

Abstract
Osteoarthritis (OA) is a degenerative joint disease characterized by articular cartilage degradation. Because OA has a multifactorial nature and complex interrelationship of the individual elements of a whole joint, there is a need for comprehensive therapeutic approaches for cartilage tissue engineering, which simultaneously address multiple aspects of disease etiology. In this work, we investigated a multifunctional carbohydrate-based drug candidate, tri-butanoylated N-acetyl-D-galactosamine analog (3,4,6-O-Bu3GalNAc) that induced cartilage tissue production by human mesenchymal stem cells (hMSCs) and human OA chondrocytes by modulating Wnt/β-catenin signaling activity. The dual effects promoted chondrogenesis of human MSC and reduced inflammation of human OA chondrocytes in in vitro cultures. Translating these findings in vivo, we evaluated therapeutic effect of 3,4,6-O-Bu3GalNAc on the rat model of posttraumatic OA when delivered via local intra-articular sustained-release delivery using microparticles and found this method to be efficacious in preventing OA progression. These results show that 3,4,6-O-Bu3GalNAc, a disease modifying OA drug candidate, has promising therapeutic potential for articular cartilage repair.
Keywords: Tissue engineering, osteoarthritis, drug delivery, mesenchymal stem cells, disease-modifying OA drug, carbohydrates
1. Introduction
Osteoarthritis (OA) is the most common cause of joint pain and physical disability in the elderly. The characteristics of OA include an irreversible loss of articular cartilage, subchondral bone thickening, osteophyte formation, and local joint inflammation [1]. Current pharmacologic OA treatment options are analgesics, non-steroidal anti-inflammatory drugs, and viscosupplementation with intra-articular injections. These treatments focus on short-term symptomatic pain relief and care of joint function [2], however with advances in the understanding of the OA pathogenesis there is a focus now on disease-modifying OA drugs (DMOADs) designed to block or reverse OA progression by targeting specific OA pathophysiological networks [3].
A new class of DMOADs, small molecules that stimulate mesenchymal stem cells (MSCs) residing in articular cartilage to differentiate into chondrocytes, has shown promise for cartilage repair [4–6]. However, the inflammatory environment in an OA joint limits the efficacy of therapeutics by disrupting joint homeostasis and reducing its capacity for regeneration [7]. The onset and progression of OA result from dominating inflammatory cytokines and catabolic regulators within all the joint tissues including bone, synovium, and cartilages [8]. The inflammatory cytokines present in an OA joint can disrupt the chondrogenic differentiation capabilities of the MSCs and reduce therapeutic efficacy of the DMOADs [9–11]. Therefore, there is a critical need to not only reduce inflammation in the local joint environments but also rebuild cartilage tissue [12].
OA is a localized disease that primarily impacts the articular joint space [13]. Because of this, the intra-articular (IA) injection of therapeutics would be suitable for local drug delivery without the risk of systemic side effects [14]. Clinically, glucocorticoids, platelet rich plasma, and hyaluronic acid are all delivered through IA injection [15]. Unfortunately, small molecular weight and larger therapeutics have a limited lifetime within the articular joint due to the clearance of IA injected drugs through draining lymphatic system of the synovial fluid [16]. Therefore, a number of controlled release formulations have been tested at the preclinical and clinical levels. It has been reported that various microspheres and nanospheres including poly(lactic-co-glycolic acid) (PLGA), chitosan, and poly(propylene sulphide) can be used for sustained drug release in OA treatment [17–20]. Clinically, glucocorticoids encapsulated in PLGA microspheres have been tested through Phase 3 [13]. Thus today there are now examples of long term release of molecules in the articular joint space. However, the therapeutic quality of these molecules in addressing the underlying disease and rebuilding tissue is still limited.
Carbohydrates, sugar molecules in the form of mono-, oligo-, and poly-saccharides, are ubiquitously present in all classes of life where they serve functions including energy storage, metabolic activity, structural integrity, and providing a carbon source. Carbohydrates are now increasingly being recognized as excellent scaffolds for small molecule drug design based on their stability, rigid core, and ability to position a wide range of functional groups in three-dimensional space to broadly engage biological targets [21]. Based on the emerging paradigm that sugars can be molded into effective drugs, combined with our previous finding that certain hexosamine analogs had anti-inflammatory properties [22, 23], we reasoned that monosaccharides provide an attractive drug development platform to target inflammation that contributes to OA progression. Moreover, such drugs could be designed to simultaneously augment production of glycosoaminoglycans (GAGs) important for tissue homeostasis and regeneration, thus creating OA drug candidates that address both pain and tissue degeneration.
We now propose that these properties establish a carbohydrate-based drug candidate, 3,4,6-O-Bu3GalNAc (Fig. 1A), as an attractive lead candidate for a new class of human OA therapeutics, in part because this compound possesses anti–NF-κB activity [24] and can increase cartilage production in an OA environment in 3D bovine tissue culture models. We hypothesized that 3,4,6-O-Bu3GalNAc could also modulate these endpoints in human disease and, by employing in vitro and in vivo disease models, we found that not only does it reduce inflammation by acting on NF-κB, but it also engages a second pathway implicated in OA: Wnt/β-catenin. By inhibiting Wnt/β-catenin signaling 3,4,6-O-Bu3GalNAc enhanced chondrogenic differentiation of human MSCs, improving cartilage tissue production in 3D cultures in vitro. Based on these promising in vitro results highlighting a novel two-pronged effect on two important signaling pathways implicated in OA disease progression, we tested the therapeutic efficacy of 3,4,6-O-Bu3GalNAc in vivo in an OA rat model. Toward clinical translation, we also formulated 3,4,6-O-Bu3GalNAc in a sustained-release delivery system to compare with repeated injections and found that it relieved cartilage degradation in the articular joints. In summary, 3,4,6-O-Bu3GalNAc has multifaceted biological activity acting on both endogenous MSCs and diseased OA chondrocytes, demonstrating promise as a new carbohydrate-based disease-modifying OA drug.
Fig. 1.

3,4,6-O-Bu3GalNAc promoted chondrogenesis of hMSCs. Cells were treated without (0 μM) or with 3,4,6-O-Bu3GalNAc (100 μM) in chondrogenic medium for two days. A. Chemical structure of 3,4,6-O-Bu3GalNAc. B. Gene expression level of chondrogenic markers SOX9 and AGGRECAN. C. Expression levels of Wnt-related genes AXIN2 and β-CATENIN. Data are averages of three individual samples relative to β-ACTIN mRNA. The relative expression level to a non-treated (0 μM) control is shown (* P<0.05, *** P<0.001, t-test). D. Protein expression of β-catenin in cytoplasmic and nuclear compartments was confirmed by Western blotting. β-Actin and β-tubulin were used as respective loading controls. Data are representative of 3 independent experiments and fold change values of 3,4,6-O-Bu3GalNAc treated group, relative to control, were obtained by using densitometry and are shown below individual lanes (0.64 ± 0.04 (P<0.05, t-test) for cytoplasmic β-catenin and 0.32±0.03 (P<0.001, t-test) for nuclear β-catenin). E. Immunofluoresence staining for cytoplasmic β-catenin. Nuclei were stained with DAPI (scale bars: 50 μm).
2. Materials and Methods
2.1. Cell isolation and culture
Human articular cartilage samples explanted from OA patients undergoing total knee arthroplasty were received from the National Disease Resource Institution (Philadelphia, PA, USA) according to an IRB-approved protocol. The cartilage tissue was cut into 1 mm3 pieces, washed 3 times with phosphate-buffered saline (PBS) supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin (Pen/Strep), and digested on an orbital shaker for 16 h at 37 °C with 0.17% (w/v) type II collagenase (Worthington Biochemical, USA) in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; In-vitrogen™, Life Technologies, USA) with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, USA). After the digestion, the filtrate was passed through a 70 μm strainer and cells were rinsed 3 times with the DMEM supplemented with Pen/Strep and 10 % FBS.
Human MSCs (hMSCs) were obtained from Dr. Arnold Caplan (Case Western Reserve University) and maintained in MSC medium with FBS from a lot defined by the Caplan Lab as supporting MSC growth. The human MSCs were maintained in growth medium composed of low-glucose DMEM supplemented with 2 mM L-glutamine, Pen/Strep, 10% defined FBS, and 1.0 ng/mL basic fibroblast growth factor (FGF; PeproTech, USA). Passage 3–5 cells were used for all experiments.
2.2. Mesenchymal stem cell differentiation
For chondrogenic differentiation (monolayer, pellet, and 3D hydrogel cultures), hMSCs were induced in chondrogenic medium consisting of high-glucose DMEM, Pen/Strep, ITS premix (6.25 μg/mL insulin, 6.25 μg/mL transferrin, 6.25 ng/mL selenous acid, 1.25 mg/mL BSA, 5.35 μg/mL linoleic acid (BD Bioscience, USA)), 100 mM sodium pyruvate (Life Technologies, USA), 40 mg/mL L-proline (Sigma-Aldrich, USA), 50 mM ascorbic acid-2-phosphate, and 10 ng/mL of transforming growth factor-β1 (TGF-β1 (Fitzgerald Industries International, USA). For osteogenic differentiation in monolayer cell culture studies, human MSCs were plated and cultured in the growth medium until they were confluent. Differentiation toward an osteogenic phenotype was then achieved through culture in osteogenic medium comprised of high-glucose DMEM supplemented with 10% FBS, Pen/Strep, 100 nM dexamethasone, 10 mM β-glycerophosphate, and 0.1 mg/mL ascorbic acid-2-phosphate.
2.3. Surgically induced OA rat model
Six-week old male Sprague-Dawley rats (Charles River, Germantown, MD) weighing approximately 240 g were used in the work. Rats were anesthetized with 3–5% isoflurane, and the hair was shaved from the medial aspect of the right femoro-tibial joint followed by a surgical scrub alternating between chlorhexidine and alcohol for aseptic surgery. The knee joint was exposed by a 1 cm longitudinal incision of skin and then joint capsule opened with a #10 scalpel. The medial collateral ligament (MCL) was transected to visualize the medial meniscus. The medial meniscus was fully transected and reflected the meniscus toward the femur. The joint capsule overlying muscle and then skin were closed with 4 Vicryl sutures. All procedures were performed according to the Institutional Animal Care and Use Committee at Johns Hopkins University School of Medicine.
After the OA surgery, the rats of the Multi-GalNAc group (n=3) were given IA injections with 500 μg of 3,4,6-O-Bu3GalNAc in 100 μL of vehicle (5 % DMSO in PBS) on days 7, 14 and 21. The rats of the Single-3,4,6-O-Bu3GalNAc group (n=3) were given IA injections with 1.5 mg of 3,4,6-O-Bu3GalNAc in 100 μL of the vehicle once on days 7. The rats of the PLGA-3,4,6-O-Bu3GalNAc group (n=4) were given IA injections with 15 mg of PLGA-3,4,6-O-Bu3GalNAc (loading 1.5 mg of 3,4,6-O-Bu3GalNAc) in 100 μL of PBS once on days 7. On day 28, the rats were sacrificed and the cartilage was collected for histological assessment of the medial tibial plateau joint through blinded graded observations by 2 observers, according to OARSI Scoring System.
3. Results
3.1. 3,4,6-O-Bu3GalNAc treatment induces human MSC chondrogenic differentiation
The sugar analog 3,4,6-O-Bu3GalNAc (Fig. 1A, Scheme S1) was synthesized by n- butyrylation of a GalNAc core, as described previously [22]. We first evaluated the potential chondrogenic effect of 3,4,6-O-Bu3GalNAc on human MSCs (hMSCs) in monolayer culture in vitro. 3,4,6-O-Bu3GalNAc (100 μM) exposure did not affect cell viability (Fig. S1). mRNA isolated from the cells in culture revealed increased expression of two genes associated with chondrogenic markers after treatment with 3,4,6-O-Bu3GalNAc, SOX9 and AGGRECAN (Fig. 1B).
We then investigated the involvement of Wnt genes and proteins because the Wnt/β-catenin signaling pathway influences chondrogenic differentiation of stem cells [25]. RT-PCR analysis showed that 3,4,6-O-Bu3GalNAc down-regulated β-CATENIN, the regulator of Wnt, as well as the Wnt target gene, AXIN2 (Fig. 1C). β-Catenin levels decreased in both nuclear and cytoplasmic compartments of hMSCs when treated with 3,4,6-O-Bu3GalNAc (Fig. 1D and 1E).
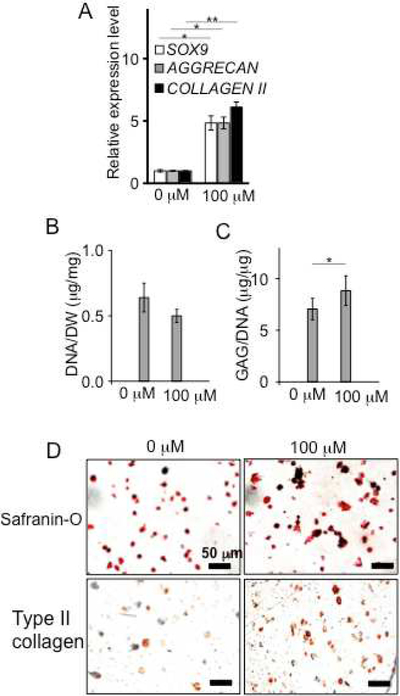
Next, we evaluated the chondrogenic effect on 3D pellet cultures of hMSCs. After treatment with 3,4,6-O-Bu3GalNAc for three weeks, the differentiated MSC pellets expressed genes associated with cartilage, including SOX9, TYPE II COLLAGEN, and AGGRECAN (Fig. 2A). Biochemical analysis of the pellets demonstrated that the amount of GAG per DNA content significantly increased while DNA levels remained similar to untreated controls (Fig. 2, B and C). The increased proteoglycan deposition was further confirmed via histological staining with Safranin-O (Fig. 2D). In addition, immunohistochemical staining of the hMSC pellets revealed up-regulation of type II collagen, but down-regulation of a hypertrophic chondrocyte-specific marker, type X collagen, and an osteogenic marker, type I collagen, indicating retention of the cartilage phenotype (Fig. 2D) [5].
Fig. 2.

Treatment of 3,4,6-O-Bu3GalNAc increased chondrogenic markers of 3D pellets of hMSCs and cartilage ECM production. Pellets were treated without (0 μM) or with 3,4,6-O-Bu3GalNAc (100 μM) in the chondrogenic medium for 21 days. A. Gene expression level of chondrogenic markers, SOX9, AGGRECAN, and TYPE II COLLAGEN (COLLAGEN II). Data are averages of three individual samples relative to β-ACTIN mRNA. The relative expression level to a non-treated (0 μM) control is shown (* P<0.05, ** P<0.01, t-test). B. DNA contents normalized to dry weight. C. sGAG contents normalized to the DNA content. Data in (B and C) are averages of three individual samples (* P<0.05, t-test). D. Safranin-O and immunohistochemical staining of pellets (type I, type II, and type X collagen). Scale bar: 500 μm.
We also sought to evaluate the effects of 3,4,6-O-Bu3GalNAc on the chondrogenic differentiation of MSCs in a 3D hydrogel culture because it closely mimics the in vivo articular cartilage environment and maintains the chondrocyte phenotype better than monolayer culture [26]. After three weeks of culture and exposure, there was a significant up-regulation of chondrogenic markers, SOX9, AGGRECAN, and TYPE II COLLAGEN (Fig. 3A). The 3,4,6-O-Bu3GalNAc also increased GAG content in the 3D hydrogel constructs while GAG contents per dry weight levels remained similar to untreated controls (Fig. 3B and C) and extracellular matrix (ECM) accumulation increased as demonstrated respectively by Safranin-O staining for proteoglycans and immunohistochemistry for type II collagen (Fig. 3D).
Fig. 3.
3,4,6-O-Bu3GalNAc treatment increases chondrogenic expression and cartilage ECM deposition by hMSCs encapsulated in 3D hydrogels. The hydrogels were incubated without (0 μM) or with 3,4,6-O-Bu3GalNAc (100 μM) in chondrogenic medium for 21 days. A. Gene expression levels of chondrogenic markers SOX9, AGGRECAN, and TYPE II COLLAGEN (COLLAGEN II). Data are averages normalized to GAPDH. The relative expression level to a non-treated (0 μM) control is shown (n = 3, * P<0.05, ** P<0.01, t-test). B. Biochemical analysis of DNA content, normalized to dry weight. C. GAG content, normalized to the DNA content. Data in (B and C) are averages of three individual samples (* P<0.05, t-test). D. Safranin-O and immunohistochemical staining (type II collagen) of cross-section of the hydrogel (Scale bar: 50 μm).
3.2. 3,4,6-O-Bu3GalNAc inhibits osteogenesis of hMSC
Wnt/β-catenin signaling plays a critical role in bone development and homeostasis [27]. In particular and relevant to OA, disruption of Wnt/β-catenin has been shown to impair MSC osteogenesis in vitro [28, 29]. To further examine the role of 3,4,6-O-Bu3GalNAc in Wnt/β-catenin signaling, we examined the effect of 3,4,6-O-Bu3GalNAc on hMSC osteogenic differentiation in vitro. Osteogenesis was attenuated upon treatment with GalNAc-a, as measured by alkaline phosphatase (ALP) activity (Fig. 4A). 3,4,6-O-Bu3GalNAc also down-regulated expression of β-CATENIN, the osteogenic transcriptional factor RUNX2, and the Wnt transcription cofactor LEF1 (Fig. 4B).
Fig. 4.

3,4,6-O-Bu3GalNAc reduces osteogenic capacity of hMSCs. Cells on monolayer and 3D hydrogel cultures were incubated without (0 μM) or with 3,4,6-O-Bu3GalNAc (100 μM) in the osteogenic medium. A. Alkaline phosphatase (ALP) activity (scale bar: 500 μm), Alizarin red (AR, scale bar: 500 μm), and Oil red O (ORO, scale bar: 100 μm). B. RT-PCR analysis of gene expression after 5 days osteogenic induction of hMSCs. C. RT-PCR analysis of osteogenic markers at 10 days of the osteogenic induction. D. RT-PCR analysis of adipogenic markers at 15 days of the osteogenic induction. For RT-PCR analysis, β-ACTIN mRNA was used as a control and three individual samples were collected and averaged. The relative expression level to a non-treated (0 μM) control is shown (* P<0.05, ** P<0.01, *** P<0.001, t-test). E. Alizarin red staining (scale bar: 50 μm) of cross section of hydrogel. F. RT-PCR analysis of osteogenic markers after 21 days osteogenic induction of hMSCs encapsulated in the hydrogels (Endogenous mRNA control: GAPDH, n = 3, *** P<0.001, t-test).
After 10 days osteogenic induction, Alizarin red staining for calcium deposition revealed decreased mineralization in 3,4,6-O-Bu3GalNAc treated hMSCs (Fig. 4A). The ability of 3,4,6-O-Bu3GalNAc to inhibit osteogenesis in culture was further confirmed by significant decreases in the expression of COLLAGEN X and bone matrix markers osteocalcin (OCN) and osteopontin (OPN) (Fig. 4C) while the expression of RUNX2 remained at the basal level (Fig. S2A). By day 15 of osteogenic induction, calcium deposition and expression of RUNX2, OCN, and OPN showed no significant difference between control and 3,4,6-O-Bu3GalNAc treatment groups (Fig. S2B and C), indicating that 3,4,6-O-Bu3GalNAc attenuated osteogensis of hMSCs but did not completely inhibit it.
Interestingly, hMSCs treated with 3,4,6-O-Bu3GalNAc exhibited lipid droplet accumulations, which were visualized by oil red O staining (Fig. 4A). The intracellular lipid accumulation is commonly observed in adipocyte cells and adipogenic lineage of MSC [30]. RT-PCR further confirmed up-regulation of several adipogenic-related genes, including fatty-acid-binding proteins (FABPs), lipoprotein lipase (LPL), and CCAAT/enhancer-binding protein α (CEBPA) (Fig. 4D). Similar to monolayer culture, the inhibition of hMSC osteogenesis by 3,4,6-O-Bu3GalNAc was observed in a 3D hydrogel system as evidenced by reduced calcium accumulation (Fig. 4E) and significant down-regulation of RUNX2, OCN, and COLLAGEN I (Fig. 4F).
3.3. Anti-inflammatory effect of 3,4,6-O-Bu3GalNAc on human OA chondrocytes
Next, we investigated the anti-inflammatory effects of 3,4,6-O-Bu3GalNAc on primary human chondrocytes from OA patients undergoing total knee arthroplasty. Chondrocytes were grown in the presence of IL-1β to maintain OA disease phenotype [31]. We examined expression of genes transcriptionally regulated by NF-κB specifically, NFKB1 and IκBα, as well as IL-1β for pro-inflammatory cytokine expression. 3,4,6-O-Bu3GalNAc treatment in monolayer culture significantly reduced expression of NFKB1, IκBα and IL-1β compared with control cells that were not treated with 3,4,6-O-Bu3GalNAc (Fig. 5A).
Fig. 5.

3,4,6-O-Bu3GalNAc exerts an anti-inflammatory effect on human OA chondrocytes. Primary human OA chondrocytes were cultured in chondrogenic medium supplemented with IL-1β for 24 h and then further incubated without (0 μM) or with 3,4,6-O-Bu3GalNAc (100 μM) in the presence of IL-1β for another 24 h. A. Gene expression of inflammatory markers and B. Wnt signaling genes. Data are averages normalized to β-ACTIN. The relative expression level to a non-treated (0 μM) control is shown (n = 3, * P<0.05, ** P<0.01, *** P<0.001, t-test). C. β-Catenin protein expression confirmed by Western blotting. Data are representative of 3 independent experiments and fold change values of 3,4,6-O-Bu3GalNAc treated group, relative to control, were obtained by using densitometry and are shown below individual lanes (0.56 ± 0.06 (P<0.05, t-test) for cytoplasmic β-catenin and 0.42 ± 0.11 (P<0.05, t-test) for nuclear β-catenin). D. immunofluorescence staining (scale bars: 50 μm).
Because pro-inflammatory cytokine-induced cartilage degradation in OA is associated with increased Wnt/β-catenin signaling [32, 33], we next evaluated the involvement of Wnt genes and proteins. 3,4,6-O-Bu3GalNAc down-regulated the expression of Wnt-related genes, β-CATENIN, LEF1, TCF1, AXIN2, and C-JUN, compared with untreated controls (Fig. 5B). β-Catenin protein levels were decreased in both nuclear and cytoplasmic chondrocyte compartments (Fig. 5C and D).
We further evaluated the inhibitory effects of 3,4,6-O-Bu3GalNAc on the human OA chondrocytes in the 3D hydrogel culture system after treatment for 21 days in the presence of IL-1β. RT-PCR confirmed down-regulation of NFKB1, IκBα, IL-1β, and the cartilage matrix–degrading enzyme MMP13 (Fig. S3). Similarly, expression of Wnt-related genes, C-JUN and β-CATENIN, was also reduced by treatment with 3,4,6-O-Bu3GalNAc (Fig. S3). The anti-inflammatory effect that accompanies inhibition of Wnt/β-catenin signaling has been shown to induce cartilage anabolism [34]. To this end, we confirmed up-regulated gene expression of AGGRECAN and TYPE II COLLAGEN, indicating the anabolic effect of 3,4,6-O-Bu3GalNAc (Fig. S3). Additionally, 3,4,6-O-Bu3GalNAc exposure increased GAG content and ECM accumulation in the 3D hydrogel constructs compared with controls (Fig. S3).
3.4. 3,4,6-O-Bu3GalNAc relives cartilage degradation in a rat model of OA
In monolayer and 3D cultures, 3,4,6-O-Bu3GalNAc exerted both chondrogenic and anti-inflammatory effects on human MSCs and OA chondrocytes, respectively. Thus, we investigated the therapeutic efficacy of 3,4,6-O-Bu3GalNAc in vivo in the medial meniscal transection (MMT) OA rat model [35]. After MMT was performed, 3,4,6-O-Bu3GalNAc (5 μg/μL) was injected weekly into the knee joint for three weeks. There were no observable post-surgical complications or adverse systemic effects (weight loss or significant swelling). Four weeks after the surgery induction of OA, the rats were sacrificed and histological assessments according to Safranin-O staining were carried out on the medial tibial plateau where the majority of the lesions were located on (Fig. 6A). Grading of the histological assessments based on OARSI (Osteoarthritis Research Society International) score [36] showed a 74% reduction in the score of the rats treated with three doses of 3,4,6-O-Bu3GalNAc (Multi-GalNAc, 500 μg per injection) compared with vehicle-treated rats (OA surgery, n=6) (Fig. 6B).
Fig. 6.

Prevention of cartilage degradation in OA induced rats by intra-articular injection of 3,4,6-O-Bu3GalNAc. A. Representative safranin-O stained histological images of the tibial plateau to evaluate the pathological changes 4 weeks after medial meniscal transection (scale bar: 200 μm). The Multi-GalNAc group was given IA injections of 3,4,6-O-Bu3GalNAc on days 7, 14, and 21 after the OA surgery. The Single-GalNAc, PLGA, and PLGA-GalNAc groups were given IA injections on days 7 after the surgery. B. Comparison of the tibial plateau joint based on the OARSI scoring system (The number of animals: 3~6, ** P<0.01, *** P<0.001, ANOVA). C. The size distribution of PLGA-GalNAc and scanning electron microscopic image of fabricated PLGA-GalNAc microspheres (scale bar: 5 μm). D. The cumulative in vitro release profile showed sustained release of 3,4,6-O-Bu3GalNAc from PLGA-GalNAc microspheres for over a month.
The local intra-articular (IA) injection of therapeutics is feasible for OA treatment, but rapid clearance of the IA injected drugs in the joint would limit their prolonged residence time from the target tissues and ultimately efficacy [13, 16]. Therefore, we evaluated sustained-release drug delivery of 3,4,6-O-Bu3GalNAc by using poly(lactic-co-glycolic acid) (PLGA) microspheres that can be used for local joint delivery [14]. We fabricated PLGA-GalNAc microspheres (diameter: 3.8 ± 0.4 μm) and showed sustained in vitro release profiles of 3,4,6-O-Bu3GalNAc from the microspheres for more than one month (Fig. 6 C and D). 15mg of PLGA-GalNAc microspheres (loading 1.5 mg of 3,4,6-O-Bu3GalNAc) were then intra-articularly injected one week after surgical induction of OA. The OARSI score showed relief of cartilage degradation in the PLGA-GalNAc treatment group, comparable to the Multi-GalNAc (Fig. 6B). Neither a single 3,4,5-O-Bu3GalNAc (1.5mg) injection nor unloaded PLGA microspheres (n=4) resolved OA disease in control rats.
4. Discussion
OA is now well recognized as a whole joint disease [8]. The pathological changes of OA result from an imbalance in the dynamic equilibrium between the breakdown and repair of all the articular joint tissues including bone, synovium, and cartilages. At the molecular level, they are mediated by anabolic and catabolic factors that participate in cross-talk and feedback between the tissues in order to regulate their expression [37]. Owing to this complex interrelationship, a single-target pharmacologic approach of DMOADs would provide limited therapeutic efficacy on OA [38]. In fact, the most effective DMOADs in development act on OA through a combination of several therapeutic targets. For example, strontium ranelate exhibits OA structure-modifying activity by modulating bone turnover and exerting an anabolic (cartilage formation) effect on chondrocytes [39]. Other drugs candidates, including calcitonin, have been shown to have beneficial metabolic effects on both bone and cartilage, promoting the growth of cartilage tissues [40] while bone morphogenetic protein 7 (BMP-7) produced strong anabolic and anti-catabolic activities on cartilage matrix, inhibiting OA progression [41]. Therefore, a comprehensive approach to target multiple elements would be desirable in the development of new DMOADs [42].
In this work, we investigated a new sugar-based therapeutic, as a small molecule inducer of chondrogenic differentiation of human MSCs and anti-inflammatory for OA treatment. Carbohydrates, including amino sugars, are emerging as a versatile class of therapeutics, as their chemical composition is easily reengineered—synthesis is cost-effective and easy to scale up, and they are structurally stable. Several studies have shown the promise of hexosamine derivatives for modulating metabolism [35, 43]. We synthesized a drug platform comprising the N-acetylated galactosamine modified with ester-linked butyrate groups, which improve amino sugar uptake in cells [44]. Upon cellular uptake, butanoylated sugars are metabolized by intracellular esterases to their natural byproducts, which in the case of 3,4,6-O-Bu3GalNAc are GalNAc and three equivalents of butyrate. The hexosamine core is a metabolic “building block” for biosynthesis of key molecules in cartilage such as proteoglycans while butyrate molecules can modulate signaling pathways [45, 46].
Towards developing a new class of sugar-based OA drugs, we previously screened monosaccharide analogs with varying core scaffolds and substitution arrangements for their ability to reduce expression of inflammatory-related genes and increase expression of cartilage specific matrix molecules [24]. These studies identified that 3,4,6-O-Bu3GalNAc efficiently addressed the excess inflammation and reduced tissue production found in animal and human arthritic cells and was therefore selected for further study and translation in this work. An important aspect of our previous work was that, although sugar analogs are enzymatically hydrolyzed intracellularly to their constituent sugar and butyrate moieties over one or two days [45] and although both of these metabolites have potential biological activity, the critical disease-modifying action of 3,4,6-O-Bu3GalNAc and 3,4,6-O-Bu3GlcNAc are not due to these metabolites. Instead, isomers such as 1,3,4-O-Bu3GlcNAc that lack a butyrate substituent at the C6-OH position also lack the ability to suppress NF-κB and fail to show protective anti-OA effects in 3D bovine tissue culture models despite generating identical metabolites [23, 47]. Therefore, going into the current study, we had established that the specific butyrate substitutions confer unique biological activity to this molecule that acts on several interrelated pathophysiological and inflammatory signaling pathways within the articular joint (Fig. 1A).
The exact biochemical mechanisms underlying the disease-modifying ability of 3,4,6-butanoylated hexosamines remain unclear. To elaborate briefly, upon discovery of the ability of 3,4,6-O-Bu3ManNAc to inhibit NF-κB, we systematically ruled out that any extant mechanism was responsible for this effect and instead hypothesized that direct binding of the analog to NF-κB proteins such as NFKB1 had a contributing role [22]. Although the binding affinity was low compared to conventional drugs, this is presumably based on the entropic penalty suffered when the numerous rotatable bonds of a tributanoylated hexosamine are constrained upon binding to a receptor. This constrained flexibility, however, has a positive aspect insofar as the molecule has the potential to achieve an induced fit to other receptors, which in this study is manifest through complementary regulation of Wnt signaling. Although key mechanistic aspects of our drug candidates remain to be unraveled, their enticing ability to elicit multiple complementary biological activities that in particular have a beneficial impact on chondrogenic MSCs – provided impetus to move ahead towards in vivo evaluation.
Regulating cell fate, state, and function of endogenous MSCs using small molecules provides a new tool for stem cell-focused therapies [48] and adds a new strategy to treating OA—a disease currently with few options. We first confirmed, through gene expression studies, that 3,4,6-O-Bu3GalNAc enhanced human MSC chondrogenesis and decreased β-catenin signaling—a pathway that, when inactivated, stimulates chondrocyte differentiation [49]. Similarly, inhibiting Wnt signaling in MSCs promotes chondrogenesis [50, 51]. In line with these previous studies, our results indicate that 3,4,6-O-Bu3GalNAc promotes chondrogenesis of hMSCs by interfering with the Wnt/β-catenin signaling activity. Disruption of the Wnt signaling pathway has been shown to not only improve chondrogenesis but also impair the osteogenic capacity of MSCs [52]. We confirmed that 3,4,6-O-Bu3GalNAc inhibited the osteogenic differentiation of hMSCs. Interestingly, adipocyte features were observed after 15 days of osteogenic induction supplemented with 3,4,6-O-Bu3GalNAc. The inverse relationship between osteogenic and adipogenic differentiation has been reported both in vitro and in vivo [53]. Given our data that suggest action of 3,4,6-O-Bu3GalNAc on the Wnt pathway, we believe that the adipocyte phenotype occurred at the expense of reduced osteogenesis.
The stimulation of endogenous MSCs into the chondrogenic lineage by small molecules has promise as a cartilage regeneration strategy. However, chronic inflammatory conditions in an OA joint may provide a sub-optimal environment for the small molecules to function [54]. A large number of inflammatory cytokines present in an OA joint that disrupt the regeneration capacity of MSCs could also limit the therapeutic potential of the small molecules. Reducing inflammation could therefore create favorable conditions for drugs to act on endogenous MSCs [12]. Chondrocytes in the OA joint are one of the primary sources of inflammatory cytokines and mediators. Here, we demonstrated that 3,4,6-O-Bu3GalNAc treatment of primary human osteoarthritic chondrocytes under OA pathophysiological conditions down-regulated expression of inflammatory gene targets involved in OA in addition to stimulating new tissue production. Recently, it was found that Wnt/β-catenin signaling in osteoarthritic chondrocytes is more activated than normal chondrocytes and linked with inflammatory cytokine-induced cartilage degradation in OA [32, 33]. In line with these findings, inhibition of Wnt/β-catenin signaling in cytokine-stimulated chondrocytes reduces cartilage degradation [34]. Our results similarly demonstrated the involvement of Wnt-related genes and proteins in the anti-inflammatory effect of 3,4,6-O-Bu3GalNAc.
In the final part of this study, we took steps towards evaluating the clinical translational potential of 3,4,6-O-Bu3GalNAc as a disease-modifying intervention by testing this compound in a surgically-induced OA rat model; encouragingly, these results showed that weekly treatments prevented cartilage damages in the OA joints (Fig 6A). These results were particularly impressive because drugs injected into the knee joint typically are rapidly cleared (within 1–5 h) through the draining lymphatic system of the synovial fluid [16]. In light of the probable rapid clearance of 3,4,6-O-Bu3GalNAc, this compound was encapsulated in PLGA microspheres for prolonged and controlled release to further improve efficacy. Our results showed sustained release profiles of 3,4,6-O-Bu3GalNAc from the microspheres over one month in vitro, without showing initial burst drug release, presumably due to the stability of the carbohydrate-based drug encapsulation [55]. The controlled release strategy proved successful considering that a single intra-articular injection of the 3,4,6-O-Bu3GalNAc-laden PLGA microspheres one week after OA induction prevented cartilage degradation, comparable to multiple treatments of free 3,4,6-O-Bu3GalNAc (Fig 6B). Despite the chondroprotective effect of 3,4,6-O-Bu3GalNAc on the young OA animal model, further in vivo studies will be clearly needed to optimize dosing regimens to translate 3,4,6-O-Bu3GalNAc for the clinical treatment of late stages of OA. Notwithstanding this caveat, we emphasize that the in vitro and in vivo results presented here provide ground-breaking support for the disease-modifying anabolic and capacity of 3,4,6-O-Bu3GalNAc - a lead drug candidate exemplifying a recent surge in interest of non-conventional carbohydrate-based drugs – to prevent OA progression and limit inflammation in diseased cells and in the articular joint.
Supplementary Material
Acknowledgements
This research was supported by the National Institute of Health (R01AR054005) and the Maryland Technology Development Corporation (TEDCO) Stem Cell Research Fund for postdoctoral fellows (Chaekyu Kim).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Wieland HA, Michaelis M, Kirschbaum BJ, Rudolphi KA. Osteoarthritis - an untreatable disease? Nat Rev Drug Discov 4 (2005) 331–344. [DOI] [PubMed] [Google Scholar]
- [2].Blanco FJ, Ruiz-Romero C. New targets for disease modifying osteoarthritis drugs: chondrogenesis and Runx1. Ann Rheum Dis 72 (2013) 631–634. [DOI] [PubMed] [Google Scholar]
- [3].Pelletier J-P. Rationale for the use of structure-modifying drugs and agents in the treatment of osteoarthritis. Osteoarthritis Cartilage 12 (2004) 63–68. [DOI] [PubMed] [Google Scholar]
- [4].Cho T-J, Kim J, Kwon S-K, Oh K, Lee J.-a., Lee D-S, et al. A potent small-molecule inducer of chondrogenic differentiation of human bone marrow-derived mesenchymal stem cells. Chem Sci 3 (2012) 3071–3075. [Google Scholar]
- [5].Johnson K, Zhu S, Tremblay MS, Payette JN, Wang J, Bouchez LC, et al. A stem cell-based approach to cartilage repair. Science 336 (2012) 717–721. [DOI] [PubMed] [Google Scholar]
- [6].Yano F, Hojo H, Ohba S, Fukai A, Hosaka Y, Ikeda T, et al. A novel disease-modifying osteoarthritis drug candidate targeting Runx1. Ann Rheum Dis 72 (2013) 748–753. [DOI] [PubMed] [Google Scholar]
- [7].Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol 7 (2011) 33–42. [DOI] [PubMed] [Google Scholar]
- [8].Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum 64 (2012) 1697–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wehling N, Palmer GD, Pilapil C, Liu F, Wells JW, Muller PE, et al. Interleukin-1beta and tumor necrosis factor alpha inhibit chondrogenesis by human mesenchymal stem cells through NF-kappaB-dependent pathways. Arthritis Rheum 60 (2009) 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Joos H, Wildner A, Hogrefe C, Reichel H, Brenner RE. Interleukin-1 beta and tumor necrosis factor alpha inhibit migration activity of chondrogenic progenitor cells from non-fibrillated osteoarthritic cartilage. Arthritis Res Ther 15 (2013) R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ousema PH, Moutos FT, Estes BT, Caplan AI, Lennon DP, Guilak F, et al. The inhibition by interleukin 1 of MSC chondrogenesis and the development of biomechanical properties in biomimetic 3D woven PCL scaffolds. Biomaterials 33 (2012) 8967–8974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mobasheri A. The future of osteoarthritis therapeutics: emerging biological therapy. Curr Rheumatol Rep 15 (2013) 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Evans CH, Kraus VB, Setton LA. Progress in intra-articular therapy. Nat Rev Rheumatol 10 (2014) 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kang ML, Im GI. Drug delivery systems for intra-articular treatment of osteoarthritis. Expert Opin Drug Deliv 11 (2014) 269–282. [DOI] [PubMed] [Google Scholar]
- [15].Zhang Z, Huang G. Micro- and Nano-Carrier Mediated Intra-Articular Drug Delivery Systems for the Treatment of Osteoarthritis. Journal of Nanotechnology 2012 (2012) 1–11. [Google Scholar]
- [16].Owen SG, Francis HW, Roberts MS. Disappearance kinetics of solutes from synovial fluid after intra-articular injection. Br J Clin Pharmacol 38 (1994) 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang Z, Huang G. Intra-articular lornoxicam loaded PLGA microspheres: enhanced therapeutic efficiency and decreased systemic toxicity in the treatment of osteoarthritis. Drug Deliv 19 (2012) 255–263. [DOI] [PubMed] [Google Scholar]
- [18].Kang ML, Ko JY, Kim JE, Im GI. Intra-articular delivery of kartogenin-conjugated chitosan nano/microparticles for cartilage regeneration. Biomaterials 35 (2014) 9984–9994. [DOI] [PubMed] [Google Scholar]
- [19].Ko JY, Choi YJ, Jeong GJ, Im GI. Sulforaphane-PLGA microspheres for the intra-articular treatment of osteoarthritis. Biomaterials 34 (2013) 5359–5368. [DOI] [PubMed] [Google Scholar]
- [20].Rothenfluh DA, Bermudez H, O’Neil CP, Hubbell JA. Biofunctional polymer nanoparticles for intra-articular targeting and retention in cartilage. Nat Mater 7 (2008) 248–254. [DOI] [PubMed] [Google Scholar]
- [21].Meutermans W, Le GT, Becker B. Carbohydrates as scaffolds in drug discovery. ChemMedChem 1 (2006) 1164–1194. [DOI] [PubMed] [Google Scholar]
- [22].Elmouelhi N, Aich U, Paruchuri VDP, Meledeo MA, Campbell CT, Wang JJ, et al. Hexosamine Template. A Platform for Modulating Gene Expression and for Sugar-Based Drug Discovery. J Med Chem 52 (2009) 2515–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Coburn JM, Bernstein N, Bhattacharya R, Aich U, Yarema KJ, Elisseeff JH. Differential response of chondrocytes and chondrogenic-induced mesenchymal stem cells to C1-OH tributanoylated N-acetylhexosamines. PLoS One 8 (2013) e58899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Campbell CT, Aich U, Weier CA, Wang JJ, Choi SS, Wen MM, et al. Targeting Pro-Invasive Oncogenes with Short Chain Fatty Acid-Hexosamine Analogues Inhibits the Mobility of Metastatic MDA-MB-231 Breast Cancer Cells. J Med Chem 51 (2008) 8135–8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Augello A, De Bari C. The regulation of differentiation in mesenchymal stem cells. Hum Gene Ther 21 (2010) 1226–1238. [DOI] [PubMed] [Google Scholar]
- [26].Nicodemus GD, Bryant SJ. Cell encapsulation in biodegradable hydrogels for tissue engineering applications. Tissue Eng Part B Rev 14 (2008) 149–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].James AW. Review of Signaling Pathways Governing MSC Osteogenic and Adipogenic Differentiation. Scientifica (Cairo) 2013 (2013) 684736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Glass DA 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell 8 (2005) 751–764. [DOI] [PubMed] [Google Scholar]
- [29].Holmen SL, Zylstra CR, Mukherjee A, Sigler RE, Faugere MC, Bouxsein ML, et al. Essential role of beta-catenin in postnatal bone acquisition. J Biol Chem 280 (2005) 21162–21168. [DOI] [PubMed] [Google Scholar]
- [30].Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science 284 (1999) 143–147. [DOI] [PubMed] [Google Scholar]
- [31].Lotz M, Blanco FJ, von Kempis J, Dudler J, Maier R, Villiger PM, et al. Cytokine regulation of chondrocyte functions. J Rheumatol Suppl 43 (1995) 104–108. [PubMed] [Google Scholar]
- [32].Hwang SG, Yu SS, Lee SW, Chun JS. Wnt-3a regulates chondrocyte differentiation via c-Jun/AP-1 pathway. FEBS Lett 579 (2005) 4837–4842. [DOI] [PubMed] [Google Scholar]
- [33].Corr M. Wnt-beta-catenin signaling in the pathogenesis of osteoarthritis. Nat Clin Pract Rheumatol 4 (2008) 550–556. [DOI] [PubMed] [Google Scholar]
- [34].Landman EB, Miclea RL, van Blitterswijk CA, Karperien M. Small molecule inhibitors of WNT/beta-catenin signaling block IL-1beta- and TNFalpha-induced cartilage degradation. Arthritis Res Ther 15 (2013) R93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gibson M, Li H, Coburn J, Moroni L, Nahas Z, Bingham C 3rd, et al. Intra-articular delivery of glucosamine for treatment of experimental osteoarthritis created by a medial meniscectomy in a rat model. J Orthop Res 32 (2014) 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Pritzker KP, Gay S, Jimenez SA, Ostergaard K, Pelletier JP, Revell PA, et al. Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage 14 (2006) 13–29. [DOI] [PubMed] [Google Scholar]
- [37].Martel-Pelletier J, Wildi LM, Pelletier JP. Future therapeutics for osteoarthritis. Bone 51 (2012) 297–311. [DOI] [PubMed] [Google Scholar]
- [38].Hunter DJ. Pharmacologic therapy for osteoarthritis--the era of disease modification. Nat Rev Rheumatol 7 (2011) 13–22. [DOI] [PubMed] [Google Scholar]
- [39].Lafeber FP, van Laar JM. Strontium ranelate: ready for clinical use as disease-modifying osteoarthritis drug? Ann Rheum Dis 72 (2013) 157–161. [DOI] [PubMed] [Google Scholar]
- [40].Karsdal MA, Sondergaard BC, Arnold M, Christiansen C. Calcitonin affects both bone and cartilage: a dual action treatment for osteoarthritis? Ann N Y Acad Sci 1117 (2007) 181–195. [DOI] [PubMed] [Google Scholar]
- [41].Hunter DJ, Pike MC, Jonas BL, Kissin E, Krop J, McAlindon T. Phase 1 safety and tolerability study of BMP-7 in symptomatic knee osteoarthritis. BMC Musculoskelet Disord 11 (2010) 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mobasheri A. The future of osteoarthritis therapeutics: targeted pharmacological therapy. Curr Rheumatol Rep 15 (2013) 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Varghese S, Theprungsirikul P, Sahani S, Hwang N, Yarema KJ, Elisseeff JH. Glucosamine modulates chondrocyte proliferation, matrix synthesis, and gene expression. Osteoarthritis Cartilage 15 (2007) 59–68. [DOI] [PubMed] [Google Scholar]
- [44].Kim EJ, Sampathkumar SG, Jones MB, Rhee JK, Baskaran G, Goon S, et al. Characterization of the metabolic flux and apoptotic effects of O-hydroxyl- and N-acyl-modified N-acetylmannosamine analogs in Jurkat cells. J Biol Chem 279 (2004) 18342–18352. [DOI] [PubMed] [Google Scholar]
- [45].Mathew MP, Tan E, Shah S, Bhattacharya R, Adam Meledeo M, Huang J, et al. Extracellular and intracellular esterase processing of SCFA-hexosamine analogs: implications for metabolic glycoengineering and drug delivery. Bioorg Med Chem Lett 22 (2012) 6929–6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sampathkumar SG, Jones MB, Meledeo MA, Campbell CT, Choi SS, Hida K, et al. Targeting glycosylation pathways and the cell cycle: sugar-dependent activity of butyrate-carbohydrate cancer prodrugs. Chem Biol 13 (2006) 1265–1275. [DOI] [PubMed] [Google Scholar]
- [47].Coburn JM, Wo L, Bernstein N, Bhattacharya R, Aich U, Bingham CO 3rd, et al. Short-chain fatty acid-modified hexosamine for tissue-engineering osteoarthritic cartilage. Tissue Eng Part A 19 (2013) 2035–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lairson LL, Lyssiotis CA, Zhu S, Schultz PG. Small molecule-based approaches to adult stem cell therapies. Annu Rev Pharmacol Toxicol 53 (2013) 107–125. [DOI] [PubMed] [Google Scholar]
- [49].Akiyama H, Lyons JP, Mori-Akiyama Y, Yang X, Zhang R, Zhang Z, et al. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev 18 (2004) 1072–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Luo S, Shi Q, Zha Z, Yao P, Lin H, Liu N, et al. Inactivation of Wnt/beta-catenin signaling in human adipose-derived stem cells is necessary for chondrogenic differentiation and maintenance. Biomed Pharmacother 67 (2013) 819–824. [DOI] [PubMed] [Google Scholar]
- [51].Im GI, Quan Z. The effects of Wnt inhibitors on the chondrogenesis of human mesenchymal stem cells. Tissue Eng Part A 16 (2010) 2405–2413. [DOI] [PubMed] [Google Scholar]
- [52].Gustafson B, Eliasson B, Smith U. Thiazolidinediones increase the wingless-type MMTV integration site family (WNT) inhibitor Dickkopf-1 in adipocytes: a link with osteogenesis. Diabetologia 53 (2010) 536–540. [DOI] [PubMed] [Google Scholar]
- [53].Zhang Y, Khan D, Delling J, Tobiasch E. Mechanisms underlying the osteo- and adipo-differentiation of human mesenchymal stem cells. ScientificWorldJournal 2012 (2012) 793823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mastbergen SC, Saris DB, Lafeber FP. Functional articular cartilage repair: here, near, or is the best approach not yet clear? Nat Rev Rheumatol 9 (2013) 277–290. [DOI] [PubMed] [Google Scholar]
- [55].Anton N, Jakhmola A, Vandamme TF. Trojan microparticles for drug delivery. Pharmaceutics 4 (2012) 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.