

Abstract
Endocrine resistance (EnR) in advanced prostate cancer (APC) is fatal. EnR can be mediated by androgen receptor splice variants (AR-SV), with AR-V7 arguably the most clinically important variant. In this study, we determined proteins key to generating AR-V7, validated our findings using clinical samples, and studied splicing regulatory mechanisms in PC models. Triangulation studies identified JMJD6 as a key regulator of AR-V7, as evidenced by its upregulation with in vitro EnR, its downregulation alongside AR-V7 by bromodomain inhibition, and its identification as a top hit of a targeted siRNA screen of spliceosome-related genes. JMJD6 protein levels increased (p<0.001) with castration resistance and were associated with higher AR-V7 levels and shorter survival (p=0.048). JMJD6 knockdown reduced PC cell growth, AR-V7 levels, and recruitment of U2AF65 to AR pre-mRNA. Mutagenesis studies suggested that JMJD6 activity is key to generation of AR-V7, with the catalytic machinery residing within a druggable pocket. Taken together, these data highlight the relationship between JMJD6 and AR-V7 in APC and support further evaluation of JMJD6 as a therapeutic target in this disease.
Keywords: JMJD6, AR-V7, Prostate Cancer, Endocrine Resistance, 2-Oxoglutarate oxygenase
Introduction
Prostate cancer (PC) is a leading cause of male cancer mortality globally. PC progression to metastatic castration-resistant PC (mCRPC) is commonly driven by persistent androgen receptor (AR) signaling [1, 2]. Abiraterone and enzalutamide, which target the AR signaling axis, are standards of care, improving both progression free (PFS) and overall survival (OS) [3, 4] for mCRPC and castration-sensitive PC (CSPC) [5]. However, some mCRPCs never respond to these therapies, while all eventually acquire resistance and invariably fatal disease [6], in part, due to constitutively active alternatively spliced AR variants (AR-SVs) that are truncated and lack the regulatory AR ligand-binding domain targeted by current AR directed therapies [7–9]. Of the many AR-SVs reported, AR splice variant 7 (AR-V7) appears most prevalent and has been associated with resistance to AR targeting therapies and poorer OS [8, 10]. Efforts to target AR-SVs directly have proved challenging due to the inherently disordered nature of the AR N-terminal domain [9]. There remains an unmet need for novel therapeutic strategies to overcome AR-SVs and improve outcome from lethal PC.
One strategy to abrogate AR-V7-mediated resistance is to target epigenetic processes regulating proteins involved in AR-V7 generation and/or stabilization. Members of the bromodomain and extra-terminal (BET) motif protein family are of particular interest as they are reported to modulate AR signaling [11]; BET inhibition downregulates AR-V7 protein expression and reduces enzalutamide-resistant patient-derived PC model growth [11]. However, BET proteins have pleiotropic roles and regulate many signaling pathways, perhaps explaining why despite extensive efforts no BET inhibitors have shown clinical utility [12].
Herein, we describe results arising from the hypothesis that key spliceosome-related proteins driving AR-V7 generation can be identified by a triangulation approach, analyzing: 1) RNA sequencing (RNA-seq) changes induced by BET inhibition that downregulates AR-V7; 2) adaptations in PC cells as they become resistant to androgen deprivation by RNA-seq studies; 3) the top hits from a targeted siRNA screen of spliceosome-related genes. We hypothesized that by directly targeting identified key regulators driving AR-V7 splicing, we could replicate the encouraging preclinical effects seen with BET inhibition, while mitigating the adverse effects associated with these agents.
Materials and Methods
Patients and tissue samples
All patients had mCRPC treated at the Royal Marsden Hospital (RMH) and provided written informed consent, being enrolled into protocols approved by the RMH ethics review committee (reference no. 04/Q0801/60). Patient clinical data were retrospectively collected from the RMH electronic patient record system.
ICR/RMH cohort.
74 previously collected biopsies were identified as having sufficient formalin-fixed, paraffin embedded (FFPE) mCRPC tissue for assessment (Bone, n = 41; Lymph node, n = 21; Liver, n = 4; Other, n = 8). Of these, 64 also had matched, same-patient diagnostic CSPC biopsies available (Supplementary Figure 1). All biopsy blocks were freshly sectioned and only considered for immunohistochemistry analyses if adequate material was present (≥50 tumor cells). All CSPC biopsies demonstrated adenocarcinoma.
International Stand Up To Cancer/Prostate Cancer Foundation (SU2C/PCF) cohort.
Due to the low level of AR-V7 expression at diagnosis of castration-sensitive PC [13], the bioinformatic analyses of patient sequencing data presented in this study were intentionally performed using publicly accessible data obtained from only mCRPC patients. Whole exome (n=231) and transcriptome (n=159) sequencing data from mCRPC patients generated by the SU2C/PCF Prostate Cancer Dream Team were downloaded and reanalyzed [2].
Antibody validation
Antibody specificity was determined by Western blot (WB) analyses (Supplementary Figure 2) comparing detection of JMJD6 protein levels in LNCaP95 whole cell lysates cultured with either non-targeting control siRNA or ON-TARGETplus pooled JMJD6 siRNA (Dharmacon; GE healthcare). AR-V7 antibody validation was performed as previously described [13].
Immunohistochemistry (IHC)
JMJD6 IHC was performed using a mouse anti-JMJD6 antibody (Santa Cruz Biotechnology; sc-28348; 200ug/ml stock). Antigen retrieval was achieved by microwaving slides in pH 6 Antigen Retrieval Buffer (HDS05–100; TCS Biosciences) for 18-min at 800 W prior to incubation with anti-JMJD6 antibody (1:50 dilution) for 1-hour at room temperature. The reaction was visualized using the EnVision system (K4061; DAKO). Antibody specificity was confirmed from LNCaP95 cell pellets following treatment with ON-TARGETplus pooled JMJD6 siRNA, compared to non-targeting control siRNA. AR-V7 IHC was performed as previously described [13]. JMJD6 and AR-V7 quantification was determined by a pathologist blinded to clinical data using the modified H score (HS) method [14]; [(% of weak staining) × 1] + [(% of moderate staining) × 2] + [(% of strong staining) × 3], to determine overall percentage JMJD6 positivity across the stained tumor sample (range: 0 to 300).
Cell lines and cultures
All cell lines were purchased from LGC Standards/ATCC unless otherwise specified and grown in recommended media (Supplementary Table 1) at 37°C in 5% CO2. Short tandem repeat profiling was performed using the Cell Authentication Service by Eurofins Medigenomix to ensure the quality and integrity of the cell lines used. Cells lines were tested for mycoplasma after thawing, then regularly every 6–8 weeks during culture using the Venor®GeM Advance Mycoplasma Detection Kit (Minerva Biolabs). Early passages were thawed every 3-months (after approximately 15–20 passages).
Small interfering RNA (siRNA):
All siRNAs were ONTARGETplus pools (Dharmacon; GE heathcare), as listed in Supplementary Table 2, and used in combination with 0.4% RNAiMax transfection reagent (ThermoFisher Scientific) as per manufacturer’s instructions. siRNA experiments were performed at 50 nM, unless otherwise specified, for 72-hours.
JMJD6 plasmid overexpression:
Wild-type pcDNA3-JMJD6-WT (JMJD6WT) and the catalytically inactive mutants pcDNA3-JMJD6-ASM2 (MUT1) and pcDNA3-JMJD6-BM1 (MUT2) JMJD6 expression constructs were kindly donated by Dr. A. Böttger [15, 16] and transfected into 22Rv1 and VCaP cell lines using Lipofectamine 3000 (Invitrogen, Carlsbad, CA). All treatments were performed using 1μg of total plasmid. For experiments requiring lower concerntrations, the empty vector control plasmid (pcDNA3) was added to JMJD6WT, MUT1 or MUT2, respectively, to make up the difference (e.g. 0.5μg JMJD6WT + 0.5μg empty vector control plasmid = 1μg total plasmid input). All plasmid overexpression experiments were performed in 2mls total volume.
Drugs:
Enzalutamide was from Selleckchem (S1250). Dimethyl sulfoxide (DMSO) was from Fisher Scientific (BP231–1). 2,4-Pyridinedicarboxylic acid (2,4-PDCA) was purchased from Sigma-Aldrich (04473).
Growth assays
Cells were plated in 48-well tissue culture plates and treated as indicated the following day, then grown for 6-days or until 80–90% confluence. To quantify growth of LNCaP, LNCaP95, 22Rv1 and PNT2 cell lines, cells were fixed with 10% (w/v) aqueous trichloroacetic acid and incubated at 4°C for 30-minutes prior to washing and air-drying. Subsequently cells were stained with sulforhodamine B (SRB) for 30-minutes prior to excess dye removal with 1% (v/v) aqueous acetic acid and further air-drying. Following this, protein-bound dye was dissolved in 10mM Tris base solution, transferred to a 96-well plate, and optical density determined at 510nm using the Synergy HT microplate reader (BioTek). VCaP cell growth assays were analyzed using CellTiter-Glo® Luminescent Cell Viability Assay (Promega) as per manufacturer instructions, and luminescence quantified using the Synergy HT microplate reader (BioTek).
Western blot (WB)
Subsequent to cell lysis with RIPA buffer (Pierce) supplemented with cOmplete™ EDTA-free Protease Inhibitor Cocktail (Roche), protein extracts (20 μg) were separated by electrophoresis on 4–12% NuPAGE® Bis-Tris gel plates (Invitrogen) prior to transfer onto Immobilon-P™ PVDF membranes of 0.45μm pore size (Millipore). Membranes were then incubated sequentially with primary and then secondary antibody in 5% milk and tris-buffered saline (TBS) and Tween®20 (Sigma Aldrich) as indicated (Supplementary Table 3). Chemiluminescence was then detected using the Chemidoc Touch imaging system (BioRad).
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
The RNeasy Plus Mini kit (Qiagen) was used to extract cellular RNA as per manufacturer’s instructions. Following cDNA synthesis with the First Strand cDNA Synthesis Kit (Roche), qRT-PCR was performed using the ViiA™ 7 System Real-Time PCR System (Life Technologies) and the TaqMan Universal PCR Master Mix (Applied Biosystems) and probes (ThermoFisher Scientific) as listed in Supplementary Table 4 [11]. The fold change in mRNA expression levels was calculated by the comparative Ct method, using the formula 2-(-(ΔΔCt) [17].
RNA immunoprecipitation (RIP) assays
Cells were transfected with either 25 nM non-targeting control siRNA (Dharmacon) or 25 nM JMJD6 siRNA (Dharmacon) using Lipofectamin RNAiMax (Invitrogen) and OPTI-MEM media (Gibco) as per manufacturer’s instructions. After 72-hours, cells were cross-linked with 0.3% (v/v) aqueous formaldehyde (Thermo Scientific). RIP assays were performed using the EZ-Magna RIP (Cross-linked) Nuclear RNA-binding Protein Immunoprecipitation Kit (Millipore; 17–10521) following the manufacturer’s protocol, and immunoprecipitated with 4μg of U2AF65 antibody (Sigma Aldrich). RNA purification and DNAse I treatment was performed using RNeasy Plus Universal Mini Kit (Qiagen). The resultant RNAs were subjected to cDNA synthesis and RT-qPCR analysis. RIP data were derived from two independent experiments (Supplementary Table 5).
RNA-seq and analysis of alternative splicing events
RNA-seq analysis comparing (1) LNCaP and LNCaP95 PC cells, and (2) LNCaP95 PC treated with either I-BET151 or vehicle (DMSO 0.1%), were performed as previously described [11]. Analyses compared impact of I-BET151 at concentrations of 500nM and 2μM for 8- and 48-hours which downregulate AR-V7 [11]) and equivalent vehicle (DMSO 0.1% for 8 and 48-hours). Only genes with baseline expression, as measured by Fragments Per Kilobase of transcript per Million mapped reads (FPKM), greater than the median expression level of all 315 spliceosome related genes at baseline across both experiments were included for analysis, with the top 15 genes most differentially expressed (FPKM) in each experiment (up- or down-regulated) being considered as genes of interest. For RNA-seq analyses of LNCaP95 PC cells treated with JMJD6 siRNA compared to non-targeting control siRNA, cellular RNA was extracted using the RNeasy Plus Mini Kit (Qiagen) as per manufacturer’s instructions. RNA quality was analyzed using the Agilent RNA Screentape assay; 100ng of total RNA from each sample was used for Agilent SureSelect library prep kit. Library quality was confirmed using the Agilent Bioanalyzer High Sensitivity DNA screentape Assay. The libraries were quantified and normalized by qPCR using Qiagen Generead Quantification Kit (Roche). Library clustering was performed on a cBot with Illumina HiSeq PE Cluster kit v3. The libraries were sequenced as paired-end 101-base-pair reads on an Illumina HiSeq 2500 membrane with an Illumina HiSeq SBS kit v3. Base-calling and quality scoring were performed using Real-Time Analysis (version 1.18.64) and FASTQ file generation and de-multiplexing using BCL2FASTQ. Paired end raw reads in FASTQ format were aligned to the reference human genome (hg19) using RNA-seq spliced read mapper TopHat (v2.0.7), with default settings [18]. The library and mapping quality were estimated using Picard tools (http://broadinstitute.github.io/picard). Alternative splicing events (skipped exons, alternative 5′ splice sites, alternative 3′ splice sites, mutually exclusive exons and retained introns) based on Ensembl v61 annotation were accessed using MATS v3.0.8 [19].
Spliceosome related gene set
The list of genes relating to the spliceosome utilized to conduct this study was determined through interrogation and amalgamation of search results from two publicly accessible databases: 1) The Gene Ontology (GO) Resource [20–22]; search-term “spliceosome” with filters “Homo sapiens” and “UniProtKB”, and 2) The Molecular Signatures Database [23, 24]; search-term “SPLICING/SPLICEOSOME/SPLICEOSOMAL”. Included genes are presented in Supplementary Table 6.
AR activity, AR-V7 activity, and gene expression evaluation
Paired-end transcriptome sequencing reads were aligned to the human reference genome (GRCh37/hg19) using Tophat2 (v2.0.7). Gene expression levels, as measured by FPKM, were calculated using Cufflinks [26]. AR signaling activity was established by determining expression levels of either (1) 43-genes regulated by AR in PC cell line and metastatic prostate cancer RNA-seq datasets, as previously described [11] (AR signature; Supplementary Table 7), or (2) the HALLMARK_ANDROGEN_RESPONSE gene set from the MSidDB (M5908 [25]; Androgen response (H); Supplementary Table 8). AR-V7 signaling activity was determined using the previously published AR-V7-associated signature based on the expression levels of 59-genes associated with AR-V7 expression in mCRPC (AR-V7 signature; Supplementary Table 9) [13].
Liquid Chromatography Mass Spectrometry (LC-MS) assays for JMJD6 inhibition by 2,4-PDCA
Hydroxylation of a 12-mer peptide substrate (NPKRSRSREHRR, prepared with a C-terminal amide) of the LUC7L2 pre-mRNA splicing factor by JMJD6 (1–362, prepared as reported) [26, 27] was monitored by liquid chromatography mass spectrometry (LC-MS) using an Agilent 1290 infinity II LC system equipped with an Agilent 1290 infinity binary pump and coupled to an Agilent 6550 Accurate Mass Quadrupole Time of Flight (Q-TOF) mass spectrometer. Note this construct has hydroxylation but not demethylation activity [26]. All JMJD61–362 enzyme reactions were performed in 50 mM Tris.Cl pH 7.5 (prepared fresh each day) at 37°C. L (+)-Ascorbic acid sodium salt (code 11140), ferrous ammonium sulphate (FAS) as ammonium iron (II) sulphate hexahydrate (215406), and 2OG were from Sigma Aldrich (Poole, Dorset). The LUC7L2 peptide substrate was synthesized to >95% purity (LC-MS) by GL-Biochem (Shanghai, China). L-Ascorbic Acid (50 mM in deionized water), 2OG (10 mM in deionized water) and iron (II) sulphate (400 mM in 10 mM HCl) solutions were prepared freshly each day. JMJD61–362 (10 μM) was pre-incubated with an 8-point and 3-fold serial dilution of 2, 4-PDCA (100 – 0.046 μM) for 15 minutes and the enzyme reaction initiated by addition of LUC7L2 substrate (100 μM LUC7L2, 400 μM L-ascorbate, 100 μM FAS, 500 μM 2OG final concentrations). The enzyme reaction was progressed for 2 hours at 37°C, then stopped by addition of formic acid to a final concentration of 1.0 % (v/v). The quenched enzyme reaction was injected (6 μl injections) onto a Proswift RP-4H 1×50 mm LC column (Thermo) and the LUC7L2 and LUC7L2-hydroxylated peptides were fractionated using a linear gradient of Solvent A (0.1% (v/v) formic acid in LCMS water) and Solvent B (0.1% (v/v) formic acid in 100% LCMS grade acetonitrile). Details of the gradient conditions, flow rates and maximum pressure limits are summarized in Supplementary Table 10. Peptide ionization was monitored in the positive ion electrospray ionisation (ESI) mode with a drying gas temperature of 280°C, a drying gas flow rate of 13 L/minute, nebulizer gas pressure of 40 PSI, sheath gas temperature of 350°C, sheath gas flow rate of 12 L/min and a nozzle voltage of 1000V. Ion chromatogram data for the +2 charge state of both the non-hydroxylated and hydroxylated peptides were extracted and integrated using MassHunter qualitative software (Agilent). The % conversion of the peptide substrate to the +16 hydroxylated peptide was calculated using the equation: % conversion = 100 × hydroxylated / (hydroxylated + non-hydroxylated peptide). The IC50 for 2, 4-PDCA was determined from non-linear regression curve fitting using GraphPad prism 6.0.
Statistical analysis
All statistical analyses were performed using Stata v13.1 or GraphPad Prism v7 and are indicated within all figures and tables. Spearman’s correlation was used to determine the association between JMJD6 and U2AF65 mRNA levels and other characteristics such as Androgen response (H), AR signature and AR-V7 signature. H-Scores are reported as median values and interquartile ranges. Comparison of JMJD6 expression levels between CSPC and mCRPC tissue samples, and correlations with next generation sequencing (NGS) data, were determined using the Wilcoxon matched-pair signed rank test. Comparisons between JMJD6 and AR-V7 expression levels in mCRPC tissue samples made using Mann-Whitney test. OS from CRPC biopsy was defined as time from CRPC biopsy to date of death. Survival analyses were estimated using the Kaplan-Meier method.
Results
Orthogonal analyses identify the 2OG-dependent dioxygenase JMJD6 as a regulator of AR-V7 expression.
To identify proteins downregulated by BET inhibition that are critical to the regulation of AR-V7 splicing, an orthogonal three-stage investigative triangulation approach was employed (figure 1A; Supplementary Figure 3). First, RNA-seq data from hormone-sensitive LNCaP cells (that do not produce AR-V7 protein) and their derivative, androgen deprivation resistant LNCaP95 cells (that do produce AR-V7 protein), were interrogated to identify which genes with roles relating to the spliceosome, as determined by GO annotations and The Molecular Signatures Database (Spliceosome related gene set; Supplementary Table 6), are significantly upregulated in LNCaP95 cells relative to LNCaP cells. These results were subsequently aligned with RNA-seq analyses comparing LNCaP95 PC cells treated with either a BET inhibitor (GSK1210151A; I-BET151) or vehicle (DMSO 0.1%), to investigate which of the spliceosome related gene set were also significantly downregulated by BET inhibition, which we, and others, have previously reported downregulates AR-V7 expression [11, 28]. To identify spliceosome related proteins that preferentially regulate AR-V7 generation, these transcriptomic data were amalgamated with the results of a targeted siRNA screen where all 315 genes in the spliceosome related gene set were individually silenced in the castration-resistant AR-V7 expressing PC cell lines LNCaP95 and 22Rv1 to determine their impact on AR-V7 protein levels relative to full-length AR (AR-FL) by WB. Genes were ranked in an order determined by the degree of AR-V7 downregulation relative to AR-FL averaged across both cell lines, with proteins causing the greatest reduction in AR-V7:AR-FL ratio being ranked highest. Only genes that were (1) significantly upregulated in LNCaP95 cells relative to LNCaP cells, (2) significantly downregulated following BET inhibition, and (3) associated with a >50% reduction in AR-V7 protein expression relative to AR-FL, were considered to be of further interest. Strikingly, these three independent lines of investigation identified the 2OG-dependent dioxygenase JMJD6 as the only gene to meet all three criteria, indicating that it may be an important regulator of AR-V7 protein expression (figure 1B).
Figure 1: Orthogonal analyses identify the 2OG-dependent dioxygenase JMJD6 as a potential regulator of AR-V7.
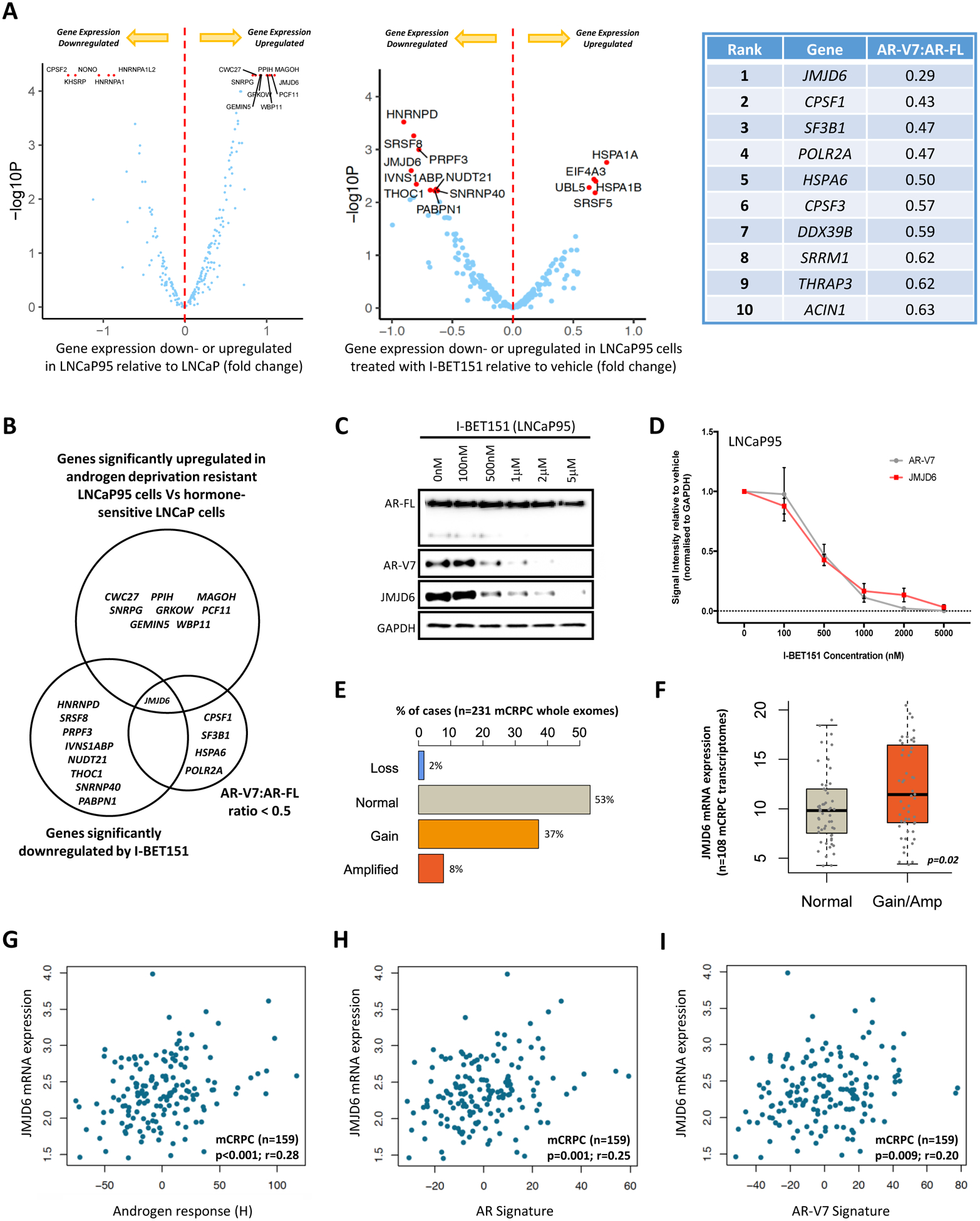
(A) Volcano plots illustrating differential mRNA expression of 315 genes relating to the spliceosome (spliceosome related gene set; supplementary table 6), as determined by RNA-seq, between hormone-sensitive LNCaP (no AR-V7 protein) and androgen deprivation resistant LNCaP95 (detectable AR-V7 protein) prostate cancer (PC) cell lines, and LNCaP95 PC cells treated with either a BET inhibitor (I-BET151) or vehicle (DMSO 0.1%). Blue dots represent genes with baseline expression (FPKM) greater than the median expression level of all 315 genes at baseline across both experiments. Top 15 genes most differentially expressed (FPKM) in each experiment (up- or down-regulated) indicated by red dots. Top 10 hits identified in targeted siRNA screen shown in accompanying table; all 315 genes in the spliceosome related gene set were individually inhibited by siRNA in 22Rv1 and LNCaP95 PC cell lines. Changes in AR-V7 protein levels relative to AR-FL were quantified by western blot (WB) densitometry. AR-V7 downregulation averaged across both cell lines with genes ranked in order of the degree of AR-V7 downregulation relative to AR-FL. (B) Venn diagram amalgamating RNA-seq analyses with siRNA screen results. Genes of interest pre-defined as being upregulated in LNCaP95 cells relative to LNCaP cells, downregulated following BET inhibition, and associated with a > 50% reduction in AR-V7 protein expression (WB) relative to AR-FL following siRNA knockdown. JMJD6 was the only gene to meet all three criteria. (C) WB demonstrating that I-BET151 treatment (48 hours) in LNCaP95 PC cells downregulates both AR-V7 and JMJD6 protein expression in a dose-dependent manner. Single representative WB shown from four separate experiments. (D) Densiometric quantification of JMJD6 (red line) and AR-V7 (grey line) protein levels (n=4; densitometry for each biological replicate normalized to GAPDH and vehicle). Demonstrates that protein levels of both JMJD6 and AR-V7 decrease in a dose-dependent manner following BET inhibition with I-BET151. (E) Whole exome analysis (n=231) shows alterations of the JMJD6 gene locus were detected in 47% of mCRPC biopsies (SU2C/PCF cohort), with these being predominately gains (37%; n=86) or amplifications (8%; n=18). (F) Whole exome analysis of mCRPC patients with matched transcriptome data from SU2C/PCF cohort (n=108) shows that JMJD6 copy number gain and amplification (Amp) associated with an increase in JMJD6 mRNA expression in mCRPC biopsies compared to samples without JMJD6 copy number gain/amplification (p=0.02; Wilcoxon test). (G-I) Scatter plots of transcriptome analysis in 159 mCRPC biopsies (SU2C/PCF cohort) showing correlations between JMJD6 mRNA expression and (G) androgen response (Hallmark; H), (H) AR signature (derived from 43 AR regulated transcripts) and (I) AR-V7 signature (derived from 59 genes associated with AR-V7 expression in mCRPC). JMJD6 mRNA expression shown as log FPKM. r-values and p-values are shown and were calculated using Spearman’s correlation.
To investigate the nature of the relationship between BET inhibition, JMJD6 and AR-V7, WB analyses were performed using LNCaP95 cells treated with I-BET151 for 48 hours. I-BET151 treatment led to a concurrent dose-dependent reduction in both JMJD6 and AR-V7 protein expression, with these both occurring to a similar extent, and at the same concentrations of I-BET151 (figure 1C–D).
Having identified JMJD6 as a protein of interest with respect to AR-V7 regulation in vitro, publicly accessible patient data repositories were then interrogated to establish its potential clinical relevance. Analysis of whole exome sequencing data from 231 mCRPC patient biopsies (SU2C/PCF) revealed JMJD6 genomic alterations in 47% (n=108/231) of evaluated samples, with these being predominately gains (37%; n=86/231) or amplifications (8%; n=18/231) (figure 1E). Importantly, analysis of available corresponding transcriptome data from these 231 mCRPC patient biopsies (n=108) found that JMJD6 gene gain/amplification correlated with an increase in JMJD6 mRNA expression compared to samples without JMJD6 copy number gain/amplification (figure 1F; p=0.02). Furthermore, when all available transcriptome sequencing data were evaluated (n=159; SU2C/PCF), JMJD6 mRNA expression levels correlated significantly with androgen response (H) (r=0.28, p<0.001), AR signature (r=0.25, p=0.001), and a previously reported AR-V7 signature (r=0.20, p=0.009), in mCRPC biopsies (figure 1G–I). Taken together, these results indicated that the JMJD6 gene is expressed in mCRPC and that its presence is associated with both AR and AR-V7 signaling activity, supporting further evaluation of JMJD6 as a gene of interest in mCRPC.
JMJD6 correlates with AR-V7 protein levels and a worse prognosis in mCRPC.
To further investigate the clinical significance of JMJD6 in lethal PC, we next validated an immunohistochemical assay for JMJD6 using whole cell lysates of LNCaP95 PC cells treated with either a non-targeting control siRNA or a JMJD6 specific siRNA (figure 2A–C), and then evaluated JMJD6 and AR-V7 protein levels in 74 mCRPC patient tissue biopsies (figure 2D; Supplementary Table 11). Of these 74 patients, 64 patients also had sufficient matched, same patient, diagnostic, CSPC tissue available for analysis (Supplementary Figure 1). Nuclear JMJD6 protein expression increased significantly (p<0.001) as patients progressed from CSPC (median H-score 12.5, IQR [0.0–67.5]) to CRPC (80 [20.0–130.0]) (figure 2E). In addition, patients with higher nuclear JMJD6 expression (≥median H-score) had significantly (p=0.036) higher nuclear AR-V7 expression (100 [22.5–147.5]; n = 41) than those patients with low nuclear JMJD6 expression (<median H-score; 50 [0.0–105.0]; n = 33) (figure 2F). Finally, those patients with higher (≥75th percentile) nuclear JMJD6 expression had a significantly shorter survival than those patients with lower (<25th percentile) nuclear JMJD6 expression (14 months [n=16] vs 8 months [n=19]; hazard ratio 2.15; 95% confidence interval 1.19 – 5.92; p = 0.017) (figure 2G).
Figure 2: JMJD6 associates with AR-V7 expression and a worse prognosis in mCRPC.
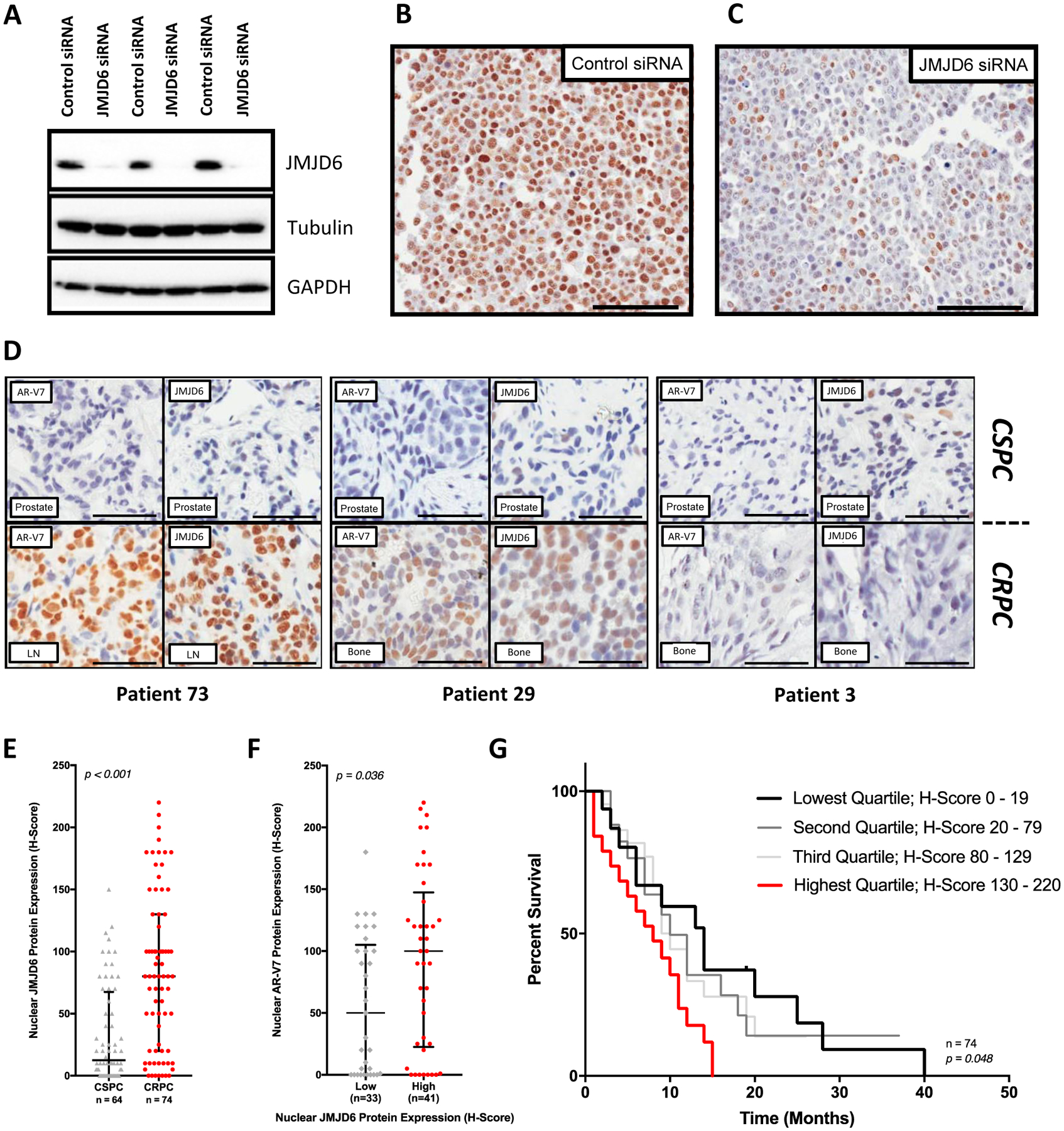
(A) Antibody specificity confirmed by detection of a single band in LNCaP95 whole cell lysates by WB, with downregulation following treatment with pooled JMJD6 siRNA compared to non-targeting control siRNA. (B) Micrograph of LNCaP95 PC cells treated with non-targeting control siRNA demonstrating positive brown nuclear staining for JMJD6. (C) Micrograph of LNCaP95 PC cells treated with pooled JMJD6 siRNA. Demonstrates a marked reduction in JMJD6 protein, with predominately blue, negative staining for JMJD6. (D) Micrographs of IHC analyses for AR-V7 (left) and JMJD6 (right) protein levels in matched, same-patient, diagnostic castration-sensitive (CSPC) (top) and mCRPC (bottom) tissue samples from three different patients (RMH/ICR patient cohort). Scale bars set to 100μm. JMJD6 protein levels in presented tissue samples are similar to AR-V7 levels in mCRPC. (E) Box and whisker plot demonstrating a significant increase (p<0.001) in JMJD6 protein levels (IHC H-Score) in mCRPC biopsies (median H-score [IQR]; CSPC (n=64) 12.5 [0.0–67.5] vs CRPC (n=74) 80 [20.0–130.0]; Wilcoxon rank-sum analysis). (F) Levels of AR-V7 protein significantly higher (p=0.036) in mCRPC tissue samples from patients with high (JMJD6 H-Score ≥ median) mCRPC JMJD6 protein levels (Low 50 [0.0–105.0; n = 33] vs High 100 [22.5–147.5; n = 41]; Mann-Whitney test). (G) Median OS from the time of CRPC tissue biopsy significantly worse in patients with the highest levels of JMJD6 (H-Score > 75th percentile) in their mCRPC tissue sample (n=74, p=0.048; Log-rank test).
Taken together, these data indicated that JMJD6 protein is produced in PC cells, that the level of JMJD6 increases significantly with the emergence of castration-resistant disease, and that this upregulation of JMJD6 correlates with a higher level of AR-V7. Whilst we appreciate that the heterogeneous nature and relatively limited size of the patient cohort presented makes definitive inferences on the impact of JMJD6 expression on survival challenging, in keeping with knowledge that AR-V7 expression is associated with a shorter OS, our results suggest that higher JMJD6 levels in mCRPC cells likely correlate with a worse prognosis. Overall, these data indicated that JMJD6 is a clinically relevant protein in mCRPC that merits further evaluation.
JMJD6 is important for PC cell growth and regulates AR-V7 expression.
We next evaluated the impact of JMJD6 on PC cell growth and AR-V7 expression. Treatment with JMJD6 siRNA (25nM) resulted in a significant reduction in the growth of the castration-resistant AR-V7-expressing PC cell lines LNCaP95 and 22Rv1, as evidence by a reduction in cell number, compared to treatment with non-targeting control siRNA (25nM) (figure 3A). The growth of androgen-sensitive LNCaP cells was also significantly inhibited by JMJD6 siRNA knockdown. Interestingly however, the reduction in growth of LNCaP PC cells, which do not produce detectable levels of AR-V7 protein, was less than that seen with either its androgen-deprivation-resistant derivative LNCaP95, or 22Rv1 PC cells. PNT2 cells, which are an immortalized model of normal prostatic epithelium, were relatively unaffected. Notably, JMJD6 knockdown by siRNA (25nM) for 72 hours downregulated both AR-V7 protein and mRNA levels (figure 3B–C). The effect of JMJD6 knockdown was also evaluated in the hormone-sensitive VCaP PC cell line, which contains the TMPRSS2/ERG rearrangement that is found in 30–40% of APCs, and which possesses a high copy gene amplification of AR. Furthermore, VCaP cells upregulate the expression of AR-V7 in response to androgen-deprivation in vitro [29, 30]. VCaP Cells were treated with either a JMJD6 siRNA (25 nM) or a non-targeting control siRNA (25 nM), both with (Enzalutamide 10μM) and without (DMSO 0.1%) AR blockade, and the effect on growth was determined after 5-days. As shown in figure 3D, JMJD6 siRNA knockdown reduced VCaP PC cell viability when compared to non-targeting control siRNA, as did treatment with enzalutamide alone. Importantly, however, combination treatment with JMJD6 siRNA and enzalutamide had a substantially more profound effect and inhibited VCaP cell viability more than either JMJD6 siRNA alone or enzalutamide treatment alone. To investigate this, RNA and WB analyses were performed using VCaP cells following 72 hour treatment with either non-targeting control siRNA or JMJD6 siRNA (25nM), both with (Enzalutamide 10μM) and without (DMSO 0.1%) AR blockade (figure 3E–G). JMJD6 knockdown downregulated AR-V7 RNA and protein levels, as previously observed in LNCaP95 and 22Rv1 cell lines (figure 3B–C); moreover, and critically, the upregulation of AR-V7 seen in response to AR blockade was also significantly attenuated by JMJD6 knockdown (figure 3H). Taken together, these data demonstrate that JMJD6 is important for PC cell viability and proliferation, and is required for the expression of AR-V7 in in vitro models of lethal PC.
Figure 3: JMJD6 is important for PC cell growth and regulates AR-V7 expression.
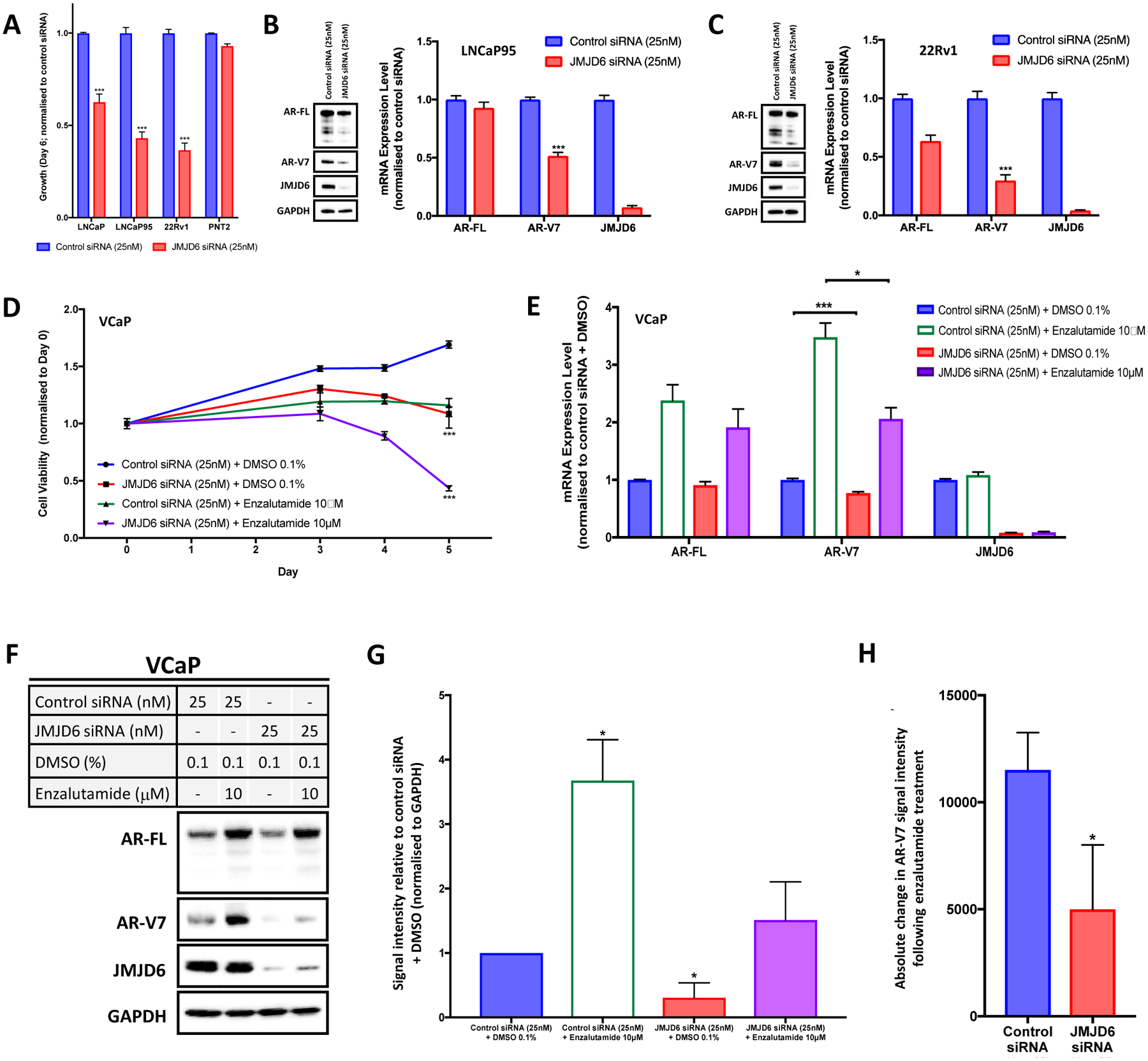
(A) JMJD6 siRNA knockdown (25nM; red bars) significantly reduces the growth (cell number; sulforhodamine B (SRB) assay) of LNCaP, LNCaP95 and 22Rv1 PC cells compared to non-targeting control siRNA (25nM; blue bars), while PNT2 cells (immortalized normal prostatic epithelial cells) were relatively unaffected. Mean cell growth (normalized to control siRNA at same concentration) shown with standard error of the mean; n ≥ 4 data points (at least 2 biological replicates with 2 technical replicates). (B-C) JMJD6 siRNA knockdown downregulates AR-V7 mRNA (qPCR) and protein (WB) levels in LNCaP95 and 22Rv1 PC cell lines. Mean RNA expression (normalized to housekeeping genes (B2M and GAPDH) and control siRNA at equivalent concentration; defined as 1.0) with standard error of the mean from three experiments is shown. (D) Line graph illustrating the impact of JMJD6 siRNA knockdown (25nM) +/− enzalutamide (10μM) on the viability of hormone-sensitive, AR amplified and AR-V7 producing VCaP PC cells compared to controls after five days, as determined using the CellTiter-Glo® Luminescent Cell Viability Assay. JMJD6 siRNA knockdown (red line) significantly reduced VCaP PC cell viability compared to control siRNA (blue line). Combination treatment with enzalutamide (purple line) resulted in a significantly more profound reduction of VCaP cell viability than either JMJD6 siRNA alone (red) or enzalutamide alone (green). n=3; mean cell viability (normalized to control siRNA at same concentration + DMSO 0.1%) shown with standard error of the mean. (E) JMJD6 knockdown downregulated baseline AR-V7 mRNA (qPCR) levels in VCaP cells. JMJD6 knockdown also resulted in a significantly lower increase in AR-V7 mRNA expression in response to AR blockade (enzalutamide 10μM; purple bar) compared to non-targeting control siRNA (green bar). Mean RNA expression (normalized to housekeeping genes (B2M, GAPDH and CDC73), and control siRNA at equivalent concentration + DMSO 0.1%; defined as 1.0) with standard error of the mean from three experiments is shown. (F-G) Single representative WB shown from three separate experiments with corresponding densiometric quantification of AR-V7 protein levels (n=3; densitometry for each biological replicate normalized to GAPDH and vehicle). JMJD6 siRNA knockdown reduces AR-V7 protein levels in VCaP PC cells. Furthermore, while AR-V7 protein levels increase significantly with AR blockade (enzalutamide 10μM), AR-V7 protein levels do not significantly change when JMJD6 is knocked down by siRNA (25nM) at the time of treatment with enzalutamide (10μM). (H) Bar chart showing absolute change in AR-V7 WB signal intensity following AR blockade (enzalutamide 10μM) compared to vehicle (DMSO) with either a non-targeting control siRNA, or a JMJD6 specific siRNA. Demonstrates that the upregulation of AR-V7 in response to enzalutamide was significantly less when JMJD6 was knocked down compared to control siRNA. p values (*, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001) were calculated for each condition compared to control (at equivalent concentration) using the mean value of technical replicates with unpaired Student’s t tests.
JMJD6 regulates AR-V7 transcription in part through recruitment of U2AF65 to AR-V7 specific splice sites in in vitro models of CRPC.
We next investigated the mechanism through which JMJD6 regulates AR-V7 production in preclinical models of CRPC. JMJD6 has been previously reported in the literature to interact with a number of proteins involved in RNA processing [15, 26, 27, 31]. Perhaps the best described example of this is its interaction with the splicing factor U2AF65, which has been demonstrated to be lysyl-5-hydroxylated by JMJD6 at residues in its arginine-serine rich region, including K15, K38 and K276 [27]. Importantly, U2AF65 is reported to play a critical role in the expression of AR-V7, having been shown to be recruited to AR-V7 specific splice sites in response to androgen deprivation therapy (ADT) [32]. Accordingly, as we observed for JMJD6, U2AF65 mRNA expression levels correlated significantly with androgen response (H) (r=0.41, p<0.001), AR signature (r=0.43, p<0.001), and AR-V7 signature (r=0.45, p<0.001) in mCRPC biopsies (Figure 4A–C). We therefore hypothesized that JMJD6-mediated regulation of AR-V7 expression occurs through either the regulation of U2AF65 levels and/or its recruitment to AR-V7 specific splice sites. To determine the relationship between JMJD6, U2AF65 and AR-V7, we studied the impact of JMJD6 and U2AF65 protein depletion (both individually and concurrently) on the level of AR-V7, as well as on the levels of both JMJD6 and U2AF65 themselves, in 22Rv1 PC cells. JMJD6 siRNA (25nM) and U2AF65 siRNA (25nM) both decreased AR-V7 protein levels (figure 4D). JMJD6 siRNA had minimal impact on U2AF65 protein levels, and U2AF65 knockdown had no impact on JMJD6 expression, in keeping with reported data [31]. Having seen no effect of JMJD6 knockdown on U2AF65 expression, RIP analyses were performed to quantify the amount of U2AF65 bound to AR-V7 specific splice sites following JMJD6 siRNA knockdown (25nM) compared to a non-targeting control siRNA, as per previously published protocols [32]. Antibodies against U2AF65, but not control IgG, precipitated AR pre-mRNA at the P1 (containing the 5’ splice site for both AR and AR-V7) and P2 (containing the 3’ splice site for AR-V7) regions in 22Rv1 cells treated with control siRNA; this effect being significantly reduced with JMJD6 siRNA (figure 4E). Taken together, these results indicate that JMJD6 regulates the recruitment of U2AF65 to AR-V7-specific splice sites.
Figure 4: JMJD6 regulates AR-V7 transcription, in part, through recruitment of splicing factor U2AF65 to AR-V7 specific splice sites in in vitro models of CRPC.
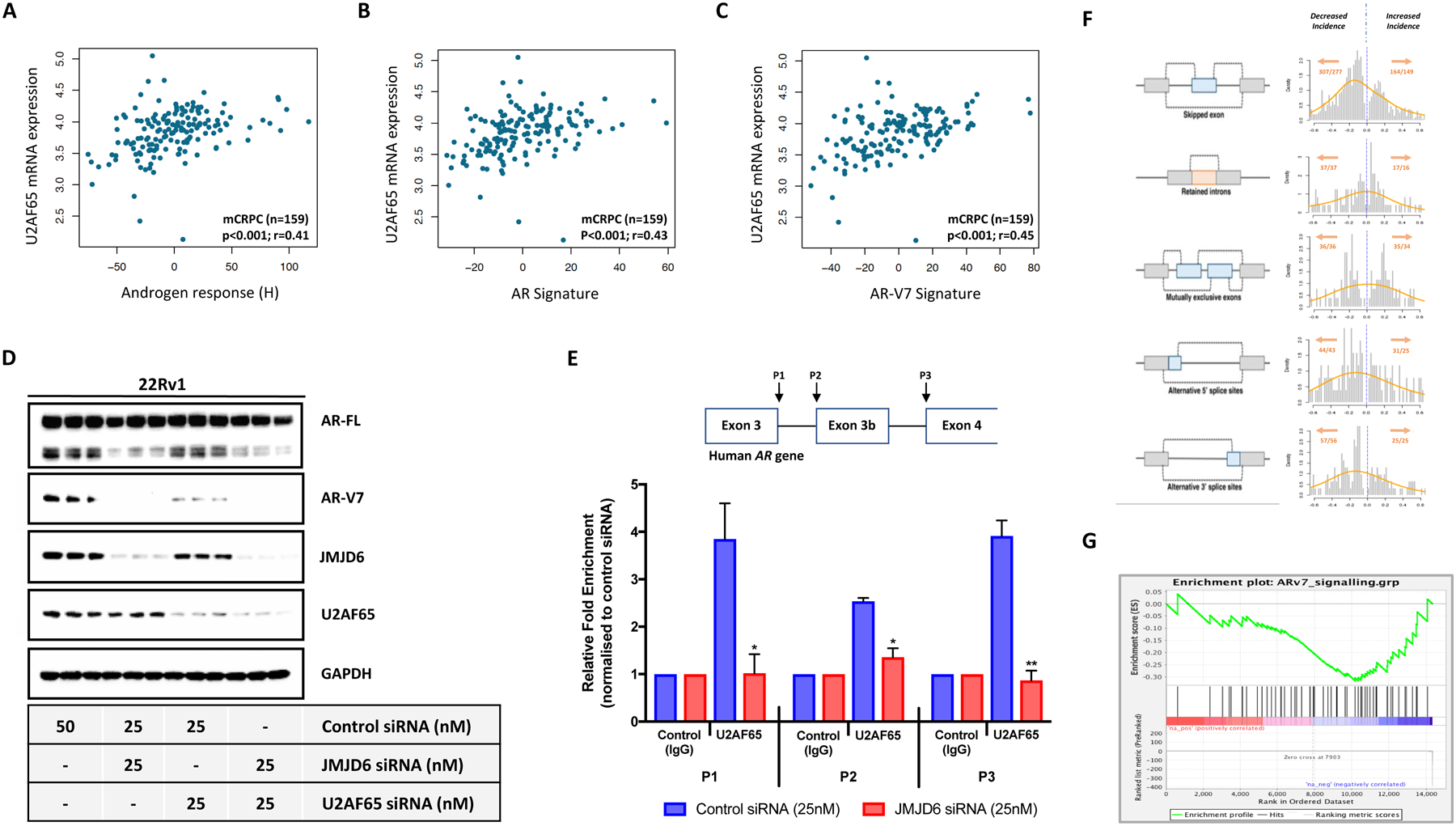
(A-C) Scatter plots showing correlations between JMJD6 mRNA expression and (A) androgen response (Hallmark; H), (B) AR signature (derived from 43 AR regulated transcripts) and (C) AR-V7 signature (derived from 59 genes associated with AR-V7 expression in mCRPC) in 159 mCRPC biopsies (SU2C/PCF cohort). U2AF65 mRNA expression shown as log FPKM. r-values and p-values are shown and were calculated using Spearman’s correlation. (D) Single WB in technical triplicate demonstrating reduction in AR-V7 protein levels with both JMJD6 and U2AF65 siRNA in 22Rv1 PC cells. JMJD6 siRNA had minimal impact on U2AF65 protein levels. (E) Schematic diagram of the human AR gene illustrating the regions targeted in RNA immunoprecipitation (RIP) assay with accompanying summary bar chart. Shows a reduction in detectable U2AF65 at the AR-V7 specific splice sites P1 (containing the 5’ splice site for both AR and AR-V7) and P2 (containing the 3’ splice site for AR-V7) in 22Rv1 PC cells treated with JMJD6 siRNA compared to non-targeting control siRNA. Indicates that JMJD6 regulates recruitment of the splicing factor U2AF65 to AR-V7 splice sites. RIP data derived from two independent experiments conducted in triplicate. p values (*, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001) were calculated for each condition compared to control (at equivalent concentration) using the mean value of technical replicates with unpaired Student’s t tests. (F) Schematic representation of alternative splicing events alongside corresponding histogram of alternative splicing mean differences between non-targeting control siRNA (blue dotted line; defined as 0.0) and JMJD6 siRNA in LNCaP95 PC cells. Left shift denotes decrease in splicing events. Total number of alternative splicing events (x) occurring in total number of genes (y) shown in orange (x/y). JMJD6 knockdown led to substantial changes in 753 alternative splicing events, with the majority of these occurring less frequently. (G) JMJD6 knockdown in LNCaP95 PC cells associated with a reduction in AR-V7 activity (derived from 59 genes associated with AR-V7 expression in mCRPC); Enrichment Score (ES) = −0.32.
To explore how JMJD6 regulates alternative splicing events in CRPC cells more broadly, RNA-seq analyses were performed of LNCaP95 PC cells prior to, and after, treatment with either JMJD6 siRNA or non-targeting control siRNA. Overall, JMJD6 knockdown led to substantial changes (determined by normalized-read count fold change >2 or <1/2 and false discovery rate <0.05) in 753 alternative splicing events involving 698 genes (figure 4F; Supplementary Table 12), with the majority of these occurring less frequently. Consistent with its assigned role in serine and arginine-rich (SR) protein modification and associated studies [26], these results indicate that JMJD6 knockdown reduces the overall incidence of alternative splicing events. Furthermore, in keeping with our previous results showing that JMJD6 knockdown downregulated AR-V7 expression (figure 3B–C and 3E–H), JMJD6 knockdown was found to reduce the mean AR-V7 signature score (figure 4G).
JMJD6-mediated AR-V7 generation is dependent on JMJD6 catalytic activity, which can be chemically inhibited to downregulate AR-V7 protein expression.
Having determined that JMJD6 regulates U2AF65 recruitment to AR-V7-specific splice sites, and given that JMJD6 has been previously demonstrated to hydroxylate U2AF65 [27], we next investigated the importance of a functional JMJD6 active site on AR-V7 levels. 22Rv1 PC cells were transfected with a JMJD6 wild-type (WT) plasmid (JMJD6WT) for 72 hours; WB and RNA analyses demonstrated increased expression of both AR-V7 protein and mRNA with JMJD6 overexpression (figure 5A). Conversely, transfection with inactivating mutations of active site residues in the JMJD6 catalytic domain by pcDNA3-JMJD6-ASM2 (MUT1; D189A and H187A) [16] and pcDNA3-JMJD6-BM1 (MUT2; N287A and T285A) markedly decreased AR-V7 protein levels (figure 5B). To validate these findings, both JMJD6WT, and the catalytically inactive mutant JMJD6MUT1, were next transfected into the VCaP PC cell line; AR-V7 expression was induced by JMJD6WT, but not by JMJD6MUT1 (figure 5C). Taken together, these results support the hypothesis that JMJD6-mediated increased expression of AR-V7 requires JMJD6 catalytic activity. Interestingly, the extent of AR-V7 upregulation in both 22Rv1 and VCaP PC cell lines was greater following transfection of lower concentrations of JMJD6WT compared to higher concentrations of JMJD6WT.
Figure 5: Evidence JMJD6-mediated AR-V7 generation is dependent on JMJD6 catalysis, which can be chemically inhibited to downregulate AR-V7 protein levels.
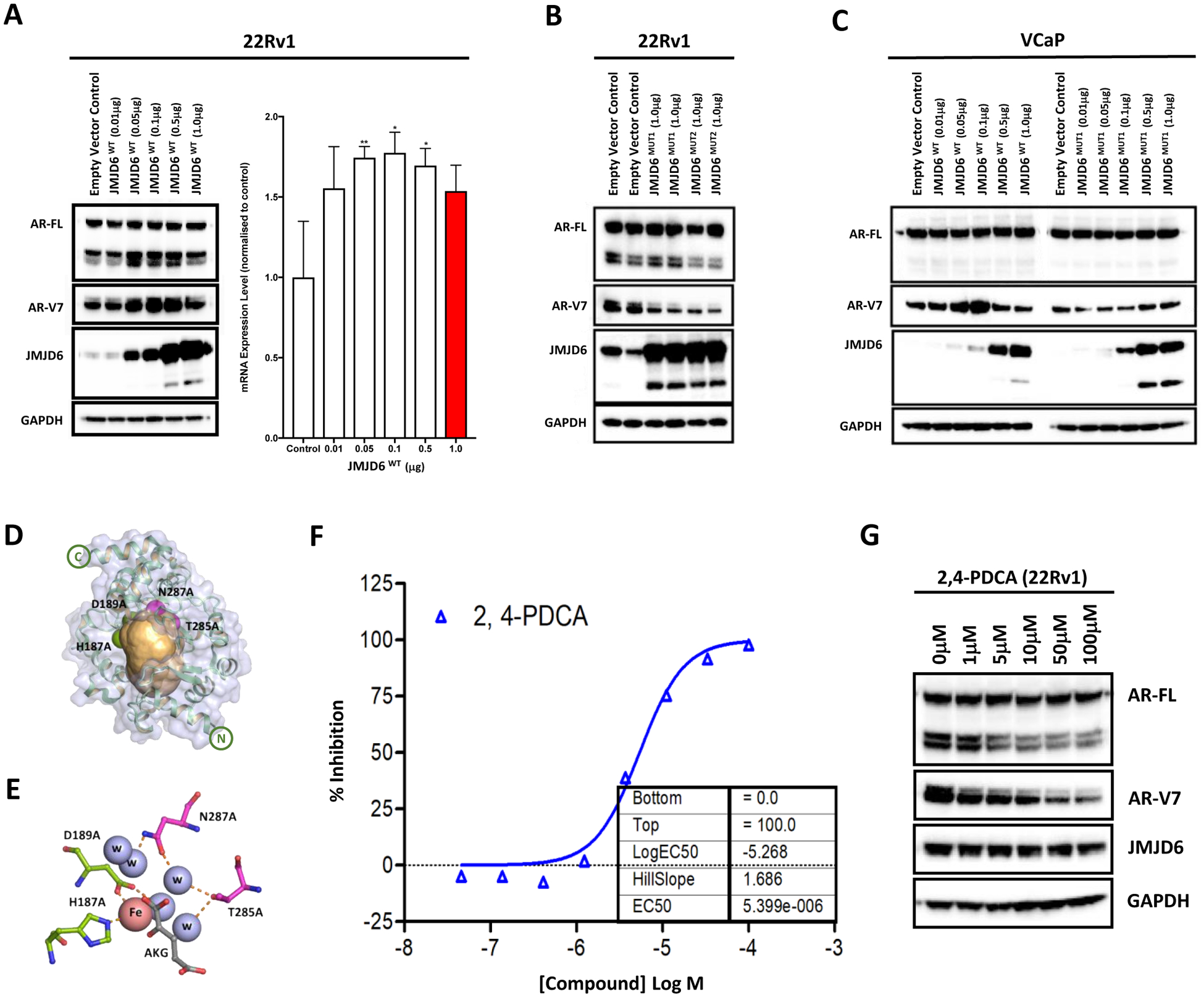
(A) Transfection of a JMJD6 wild-type (JMJD6WT) plasmid at increasing concentrations (all receiving 1μg of plasmid in total, with empty vector control added to make up the difference) into 22Rv1 PC cells led to an increase in AR-V7 protein (WB) and mRNA (qPCR) levels. Mean mRNA levels were normalized to housekeeping genes (B2M and GAPDH), and to studies with an empty vector control plasmid at equivalent concentration; the empty vector control data were defined as 1.0 with standard error of the mean from three experiments shown. p values (*, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001) were calculated for each condition compared to control (at equivalent concentration), using the mean value of technical replicates with unpaired Student’s t tests. (B) Conversely, transfection with inactivating mutations of active site residues in the JMJD6 catalytic domain by JMJD6MUT1 (D189A and H187A) and JMJD6MUT2 (N287A and T285A) decreased AR-V7 protein levels (empty vector control, JMJD6MUT1 and JMJD6MUT2 = 1μg of total plasmid). (C) AR-V7 expression was induced by JMJD6WT but not by JMJD6MUT1 in VCaP PC cells, suggesting that JMJD6-mediated AR-V7 expression requires active JMJD6. Singleton WB validating findings presented in (B) in an alternative cell line model. (D-E) Graphic representation of JMJD6 tertiary structure [58]. The inactivating substitutions of active site residues in the JMJD6 catalytic domain by JMJD6MUT1 (D189A and H187A; green spheres) and JMJD6MUT2 (N287A and T285A; magenta spheres) reside within a predicted druggable pocket (shown in orange), identified by the canSAR knowledgebase [33, 34]. (F) Liquid chromatography-mass spectrometry (LC-MS) analysis demonstrating that the 2OG mimic pyridine-2,4-dicarboxylic acid (2,4-PDCA) resulted in a dose-dependent reduction in isolated JMJD6-mediated lysyl-5-hydroxylation of its known target LUC7L; indicating that 2,4-PDCA is an inhibitor of JMJD6 lysyl hydroxylase catalytic activity. (G) WB showing that 2,4-PDCA caused a dose-dependent reduction of AR-V7 protein levels in 22Rv1 PC cells. Single representative WB shown from two separate experiments.
Importantly, studies of the physicochemical and geometric properties of JMJD6 with known drug targets such as protein kinases, interrogated using the canSAR drug discovery platform [33, 34], indicated that JMJD6 contains a ‘druggable’ pocket within its tertiary structure (defined as sites that harbor physiochemical and geometric properties consistent with binding orally-bioavailable small molecules [34]; (figure 5D–E; supplementary figure 4). Analogous pockets have been targeted in other 2OG oxygenases, in some cases leading to clinically approved drugs [35, 36]. Furthermore, consistent with crystallographic studies of JMJD6 [36, 37], these analyses demonstrated that the amino acids D189, H187A, N287 and T285, important for JMJD6 catalytic activity, lie within this druggable cavity.
To identify small molecule inhibitors of JMJD6 that would not disrupt its active site, liquid chromatography-mass spectrometry (LC-MS) analyses were performed. These identified the 2OG mimic pyridine-2,4-dicarboxylic acid (2,4-PDCA) as a JMJD6 inhibitor; 2,4-PDCA is a broad-spectrum, active site binding, 2OG-dependent oxygenase inhibitor [35, 38, 39]. 2,4-PDCA caused dose-dependent reduction in isolated JMJD6-mediated lysyl-5-hydroxylation of the known downstream target LUC7-Like (LUC7L) [15, 26] (figure 5F). Having confirmed 2,4-PDCA to be an inhibitor of JMJD6 lysyl hydroxylase catalytic activity, we subsequently treated 22Rv1 PC cells with 2,4-PDCA for 48 hours. As shown in figure 5G, 2,4-PDCA resulted in a dose-dependent reduction in AR-V7 protein levels, supporting our previous siRNA and mutagenesis experiments. Taken together, these results support the proposal that a functional JMJD6-active site is required for AR-V7 protein production, and show that the JMJD6 active site is druggable. Thus, JMJD6 is a viable therapeutic target for drug discovery efforts to abrogate oncogenic AR-V7 signaling.
Discussion
Resistance to PC endocrine therapies including abiraterone and enzalutamide is inevitable and invariably fatal, and at least in part driven by constitutively active AR-SVs that remain undruggable. We report that the 2OG-dependent dioxygenase JMJD6, which associates with worse prognosis and disease aggressiveness in multiple tumor types [40–43], plays an important role in PC biology, implicating JMJD6 in AR-V7 production. We demonstrate that JMJD6 is expressed in PC and increases significantly at castration-resistance, with this increase associating with AR-V7 protein overexpression in mCRPC biopsies and poorer survival. Our orthogonal investigations reveal JMJD6 to be critical for PC growth and a key regulator of AR-V7 expression. JMJD6 knockdown inhibits the upregulation of AR-V7 protein in response to AR blockade in hormone-sensitive VCaP PC cells. This is likely of therapeutic importance because for AR-V7 targeting to be successful, novel therapies are needed that can block AR-V7 generation rather than just counteract its oncogenic effects once EnR is established [13]. Moreover, the reduction in AR-V7 levels and PC cell growth seen following JMJD6 siRNA knockdown suggests limited functional redundancy, which is particularly striking given that recently two other 2OG-dependent JmjC-domain containing oxygenases, JMJD1A/KDM3A [44] and KDM4B [45], have also been reported to regulate AR-V7 generation. However, while JMJD1A/KDM3A and KDM4B are assigned as N-methyl lysine demethylases [46, 47], like other JmjC KDMs, other roles for them including N-methyl arginine demethylation are possible [48]. Given their roles in histone modification it is thus unclear as to what extent KDM4B/JMJD1A directly regulate AR splicing. Therefore, although it is likely that other 2OG-dependent JmjC-domain containing proteins play a role in the overall activity of the spliceosome machinery and AR splicing, albeit probably through alternative mechanisms, our results demonstrate that targeting the 2OG-dependent catalytic activity of JMJD6 is a promising PC drug discovery strategy. A better understanding of the interplay between these different proteins and the spliceosome machinery is now required.
Our in vitro results indicate that JMJD6 regulates the expression of AR-V7, at least in part, by modulating the recruitment of the splicing factor U2AF65 to AR-V7 specific pre-mRNA splice sites, which we have previously shown to be critical for the expression of AR-V7 [32]. Moreover, our evidence implies that the JMJD6-mediated regulation of AR-V7 expression is dependent on an intact JMJD6 catalytic site, which is in keeping with previous reports that JMJD6 lysyl-5-hydroxylates U2AF65 [27], and in doing so regulates U2AF65-mediated alternative splicing events [31]. Interestingly, however, the degree of AR-V7 upregulation in our studies was greater following transfection of lower concentrations of JMJD6WT compared to higher concentrations of JMJD6WT. This observation is consistent with, though does not prove, a role for JMJD6 induced catalysis in the increased production of AR-V7, as opposed to this occurring through a protein scaffold function of JMJD6 which would be expected to increase AR-V7 levels in line with JMJD6 levels. Given that JMJD6 is an Fe(II) and 2OG-dependent oxygenase, it is possible that the apparent dip in AR-V7 production despite the higher levels of JMJD6 reflects an inability of the cell to maintain optimal JMJD6 activity when it is overexpressed beyond a certain point due to a lack of Fe(II) and/or 2OG/dioxygen. Further work is required to investigate this intriguing potential link between cellular metabolism and transcriptional regulation. Importantly, our analyses reveal that the JMJD6 catalytic site resides within a druggable pocket, and we demonstrate that the known 2OG oxygenase inhibitor 2,4-PDCA, which we show to inhibit JMJD6 lysyl-5-hydroxylation, downregulates AR-V7 protein levels in castration-resistant PC cells. Taken together these findings point towards a JMJD6/U2AF65/AR-V7 regulatory pathway, wherein JMJD6 enzymatic activity, most likely through hydroxylation of U2AF65, and/or other SR proteins, regulates U2AF65 recruitment to AR-V7 specific splice sites, which then facilitates the generation of AR-V7 through interaction with the spliceosome. Given that JMJD6 has the potential to hydroxylate/interact with SR proteins other than U2AF65 [15, 27, 49, 50], and may have other cellular functions, its biological roles are likely widespread and context-dependent. However, in light of the crucial role of AR-SVs, especially AR-V7 in CRPC, therapeutic modulation of its spliceosome regulatory roles may be particularly suited to PC treatment. Our demonstration that inhibition of JMJD6 by a broad-spectrum 2OG oxygenase inhibitor downregulates AR-V7 levels should promote the pursuit of more potent and selective JMJD6 inhibitors in future drug discovery efforts.
Our conclusions may, however, depend on the molecular characteristics of the various models used. This is particularly relevant given the apparent pleiotropic roles of JMJD6 [49, 51]. However, it should be noted that this does not preclude therapeutically useful targeting of 2OG oxygenases, as shown by the clinical approvals of HIF prolyl hydroxylase inhibitors [52]. Aside from the likelihood of its multiple context dependent substrates and partners [15, 26], the activity of JMJD6 could be limited by (local or global) iron, dioxygen or 2OG availability, as is the case for some, but not all, 2OG oxygenases including the hypoxia inducible factor prolyl hydroxylases [53]. 2OG is a vital intermediate in the TCA cycle and is generated by processes such as glutaminolysis. 2OG levels vary depending on cell replication rate, hypoxia, androgen deprivation, and genomic aberrations (e.g. PTEN loss) common in PC [2], therefore it is possible that variations in 2OG levels impact JMJD6 activity and hence AR-V7 levels. Nevertheless, the results reported here have been replicated in a number of different cell lines with differing genomic backgrounds, supporting in vivo studies on the role of JMJD6 in PC.
Despite animal work demonstrating the importance of JMJD6 in development [49, 50], and extensive cellular studies, the lack of a validated downstream in vitro ‘read-out’ of physiologically relevant effects of JMJD6 catalysis is a significant obstacle in JMJD6 research. The effects of JMJD6 on AR-V7 levels are thus of general interest with resepct to the role of JMJD6 in splicing. However, whilst the plasmids and methods used herein have been previously characterized [16, 27], without an established, quantifiable marker of JMJD6 catalysis in our models other than AR-V7, it is not possible to definitively state that the changes on AR-V7 levels observed are dependent solely on catalysis by JMJD6. This is of particular relevance when considering our overexpression and mutagenesis experiments; we are unable to ascertain the level of functionality of the expressed JMJD6WT, nor that our mutants are completely inactive in cells where endogenous JMJD6 is present; this limits the strength with which inferences can be made on the importance of JMJD6 catalytic activity for AR-V7 expression. Thus, we cannot rule out that JMJD6-mediated regulation of AR-V7 involves a stoichiometric protein scaffold type interaction, which may or may not be linked to lysine-hydroxylation (or other JMJD6 catalyzed reaction). Indeed, a stoichiometric mechanism has been proposed for the AT hook domain of JMJD6 with respect to its role in adipogenesis in a manner independent of catalysis [54]. To investigate the role of JMJD6 catalysis in regulating AR-V7 levels therefore, we also employed inhibition of JMJD6 with the small molecule 2,4-PDCA, which we found inhibits JMJD6 lysyl-5-hydroxylation and downregulates AR-V7 levels, supporting the proposal that catalysis by JMJD6 is implicated in AR-V7 upregulation. However, we employed 2,4-PDCA as a tool to provide ‘proof-of-principle’ evidence that PC cell inhibition of JMJD6 is possible and impacts AR-V7 protein levels. At least in some cell types, the permeability of 2,4-PDCA is low, with high concentrations being required to elicit its effects in vitro [55, 56]. 2,4-PDCA itself is thus unlikely to be useful for in vivo studies, which is a limitation of our work. Furthermore, 2,4-PDCA is a broad-spectrum 2OG dioxygenase inhibitor and may inhibit other 2OG oxygenases, including JmjC-domain containing proteins. Therefore, although JMJD6 represents a ‘druggable’ target of considerable interest in PC, additional in vivo work employing potent and selective inhibitors of JMJD6 is required to definitively demonstrate that JMJD6 inhibition is a viable therapeutic strategy. Very recent studies describe more drug-like JMJD6 inhibitors with in vivo activity and minimal toxicity that merit study in PC models [57].
In conclusion, through orthogonal analyses we identify JMJD6 as being critical to PC cell growth and an important regulator of AR-V7 protein levels in preclinical models of CRPC. Furthermore, JMJD6 inhibition has potential to overcome oncogenic AR-V7 signaling, and is an eminently tractable new therapeutic target for mCRPC that merits further evaluation in in vivo studies.
Supplementary Material
Statement of significance.
We identify JMJD6 as being critical for the generation of AR-V7 in prostate cancer, where it may serve as a tractable target for therapeutic intervention.
Acknowledgments
We acknowledge Dr A. Böttger (Department of Biology, Ludwig Maximillians University, Munich, Germany), for kindly donating the plasmids with cDNA encoding pcDNA3-JMJD6-WT, pcDNA3-JMJD6-ASM2, and pcDNA3-JMJD6-BM1. Research Supported by an Stand Up To Cancer-Prostate Cancer Dream Team Translational Research Grant (SU2C-AACR-DT0712). Stand Up to Cancer is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C.
Funding
This research was funded by Cancer Research UK. Prostate Cancer UK Research Innovation Award (RIA17-ST2-014). AA was supported by European Research Council Consolidator Grant [683136]; Swiss Cancer League [4267]; Swiss National Science Foundation [176045]; Prostate Cancer Foundation [19CHAL08]; and Italian Association for Cancer Research Investigator Grant [22030]. CJS and AK thank CRUK (C8717/A18245) and the Wellcome Trust (106244/Z/14/Z) for funding. SRP gratefully acknowledges DOD Grants W81XWH-17-0323 and W81XWH-20-0146 (SRP and CTS) and DOD W81XWH-17-0414 (SRP) and Veterans Affairs Research Service Merit Award and Senior Clinician Scientist Research Award. AS also gratefully acknowledges previous research funding from the Academy of Medical Sciences, the Medical Research Council, and Prostate Cancer UK, and current funding from the Prostate Cancer Foundation and Wellcome Trust. JdB also gratefully acknowledges research funding from Prostate Cancer UK and the Movember Foundation through the London Movember Centre of Excellence (CEO13_2-002), the Prostate Cancer Foundation, Stand Up To Cancer, the UK Department of Health through an Experimental Cancer Medicine Centre grant, and the US Department of Defense. Professor Johann de Bono is a National Institute for Health Research (NIHR) Senior Investigator. The views expressed in this article are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Footnotes
Conflicts of interest
AP, JW, AN, WY, IF, RP, AF, RR, DNR, PDM, BG, RC, VG, LB, MC, SM, MBL, SC, NT, BAL, AS and JdB are all employees of The Institute of Cancer Research (ICR), which has a commercial interest in abiraterone. The ICR operates a Rewards to Inventors scheme through which employees of the ICR may receive financial benefit following commercial licensing. BAL is currently or has been a consultant and received fees from to Astex Pharmaceuticals, GSK and Difiniens AG (member of Astra Zeneca group) and received speakers honorarium from Astellas Pharma. BAL is a former employee of Inpharmatica Ltd. SRP is President of ESSA Pharma (Consultant), ProsTech Inc. AS has received travel support from Sanofi and Roche-Genentech, and speakers honorarium from Astellas Pharma. JdB has served on advisory boards and received fees from many companies including Astra Zeneca, Astellas, Bayer, Boehringer Ingelheim, Cellcentric, Daiichi, Genentech/Roche, Genmab, GSK, Janssen, Merck Serono, Merck Sharp & Dohme, Menarini/Silicon Biosystems, Orion, Pfizer, Qiagen, Sanofi Aventis, Sierra Oncology, Taiho, Vertex Pharmaceuticals. He is an employee of The ICR, which have received funding or other support for his research work from AZ, Astellas, Bayer, Cellcentric, Daiichi, Genentech, Genmab, GSK, Janssen, Merck Serono, MSD, Menarini/Silicon Biosystems, Orion, Sanofi Aventis, Sierra Oncology, Taiho, Pfizer, Vertex, and which has a commercial interest in abiraterone, PARP inhibition in DNA repair defective cancers and PI3K/AKT pathway inhibitors (no personal income). JdB was named as an inventor, with no financial interest, for patent 8,822,438. He has been the CI/PI of many industry sponsored clinical trials.
References
- 1.Visakorpi T, et al. , In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet, 1995. 9(4): p. 401–6. [DOI] [PubMed] [Google Scholar]
- 2.Robinson D, et al. , Integrative clinical genomics of advanced prostate cancer. Cell, 2015. 161(5): p. 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Bono JS, et al. , Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med, 2011. 364(21): p. 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scher HI, et al. , Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med, 2012. 367(13): p. 1187–97. [DOI] [PubMed] [Google Scholar]
- 5.Fizazi K, et al. , Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N Engl J Med, 2017. 377(4): p. 352–360. [DOI] [PubMed] [Google Scholar]
- 6.Scher HI and Sawyers CL, Biology of Progressive, Castration-Resistant Prostate Cancer: Directed Therapies Targeting the Androgen-Receptor Signaling Axis. Journal of Clinical Oncology, 2005. 23(32): p. 8253–8261. [DOI] [PubMed] [Google Scholar]
- 7.Dehm SM, et al. , Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res, 2008. 68(13): p. 5469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antonarakis E, et al. , Androgen receptor variant-driven prostate cancer: clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis, 2016. 19(3): p. 231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paschalis A, et al. , Alternative splicing in prostate cancer. Nat Rev Clin Oncol, 2018. [DOI] [PubMed] [Google Scholar]
- 10.Antonarakis ES, et al. , Clinical Significance of Androgen Receptor Splice Variant-7 mRNA Detection in Circulating Tumor Cells of Men With Metastatic Castration-Resistant Prostate Cancer Treated With First- and Second-Line Abiraterone and Enzalutamide. J Clin Oncol, 2017. 35(19): p. 2149–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Welti J, et al. , Targeting bromodomain and extra-terminal (BET) family proteins in castration resistant prostate cancer (CRPC). Clin Cancer Res, 2018. [DOI] [PubMed] [Google Scholar]
- 12.Pervaiz M, Mishra P, and Gunther S, Bromodomain Drug Discovery - the Past, the Present, and the Future. Chem Rec, 2018. 18(12): p. 1808–1817. [DOI] [PubMed] [Google Scholar]
- 13.Sharp A, et al. , Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J Clin Invest, 2019. 129(1): p. 192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Detre S, Saclani Jotti G, and Dowsett M, A “quickscore” method for immunohistochemical semiquantitation: validation for oestrogen receptor in breast carcinomas. J Clin Pathol, 1995. 48(9): p. 876–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heim A, et al. , Jumonji domain containing protein 6 (Jmjd6) modulates splicing and specifically interacts with arginine-serine-rich (RS) domains of SR- and SR-like proteins. Nucleic Acids Res, 2014. 42(12): p. 7833–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mantri M, et al. , Crystal structure of the 2-oxoglutarate- and Fe(II)-dependent lysyl hydroxylase JMJD6. J Mol Biol, 2010. 401(2): p. 211–22. [PubMed] [Google Scholar]
- 17.Winer J, et al. , Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem, 1999. 270(1): p. 41–9. [DOI] [PubMed] [Google Scholar]
- 18.Kim D and Salzberg SL, TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol, 2011. 12(8): p. R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen S, et al. , MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res, 2012. 40(8): p. e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashburner M, et al. , Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics, 2000. 25(1): p. 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abida W, et al. , Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A, 2019. 116(23): p. 11428–11436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carbon S, et al. , AmiGO: online access to ontology and annotation data. Bioinformatics, 2009. 25(2): p. 288–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramanian A, et al. , Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences, 2005. 102(43): p. 15545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liberzon A, et al. , Molecular signatures database (MSigDB) 3.0. Bioinformatics, 2011. 27(12): p. 1739–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liberzon A, et al. , The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst, 2015. 1(6): p. 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Islam MS, et al. , Biochemical and structural investigations clarify the substrate selectivity of the 2-oxoglutarate oxygenase JMJD6. J Biol Chem, 2019. 294(30): p. 11637–11652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Webby CJ, et al. , Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science, 2009. 325(5936): p. 90–3. [DOI] [PubMed] [Google Scholar]
- 28.Asangani IA, et al. , BET Bromodomain Inhibitors Enhance Efficacy and Disrupt Resistance to AR Antagonists in the Treatment of Prostate Cancer. Mol Cancer Res, 2016. 14(4): p. 324–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Bokhoven A, et al. , Molecular characterization of human prostate carcinoma cell lines. Prostate, 2003. 57(3): p. 205–25. [DOI] [PubMed] [Google Scholar]
- 30.Yu Z, et al. , Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin Cancer Res, 2014. 20(6): p. 1590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yi J, et al. , JMJD6 and U2AF65 co-regulate alternative splicing in both JMJD6 enzymatic activity dependent and independent manner. Nucleic Acids Res, 2017. 45(6): p. 3503–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu LL, et al. , Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene, 2014. 33(24): p. 3140–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bulusu KC, et al. , canSAR: updated cancer research and drug discovery knowledgebase. Nucleic Acids Res, 2014. 42(Database issue): p. D1040–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tym JE, et al. , canSAR: an updated cancer research and drug discovery knowledgebase. Nucleic Acids Res, 2016. 44(D1): p. D938–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rose NR, et al. , Inhibition of 2-oxoglutarate dependent oxygenases. Chem Soc Rev, 2011. 40(8): p. 4364–97. [DOI] [PubMed] [Google Scholar]
- 36.Yeh TL, et al. , Molecular and cellular mechanisms of HIF prolyl hydroxylase inhibitors in clinical trials. Chem Sci, 2017. 8(11): p. 7651–7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leung IK, et al. , Structural and mechanistic studies on gamma-butyrobetaine hydroxylase. Chem Biol, 2010. 17(12): p. 1316–24. [DOI] [PubMed] [Google Scholar]
- 38.Bonnici J, et al. , Inhibitors of both the N-methyl lysyl- and arginyl-demethylase activities of the JmjC oxygenases. Philos Trans R Soc Lond B Biol Sci, 2018. 373(1748). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thalhammer A, et al. , Inhibition of the histone demethylase JMJD2E by 3-substituted pyridine 2,4-dicarboxylates. Organic & Biomolecular Chemistry, 2011. 9(1): p. 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aprelikova O, et al. , The epigenetic modifier JMJD6 is amplified in mammary tumors and cooperates with c-Myc to enhance cellular transformation, tumor progression, and metastasis. Clin Epigenetics, 2016. 8: p. 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang F, et al. , JMJD6 promotes colon carcinogenesis through negative regulation of p53 by hydroxylation. PLoS Biol, 2014. 12(3): p. e1001819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, et al. , High expression of JMJD6 predicts unfavorable survival in lung adenocarcinoma. Tumour Biol, 2013. 34(4): p. 2397–401. [DOI] [PubMed] [Google Scholar]
- 43.Lee YF, et al. , JMJD6 is a driver of cellular proliferation and motility and a marker of poor prognosis in breast cancer. Breast Cancer Res, 2012. 14(3): p. R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan L, et al. , Histone demethylase JMJD1A promotes alternative splicing of AR variant 7 (AR-V7) in prostate cancer cells. Proc Natl Acad Sci U S A, 2018. 115(20): p. E4584–e4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duan L, et al. , Histone lysine demethylase KDM4B regulates the alternative splicing of the androgen receptor in response to androgen deprivation. Nucleic Acids Res, 2019. 47(22): p. 11623–11636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilson C and Krieg AJ, KDM4B: A Nail for Every Hammer? Genes (Basel), 2019. 10(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamane K, et al. , JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell, 2006. 125(3): p. 483–95. [DOI] [PubMed] [Google Scholar]
- 48.Walport LJ, et al. , Arginine demethylation is catalysed by a subset of JmjC histone lysine demethylases. Nat Commun, 2016. 7: p. 11974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bottger A, et al. , The oxygenase Jmjd6--a case study in conflicting assignments. Biochem J, 2015. 468(2): p. 191–202. [DOI] [PubMed] [Google Scholar]
- 50.Kwok J, et al. , Jmjd6, a JmjC Dioxygenase with Many Interaction Partners and Pleiotropic Functions. Front Genet, 2017. 8: p. 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kwok J, et al. , Jmjd6, a JmjC Dioxygenase with Many Interaction Partners and Pleiotropic Functions. Front Genet, 2017. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen R and Forsyth N, Editorial: The Development of New Classes of Hypoxia Mimetic Agents for Clinical Use. Frontiers in Cell and Developmental Biology, 2019. 7: p. 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mahon PC, Hirota K, and Semenza GL, FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev, 2001. 15(20): p. 2675–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reyes-Gutierrez P, Carrasquillo-Rodriguez JW, and Imbalzano AN, Promotion of adipogenesis by JMJD6 requires the AT hook-like domain and is independent of its catalytic function. PLoS One, 2019. 14(8): p. e0216015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tschank G, et al. , Pyridinedicarboxylates, the first mechanism-derived inhibitors for prolyl 4-hydroxylase, selectively suppress cellular hydroxyprolyl biosynthesis. Decrease in interstitial collagen and Clq secretion in cell culture. Biochem J, 1987. 248(3): p. 625–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kristensen LH, et al. , Studies of H3K4me3 demethylation by KDM5B/Jarid1B/PLU1 reveals strong substrate recognition in vitro and identifies 2,4-pyridine-dicarboxylic acid as an in vitro and in cell inhibitor. Febs j, 2012. 279(11): p. 1905–14. [DOI] [PubMed] [Google Scholar]
- 57.Zheng H, et al. , Jumonji domain-containing 6 (JMJD6) identified as a potential therapeutic target in ovarian cancer. Signal Transduct Target Ther, 2019. 4: p. 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berman HM, et al. , The Protein Data Bank. Nucleic Acids Res, 2000. 28(1): p. 235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.