

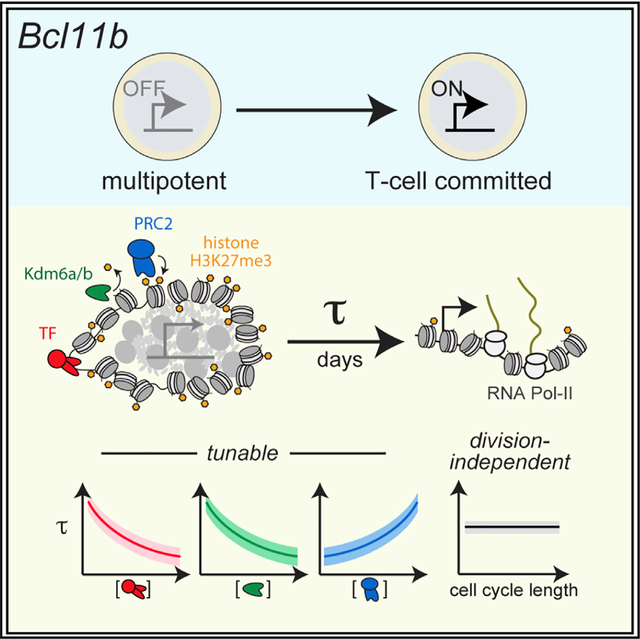
SUMMARY
During development, progenitors often differentiate many cell generations after receiving signals. These delays must be robust yet tunable for precise population size control. Polycomb repressive mechanisms, involving histone H3 lysine-27 trimethylation (H3K27me3), restrain the expression of lineage-specifying genes in progenitors and may delay their activation and ensuing differentiation. Here, we elucidate an epigenetic switch controlling the T cell commitment gene Bcl11b that holds its locus in a heritable inactive state for multiple cell generations before activation. Integrating experiments and modeling, we identify a mechanism where H3K27me3 levels at Bcl11b, regulated by methyltransferase and demethylase activities, set the time delay at which the locus switches from a compacted, silent state to an extended, active state. This activation delay robustly spans many cell generations, is tunable by chromatin modifiers and transcription factors, and is independent of cell division. With their regulatory flexibility, such timed epigenetic switches may broadly control timing in development.
Graphical abstract
In brief
H3K27me3 is found on repressed lineage-specifying gene loci in stem cells and may delay their activation in response to signals. Pease et al. identify an adjustable timer mechanism, where H3K27me3 levels at the T cell commitment gene Bcl11b, set by opposing writer and eraser enzymes, modulate the timing of gene locus decompaction and activation.
INTRODUCTION
During multicellular development, stem and progenitor cells often differentiate many days and cell divisions after receiving instructive signals. These differentiation delays must be robust yet tunable over timescales spanning multiple cell generations for precise control over differentiated population sizes. In diverse contexts, cell differentiation delays are generated by timing mechanisms operating autonomously in single cells. In classic studies of oligodendrocyte differentiation, precursor cells exposed to signals delayed their differentiation by up to eight cell divisions, due to a cell-autonomous timing mechanism (Gao et al., 1997; Temple and Raff, 1986). Such autonomous timing control is seen in diverse systems, from brain and muscle development to adaptive immunity (Burton et al., 1999; Heinzel et al., 2017; Otani et al., 2016).
Although timing delays in some embryonic systems are generated by mechanisms that count cell divisions (Amodeo et al., 2015; Newport and Kirschner, 1982), differentiation delays during later vertebrate development or in adult stem cells are often uncoupled from cell cycle progression, such that changes to rates of cell division do not affect delay duration (Burton et al., 1999; Gao et al., 1997; Heinzel et al., 2017; Li et al., 2019; Okamoto et al., 2016; Osmond, 1991; Otani et al., 2016). A mechanism for setting the elapsed time to differentiation apart from cell division could provide functional advantages to cells, including operation in non-dividing cells and an ability to modulate cell expansion while maintaining a constant temporal schedule for differentiation. However, it is unknown how division-independent timing control is implemented on a molecular level.
Polycomb repressive mechanisms, involving histone H3 lysine-27 trimethylation (H3K27me3), are important for differentiation timing control. H3K27me3 modifications are often found at the loci of lineage-specifying genes in stem cells, where they restrain the expression of these genes and resultant differentiation (Boyer et al., 2006; Lee et al., 2006). During differentiation, instructive signals activate transcription factors that bind to lineage-specifying genes and initiate H3K27me3 removal. However, although transcription factors usually bind rapidly upon signal exposure within minutes, H3K27me3 loss and gene activation often occur much more slowly, such that gene loci can heritably maintain a silent state over multiple cell generations prior to activation (Berry et al., 2017; Kaikkonen et al., 2013; Bintu et al., 2016; Mayran et al., 2018). In a prevailing view, this epigenetic maintenance of the silent state before activation results from the passive dilution of H3K27me3 modified histones through serial cell division (Gaydos et al., 2014; Jadhav et al., 2020; Sun et al., 2014). However, although passive dilution mechanisms can delay differentiation over multiple cell divisions, these delays would depend on cell cycle duration and be inconsistent with the division-independent timers found in vertebrates. Alternatively, H3K27me3 loss may be actively controlled by the opposing polycomb repressive complex 2 (PRC2) methyltransferase and Kdm6a/b demethylase activities (Li et al., 2014; Park et al., 2014; Seenundun et al., 2010). However, it is unclear whether such active mechanisms could generate differentiation delays spanning many cell divisions and whether these delays could be both tunable and cell-division independent.
To address these questions, we investigated the mechanism of a time-delayed epigenetic switch controlling the activation of Bcl11b, a transcription factor essential for T cell lineage commitment and identity (Hosokawa et al., 2018; Ikawa et al., 2010; Li et al., 2010; Figure 1A). In early T cell progenitors, Notch signals activate Bcl11b, both directly (Ikawa et al., 2010; Li et al., 2010) and indirectly, by activating its upstream regulators Gata3 and TCF-1 (García-Ojeda et al., 2013; Germar et al., 2011; Zhou et al., 2019). However, although these upstream regulators become active shortly after thymic entry, Bcl11b activation and T cell lineage commitment occur ~5–10 days later, during which progenitors proliferate 1,000-fold (Manesso et al., 2013; Porritt et al., 2003; Zhou et al., 2019). Using a dual-allele Bcl11b reporter strain, where each endogenous gene copy is tagged with distinguishable fluorescent protein reporters (Ng et al., 2018), we found that this long delay in Bcl11b activation arises partly because of an epigenetic switch acting at individual Bcl11b loci, in cis. This epigenetic switch activates probabilistically, with a multi-day time constant that is controlled by Gata3 and TCF-1 (Kueh et al., 2016), along with a distal enhancer to which these factors bind (Ng et al., 2018). However, our previous studies did not clarify the mechanism of the timed epigenetic switch itself or its basis for generating controllable timing delays in gene activation. We address these outstanding questions here.
Figure 1. A timed epigenetic switch delays Bcl11b activation and T cell lineage commitment.
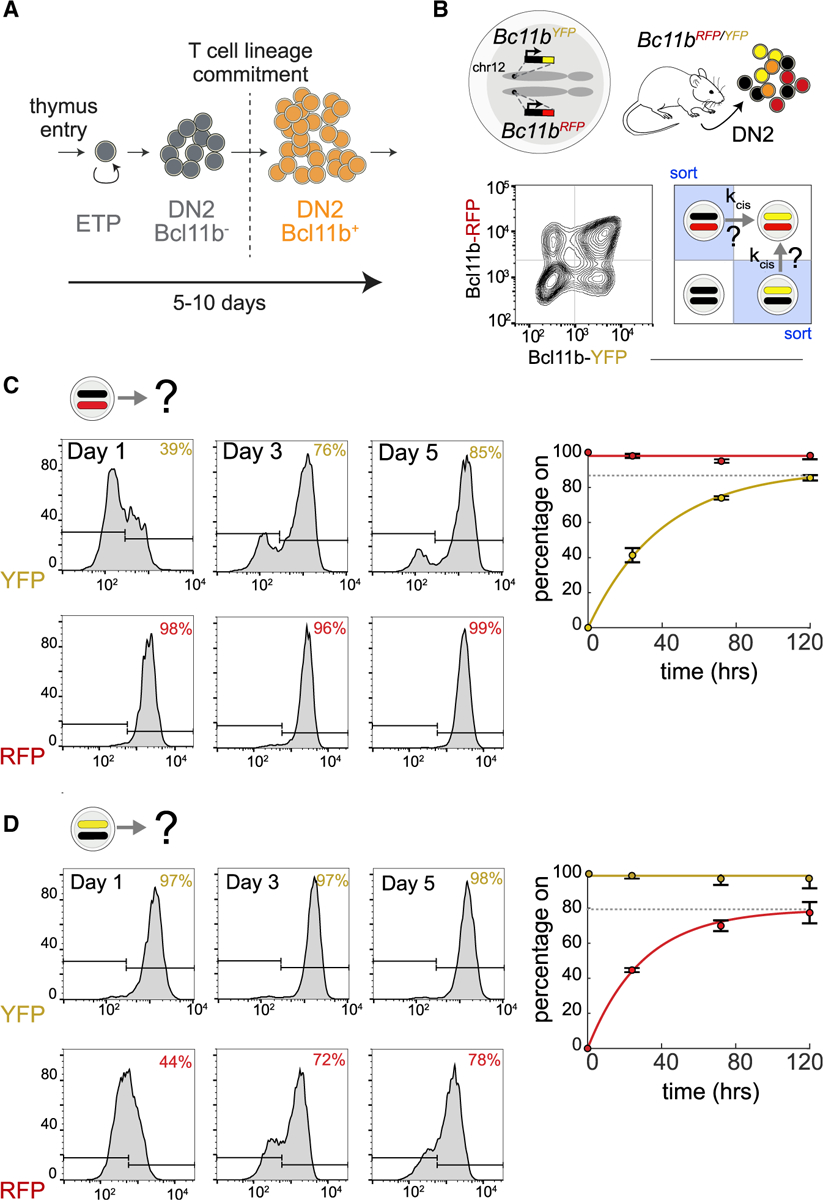
(A) Bcl11b, a transcription factor that drives T cell lineage commitment, turns on with a multi-day time delay.
(B) Dual-allelic Bcl11b reporter mouse (top), along with flow cytometry plot showing levels of each Bcl11b allele in DN2 progenitors (bottom left) and with strategy to purify Bcl11b monoallelic expressing progenitors for live-cell analysis of epigenetic switch timing, kcis.
(C and D) DN2 monoallelic Bcl11b-expressing progenitors were purified, cultured on OP9-DL1 feeders with 5 ng/mL interleukin-7 (IL-7) and Flt3L, and analyzed by flow cytometry.
Data represent means and 95% confidence intervals for n = 3 independent experiments. Curves represent fits to the equation y = F(1 − e−kt), where F is the final percentage of cells positive for assayed allele (represented by the dotted gray lines); k = 0.025 h−1 ± 0.005 for YFP activation and k = 0.034 h−1 ± 0.009 for RFP activation.
RESULTS
A timed epigenetic switch delays Bcl11b activation and T cell lineage commitment
To study the cis-acting epigenetic event controlling Bcl11b activation timing in isolation from other events occurring in trans, we analyzed Bcl11b locus activation dynamics in progenitors that already have one Bcl11b allele active and must therefore contain all trans-factors necessary for expression (Figure 1B). Using fluorescence-activated cell sorting (FACS), we purified monoallelic Bcl11b expressing DN2 progenitors from dual-allelic reporter mice and analyzed activation of the silent allele by co-culture with OP9-DL1 cells, an in vitro system that recapitulates early transitions in T cell development (Holmes and Zuniga-Pflucker, 2009). Inactive Bcl11b alleles turned on after a long time delay such that the fraction of biallelically expressing cells increased progressively over the course of 5 days (Figures 1C and 1D). Activation kinetics were similar for both YFP (mCitrine yellow fluorescent protein) and RFP (mCherry red fluorescent protein) alleles and were well described by a single exponential curve, consistent with activation being controlled by a single stochastic event occurring with equal likelihood at each allele.
H3K27me3 levels at the Bcl11b locus tune activation timing
The repressive histone modification H3K27me3 is highly enriched at silent Bcl11b loci in hematopoietic progenitor cells, but not in committed T cells where Bcl11b is expressed (Zhang et al., 2012). Therefore, H3K27me3 removal could regulate the epigenetic event controlling Bcl11b activation timing. To test this possibility, we first determined whether H3K27me3 marks are removed from the Bcl11b locus at the same time it turns on. To pinpoint when H3K27me3 loss occurs relative to locus activation, we measured H3K27me3 levels in three progenitor populations having different numbers of active Bcl11b loci. In bone marrow progenitors, where both Bcl11b alleles are inactive, there was an abundance of H3K27me3 across the 5′ end of Bcl11b (Figure 2A). These broad H3K27me3 peaks were roughly halved in monoallelic Bcl11b expressing DN2 progenitors and were almost completely absent in biallelic Bcl11b DN2 progenitors (Figure 2A). These results show H3K27me3 is lost from the Bcl11b locus concurrently with its activation.
Figure 2. H3K27me3 levels, set by PRC2 and Kdm6a/b demethylases, modulate Bcl11b activation timing.
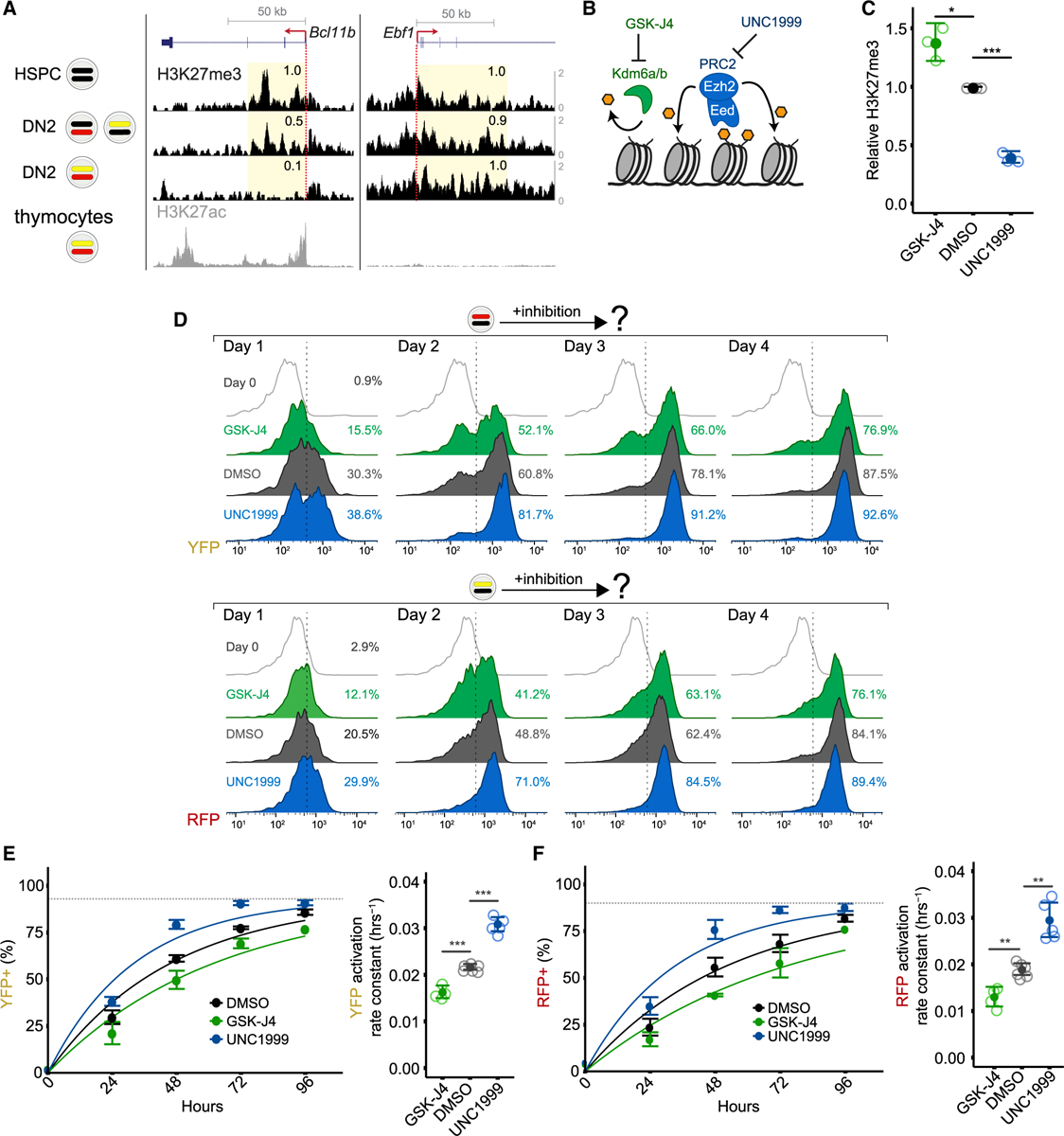
(A) H3K27me3 distributions were profiled by CUT&RUN in Lin− bone marrow progenitors (hematopoietic stem and progenitor cells [HSPCs]), as well as purified DN2 monoallelic and biallelic Bcl11b-expressing cells with UCSC Genome Browser tracks showing H3K27me3 densities at Bcl11b, as well as at Ebf1, a B cell regulator that is repressed during T cell development. Relative read densities of shaded areas are shown. H3K27ac levels in thymocytes, obtained from ENCODE accession number ENCSR000CCH (Davis et al., 2018), demarcate transcribed region. Data are representative of two independent experiments.
(B) Schematic depicting inhibition of H3K27 demethylases Kdm6a/b or H3K27 methyltransferase PRC2.
(C) DN2 monoallelic progenitors treated with the indicated inhibitors were sorted for anti-H3K27me3 CUT&RUN followed by qPCR at the Bcl11b promoter. Mean values are shown for n = 3 independent experiments (two-sample t test, one-tailed: *p < 0.05; ***p < 0.001).
(D) Purified DN2 monoallelic expressing cells were re-cultured with the indicated inhibitors and analyzed by flow cytometry. Histograms show results from one representative experiment.
(E and F) (Left) Mean activation percentages and 95% confidence intervals are plotted with curves representing fits to the equation y = F(1 − e−kt), where F = maximum percentage of cells positive for assayed allele (represented by the dotted gray lines). (Right) Data represent mean rate constants, k, with 95% confidence intervals (two-sample t test, one-tailed: **p < 0.01; ***p < 0.001; n = 4–6 independent experiments).
See also Figure S1.
H3K27me3 may modulate the timing of Bcl11b activation; alternatively, its loss may simply be a consequence of gene activation due to clearance of methylated nucleosomes by active transcription (Hosogane et al., 2016; Kraushaar et al., 2013). To determine whether H3K27me3 modifications play a causal role in controlling Bcl11b activation timing, we cultured monoallelic Bcl11b expressing DN2 progenitors with small-molecule inhibitors targeting H3K27me3-modifying enzymes and analyzed the effects on activation of the silent Bcl11b allele. These inhibitors, which target either the PRC2 methyltransferase subunit Ezh2 (UNC1999) or the H3K27 demethylases Kdm6a/b (GSKJ4; Figure 2B), resulted in an ~60% decrease and ~40% increase, respectively, in H3K27me3 abundance at the Bcl11b promoter in monoallelic Bcl11b expressing DN2 progenitors (Figure 2C), indicating that they actively modulate H3K27me3 levels at inactive Bcl11b loci.
To determine whether H3K27me3 levels regulate Bcl11b activation timing, we assayed the expression of inactive Bcl11b alleles in DN2 progenitors over the course of 4 days. In the absence of any inhibitors, the silent Bcl11b alleles in monoallelic progenitors activated at an average rate of 0.022 h−1 and 0.019 h−1 for YFP and RFP alleles, respectively (Figures 2D–2F). Kdm6a/b demethylase inhibition decreased the activation rate for each silent allele in a graded manner (27%–37%). Conversely, Ezh2 inhibition increased the activation rate for each silent allele (41%–58%). Similar graded decreases or increases in Bcl11b activation probabilities were observed for another structurally unrelated Kdm6a/b inhibitor, IOX-1, and other Ezh2 inhibitors, GSK126 and GSK-343 (Figure S1A). Furthermore, short-hairpin-RNA-mediated knockdown of another essential PRC2 subunit, Eed, also increased the activation rate of silent Bcl11b alleles (Figures S1A, S1C, and S1D), arguing against non-specific pharmacological effects. Notably, all H3K27me3 perturbations tested altered the fraction of activated cells without altering expression levels in activated cells (Figure S1A), indicating a specific role of these modifications in tuning gene activation timing. Importantly, perturbing H3K27 methylation rates had no effect on the apoptosis frequency among different populations, indicating that these changes in activation fractions are not due to differential cell death in these populations (Figure S1E). Taken together, these results show that H3K27me3 levels at the Bcl11b locus, set by opposing PRC2 methyltransferase and Kdm6a/b demethylase activities, control the timing of Bcl11b activation.
Bcl11b activation timing is regulated independently of cell division
Activation delays of polycomb-repressed genes have been proposed to result from the passive dilution of H3K27me3 modified histones with cell division (Coleman and Struhl, 2017; Jiang and Berger, 2017; Strome et al., 2014; Sun et al., 2014). However, the observed regulation of Bcl11b activation timing by H3K27 demethylases that counteract PRC2-mediated methylation (Figure 2) suggests an active mechanism is involved that could potentially operate independently of cell division. To determine whether Bcl11b activation timing depends on cell division, we accelerated the cell division rate in DN2 RFP+/YFP− progenitors by transducing them with the proto-oncogene c-Myc and used quantitative live-cell imaging to measure the activation kinetics of the silent YFP allele (Figure 3A). As expected, c-Myc overexpression resulted in an ~2-fold increase in the cell division rate (Figure 3B; see also Figure S2, Table S1, and Quantitative and statistical analysis). However, despite accelerating cell division, c-Myc overexpression did not change Bcl11b activation timing, with control and accelerated progenitors activating the silent YFP allele with the same exponential time constant (~136 h; Figure 3C). This lack of change in activation timing following cell division acceleration was also observed by flow cytometry for silent RFP alleles (Figure 3D). Therefore, in contrast to the passive dilution paradigm, these results show that the epigenetic switch controlling Bcl11b activation generates time delays in gene activation that are independent of cell division.
Figure 3. Bcl11b activation timing is independent of cell-division speed.
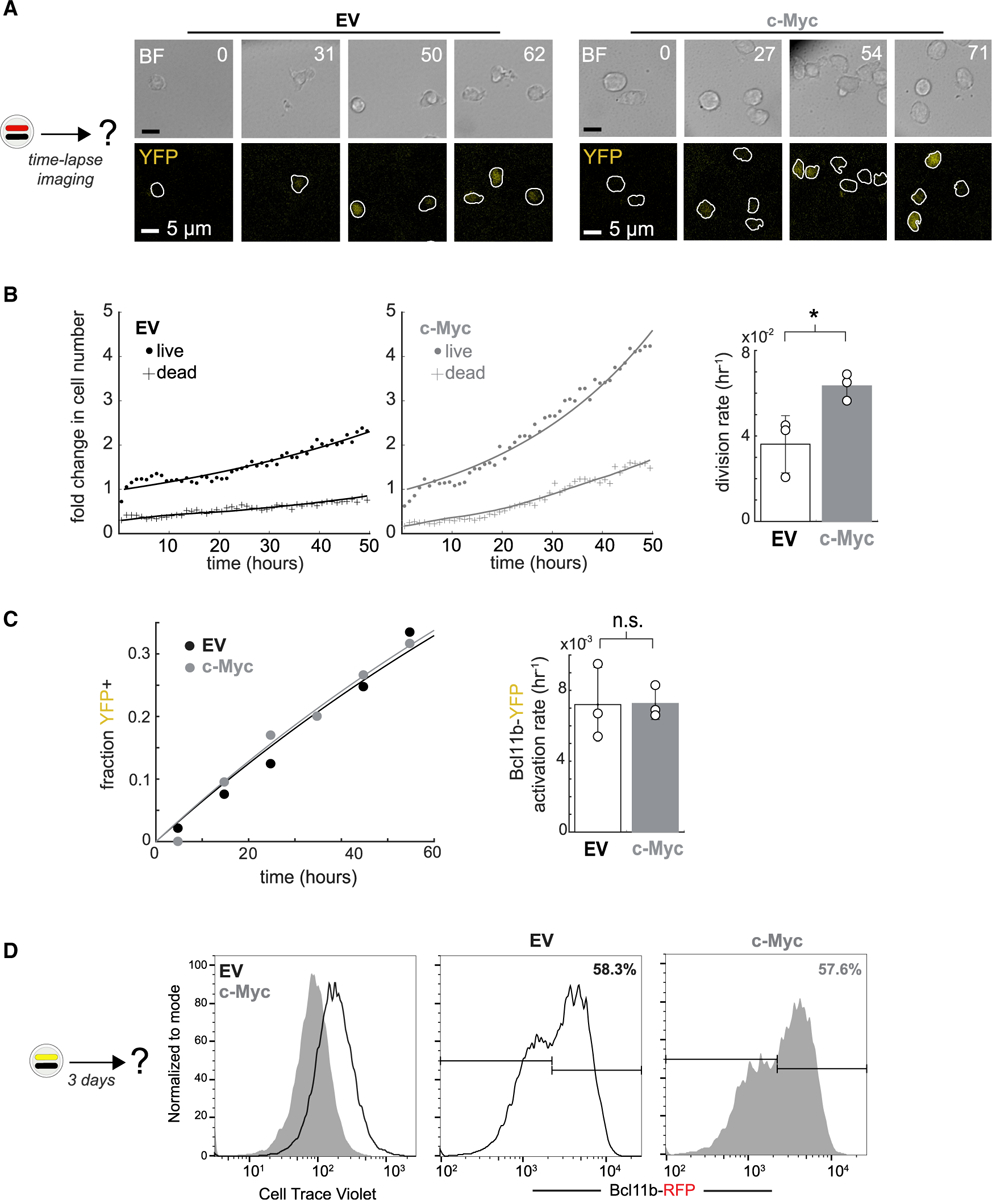
Bcl11bYFP−/RFP+ DN2 progenitors transduced with either an empty vector (EV) or c-Myc overexpressing retroviral vector were purified, re-cultured on DL1-coated plates, and monitored by time-lapse imaging.
(A) Time-lapse images. White boundaries show automated cell segmentation. Numbers show elapsed time in hours.
(B) (Left) Time evolution of live and dead cell numbers. Data were fitted to a population dynamics model as shown in Figure S2. (Right) Data represent mean and standard deviation of cell division rates for n = 3 independent experiments (paired two-sample t test, one-tailed; *p < 0.025).
(C) (Left) Fraction of YFP+ cells over time. (Right) Data represent mean and standard deviation of Bcl11b-YFP activation rates for n = 3 independent experiments (paired two-sample t test, one-tailed; n.s., not significant).
(D) Bcl11bRFP+/YFP− DN2 progenitors transduced with either EV or c-Myc were re-cultured on OP9-DL1 stromal monolayers for 3 days before analyzing by flow cytometry.
A methylation-compaction mechanism for tunable, division-independent timing control
The molecular mechanism underlying the timed Bcl11b epigenetic switch must account for its observed emergent properties, namely, (1) its ability to robustly set time delays that span multiple cell generations, (2) its stochastic nature, (3) its tunability by histone-modifying enzyme activities, and (4) its cell division independence. To identify mechanisms with these emergent properties, we used mathematical modeling to analyze a series of candidate mechanisms. H3K27me3 can bind PRC2 at an allosteric site and activate its methyltransferase activity (Margueron et al., 2009), allowing these modifications to spread to neighboring nucleosomes and be maintained across cell division. Previous work has shown that such positive feedback, mediated by a read-write mechanism for histone methylation, can generate bistable switching that occurs over timescales spanning many cell generations (Dodd et al., 2007; Zhang et al., 2014). Therefore, we first analyzed a simple model of this positive feedback mechanism. In this methylation read-write (M) model, the Bcl11b locus comprises a linear array of N nucleosomes, each of which can be methylated or demethylated (Figure 4A; see Methods S1). Each nucleosome is methylated at a rate that increases with the number of nearby methylated nucleosomes and is demethylated at a first-order rate. During DNA replication, nucleosomes randomly segregate to the two daughter strands, resulting in half of the methylated nucleosomes being replaced by demethylated ones (Coleman and Struhl, 2017).
Figure 4. A methylation-compaction switching mechanism generates tunable, division-independent delays in gene activation.
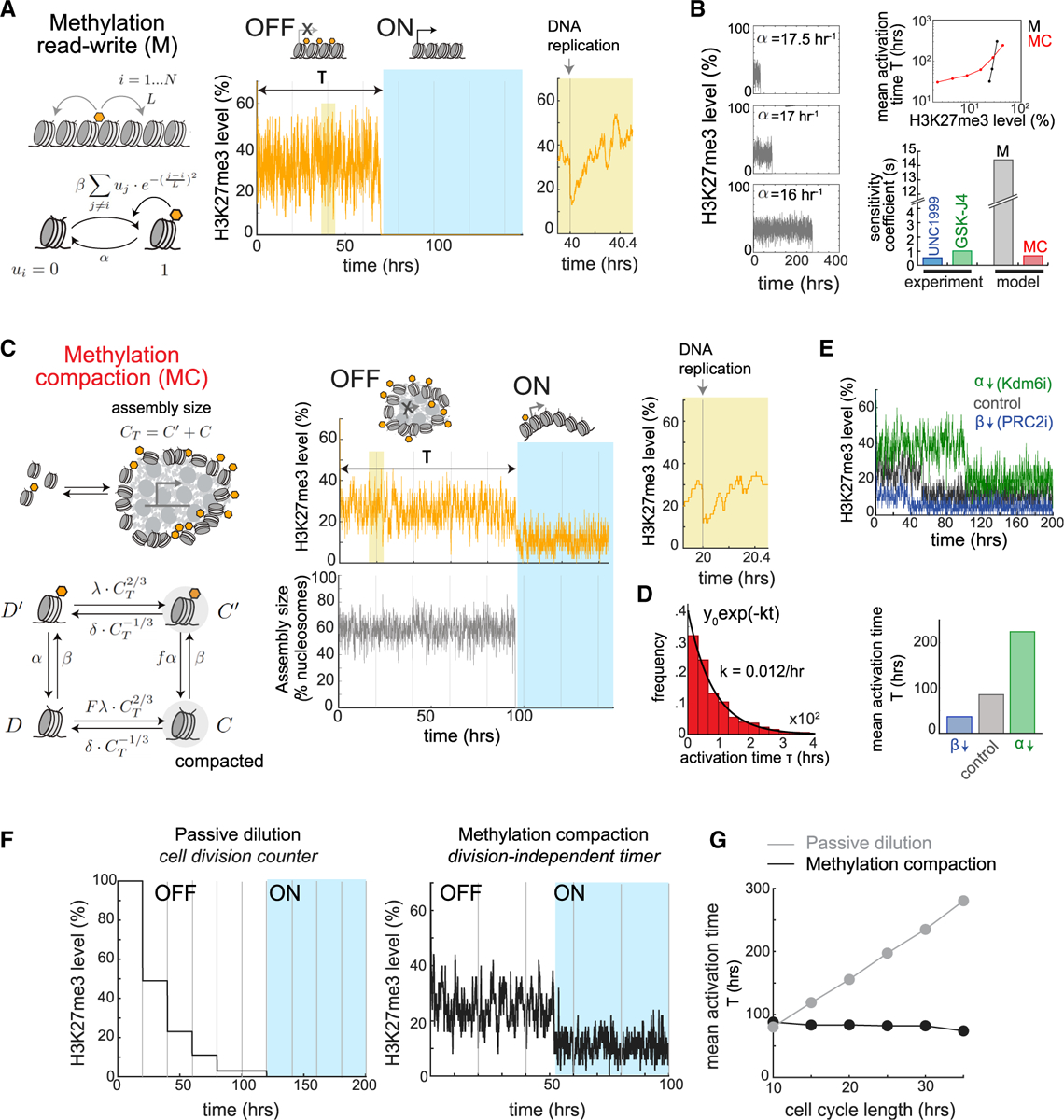
(A) Methylation read-write (M) model (left), along with representative simulation (right).
(B) Representative simulations of M model with different demethylation rates α (left). Mean activation times against H3K27me3 levels (top right) and sensitivity coefficients for this relationship (bottom right) are shown.
(C) Methylation compaction (MC) model (left), along with representative simulation (right).
(D) Histogram shows distribution of activation times, along with exponential fit.
(E) Representative simulations of MC model, simulating PRC2 or Kdm6a/b inhibition (top), along with mean activation times (bottom).
(F) Simulations of passive dilution and MC models. Vertical lines indicate DNA replication events.
(G) Mean activation times as a function of cell cycle length.
See also Figures S3–S7.
Our simulations revealed that single-nucleosome arrays could switch from a repressed H3K27 methylated state to a demethylated state with stochastic time delays spanning multiple cell divisions (Figure 4A, center), consistent with previous work (Dodd et al., 2007; Zhang et al., 2014). However, in our simulations, activation timing was extremely sensitive to H3K27 methylation levels in the silent state, with minor changes in methylation levels (~10%) causing dramatic changes activation timing (~300-fold; Figure 4B). This extreme sensitivity far exceeded the sensitivity coefficient observed experimentally (Figures 2C–2F and 4B) across a wide range of methyltransferase reach lengths (Figures S3A and S3B) and was also seen in other studies (Dodd et al., 2007; Zhang et al., 2014), indicating that it represents a general feature of positive-feedback-mediated switching and not a specific aspect of our model. By analyzing this system using a transition state theory framework (Figures S3C–S3E; see also Methods S1), we found that switching times scale exponentially with methylation or demethylation rates, thus explaining the observed extreme sensitivity. Thus, models that consider histone modification dynamics alone are inconsistent with the tunable control of activation by H3K27me3-modifying enzymes observed experimentally (Figures 2C–2F).
H3K27me3 modifications repress gene expression by promoting the formation of compacted chromatin assemblies inaccessible to the transcription machinery. Compaction may occur through H3K27me3-dependent recruitment of PRC1, which can self-associate through intrinsically disordered domains on its Cbx2 subunit to promote chromatin condensation (Plys et al., 2019; Tatavosian et al., 2019); alternatively, compaction may involve direct interactions between nucleosomes that are modulated by the H3K27me3 state (Gibson et al., 2019; Sanulli et al., 2019). In both cases, weak multivalent interactions between nucleosomes drive chromatin condensation through phase separation.
In light of these recent findings, we developed a second model, where H3K27me3 does not directly repress transcription per se but instead enhances the strength of nucleosomal interactions to keep the locus compacted and restrain its activation (Figure 4C; see Methods S1). In this methylation-compaction (MC) model, the Bcl11b locus consists of an ensemble of nucleosomes that can exist in methylated or demethylated states and can be included in or excluded from a compacted nucleosome assembly. Methylation and demethylation of nucleosomes occur with first-order rates, whereas association and dissociation of nucleosomes with the compacted chromatin assembly occur at rates proportional to its surface area, which we assume scales with the two-thirds power of the number of nucleosomes in the assembly CT. This rate dependency assumes that nucleosomes enter (or exit) the compacted assembly only by formation (or breakage) of weak multivalent interactions with nucleosomes on the assembly surface. Methylation and compaction are coupled such that the methylation state of a nucleosome affects its rate of association with a compacted assembly and vice versa. Below a threshold number of nucleosomes, the chromatin assembly is unstable and dissolves, leading to locus decompaction and consequent gene expression. This stability threshold reflects a minimum nucleus size needed to maintain a phase-separated condensate.
Simulations of the methylation-compaction model revealed that the gene locus can maintain a H3K27 methylated and compacted state for multiple cell divisions before switching in an all-or-none manner to a decompacted, low-methylation state (Figure 4C, center). As with the methylation-only model, the time delay in switching is well described by a first-order stochastic process, with a constant probability of activation per unit time (Figure 4D). However, in contrast with the methylation read-write model but in concordance with our experimental results (Figure 2), changing H3K27me3 levels by varying methylation or demethylation rates changed gene activation timing in a much more graded manner (Figures 4B and 4E). This tunability was robust over different parameter ranges (Figure 4B, top right), different degrees of cooperativity for H3K27 methylation (Figure S4), and different degrees of assembly disruption after DNA replication (Figures S5A–S5E). A transition state theory analysis (see Methods S1 and Figures S3C–S3E) showed that, in order for gene activation timing to be finely tunable, nucleosomes must be able to associate with each other even without H3K27me3 modifications, such that methylated and demethylated nucleosomes can associate with each other with comparable affinities. Consistent with this idea, there are multiple mechanisms for nucleosomal interactions that work independently of H3K27me3 modifications (Francis et al., 2004; Gibson et al., 2019; Larson et al., 2017; Sanulli et al., 2019; Strom et al., 2017).
As cell division rate did not affect timing delays in Bcl11b activation (Figure 3), we tested whether the methylation-compaction mechanism also generates cell-division-independent activation delays. Indeed, in contrast to a passive dilution model for H3K27me3 loss (Figure 4F), the methylation-compaction model generated activation delays that were constant over a range of cell cycle speeds (Figures 4G and S5A–S5E). We note that this cell division independence was lost when histone methylation and demethylation rates were reduced to be slower than the cell division rates (Figure S5F), implying a need for rapid histone methylation dynamics for upholding cell cycle independence. Indeed, histone methylation and nucleosome compaction dynamics occur with reported timescales of minutes and seconds (Kristensen et al., 2011; Larson et al., 2017; Sneeringer et al., 2010), respectively, which are far faster than the typical cell division rates which range from hours to days. This explains why the epigenetic state recovers rapidly after DNA replication as observed in our simulations (Figures 4A and 4B, right).
These modeling results suggest that timing control by H3K27me3 has the following characteristics: first, H3K27me3 loss does not directly result in gene activation but instead modulates a separate process that acts as the gatekeeper for gene transcription. Given recent insights into how nucleosomes can interact to form phase-separated structures (Gibson et al., 2019), and how these interactions can be modulated by histone tail modifications and/or binding proteins (Larson et al., 2017; Plys et al., 2019; Strom et al., 2017; Tatavosian et al., 2019), we propose that H3K27me3 loss weakens nucleosomal interactions at the Bcl11b locus, promoting gene locus decompaction and gene activation. Second, the compaction process itself must be partially independent of H3K27me3 such that the compacted nucleosome assembly can be maintained by other chromatin-associated proteins (Francis et al., 2004; Larson et al., 2017; Strome et al., 2014).
The Bcl11b locus switches to an extended conformation with activation
The methylation-compaction mechanism above assumes that the Bcl11b locus is compacted prior to activation but switches to an extended, decompacted state during activation. To validate this assumption, we measured the end-to-end distances between genomic regions at the Bcl11b locus using DNA fluorescence in situ hybridization (FISH), an established approach to estimate the degree of chromatin compaction at a gene locus (Eskeland et al., 2010; Giorgetti et al., 2015). To estimate the degree of compaction independently from RNA polymerase II (RNAPII)-mediated DNA looping at the Bcl11b locus (Hu et al., 2018; Zheng et al., 2019), we designed a pair of FISH probe sets that flank a 100-kb region upstream of the promoter. This upstream region begins at the Bcl11b promoter and resides at the edge of the putative heterochromatin compaction domain that encompasses Bcl11b (Figure 5A). We performed DNA-FISH in early T cell progenitors (DN1) and DN2 progenitors before or after Bcl11b activation (RFP−/YFP− versus RFP+/YFP+) and measured end-to-end distances using three-dimensional imaging.
Figure 5. The Bcl11b locus switches to an extended conformation with activation.
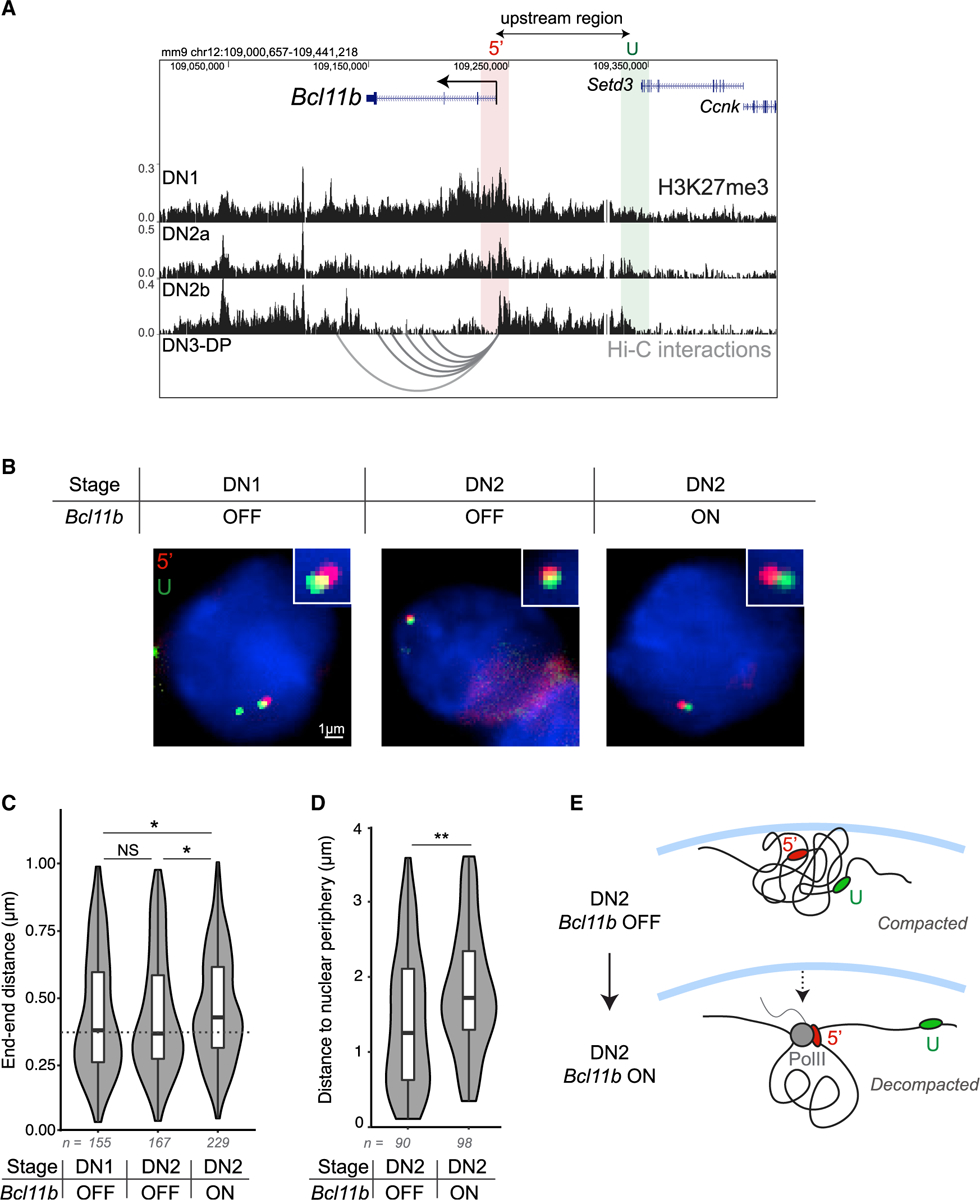
(A) UCSC Genome Browser view of H3K27me3 chromatin immunoprecipitation sequencing (ChIP-seq) results in T cell progenitors (Zhang et al., 2012) and Hi-C representation maps of interactions between the Bcl11b transcription start site and other DNaseI hypersensitivity sites in Bcl11b+ DN3-DP T cell progenitors (Hu et al., 2018).
(B) Representative images for each condition. Centroids for each foci pair shown exist in the same z-plane and thus provide visual representation of the Euclidean distance.
(C) Violin plots show the results from 3D Euclidean distance measurements between each probe pair (Mann Whitney U test significance: *p < 0.05; n = number of foci pairs). T cell progenitors were sorted based on cell surface markers and Bcl11b reporter expression (either Bcl11b OFF [RFP−/YFP−] or Bcl11b ON [RFP+/YFP+]) before performing DNA-FISH with upstream end-end probes above.
(D) Violin plots show the results from 2D measurements between the Bcl11b promoter and the nuclear periphery (Mann Whitney U test significance: **p < 0.01; n = number of foci).
(E) Schematic depicting decompaction model. In the OFF state, Bcl11b exists in a compacted conformation residing at the nuclear lamina. In the ON state, the locus moves away from the nuclear periphery and becomes decompacted, resulting in increased distance between the promoter, 5′, and the upstream, U, regions.
These experiments showed that the Bcl11b locus showed similar end-to-end distances in RFP−/YFP− DN1 and DN2 progenitors; however, in RFP+/YFP+ DN2 progenitors, where Bcl11b first turns on, this end-to-end distance increased significantly, consistent with this locus maintaining a condensed state through early T cell development but switching abruptly to an extended state during Bcl11b activation (Figures 5B and 5C). Repressed heterochromatic regions of the genome frequently reside at the nuclear periphery, where interactions between nuclear lamina proteins and nucleosomes are thought to facilitate chromatin compaction (van Steensel and Belmont, 2017; Ulianov et al., 2019). Therefore, we examined whether Bcl11b moves from the nuclear periphery to the interior as it decompacts and turns on, as suggested from previous studies (Isoda et al., 2017). Indeed, the distance between the Bcl11b promoter and the nuclear periphery was higher in Bc111b-expressing DN2 progenitors compared to Bcl11b-non-expressing progenitors at the same DN2 stage, consistent with a transition from the nuclear periphery to the interior upon gene activation (Figure 5D). This finding, in conjunction with the observed increase in the end-end distance of the Bcl11b locus, suggests that Bcl11b transitions from a condensed heterochromatin-associated state to an accessible euchromatin-associated state during transcriptional activation (Figure 5E), consistent with the methylation-compaction model.
H3K27me3-independent regulation of Bcl11b epigenetic switch timing
The methylation-compaction model predicts that, in order for activation time delays to be tunable, nucleosomes must retain an ability to interact independently of H3K27me3. H3K9me2/3 is also associated with repressive heterochromatin domains and can serve as a binding site for HP1α, which can facilitate nucleosome adhesion in the absence of H3K27me3 (Poleshko et al., 2013; Sanulli et al., 2019; Wang et al., 2019). H3K9me3 is enriched at the Bcl11b locus in non-T-cell lineages (Figure S6A); therefore, we tested whether H3K9 methylation also regulates Bcl11b activation timing. To do so, we sorted DN2 progenitors with one active Bcl11b allele, re-cultured them with inhibitors targeting H3K9me3-modifying enzymes, and quantified the Bcl11b activation state after 3 days. We found that inhibition of Lsd1, an H3K9 demethylase, decreased the fraction of biallelic Bcl11b-expressing cells, whereas inhibition of G9a, an H3K9 methyltransferase, increased the fraction of biallelic Bcl11b-expressing cells (Figures S6B and S6C). The dose-dependent yet moderate degree to which H3K9 methylation perturbations altered Bcl11b activation rate was similar to that for H3K27 methylation perturbations, as seen in Figure 2D. Thus, consistent with the methylation-compaction model, multiple histone modifications work together to modulate Bcl11b activation timing, conceivably by working together to modulate nucleosomal interactions at the gene locus.
Transcription factors can tune gene activation time delays
Bcl11b activation timing is tunable not only by chromatin-modifying enzymes, as shown above (Figure 2), but also by two transcription factors, Gata3 and TCF-1, through a distal enhancer that physically interacts with the Bcl11b promoter before activation (Hu et al., 2018; Isoda et al., 2017; Kueh et al., 2016; Ng et al., 2018). These findings are consistent with broader literature showing that transcription factors and their binding sequences can modulate target gene activation probabilities (Dufourt et al., 2018; Walters et al., 1995; Weintraub, 1988), though it remains unknown how they achieve such tunable timing control. Here, we tested whether the methylation-compaction mechanism could integrate information about transcription factor levels to control activation timing. We first considered a scheme, where transcription factors bind to nucleosomes and block their association with other nucleosomes in the compacted assembly (Figure S7A). Indeed, both Gata3 and TCF-1 have been identified as pioneer factors, which possess affinity to nucleosomes in addition to specific DNA sequences (Fernandez Garcia et al., 2019; Johnson et al., 2018; Meers et al., 2019; Zaret and Carroll, 2011). Such disruption of compaction could also occur via the activation of gene or non-coding RNA transcription (Rinn et al., 2007; Tu et al., 2017) or by recruitment of factors that disrupt interactions between nucleosomes (Kraushaar et al., 2013; Talbert and Henikoff, 2017; Zhou et al., 2016).
Our simulations revealed that the rate constant for gene activation varied with both transcription factor concentration and the number of transcription factor binding sites (Figure S7B). Increasing the number of bound nucleosomes NB increased the activation rate in a synergistic manner, with a progressive increase in the maximal activation rate with addition of each binding site (Figure S7B, top right). Interestingly, increasing the number of binding sites also increased the transcription factor concentration at which half-maximal activation occurred (Figure S7B, bottom right). These findings suggest that transcription factors can tunably control the timing of gene activation by blocking internucleosomal interactions and chromatin compaction.
Besides blocking nucleosomal interactions, transcription factors may also promote activation by inducing histone demethylation. Histone demethylation could occur by direct recruitment of Kdm6a/b demethylases (Estarás et al., 2013; Seenundun et al., 2010; Williams et al., 2014) or through PRC2 eviction by chromatin-remodeling enzymes (Kadoch et al., 2017). Therefore, we considered a second mechanism, where transcription factors induce the demethylation of NR nucleosomes around their binding vicinities (Figure S7C). Our simulations revealed that transcription factors could modulate activation timing, as above, but only if they could induce demethylation of a large number of nucleosomes in their binding vicinity (Figure S7D). When a smaller number of nucleosomes were impacted, activation rates shifted only slightly when transcription factor levels increased (Figure S7D). Consistent with this idea, transcription factors bound to a single binding site can often induce chromatin changes over a significant area spanning many nucleosomes (Hass et al., 2015; Heinz et al., 2010).
DISCUSSION
During development, progenitor cells differentiate, with time delays spanning multiple days and cell generations. These delays are often independent of cell division and are tunably controlled by regulatory inputs. Here, we elucidated the mechanism of a time-delayed epigenetic switch controlling the activation of the T cell specifying gene Bcl11b and developed a mathematical model to explain its emergent properties. We show that H3K27me3 levels at the Bcl11b locus, set by opposing methyltransferase and demethylase activities, modulate the Bcl11b activation time delay by controlling its switch from a compacted, silent state to an extended, actively expressing state. Activation delays generated by this methylation-compaction mechanism robustly span multiple cell generations, can be tunably modulated by both histone-modifying enzymes and transcription factors, and are set independently from cell division.
In contrast to previous epigenetic switching models, which only consider the dynamics of histone modification (Dodd et al., 2007; Zhang et al., 2014), the methylation-compaction model we propose couples histone methylation to nucleosomal interactions. Consistent with this idea, both H3K27me3 and H3K9me2/3 can promote nucleosomal self-association though histone-tail interactions (Gibson et al., 2019) or by recruiting protein complexes that self-associate to form phase-separated condensates (Plys et al., 2018; Sanulli et al., 2019; Tatavosian et al., 2019; Wang et al., 2019). Importantly, in order for the activation times to be tunable, nucleosomes must retain some self-interaction affinity without H3K27me3, such that methylation promotes but is not strictly necessary for nucleosomal association. The concept that modification states of proteins modulate their interaction affinities is well established in the study of cytoskeletal polymers (Howard, 2001; Mitchison, 1992; Phillips et al., 2012) but could provide a fresh perspective on the relationship between chromatin modifications and chromatin structure. Further testing of the methylation-compaction model will require direct interrogation of chromatin states at individual gene loci in single cells, work that will be aided by new methods to simultaneously visualize histone modification states and chromatin folding at single gene loci in single cells (Kundu et al., 2017; Xu et al., 2018; Woodworth et al., 2020).
The methylation-compaction switching mechanism could underlie diverse cell-autonomous timers that have been observed to work independently of cell division (Burton et al., 1999; Gao et al., 1997; Heinzel et al., 2017; Li et al., 2019; Okamoto et al., 2016; Osmond, 1991; Otani et al., 2016). Measuring elapsed time independently of cell division could enable unique functions, including operation in non-dividing cells and constancy amid changes to cell proliferation, which could allow for tunable population size control, an idea we explore in a separate study (Nguyen et al., 2019). Our simulations revealed that such division-independent timing control requires active turnover of H3K27me3 and nucleosome compaction dynamics to be rapid compared to the cell cycle length (Methods S1; Figure S5F). We currently lack methods to measure H3K27me3 turnover kinetics at specific genomic loci in vivo; however, the active roles of PRC2 and Kdm6a/b demethylases in modulating H3K27me3 levels at the Bcl11b locus (Figure 2), their fast catalysis rates (Kristensen et al., 2011; Sneeringer et al., 2010), along with the observation that nucleosomes within polycomb domains are replaced with kinetics much faster than that of cell division (1.5 h versus 20 h; Deal et al., 2010) suggest that H3K27me3 indeed turns over at a much faster timescale than that of cell division.
Cell type specification during multicellular development is controlled by gene-regulatory networks whose dynamics unfold over timescales spanning many cell generations. The division-independent timed epigenetic switch we describe here is uniquely tunable at multiple levels of gene regulation, including histone modifications, transcription factors, and non-coding cis-regulatory elements. Thus, it could serve as a modular building block for gene-regulatory networks that enables robust, adjustable control of developmental timing as well as organism size and form.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hao Yuan Kueh (kueh@uw.edu).
Materials availability
The pMSCV-mTagBFP2-shEed plasmid is available from the Lead Contact upon request.
Data and code availability
The accession number for the H3K27me3 CUT&RUN data reported in this paper is NCBI Gene Expression Omnibus: GSE134749. Scripts used for imaging analysis and simulations of mathematical models have been deposited to GitHub: https://github.com/KuehLabUW/Pease_et_al.2021. All other data supporting the findings of this paper will be available from the Lead Contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Models
C57BL/6 Bcl11bRFP/YFP mice were generated as described before (Ng et al., 2018). Briefly, Bcl11bYFP/YFP mice were generated by inserting an IRES-H2B-mCitrine-neo cassette into the 3′ UTR of Bcl11b and Bcl11bRFP/RFP mice were generated by inserting an IRES-H2B-mCherrry-neo cassette into the same location. Dual allelic Bcl11bRFP/YFP mice with identical Bcl11b alleles except for fluorescent protein reporters were generated by breeding Bcl11bRFP/RFP mice to Bcl11bYFP/YFP mice. Bone marrow derived from F1 Bcl11bRFP/YFP mice at 2–4 months of age were used for all in vitro T cell development assays. Sex was determined not to be influential for these studies, thus male and female bone marrow were combined and analyzed together. All animals were bred and maintained at the University of Washington. All animal protocols were reviewed and approved by the Institute Animal Care and Use Committee at the University of Washington (Protocol No: 4397-01).
Cell Line Culture
Primary cells isolated from bone marrow were cultured on a OP9-DL1 monolayer stromal cells (Holmes and Zuniga-Pflucker, 2009) at 37°C in 5% CO2 conditions with standard culture medium [80% aMEM (GIBCO), 20% Fetal Bovine Serum (Corning), Pen-Strep-Glutamine (GIBCO)] supplemented with appropriate cytokines (described in Method Details). Phoenix-Eco cells were cultured at 37°C in 5% CO2 with standard culture medium [90% DMEM (GIBCO), 10% Fetal Bovine Serum (Corning), Pen-Strep-Glutamine (GIBCO)] All cell lines were tested and found to be negative for mycoplasma contamination.
METHOD DETAILS
Cell purification
To isolate hematopoietic stem and progenitor cells (HSPCs) for in vitro differentiation or CUT&RUN experiments, bone marrow cells were harvested from femurs and tibias of 2 to 4 month-old Bcl11bRFP/YFP mice. CD117 MicroBeads (Miltenyi Biotec) were used to enrich HPSCs which were frozen in 90% FBS and 10% DMSO at 106 cells/mL. For CUT&RUN experiments, HPSCs were further purified by staining with anti-CD117 APC-eFluor780 (ThermoFisher Scientific) and with biotinylated antibodies against a panel of bone marrow lineage markers (CD19, CD11b, CD11c, NK.1.1, Ter119, CD3ε, Gr-1 and B220 (BioLegend)). Cells were then washed with HBH (Hank Balanced Salt Solution (HBSS) with 0.1% bovine serum albumin and 10mM HEPES) and stained with streptavidin-PerCP/Cy5.5 (BioLegend).
In vitro differentiation of T cell progenitors
To generate double-negative (DN) T cells in vitro, thawed CD117-enriched bone marrow progenitors were cultured on OP9-DL1 stromal cell monolayers as described before using standard culture medium [80% αMEM (GIBCO), 20% Fetal Bovine Serum (Corning), Pen-Strep-Glutamine (GIBCO)], grown at 37°C in 5% CO2 conditions]. All in vitro T cell generation cultures were supplemented with 5ng/mL Flt3-L and 5 ng/mL IL-7 (Peprotech), and were sorted after 6 to 8 days of culture before transducing with retroviral vectors or treating with small molecule inhibitors. DN2 cells were re-cultured in the same conditions following all cell sorting experiments.
Flow cytometry and cell sorting
Fluorescence activated cell sorting (FACS) was used to isolate DN2 cells of interest with the following protocol. Bone marrow derived cell cultures were scraped and incubated in 2.4G2 Fc blocking solution and stained with anti-CD25 APC-eFluor 780 (Clone PC61.5, eBioscience) and with biotinylated antibodies against a panel of lineage markers (CD19, CD11b, CD11c, NK.1.1, Ter119, CD3ε, Gr-1 and B220 (BioLegend)). Stained cells were washed with HBH (Hank Balanced Salt Solution (HBSS) with 0.1% bovine serum albumin (BSA) and 10mM HEPES and stained with streptavidin-PerCP/Cy5.5 (BioLegend). Stained cells were washed, resuspended in HBH, and filtered through a 40-um nylon mesh for sorting with a BD FACS Aria III (BD Biosciences) with assistance from the University of Washington Pathology Flow Cytometry Core Facility. A benchtop MacsQuant VYB flow cytometer (Miltenyi Biotec) and a benchtop Attune NxT Flow Cytometer (ThermoFisher Scientific) were used to analyze time course and perturbation experiments and acquired data were analyzed with FlowJo software (Tree Star).
Retroviral construct and transduction
Overexpression of c-Myc was achieved using cMyc H2B-mCerulean MSCV retroviral vector which was described previously (Kueh et al., 2016). Retroviral mir30-based constructs (a gift from J. Zuber) were used as a backbone for delivering short hairpin RNA (Fellmann et al., 2013). pBAD-mTagBFP2 (a gift from V. Verkhusha, Addgene plasmid #34632) was used to substitute mTagBFP2 for the existing GFP using PCR cloning with the restriction enzymes NcoI and SalI. The pMSCV-mTagBFP2-shEed retroviral construct was generated by PCR cloning as previously described (Fellmann et al., 2013) using the mir-30-shEed PCR template sequence and pMSCV-mir-30 backbone described in the Key resources table.
Retroviral particles were generated using the Phoenix-Eco packaging cell line. Viral supernatants were collected at 2 and 3 days after transfection and immediately frozen at −80°C. To infect bone marrow derived T cell progenitors, 33 μg/mL retronectin (Clontech) and 2.67 μg/mL of DL1-extracellular domain fused to human IgG1 Fc protein (a gift from I. Bernstein) were added in a volume of 250 μL per well in 24-well tissue culture plates (Costar, Corning) and incubated overnight. Viral supernatants were added the next day into coated wells and centrifuged at 2000 rcf. for 2 hours at 32°C. Bone marrow derived derived T cell progenitors used for viral transduction were cultured for 6–7 days according to conditions described above, disaggregated, filtered through a 40-μm nylon mesh, and 106 cells were transferred onto each retronectin/DL1-coated virus-bound well supplemented with 5 ng/mL SCF (Peprotech), 5 ng/mL Flt3-L, and 5 ng/mL IL-7.
CUT&RUN H3K27me3 profiling
CUT&RUN experiments were carried out as previously described (Skene et al., 2018) with the following modifications: 1–2.5×105 cells were isolated by FACS as described in sections above, bound to Concanavalin A coated magnetic beads (Bangs Laboratories), and permeabilized with 0.025% (wt/vol) digitonin. Permeabilized cells were incubated overnight at 4°C with 5ug of anti-H3K27me3 (Active Motif) and then washed before incubating with protein A-MNase fusion protein (a gift from S. Henikoff) for 15 minutes at room temperature. After washing, cells were incubated in CaCl2 to induce MNase cleavage activity for 30 minutes at 0°C. The reaction was stopped with 2xSTOP buffer (200 mM NaCl, 20 mM EDTA, 4 mM EGTA, 50 μg/mL RNase A, 50 μg/mL glycogen, and 2pg/mL of yeast spike-in DNA). Histone-DNA complexes were isolated from insoluble nuclear chromatin by centrifugation and DNA was extracted with a NucleoSpin PCR Clean-up kit (Macherey-Nagel). For CUT&RUN quantitative PCR, human Kasumi-1 cell line (ATCC CRL-2724) were added before binding the cells to Concanavalin A beads for internal standard instead of yeast spike-in DNA.
CUT&RUN library preparation and sequencing
Library preparation from CUT&RUN products was completed with KAPA Hyper Prep Kit (KAPA Biosystems) following standard protocol with PCR amplification settings adjusted so that annealing and extension steps are combined into one step at 60°C for 10 s. Library products were size selected to be within 200 – 300 bp range using AMPure beads (Agencourt). Libraries were sequenced using an Illumina MiSeq system with paired-end 25 bp sequencing read length and TruSeq primer standard for approximately 5 millions reads per sample.
CUT&RUN sequencing analysis
Paired-end sequencing reads were aligned separately to mouse (NCBI37/mm9) and yeast (SacCer_Apr2011/sacCer3) genomes using Bowtie2 (Langmead and Salzberg, 2012) with the following setting:–local–very-sensitive-local–no-unal–no-mixed–no-discordant -I 10 -X 700 as suggested for mapping CUT&RUN sequencing data (Skene et al., 2018). The alignment setting was designed to specifically search with high stringency for only appropriately paired reads with the proper orientation. The resulting alignments were converted to BAM files with SAMtools (Li et al., 2009) and then converted to BED files with BEDTools (Quinlan and Hall, 2010). Reads were sorted and filtered to remove random chromosomes. BEDTools genomecov was used to generate histograms for the mapped reads using a scaling factor that is the product of the number of spiked-in yeast reads and the number of input cells. The resulting bedGraph files were visualized using the UCSC Genome Browser (Davis et al., 2018; Kent et al., 2002).
CUT&RUN qPCR
Extracted DNA from CUT&RUN samples was size selected with Ampure XP magnetic beads (Beckman Coulter) to remove fragments > 800bp. Primers were designed to detect the mouse Bcl11b promoter (see Key resources table for sequences). PowerUp SYBR Green Master Mix (ThermoFisher Scientific) and CFX96 Real-Time PCR Detection System (Bio-Rad) were used for quantitative PCR. Since Kasumi-1 cells were used as internal standard, relative enrichment of H3K27me3 at Bcl11b was quantified by the ΔΔCq method using the human PAX5 promoter for normalization to account for differences in efficiency and sample loss during processing.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse Ter119 Biotin (clone TER-119) | eBioscience | Cat#13-5921-85; RRID:AB_466798 |
| Anti-mouse NK1.1 Biotin (clone PK136) | eBioscience | Cat#13-5941-85; RRID:AB_466805 |
| Anti-mouse Gr-1 Biotin (clone RB6–8C5) | eBioscience | Cat#13-5931-86; RRID:AB_466802 |
| Anti-mouse CD11c Biotin (clone N418) | eBioscience | Cat#13-0114-85; RRID:AB_466364 |
| Anti-mouse CD11b Biotin (clone M1/70) | eBioscience | Cat#13-0112-86; RRID:AB_466361 |
| Anti-mouse CD19 Biotin (clone 1D3/6D5) | eBioscience | Cat#13-0193-85; RRID:AB_657658 |
| Anti-mouse CD3e Biotin (clone 145-2 C11) | eBioscience | Cat#13-0031-85; RRID:AB_466320 |
| Anti-human/mouse B220 Biotin (clone RA3-6B2) | eBioscience | Cat#13-0452-85; RRID:AB_466450 |
| Anti-human/mouse CD44 eFluor 450 (clone IM7) | eBioscience | Cat#48-0441-82; RRID:AB_1272246 |
| Anti-mouse CD25 APC-eFluor 780 (clone PC61.5) | eBioscience | Cat#47-0251-82; RRID:AB_1272179 |
| Streptavidin PerCP-Cyanine5.5 | Biolegend | Cat#405214; RRID:AB_2716577 |
| APC Annexin V | Biolegend | Cat#640919 |
| Bacterial and virus strains | ||
| Stable Competent E.coli | NEB | Cat#C3040 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human Flt3-Ligand | PreproTech | Cat#300–19 |
| Recombinant Human IL-7 | PreproTech | Cat#200–07 |
| Recombinant Human Stem Cell Factor (SCF) | PreproTech | Cat#300–07 |
| Anti-H3K27me3 antibody | Active Motif | Cat#39156 |
| Retronectin | Takara | Cat#T100B |
| DL1-ext IgG Protein | Gift from Irwin Bernstein (Varnum-Finney et al., 2000) | N/A |
| FuGENE 6 Transfection Reagent | Promega | Cat#E2691 |
| UNC1999 | Caymen Chemical | Cat#14621 |
| GSKJ4 | Caymen Chemical | Cat#12073 |
| IOX1 | Caymen Chemical | Cat#11572 |
| GSK126 | Caymen Chemical | Cat#15415 |
| GSK343 | Caymen Chemical | Cat#14094 |
| Concavalin A | Bangs Laboraatories | Cat#BP531 |
| protein A-MNase | gift from Steven Henikoff (Skene et al., 2018) | N/A |
| Deposited data | ||
| T cell progenitor H3K27me3 CUT&RUN | This paper | Gene Expression Omnibus GSE134749 |
| Thymocyte H3K27ac ChIP-sequencing | Davis et al., 2018 | ENCODE accession ENCSR000CCH |
| Erythroblast H3K9me3 ChIP-sequencing | Davis et al., 2018 | ENCODE accession ENCSR000DHN |
| T cell progenitor H3K27me3 ChIP-sequencing | Zhang et al., 2012 | Gene Expression Omnibus GSE31235 |
| DN3-DP thymocyte Hi-C | Hu etal., 2018 | Gene Expression Omnibus GSE79875 |
| Experimental models: cell lines | ||
| OP9-DL1-GFP | Holmes and Zúñiga-Pflücker, 2009 | N/A |
| Kasumi-1 | ATCC | Cat#CRL-2724; RRID:CVCL_0589 |
| Pheonix-Eco | ATCC | Cat#CRL-3214, RRID:CVCL_H717 |
| Experimental models: organisms/strains | ||
| Mouse: Bcl11bRFP/YFP | Ng et al., 2018 | N/A |
| Oligonucleotides | ||
|
Bcl11b forward primer: TCCACCTACCAGACCCCGAA |
IDT | N/A |
|
Bcl11b reverse primer: CTTCTTCAAAGTGCTTGGCCTC |
IDT | N/A |
|
PAX5 forward primer: CCAGGATGTGCTGCTGTCCCAG |
IDT | N/A |
|
PAX5 reverse primer: CTCCCTGGTGCTGTGCACTGA |
IDT | N/A |
| mir-30-shEed template: TGCTGTTGACAGTGAGCGAAGGCATTATAAGAATAATTAATAGTGAAGCCACAGATGTATTAATTATTCTTATAATGCCTCTGCCTACTGCCTCGGA |
IDT | N/A |
| Recombinant DNA | ||
| pBAD-mTagBFP2 | Addgene | Cat #34632 |
| pMSCV-miR-30 | Gift from Johannes Zuber (Fellmann et al., 2013) | N/A |
| pMSCV-c-Myc-H2B-mCerulean | Kueh et al., 2016 | N/A |
| pBanshee-CFP | Kueh et al., 2016 | N/A |
| pCL-Eco | Imgenex | Cat#NBP2-29540 |
| Software and algorithms | ||
| Software: FlowJo (v10.0.8) | Tree Star | https://www.flowjo.com/ |
| Software: MATLAB (R2018b) | MathWorks | https://www.mathworks.com/products/matlab.html |
| Software: R (v3.9) | N/A | https://www.r-project.org/ |
| Rstudio (v1.2.5042) | N/A | https://rstudio.com/ |
| Ggplot2 (v.3.3) | N/A | https://ggplot2.tidyverse.org/ |
| Bedtools (v2.17.0) | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| Samtools (v0.1.19-96b5f2294a) | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Fiji (v2.0.0) | Schindelin et al., 2012 | https://imagej.net/Downloads |
| BUnwarpJ | Arganda-Carreras et al., 2006 | https://imagej.net/BUnwarpJ |
| Python(v3.6) | N/A | https://www.python.org |
| Other | ||
| BD FACS Aria III Cell Sorter | BD Biosciences | N/A |
| NucleoSpin PCR Clean-up | Macherey-Nagel | Cat#740609.50 |
| Illumina MiSeq | Illumina | N/A |
| Attune NxT Acoustic Focusing Cytometer | ThermoFisher Scientific | N/A |
| KAPA HyperPrep Kit | Roche | Cat#07962312001 |
| PowerUp SYBR Green Master Mix | ThermoFisher Scientific | Cat#A25741 |
| Ampure XP magnetic beads | Beckman Coulter | Cat#A63880 |
| CFX96 Real-Time PCR Detection System | Bio-Rad | Cat#1855196 |
| CD117 Microbeads | Miltenyi Biotec | Cat#130-091-224 |
| LS Columns | Miltenyi Biotec | Cat#130-042-401 |
| 250mm-diameter PDMS circular micromesh arrays | Microsurfaces Pty Ltd | Cat#MMA-0250-100-08-01 |
| Custom scripts for imaging analysis and simulations | This paper | https://github.com/KuehLabUW/Pease_et_al.2021 |
Cell preparation for time-lapse imaging
T cell progenitors derived from the in vitro differentiation protocol above were harvested and infected with either a MSCV empty vector or c-Myc overexpression vector harboring an IRES-H2B-mCerulean reporter cassette. 16–24 hours later CFP-positive cells were purified by FACS and seeded onto PDMS micromesh (250 μm hole diameter, Microsurfaces) mounted on top of a 24-well glass bottom plate (Mattek). To prepare the stromal-free differentiation system, which facilitates cell identification during imaging, the top face of PDMS micromesh was first blocked by incubating in solution of 130 μg/ml BSA while mounted on top of a 24-well plate overnight at 4°C. This step prevents subsequent binding of retronectin to the side of the micromesh walls. Blocked micromeshes was then transferred to a clean 24-well glass bottom plate. The well and mesh constructs were incubated in a solution of 10 μg/ml retronection and 3 μg/ml DL-1 overnight at 4°C. The well was then washed with PBS, and culture media [80% αMEM (GIBCO), 20% Fetal Bovine Serum (Corning), Pen-Strep-Glutamine (GIBCO), 5 ng/ml IL-7 (Clontech), 5 ng/ml Flt-3 (Clontech), 50 ng/ml mSCF (Clontech), 50 μM beta-mercaptoethanol (Sigma) grown at 37°C in 5% CO2 conditions] was added, and sorted cells were introduced at a concentration of 5–10 cells per microwell. This stromal-free system enables a greater fold enhancement of cell division rate by cMyc transduction and better resolution for imaging as well as recapitulating Bcl11b activation and T cell lineage commitment, but supports a lower baseline rate of proliferation in unmodified cells compared to the OP9-DL1 system.
Oligopaint DNA-FISH
The OligoMiner pipeline was used to design Oligopaint libraries (Beliveau et al., 2018). 35–52bp probes were designed to target 20kb regions at a density of approximately 12–14 probes per kilobase. Bone marrow progenitors were grown on OP9-DL1 stromal cells for 8 days under normal growth conditions. Cells were filtered through a 70uM filter and incubated with 2.4G2 blocking buffer before staining with anti-CD25 APC-eFluor 780 (Clone PC61.5, eBioscience), anti-CD44 APC (Clone IM7, eBioscience), and biotinylated antibodies against a panel of lineage markers (CD19, CD11b, CD11c, NK.1.1, Ter119, CD3ε, Gr-1 and B220 (BioLegend)). DN1 progenitors (CD25−/CD44+/Bcl11bRFP−/YFP−), DN2a progenitors (CD25+/CD44+/Bcl11bRFP−/YFP−) and DN2b progenitors (CD25+/CD44+/Bcl11bRFP+/YFP+) were purified by FACS and centrifuged on top of poly-L-lysine coated 18-well chambered glass coverslips (Ibidi). Cells were then fixed with 4% paraformaldehyde for 10 minutes and permeabilized for 10 minutes in 0.1% Triton X-100 before performing the Oligopaint DNA-FISH protocol (Beliveau et al., 2017). Permeabilized cells were incubated in 0.1N HCl for 5 minutes, followed by RNaseA (100ug/ml) for 1 hour at 37°C. Cells were then washed with 2x SSCT (2x saline sodium citrate + 0.1% Tween-20) and incubated with 2x SSCT + 50% formamide for 20 minutes at 60°C. A hybridization mixture was prepared containing 50% formamide, 2x SSCT, 3mM sodium azide, 10% dextran sulfate, 100nM of adaptor oligos, 100nM of fluorescently labeled reporter oligos, and 500nM of probes. The hybridization mixture was added and incubated for 3 minutes at 78°C before incubating overnight in a humidifier chamber at 37°C. Approximately 18 hours later, cells were washed with pre-heated 2x SSCT for 5 minutes at 60°C. This step was repeated four times before performing the final wash at room temperature. Cells were then stained with 10ug/mL Hoechst 33342 (ThermoFisher) for 15 minutes before washing with PBS and imaging in with a photoprotective buffer (10% glucose, 200mM Tris, glucose oxidase (GLOX), catalase, 1mM methyl viologen hydrate, 1mM ascorbic acid).
FISH imaging and analysis
Cells were imaged with an inverted widefield fluorescence microscope (Leica DMi8) using a 100X oil objective, using an sCMOS camera (Photometrics Prime 95B) and a motorized stage (ASI MS-2000). Z sections were collected at a step size of 100nm. Chromatic aberrations were corrected for using Fiji (Schindelin et al., 2012) and BUnwarpJ (Arganda-Carreras et al., 2006) as described previously (Giorgetti et al., 2015). After nuclei segmentation, the z slice with the maximum intensity for each foci was chosen as the z coordinate. Each selected z slice was then fit to a two-dimensional Gaussian to determine the xy coordinates for the centroid of the foci. Euclidean distances between each pair of foci and between each labeled promoter focus and the nearest nuclear edge were calculated.
QUANTIFICATION AND STATISTICAL ANALYSIS
The following statistical tests were used in this study: two-sample, one-tailed t test (Figures 2E, 2F, 3B, 3C, and S1D); and the Mann Whitney U-test (Figures 5C and 5D). Details for statistical tests performed are described in the indicated figure legends. All statistical tests were performed using MATLAB or R.
Modeling simulations
All models were simulated using the Gillespie algorithm provided in the Tellurium package in Python 2.7 (Choi et al., 2018). Plotting of simulation results was done in MATLAB. A detailed description of the models can be found in the mathematical appendix (see Methods S1: Mathematical Appendix, related to STAR Methods).
Image analysis of time-lapse movies
Image segmentation
Cell segmentation was performed in MATLAB (Mathworks, Natick, MA) using custom scripts described previously (Kueh et al., 2016; Ng et al., 2018). The segmentation algorithm was performed on CFP fluorescent signals as all transduced cells carried an H2B-mCerulean CFP reporter cassette. Briefly, images underwent (1) correction by subtraction of uneven background signal stemming from the bottom of the glass plate or the side of the PDMS microwells (2) Gaussian blur followed by pixel value saturation to fix uneven signal intensity within the nucleus of the cell and (3) Laplacian edge detection algorithm to identify the nucleus boundary. Non-cell objects were excluded via size and shape limit exclusions, and segmentation parameters were chosen such that the number of non-cell objects are < 1% of the total segmented cells.
Identification of live and dead cell population
In movies of cMyc or empty vector (EV) transduced cells, live and dead cells possessed distinct morphological features as observed in the CFP fluorescence channel. Live cell nuclei had a round, smooth oval shape while dead cell nuclei tended to be more granular, with small but very bright puncta. To provide unbiased, automated recognition of live and dead cells based on these features, we applied a Laplacian mask filter to each segmented cell to delineate the ‘smoothness’ of its signal, then applied a threshold-cutoff to identify regions with high CFP signal. The resultant list of object features were recorded for each cell object: 1) nuclei area, 2) perimeter, 3) fluorescent intensity, 4) puncta number, 5) mean puncta area, 6) mean puncta perimeter, and 7) area for puncta above the cutoff threshold. Approximately one hundred individual cell images (10% of each dataset) were then manually annotated as ‘live’ or ‘dead’. Annotations were then linked to the above feature matrix, and a decision tree supervised machine learning algorithm was then used to generate a model based on the annotated live/dead classification and matrix features of the training images (Figures S2A–S2C). Finally, built-in MATLAB model evaluation functions resubLoss and crossval were used to validate that mis-assignment error is below 15% for all datasets. This approach was utilized to provide an objective, automated method to distinguish between live and dead populations.
Bcl11b activation rate fitting
The following procedure was used to quantify Bcl11b activation rate from timelapse movies: first, the YFP and RFP signal intensity of segmented cells were calculated. Next, each cell object was classified as ‘live’ or ‘dead’, using classification prediction by trained model described in the previous section. Cells classified as ‘live’ were selected, and their YFP RFP fluorescence 2D histograms were then fitted to a two-component mixed 2D Gaussian model to obtain the fraction of YFP-OFF and YFP-ON cells in the population at a given time. To calculate background-corrected fluorescent values of the Bcl11b YFP and Bcl11b RFP signals, we calculated the pixel intensity of an annulus surrounding the segmented cell and subtracted this value from the raw signal intensity in the cell interior. This approach eliminates autofluorescence from the bottom of the glass plate as well as at the edge of the PDMS microwell.
To obtain the time evolution of Bcl11b biallelic population fractions from initial Bcl11b YFP-RFP+ population, cells were first filtered based on their ‘live/dead’ category, and only ‘live’ cells were included in subsequent calculations. We used a modified version of least-squares fit of a two-component mixed 2D Gaussian function described by Ng et al. (2018) to fit the 2D histogram of Bcl11b YFP and Bcl11b RFP fluorescence levels. Specifically, let y and r be the intensity of Bcl11b YFP and Bcl11b RFP fluorescence, respectively, the overall fit, F(r,y), is given by:
| (Equation 1) |
Each 2D Gaussian f is given by:
| (Equation 2) |
Here, i = 1,2 correspond to the red mono-allelic and biallelic populations, since all starting cells are red mono-allelic, we excluded the other two populations (non-expressing and yellow mono-allelic). Ni is the volume under the Gaussian curve when integrated over r and y and is the approximation for the number of cells in each population in Bcl11b RFP mono-allelic and biallelic states.
To fit our data to F(r,y), we followed a two-step process described previously (Ng et al., 2018): (1) We fitted Bcl11b YFP/RFP 2D histogram at an early time point (0 < t < 20) to f1(r,y)to obtain the means, standard deviations, and correlation coefficients (ur,1,σr,1,uy,1,σy,1,p1)) of the Bcl11b RFP mono-allelic population. At this early time point, cells remained inactive for the Bcl11b YFP allele. (2) Next, we fitted the 2D histograms of Bcl11b YFP/RFP levels at successive time bins of 20 hours, fixing the parameter of the first Gaussian f1(r,y), and enabling the parameters for the second Gaussian f2(r,y), to vary within bounds observed in the fluorescent distributions of Bcl11b biallelic populations. After fitting, the fraction of biallelic cell at a given time window centered on time t is given by:
| (Equation 3) |
The confident bounds for is given by:
| (Equation 4) |
Afterward, the resulting fraction of biallelic cells as a function of time window centered at time t from the mixed Gaussian fit was then fitted to the probability density function of a first order process:
| (Equation 5) |
Where λ is the rate for activation of the initially silent Bcl11b-YFP allele. We chose this function for activation rate fitting since our histone dynamics simulations suggested that Bcl11b activation can be estimated as a first order stochastic process (see Figure 4D). For this function, fitting was done using the MATLAB fit function and a 95% confidence interval for the fit was recorded.
Population dynamics model and fitting
We built a mathematical model to describe the population dynamics of progenitor cells transfected with an empty vector (EV) and c-Myc. From initial inspection of time-lapse movies (Figure 3), progenitors transduced with c-Myc appear to expand more quickly than control progenitors, as expected. Faster expansion of c-Myc-transduced cells could be due to faster cell cycling or slower cell death. To disentangle these two effects, we quantified numbers of both live and dead cells over time (Figure 3B) and fit these data to population dynamics models to obtain division and death rates:
The model includes a population of live cells (X) with a division rate kb and a death rate kd to generate the observable dead cell population (Y). This population in turn has a clearance rate δ representing the process by which CFP level degrades and dead cells become undetectable.
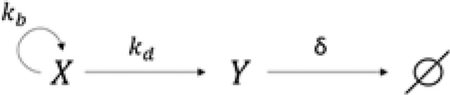
We then obtained the division rate kb and the death rate kd through a two-step curve fitting process. First the difference between these two ratesK = kb − kdwas obtained by fitting the number of live cells over time::
| (Equation 6) |
where Xo is the initial number of live cells at the start of imaging. The death rate was then obtained by fitting the number of observable dead cells over time, which, given the above transition scheme, is given by the following: :
| (Equation 7) |
Here Yois initial observed number of dead cells at the start of imaging and P(τ)gives the probability of the dead cell remaining observable in the CFP fluorescent channel a period of time τ after its first appearance. In this model, whenever a cell starts to die, its probability of being detected decreases as per function P(τ), and the number of dead cells at a given time T is the sum of all the still-detectable dead cells generated since the start of imaging up until T. This decrease in detection probability arises because progressive dimming of CFP fluorescence, together with morphological changes following death results in the failure of the cells to be segmented.
We determined P(τ) empirically for EV and cMyc population separately by manually following 30 different dead cells and recording the time period in which it was detected and undetected until complete disappearance. We then calculated the fraction of dead cells that remained detectable after a given time had elapsed. An exponential function decay function was used to fit this ‘fraction detected’ curve and to estimate value for clearance rate (Figures S2B and S2C):
| (Equation 8) |
Here, P(τ) is the probability of a given dead cell to be detected under the CFP fluorescent channel after a period of time since its initial death. δ is the clearance rate of this process.
To fit imaging data to Equation 6, we classified segmented cell objects as live or dead using a trained machine learning model as described in ‘Image analysis of time-lapse movies’ section. Number of live cells as a function of time was fitted to Equation 6 using a MATLAB fit function and 95% confidence interval for the fit was recorded.
To fit imaging data to Equation 7, we tested a series of candidate kd,ivalues; for each kd,i, a predicted Yp,i(t)curve was generated based on Equation 7 where T = t1,t2,t3,…with ti being the time point at which experimental measurement took place. Yp,i(T)is then compared to the experimentally observed dead cell number Yexp(T)using sum square error method:
| (Equation 9) |
The best fit kdvalue is chosen to be the value kd,i, whose sseiis the smallest.
In order to calculate the confidence bound of the fit, we first performed a nonlinear regression by calculating the residuals of the model’s predicted values Yp(ti):
| (Equation 10) |
We then calculated to the Jacobian of the model function to estimate the covariance at each time point, given by:
| (Equation 11) |
These inputs were used to estimate 95% confidence interval using MATLAB ‘Nonlinear regression parameter confidence intervals’ function nlparci.
A summary of results from data fitting are tabulated in Table S1.
Supplementary Material
Highlights.
Timed epigenetic switch delays Bcl11b activation over multiple cell cycles
H3K27me3 levels, set by PRC2 and Kdm6a/b, tunably set this activation delay
This activation time delay is set independently from cell cycle length
Quantitative methylation-compaction model explains tunable, division-independent delays
ACKNOWLEDGMENTS
We thank members of the Kueh lab for discussions and also thank Steve Henikoff, James Briscoe, Ellen Rothenberg, Michael Elowitz, and Joe Levine for discussions and feedback on this work. We also thank Steve Henikoff for the generous gift of pA-MNase for CUT&RUN experiments. This work was funded by a Pathway to Independence Award 5R00HL119638 (to H.Y.K.) from the National Institutes of Health from the United States; grant R01HL146478 (to H.Y.K.) from the National Heart, Lung, and Blood Institute of the United States; URoL EF-2021552 (to H.Y.K.) from the National Science Foundation of the United States; a Stem Cell Scientist Award (to H.Y.K.) from the Tietze Foundation of the United States; a fellowship (to N.A.P.) from the University of Washington Institute of Stem Cell and Regenerative Medicine of the United States; and F31 Fellowships F31 HL151090 (to N.A.P.) and F31 HL142132 (to M.A.W.) from the National Heart, Lung, and Blood Institute of the United States.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108888.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following references appear in the supplemental information: Agger et al. (2007); Alabert et al. (2015); Angel et al. (2011); Arbona et al. (2017); Erickson and Pantaloni (1981); Gray et al. (2016); Isono et al. (2013); Jackson and Berkowitz (1980); Kahn et al. (2016); Kramers (1940); Ladoux et al. (2000); MacPherson et al. (2018); Nuebler et al. (2018).
REFERENCES
- Agger K, Cloos PAC, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, and Helin K (2007). UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449, 731–734. [DOI] [PubMed] [Google Scholar]
- Alabert C, Barth TK, Reverón-Gómez N, Sidoli S, Schmidt A, Jensen ON, Imhof A, and Groth A (2015). Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev. 29, 585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amodeo AA, Jukam D, Straight AF, and Skotheim JM (2015). Histone titration against the genome sets the DNA-to-cytoplasm threshold for the Xenopus midblastula transition. Proc. Natl. Acad. Sci. USA 112, E1086–E1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angel A, Song J, Dean C, and Howard M (2011). A polycomb-based switch underlying quantitative epigenetic memory. Nature 476, 105–108. [DOI] [PubMed] [Google Scholar]
- Arbona J-M, Herbert S, Fabre E, and Zimmer C (2017). Inferring the physical properties of yeast chromatin through Bayesian analysis of whole nucleus simulations. Genome Biol. 18, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arganda-Carreras I, Sorzano COS, Marabini R, Carazo JM, Ortizde-Solorzano C, and Kybic J (2006). Consistent and elastic registration of histological sections using vector-spline regularization. In Computer Vision Approaches to Medical Image Analysis, Beichel RR and Sonka M, eds. (Springer; Berlin Heidelberg: ), pp. 85–95. [Google Scholar]
- Beliveau BJ, Boettiger AN, Nir G, Bintu B, Yin P, Zhuang X, and Wu C-T (2017). In situ super-resolution imaging of genomic DNA with Oligo-STORM and OligoDNA-PAINT. Methods Mol. Biol 1663, 231–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliveau BJ, Kishi JY, Nir G, Sasaki HM, Saka SK, Nguyen SC, Wu CT, and Yin P (2018). OligoMiner provides a rapid, flexible environment for the design of genome-scale oligonucleotide in situ hybridization probes. Proc. Natl. Acad. Sci. USA 115, E2183–E2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry S, Dean C, and Howard M (2017). Slow chromatin dynamics allow polycomb target genes to filter fluctuations in transcription factor activity. Cell Syst. 4, 445–457.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bintu L, Yong J, Antebi YE, McCue K, Kazuki Y, Uno N, Oshimura M, and Elowitz MB (2016). Dynamics of epigenetic regulation at the single-cell level. Science 351, 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. (2006). Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353. [DOI] [PubMed] [Google Scholar]
- Burton PBJ, Raff MC, Kerr P, Yacoub MH, and Barton PJR (1999). An intrinsic timer that controls cell-cycle withdrawal in cultured cardiac myocytes. Dev. Biol 216, 659–670. [DOI] [PubMed] [Google Scholar]
- Choi K, Medley JK, König M, Stocking K, Smith L, Gu S, and Sauro HM (2018). Tellurium: an extensible python-based modeling environment for systems and synthetic biology. Biosystems 171, 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RT, and Struhl G (2017). Causal role for inheritance of H3K27me3 in maintaining the OFF state of a Drosophila HOX gene. Science 356, eaai8236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, Hilton JA, Jain K, Baymuradov UK, Narayanan AK, et al. (2018). The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 46 (D1), D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal RB, Henikoff JG, and Henikoff S (2010). Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 328, 1161–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd IB, Micheelsen MA, Sneppen K, and Thon G (2007). Theoretical analysis of epigenetic cell memory by nucleosome modification. Cell 129, 813–822. [DOI] [PubMed] [Google Scholar]
- Dufourt J, Trullo A, Hunter J, Fernandez C, Lazaro J, Dejean M, Morales L, Nait-Amer S, Schulz KN, Harrison MM, et al. (2018). Temporal control of gene expression by the pioneer factor Zelda through transient interactions in hubs. Nat. Commun 9, 5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson HP, and Pantaloni D (1981). The role of subunit entropy in cooperative assembly. Nucleation of microtubules and other two-dimensional polymers. Biophys. J 34, 293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskeland R, Leeb M, Grimes GR, Kress C, Boyle S, Sproul D, Gilbert N, Fan Y, Skoultchi AI, Wutz A, and Bickmore WA (2010). Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol. Cell 38, 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estarás C, Fueyo R, Akizu N, Beltrán S, and Martínez-Balbás MA (2013). RNA polymerase II progression through H3K27me3-enriched gene bodies requires JMJD3 histone demethylase. Mol. Biol. Cell 24, 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai DY, Barbosa IAM, Kwon JS, Guan Y, et al. (2013). An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 5, 1704–1713. [DOI] [PubMed] [Google Scholar]
- Fernandez Garcia M, Moore CD, Schulz KN, Alberto O, Donague G, Harrison MM, Zhu H, and Zaret KS (2019). Structural features of transcription factors associating with nucleosome binding. Mol. Cell 75, 921–932.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis NJ, Kingston RE, and Woodcock CL (2004). Chromatin compaction by a polycomb group protein complex. Science 306, 1574–1577. [DOI] [PubMed] [Google Scholar]
- Gao F-B, Durand B, and Raff M (1997). Oligodendrocyte precursor cells count time but not cell divisions before differentiation. Curr. Biol 7, 152–155. [DOI] [PubMed] [Google Scholar]
- García-Ojeda ME, Klein Wolterink RGJ, Lemaître F, Richard-Le Goff O, Hasan M, Hendriks RW, Cumano A, and Di Santo JP (2013). GATA-3 promotes T-cell specification by repressing B-cell potential in pro-T cells in mice. Blood 121, 1749–1759. [DOI] [PubMed] [Google Scholar]
- Gaydos LJ, Wang W, and Strome S (2014). Gene repression. H3K27me and PRC2 transmit a memory of repression across generations and during development. Science 345, 1515–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germar K, Dose M, Konstantinou T, Zhang J, Wang H, Lobry C, Arnett KL, Blacklow SC, Aifantis I, Aster JC, and Gounari F (2011). T-cell factor 1 is a gatekeeper for T-cell specification in response to Notch signaling. Proc. Natl. Acad. Sci. USA 108, 20060–20065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Doolittle LK, Schneider MWG, Jensen LE, Gamarra N, Henry L, Gerlich DW, Redding S, and Rosen MK (2019). Organization of chromatin by intrinsic and regulated phase separation. Cell 179, 470–484.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgetti L, Piolot T, and Heard E (2015). High-resolution 3D DNA FISH using plasmid probes and computational correction of optical aberrations to study chromatin structure at the sub-megabase scale. In Nuclear Bodies and Noncoding RNAs, Nakagawa S and Hirose T, eds. (Springer; New York: ), pp. 37–53. [DOI] [PubMed] [Google Scholar]
- Gray F, Cho HJ, Shukla S, He S, Harris A, Boytsov B, Jaremko Ł, Jaremko M, Demeler B, Lawlor ER, et al. (2016). BMI1 regulates PRC1 architecture and activity through homo- and hetero-oligomerization. Nat. Commun 7, 13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hass MR, Liow H-H, Chen X, Sharma A, Inoue YU, Inoue T, Reeb A, Martens A, Fulbright M, Raju S, et al. (2015). SpDamID: marking DNA bound by protein complexes identifies Notch-dimer responsive enhancers. Mol. Cell 59, 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel S, Binh Giang T, Kan A, Marchingo JM, Lye BK, Corcoran LM, and Hodgkin PD (2017). A Myc-dependent division timer complements a cell-death timer to regulate T cell and B cell responses. Nat. Immunol 18, 96–103. [DOI] [PubMed] [Google Scholar]
- Holmes R, and Zuniga-Pflucker JC (2009). The OP9-DL1 system: generation of T-lymphocytes from embryonic or hematopoietic stem cells in vitro. Cold Spring Harb. Protoc 2009, pdb.prot5156. [DOI] [PubMed] [Google Scholar]
- Hosogane M, Funayama R, Shirota M, and Nakayama K (2016). Lack of transcription triggers H3K27me3 accumulation in the gene body. Cell Rep. 16, 696–706. [DOI] [PubMed] [Google Scholar]
- Hosokawa H, Romero-Wolf M, Yui MA, Ungerbäck J, Quiloan MLG, Matsumoto M, Nakayama KI, Tanaka T, and Rothenberg EV (2018). Bcl11b sets pro-T cell fate by site-specific cofactor recruitment and by repressing Id2 and Zbtb16. Nat. Immunol 19, 1427–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard J (2001). Mechanics of Motor Proteins and the Cytoskeleton (Sinauer Associates; ). [Google Scholar]
- Hu G, Cui K, Fang D, Hirose S, Wang X, Wangsa D, Jin W, Ried T, Liu P, Zhu J, et al. (2018). Transformation of accessible chromatin and 3D nucleome underlies lineage commitment of early T cells. Immunity 48, 227–242.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa T, Hirose S, Masuda K, Kakugawa K, Satoh R, Shibano-Satoh A, Kominami R, Katsura Y, and Kawamoto H (2010). An essential developmental checkpoint for production of the T cell lineage. Science 329, 93–96. [DOI] [PubMed] [Google Scholar]
- Isoda T, Moore AJ, He Z, Chandra V, Aida M, Denholtz M, Piet van Hamburg J, Fisch KM, Chang AN, Fahl SP, et al. (2017). Non-coding transcription instructs chromatin folding and compartmentalization to dictate enhancer-promoter communication and T cell fate. Cell 171, 103–119.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isono K, Endo TA, Ku M, Yamada D, Suzuki R, Sharif J, Ishikura T, Toyoda T, Bernstein BE, and Koseki H (2013). SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev. Cell 26, 565–577. [DOI] [PubMed] [Google Scholar]
- Jackson MB, and Berkowitz SA (1980). Nucleation and the kinetics of microtubule assembly. Proc. Natl. Acad. Sci. USA 77, 7302–7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav U, Manieri E, Nalapareddy K, Madha S, Chakrabarti S, Wucherpfennig K, Barefoot M, and Shivdasani RA (2020). Replicational dilution of H3K27me3 in mammalian cells and the role of poised promoters. Mol. Cell 78, 141–151.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, and Berger F (2017). DNA replication-coupled histone modification maintains polycomb gene silencing in plants. Science 357, 1146–1149. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Georgakilas G, Petrovic J, Kurachi M, Cai S, Harly C, Pear WS, Bhandoola A, Wherry EJ, and Vahedi G (2018). Lineage-determining transcription factor TCF-1 initiates the epigenetic identity of T cells. Immunity 48, 243–257.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Williams RT, Calarco JP, Miller EL, Weber CM, Braun SMG, Pulice JL, Chory EJ, and Crabtree GR (2017). Dynamics of BAF-polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat. Genet 49, 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn TG, Dorafshan E, Schultheis D, Zare A, Stenberg P, Reim I, Pirrotta V, and Schwartz YB (2016). Interdependence of PRC1 and PRC2 for recruitment to polycomb response elements. Nucleic Acids Res. 44, 10132–10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaikkonen MU, Spann NJ, Heinz S, Romanoski CE, Allison KA, Stender JD, Chun HB, Tough DF, Prinjha RK, Benner C, and Glass CK (2013). Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol. Cell 51, 310–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramers HA (1940). Brownian motion in a field of force and the diffusion model of chemical reactions. Physica 7, 284–304. [Google Scholar]
- Kraushaar DC, Jin W, Maunakea A, Abraham B, Ha M, and Zhao K (2013). Genome-wide incorporation dynamics reveal distinct categories of turnover for the histone variant H3.3. Genome Biol. 14, R121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen JBL, Nielsen AL, Jørgensen L, Kristensen LH, Helgstrand C, Juknaite L, Kristensen JL, Kastrup JS, Clausen RP, Olsen L, and Gajhede M (2011). Enzyme kinetic studies of histone demethylases KDM4C and KDM6A: towards understanding selectivity of inhibitors targeting oncogenic histone demethylases. FEBS Lett. 585, 1951–1956. [DOI] [PubMed] [Google Scholar]
- Kueh HY, Yui MA, Ng KKH, Pease SS, Zhang JA, Damle SS, Freedman G, Siu S, Bernstein ID, Elowitz MB, and Rothenberg EV (2016). Asynchronous combinatorial action of four regulatory factors activates Bcl11b for T cell commitment. Nat. Immunol 17, 956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu S, Ji F, Sunwoo H, Jain G, Lee JT, Sadreyev RI, Dekker J, and Kingston RE (2017). Polycomb repressive complex 1 generates discrete compacted domains that change during differentiation. Mol. Cell 65, 432–446.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladoux B, Quivy J-P, Doyle P, du Roure O, Almouzni G, and Viovy J-L (2000). Fast kinetics of chromatin assembly revealed by single-molecule videomicroscopy and scanning force microscopy. Proc. Natl. Acad. Sci. USA 97, 14251–14256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, and Narlikar GJ (2017). Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, et al. (2006). Control of developmental regulators by polycomb in human embryonic stem cells. Cell 125, 301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Leid M, and Rothenberg EV (2010). An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science 329, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Wang HY, Chepelev I, Zhu Q, Wei G, Zhao K, and Wang R-F (2014). Stage-dependent and locus-specific role of histone demethylase Jumonji D3 (JMJD3) in the embryonic stages of lung development. PLoS Genet. 10, e1004524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Natarajan A, Ezike J, Barrasa MI, Le Y, Feder ZA, Yang H, Ma C, Markoulaki S, and Lodish HF (2019). Rate of progression through a continuum of transit-amplifying progenitor cell states regulates blood cell production. Dev. Cell 49, 118–129.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson Q, Beltran B, and Spakowitz AJ (2018). Bottom-up modeling of chromatin segregation due to epigenetic modifications. Proc. Natl. Acad. Sci. USA 115, 12739–12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manesso E, Chickarmane V, Kueh HY, Rothenberg EV, and Peterson C (2013). Computational modelling of T-cell formation kinetics: output regulated by initial proliferation-linked deferral of developmental competence. J. R. Soc. Interface 10, 20120774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, et al. (2009). Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461, 762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayran A, Khetchoumian K, Hariri F, Pastinen T, Gauthier Y, Balsalobre A, and Drouin J (2018). Pioneer factor Pax7 deploys a stable enhancer repertoire for specification of cell fate. Nat. Genet 50, 259–269. [DOI] [PubMed] [Google Scholar]
- Meers MP, Janssens DH, and Henikoff S (2019). Pioneer factor-nucleosome binding events during differentiation are motif encoded. Mol. Cell 75, 562–575.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison TJ (1992). Compare and contrast actin filaments and microtubules. Mol. Biol. Cell 3, 1309–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newport J, and Kirschner M (1982). A major developmental transition in early Xenopus embryos: I. characterization and timing of cellular changes at the midblastula stage. Cell 30, 675–686. [DOI] [PubMed] [Google Scholar]
- Ng KK, Yui MA, Mehta A, Siu S, Irwin B, Pease S, Hirose S, Elowitz MB, Rothenberg EV, and Kueh HY (2018). A stochastic epigenetic switch controls the dynamics of T-cell lineage commitment. eLife 7, e37851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PHB, Pease NA, Ng KKH, Irwin B, and Kueh HY (2019). Temporal scaling in developmental gene networks by epigenetic timing control (preprint). bioRxiv. 10.1101/752170. [DOI] [Google Scholar]
- Nuebler J, Fudenberg G, Imakaev M, Abdennur N, and Mirny LA (2018). Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. USA 115, E6697–E6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto M, Miyata T, Konno D, Ueda HR, Kasukawa T, Hashimoto M, Matsuzaki F, and Kawaguchi A (2016). Cell-cycle-independent transitions in temporal identity of mammalian neural progenitor cells. Nat. Commun 7, 11349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmond DG (1991). Proliferation kinetics and the lifespan of B cells in central and peripheral lymphoid organs. Curr. Opin. Immunol 3, 179–185. [DOI] [PubMed] [Google Scholar]
- Otani T, Marchetto MC, Gage FH, Simons BD, and Livesey FJ (2016). 2D and 3D stem cell models of primate cortical development identify species-specific differences in progenitor behavior contributing to brain size. Cell Stem Cell 18, 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DH, Hong SJ, Salinas RD, Liu SJ, Sun SW, Sgualdino J, Testa G, Matzuk MM, Iwamori N, and Lim DA (2014). Activation of neuronal gene expression by the JMJD3 demethylase is required for postnatal and adult brain neurogenesis. Cell Rep. 8, 1290–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R, Kondev J, Theriot J, and Garcia H (2012). Physical Biology of the Cell (Garland Science; ). [Google Scholar]
- Plys AJ, Davis CP, Kim J, Rizki G, Keenen MM, Marr SK, and Kingston RE (2018). Phase separation and nucleosome compaction are governed by the same domain of polycomb repressive complex 1. BioRxiv, 467316. [Google Scholar]
- Plys AJ, Davis CP, Kim J, Rizki G, Keenen MM, Marr SK, and Kingston RE (2019). Phase separation of polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev. 33, 799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poleshko A, Mansfield KM, Burlingame CC, Andrake MD, Shah NR, and Katz RA (2013). The human protein PRR14 tethers heterochromatin to the nuclear lamina during interphase and mitotic exit. Cell Rep. 5, 292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porritt HE, Gordon K, and Petrie HT (2003). Kinetics of steady-state differentiation and mapping of intrathymic-signaling environments by stem cell transplantation in nonirradiated mice. J. Exp. Med 198, 957–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, and Chang HY (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanulli S, Trnka MJ, Dharmarajan V, Tibble RW, Pascal BD, Burlingame AL, Griffin PR, Gross JD, and Narlikar GJ (2019). HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 575, 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seenundun S, Rampalli S, Liu Q-C, Aziz A, Palii C, Hong S, Blais A, Brand M, Ge K, and Dilworth FJ (2010). UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogenesis. EMBO J. 29, 1401–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff JG, and Henikoff S (2018). Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc 13, 1006–1019. [DOI] [PubMed] [Google Scholar]
- Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, and Copeland RA (2010). Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. USA 107, 20980–20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, and Karpen GH (2017). Phase separation drives heterochromatin domain formation. Nature 547, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strome S, Kelly WG, Ercan S, and Lieb JD (2014). Regulation of the X chromosomes in Caenorhabditis elegans. Cold Spring Harb. Perspect. Biol 6, a018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Looi L-S, Guo S, He Z, Gan E-S, Huang J, Xu Y, Wee W-Y, and Ito T (2014). Timing mechanism dependent on cell division is invoked by Polycomb eviction in plant stem cells. Science 343, 1248559. [DOI] [PubMed] [Google Scholar]
- Talbert PB, and Henikoff S (2017). Histone variants on the move: substrates for chromatin dynamics. Nat. Rev. Mol. Cell Biol 18, 115–126. [DOI] [PubMed] [Google Scholar]
- Tatavosian R, Kent S, Brown K, Yao T, Duc HN, Huynh TN, Zhen CY, Ma B, Wang H, and Ren X (2019). Nuclear condensates of the polycomb protein chromobox 2 (CBX2) assemble through phase separation. J. Biol. Chem 294, 1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple S, and Raff MC (1986). Clonal analysis of oligodendrocyte development in culture: evidence for a developmental clock that counts cell divisions. Cell 44, 773–779. [DOI] [PubMed] [Google Scholar]
- Tu S, Yuan G-C, and Shao Z (2017). The PRC2-binding long non-coding RNAs in human and mouse genomes are associated with predictive sequence features. Sci. Rep 7, 41669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulianov SV, Doronin SA, Khrameeva EE, Kos PI, Luzhin AV, Starikov SS, Galitsyna AA, Nenasheva VV, Ilyin AA, Flyamer IM, et al. (2019). Nuclear lamina integrity is required for proper spatial organization of chromatin in Drosophila. Nat. Commun 10, 1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and Belmont AS (2017). Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum-Finney B, Wu L, Yu M, Brashem-Stein C, Staats S, Flowers D, Griffin JD, and Bernstein ID (2000). Immobilization of Notch ligand, Delta-1, is required for induction of notch signaling. J. Cell Sci 113, 4313–4318. [DOI] [PubMed] [Google Scholar]
- Walters MC, Fiering S, Eidemiller J, Magis W, Groudine M, and Martin DI (1995). Enhancers increase the probability but not the level of gene expression. Proc. Natl. Acad. Sci. USA 92, 7125–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Gao Y, Zheng X, Liu C, Dong S, Li R, Zhang G, Wei Y, Qu H, Li Y, et al. (2019). Histone modifications regulate chromatin compartmentalization by contributing to a phase separation mechanism. Mol. Cell 76, 646–659.e6. [DOI] [PubMed] [Google Scholar]
- Weintraub H (1988). Formation of stable transcription complexes as assayed by analysis of individual templates. Proc. Natl. Acad. Sci. USA 85, 5819–5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K, Christensen J, Rappsilber J, Nielsen AL, Johansen JV, and Helin K (2014). The histone lysine demethylase JMJD3/KDM6B is recruited to p53 bound promoters and enhancer elements in a p53 dependent manner. PLoS ONE 9, e96545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodworth MA, Ng KKH, Halpern AR, Pease NA, Nguyen PHB, Kueh HY, and Vaughan JC (2020). Multiplexed single-cell profiling of chromatin states at genomic loci by expansion microscopy (preprint). bioRxiv. 10.1101/2020.11.17.385476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Ma H, Jin J, Uttam S, Fu R, Huang Y, and Liu Y (2018). Super-resolution imaging of higher-order chromatin structures at different epigenomic states in single mammalian cells. Cell Rep. 24, 873–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS, and Carroll JS (2011). Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 25, 2227–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JA, Mortazavi A, Williams BA, Wold BJ, and Rothenberg EV (2012). Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell 149, 467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Tian X-J, Mukhopadhyay A, Kim KS, and Xing J (2014). Statistical mechanics model for the dynamics of collective epigenetic histone modification. Phys. Rev. Lett 112, 068101. [DOI] [PubMed] [Google Scholar]
- Zheng M, Tian SZ, Capurso D, Kim M, Maurya R, Lee B, Piecuch E, Gong L, Zhu JJ, Li Z, et al. (2019). Multiplex chromatin interactions with single-molecule precision. Nature 566, 558–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou CY, Johnson SL, Gamarra NI, and Narlikar GJ (2016). Mechanisms of ATP-dependent chromatin remodeling motors. Annu. Rev. Biophys 45, 153–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Yui MA, Williams BA, Yun J, Wold BJ, Cai L, and Rothenberg EV (2019). Single-cell analysis reveals regulatory gene expression dynamics leading to lineage commitment in early T cell development. Cell Syst. 9, 321–337.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the H3K27me3 CUT&RUN data reported in this paper is NCBI Gene Expression Omnibus: GSE134749. Scripts used for imaging analysis and simulations of mathematical models have been deposited to GitHub: https://github.com/KuehLabUW/Pease_et_al.2021. All other data supporting the findings of this paper will be available from the Lead Contact.