

Abstract
Motivation
Population-scale sequenced cohorts are foundational resources for genetic analyses, but processing raw reads into analysis-ready cohort-level variants remains challenging.
Results
We introduce an open-source cohort-calling method that uses the highly accurate caller DeepVariant and scalable merging tool GLnexus. Using callset quality metrics based on variant recall and precision in benchmark samples and Mendelian consistency in father-mother-child trios, we optimize the method across a range of cohort sizes, sequencing methods and sequencing depths. The resulting callsets show consistent quality improvements over those generated using existing best practices with reduced cost. We further evaluate our pipeline in the deeply sequenced 1000 Genomes Project (1KGP) samples and show superior callset quality metrics and imputation reference panel performance compared to an independently generated GATK Best Practices pipeline.
Availability and implementation
We publicly release the 1KGP individual-level variant calls and cohort callset (https://console.cloud.google.com/storage/browser/brain-genomics-public/research/cohort/1KGP) to foster additional development and evaluation of cohort merging methods as well as broad studies of genetic variation. Both DeepVariant (https://github.com/google/deepvariant) and GLnexus (https://github.com/dnanexus-rnd/GLnexus) are open-source, and the optimized GLnexus setup discovered in this study is also integrated into GLnexus public releases v1.2.2 and later.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Sequencing a single individual can identify variants informative for diseases (Yang et al., 2013), traits (Welter et al., 2014) and ancestry (The 1000 Genomes Project Consortium, 2010). Jointly using sequence data of multiple individuals can discover rare genetic diseases (Ramoni et al., 2017). Population-scale sequencing generates annotation resources for clinical sequencing, such as dbSNP (Sherry, 2001), ExAC (Lek et al., 2016), DiscovEHR (Dewey et al., 2016), TOPMed (Taliun et al., 2019) and gnomAD (Karczewski et al., 2020), and enables well-powered association studies (Ozaki et al., 2002) in large datasets of sequenced and phenotyped individuals (Bycroft et al., 2018).
Single-sample variant calling methods (Garrison and Marth, 2012; Kim et al., 2018; Luo et al., 2019; McKenna et al., 2010; Poplin et al., 2018b) use sequence reads mapped to a reference genome to identify and genotype positions which differ from the reference. Many variant callers support the generation of Genome Variant Call Format (gVCF) outputs, which supplement the variant calls with block records of non-variant regions annotated with confidence estimates that the regions match the reference genome. Joint genotyping tools such as GATK GenotypeGVCFs (Poplin et al., 2018a) and GLnexus (Lin et al., 2018) transform a cohort of gVCFs into a project-level VCF that contains a complete matrix of every variant in a cohort with a call for each sample. Compared to a full joint-calling strategy, joint genotyping both substantially reduces the size of required input data and avoids the need to fully reprocess all samples when adding samples to an existing cohort.
Joint genotyping of large cohorts introduces unique challenges. Harmonizing the representation of overlapping alleles is algorithmically intricate, and the number of overlapping alleles increases with cohort size. In addition, even with high single-sample variant calling accuracy, many samples will aggregate a large number of total errors. At the same time, large cohorts present unique opportunities to increase accuracy. Cross-referencing genotype likelihoods across a cohort can help refine calls and filter errors, for example by identifying recurrent artifacts that violate Hardy-Weinberg equilibrium (HWE) (Hardy, 1908).
Here, we introduce a framework to generate highly accurate and scalable cohort callsets with DeepVariant, using its superior calibration of variant confidences and high single-sample accuracy (Poplin et al., 2018b). We adapt the scalable joint genotyper GLnexus (Lin et al., 2018) to DeepVariant gVCFs and tune filtering and genotyping parameters to optimize performance for whole-genome sequences (WGS) and whole-exome sequences (WES) across a range of sequence coverages and cohort sizes. We compare the resulting callsets to analogous callsets generated by the broadly used GATK Best Practices (DePristo et al., 2011) which serve as current state-of-the-art benchmarks. Finally, we apply the optimized method to the recent deep sequencing of the 1000 Genomes Project (1KGP) phase 3 samples (The 1000 Genomes Project Consortium, 2015). We evaluate the resulting callset across multiple quality metrics and performance as an imputation reference panel against a callset independently generated using a GATK Best Practices pipeline.
2 Materials and methods
2.1 Data acquisition and preparation
Throughout this study we use the human GRCh38 reference genome with no ALT contigs. We use 7 WGS samples (HG001-HG007), 2 WES samples (HG001, HG002) from Genome in a Bottle (GIAB) project (Zook et al., 2014, 2019), 929 WGS and 346 WES samples from Clinical Sequencing Evidence-Generating Research (CSER) (Amendola et al., 2018), and finally 313 WGS samples from Population Architecture Using Genomics and Epidemiology (PAGE) (Matise et al., 2011). All WGS samples are deeply sequenced at 40–50× coverage. We identify 249 disjoint trios in CSER WGS samples, among which we randomly selected five WGS trios and five WES trios (Supplementary Table S3) among non-outliers to use for Mendelian violation rate estimation during callset evaluation. To identify the outlier samples, we examined six variant summary statistics for each sample: the number of total records, SNPs and indels, the Ti:Tv ratio, the mean SNP quality and the mean indel quality. Non-outliers are defined as the samples whose z-scores for all six statistics are at most one.
We created custom WGS cohorts of size 3, 100, 333 and 1247 and a WES cohort of size 346 using the above samples (Supplementary Tables S2 and S8) for which both GIAB concordance and Mendelian violation rate could be evaluated. We used the GIAB benchmark variant v3.3.2 to evaluate concordance. Finally, we created 15× autosomal coverage BAMs from all BAM files from GIAB, CSER and PAGE datasets by downsampling the full BAMs with samtools (Li et al., 2009) (‘samtools view -s’).
For our independent evaluation and demonstration with 1KGP, we used the recent deep sequencing (~30× coverage) of the 1KGP phase 3 cohort by New York Genome Center. We used third party tools such as samtools (Li et al., 2009), BWA-MEM (Li, 2013), samblaster (Faust and Hall, 2014), etc. for data processing steps outlined above. Full details can be found in Supplementary Note S2.
2.2 DeepVariant and GLnexus
We used DeepVariant v0.8.0 and the publicly released WGS model v0.8.0 to generate the single-sample variant calls for all samples in GIAB, CSER and PAGE. A single-line command to run DeepVariant in a pre-built docker container is available on the DeepVariant public repository (https://github.com/google/deepvariant). The DeepVariant calls for the sample from 1000 Genomes Project were generated using a custom model trained exclusively for the NovaSeq platform. Both the custom model and all single-sample DeepVariant calls generated by it are publicly available, as described in ‘Availability and Implementation’ in the Abstract.
To merge and evaluate the multiple cohorts in parallel, we deployed the open-source GLnexus algorithm using Apache Beam (https://beam.apache.org) on Google internal compute clusters. The Beam-based pipeline abstracts away the need to specify multi-threading on a single machine (as is done in the open-source GLnexus), and deploys hundreds of different parameter configurations on thousands of CPUs. The pipeline produces identical scientific results to the open-source GLnexus v1.2.2 when run with the same parameters. To both ensure our train/test dataset split is non-overlapping and limit computational costs of this study, we used separate individual chromosomes for pipeline optimization and evaluation. For consistency with previous studies (Lin et al., 2018; Poplin et al., 2018b), we used chromosome 2 to optimize the pipeline, and computed final performance benchmarks separately on chromosome 20. The optimized DeepVariant parameters from this study, which are discussed in detail in Results, are included in open-source GLnexus v1.2.2 or later versions in two presets: –config DeepVariantWGS for WGS and –config DeepVariantWES for WES. After installing the GLnexus command line tool, users can merge DeepVariant calls in these optimized setups using a single command like
$ glnexus_cli –config DeepVariantWGS \
deepvariant.*.g.vcf.gz > cohort.bcf
In addition to parameter optimization, we modified the internals of both DeepVariant and GLnexus for better communication between the tools and to improve the joint-calling process. All modifications were incorporated into open-sourced DeepVariant (v0.8.0 or later) and GLnexus (v1.2.2 or later).
2.3 GATK
We followed GATK Best Practices v4.1.2.0 to establish the baseline performance of each callset generated from GIAB, CSER and PAGE data. Starting from the BAM files, prepared as described above, we ran HaplotypeCaller in GVCF mode to call single-sample variants, followed by GenomicsDBImport and GenotypeGVCFs to consolidate and jointly genotype the cohort, and finally VariantRecalibrator and ApplyVQSR for variant quality score recalibration (VQSR) (see Supplementary Note S3 for full details). We performed the steps on each chromosome separately in parallel and combined calls at the end to speed up the process. Cost benchmarking was performed on chromosome 20. For the 1KGP samples, we downloaded the GATK cohort callset independently generated by the New York Genome Center using samtools v1.3.1, Picard v2.4.1 and GATK v3.5. The complete description of the pipeline used to generate this callset is available on the European Bioinformatics Institute’s FTP (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20190425_NYGC_GATK/1000G_README_2019April10_NYGCjointcalls.pdf).
2.4 Single-sample variant call statistics
We used bcftools v1.9 (samtools.github.io/bcftools) and hap.py v0.3.9 (Krusche et al., 2019) (github.com/illumina/hap.py) to generate basic call statistics from single-sample VCFs in the GIAB, PAGE and CSER datasets.
2.5 GLnexus parameter optimization
We used Google Vizier (Golovin et al., 2017), a Google-internal service for performing black-box optimization, for optimizing the configurable parameters of GLnexus (Supplementary Table S4). We constructed an optimization objective function incorporating both GIAB benchmark call concordance and the rate of Mendelian violations, and optimized the parameters for this objective in two steps, first using Pareto-optimal search algorithm to reduce the search space and then performing an exhaustive grid search within the reduced parameter space. Complete details of this work can be found in Results and in Supplementary Note S4.
2.6 1KGP imputation reference panel creation
We generated the 1KGP reference panels from DV-GLN-OPT and GATK-VQSR callsets (see Results) by applying identical minimal transformations to them and phasing them with Eagle2 (Loh et al., 2016). We followed a standard pipeline for generating a reference panel recommended in Eagle’s website. More details and a script used for these steps are included in Supplementary Notes S2 and S5.
2.7 Imputing pseudo-microarray variant calls
To evaluate 1KGP imputation reference panels, we first generated pseudo-microarray variant calls from GIAB benchmark variants of HG002 and HG005 in the benchmark regions by extracting the variant sites used by a popular commercial microarray kit (Illumina Infinium OmniExpress-24). We obtained the microarray sites from the CSV manifest file (GRCh38 version) on Illumina’s official website (ftp://webdata2:webdata2@ussd-ftp.illumina.com/downloads/productfiles/humanomniexpress-24/v1-2/infinium-omniexpress-24-v1-2-manifest-file-csv.zip) and converted it to VCF format using Illumina’s GTCtoVCF tool (github.com/Illumina/GTCtoVCF).
Starting from GIAB v3.3.2 benchmark variants for HG002 and HG005, we removed all existing phasing information from the VCF, extracted the high-confidence variants in the microarray sites using bcftools v1.9, and added homozygous-reference genotypes to all microarray sites not present in GIAB VCFs. We phased the resulting pseudo-microarray variants with Eagle v2.4.1 using the reference panel to evaluate (DV-GLN-OPT or GATK-VQSR, see Results) and the hg38 genetic map file released with Eagle.
Finally, we imputed the phased pseudo-microarray variants with Beagle v5.0 (Browning et al., 2018) using the same reference panel used in the phasing step. A complete script for running Beagle can be found in Supplementary Note S6.
3 Results
3.1 Cohort variant call evaluation strategies
In contrast to single-sample variant calling, for which the Genome in a Bottle (GIAB) (Zook et al., 2014, 2019) dataset enables broadly accepted accuracy metrics to benchmark and compare tools and methods (https://precision.fda.gov/challenges/truth and https://precision.fda.gov/challenges/10), for cohort variant calls there is no existing standard for comparison. Here we use four different measures of variant calling accuracy to optimize and evaluate cohort variant calls, for single nucleotide variants, small indels (excluding structural variants), and homozygous reference regions. For both optimization and evaluation, we examined two accuracy metrics: (i) we computed concordance of the Genome in a Bottle (GIAB) HG001-HG007 benchmark samples to directly measure variant accuracy within the well-characterized 83% of the genome in the GRCh38 GIAB v3.3.2 benchmark regions, and (ii) we computed Mendelian violation rates in trios to indirectly measure variant accuracy genome-wide. For evaluation only, we included two additional indirect measures of callset quality: (iii) we computed the ratio of transitions to transversions (Ti:Tv ratio) of single nucleotide polymorphisms (SNPs) for all samples to measure deviations from the expected genome-wide ratio of approximately 2.0–2.1 (Bainbridge et al., 2011), as random genotyping errors would reduce this ratio, and (iv) we computed deviations from HWE at the cohort level, which can be indicative of recurrent artifacts in a variant calling algorithm (Graffelman et al., 2017).
3.2 Cohorts used in development and evaluation
Four distinct data sources were used to optimize and evaluate cohort variant calls. The first data source is the aforementioned GIAB consortium which provides a well-characterized set of truth variants. To maximize the diversity of samples and sites used for evaluation, other trios in the cohort were used to compute Mendelian violation rates (with the sole exception being the three person HG002-HG003-HG004 cohort) and the children of GIAB trios were excluded from the GIAB metrics calculations. The second data source is the Clinical Sequencing Evidence-Generating Research (CSER) consortium (Amendola et al., 2018), which contains 929 WGS and 344 WES samples, including 249 WGS trios and 112 WES trios. The third data source is the Population Architecture using Genomics and Epidemiology (PAGE) consortium (Matise et al., 2011), which contains 313 WGS. The final data source is the recent 30× WGS of 2504 individuals from 1KGP. We fully withheld the 1KGP cohort from development, only using it as a final independent evaluation set. Within this cohort, a single cryptic trio (Roslin et al., 2016) was used to compute Mendelian violation rates.
We performed analyses both at full sequence coverage as well as in the same cohorts downsampled to 15× coverage. We targeted robust performance across the diversity of sequencing projects by representing cohorts of high and low coverage, WES and WGS, those sequenced by various groups, on various instruments, and across a wide array of ancestries.
3.3 Quality properties of single-sample variant calls
We first investigated variant call properties of 1248 individuals from the GIAB (n = 6), CSER (n = 929) and PAGE (n = 313) cohorts. The total number of SNPs reported by DeepVariant is lower than GATK4 HaplotypeCaller for nearly all individuals, and the number of indels is also lower for most individuals. However, the Ti: Tv ratio measured in all individuals, and both precision and recall computed separately for SNPs and indels in the six GIAB individuals, are all higher in DeepVariant than GATK4 HaplotypeCaller (Supplementary Fig. S1).
To illustrate why different single-sample variant callers require separate calibration during joint genotyping, we compared Genotype Quality (GQ) scores estimated by each caller, defined as the Phred-scaled conditional probability that a genotype is incorrect, to GQ scores derived empirically from GIAB ground truth (Fig. 1). In detail, we first binned the variants by their GQ values estimated by each caller, computed the empirical error rate for variants in each bin against the GIAB truth, and then converted the empirical error rate to Phred-scale to obtain the empirical GQ for each bin. DeepVariant shows markedly better GQ calibration than GATK4 HaplotypeCaller at all reported GQ scores (Fig. 1A, B). DeepVariant is well-calibrated both across sequence coverages and when stratified by variant type, with a slight bias toward overconfidence in homozygous alternate SNPs (Supplementary Fig. S2). To rigorously quantify GQ calibration, we computed the Brier score (Brier, 1950) and Spiegelhalter’s Z statistic (Spiegelhalter, 1986) by converting the Phred-scaled GQ into the caller’s estimated probability of correctly calling the genotype, and comparing it to the GIAB truth (Supplementary Table S1). Substantially lower Brier scores (0.000918 for DeepVariant and 0.005371 for GATK, averaged over all samples) and Spiegelhalter’s Z values (0.52 for DeepVariant and 1141.03 for GATK, averaged over all samples) confirm DeepVariant’s superior calibration.
Fig. 1.
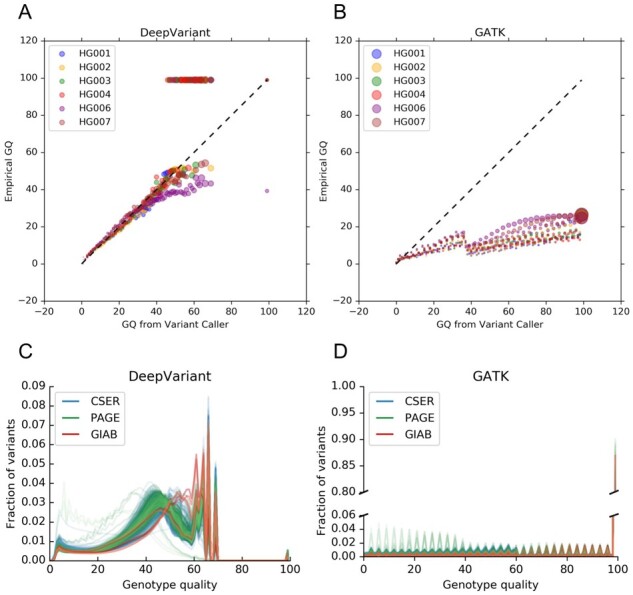
Genotype quality (GQ) distribution properties of PASS variants. (A) Genotype quality calibration for DeepVariant v0.8.0. Reported GQ is plotted against the empirical GQ calculated using genome-wide GIAB benchmark variant calls at 40× coverage. Each data point is a set of variant calls with the same GQ (x-axis), and the y-axis value is the empirical error rate calculated from the GIAB truth set. Both axes are in Phred-scale. Marker areas are proportional to the square root of the number of variants. The dotted line represents perfect calibration. (B) Genotype quality calibration for GATK4 HaplotypeCaller, analogous to (A). (C) Distributions of reported GQ for DeepVariant v0.8.0 in all 1248 samples computed genome-wide. (D) Distributions of reported GQ for GATK4 HaplotypeCaller in all 1248 samples computed on chromosome 2 only. Note the broken y-axis and different scales. See also Supplementary Figure S4
The overall distribution of GQ scores within a sample determines the information content of the field. Substantial variant fractions occur across the GQ spectrum for DeepVariant calls (Fig. 1C), and the DeepVariant GQ score distribution shifts smoothly toward higher qualities as sequence coverage increases (Supplementary Fig. S3). In contrast, GATK4 HaplotypeCaller both produces a consistently oscillating GQ distribution for variants with GQ < 99 (Fig. 1D) and frequently reports most variants as GQ = 99 (Supplementary Fig. S4A). When considered in conjunction with the GQ calibration comparisons (Supplementary Table S1), these results suggest that joint genotyping algorithms may be able to more accurately refine individual genotypes produced by DeepVariant since joint genotyping algorithms refine individuals’ variant calls based on observed allele frequencies in the rest of the cohort, using GQ as a prior on genotype.
The large-scale analysis of genomes and exomes in gnomAD identified variant quality normalized by read depth (QD) as the most important feature for discriminating true variants from artifacts in GATK calls (Karczewski et al., 2020). Consistent with that observation, GATK single-sample QD is less uniformly biased than GQ (Supplementary Fig. S4B) and has a more informative distribution across the cohorts (Supplementary Fig. S4C).
3.4 Optimized parameters for joint calling
We adapted GLnexus (Lin et al., 2018) for merging DeepVariant gVCFs because of its computational scalability to large cohorts, access to relevant parameters, performance on allele normalization and open-source license. To identify optimal parameters for merging DeepVariant gVCFs, we created four custom WGS cohorts of 3, 100, 333 and 1247 samples at both high coverage (40–50×) and low coverage (15×) on chromosome 2, resulting in eight total cohorts (Supplementary Table S2). The cohorts contain five mutually non-descendant GIAB samples used to evaluate benchmark calls and five non-GIAB trios used to compute Mendelian violation rates (Supplementary Tables S2B, C and S3).
We focused on the four tunable GLnexus parameters (Supplementary Table S4) most crucial to optimize: ‘min_’, the quality threshold applied to each discovered allele; ‘min_’, the quality threshold applied to alleles whose copy number is at least two; ‘min_GQ’, the minimum genotype quality to be used for copy number estimates for the alleles; and ‘revise_genotypes’, a boolean switch indicating whether to use cohort information to re-genotype low quality genotype calls.
We extensively explored parameter configurations using Google Vizier (Golovin et al., 2017) to optimize a multiple metric objective function. Minimization of Mendelian violation rate in trio samples encourages precision genome-wide. Maximization of concordance with GIAB samples, measured as the harmonic mean of SNP and indel F1 scores, encourages both precision and recall in the well-characterized subset of the genome. Together, the joint metric discourages strategies which improve Mendelian violation rate at the expense of genotype errors (for example, by filtering true variant sites). We first performed a Pareto-optimal search using Vizier’s default Bayesian hyperparameter selection algorithm to reduce the problem space, and then explored the reduced space using an exhaustive grid search. Many configurations simultaneously improve both Mendelian violation rate and concordance with GIAB when compared to the GLnexus configuration that performs no variant modification (Fig. 2, Supplementary Fig. S5).
Fig. 2.
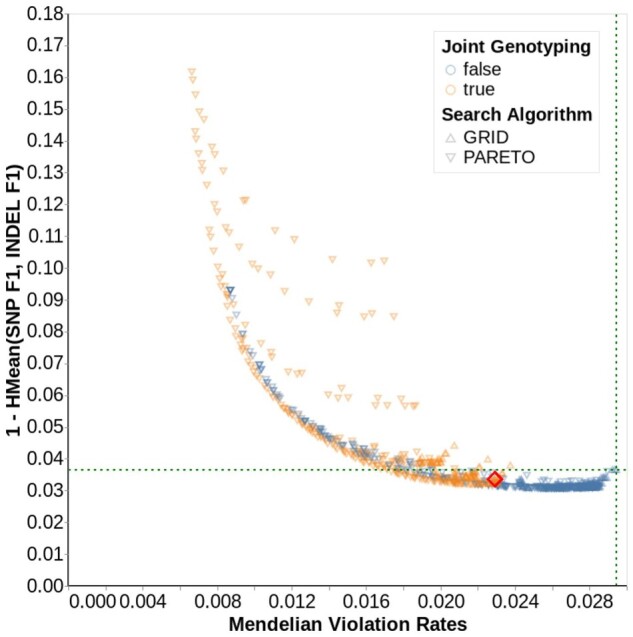
Parameter search for n = 1247, 15× cohort. Each data point represents a unique parameter combination explored by Vizier. The color indicates whether the GLnexus parameter to revise genotypes was true (orange) or false (blue), and the shape represents the search algorithm. The x-axis indicates Mendelian violation rate. The y-axis indicates errors on GIAB through the harmonic mean of SNP F1 and indel F1 (lower is more accurate). Points toward the lower left are more accurate on both metrics. The intersection of the green horizontal and vertical dotted lines indicates the performance using GLnexus with no variant modification (Supplementary Table S4). Supplementary Figure S5 shows the results for all cohort sizes and coverages. The red diamond indicates the parameter set we selected for the optimized DeepVariant+GLnexus pipeline
The smooth Pareto-optimal boundary (Fig. 2, Supplementary Fig. S5) indicates that the tradeoff between recall and precision can be tuned in an application-specific manner. We investigated the extent to which parameter settings are applicable across cohort sizes and sequence coverage by summing the rate of error reduction across five metrics over the ‘no modification’ parameter setting (see Supplementary Note S4 for details). We selected the best-performing parameter configuration as the ‘optimized’ setting after verifying its strong performance across cohort sizes and sequence coverages (Supplementary Figs S6–S8).
Next, we compared four variant calling and merging methods across all 8 cohorts. The first and second methods use the GATK4 Best Practices (DePristo et al., 2011; McKenna et al., 2010; Poplin et al., 2018a) pipeline and either retain all variants (‘GATK-Joint’) or retain only variants that pass variant quality score recalibration (‘GATK-VQSR’). The third method uses DeepVariant for single-sample calling and GLnexus to merge the calls, with DeepVariant run with default parameters and GLnexus run in a setting to avoid single-sample variant modification (‘DV-GLN-NOMOD’) (Supplementary Table S4). The final method uses the optimized version of the DeepVariant+GLnexus pipeline (‘DV-GLN-OPT’) (Supplementary Note S1). After verifying qualitatively similar callset properties on distinct chromosomes (Supplementary Fig. S9), we generated all evaluation callsets on chromosome 20 to avoid overlap with the training data from chromosome 2.
The callsets were evaluated on five measures of quality: SNP and indel false discovery rates, false negative rates and total Mendelian violation rate, for each cohort size and at both 15× and 40× sequence coverage. DV-GLN-OPT equals or exceeds both GATK-based methods in 38 of the 40 measured metrics (Fig. 3, Supplementary Table S5), with only SNP false discovery rates in 15× coverage callsets not uniformly stronger. In the cohort of 1247 individuals at 40× coverage, DV-GLN-OPT has a 3.0-fold lower Mendelian violation rate (1.7% versus 5.0%), 17.6-fold lower SNP F1 error (0.07% versus 1.23%) and 2.6-fold lower indel F1 error (1.14% versus 2.92%) than GATK-VQSR. DV-GLN-OPT generally, though not strictly, also outperforms DV-GLN-NOMOD.
Fig. 3.
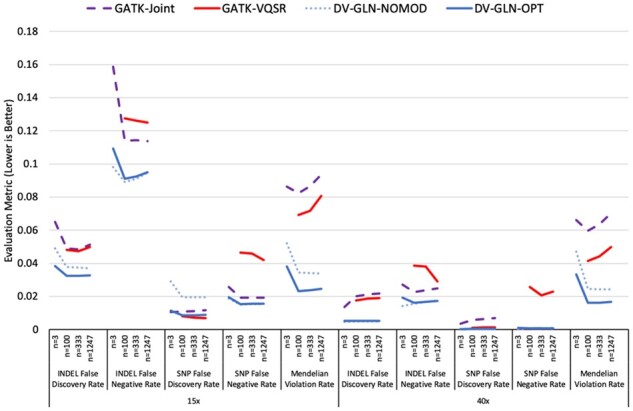
Comparison of four cohort callset creation methods. Four calling and merging pipelines are applied at both 15× and 40× sequence coverage for WGS cohorts of size n = 3, 100, 333 and 1247. Five evaluation metrics are presented: Mendelian Violation Rate, SNP False Discovery Rate (1-Precision), SNP False Negative Rate (1-Recall), indel False Discovery Rate and indel False Negative Rate. In all cases, lower values are better. All evaluation metrics are computed on chr20. See Supplementary Table S5 for the precise values and the variances of each metric
We repeated the parameter search technique described above in a single WES cohort of 346 samples (Supplementary Table S2B). Similarly to WGS, there exist many configurations that strictly outperform the ‘no modification’ parameter setting (Supplementary Fig. S10) and the WES-optimized DV+GLnexus pipeline outperforms the GATK4 Best Practices in all metrics (Supplementary Table S5C).
3.5 Evaluation on deeply sequenced 1000 Genomes Project phase 3
To evaluate DV-GLN-OPT in an independent dataset, we generated a cohort callset for high-coverage sequencing reads of the 2504 samples in 1KGP (The 1000 Genomes Project Consortium, 2015), and made the cohort callset and all DeepVariant single-sample calls publicly available (see ‘Availability and Implementation’ in Abstract). We compared the single-sample variant calls from DeepVariant with those of GATK HaplotypeCaller, and the DV-GLN-OPT callset with that of an independently generated GATK-VQSR pipeline (see Section 2.3). Each sample has a higher Ti:Tv ratio in the DeepVariant-based variant calls in both cases (Fig. 4A, in light green and blue, respectively). Taken together, these results provide indirect evidence that the DeepVariant calls are of higher quality.
Fig. 4.
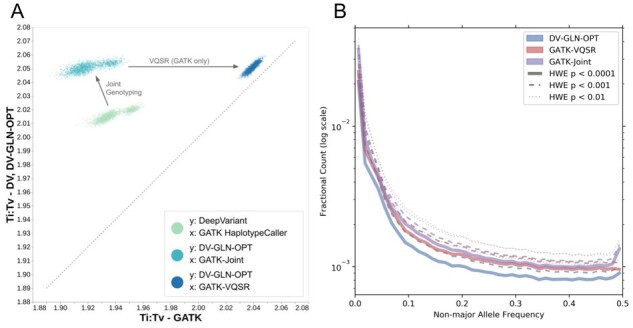
1KGP cohort callset quality. (A) Ti:Tv ratios of 1KGP samples, from single-sample SNPs and joint-called SNPs, generated by DV-GLN-OPT and GATK pipeline. Each point represents the ratio in one of the 2504 samples across the whole genome. Each point cloud compares the Ti:Tv ratios in variant calls from the two systems, after equivalent steps are performed. The first cloud (in light green) compares the Ti:Tv ratios from DeepVariant (y-axis) and GATK HaplotypeCaller (x-axis) single sample calls. The second cloud (in turquoise) compares Ti:Tv after joint-genotyping is performed (optimized GLnexus for DeepVariant, and GenomicsDBImport+GenotypeGVCFs for GATK HaplotypeCaller). Finally the third cloud (in blue) compares the final outputs from the two systems, after VQSR is performed for GATK (x-axis), while no additional operation is performed for the optimized DeepVariant-GLnexus calls. (B) Fractional counts of autosomal variants with low HWE p-values, binned by non-major allele frequency in DV-GLN-OPT, GATK-VQSR and GATK-Joint. The major allele is the allele with the largest allele count in a given variant within the callset. The variants are aggregated in non-major-allele-frequency bins of size 0.0125, and the frequency is clipped at 0.5 for visualization purposes (for all methods the fractional counts in bins after 0.5 are less than )
Overall callset composition depends on the filtering methods applied. The GATK-VQSR callset contains substantially fewer total variants and rare variants compared to both DV-GLN-OPT and GATK-Joint (Supplementary Fig. S11). To identify recurrent variant calling and genotyping artifacts, we quantified the sites which violate HWE in each callset at various p-value thresholds (Fig. 4B). Only 6.62% of autosomal sites in the DV-GLN-OPT callset have HWE p < (7 443 684 of 112 451 553 total autosomal sites), compared to 8.05% in GATK-VQSR (8 276 874 of 102 804 074) and 9.77% in GATK-Joint (11 724 367 of 120 046 355). Finally, we observed that the GATK-based callsets limit the maximum number of alleles at any position to six, and thus exclude a number of alleles present at highly variable sites (Supplementary Fig. S11). Manual inspection confirmed that most highly multiallelic variants are short tandem repeats of varying lengths, with the bulk of calls attributable to few common alleles but a long tail of additional alleles (Supplementary Fig. S12).
To further assess 1KGP callset quality, we evaluated Mendelian violation rate within a single cryptic trio present in the cohort (Roslin et al., 2016). We first verified the trio’s relatedness (NA20882: mother, NA20891: father, NA20900: child, all of Gujarati Indian ancestry), and used this trio to compute Mendelian violations in DV-GLN-OPT, GATK-VQSR and GATK-Joint (Fig. 5).
Fig. 5.
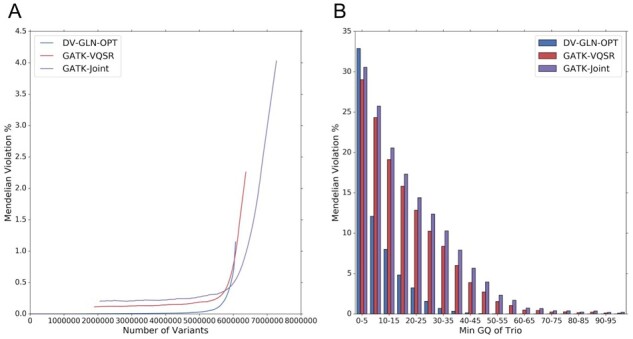
Mendelian violations in autosomes of a cryptic trio in 1KGP. (A) The percentage of variants that violate Mendelian inheritance in the trio NA20900-NA20891-NA20882 as a function of the number of variants considered. Variants are ranked by the minimum GQ within the trio. Callset variants with homozygous reference calls for all three trio samples, and those have indeterminate violation status due to missing genotype calls in the trio, are ignored. (B) Mendelian violation percentages of the same trio binned by minimum GQ in the trio using bin size 5
To quantify the improvement to Mendelian violation rate and GQ calibration, we sorted variant calls from most to least confident using the minimum GQ in the trio, independently for each callset. Importantly, variant-level metrics such as QUAL were not used for this analysis because the call qualities for three samples in a trio may differ substantially from a variant quality metric computed over all 2504 samples. DV-GLN-OPT calls have a lower overall Mendelian violation rate, as evidenced by cumulative Mendelian violation rate plotted as a function of variants retained (Fig. 5A). While all callsets show decreased Mendelian violation rates as the minimum GQ of the trio is increased (Fig. 5B), the broader GQ distribution of DeepVariant (Fig. 1) enables better separation of true and false calls. Remarkably, applying the maximally stringent GQ = 99 filter to the GATK-VQSR callset retains only 1.9 million sites (29.8%) at a Mendelian violation rate of 0.11%, whereas the DV-GLN-OPT callset can retain 5.5 million sites (90.6%) at a lower Mendelian violation rate of < 0.1%.
Finally, we annotated variants discovered in 1KGP using the Ensembl Variant Effect Predictor (VEP) (McLaren et al., 2016) to analyze their coding consequences. We analyzed variants in three groups: variants called by both DV-GLN-OPT and GATK-VQSR, by DV-GLN-OPT exclusively, and by GATK-VQSR exclusively (Supplementary Fig. S13). Among the annotations with the highest potential impact (‘stop gained’, ‘frameshift’, ‘stop lost’, ‘start lost’), we found more variants discovered exclusively by DV-GLN-OPT than by GATK-VQSR. We also analyzed the same groups of variants when restricted to variants detected exclusively in each 1KGP superpopulation (African, American, East Asian, European, South Asian). While the total number of variants exclusive to each superpopulation varied, the overall distribution of the annotations and the three groups of variants seems broadly consistent across populations.
3.6 Evaluation of 1KGP callsets as imputation reference panels
The 1KGP dataset is frequently used for population-based downstream applications, such as genotype phasing and imputation, due to its genetic diversity and large sample size (Delaneau et al., 2014; Huang et al., 2012; Nikpay et al., 2015; Shaikho et al., 2017). To illustrate how the accuracy of the DV-GLN-OPT callset translates into improved results for these downstream analyses, we assessed the performance of imputing variants using it as a reference panel. We first created reference panels from the deeply sequenced DV-GLN-OPT and GATK-VQSR 1KGP callsets described in the previous section, by applying identical minimal transformations to both cohort VCFs (see Materials and Methods) and phasing the callsets with Eagle2 (Loh et al., 2016).
The DV-GLN-OPT panel contains 4.69% more variant sites than the GATK-VQSR panel generated from the same source. More than 99% of the GATK-VQSR panel sites are present in the DV-GLN-OPT panel, while fewer than 95% of the DV-GLN-OPT panel sites are present in the GATK-VQSR panel (Fig. 6A).
Fig. 6.
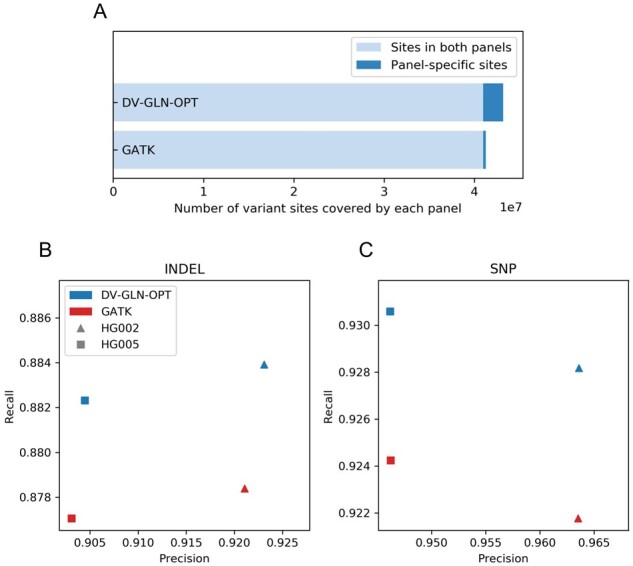
Imputation accuracy of 1KGP reference panel. (A) Variant sites covered by DV-GLN-OPT and GATK panel. The DV-GLN-OPT reference panel generated from 1KGP samples covers 43 181 562 variant sites, while the GATK panel from the same samples covers 41 247 330 sites. The intersection of the two panel regions (marked in light blue) covers 40 972 007 sites, which is 94.88% of the DV-GLN-OPT panel and 99.33% of the GATK panel. (B) Imputed genotype accuracy for indels. The accuracy of the imputed variants are measured by computing concordance with the GIAB benchmark calls using hap.py. Blue colored markers are from DV-GLN-OPT panel while the red markers are from GATK panel. The shaped markers show precision and recall computed across the GIAB evaluation region for two samples. (C) Imputed genotype accuracy for SNPs. Shapes and colors as in (B)
To evaluate the imputation quality of the two reference panels, we extracted variant calls for the approximately 710k sites assayed by the Illumina Infinium OmniExpress-24 microarray for two GIAB child samples (HG002 and HG005, of Ashkenazi Jewish and Han Chinese ancestry, respectively) in their benchmark regions. For each of the DV-GLN-OPT and GATK-VQSR reference panels, we phased the pseudo-microarray variants with Eagle2 and imputed the phased variants into the panel with Beagle 5.0 (Browning et al., 2018).
The imputed variant calls were scored in two evaluation regions (Supplementary Table S6). The first evaluation region, hereafter the ‘GIAB evaluation region,’ comprises the GIAB benchmark regions common to both HG002 and HG005, agnostic to either reference panel. This measures both the accuracy of the imputed genotypes and the number of benchmark variants absent in the reference panel. The second evaluation region, hereafter the ‘panel evaluation region,’ comprises a subset of the GIAB evaluation region additionally present in both the DV-GLN-OPT and the GATK-VQSR reference panels. This allows a direct comparison of variants, but provides limited information about overall individual panel quality.
The DV-GLN-OPT panel outperforms the GATK panel in F1 score in all eight experiments (two samples, two variant types and two evaluation regions). Of note, DV-GLN-OPT produces substantially higher recall than GATK-VQSR for both indels and SNPs when evaluated in the GIAB evaluation region (Fig. 6B, C). The DV-GLN-OPT panel produces on average 4.41% fewer false negative indels and 8.28% fewer false negative SNPs, while maintaining superior indel precision and indistinguishable SNP precision. As expected, evaluation metric differences are more subtle in the panel evaluation region, but the DV-GLN-OPT panel produces higher F1 scores for both samples and for both indels and SNPs (Supplementary Table S6).
3.7 Cost benchmarking
In large-scale sequencing projects, the temporal and financial cost of running bioinformatics tools can be prohibitively large. To compare the computational cost of the DeepVariant-GLnexus and GATK pipelines, we reanalyzed chromosome 22 in the 2504 1KGP samples. Starting from the aligned sequencing reads, we ran DeepVariant and GATK HaplotypeCaller to produce gVCFs on a separate virtual machine with a fixed machine type for each sample, using the Docker images published by the respective tool developers.
Using machines with 8 virtual CPUs (vCPUs) each, DeepVariant finished each chr22 sample using 40% less average time elapsed (20.0 minutes) without GPU/TPU acceleration, than GATK HaplotypeCaller (33.2 minutes) (Fig. 7A). The difference is mainly attributable to DeepVariant’s efficient internal multithreading (Fig. 7B, Supplementary Table S7). This implies that one can easily assign more vCPUs to each cloud machine to get a speedup almost proportional to the increased resources (Supplementary Fig. S14), without requiring an external workflow that splits the chromosome into smaller shards. Note that the cost difference between the two callers would expand significantly if the Base Quality Score Recalibration (BQSR) preprocessing step were included for GATK, which is part of GATK Best Practices but not recommended for DeepVariant.
Fig. 7.
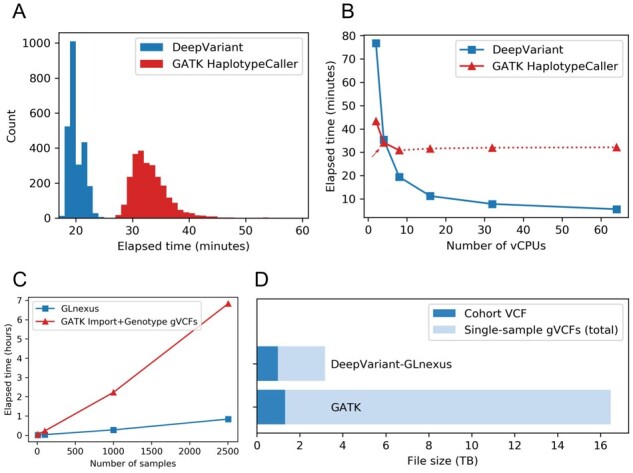
Cost benchmarking DeepVariant-GLnexus and GATK pipeline. (A) Distribution of elapsed real times to generate single-sample gVCF (chr22 only) from aligned reads across n = 2504 1KGP samples, using DeepVariant and GATK HaplotypeCaller (BQSR not included) in 8-vCPU machine. GPU/TPU acceleration was not used for DeepVariant. (B) Elapsed real times to generate gVCF (chr22 only) of one sample (NA12878) using a cloud machine with varying number of vCPUs, with DeepVariant and GATK HaplotypeCaller (excluding BQSR). The default value for HaplotypeCaller’s HMM multithreading flag (–native-pair-hmm-threads) is 4 (red arrow) and it was practically ineffectual for 16 vCPUs and more (red dotted lines). (C) Elapsed real times to merge the chr22 gVCF files from (A) into a cohort VCF for nested subsets of the 1KGP samples, using GLnexus (for DeepVariant gVCFs) and GATK GenomicsDBImport + GenotypeGVCFs (for HaplotypeCaller gVCFs). GATK VQSR step was not included. (D) The file sizes of the whole-genome cohort VCFs and the single-sample gVCFs of 1KGP samples from DeepVariant-GLnexus and GATK pipeline
Next, we processed the n = 2504 sample chromosome 22 gVCF files to produce cohort VCF files using GLnexus (from DeepVariant gVCFs), on the one hand, and GATK’s GenomicsDBImport and GenotypeGVCFs tools (from HaplotypeCaller gVCFs), on the other. While GLnexus supports internal multithreading, the two GATK tools are effectively single-threaded and require an external parallelization workflow to achieve practical runtimes, which we reproduced based on the developers’ specifications (subdividing the length of chromosome 22 to scatter across processes). Still, using a single 32-vCPU virtual machine, GLnexus is 8 times faster (0.84 hours) than the equivalent GATK tools (6.83 hours), with superior scalability to larger cohorts (Fig. 7C). For this benchmark we did not run Variant Quality Score Recalibration (VQSR) for GATK, which is a recommended step after GenotypeGVCFs in its Best Practices and will add additional runtime to its pipeline. The runtime scalability of GLnexus up to 50 000 exomes compared to GATK can be found in Lin et al. 2018 (Fig. 4).
Another relevant cost to users of these pipelines is the cost of storing the artifacts from them. In the standard block-compressed variant call format (Danecek et al., 2011; Li et al., 2009), the total size of the DeepVariant gVCFs for all chromosomes of 1KGP samples is 7 times smaller (total 2.20TB, average 878MB/sample) than GATK HaplotypeCaller gVCFs (total 15.16TB, average 6053 MB/sample) (Fig. 7D), which is a result of DeepVariant’s efficient quantization of the reference records. Moreover, the final cohort VCF from DeepVariant-GLnexus pipeline is also 26% smaller (0.97TB) than the one from GATK pipeline (1.32TB). This reduction in file sizes directly translates to a similar ratio of cost savings in cloud storage services. To further reduce the sizes of the cohort VCF, one may consider using the BCF (binary VCF) format or other data formats designed for a large number of samples (Danek and Deorowicz, 2018; Kelleher et al., 2019; Layer et al., 2016; Li, 2016; Lin et al., 2020; Zheng et al., 2017).
4 Discussion
Population-scale sequenced cohorts are foundational resources for many genetic analyses, including genotype-phenotype discovery, variant interpretation and genotype imputation. As sequencing projects have grown to include hundreds of thousands of samples, the need for highly accurate variant calls and computationally efficient merging algorithms is increasingly acute. By optimizing GLnexus to merge single-sample DeepVariant calls, we demonstrated that the superior accuracy (Poplin et al., 2018b) and generalizability across sequencing methods (Wenger et al., 2019) of DeepVariant can generate more accurate cohort callsets at large scale at lower cost. The callset quality metrics of the optimized pipeline consistently outperformed the GATK Best Practices across a range of cohort sizes and sequence coverages. In addition, we showed that variant confidences are well calibrated to Mendelian violation rate, allowing tuning of callsets for very high precision or for high recall.
When optimizing callset creation, we investigated callset quality stratified by both sequencing coverage and cohort size. Results within a given sequencing coverage were qualitatively similar regardless of cohort size, suggesting that a major driver of parameter sensitivity is the distribution of individual call confidence estimates. Even so, when optimizing equally between benchmark callset accuracy and Mendelian violation rate, we observed a single parameter set that provides strong performance across the range of WGS cohorts analyzed.
Although we demonstrated the strength of the DeepVariant+GLnexus method, there are multiple areas for future improvement. At both 15× and 40× coverage, the precision-recall curves of SNPs versus indels is markedly different. As expected, parameter variation can tune SNPs to improve recall at the expense of precision or vice versa. In contrast, indels appear to have nearly globally optimal parameters, suggesting that distinct handling of the two variant classes may further improve callset quality. Additionally, the tunable GLnexus parameters affect allele harmonization and genotyping, but do not apply any hard filtering to output calls. We observed an overrepresentation of Mendelian violations at very low GQ values, indicating that direct omission of low quality sites or genotypes also may improve callset quality. Finally, a small fraction (0.4%) of autosomal sites contain seven or more total alleles, and typically represent short tandem repeats (Fan and Chu, 2007). While these sites likely capture some of the known hypervariability of these regions, this benefit is weighed against the practical difficulty of representing and analyzing these sites in downstream applications.
Comparison of cohort callsets is a less mature field than comparison of single-sample callsets. Here we focused on four evaluation metrics (accuracy of GIAB sample calls, Mendelian concordance, Ti:Tv ratio and HWE p-value distributions) to incorporate direct variant accuracy measures where possible, but also include indirect signals of quality genome-wide. We used the recent deep sequencing of 1KGP to perform an orthogonal analysis on a publicly available dataset. The resulting optimized DeepVariant+GLnexus callset possesses superior metrics to a GATK Best Practices callset generated independently, including a 32% reduction in sites violating HWE at p < . Moreover, an imputation reference panel derived from the DeepVariant+GLnexus callset results in higher imputation accuracy, which shows that improving cohort-level variant calls yields improved performance in a common downstream application. Both the cohort callset and all DeepVariant single-sample calls are freely available at Google Cloud Storage (https://console.cloud.google.com/storage/browser/brain-genomics-public/research/cohort/1KGP).
To our knowledge, this is the most accurate 1KGP callset currently available as measured by the above metrics, and as such has substantial utility within the genomics community for studies of genetic variation. Furthermore, we hope this resource spurs additional innovation in the development and evaluation of population-scale cohorts.
Supplementary Material
Acknowledgements
The authors thank Babak Alipanahi, Thomas Colthurst, Greg Corrado and Mark DePristo for helpful discussions and feedback, and the New York Genome Center and its sponsors for the deep resequencing of the 1KGP samples.
PAGE dataset: Samples and data of The Charles Bronfman Institute for Personalized Medicine (IPM) BioMe BioBank used in this study were provided by The Charles Bronfman Institute for Personalized Medicine at the Icahn School of Medicine at Mount Sinai (New York). Phenotype data collection was supported by The Andrea and Charles Bronfman Philanthropies. Funding support for genotyping, which was performed at The Center for Inherited Disease Research (CIDR), was provided by the NIH (U01HG007417). The datasets used for the analyses described in this manuscript were obtained from dbGaP at http://www.ncbi.nlm.nih.gov/gap through dbGaP accession number phs000925.v1.p1.
CSER dataset: These results are in whole or part based upon data generated by the Clinical Sequencing Exploratory Research (CSER) consortium established by the NHGRI. Funding support was provided through cooperative agreements with the NHGRI and NCI through grant numbers U01 HG007301 (Genomic Diagnosis in Children with Developmental Delay). Information about CSER and the investigators and institutions who comprise the CSER consortium can be found at http://www.genome.gov/27846194.
The 1000 Genomes Project high-coverage sequencing dataset: These data were generated at the New York Genome Center with funds provided by NHGRI Grant 3UM1HG008901-03S1.
Funding
All compute resources used in this work were provided by Google LLC. M.F.L. received compensation for contributions to this work under a consulting engagement with Google LLC.
Conflict of Interest: T.Y., H.L., P-C.C., A.C., and C.Y.M. are employees and equity owners of Google LLC. M.F.L. has consulting engagements with Google LLC and DNAnexus Inc., and equity in the latter.
Contributor Information
Taedong Yun, Google Health, Cambridge, MA 02142, USA.
Helen Li, Google Health, Palo Alto, CA 94304, USA.
Pi-Chuan Chang, Google Health, Palo Alto, CA 94304, USA.
Michael F Lin, mlin.net LLC, Honolulu, HI 96816, USA.
Andrew Carroll, Google Health, Palo Alto, CA 94304, USA.
Cory Y McLean, Google Health, Cambridge, MA 02142, USA.
References
- Amendola L.M. et al. (2018) The Clinical Sequencing Evidence-Generating Research Consortium: integrating genomic sequencing in diverse and medically underserved populations. Am. J. Hum. Genet., 103, 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge M.N. et al. (2011) Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol., 12, R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brier G.W. (1950) Verification of forecasts expressed in terms of probability. Mon. Weather Rev., 78, 1–3. [Google Scholar]
- Browning B.L. et al. (2018) A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet., 103, 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft C. et al. (2018) The UK Biobank resource with deep phenotyping and genomic data. Nature, 562, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P. et al. ; 1000 Genomes Project Analysis Group. (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danek A., Deorowicz S. (2018) GTC: how to maintain huge genotype collections in a compressed form. Bioinformatics, 34, 1834–1840. [DOI] [PubMed] [Google Scholar]
- Delaneau O. et al. ; The 1000 Genomes Project Consortium. (2014) Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat. Commun., 5, 3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo M.A. et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet., 43, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey F.E. et al. (2016) Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science, 354, aaf6814. [DOI] [PubMed] [Google Scholar]
- Fan H., Chu J.Y. (2007) A brief review of short tandem repeat mutation. Genomics Proteomics Bioinf., 5, 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust G.G., Hall I.M. (2014). SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics, 30, 2503–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E., Marth G. (2012) Haplotype-based variant detection from short-read sequencing. arXiv, arXiv: 1207.3907. [Google Scholar]
- Golovin D. et al. (2017) Google vizier: A service for black-box optimization. In Proceedings of the ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Vol. Part F1296 of KDD ’17, ACM. pp. 1487–1496.
- Graffelman J. et al. (2017) A genome-wide study of Hardy–Weinberg equilibrium with next generation sequence data. Hum. Genet., 136, 727–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy G.H. (1908) Mendelian proportions in a mixed population. Science, 28, 49–50. [DOI] [PubMed] [Google Scholar]
- Huang J. et al. (2012) 1000 Genomes-based imputation identifies novel and refined associations for the Wellcome Trust Case Control Consortium phase 1 Data. Eur. J. Hum. Genet., 20, 801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski K.J. et al. ; Genome Aggregation Database Consortium. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher J. et al. (2019) Inferring whole-genome histories in large population datasets. Nat. Genet., 51, 1330–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. et al. (2018) Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods, 15, 591–594. [DOI] [PubMed] [Google Scholar]
- Krusche P. et al. ; The Global Alliance for Genomics and Health Benchmarking Team. (2019) Best practices for benchmarking germline small-variant calls in human genomes. Nat. Biotechnol., 37, 555–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer R.M. et al. ; Exome Aggregation Consortium. (2016) Efficient genotype compression and analysis of large genetic-variation data sets. Nat. Methods, 13, 63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M. et al. ; Exome Aggregation Consortium. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, arXiv: 1303.3997. [Google Scholar]
- Li H. (2016) BGT: efficient and flexible genotype query across many samples. Bioinformatics, 32, 590–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. ; 1000 Genome Project Data Processing Subgroup. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M.F. et al. (2018) GLnexus: joint variant calling for large cohort sequencing. bioRxiv, 343970. doi: 10.1101/343970. [Google Scholar]
- Lin M.F. et al. (2020) Sparse Project VCF: efficient encoding of population genotype matrices. Bioinformatics, doi: 10.1093/bioinformatics/btaa1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh P.R. et al. (2016) Reference-based phasing using the haplotype reference consortium panel. Nat. Genet., 48, 1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R. et al. (2019) A multi-task convolutional deep neural network for variant calling in single molecule sequencing. Nat. Commun., 10, 998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise T.C. et al. (2011) The next PAGE in understanding complex traits: design for the analysis of population architecture using genetics and epidemiology (PAGE) study. Am. J. Epidemiol., 174, 849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A. et al. (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W. et al. (2016) The ensembl variant effect predictor. Genome Biol., 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikpay M. et al. (2015) A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet., 47, 1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki K. et al. (2002) Functional SNPs in the lymphotoxin- gene that are associated with susceptibility to myocardial infarction. Nat. Genet., 32, 650–654. [DOI] [PubMed] [Google Scholar]
- Poplin R. et al. (2018a) Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv, 201178. doi: 10.1101/201178. [Google Scholar]
- Poplin,R. et al. (2018b) A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol., 36, 983–987. [DOI] [PubMed] [Google Scholar]
- Ramoni R.B. et al. (2017) The undiagnosed diseases network: accelerating discovery about health and disease. Am. J. Hum. Genet., 100, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roslin N. et al. (2016) Quality control analysis of the 1000 Genomes Project Omni2.5 genotypes. bioRxiv, 078600. doi: 10.1101/078600. [Google Scholar]
- Shaikho E.M. et al. (2017) A phased SNP-based classification of sickle cell anemia HBB haplotypes. BMC Genomics, 18, 608–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry S.T. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res., 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelhalter D.J. (1986) Probabilistic prediction in patient management and clinical trials. Stat. Med., 5, 421–433. [DOI] [PubMed] [Google Scholar]
- Taliun D. et al. (2019) Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. bioRxiv, 563866. doi: 10.1101/563866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. (2010) A map of human genome variation from population-scale sequencing. Nature, 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter D. et al. (2014) The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res., 42, D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger A.M. et al. (2019) Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol., 37, 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. et al. (2013) Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N. Engl. J. Med., 369, 1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X. et al. (2017) SeqArray-a storage-efficient high-performance data format for WGS variant calls. Bioinformatics, 33, 2251–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook J.M. et al. (2014) Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol., 32, 246–251. [DOI] [PubMed] [Google Scholar]
- Zook J.M. et al. (2019) An open resource for accurately benchmarking small variant and reference calls. Nat. Biotechnol., 37, 561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.