD (n=8995), use of lipid- or blood pressure–lowering medication (n=69 934), or missing values at baseline on at least 1 of the control covariates listed in the Figure legend (n=70 690).
Figure.
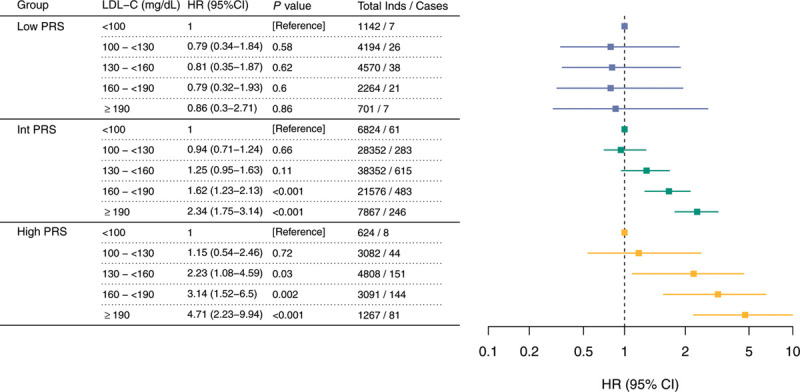
Association of PRS groups and LDL-C bins with CAD. Association of LDL-C levels with incident CAD in low PRS (first decile of PRS distribution), Int PRS (between the second and the ninth decile of PRS distribution), and high PRS (10th decile of PRS distribution). In each PRS group, the relative risk of CAD (HR) was calculated relative to the <100 mg/dL LDL-C reference level in a Cox proportional hazards model. The Cox proportional hazards model used time since study entry as the time scale (median follow-up period, 8.18 years) and was adjusted for the following control covariates: age, sex, genotyping array, the first 4 principal components of ancestry, Townsend deprivation index, diabetes, current smoking status, family history of heart disease, systolic blood pressure, glycohemoglobin, triglycerides, body mass index, C-reactive protein, lipoprotein(a), high-density lipoprotein cholesterol, and dietary antioxidant intake (total servings per day of fruits and vegetables). CAD indicates coronary artery disease; HR, hazard ratio; Int, intermediate; LDL-C, low-density lipoprotein cholesterol; and PRS, polygenic risk score.
We divided individuals into 3 groups on the basis of their polygenic risk: high PRS (top decile of the PRS distribution, n=12 872), intermediate PRS (within the second and ninth PRS deciles, n=102 970), and low PRS (bottom PRS decile, n=12 872). In each PRS group, we binned individuals according to baseline measurements of LDL-C. We used Cox proportional hazard models to calculate the hazard ratio for each LDL-C bin relative to reference populations with LDL-C <100 mg/dL and adjusting for major atherosclerotic clinical and lifestyle risk factors (see the Figure legend). An additional Cox model tested for the presence of an interaction between continuous LDL-C and PRS.
CAD risk conferred by LDL-C was modulated by the PRS (Pinteraction=0.0010). For individuals in the high PRS group, the increase in the point estimate of the hazard ratio for each LDL-C bin was almost double compared with individuals in the intermediate PRS group. Individuals with high PRS and LDL-C 130 to <160 mg/dL showed comparable increased risk (hazard ratio, 2.23 [95% CI, 1.08–4.59]) to those with intermediate PRS and LDL-C ≥190 mg/dL (hazard ratio, 2.34 [95% CI, 1.75–3.14]). Conversely, CAD risk in individuals in the low PRS group did not follow the stepwise increase observed in the other PRS groups (Figure).
Here, we show that CAD risk conferred by LDL-C is modified by an individual’s polygenic background. This study supports those reporting greater benefit from LDL-C–lowering drugs in European individuals with high PRS.5 An important next step will be understanding how these results generalize in other prospective cohorts and in populations of different ancestries. Because PRS can be quantified from a young age, it can provide information to mitigate the elevated lifetime risk that even moderate LDL-C exposure causes in individuals with high CAD PRS.
Acknowledgments
The authors thank the volunteers and managers of the UK Biobank project; and Prof Antonio Vittorino Gaddi, MD; Prof Gemma Figtree, MD; Alessandro Boccanelli, MD; and Gualtiero Colombo, MD, for their comments on the article.
Sources of Funding
This work was supported by private funding from Allelica Srl. Dr Pastorino received funding from Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy.
Disclosures
Dr Bolli, P. Di Domenico, and Dr Bottà are employees of Allelica Srl. Dr Busby consults for and has received honoraria from Allelica Srl. Dr Bottà, P. Di Domenico, and Dr Busby hold equity in Allelica Srl. Dr Pastorino reports no conflicts or other disclosures.
Footnotes
Contributor Information
Alessandro Bolli, Email: aless.bolli@gmail.com.
Paolo Di Domenico, Email: paolo@allelica.com.
Roberta Pastorino, Email: roberta.pastorino@unicatt.it.
George B. Busby, Email: george.busby@gmail.com.
References
- 1.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Inouye M, Abraham G, Nelson CP, Wood AM, Sweeting MJ, Dudbridge F, Lai FY, Kaptoge S, Brozynska M, Wang T, et al. ; UK Biobank CardioMetabolic Consortium CHD Working Group. Genomic risk prediction of coronary artery disease in 480,000 adults: implications for primary prevention. J Am Coll Cardiol. 2018;72:1883–1893. doi: 10.1016/j.jacc.2018.07.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Privé F, Vilhjálmsson BJ, Aschard H, Blum MGB. Making the most of clumping and thresholding for polygenic scores. Am J Hum Genet. 2019;105:1213–1221. doi: 10.1016/j.ajhg.2019.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–1224. doi: 10.1038/s41588-018-0183-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Damask A, Steg PG, Schwartz GG, Szarek M, Hagström E, Badimon L. Patients with high genome-wide polygenic risk scores for coronary artery disease may receive greater clinical benefit from alirocumab treatment in the Odyssey Outcomes Trial. Circulation. 2020;141:624–636. doi: doi.org/10.1161/CIRCULATIONAHA.119.044434 [DOI] [PubMed] [Google Scholar]