

Abstract
The wild-type p53 induced phosphatase 1, Wip1 (PP2Cδ), is a PP2C family Ser/Thr phosphatase that negatively regulates the function of multiple proteins such as p53, ATM, Chk1, Chk2, Mdm2 and p38 MAPK involved in a DNA damage response. Wip1 dephosphorylates and inactivates its protein targets which are critical for cellular stress responses. Additionally, Wip1 frequently amplified and overexpressed in several human cancer types. Because of its negative role in regulating the function of essential proteins, Wip1 has been identified as a potential therapeutic target in various types of cancers. Based on a recently reported Wip1 inhibitor (G-1), we performed an extensive structure-activity relationship (SAR) analysis. This led us to interesting findings in SAR trends and to the discovery of new chemical analogues with good specificity and bioavailability.
Keywords: Wip1, Inhibitors, Enzyme kinetics, ELISA assay
Introduction
Wip1 (PPM1D or PP2Cδ), a member of the protein phosphatase 2C (PP2C) family, was first identified as the product of a gene induced by wild-type p53 after DNA damage.[1] Similar to the other members of the PP2C family, Wip1 is monomeric, insensitive to oakadaic acid, and dependent on millimolar concentrations of Mn2+ or Mg2+ for activity in vitro.[2] Wip1 inactivates a wide range of proteins, such as p38 MAPK,[3] Chk1,[4] Chk2,[5] ATM,[6] and p53,[4] by dephosphorylating phosphothreonine (pT) or phosphoserine (pS) residues. Wip1 is amplified or overexpressed in numerous human cancers including breast cancer,[7] ovarian clear cell carcinoma,[8] gastric cancer,[9] pancreatic adenocarcinoma,[10] medulloblastoma,[11] and neuroblastoma.[12] Moreover, Ppm1d-null mice show a tumor-resistant phenotype, suggesting Wip1 has a critical role in controlling cell cycle checkpoints in response to DNA damage.[13] Thus, developing new strategies for inhibiting Wip1 activity may be beneficial in the treatment of a number of human cancers.[14]
The Wip1 inhibitors identified to date have been discovered through the use of extensive chemical library screening approaches [15] or substrate-based design of phosphopeptides.[16] Based on biochemical and biophysical screening of small molecule and DNA-encoded compound libraries, an allosteric inhibitor of Wip1, GSK2830371, was recently reported.[17] It was proposed that the basis for the selectivity of this inhibitor for Wip1 over other PP2C family members was due to its interaction with the Flap of Wip1. The Flap is a sub-domain located near the catalytic site that is important for substrate recognition and exhibits substantial amino acid sequence variability among PP2C family members. Although GSK2830371 is a selective and potent inhibitor of Wip1 activity in cells, it does not exhibit favorable pharmacokinetics.[12a, 17–18]
As the Flap subdomain provides the key structural linkage between the activity and the substrate selectivity of PP2C phosphatases, we believe it is important to understand the structure-activity relationships (SAR) in Flap subdomain-based inhibitors. As the starting point of our investigation, we chose one of the initial compounds identified in the biochemical high throughput screen (HTS) reported by Gilmartin et al. [17]. Very limited SAR is available for these classes of compounds as the manuscript of Gilmartin et al. only discloses a small number of molecules. We therefore performed a more thorough SAR study and developed analogues that exhibit good affinity and bioavailability. Our study demonstrates interesting SAR trends and also provides critical insights into designing new compounds that target the Flap subdomain of Wip1.
Results and Discussion
We performed detailed biochemical studies of analogs based on the initial hit of the biochemical screen described by Gilmartin et al.,[17] hereafter referred to as compound G-1, (Figure 1A). To facilitate the SAR study, we synthesized variants of G-1 with alterations in functional groups at either end as well as a central linker between the ends that could be altered to adjust the distance.
Figure 1.

Initial Wip1 inhibitors. (A) Structures of compound G-1 and 1. (B) Inhibition of Wip1 phosphatase activity toward the ATM (1981pS) phosphopeptide by compounds G-1 and 1.
At the outset, the central 3-cyclohexyl-L-alanine moiety of G-1 was retained and 2-carboxythiophene was used to form the N-terminal amide. To introduce a linker, an amide was formed with the ε amine sidechain of Lysine. The α-amine of Lysine was attached to a 3-hydroxybenzoyl group that serves as the C-terminal cap (Figure 1A). Compound 1 was synthesized via amino acid coupling reactions using 2-thiophenecarboxylic acid, 3-cyclohexyl-L-alanine, L-Lysine and 3-hydroxybenzoic acid as shown in Scheme 1. Importantly, we chose to test the activity of these analogs to inhibit the phosphatase activity of Wip1 using the ATM (1891pS) phosphopeptide, which is a physiological substrate,[6, 19] rather than fluorescein diphosphate which is an artificial substrate. When using the ATM phosphopeptide substrate in assays, compound G-1 had an IC50 of 4.9 μM, which is about ten times weaker compared to inhibition tests using fluorescein diphosphate as the substrate. Under the same testing conditions, 1 had an IC50 of 0.92 μM, approximately five-fold better that G-1 (Figure 1B). To better understand the SAR of these types of molecules, we identified three regions of 1 for further modifications (Figure 2).
Scheme 1.

General synthesis protocol for Compound 1; Reagents and conditions: (i) EDC, HOBt, NMM, DMF, RT, overnight, (ii) H2/Pd/C, MeOH, RT, 6 h; (iii) LiOH, MeOH:H2O (4:1), 5 h.
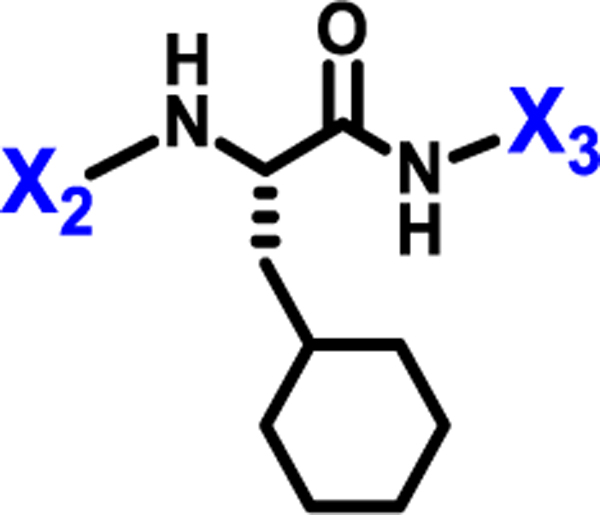
Figure 2.

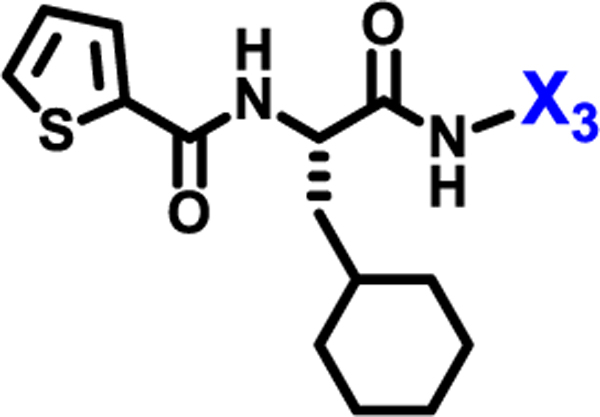
X1, X2, and X3 positions of compound 1.
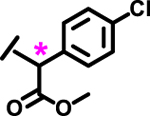
We initially explored variation of the 3-cyclohexyl-L-alanine moiety at position X1 as well as variations at X3 (Table 1). Replacement of the 3-cyclohexyl-L-alanine with L-leucine (2) worsened the IC50 by more than a factor of 250. The corresponding L-Tyrosine (3) and L-Serine (4) analogs showed only weak 25% and 20% inhibition at 50 μM concentration, respectively. Modification with L-Phenylalanine (5) or (S)-2-(4-pentenyl)alanine (6) resulted in a complete loss of activity.
Table 1.
SAR at position X1
| Compound | Structure | IC50 (μM) | inhibition† |
|---|---|---|---|
| 1 | 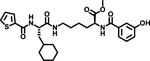 |
0.92 | |
| 2 | 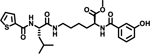 |
>100 | |
| 3 | 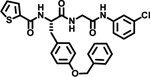 |
25% | |
| 4 | 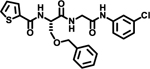 |
20% | |
| 5 | 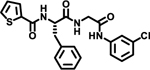 |
ND | |
| 6 | 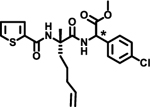 |
ND | |
| 7 | 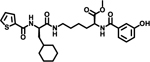 |
ND | |
| 8 | 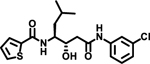 |
ND | |
| 9 | 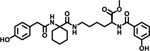 |
ND | |
| 10 | 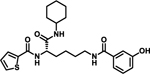 |
ND | |
Racemic compound,
Inhibition at 50 μM compared with DMSO; ND-not detectable
Notably, changing the stereochemical configuration of 3-cyclohexyl-L-alanine to the D isomer (7) also led to the complete loss of inhibitory activity. Furthermore, substitution with L-statine (8), substitution with racemic 1-aminocyclohexanecarboxyl (9), or addition of an amide linkage to the cyclohexyl moiety (10) did not improved activity, suggesting critical roles of both the stereogenic center and the cyclohexyl ring.
The loss in inhibition observed upon substitution at position X1 indicates that the 3-cyclohexyl-L-alanine is highly constrained, plays a vital role in binding to the Wip1 protein and hence cannot be changed.
Next, we explored variations at the X2 and X3 positions while retaining the central cyclohexyl-L-alanine (Table 2). Replacement of the 2-thiophenecarboxyl moiety with 3-aminobenzoyl at the X2 position and varying X3 with differently substituted aromatic moieties produced moderate effects on the activity (11–14). However, reducing the distance between 3-cyclohexyl-L-alanine and a 3-chloro substituted aromatic ring at X3 (compounds 11 and 13) resulted in a five-fold improvement in activity. Within position X2, N-terminal extension of the 3-amino benzoyl moiety with acetyl (15), orotyl (16), 4-hydroxyphenylacetyl (17), or 2-nitrophenyl sulfonyl (18) (with reduced distance between X1 and X3 on the substituted aromatic ring) led to activities similar to those of compounds 11–14. We next explored various substitutions at X2, such as N-acetyl proline (19), 1H-tetrazole-5-acetyl (21), Cbz-Lysine (22), and orotyl (23), all of which led to complete loss of activity except for weak activity exhibited by compound 20 (IC50 =167 μM). We also tested some phosphorylated serine analogues (24–26) at position X2, but these compounds exhibited little to no inhibitory activity. Furthermore, we synthesized several compounds (data not shown) with di- and tri- substituted thiophenecarboxyl derivatives at the X2 position of compound 1, and these showed no improvement in activity or resulted into very weak inhibitors.
Table 2.
Structural modifications at positions X2 and X3.
 | ||||
|---|---|---|---|---|
| Compound | X2 | X3 | IC50 (μM)† | inhibition‡ |
| 11 |  |
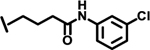 |
11 | |
| 12 | 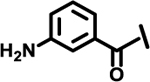 |
 |
4.3 | |
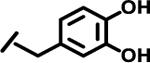
| 13 | 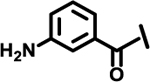 |
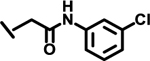 |
2.2 | |
| 14 | 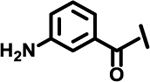 |
*
|
1.7 | |
| 15 | 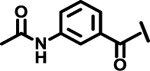 |
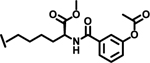 |
12.9 | |
| 16 | 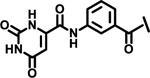 |
4.5 | ||
| 17 | 3.3 | |||
| 18 | 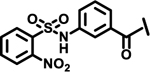 |
 |
1.0 | |
| 19 | 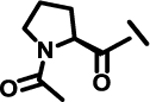 |
 |
ND | |
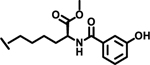
| 20 | 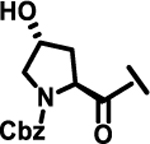 |
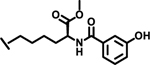 |
>100 | |
| 21 | 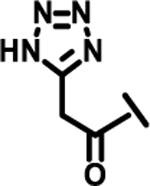 |
 |
ND | |
| 22 | 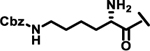 |
 |
ND | |
| 23 | 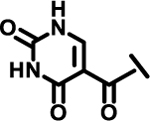 |
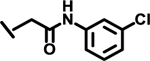 |
ND | |
| 24 | 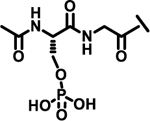 |
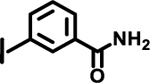 |
ND | |
| 25 | 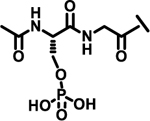 |
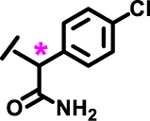 |
7%† | |
| 26 | 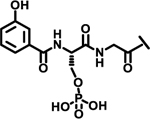 |
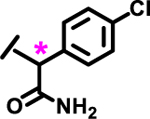 |
8%† | |
Racemic compound.
ND: not detectable.
Inhibition at 50 μM compared with DMSO.
These results suggest that SAR at position X2 is highly constrained. Most of the modifications we tested at position X2 led to significant or complete loss of inhibitory potency for Wip1. Only, inhibitor 18 showed a potency similar to that of the inhibitor 1, with an IC50 of 1 μM. Although some modifications at position X2 resulted in moderate improvements (11–26), the SAR appears to be largely constrained to the original 2-thiophenecarboxamide. Based on the overall results, any modification at X1 is highly disfavored and only slight alterations at X2 are tolerated.
To evaluate the effects of modifying position X3, we retained the L-cyclohexyl alanine and carboxy thiophene moieties at positions X1 and X2, respectively. We modified X3 as depicted in Table 3. Initially we acetylated the α-amino group of L-lysine with free carboxy acid (27) or methyl ester (28), each of which showed moderate activity. Further extension of these acetyl analogous with substituted-benzene derivatives (compounds 29 and 30) resulted in 3.5 and 7-fold improved activity, respectively, compared with compound 28. Furthermore, compound 30 showed the same activity as compound 18. On the other hand, replacing the 3-hydroxybenzoyl group with 4-hydroxyphenylacetyl (31), 6-bromo-3-carboxy pyridine (32), or 4-Bromo-α-hydroxyphenylacetyl (33) resulted in ≥ 3-fold reduced activity compared with compound 30, suggesting that substitution with electron donating or electron withdrawing groups (compound 29–33) at X3 has very little or no effect on the activity. In addition, replacing the aromatic ring (compound 29–33) with the aliphatic diethylphosphonoacetyl moiety (34) led to an 18-fold drop in activity. Substitution of the aromatic ring of X3 position, with 3-hydroxybenzyol at the α-amino group of L-lysine, restored inhibition (35), indicating that proper substitution of the six-membered aromatic ring in X3 is essential for activity. Improvements in activity were observed with compounds 36, 37, and 38 with modest changes in the arrangement of functional groups and carbon chain lengths. Interestingly, we found a moderate effect of the carbon chain length between the X1 and X3 positions in comparing compounds 38 and 39. We found that compound 39, which consists of 2-carboxythiophene, 3-cyclohexyl-L-alanine, and glycine-linked 3-chloroaniline at X2, X1, and X3 positions respectively, was the most active inhibitor in this study.
Table 3.
SAR at X3 position
 | ||
|---|---|---|
| Compound | X3 | IC50 (μM) |
| 27 | 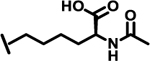 |
9.0 |
| 28 | 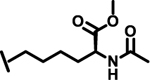 |
6.9 |
| 29 | 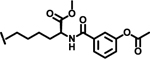 |
1.9 |
| 30 | 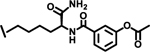 |
1.0 |
| 31 | 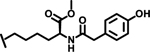 |
3.8 |
| 32 | 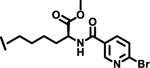 |
3.9 |
| 33 | 3.0 | |
| 34 | 17.9 | |
| 35 | 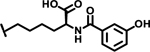 |
5.4 |
| 36 | 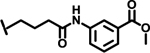 |
2.6 |
| 37 | 1.5 | |
| 38 | 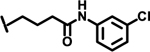 |
1.0 |
| 39 | 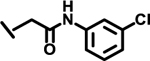 |
0.65 |
Racemic compound
In summary, slight modifications at X1 led to complete losses in activity. At position X1, it is important to maintain the correct type of cyclic ring (cyclohexane) as well as the correct chirality (S). Slight modifications are tolerated at positions X2 and X3. For instance, compounds 18 (with modification at X2) and 38 (with modification at X3) retained activities similar to that of compound 1. A small reduction in carbon chain length between positions X1 and X3 led to slightly improved activity (compounds 38 and 39).
Taken together, these results suggest that the SAR for the class of compound is not permissive for a wide range of substitutions. Compound 39 was identified as the best analog from the series of molecules, and was therefore selected for additional studies.
As specificity within the same family of PP2C phosphatases is a critical issue in drug development, we assessed whether compound 39 exhibited inhibitory activity toward human PPM1A, a closely related PP2C phosphatase. As shown in Figure 3A, compound 39 potently inhibited Wip1 phosphatase activity while remaining inactive with PPM1A. This result is consistent with the high specificity of the inhibitors reported by Gilmartin et al. [17]
Figure 3.

Selectivity and mechanism of inhibition of compound 39 over PPM1A.(A) Inhibition of Wip1 activity toward the ATM (1981pS) phosphopeptide by compound 39. (B) Mixed Mode Inhibition Scheme. (C) Dependence of Wip1 phosphatase activity on concentrations of [S1981pS] human ATM (1976–1986)-GY peptide in absence ( ) and presence of 0.4 μM (
) and presence of 0.4 μM ( ) and 2.0 μM (
) and 2.0 μM ( ) compound 39.
) compound 39.
Next, we investigated the mechanism of inhibition. In the Mixed Inhibition Model (Figure 3B), the inhibitor can bind to both the free enzyme, E and the enzyme-substrate complex, ES, inhibiting catalytic activity. The dependence of the initial rates of reaction on the concentrations of substrate and compound 39 were globally fitted to the Mixed Inhibition Model (Figure 3C). The data were well described by the model, indicating that compound 39 inhibits Wip1 activity toward phosphopeptide substrates by an allosteric mechanism with parameter estimates of kcat = 7.45 ± 0.25 s−1, Ki = 2.2 ± 0.7 μM and Km = 76 ± 7 μM with α = 1.41. As the value of α is greater than unity, the binding of the inhibitor is stronger to the free enzyme than to the enzyme-substrate complex.
We chose to test the bioavailability and inhibitory activity of compounds 1 and 39 on Wip1 phosphatase activity in the human breast adenocarcinoma cell line MCF-7. The S139-phosphorylated form of the histone variant H2AX (γH2AX) is an important component of the DNA double strand break-induced DNA damage response (DDR) signaling pathway. Phosphopeptides containing H2AX pS139 are directly dephosphorylated by Wip1 and dephosphorylation of γH2AX by Wip1 is important for the recovery of cells following the induction of DNA damage.[19–20] γH2AX levels following the induction of DNA double-strand breaks by ionizing radiation or chemicals have been used as an indicator of Wip1 activity in human cells.[21]
To assess the bioavailability of our Wip1 inhibitors in MCF7 cells, we adapted a recently reported ELISA method for quantitative determination of ƴH2AX levels.[22] In agreement with previously reported qualitative results,[17] pre-treatment of MCF7 cells with GSK2830371 resulted in a dose-dependent increase in γH2AX levels 75’ min after exposure to 10 Gy ionizing radiation (IR) (Figure S1, EC50 = 0.20 ± 0.12 μM, n = 3). Similarly, pre-treatment of MCF7 cells with compound 1 or compound 39 resulted in a dose-dependent increase in γH2AX levels 75’ min after exposure to 10 Gy IR (Figure 4). Importantly, treatment of MCF7 cells with GSK2830371, compound 1 or compound 39 did not produce a detectable increase in ƴH2AX levels in the absence of exposure to IR (Figure S2). These results show that compounds 1 and 39 are (i) bioavailable in MCF7 cells, (ii) do not induce H2AX phosphorylation by themselves and (iii) can suppress Wip1 enzymatic activity towards H2AX phospho-Ser139.
Figure 4.

Dose-dependent inhibition of Wip1-induced dephosphorylation of γH2AX in human breast cancer cells during recovery from following exposure to ionizing radiation (IR). MCF7 cells were incubated with various concentrations of compound 1 (left panel) or compound 39 (right panel) for 1 h prior to exposure to 10 Gy IR and continuing through a 75-min recovery period. γH2AX levels were determined by quantitative ELISA. The dose response was fitted by a four-parameter logistical model. Curves shown are representative. Compound 1: EC50 = 56 ± 5μM, n = 3. Compound 39: EC50 = 24 ± 6 μM, n = 3.
Conclusions
In summary, we performed an extensive SAR study based on compound G-1, an initial hit from a screen that was used for development of a novel Wip1 inhibitor.[17] In the present study, we found that the inhibitory activity is highly dependent on the carbocyclic ring and chirality at the central position X1. All tested modifications at this position resulted in complete loss of inhibitory activity. Some modifications at positions X2 and X3 positions were tolerated, but most did not lead to significant improvements in activity. All three parts of the scaffold investigated in the SAR study are crucial for activity. These studies led to the development of compound 39, which exhibited improved inhibitory compared with that of compound G-1 in assays that are designed to test activity in the presence of physiologically-relevant substrates. Furthermore compounds 1 and 39 were found to suppress Wip1 enzymatic activity towards H2AX phospho-Ser139 in MCF-7 cells, demonstrating their bioavailability to cells. Further work to identify new classes of Wip1 Flap-targeted inhibitors will continue, using the assays described in this study.
Experimental Section
Fmoc or Boc-protected amino acids were purchased from Novabiochem (San Diego CA). All chemicals and solvents were purchased from Sigma-Aldrich (St. Louis, MO) and AnaSpec (Fremont, CA). All reagents and solvents were used as received from commercial sources without further purification.
Final compounds were purified using reversed-phase high-performance liquid chromatography (RP-HPLC) on a preparative C4 column (BioAdvantage Pro 300, Thomson Liquid Chromatography) with a binary solvent system: a linear gradient of CH3CN in 0.04 % trifluoroacetic acid (TFA) and water in 0.05% TFA with a flow rate 7 mL/min and detected at 220 nm. Analytical HPLC was performed using a C4 reverse phase column with a binary solvent system: linear gradient of 0.04 % TFA:CH3CN 10–900% in 0.05% aqueous TFA in 30 min at a flow rate of 1 mL/min, detected at 230 nm. The purity of the compounds was found to be >94% using analytical HPLC based on peak area percentage (For compounds 1 and 39 which were tested in cell based assay the purity was >98%). Mass spectra were obtained using a Waters MALDI micro MX (MALDI-TOF) with α-cyano-4-hydroxy cinnamic acid a matrix. Proton NMR spectra were recorded at 400 MHz on a Bruker 400 spectrometer. For NMR spectra, chemical shifts are expressed in parts per million (ppm) downfield from internal tetramethylsilane (δ 0) in DMSO-d6 as a solvent.
General procedure for synthesis of compounds 1–23 and 27–39:
1. General procedure for peptide coupling:
The acid (1 mmol) was dissolved in DMF (2 mL) and cooled to 0 °C with an ice bath. HOBt. H2O (1.2 mmol), EDC.HCl (1.2 mmol), and N-methylmorpholine (3 mmol) were added sequentially. The resulted mixture was stirred for additional 5 min before amine (1 mmol) was added. After stirring at 0 °C for 30 min, the reaction mixture was allowed to attain room temperature and stirred overnight. The reaction mixture was diluted with ethyl acetate (15 mL) and 10% citric acid (10 mL), the aqueous layer was further extracted with ethyl acetate (2 × 10 mL). The combined organic extracts were washed with water, sat. NaHCO3, brine, dried over anhydrous Na2SO4 and concentrated in vacuo to give the crude product which was used in the next step without purification. In case of final compounds, a small portion of the product was purified by preparative HPLC. The desired fractions were collected and immediately lyophilized to afford amorphous powders.
2. General procedure for N-Boc-deprotection with 4N HCl in dioxane
4 N HCl in 1,4-dioxane was added to the afforded Boc-protected peptide/amine (ca. 1.0 mmol) at 0 °C and stirred for 1 h at room temperature. The solvent was removed under vacuum to afford the product as the corresponding HCl salt and used directly to the next step without purification.
3. General procedure for ester hydrolysis
To a solution of methyl ester (1 mmol) in 1:1 (THF: H2O, 5 mL) was added lithium hydroxide monohydrate (1.5–2 mmol) at 0 °C and stirred for 6 h at room temperature. The solvent was evaporated under vacuum, the residue was taken into water (2–3 mL) and acidified to pH = 2 with 1N HCl and extracted with ethyl acetate (3 × 20 mL). The organic layer was washed with brine, dried over Na2SO4 and filtered. The filtrate was evaporated under vacuum to afford corresponding acid.
4. General procedure for hydrogenation
To a solution of Cbz-protected amine (1 mmol) in MeOH (10 mL) was added 10% Pd/C (35 mg). The suspension was stirred under 1 atm of H2 for 5–6 h and then vacuum filtered through a bed of Celite, which was washed with Methanol. Evaporation under vacuum to afford the free amine which was directly used for next step without purification.
5. Spectral data for Methyl-N6-((S)-3-cyclohexyl-2-(thiophene-2-carboxamido)propanoyl)-N2-(3-hydroxybenzoyl)-L-lysinate (1)
Compound 1 was synthesized starting from thiophene-2-carboxylic acid according to Scheme-1. Yield: 15 mg; (79%) Purity: >98%. MALDI-TOF Mass (m/z): calcd. for C28H37N3O6S 543.24 found 545.1 for [M+2H]+, 567.1 for [M+Na]+, 583.1 for [M+K]+. 1H NMR (400 MHz, DMSO-d6) δ 8.56 (d, J = 7.3 Hz, 1H), 8.41 (dd, J = 3.8, 8.3 Hz, 1H), 7.98 (t, J = 5.7 Hz, 1H), 7.89 (dd, J = 1.1, 3.7 Hz, 1H), 7.73 (dd, J = 1.1, 5.0 Hz, 1H), 7.33–7.19 (m, 3H), 7.12 (dd, J = 3.7, 5.0 Hz, 1H), 6.90 (ddd, J = 1.1, 2.5, 7.9 Hz, 1H), 4.46–4.28 (m, 2H), 3.61 (s, 3H), 3.13–2.98 (m, 2H), 1.78–1.73 (m, 2H), 1.68 – 1.46 (m, 7H), 1.44 –1.21 (m, 5H), 1.08 (m, 3H), 0.90–0.78 (m, 2H).
Supplementary Material
Acknowledgements
This research was supported by the Intramural Research Programs of the Center for Cancer Research (CCR), National Cancer Institute (NCI) and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institutes of Health (NIH).
References
- [1].Fiscella M, Zhang H, Fan S, Sakaguchi K, Shen S, Mercer WE, Vande Woude GF, O’Connor PM, Appella E, Proc Natl Acad Sci U S A 1997, 94, 6048–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lu X, Nguyen TA, Moon SH, Darlington Y, Sommer M, Donehower LA, Cancer Metastasis Rev 2008, 27, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Takekawa M, Adachi M, Nakahata A, Nakayama I, Itoh F, Tsukuda H, Taya Y, Imai K, Embo J 2000, 19, 6517–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lu X, Nannenga B, Donehower LA, Genes Dev 2005, 19, 1162–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5] a).Fujimoto H, Onishi N, Kato N, Takekawa M, Xu XZ, Kosugi A, Kondo T, Imamura M, Oishi I, Yoda A, Minami Y, Cell Death Differ 2006, 13, 1170–1180; [DOI] [PubMed] [Google Scholar]; b) Oliva-Trastoy M, Berthonaud V, Chevalier A, Ducrot C, Marsolier-Kergoat MC, Mann C, Leteurtre F, Oncogene 2007, 26, 1449–1458; [DOI] [PubMed] [Google Scholar]; c) Yoda A, Xu XZ, Onishi N, Toyoshima K, Fujimoto H, Kato N, Oishi I, Kondo T, Minami Y, J Biol Chem 2006. [DOI] [PubMed] [Google Scholar]
- [6].Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, Timofeev ON, Dudgeon C, Fornace AJ, Anderson CW, Minami Y, Appella E, Bulavin DV, Mol Cell 2006, 23, 757–764. [DOI] [PubMed] [Google Scholar]
- [7] a).Li J, Yang Y, Peng Y, Austin RJ, van Eyndhoven WG, Nguyen KC, Gabriele T, McCurrach ME, Marks JR, Hoey T, Lowe SW, Powers S, Nat Genet 2002, 31, 133–134; [DOI] [PubMed] [Google Scholar]; b) Natrajan R, Lambros MB, Rodriguez-Pinilla SM, Moreno-Bueno G, Tan DS, Marchio C, Vatcheva R, Rayter S, Mahler-Araujo B, Fulford LG, Hungermann D, Mackay A, Grigoriadis A, Fenwick K, Tamber N, Hardisson D, Tutt A, Palacios J, Lord CJ, Buerger H, Ashworth A, Reis-Filho JS, Clin Cancer Res 2009, 15, 2711–2722; [DOI] [PubMed] [Google Scholar]; c) Ruark E, Snape K, Humburg P, Loveday C, Bajrami I, Brough R, Rodrigues DN, Renwick A, Seal S, Ramsay E, Duarte Sdel V, Rivas MA, Warren-Perry M, Zachariou A, Campion-Flora A, Hanks S, Murray A, Ansari Pour N, Douglas J, Gregory L, Rimmer A, Walker NM, Yang TP, Adlard JW, Barwell J, Berg J, Brady AF, Brewer C, Brice G, Chapman C, Cook J, Davidson R, Donaldson A, Douglas F, Eccles D, Evans DG, Greenhalgh L, Henderson A, Izatt L, Kumar A, Lalloo F, Miedzybrodzka Z, Morrison PJ, Paterson J, Porteous M, Rogers MT, Shanley S, Walker L, Gore M, Houlston R, Brown MA, Caufield MJ, Deloukas P, McCarthy MI, Todd JA, Breast C Ovarian Cancer Susceptibility, C. Wellcome Trust Case Control, Turnbull C, Reis-Filho JS, Ashworth A, Antoniou AC, Lord CJ, Donnelly P, Rahman N, Nature 2013, 493, 406–410.23242139 [Google Scholar]
- [8] a).Hirasawa A, Saito-Ohara F, Inoue J, Aoki D, Susumu N, Yokoyama T, Nozawa S, Inazawa J, Imoto I, Clin Cancer Res 2003, 9, 1995–2004; [PubMed] [Google Scholar]; b) Tan DS, Lambros MB, Rayter S, Natrajan R, Vatcheva R, Gao Q, Marchio C, Geyer FC, Savage K, Parry S, Fenwick K, Tamber N, Mackay A, Dexter T, Jameson C, McCluggage WG, Williams A, Graham A, Faratian D, El-Bahrawy M, Paige AJ, Gabra H, Gore ME, Zvelebil M, Lord CJ, Kaye SB, Ashworth A, Reis-Filho JS, Clin Cancer Res 2009, 15, 2269–2280. [DOI] [PubMed] [Google Scholar]
- [9].Fuku T, Semba S, Yutori H, Yokozaki H, Pathol Int 2007, 57, 566–571. [DOI] [PubMed] [Google Scholar]
- [10] a).Loukopoulos P, Shibata T, Katoh H, Kokubu A, Sakamoto M, Yamazaki K, Kosuge T, Kanai Y, Hosoda F, Imoto I, Ohki M, Inazawa J, Hirohashi S, Cancer Sci 2007, 98, 392–400; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wu B, Guo BM, Kang J, Deng XZ, Fan YB, Zhang XP, Ai KX, Apoptosis 2016, 21, 365–378. [DOI] [PubMed] [Google Scholar]
- [11].Castellino RC, De Bortoli M, Lu X, Moon SH, Nguyen TA, Shepard MA, Rao PH, Donehower LA, Kim JY, J Neurooncol 2008, 86, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12] a).Richter M, Dayaram T, Gilmartin AG, Ganji G, Pemmasani SK, Van Der Key H, Shohet JM, Donehower LA, Kumar R, PLoS One 2015, 10, e0115635; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Saito-Ohara F, Imoto I, Inoue J, Hosoi H, Nakagawara A, Sugimoto T, Inazawa J, Cancer Res 2003, 63, 1876–1883. [PubMed] [Google Scholar]
- [13].Demidov ON, Kek C, Shreeram S, Timofeev O, Fornace AJ, Appella E, Bulavin DV, Oncogene 2007, 26, 2502–2506. [DOI] [PubMed] [Google Scholar]
- [14] a).Goloudina AR, Kochetkova EY, Pospelova TV, Demidov ON, Oncotarget 2016, 7, 31563–31571; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pechackova S, Burdova K, Macurek L, J Mol Med (Berl) 2017, 95, 589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15] a).Belova GI, Demidov ON, Fornace AJ Jr., Bulavin DV, Cancer Biol Ther 2005, 4, 1154–1158; [DOI] [PubMed] [Google Scholar]; b) Rayter S, Elliott R, Travers J, Rowlands MG, Richardson TB, Boxall K, Jones K, Linardopoulos S, Workman P, Aherne W, Lord CJ, Ashworth A, Oncogene 2008, 27, 1036–1044. [DOI] [PubMed] [Google Scholar]
- [16] a).Hayashi R, Tanoue K, Durell SR, Chatterjee DK, Jenkins LM, Appella DH, Appella E, Biochemistry 2011, 50, 4537–4549; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yamaguchi H, Durell SR, Feng H, Bai Y, Anderson CW, Appella E, Biochemistry 2006, 45, 13193–13202. [DOI] [PubMed] [Google Scholar]
- [17].Gilmartin AG, Faitg TH, Richter M, Groy A, Seefeld MA, Darcy MG, Peng X, Federowicz K, Yang J, Zhang SY, Minthorn E, Jaworski JP, Schaber M, Martens S, McNulty DE, Sinnamon RH, Zhang H, Kirkpatrick RB, Nevins N, Cui G, Pietrak B, Diaz E, Jones A, Brandt M, Schwartz B, Heerding DA, Kumar R, Nat Chem Biol 2014, 10, 181–187. [DOI] [PubMed] [Google Scholar]
- [18].Esfandiari A, Hawthorne TA, Nakjang S, Lunec J, Mol Cancer Ther 2016, 15, 379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yamaguchi H, Durell SR, Chatterjee DK, Anderson CW, Appella E, Biochemistry 2007, 46, 12594–12603. [DOI] [PubMed] [Google Scholar]
- [20] a).Cha H, Lowe JM, Li H, Lee JS, Belova GI, Bulavin DV, Fornace AJ Jr., Cancer Res 2010, 70, 4112–4122; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Macurek L, Lindqvist A, Voets O, Kool J, Vos HR, Medema RH, Oncogene 2010, 29, 2281–2291; [DOI] [PubMed] [Google Scholar]; c) Moon SH, Nguyen TA, Darlington Y, Lu X, Donehower LA, Cell Cycle 2010, 9, 2092–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21] a).Clausse V, Goloudina AR, Uyanik B, Kochetkova EY, Richaud S, Fedorova OA, Hammann A, Bardou M, Barlev NA, Garrido C, Demidov ON, Cell Death Dis 2016, 7, e2195; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tanoue K, Miller Jenkins LM, Durell SR, Debnath S, Sakai H, Tagad HD, Ishida K, Appella E, Mazur SJ, Biochemistry 2013, 52, 5830–5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ji J, Zhang Y, Redon CE, Reinhold WC, Chen AP, Fogli LK, Holbeck SL, Parchment RE, Hollingshead M, Tomaszewski JE, Dudon Q, Pommier Y, Doroshow JH, Bonner WM, PLoS One 2017, 12, e0171582. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.