

Abstract
The human body is home to a diverse and functionally important assemblage of symbiotic microbes that varies predictably over different spatial scales, both within and across body sites. The composition of these spatially distinct microbial consortia can be impacted by a variety of stochastic and deterministic forces, including dispersal from different source communities, and selection by regionally-specific host processes for the enrichment of physiologically significant taxa. In this chapter, we review the composition, function, and assembly of the healthy human gastrointestinal, skin, vaginal, and respiratory microbiomes, with special emphasis on the regional distribution of microbes throughout the gastrointestinal tract.
Keywords: human microbiome, composition, gut, skin, vagina, respiratory tract, regional, community assembly, biogeography
1. Introduction
The gradual discovery that trillions of symbiotic microbes not only occupy sites across the human body, but are often integral players in the healthy physiology of those sites, has been one of the most significant advances in biomedicine since germ theory first revolutionized the field in the 19th century. Indeed, we have likely been evolving and coevolving with our microbes since well before the dawn of our species, and the profound interdependence between host and microbe has thrown into question the very notion of human selfhood. Some would even argue that we must reconceive of human beings (and, for that matter, most multicellular life) as “holobionts,” or superorganisms whose evolutionary trajectory can only be considered in light of the composite phenotype conferred by both host and microbial genomes.
Especially over the past decade, scientists have amassed extensive evidence that many aspects of healthy bodily function are intimately dependent upon the particular assemblage of microbes that we carry and their respective genomes, or our “microbiome.” It therefore becomes extremely clinically relevant to understand which microbes appear where in the body, and what they are doing. This has been the focus of much foundational exploratory groundwork in the microbiome field, and particularly with the advent of culture-independent methods of bacterial quantification, we have tremendously expanded our knowledge of microbiome composition, function, stability, and resilience in health and disease, across a variety of body sites. In this chapter, we will review the current state of knowledge about the composition and function of the healthy human gastrointestinal, skin, vaginal, and respiratory microbiomes, with particular emphasis on regional specificity of microbes within the gastrointestinal tract, and the ecological processes that may contribute to community assembly in each of these habitats.
2. Ecological Principles for Understanding Community Assembly
2.1. Overview
As the microbiome field has matured, researchers have advanced beyond correlative investigations of which microbes appear under what circumstances and begun to pick apart the mechanisms for how these communities assemble, with an eye towards manipulating those processes and engineering the microbiome for therapeutic gain. Before diving into the particulars of which microbes appear in which parts of the body, we will briefly review some of the theory and evidence for how human-associated microbial communities assemble in the first place. As we proceed through the chapter, we will revisit these processes as they apply to different body sites.
The forces that govern microbial assembly appear to be complex and dynamic, involving host genotype, inter-microbial interactions, and environmental factors like diet. As such, many organizing principles for describing and understanding these community assembly processes have been borrowed from macroecology, a field explicitly focused on the complex interactions within and between species, and between species and their environments. Canonically, community assembly can be broken down into four fundamental processes, which then give rise to more complex phenomena. These processes are dispersal, selection, diversification, and drift (Figure 1). We will describe each of these briefly below (for more thorough review, see Vellend or Costello).1,2
Figure 1. Summary of ecological processes that impact community assembly.
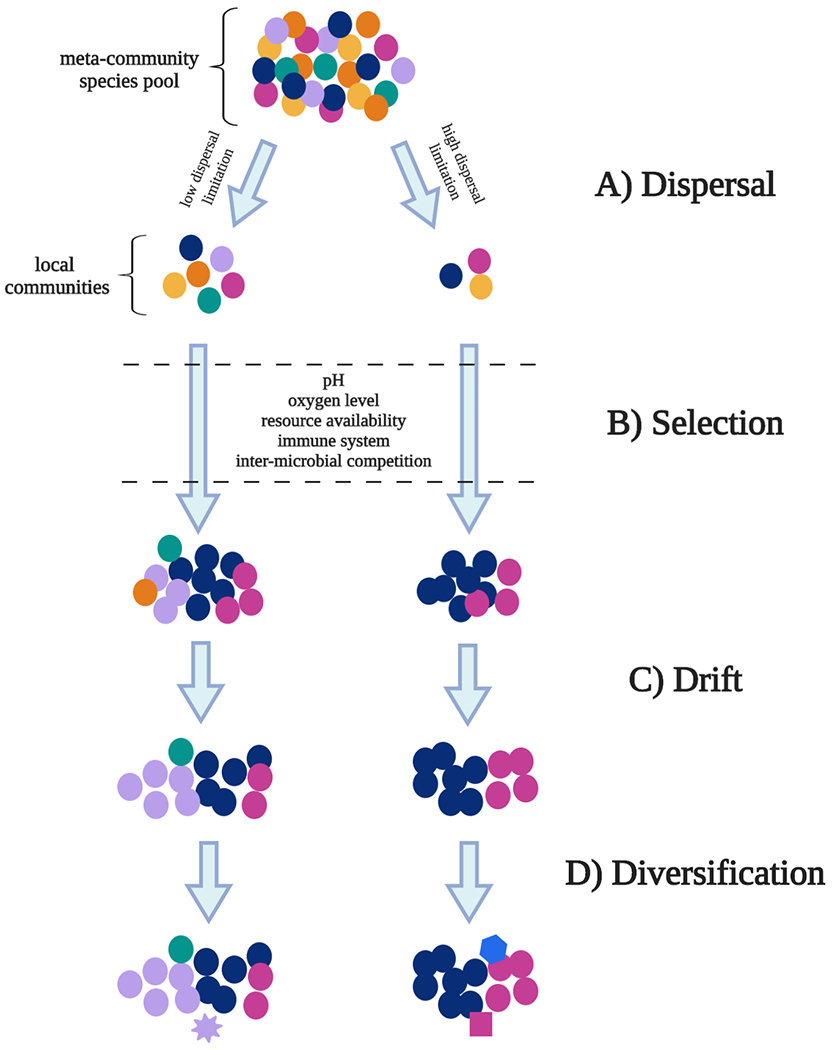
A) Dispersal refers to the immigration and emigration of microbes between local habitats. The meta-community species pool represents the collection of microbial species present across all local habitats. As species disperse between these habitats, they can be limited by factors such as motility and distance from the source community. B) Selection refers to the process whereby species better adapted to their environment tend to survive better and produce more offspring. Some potential habitat filters relevant to microbial life are listed between the dashed lines. C) Drift refers to random fluctuations in species abundances due to stochastic changes in birth and death rates over time. This can disproportionately impact low-abundance species, which can become extinct after a random dip in abundance. D) Diversification refers to the generation of new genetic variants within a population.
2.2. Dispersal
Dispersal refers to the immigration and emigration of microbes from one local habitat to another (Figure 1A).2 In the context of the human body, each individual can be thought of as an “island” or “patch” that draws its particular consortium of microbes from the broader pool of microbes available on nearby hosts and environments, or the “meta-community.” Thus, the possible diversity of microbes that could colonize our bodies is limited by the diversity of microbes in the meta-community, and by the ability of those microbes to physically move to our “patch.” For example, initial colonization of the infant gut microbiota is thought to occur largely by dispersal from the available microbes associated with either the mother or the hospital environment3. Depending on mode of delivery (vaginal birth or C-section), the neonate may be differentially exposed to maternal or environmental microbes, thereby limiting dispersal from those sources, and shaping the possible range of microbes the infant might acquire.
2.3. Selection
Selection is a deterministic evolutionary force by which better adapted variants within a population produce more offspring over their lifetime than poorly adapted competitors, and over time, displace those poorly adapted variants from the population (Figure 1B). These selective pressures often take the form of “habitat filters,” or environmental conditions (metabolic resource availability, pH, or adhesion sites, for instance) that limit which taxa are able to survive and grow in a given site. Thus, among the pool of microbes that have dispersed to a site and variants that have arisen via local diversification, those that cannot effectively utilize the available resources, and therefore cannot identify a suitable niche, will be driven from the community. Conversely, microbes that arrive in a site and utilize those resources especially effectively can experience positive selection, in which they grow more rapidly than other populations, and bloom to high levels of abundance in the community.
In the context of the microbiome, selective pressures can also be imposed by interactions with other microbes, host cells, and bacteriophage in the community, which can act to modify the available niche space. Even if a bacterial strain could theoretically survive using resources that are available in a given environment, in order for it to persist in that community, it has to be able to outcompete the other microbes that are likewise vying for those limited resources. Host cells and bacteriophage can also respond to the presence of particular microbes and act to suppress or promote certain populations directly or by modifying the environment.4,5
2.4. Drift
Ecological drift refers to random fluctuations in species abundances due to stochastic changes in birth and death rates (Figure 1C). Practically speaking, this means that low abundance species are more likely to go extinct from a community, especially in the wake of a perturbation, even if they may have been better adapted to that community than other taxa that happened to be present at higher abundance.6 Drift can play a particularly prominent role in community re-assembly after a strong perturbation like a course of antibiotics.7,8
2.5. Diversification
Diversification describes the generation of new genetic variants within a given population (Figure 1D). This could occur via mutation, recombination, or as is often the case in bacteria, by horizontal gene transfer. Notably, by contrast with larger-scale organisms, diversification in microbes occurs at a much faster rate owing to large population sizes, rapid reproductive rates, and high mutation rates.9 Recent data has shown that common human-associated bacteria like Bacteroides fragilis, for instance, can evolve specific, within-person adaptations over the course of that person’s lifetime, and horizontal gene transfer can allow for antibiotic resistant variants to arise rapidly within a community in the wake of an antibiotic perturbation.10,11 Diversification therefore provides a way for bacteria already present in a community to adapt to changing selective pressures.
2.6. Applications to the human microbiome
Within the human body, many of these forces may be at play. After all, different body sites constitute highly distinct ecological niches. The lung, for instance, is highly aerobic, whereas the GI tract is predominantly anaerobic; the vagina is comparatively acidic, and the skin is exposed to a barrage of variable conditions like humidity, salinity, and temperature. Within each of these sites, microbes also face different pressures from the host immune system, degrees of exposure to foreign microbes, and patterns of resource availability. Given this environmental variability across body sites, it stands to reason that habitat filtering and deterministic selective processes would play an important role in community assembly, and this is largely borne out by the data: evidence from the Human Microbiome Project has shown that human microbiota are more similar across individuals than across body sites.12 That is, my gut microbiome is more similar to your gut than my gut is to my elbow, suggesting that selective processes unique to the gut (or elbow) act to shape these communities in predictable ways.
Especially from a biomedical and translational perspective, it is tempting to assume that deterministic forces like selection are the primary drivers of microbiome composition. However, microbial ecologists have increasingly been making efforts to quantify and account for the extent to which more random, stochastic processes like dispersal and drift play into community assembly. For example, Sprockett et al. argue that microbiome researchers seeking to understand community assembly must account for priority effects and historical contingency, in which a community can be shifted towards an alternative stable state by chance early life exposures to a limited subset of microbes, which then occupy and/or modify the available niche space, impacting the ability of late-arriving microbes to colonize.13 This highlights the possibility that feedbacks between stochastic and deterministic processes can amplify differences in community assembly, allowing communities under similar selective pressures to undergo divergent compositional trajectories. This may explain some of the substantial variation in microbial composition that remains within body sites among healthy individuals.12
As we review the microbiome composition and function of the body sites below, we will attempt to highlight what is known about community assembly processes in each of those sites. Filling in the gaps in our understanding of these phenomena is sure to be a central focus of future research efforts.
3. Gastrointestinal Tract
3.1. Overview
While all of the body sites reviewed in this chapter are topologically external, the gastrointestinal tract (GIT) is exceptional for the sheer volume of challenges it experiences from the outside world, and for the wide diversity of functional roles it must play as it incorporates defense into the processes of digestion and absorption. Every time we eat, our GIT must identify and neutralize any potentially hazardous elements that hitchhike into the gut with the foreign matter we have ingested, while simultaneously breaking down and converting nutrients from that same matter into a form that can be easily absorbed, all the while sensing and providing feedbacks to the rest of the body. Thus, the GIT is home to not only a vast array of anatomical structures and biochemical processes that partition the work of digestion and absorption, but also sensory, neurological, and endocrinological networks, as well as complex immunological and ecological systems that are integral to defense and homeostasis.
Given the tremendous diversity of functions that must be performed by the GIT, it is perhaps unsurprising that the gut environment does not consist of a single uniform ecological niche, but rather a heterogeneous collection of distinct habitats along the rostral-caudal axis, which are home to the most abundant and diverse microbiota in the human body.12 Along this axis, there are several critical gradients: microbial load and diversity increases from the rostral to caudal end, oxygen levels decrease, mucus thickness varies, and pH dips in the stomach and increases distally from there (Figure 2).14–16 Each region of the GIT – which we will break down into oral cavity, esophagus, stomach, small intestine, and colon – performs locally specialized digestive, metabolic, immune, and endocrinological functions. This means there is local variation in nutrient bioavailability, GI transit time, and secretion of immune factors and bile acids, among other things.15,17 These distinctions impact local microbial composition in significant and often predictable ways, and the functions performed by such locally distinct microbial assemblages can then feed back into local host processes for a mutually beneficial symbiosis, or in disease states, a “dysbiotic” microbiota can contribute to impairment of host functionality.
Figure 2. Ecologically relevant spatial gradients across the gastrointestinal tract.
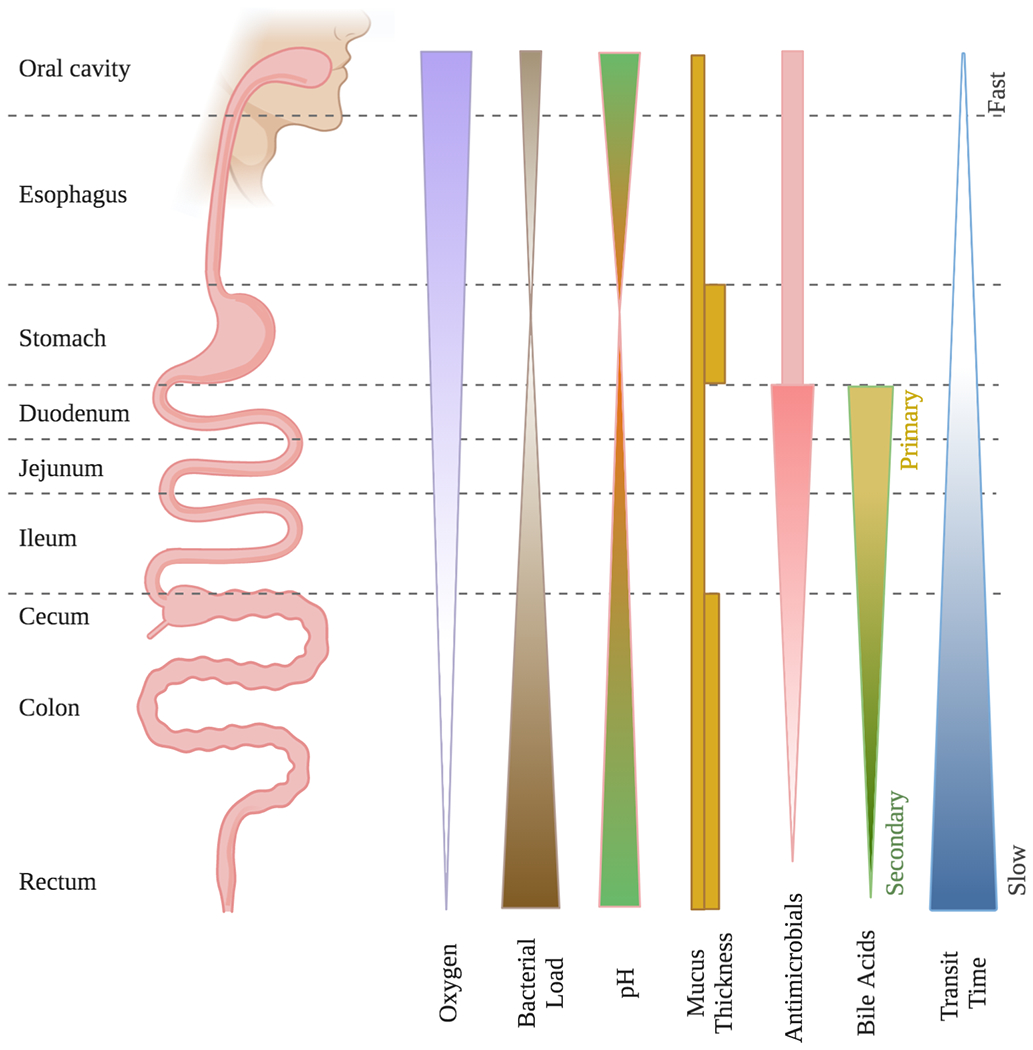
Rostrally to caudally throughout the gastrointestinal tract, a variety of physiological and biochemical features vary in accordance with the distinct digestive, absorptive, defensive, and endocrinological functional roles of each region. These factors, including oxygen level, pH, mucus thickness, antimicrobial production, bile acid production, and transit time, in turn impact the structure and function of regional microbial community membership.
Despite extensive efforts to characterize the microbiomes of healthy individuals, a concrete definition of the “healthy” gut microbiome has been elusive. In part, this is because of substantial microbial diversity even within the same body site among healthy individuals, especially at higher taxonomic resolution (that is, genus, species or strain level).12,18 Some of that variation may be attributable to idiosyncratic combinations of stochastic and deterministic assembly forces at play. Nevertheless, common definitions of the healthy gut microbiome have come to center around functional potential, rather than taxonomic composition, as well as stability over time, and resilience in the face of perturbation.19 Here, we describe some such compositional and functional characteristics of the “healthy” regional gastrointestinal microbiota.
3.2. Oral Cavity
Serving as the entry point for both the GIT and the respiratory tract, the oral cavity is responsible for a wide variety of functional roles, serving as a critical respiratory conduit, as well as the initiating site for many digestive processes. These include mastication, moistening, the first stages of enzymatic breakdown, as well as taste and other sensory reception, which can play an important role in defense against toxic or pathogenic agents. As the first opportunity for encounter between invading pathogens and the GIT, the oral cavity also plays a pivotal immunological role. Reflecting this diversity of functional roles, the oral cavity is home to a suite of anatomical and functional adaptations that create a rich assortment of spatially distinct habitats, the relative accessibility of which has paved the way for high-resolution spatial sampling of the oral microbiota. Recent work has estimated that some 200-500 unique bacterial species inhabit the mouth, reaching abundances of ~20 million individual cells.20
Anatomically, the mouth contains a variety of distinct habitats and topological features, including lips, teeth, gingiva, tongue, cheeks, hard and soft palate, and tonsils. The first key distinction between these features is whether they have shedding or non-shedding surfaces: the only non-shedding surface in the mouth is the dental enamel, while the remainder of the oral mucosa are covered in stratified squamous epithelial cells that regularly turnover and slough off. The non-shedding dental enamel, as a more permanent structure in the mouth, allows for a buildup of plaque, or bacterial biofilms. These multi-species structures are fascinating examples of microbial cooperation and successive community assembly: a base of filamentous Corynebacterium tendrils provides a scaffold upon which organized consortia of taxa including Streptococcus, Haemophilus/Aggregatibacter, Porphyromonas, Capnocytophaga, and Fusobacteria attach in “hedgehog” structures according to micron-scale biochemical gradients.21 Plaques above and below the gum line – that is, supra- and subgingival plaques, respectively – also contain distinct microbiota, in part due to the capture and accumulation of nutrients and immune molecules in the crevices between gingiva and teeth.22
Although the dominant members of supragingival plaques remain abundant across the mouth, the less abundant members of these communities vary by tooth class (incisor versus molar) and tooth aspect (buccal or lingual).23–25 Data from Proctor et al. suggests that this is because salivary flow is a major selective force in the oral cavity: as saliva flows from the back to the front of the mouth from various major and minor salivary glands, it creates biophysical gradients of moisture and pH, while also acting to physically shuttle bacteria around, release or clear metabolites from partially digested food, and stimulate secretion of mucin.26,27 These gradients select for an enrichment of taxa like Veillonella on the molars, and Streptococcus and Actinomyces on the incisors.25 Microbially-mediated diseases like periodontitis and dental caries are commonly associated with hyposalivation, suggesting that in the absence of salivary flow and associated microenvironmental gradients in the oral cavity, dysbiotic communities can assemble.26,28,29
By contrast with the non-shedding dental enamel, the soft tissues of the oral mucosa feature shedding epithelial cells that turnover approximately every two to three weeks.30 Microbial communities on keratinized (hard palate, tongue dorsum, keratinized gingiva) and non-keratinized (buccal and labial mucosa, tongue ventrum, soft palate) tissues are hypothesized to vary according to differences in adhesion mechanisms and proximity to salivary glands.26,31,32
As the initial site of ingestion, the oral cavity is also exposed to a number of pathogens, and a major selective force in the assembly of this community is constant basal activation of the immune system.33,34 Thus, given the diverse assortment of microbes that can disperse into the oral cavity, regular surveillance and basal immune activity, via the IL-17 pathway for instance, is responsible for filtering out potentially pathogenic agents.35 Symbiotic residents of the oral microbiome such as Streptococcus, Veillonella, and Granulicatella have been shown to contribute to these defensive processes, stimulating an increase in the production of immune effectors like anti-microbial peptides (AMPs) and pro-inflammatory cytokines, and increasing epithelial barrier function and mucosal thickness.36
3.3. Esophagus
By contrast with the oral cavity, the esophagus is relatively uniform in anatomy and function. Flanked by the upper and lower esophageal sphincters, the esophagus is primarily responsible for shuttling food from the oral cavity into the stomach. It is covered in non-keratinized stratified squamous epithelial cells and coated in mucus secreted by submucosal glands.
Although a multitude of microbes pass through the esophagus with ingested food in transit to the stomach, these are thought to be largely transient.37 The more stable resident population of the esophagus resides attached to the mucosal surfaces, which can only be sampled via an invasive upper endoscopy procedure with brushings or biopsy, severely limiting characterization of this body site. Nevertheless, a small number of studies have found that the esophageal mucosa are primarily colonized by ingested microbes from the oral cavity, exhibiting a high degree of compositional overlap, with representation from the same major taxa including Streptococcus, Prevotella, and Veillonella.37,38 Unlike the oral cavity, the esophagus shows a notable absence of Spirochaetes. These patterns are consistent with dispersal-limited community assembly, in which the microbes that colonize the esophagus are a subset of the meta-community present in the oral cavity.
3.4. Stomach
The stomach plays critical digestive, defensive, and endocrinological roles in the GIT. Ingested material that reaches the stomach is mechanically broken down through peristaltic churning, chemically degraded by hydrochloric acid, and enzymatically broken down by zymogen proteases like pepsinogen, which is activated in the acidic stomach environment. The stomach also senses and regulates food intake, motility, and appetite through various mechanisms including stretch mechanoreceptors and production of hormones like gastrin and ghrelin. These functions both impact and are impacted by the gastric microbiota.
These are harsh conditions for microbial life. The low pH of the stomach (~1.5 – 2.5) constitutes a fairly extreme habitat filter which, in addition to serving as a defense mechanism against ingested pathogens, significantly limits the diversity of commensal microbes that are able to survive there. Historically, this led scientists to believe that the stomach was completely sterile, until Helicobacter pylori, an infectious, acidophilic bacterium that contributes to the development of peptic ulcers, was discovered thriving in the gastric mucosa of affected individuals.39 Recently, however, culture-independent techniques have demonstrated that although overall bacterial load is relatively low in the stomach (~101 – 103 CFU/ml), there is representation of diverse phyla, including Streptococcus, Prevotella, Veillonella, and Rothia.40–42 More than 65% of the taxa identified in the stomach are members of the oral microbiota, although many of these, identified in the gastric juice, are thought to be largely transient.43
Community assembly in the stomach can be understood as a balance between dispersal from the oral cavity and the duodenum, and selection driven largely by pH, a dominant habitat filter. This is supported by evidence showing that higher pH in the stomach corresponds to overgrowth of taxa likely derived from oral and intestinal source communities.44 Acidity levels can be impacted by use of medications such as proton pump inhibitors, or even resident microbes: H. pylori, which has been documented at ~50% prevalence in asymptomatic adults in the USA, is well known to reduce stomach acidity.45,46 By breaking down urea into ammonia, a basic substance that neutralizes stomach acid, H. pylori can burrow into the gastric mucosa, where it stimulates an immune response that results in atrophy of the cells lining the stomach, further reducing the secretion of acid.43,47 As the stomach de-acidifies, it becomes more amenable to microbes that disperse from either the oral cavity or the duodenum, ultimately contributing to a “dysbiotic” disease state.44
Although H. pylori is commonly regarded as an endemic pathogen of sorts in the modern age, it has been hypothesized that, before industrialization, the pro-inflammatory IL-17 response provoked by this highly prevalent microbe served a protective role against other infectious pathogens.48,49 Since then, germ theory was born, hygiene practices have evolved, and antibiotics have been developed, causing the prevalence of such infectious agents to decrease, and rendering the pro-inflammatory impacts of H. pylori mismatched to our “clean” environment. Thus, H. pylori is now most commonly associated with peptic ulcer disease and gastric cancers (see Blaser for a fascinating review of H. pylori’s disparate roles in upper and lower gastric disease).50
3.5. Small Intestine
The small intestine is the primary site of nutrient absorption in the GIT. It features a single layer of absorptive columnar intestinal epithelial cells (IECs) interspersed with a variety of specialized cells including goblet cells, Paneth cells, microfold cells, and enteroendocrine cells that aid in digestion, defense, and secretion. The apical surface of these IECs maximizes surface area for absorption, featuring crypts, villi, and microvilli, and is coated in a single, loosely attached layer of mucus that is largely free of microbes.15,51 Compared to the large intestine, the small intestine is more aerobic, more acidic, secretes higher levels of anti-microbial molecules, and has much shorter transit time.15 Microbes that colonize these regions must not only survive these habitat filters, but must compete for nutrients with both other bacteria and host cells. Therefore, very generally, the small intestine harbors an abundance of fast-growing, facultatively anaerobic bacteria that can utilize carbohydrate energy sources, have robust AMP tolerance mechanisms, and can adhere to mucus, dietary substrate, or epithelial tissues despite rapid GI motility.15,52
To facilitate the absorption of nutrients into the IECs, the mucus coating of the small intestine is much thinner than in other regions of the GIT. Thus, the small intestine faces a trade-off between the defensive barrier functions that a thick, secondary mucus layer could provide, and the ability to effectively digest and uptake nutrients from the intestinal lumen. To combat this, the small intestine features special immunological adaptations like Paneth cells in the base of the crypts, which secrete copious amounts of AMPs like lysozyme and α-defensins into the mucus layer, and abundant plasma cells that release secretory IgA into the mucus, thereby inhibiting microbial colonization.53–55 The small intestinal epithelia overlay specialized gut-associated lymphoid tissue (GALT) and Peyer’s patches in the mucosa and submucosa that function in presentation of antigens and activation of the adaptive immune system.56 As a consequence of this extensive immune activity, some commensal microbes in the gut have been shown to possess unique antigen modifications to escape innate immune mechanisms.57
The study of the unique functional roles of the small bowel microbiota in healthy host physiology is an often-overlooked but nascent sub-field within medical microbiome research. While this is largely due to the relative inaccessibility of small intestinal sites compared to fecal sampling, recent work in animal models has begun to establish an essential role for the small bowel microbiota in host metabolism and nutrient absorption. For instance, evidence from Martinez-Guryn et al. shows that, like the well-characterized colonic microbiota, the small bowel microbiota of mice is dramatically impacted by Western-style high-fat (HF) diet, and in fact plays an indispensable role in triglyceride digestion and absorption.58 This work showed that small intestinal microbes are necessary for the release of pancreatic lipase in response to dietary lipid, indirectly aiding in host lipid digestion. Moreover, the authors showed that metabolites released by taxa like Clostridiaceae that bloom under HF conditions interact with absorptive enterocytes of the duodenum and jejunum to enhance the uptake of free fatty acids. Thus, as the host consumes more fat, the microbiome shifts and enables the host to absorb that fat. This is a striking example of the bidirectional crosstalk between host and microbe that is essential for the maintenance of healthy host physiology, especially in the face of rapidly changing conditions like variable diet.
Proximally to distally from the stomach, the small intestine can be divided into three segments, each of which features distinct functional roles, anatomical features, and microbial inhabitants: the duodenum, the jejunum, and the ileum. We will describe each of these regions below.
3.5.1. Duodenum
The duodenum comprises the first 10 - 15 inches of the small intestine. It is a site of significant secretory and digestive activity, with mucus from goblet cells protecting the intestinal epithelium from the acidic gastric chyme, bicarbonate from the pancreas bringing the pH back up into the neutral zone, and bile and pancreatic enzymes streaming in to aid in the breakdown and absorption of carbohydrates, fats, and proteins. Anatomically, the duodenal epithelia feature crypts and long villi with a microvilli “brush border” packed full of digestive enzymes for secretion and nutrient transporters for uptake. Immunologically, the duodenum features submucosal GALT, although there are fewer Peyer’s patches than in the jejunum and ileum.56
These features impact the duodenal microbiome in particular ways. First, a fairly high level of swallowed oxygen, as well as oxygen diffused through duodenal tissues, prevents the establishment of more strictly anaerobic microbes. Second, rapid motility of the duodenum may prevent many microbes that disperse from upper segments of the GI tract from having time to adhere or to establish. Thus, bacterial load is fairly low, in the range of 101 - 103 CFU/ml, and tends to be dominated by bacteria from the phyla Firmicutes, Actinobacteria, and Proteobacteria, which contain many facultative anaerobes.59,60
Primary bile acids (BAs), which are synthesized from cholesterol in the liver, are deposited via the bile duct directly into the duodenum. These amphipathic molecules act as a detergent that aids in the absorption of lipids by emulsifying fat globules. BAs, however, have been shown to exhibit strongly bactericidal activity, and therefore act as a selective pressure for more bile-tolerant microbes throughout the small intestine.15,61 Bile salt hydrolases (BSH) are enzymes that deconjugate primary BAs, and they are widespread across the microbial inhabitants of the gut.62 It is hypothesized that these BSH enzymes confer increased tolerance to primary BAs, which could be especially important in the proximal small intestine where primary BAs are most abundant.63–65
Exciting new work on extracellular electron transfer (EET) also suggests that iron, which is particularly abundant in the duodenum and jejunum, may be an important metabolic resource for gut bacteria that can utilize EET to produce energy.66 Analogous to mammalian intracellular oxidative phosphorylation, these bacteria can use extracellular minerals like iron as an electron acceptor, generating electricity and ATP, and supporting bacterial growth on non-fermentable carbon sources. EET pathway orthologues have recently been identified in the genomes of a wide variety of pathogenic and commensal Firmicutes species, suggesting that EET may be a significant competitive adaptation in the resource-limited environment of the proximal small intestine.
3.5.2. Jejunum
The jejunum harbors many of the same functional and anatomical features as the duodenum, but with less oxygen, neutral pH, longer villi, increasing numbers of Peyer’s patches, slower transit time, and an altered nutrient profile, dependent on what has already been digested or absorbed in the duodenum. Compared to the duodenum, the jejunum performs fewer digestive functions and is more specialized for absorption, serving as the primary absorption site for carbohydrates, small peptides, and vitamins and minerals like folate, calcium, magnesium, and other trace elements.67
Because the jejunum is only accessible by endoscopy in humans, studies investigating its microbial composition in healthy individuals are few and far between. Early work related to small intestinal bacterial overgrowth syndrome revealed that the typical bacterial load of the jejunum in healthy individuals is approximately 103 – 105 CFU/ml.68 Subsequent culture-independent analyses have confirmed this estimate, and revealed that compositionally, the jejunum is dominated by a number of facultatively anaerobic genera including Streptococcus, Prevotella, Veillonella, Rothia, and Fusobacterium, as well as Escherichia, and Klebsiella.69 Although mucosal and luminal jejunal samples have not been simultaneously collected from the same individuals for direct comparison, independent studies of the mucosal and luminal jejunal microbiota have identified representation of similar taxa and abundances.69–71
3.5.3. Ileum
The ileum continues along the same broader environmental gradients: lower oxygen levels, slower transit time, and shorter, broader villi with fewer digestive enzymes. Digesta that arrive in the ileum have been largely depleted of nutrients that are useable by the host, and its primary absorptive roles are uptake of vitamin B12 and reuptake of bile acids.67 While Peyer’s patches are sparse in the ileum, it has an abundance of AMP-secreting Paneth cells in its crypts, and a relatively high number of mucus-secreting goblet cells that create a patchy mucus layer of variable thickness.56,72
Here, moving distally, the gut environment begins to approach the optimal conditions for microbial growth that characterize the colon: because the host has already taken up the majority of nutrients it is capable of using, microbes that inhabit the distal ileum are released from competition with host cells for remaining resources. Moreover, the trade-off between absorptive capacity and defense has been largely relieved, as nutrient absorption is less important in the ileum, and therefore immune mechanisms that are thought to reduce microbial density in the proximal small intestine are relaxed.15 Consequently, we see an increase in microbial load by several orders of magnitude compared to the jejunum, with estimates ranging from 103 – 108 CFU/ml.56
Because the ileum is difficult to sample without invasive procedures, insights into its composition have mostly been gleaned from patient populations with ileostomies, or those undergoing radical cystectomy.52,73,74 With the caveat that such patient populations typically have underlying health conditions that may or may not impact the composition of the ileal microbiota, these studies have revealed that the ileum is home to both facultative and obligate anaerobes, including taxa from the genera Streptococcus, Granulicatella, Actinomyces, Solobacterium, Rothia, Gemella, and TM7(G-1).52,73 This is significantly different than the colonic microbiota, with less representation of strictly anaerobic families like Clostridiaceae, Peptostreptococcaceae, or Eubacteriaceae, and an enrichment for facultative anaerobes like Streptococcaceae and Lactobacillaceae, although this may be due in part to differences in oxygen tension in ileostomy patients.73
Although immune release is thought to be one reason why bacterial load in the ileum is higher than in other parts of the small intestine, mucosal immunity still plays a pivotal role in shaping microbial composition in the ileum, and vice versa.75 Abundant production of AMPs such as cathelicidins, C-type lectins, and defensins by Paneth cells create a defensive border between epithelial cells and microbial inhabitants. However, production and secretion of these immune effector molecules is dependent upon stimulation by commensal microbes: germ-free mice show reduced levels of AMPs like the Gram-positive-targeting REGIII-γ, which is restored upon conventionalization with cecal contents from specific-pathogen free mice.76 Immune stimulation by commensal microbes can play an important role in colonization resistance against certain pathogenic organisms, as well. For instance, mice lacking segmented filamentous bacterium, a microbial resident unique to the terminal ileum, fail to develop Th17 cells, and are consequently highly susceptible to infection by pathogens like Citrobacter rodentium and Salmonella typhimurium.77,78
Ileal and colonic microbes also play an important role in the maintenance of host circadian rhythmicity. Circadian rhythms refer to periodic 24-hour oscillations in gene expression across body tissues, which act to coordinate homeostatic functions including the sleep-wake cycle, feeding behavior, and glucose and lipid metabolism.79 By temporal partitioning of various catabolic and anabolic processes, circadian rhythms are able to curtail inefficiency in resource use by the body, and dysregulation of these rhythms has been associated with chronic metabolic disorders like diabetes and obesity.80 Although the central circadian clock is located in the suprachiasmatic nucleus of the brain and is primarily entrained by photic cues, oscillatory dynamics in peripheral tissues like the gut can also be entrained by feeding behaviors and microbial stimuli.81,82 In particular, Wang et al. have shown that, in the small intestine, rhythmic, circadian expression of Nfil3 is regulated by the intestinal microbiota, and that antibiotic-induced loss of Nfil3 rhythmicity is associated with increased body weight and fat storage.83
3.6. Colon
The colon is home to the most abundant assortment of microbes in the human body, containing orders of magnitude more microbes than all other body sites combined with bacterial density on the scale of 1010 – 1012 CFU/ml.59 This is promoted by the specific anatomical and functional features unique to this region.
Like the small intestine, the colon features a single layer of polarized columnar intestinal epithelial cells with divot-like crypts, but unlike the small intestine, the colonic epithelium lacks villi and microvilli. Goblet cells in the colonic epithelia secrete a thick, two-layered mucus coat, with a dense inner layer that stays largely sterile, and a loose outer layer that harbors an abundance of specialized microbes.16,51 The colon is highly anaerobic, and the digesta that pass through consist primarily of complex polysaccharides and fibers that could not be digested by host processes, as well as trace nutrients and any remaining bile acids that were not absorbed in the ileum. Motility in the colon is much slower, with typical transit time of up to 30 hours.84
These features allow microbes to thrive for a number of reasons. First, fermentative metabolism is widespread in bacteria, and the anaerobic environment and abundance of fibrous substrates in the large intestine create optimal conditions for this process. Second, the slow transit time through the colon allows microbes plenty of time to adhere, consume, multiply, and expand in physical space. Thus, because they are not being regularly flushed through the system, microbes in the colon can accumulate to higher levels. Third, the colon provides a number of spatially distinct niches that can support different microbial communities, from the colonic crypts, to the outer mucus layer, to the inter-fold regions of the lumen and central lumen. This spatial heterogeneity allows for niche partitioning of limited resources, increasing the total level of diversity that can be supported.85 Finally, as long-term, co-evolved symbionts, microbes in the colon have developed unique adaptive mechanisms of immune evasion and modulation to avoid being actively removed from the gut by host defense mechanisms.57,86
Microbial synthesis of short-chain fatty acids (SCFAs) through fermentative metabolism of complex polysaccharides is the canonical mutualistic function of the gut microbiome: this was perhaps the first major physiological function discovered to depend wholly on the gut microbial “organ.” In this catabolic process, fibrous substrates like cellulose, pectin, inulin, and high-amylose starch, or host-derived mucosal glycans, are fermented anaerobically by microbial symbionts across the Firmicutes and Bacteroides phyla, releasing SCFAs like acetate, proprionate, and butyrate.87 These SCFAs are salvaged by host tissues, contributing an estimated 5 – 15% of the total caloric requirement for humans.88 While all of these SCFAs can feed into various host and microbial metabolic pathways, butyrate in particular serves as the preferred energy source for colonocytes.89,90 These cells oxidize butyrate into CO2, thereby depleting the oxygen supply in the colon and promoting an anaerobic state that is important for pathogen resistance, immune homeostasis, and of course, the growth of butyrate-producing anaerobic microbial populations, in a classic positive feedback loop.87,90–92
Microbial composition, and therefore fermentation and metabolism, in the colon can be dramatically affected by the components of the diet consumed by the host. One early study examined human subjects who switched between an animal- and plant-based diet for a designated period of time, demonstrating that microbial composition changes rapidly and reversibly in response to dietary components.93 Additional work has extensively documented the profound impact of high-fat and low-fat diet on fecal microbial communities, with high-fat diet shifting communities towards a greater ratio of Firmicutes:Bacteroidetes taxa.94 At somewhat higher nutrient-resolution, recent work has shown that administration of specific complex polysaccharides can promote the growth of specific Bacteroides species.95,96
Whereas resource availability in the colonic lumen varies depending on dietary intake, many colonic microbes reside in and consume host-derived components of the mucosa, which may remain somewhat more stable across dietary behavioral patterns.15 The glycoprotein that makes up the majority of the colonic mucus, Mucin 2 (MUC2), is coated with a diverse assortment of O-linked glycans, oligosaccharides that can be cleaved from MUC2 and metabolized to support the growth of various specialized microbial taxa like Akkermansia muciniphila.97,98 Different microbes vary in their capacity to penetrate and adhere to the mucus layer, as well as in their tolerance to AMPs and oxygen that diffuse outward through the mucus from the underlying epithelial cells.99,100 Thus, the mucosal niche experiences unique selective pressures compared to the luminal niche in the colon, and consequently supports an enrichment of aerotolerant, asaccharolytic protein-metabolizing species from the phyla Actinobacteria and Proteobacteria compared to the lumen.100,101 Within the mucosa, as well, researchers have documented further spatial niche partitioning. For instance, Acinetobacter species are particularly effective at navigating through the mucus layer and its associated biochemical gradients to bind directly to the IECs of the colonic crypts.102
Like the mucosa, the lumen itself also harbors significant spatial heterogeneity, much of which has been overlooked by the use of fecal sampling in the majority of colonic microbiome studies. Evidence in mice demonstrates that the characteristic mucosal folds of the colonic walls create distinct “inter-fold” regions in the intestinal lumen, which are enriched for taxa in the families Lachnospiraceae and Ruminococcus. These taxa are thought to benefit both from the local accumulation of mucus from the epithelia and from an environment that is relatively protected from the flow of other luminal contents.17,103 The digesta of the central lumen, by contrast, are dominated by strictly anaerobic, saccharolytic taxa from the families Bacteroidaceae, Enterococcaceae, Prevotellaceae, and Rikenellaceae.103
Proximally to distally, the microbial inhabitants of the colon exhibit locally specialized functions that seem to broadly reflect resource availability. Because digesta entering the colon have higher concentrations of complex polysaccharides and bile acids, fermentation and bile acid metabolism are largely localized to the cecum and proximal colon.65,75,104
Bile acid metabolism extensively impacts microbial composition in the colon. Primary BAs not absorbed in the ileum enter the cecum and colon, where they can be deconjugated via BSH enzymes that are widespread among commensal microbes, and/or converted into secondary BAs by select bacterial species in the Clostridia class with 7-α-dehydroxylase enzymes.62,105 BAs are bactericidal to many taxa, and therefore the processes of deconjugation and conversion can serve as a defensive measure for microbial community members.61,106 Secondary BAs are typically harmless to the bacteria that synthesized them, but can still exert toxic effects on other microbial taxa. Thus, under conditions with high concentrations of primary BAs, as in the proximal colon, we would expect that the ability to convert primary BAs into secondary BAs would confer a species-specific growth advantage. This prediction finds support in the literature. One study in rats showed that the size of the BA pool can exert an extremely strong selective pressure on the colonic microbial community: rodents that received a dietary BA supplement exhibited dramatic, phylum-level shifts in microbial composition, particularly favoring Clostridia species that can convert primary BAs into secondary BAs.107 As high-fat diet induces the production of more bile salts for lipid absorption, these bile salts in turn impact the colonic microbiota.93,108
The composition and size of the BA pool, which both affect and are affected by microbial metabolism throughout the colon, likewise have profound impacts on host health and metabolism. For instance, deconjugated bile acids produced by the actions of microbial BSH enzymes can disrupt micelle formation and inhibit absorption of cholesterol and other lipids across intestinal membranes, with implications for cardiovascular disease.65 Secondary BAs generated by commensal microbes can activate nuclear receptors like Farnesoid X Receptor (FXR) and G-protein coupled receptors like TGR5 to communicate across host tissues and regulate primary bile acid synthesis and degradation, glucose and lipid metabolism, and energy homeostasis.109–111 Finally, microbially-synthesized bile acids are increasingly being investigated for their roles as mediators of inflammatory disease.108,112
The inflammatory state of the host, in and of itself, plays a large role in the composition and function of the colonic microbiome, reflecting the delicate homeostatic balance between the immune system and the microbiome in this densely populated region of the GIT. Microbes that colonize the healthy gut have evolved a diverse array of mechanisms to avoid triggering an immune response. For instance, many commensal strains, particularly from the Bacteroides genus, have evolved a modification to the lipopolysaccharide (LPS) on their outer membrane that renders it invisible to host-derived AMPs.57 Other commensal taxa have evolved more explicit immunomodulatory strategies. A component of the polysaccharide capsule of Bacteroides fragilis, for example, stimulates regulatory T cells to produce immunosuppressive interleukin-10, allowing B. fragilis to colonize the mucosal niche.86,113 Although it is unclear whether a dysbiotic microbiome triggers a pathologic immune response or vice versa, many studies have documented a compositionally distinct microbiome under inflammatory conditions that is broadly characterized by reduced representation of the Firmicutes and Bacteroidetes phyla, and increased abundance of Actinobacteria and Proteobacteria.114–117
4. Skin
4.1. Overview
As an enormous organ that is fully external, the skin and its associated microbiome may be exposed to the most variable conditions of any body site. Microbes can arrive on the skin from any source community that the host encounters, experiencing very low dispersal limitation, and although the skin exhibits distinct biogeography across both regional and local scales, the conditions in these sites often vary in accordance with environmental conditions and host behaviors. Generally, the skin is dry, cool, aerobic, and acidic, although across regional structures and folds, there is variation in these physicochemical parameters, as well as in the density of micro-scale topological structures. Certain regions, like the axilla, for instance, tend to be warmer and more moist, with densely packed sweat glands and hair follicles. Even this pocket, however, is subject to temporal and behavioral variability – an individual that is exercising on a hot day will have a different axillary environment than an individual that is sitting around on a cold day. And while micro-scale topological features like hair follicles or sweat glands tend to act as more consistent habitat filters, even these sites are subject to frequent perturbations due to variable host cosmetic and hygienic behaviors.
The skin, therefore, emerges as a site with 1) very low dispersal limitation, and an extensive meta-community species pool, 2) numerous regional and local habitat filters for niche differentiation, and 3) high rates of perturbation. These criteria are among the most widely cited in the ecological literature as mechanisms for maintenance of diversity.85,118,119 Such characteristics pave the way for stochastic processes like dispersal and drift to interact with the diverse selective pressures of the skin, creating a wide variety of possible community outcomes across individuals. And indeed, data from the Human Microbiome Project has shown that, although the GIT exhibits comparable alpha diversity, or within-site diversity, the skin has the highest beta diversity of all body sites, which means that it is the most variable from person to person.12,120 Furthermore, longitudinal tracking of the skin microbiome reveals that it is also the least temporally stable body site, with substantial intra-individual variation over time.120,121
4.2. Skin anatomy, physiology, and microbiome composition
Structurally, the skin consists of shedding layers of stratified, keratinized squamous epithelial cells with interspersed structures like hair follicles, pores, and sebaceous glands. An estimated 1011 microbial cells blanket the surface of the epidermis, descending into the pores, glands, and follicles.122,123 These structures are not only important sources of physical spatial heterogeneity, but their physiological functions contribute to local patchiness in moisture, acidity, and salinity across the skin, creating distinct, and often selective, ecological niches (Figure 3).124
Figure 3. The skin microbiota varies by region (adapted from Grice et al.).122.
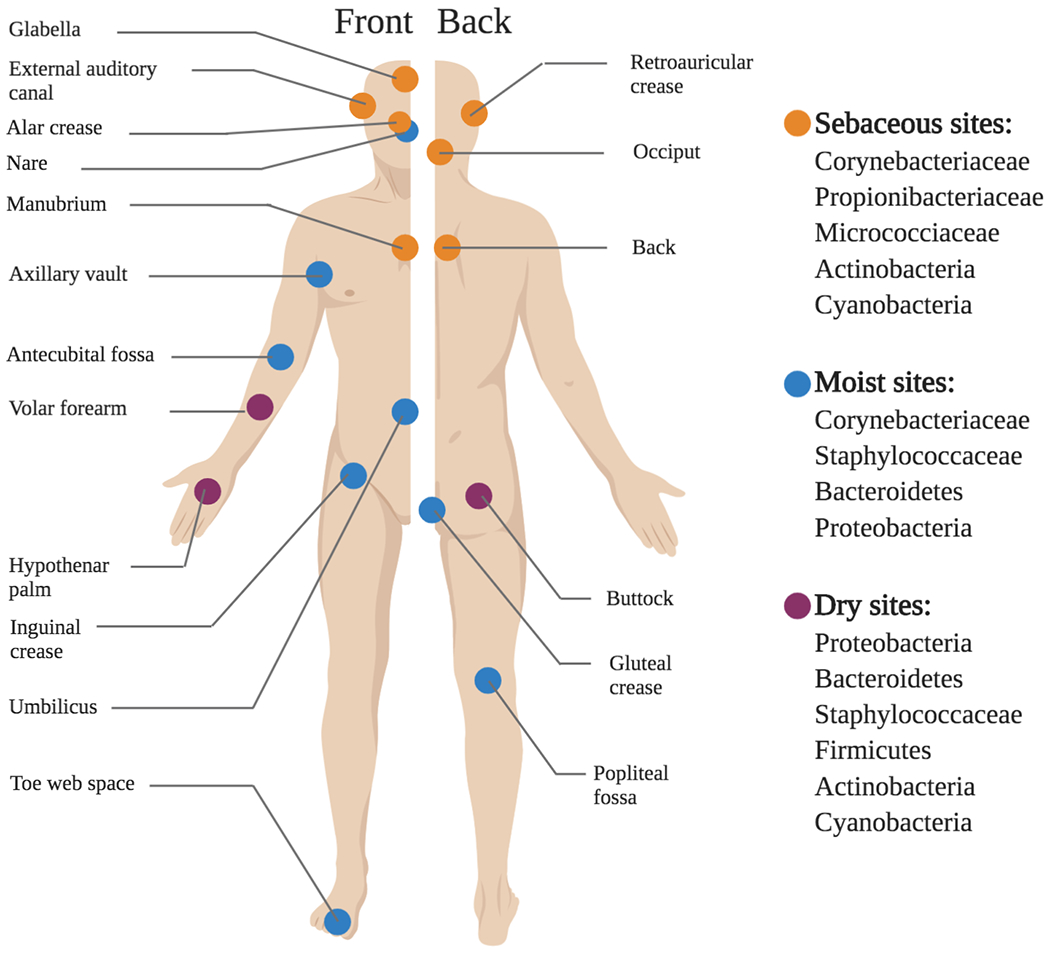
The composition of the skin microbiome varies in accordance with regional and local topographical features of the skin. Broadly, these sites can be categorized by whether they are moist, dry, or sebaceous, and each of these categories features a distinct subset of microbial taxa that are uniquely well-adapted to those conditions.
Apocrine sweat glands, for instance, recruit Corynebacteria species. In response to adrenaline, these glands release odorless steroids, acids, and other volatile secretions, which are consumed and metabolized by Corynebacteria into the characteristic malodorous compounds of sweat.125–127 Sebaceous glands, which are connected to hair follicles and secrete a lipid-rich substance called sebum that protects and lubricates the skin, are also highly selective for particular bacteria. Skin regions with dense sebaceous glands, like the face, chest, and back, show the lowest beta diversity compared to other skin sites, consistently harboring the same bacterial taxa across individuals.128 These glands recruit lipophilic species like Propionibacterium acnes, which consumes and degrades the lipids that make up sebum.129,130 This releases free fatty acids as a byproduct, contributing to the baseline acidification of the skin, a primary barrier against pathogens.131,132 Sebaceous sites are also home to fungal commensals like Malassezia, another lipophilic microorganism.133
The density of such topological structures, as well as the broader-scale anatomy of different skin regions, impacts the physiology and environment in those locations. Temperature and humidity, for example, vary dramatically across different anatomical structures. Less exposed, folded regions of the skin like the axillary vault, inguinal crease, gluteal crease, or umbilicus, tend to be both warmer and more moist than other more exposed regions of the skin, and they consequently recruit an abundance of humidity-loving bacteria like Corynebacteria and Staphylococci.121,128 Drier, more exposed regions like the limbs experience greater temperature fluctuations, as well as more frequent perturbations than other parts of the body, possibly contributing to the relatively low bacterial biomass in those regions.122,134 Despite this, such exposed regions are among the most diverse sites in the human body, with representation from the Actinobacteria, Proteobacteria, Firmicutes, and Bacteroidetes phyla.121,128,135
4.3. Functional roles of the skin microbiome
Functionally, the skin microbiome appears to be somewhat less integral to healthy physiology than the microbiomes of other body sites. Most skin symbionts are thought to be commensal, with relatively few examples of obligate mutualism in which the host depends on its microbes to fulfill a particular function. This is perhaps unsurprising in an environment as variable as the skin – the selective pressures are inconsistent, and therefore selection cannot act in a directed manner. Nevertheless, there is emerging evidence that the skin microbiota plays a role in stimulating and educating the host immune system: secretion of the AMPs like cathelicidin and β-defensins, as well as the production of complement, are induced by commensal stimulation of host innate immune receptors.136–138 Moreover, in mice, recent evidence showed that skin commensals can tune and regulate the adaptive immune response through their ability to shift IL-1 production.139,140 Finally, commensal microbes like S. epidermidis have demonstrated the ability to directly inhibit the growth of pathogenic organisms like Staphylococcus aureus and Group A Streptococcus.141
5. Vagina
5.1. Overview
The vaginal microbiome is among the least diverse human body sites, and has shown one of the tightest, most consistently mutualistic symbioses with the host. While vaginal microbial residents consume host-derived materials from sloughed cells and secretions, the host is thought to benefit from the protective role that vaginal microbes may play against colonization by an assortment of pathogens, including those that might otherwise cause yeast infections, sexually transmitted infections, and urinary tract infections. These tight associations between the host and specific microbial taxa, and mechanistically well-understood mutualisms suggest that the vaginal microbiome may be an exceptional example of host-microbe coevolution.
5.2. Vaginal anatomy, physiology, and microbiome composition
The vaginal epithelium consists of stratified, glycogen-filled, non-keratinized squamous cells that are rapidly shed, with the top layer turning over approximately once every four hours.142 It is largely anaerobic, and quite acidic (pH 3.5 – 4.5) in reproductive-age women. Although vaginal epithelial cells can produce lactic acid, substantial evidence suggests that mutualistic bacteria, predominantly Lactobacilli, are the primary source of this acidity, converting glycogen stores from shedding epithelial cells into lactic acid through anaerobic fermentation processes.143
This ecosystem, in most healthy reproductive-age women, is dominated by Lactobacilli.144 Historically, the presence of abundant Lactobacilli and concomitantly acidic pH were considered signatures of a “healthy” vaginal microbiome, whereas a more diverse microbiome with higher pH has been correlated with preterm birth, “dysbiotic” conditions like bacterial vaginosis (BV), and the presence of vaginal pathogens like Gardnerella or Trichomonas.145–148 However, broader culture-independent sampling across different demographic groups has revealed that approximately 20-30% of healthy women have a more diverse, non-Lactobacillus-dominated microbiome, once again muddling our understanding of what exactly defines “health” in this ecosystem.144,149 This work has identified five distinct “community state types” (CSTs) among healthy women, four of which are each dominated by a different species of Lactobacillus (L. crispatus, L. gasseri, L. jensenii, L. iners), and the fifth (CST-IV) by a diverse consortium of facultative and strict anaerobes, including microbes from the genera Atopobium, Corynebacterium, Anaerococcus, Peptoniphilus, Prevotella, and Gardnerella.144,150 Demographically, black and Hispanic women are more likely to have a CST-IV consortium, whereas white and Asian women are more likely to have any of the other types.144,149,151
5.3. Functional roles of the vaginal microbiome
The native vaginal microbiome is thought to contribute significantly to pathogen resistance in the vagina by a number of mechanisms. First, lactic acid produced in copious amounts by resident Lactobacilli, and in lesser amounts by host cells and other microbial inhabitants, is inhospitable or directly inhibitory to many invading pathogens including sexually-transmitted pathogens like Chlamydia trachomatis, Neisseria gonorrhoeae, and HIV.152–154 Different Lactobacillus species produce different amounts and isomers of lactic acid, contributing variably to the lowering of vaginal pH, and therefore their protective capacity.155 Second, lactic acid has been shown to exhibit protective immunomodulatory properties. Whereas a pro-inflammatory milieu is associated with BV and STI acquisition, work from Hearps et al. have demonstrated that lactic acid can induce production of the anti-inflammatory cytokine IL-1RA, and inhibit production of pro-inflammatory cytokines IL-6 and IL-8.156 Third, vaginal Lactobacilli can produce anti-microbial bacteriocins that specifically target pathogens like Klebsiella, Gardnerella, E. coli, and Enterococcus faecalis.157,158 Finally, it has been hypothesized that native vaginal microbes help to prevent colonization by pathogens in part by competitive exclusion: the native vaginal microbiota is better adapted to the vaginal environment, and can more efficiently extract the limited resources there, preventing potential invaders from growing and establishing.159
5.4. Community assembly in the vagina
It is an open question as to why certain healthy women have a Lactobacillus-dominated vaginal microbiome and others do not, and among those who do, why different Lactobacillus species come to dominance. Unlike the GI tract where it is easy to speculate about the selective pressures and habitat filters that shape community assembly, clear habitat filters have not been identified in the vagina. Many have hypothesized that Lactobacilli are selected for their ability to metabolize glycogen-derived resources, but this still fails to explain why other glycogen-metabolizing bacteria aren’t more prominent in these communities, or why only four out of over 130 known Lactobacillus species dominate the vaginal microbiota of 70-80% of healthy women.160 Functional genomic efforts to identify habitat-related traits specific to these four Lactobacilli have been unsuccessful.161 One speculative explanation for single-strain dominance could be that priority effects allow a single, early-arriving Lactobacillus species to establish, conferring a spatial and numerical growth advantage that limits the ability of late-arriving species to compete, but this possibility requires experimental investigation.
Unfortunately, there exist no established animal models for studying the vaginal microbiome: differences in reproductive strategies that often facilitate research throughput (i.e. more frequent reproduction and greater numbers of offspring) dramatically affect the anatomy and physiology of the reproductive tract and its associated microbiome. Somewhat surprisingly, no other mammals, including closely related mammals like non-human primates, display such high abundances of Lactobacilli, or an even passably human-like community composition.162 Thus, mechanistic understanding of vaginal community assembly may remain elusive until better experimental systems are established.
5.5. Vaginal microbiome stability and resilience
The physiology of the vagina is uniquely time-dependent compared to other body sites, given the cyclic nature of reproductive physiology: hormone levels oscillate with regular menstrual cycling, and pregnancy causes a dramatic series of anatomical and functional changes to unfold in an orderly progression over time. In addition to these longer-scale chronic disturbances to the vaginal microbiome, the vagina is often subject to more acute perturbations due to sexual behaviors, use of contraceptives, lubricants, and menstrual products, and other behavioral practices. Given the regularity of both chronic and acute perturbations to this community, whether or how stable the vaginal microbiome is has been a fundamentally important question. Interestingly, the healthy vagina is both remarkably stable and highly resilient. Acute behavioral disturbances to the vaginal community, as well as regular monthly menstrual cycling, seem to induce only mild shifts in microbial community membership, and most communities ultimately return to their baseline compositional state.163–165 Similarly, longitudinal studies of the vaginal microbiome throughout pregnancy have revealed that, although there is reduced representation of CST-IV among pregnant women, suggesting a pregnancy-induced shift towards Lactobacillus dominance, over the course of pregnancy, the vaginal microbial community remains exceptionally stable compared to non-pregnant women.149,166,167
6. Respiratory Tract
6.1. Overview
Historically, although the upper respiratory tract (URT) was well known to harbor large numbers of bacteria, it was thought that the lower respiratory tract (LRT) was sterile except when in a state of active disease. With the advent of culture-independent sequencing, this misconception has been revised, and we now know that there is in fact a low biomass but significant healthy LRT microbiome. The characterization of the respiratory tract microbiome and its dynamics over time and across disease states is an emerging field that is slowly bringing nuance to long-held pulmonological dogma.
6.2. Upper respiratory tract anatomy, physiology, and microbiome composition
The URT consists of the nares, nasal and oral cavities, pharynx, and upper larynx. For a description of the microbiota of the oral cavity, see the gastrointestinal tract section above.
Although the microbiota of the pharynx generally reflects the microbiota of the oral cavity, the nares and nasal cavity harbor a distinct subset of microbes. Anatomically, the nares features more skin-like features than the rest of the nasal mucosa, including keratinized, stratified squamous epithelial cells, sweat and sebaceous glands, and follicles through which coarse, specialized hairs called vibrissae emerge. Although air is moistened and warmed as it proceeds through the nasal cavity, air that enters the nasal vestibule is at ambient temperature, and therefore this habitat is cooler and drier than the rest of the nasal cavity. Microbial communities inhabiting the nasal vestibule are highly reflective of skin communities, featuring abundant Corynebacteria, Staphylococcus, and Propionibacteria.121,168
Located deeper within the nasal cavity, the nasal mucosa have several significant anatomical and physiological features that distinguish them from both the nasal vestibule and the oral cavity. Compared to the nasal vestibule, the nasal mucosa are warmer and more moist. The epithelium transitions from keratinized stratified squamous cells to pseudostratified, ciliated columnar cells that are blanketed in a flowing layer of mucus. In conjunction with this mucus, the cilia perform an up-and-out sweeping motion throughout the respiratory tract to clear particulate matter. This region is dominated by Actinobacteria, including Corynebacterium and Proprionibacterium as in the anterior nares, but had greater representation of species from Proteobacteria.169,170 This suggests that the nasal cavity may be seeded by microbes that have dispersed from the nasal vestibule, but has distinct habitat filters that select for a unique subset of these microbes.
6.3. Lower respiratory tract anatomy, physiology, and microbiome composition
The LRT comprises the trachea, bronchi, bronchioles, and alveoli. These environments are uniformly quite nutrient-poor and aerobic, although a number of significant anatomical and physiological transitions take place from the proximal to distal end. Most of these tissues, from the nasal cavity through the bronchioles, make up the “conductive” portion of the respiratory tract, responsible for bringing air into the lungs and clearing all other particulate matter, while the alveoli make up the “respiratory” portion, and are responsible for gas exchange. The epithelia of these regions largely reflect these distinct physiological roles.
The trachea and bronchi, like the nasal cavity, feature ciliated pseudostratified columnar epithelia. At the bronchioles, this epithelium transitions into simple columnar cells, which flatten into cuboidal epithelium, and eventually progress to a thin lining of squamous cells in the alveoli. An assortment of secretory cells are interspersed throughout the LRT, releasing mucus which coats the epithelial surface and traps secreted antimicrobial peptides and immunomodulatory molecules.171 Airway mucus is thickest in the proximal regions of the LRT, thins towards the distal bronchioles, and is eventually replaced by surfactant in the alveoli.171 Proximally to distally, the LRT also features gradients of oxygen tension, pH, temperature, and density of inhaled particles.172–174
Although it seems likely that this spatial heterogeneity could give rise to local differences in microbial composition across the LRT, in humans, current sampling techniques do not have the spatial resolution to investigate this possibility. Samples of the LRT microbiome are typically collected through a process called bronchoalveolar lavage (BAL), in which a bronchoscope is inserted into the bronchioles, and sterile saline is introduced and recollected with whatever microbial community members were washed into it. Microbes in different layers of the epithelial mucus, or those that might appear in the alveolus versus in the bronchiole, are all mixed into a single homogenized solution, hampering detection of distinct communities across microhabitats.
On a somewhat broader spatial scale, however, BAL data gathered from different lobes of the lung seem to suggest that, despite observed environmental gradients across this same regional scale, microbial communities are nearly indistinguishable.175 Sequencing-based analysis of samples from the lingula, right middle lobe, left and right upper lobes, and supraglottic space of the trachea all featured abundant aerotolerant Prevotella, Veillonella, and Streptococcus species.176 Samples from the LRT sites of each individual were more similar to the URT source community from that same individual than the same LRT site in other individuals, likely reflecting the unique, individual-based meta-community from which the LRT community is assembled.176 These results suggest that habitat filters may not play as important a role in community assembly in the lung as neutral, dispersal-based processes.177
6.4. Community assembly processes in the healthy and diseased lung
Given these findings, Dickson et al. have proposed a model of microbial community assembly in the lung adapted from island biogeography theory, a classic ecological model (Figure 4).118,176 In this model, Dickson et al. suggest that diversity in the lung is a balance between stochastic immigration from the meta-community species pool (the URT) and extinction events. The closer a site is to the source community, the greater the rate of immigration, and the higher diversity that site will maintain. Under this model, immigration into the LRT occurs when microbes from the URT physically migrate down the oropharynx, are inhaled as aerosolic particulate matter, or are micro-aspirated during sleep. Extinction events occur due to various forces like coughing, mucociliary sweeping, and immune clearance. Since communities at different sites are drawing from the same meta-community pool, there will be significant overlap between sites – each local community should theoretically be a subset of the same source community – but we expect decreasing species richness with distance from the source community. While higher resolution spatial data on the LRT microbiome would be invaluable in testing and refining this hypothesis, Dickson et al. found that species richness did decrease with distance between each sampling site and the URT, supporting this expectation.176
Figure 4. Adapted island theory of healthy lung microbiome assembly.
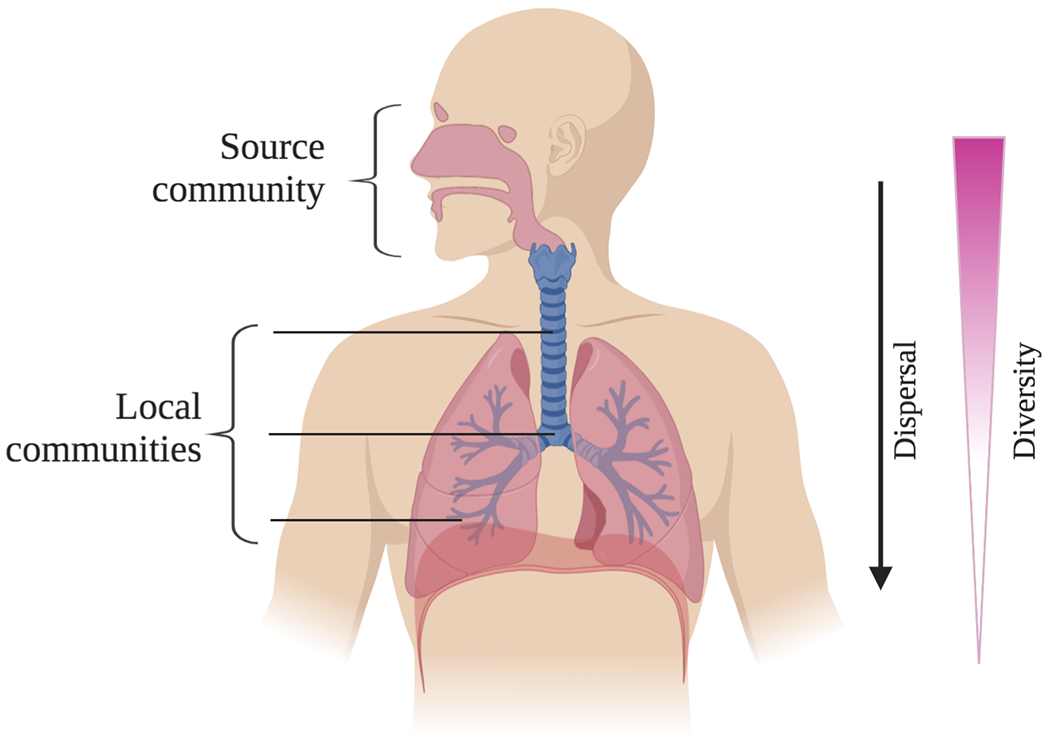
According to this model proposed by Dickson et al., the upper respiratory tract (URT), and particularly the oral cavity, constitute a source community from which microbes disperse to the lower respiratory tract (LRT). Microbes present in each local habitat of the LRT are therefore a subset of the microbes present in the URT source community. Sites deeper within the LRT face greater dispersal limitation, as they are physically farther from the source community, and therefore diversity decreases with distance from the URT.
Interestingly, as patients transition from health to disease, the relative importance of neutral and selective processes seems to shift: the lungs of patients with advanced lung disease like cystic fibrosis or COPD exhibit distinct spatial heterogeneity in microbial community structure that is absent in healthy individuals.177–179 This could be due to either the proliferation of native or invading microbes that thrive under disease conditions that are locally exacerbated, or by the increased sensitivity of existing microbes to such local conditions, and their resulting extinction. Generally, injury and inflammation of the respiratory tract lead to increased temperature and increased production of mucus, which in turn creates anaerobic pockets that can support the growth of particular community members or pathogens.180,181 Locally severe disease may create such conditions in distinct regions, allowing for these new habitat filters to shape community composition there.
7. Conclusions
It is only fitting that a body with as many specialized functions as ours features accordingly diverse, specialized microbiota across those different anatomical and functional sites. In this review, we sought to emphasize that human-associated microbial communities can be shaped by a number of different processes, like dispersal, selection, and drift, and that these processes can contribute in variable measure to the assembly and composition of different body sites. Hopefully, a stronger understanding of these forces will one day allow us to more strategically manipulate these communities towards health.
We also wanted to emphasize that, although much of the data presented here has been instrumentally defined as “healthy” due to the absence of clinical symptoms among study participants, a more precise definition of the “healthy” microbiome is necessary before the microbiome field can transition into a more translational era. Given the degree of microbial diversity even among healthy individuals, an important focus of the field should be the development of positive criteria for salient functions or characteristics of a “healthy” microbiome across body sites, rather than simply the absence of disease. As our definitions, frameworks, and mechanistic understandings of the microbiome continue to develop and refine, our capacity to engineer this complex ecological network for positive health impacts can only expand.
Acknowledgments
MK was supported by MSTP Training Grant (T32GM007281).
References
- 1.Vellend M Conceptual synthesis in community ecology. Q Rev Biol. 2010;85(2):183–206. doi: 10.1086/652373 [DOI] [PubMed] [Google Scholar]
- 2.Costello EK, Stagaman K, Dethlefsen L, Bohannan BJM, Relman DA. The application of ecological theory toward an understanding of the human microbiome. Science. 2012;336(6086):1255–1262. doi: 10.1126/science.1224203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dominguez-Bello MG, Costello EK, Contreras M, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107(26):11971–11975. doi: 10.1073/pnas.1002601107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chibani-Chennoufi S, Bruttin A, Dillmann ML, Brüssow H. Phage-host interaction: An ecological perspective. J Bacteriol. 2004;186(12):3677–3686. doi: 10.1128/JB.186.12.3677-3686.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun CL, Relman DA. Microbiota’s “little helpers”: Bacteriophages and antibiotic-associated responses in the gut microbiome. Genome Biol. 2013;14(7):127. doi: 10.1186/gb-2013-14-7-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilbert B, Levine JM. Ecological drift and the distribution of species diversity. Proc R Soc B Biol Sci. 2017;284(1855). doi: 10.1098/rspb.2017.0507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaura E, Brandt BW, de Mattos MJT, et al. Same Exposure but two radically different responses to antibiotics: Resilience of the salivary microbiome versus long-term microbial shifts in feces. MBio. 2015;6(6). doi: 10.1128/mBio.01693-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(SUPPL. 1):4554–4561. doi: 10.1073/pnas.1000087107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marais GAB, Calteau A, Tenaillon O. Mutation rate and genome reduction in endosymbiotic and free-living bacteria. Genetica. 2008;134(2):205–210. doi: 10.1007/s10709-007-9226-6 [DOI] [PubMed] [Google Scholar]
- 10.Zhao S, Lieberman TD, Poyet M, et al. Adaptive Evolution within Gut Microbiomes of Healthy People. Cell Host Microbe. 2019;25(5):656–667.e8. doi: 10.1016/j.chom.2019.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huddleston JR. Horizontal gene transfer in the human gastrointestinal tract: Potential spread of antibiotic resistance genes. Infect Drug Resist. 2014;7:167–176. doi: 10.2147/IDR.S48820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. doi: 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sprockett D, Fukami T, Relman DA. Role of priority effects in the early-life assembly of the gut microbiota. Nat Rev Gastroenterol Hepatol. 2018;15(4):197–205. doi: 10.1038/nrgastro.2017.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedman ES, Bittinger K, Esipova TV., et al. Microbes vs. chemistry in the origin of the anaerobic gut lumen. Proc Natl Acad Sci U S A. 2018;115(16):4170–4175. doi: 10.1073/pnas.1718635115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donaldson GP, Lee SM, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol. 2016;14(1):20–32. doi: 10.1038/nrmicro3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johansson ME V, Sjövall H, Hansson GC. The gastrointestinal mucus system in health and disease. Nat Rev Gastroenterol Hepatol. 2013;10(6):352–361. doi: 10.1038/nrgastro.2013.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tropini C, Earle KA, Huang KC, Sonnenburg JL. The Gut Microbiome: Connecting Spatial Organization to Function. Cell Host Microbe. 2017;21(4):433–442. doi: 10.1016/j.chom.2017.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falony G, Joossens M, Vieira-Silva S, et al. Population-level analysis of gut microbiome variation. Science (80- ). 2016;352(6285):560–564. doi: 10.1126/science.aad3503 [DOI] [PubMed] [Google Scholar]
- 19.Bäckhed F, Fraser CM, Ringel Y, et al. Defining a healthy human gut microbiome: Current concepts, future directions, and clinical applications. Cell Host Microbe. 2012;12(5):611–622. doi: 10.1016/j.chom.2012.10.012 [DOI] [PubMed] [Google Scholar]
- 20.Dewhirst FE, Chen T, Izard J, et al. The human oral microbiome. J Bacteriol. 2010;192(19):5002–5017. doi: 10.1128/JB.00542-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Welch JLM, Rossetti BJ, Rieken CW, Dewhirst FE, Borisy GG. Biogeography of a human oral microbiome at the micron scale. Proc Natl Acad Sci U S A. 2016;113(6):E791–E800. doi: 10.1073/pnas.1522149113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ledder RG, Gilbert P, Huws SA, et al. Molecular analysis of the subgingival microbiota in health and disease. Appl Environ Microbiol. 2007;73(2):516–523. doi: 10.1128/AEM.01419-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato Y, Yamagishi J, Yamashita R, et al. Inter-Individual Differences in the Oral Bacteriome Are Greater than Intra-Day Fluctuations in Individuals. Martinez-Abarca F, ed. PLoS One. 2015;10(6):e0131607. doi: 10.1371/journal.pone.0131607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simón-Soro A, Tomás I, Cabrera-Rubio R, Catalan MD, Nyvad B, Mira A. Microbial geography of the oral cavity. J Dent Res. 2013;92(7):616–621. doi: 10.1177/0022034513488119 [DOI] [PubMed] [Google Scholar]
- 25.Haffajee AD, Teles RP, Patel MR, Song X, Yaskell T, Socransky SS. Factors affecting human supragingival biofilm composition. II. Tooth position. J Periodontal Res. 2009;44(4):520–528. doi: 10.1111/j.1600-0765.2008.01155.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Proctor DiM Fukuyama JA, Loomer PM, et al. A spatial gradient of bacterial diversity in the human oral cavity shaped by salivary flow. Nat Commun. 2018;9(1):1–10. doi: 10.1038/s41467-018-02900-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dawes C, Watanabe S, Biglow-Lecomte P, Dibdin GH. Estimation of the velocity of the salivary film at some different locations in the mouth. J Dent Res. 1989;68(11):1479–1482. doi: 10.1177/00220345890680110201 [DOI] [PubMed] [Google Scholar]
- 28.Almståhl A, Wikström M, Stenberg I, Jakobsson A, Fagerberg-Mohlin B. Oral microbiota associated with hyposalivation of different origins. Oral Microbiol Immunol. 2003;18(1):1–8. doi: 10.1034/j.1399-302X.2003.180101.x [DOI] [PubMed] [Google Scholar]
- 29.Costalonga M, Herzberg MC. The oral microbiome and the immunobiology of periodontal disease and caries. Immunol Lett. 2014;162(2):22–38. doi: 10.1016/j.imlet.2014.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brogden K, Squier C. Human Oral Mucosa : Development, Structure, and Function. Wiley-Blackwell; 2011. [Google Scholar]
- 31.Mager DL, Ximenez-Fyvie LA, Haffajee AD, Socransky SS. Distribution of selected bacterial species on intraoral surfaces. J Clin Periodontol. 2003;30(7):644–654. doi: 10.1034/j.1600-051X.2003.00376.x [DOI] [PubMed] [Google Scholar]
- 32.Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. Defining the normal bacterial flora of the oral cavity. J Clin Microbiol. 2005;43(11):5721–5732. doi: 10.1128/JCM.43.11.5721-5732.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Proctor DM, Relman DA. The Landscape Ecology and Microbiota of the Human Nose, Mouth, and Throat. Cell Host Microbe. 2017;21(4):421–432. doi: 10.1016/j.chom.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsukamoto Y, Usui M, Yamamoto G, et al. Role of the junctional epithelium in periodontal innate defense and homeostasis. J Periodontal Res. 2012;47(6):750–757. doi: 10.1111/j.1600-0765.2012.01490.x [DOI] [PubMed] [Google Scholar]
- 35.Abusleme L, Moutsopoulos NM. IL-17: overview and role in oral immunity and microbiome. Oral Dis. 2017;23(7):854–865. doi: 10.1111/odi.12598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shang L, Deng D, Buskermolen JK, et al. Multi-species oral biofilm promotes reconstructed human gingiva epithelial barrier function. Sci Rep. 2018;8(1):1–12. doi: 10.1038/s41598-018-34390-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pei Z, Bini EJ, Yang L, Zhou M, Francois F, Blaser MJ. Bacterial biota in the human distal esophagus. Proc Natl Acad Sci U S A. 2004;101(12):4250–4255. doi: 10.1073/pnas.0306398101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fillon SA, Harris JK, Wagner BD, et al. Novel Device to Sample the Esophageal Microbiome-The Esophageal String Test. PLoS One. 2012;7(9). doi: 10.1371/journal.pone.0042938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;323(8390):1311–1315. doi: 10.1016/S0140-6736(84)91816-6 [DOI] [PubMed] [Google Scholar]
- 40.Bik EM, Eckburg PB, Gill SR, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A. 2006;103(3):732–737. doi: 10.1073/pnas.0506655103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X-X, Wong GL-H, To K-F, et al. Bacterial Microbiota Profiling in Gastritis without Helicobacter pylori Infection or Non-Steroidal Anti-Inflammatory Drug Use. Ahmed N, ed. PLoS One. 2009;4(11):e7985. doi: 10.1371/journal.pone.0007985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Engstrand L, Lindberg M. Helicobacter pylori and the gastric microbiota. Best Pract Res Clin Gastroenterol. 2013;27(1):39–45. doi: 10.1016/j.bpg.2013.03.016 [DOI] [PubMed] [Google Scholar]
- 43.Nardone G, Compare D. The human gastric microbiota: Is it time to rethink the pathogenesis of stomach diseases? United Eur Gastroenterol J. 2015;3(3):255–260. doi: 10.1177/2050640614566846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanduleanu S, Jonkers D, De Bruine A, Hameeteman W, Stockbrügger RW. Non-Helicobacter pylori bacterial flora during acid-suppressive therapy: Differential findings in gastric juice and gastric mucosa. Aliment Pharmacol Ther. 2001;15(3):379–388. doi: 10.1046/j.1365-2036.2001.00888.x [DOI] [PubMed] [Google Scholar]
- 45.Vesper B, Jawdi A, Altman K, Haines G III, Tao L, Radosevich J. The Effect of Proton Pump Inhibitors on the Human Microbiota. Curr Drug Metab. 2009;10(1):84–89. doi: 10.2174/138920009787048392 [DOI] [PubMed] [Google Scholar]
- 46.Graham DY, Malaty HM, Evans DG, Evans DJ, Klein PD, Adam E. Epidemiology of Helicobacter pylori in an asymptomatic population in the United States: Effect of age, race, and socioeconomic status. Gastroenterology. 1991;100(6):1495–1501. doi: 10.5555/URI:PII:001650859190644Z [DOI] [PubMed] [Google Scholar]
- 47.Kuipers EJ, Pérez-Pérez GI, Meuwissen SG, Blaser MJ. Helicobacter pylori and atrophic gastritis: importance of the cagA status. J Natl Cancer Inst. 1995;87(23):1777–1780. doi: 10.1093/jnci/87.23.1777 [DOI] [PubMed] [Google Scholar]
- 48.Kabir S The Role of Interleukin-17 in the Helicobacter pylori Induced Infection and Immunity. Helicobacter. 2011;16(1):1–8. doi: 10.1111/j.1523-5378.2010.00812.x [DOI] [PubMed] [Google Scholar]
- 49.Perry S, de Jong BC, Solnick J V., et al. Infection with Helicobacter pylori Is Associated with Protection against Tuberculosis. Pai M, ed. PLoS One. 2010;5(1):e8804. doi: 10.1371/journal.pone.0008804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blaser MJ. Hypothesis: The Changing Relationships of Helicobacter pylori and Humans: Implications for Health and Disease . J Infect Dis. 1999;179(6):1523–1530. doi: 10.1086/314785 [DOI] [PubMed] [Google Scholar]
- 51.Johansson MEV, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A. 2008;105(39):15064–15069. doi: 10.1073/pnas.0803124105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zoetendal EG, Raes J, Van Den Bogert B, et al. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012;6(7):1415–1426. doi: 10.1038/ismej.2011.212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9(5):356–368. doi: 10.1038/nrmicro2546 [DOI] [PubMed] [Google Scholar]
- 54.Meyer-Hoffert U, Hornef MW, Henriques-Normark B, et al. Secreted enteric antimicrobial activity localises to the mucus surface layer. Gut. 2008;57(6):764–771. doi: 10.1136/gut.2007.141481 [DOI] [PubMed] [Google Scholar]
- 55.Peterson DA, McNulty NP, Guruge JL, Gordon JI. IgA Response to Symbiotic Bacteria as a Mediator of Gut Homeostasis. Cell Host Microbe. 2007;2(5):328–339. doi: 10.1016/j.chom.2007.09.013 [DOI] [PubMed] [Google Scholar]
- 56.Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. 2014;14(10):667–685. doi: 10.1038/nri3738 [DOI] [PubMed] [Google Scholar]
- 57.Cullen TW, Schofield WB, Barry NA, et al. Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science (80- ). 2015;347(6218):170–175. doi: 10.1126/science.1260580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martinez-Guryn K, Hubert N, Frazier K, et al. Small Intestine Microbiota Regulate Host Digestive and Absorptive Adaptive Responses to Dietary Lipids. Cell Host Microbe. 2018;23(4):458–469.e5. doi: 10.1016/j.chom.2018.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. 2006;7(7):688–693. doi: 10.1038/sj.embor.7400731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Angelakis E, Armougom F, Carrière F, et al. A Metagenomic Investigation of the Duodenal Microbiota Reveals Links with Obesity. Covasa M, ed. PLoS One. 2015;10(9):e0137784. doi: 10.1371/journal.pone.0137784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Watanabe M, Fukiya S, Yokota A. Comprehensive evaluation of the bactericidal activities of free bile acids in the large intestine of humans and rodents. J Lipid Res. 2017;58(6):1143–1152. doi: 10.1194/jlr.M075143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gérard P Metabolism of Cholesterol and Bile Acids by the Gut Microbiota. Pathogens. 2013;3(1):14–24. doi: 10.3390/pathogens3010014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grill JP, Cayuela C, Antoine JM, Schneider F. Isolation and characterization of a Lactobacillus amylovorus mutant depleted in conjugated bile salt hydrolase activity: Relation between activity and bile salt resistance. J Appl Microbiol. 2000;89(4):553–563. doi: 10.1046/j.1365-2672.2000.01147.x [DOI] [PubMed] [Google Scholar]
- 64.Dussurget O, Cabanes D, Dehoux P, et al. Listeria monocytogenes bile salt hydrolase is a PrfA-regulated virulence factor involved in the intestinal and hepatic phases of listeriosis. Mol Microbiol. 2002;45(4):1095–1106. doi: 10.1046/j.1365-2958.2002.03080.x [DOI] [PubMed] [Google Scholar]
- 65.Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes. 2016;7(1):22–39. doi: 10.1080/19490976.2015.1127483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Light SH, Su L, Rivera-Lugo R, et al. A flavin-based extracellular electron transfer mechanism in diverse Gram-positive bacteria. Nature. 2018;562(7725):140–157. doi: 10.1038/s41586-018-0498-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thomson ABR, Drozdowski L, Iordache C, et al. Small bowel review: Normal physiology, part 1. Dig Dis Sci. 2003;48(8):1546–1564. doi: 10.1023/a:1024719925058 [DOI] [PubMed] [Google Scholar]
- 68.Quigley EMM, Abu-Shanab A. Small Intestinal Bacterial Overgrowth. Infect Dis Clin North Am. 2010;24(4):943–959. doi: 10.1016/j.idc.2010.07.007 [DOI] [PubMed] [Google Scholar]
- 69.Sundin OH, Mendoza-Ladd A, Zeng M, et al. The human jejunum has an endogenous microbiota that differs from those in the oral cavity and colon. BMC Microbiol. 2017;17(1):160. doi: 10.1186/s12866-017-1059-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang M, Ahrné S, Jeppsson B, Molin G. Comparison of bacterial diversity along the human intestinal tract by direct cloning and sequencing of 16S rRNA genes. FEMS Microbiol Ecol. 2005;54(2):219–231. doi: 10.1016/j.femsec.2005.03.012 [DOI] [PubMed] [Google Scholar]
- 71.Dlugosz A, Winckler B, Lundin E, et al. No difference in small bowel microbiota between patients with irritable bowel syndrome and healthy controls. Sci Rep. 2014;5:8508. doi: 10.1038/srep08508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Szentkuti L, Lorenz K. The thickness of the mucus layer in different segments of the rat intestine. Histochem J. 1995;27(6):466–472. doi: 10.1007/bf00173712 [DOI] [PubMed] [Google Scholar]
- 73.Villmones HC, Haug ES, Ulvestad E, et al. Species Level Description of the Human Ileal Bacterial Microbiota. Sci Rep. 2018;8(1):1–9. doi: 10.1038/s41598-018-23198-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Booijink CCGM, El-Aidy S, Rajilić-Stojanović M, et al. High temporal and inter-individual variation detected in the human ileal microbiota. Environ Microbiol. 2010;12(12):3213–3227. doi: 10.1111/j.1462-2920.2010.02294.x [DOI] [PubMed] [Google Scholar]
- 75.Martinez-Guryn K, Leone V, Chang EB. Regional Diversity of the Gastrointestinal Microbiome. Cell Host Microbe. 2019;26(3):314–324. doi: 10.1016/j.chom.2019.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hooper L V, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336(6086):1268–1273. doi: 10.1126/science.1223490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ivanov II, Atarashi K, Manel N, et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell. 2009;139(3):485–498. doi: 10.1016/j.cell.2009.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stockinger B, Omenetti S. The dichotomous nature of T helper 17 cells. Nat Rev Immunol. 2017;17(9):535–544. doi: 10.1038/nri.2017.50 [DOI] [PubMed] [Google Scholar]
- 79.Huang W, Ramsey KM, Marcheva B, Bass J. Circadian rhythms, sleep, and metabolism. J Clin Invest. 2011;121(6):2133–2141. doi: 10.1172/JCI46043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Turek FW, Joshu C, Kohsaka A, et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005;308(5724):1043–1045. doi: 10.1126/science.1108750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leone V, Gibbons SM, Martinez K, et al. Effects of diurnal variation of gut microbes and high-fat feeding on host circadian clock function and metabolism. Cell Host Microbe. 2015;17(5):681–689. doi: 10.1016/j.chom.2015.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thaiss CA, Zeevi D, Levy M, et al. Transkingdom Control of Microbiota Diurnal Oscillations Promotes Metabolic Homeostasis. Cell. 2014;159(3):514–529. doi: 10.1016/j.cell.2014.09.048 [DOI] [PubMed] [Google Scholar]
- 83.Wang Y, Kuang Z, Yu X, Ruhn KA, Kubo M, Hooper LV. The intestinal microbiota regulates body composition through NFIL3 and the circadian clock. Science (80- ). 2017;357(6354):912–916. doi: 10.1126/science.aan0677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Metcalf AM, Phillips SF, Zinsmeister AR, MacCarty RL, Beart RW, Wolff BG. Simplified assessment of segmental colonic transit. Gastroenterology. 1987;92(1):40–47. doi: 10.1016/0016-5085(87)90837-7 [DOI] [PubMed] [Google Scholar]
- 85.Chesson P Mechanisms of Maintenance of Species Diversity. Annu Rev Ecol Syst. 2000;31(1):343–366. doi: 10.1146/annurev.ecolsys.31.1.343 [DOI] [Google Scholar]
- 86.Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107(27):12204–12209. doi: 10.1073/pnas.0909122107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sommer F, Bäckhed F. Know your neighbor: Microbiota and host epithelial cells interact locally to control intestinal function and physiology. BioEssays. 2016;38(5):455–464. doi: 10.1002/bies.201500151 [DOI] [PubMed] [Google Scholar]
- 88.Bergman EN. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol Rev. 1990;70(2):567–590. doi: 10.1152/physrev.1990.70.2.567 [DOI] [PubMed] [Google Scholar]
- 89.Donohoe DR, Wali A, Brylawski BP, Bultman SJ. Microbial Regulation of Glucose Metabolism and Cell-Cycle Progression in Mammalian Colonocytes. Bereswill S, ed. PLoS One. 2012;7(9):e46589. doi: 10.1371/journal.pone.0046589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hamer HM, Jokners D, Venema K, Vanhoutvin S, Troost FJ, Brummer R- J. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2007;27(2):104–119. doi: 10.1111/j.1365-2036.2007.03562.x [DOI] [PubMed] [Google Scholar]
- 91.Kelly CJ, Zheng L, Campbell EL, et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe. 2015;17(5):662–671. doi: 10.1016/j.chom.2015.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rivera-Chávez F, Zhang LF, Faber F, et al. Depletion of Butyrate-Producing Clostridia from the Gut Microbiota Drives an Aerobic Luminal Expansion of Salmonella. Cell Host Microbe. 2016;19(4):443–454. doi: 10.1016/j.chom.2016.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–563. doi: 10.1038/nature12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-Induced Obesity Is Linked to Marked but Reversible Alterations in the Mouse Distal Gut Microbiome. Cell Host Microbe. 2008;3(4):213–223. doi: 10.1016/j.chom.2008.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sonnenburg ED, Zheng H, Joglekar P, et al. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell. 2010;141(7):1241–1252. doi: 10.1016/j.cell.2010.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kashyap PC, Marcobal A, Ursell LK, et al. Genetically dictated change in host mucus carbohydrate landscape exerts a diet-dependent effect on the gut microbiota. Proc Natl Acad Sci U S A. 2013;110(42):17059–17064. doi: 10.1073/pnas.1306070110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Larsson JMH, Karlsson H, Sjövall H, Hansson GC. A complex, but uniform O-glycosylation of the human MUC2 mucin from colonic biopsies analyzed by nanoLC/MSn. Glycobiology. 2009;19(7):756–766. doi: 10.1093/glycob/cwp048 [DOI] [PubMed] [Google Scholar]
- 98.Png CW, Lindén SK, Gilshenan KS, et al. Mucolytic Bacteria With Increased Prevalence in IBD Mucosa Augment In Vitro Utilization of Mucin by Other Bacteria. Am J Gastroenterol. 2010;105(11):2420–2428. doi: 10.1038/ajg.2010.281 [DOI] [PubMed] [Google Scholar]
- 99.Swidsinski A, Sydora BC, Doerffel Y, et al. Viscosity gradient within the mucus layer determines the mucosal barrier function and the spatial organization of the intestinal microbiota. Inflamm Bowel Dis. 2007;13(8):963–970. doi: 10.1002/ibd.20163 [DOI] [PubMed] [Google Scholar]
- 100.Albenberg L, Esipova TV, Judge CP, et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology. 2014;147(5):1055–1063.e8. doi: 10.1053/j.gastro.2014.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Eckburg PB, Bik EM, Bernstein CN, et al. Microbiology: Diversity of the human intestinal microbial flora. Science (80- ). 2005;308(5728):1635–1638. doi: 10.1126/science.1110591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pédron T, Mulet C, Dauga C, et al. A crypt-specific core microbiota resides in the mouse colon. MBio. 2012;3(3). doi: 10.1128/mBio.00116-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nava GM, Friedrichsen HJ, Stappenbeck TS. Spatial organization of intestinal microbiota in the mouse ascending colon. ISME J. 2011;5(4):627–638. doi: 10.1038/ismej.2010.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang Y, Devkota S, Musch MW, et al. Regional Mucosa-Associated Microbiota Determine Physiological Expression of TLR2 and TLR4 in Murine Colon. Yang C-H, ed. PLoS One. 2010;5(10):e13607. doi: 10.1371/journal.pone.0013607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47(2):241–259. doi: 10.1194/jlr.R500013-JLR200 [DOI] [PubMed] [Google Scholar]
- 106.Begley M, Gahan CGM, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev. 2005;29(4):625–651. doi: 10.1016/j.femsre.2004.09.003 [DOI] [PubMed] [Google Scholar]
- 107.Islam KBMS, Fukiya S, Hagio M, et al. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology. 2011;141(5):1773–1781. doi: 10.1053/j.gastro.2011.07.046 [DOI] [PubMed] [Google Scholar]
- 108.Devkota S, Wang Y, Musch MW, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature. 2012;487(7405):104–108. doi: 10.1038/nature11225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Makishima M, Okamoto AY, Repa JJ, et al. Identification of a nuclear receptor for bite acids. Science (80- ). 1999;284(5418):1362–1365. doi: 10.1126/science.284.5418.1362 [DOI] [PubMed] [Google Scholar]
- 110.Kawamata Y, Fujii R, Hosoya M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278(11):9435–9440. doi: 10.1074/jbc.M209706200 [DOI] [PubMed] [Google Scholar]
- 111.Li T, Chiang JYL. Bile acids as metabolic regulators. Curr Opin Gastroenterol. 2015;31(2):159–165. doi: 10.1097/MOG.0000000000000156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sinha SR, Haileselassie Y, Nguyen LP, et al. Dysbiosis-Induced Secondary Bile Acid Deficiency Promotes Intestinal Inflammation. Cell Host Microbe. 2020;27(4):659–670.e5. doi: 10.1016/j.chom.2020.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Round JL, Lee SM, Li J, et al. The toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science (80- ). 2011;332(6032):974–977. doi: 10.1126/science.1206095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Halfvarson J, Brislawn CJ, Lamendella R, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol. 2017;2(5):1–7. doi: 10.1038/nmicrobiol.2017.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sokol H, Pigneur B, Watterlot L, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105(43):16731–16736. doi: 10.1073/pnas.0804812105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Willing B, Halfvarson J, Dicksved J, et al. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflamm Bowel Dis. 2009;15(5):653–660. doi: 10.1002/ibd.20783 [DOI] [PubMed] [Google Scholar]
- 117.Rajca S, Grondin V, Louis E, et al. Alterations in the intestinal microbiome (Dysbiosis) as a predictor of relapse after infliximab withdrawal in Crohn’s disease. Inflamm Bowel Dis. 2014;20(6):978–986. doi: 10.1097/MIB.0000000000000036 [DOI] [PubMed] [Google Scholar]
- 118.MacArthur RH, Wilson EO. The Theory of Island Biogeography. Princeton University Press; 2001. [Google Scholar]
- 119.Wilkinson DM. The Disturbing History of Intermediate Disturbance. Oikos. 1999;84(1):145. doi: 10.2307/3546874 [DOI] [Google Scholar]
- 120.Caporaso JG, Lauber CL, Costello EK, et al. Moving pictures of the human microbiome. Genome Biol. 2011;12(5):R50. doi: 10.1186/gb-2011-12-5-r50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science (80- ). 2009;326(5960):1694–1697. doi: 10.1126/science.1177486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol. 2011;9(4):244–253. doi: 10.1038/nrmicro2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sender R, Fuchs S, Milo R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLOS Biol. 2016;14(8):e1002533. doi: 10.1371/journal.pbio.1002533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Leeming JP, Holland KT, Cunliffe WJ. The microbial ecology of pilosebaceous units isolated from human skin. J Gen Microbiol. 1984;130(4):803–807. doi: 10.1099/00221287-130-4-803 [DOI] [PubMed] [Google Scholar]
- 125.Decréau RA, Marson CM, Smith KE, Behan JM. Production of malodorous steroids from androsta-5,16-dienes and androsta-4,16-dienes by Corynebacteria and other human axillary bacteria. J Steroid Biochem Mol Biol. 2003;87(4-5):327–336. doi: 10.1016/j.jsbmb.2003.09.005 [DOI] [PubMed] [Google Scholar]
- 126.Natsch A, Gfeller H, Gygax P, Schmid J, Acuna G. A specific bacterial aminoacylase cleaves odorant precursors secreted in the human axilla. J Biol Chem. 2003;278(8):5718–5727. doi: 10.1074/jbc.M210142200 [DOI] [PubMed] [Google Scholar]
- 127.Emter R, Natsch A. The sequential action of a dipeptidase and a β-lyase is required for the release of the human body odorant 3-methyl-3-sulfanylhexan-1-ol from a secreted Cys-Gly-(S) conjugate by Corynebacteria. J Biol Chem. 2008;283(30):20645–20652. doi: 10.1074/jbc.M800730200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Grice EA, Kong HH, Conlan S, et al. Topographical and temporal diversity of the human skin microbiome. Science (80- ). 2009;324(5931):1190–1192. doi: 10.1126/science.1171700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Brüggemann H, Henne A, Hoster F, et al. The complete genome sequence of Propionibacterium acnes, a commensal of human skin. Science (80- ). 2004;305(5684):671–673. doi: 10.1126/science.1100330 [DOI] [PubMed] [Google Scholar]
- 130.Marples RR, Downing DT, Kligman AM. Control of free fatty acids in human surface lipids by Corynebacterium acnes. J Invest Dermatol. 1971;56(2):127–131. doi: 10.1111/1523-1747.ep12260695 [DOI] [PubMed] [Google Scholar]
- 131.Roth RR, James WD. Microbial Ecology of the Skin. Annu Rev Microbiol. 1988;42(1):441–464. doi: 10.1146/annurev.mi.42.100188.002301 [DOI] [PubMed] [Google Scholar]
- 132.Elias PM. The skin barrier as an innate immune element. Semin Immunopathol. 2007;29(1):3–14. doi: 10.1007/s00281-007-0060-9 [DOI] [PubMed] [Google Scholar]
- 133.Jo JH, Kennedy EA, Kong HH. Topographical and physiological differences of the skin mycobiome in health and disease. Virulence. 2017;8(3):324–333. doi: 10.1080/21505594.2016.1249093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Marples MJ. The Ecology of the Human Skin. Springfield, IL: Charles C. Thomas; 1965. [Google Scholar]
- 135.Gao Z, Perez-Perez GI, Chen Y, Blaser MJ. Quantitation of major human cutaneous bacterial and fungal populations. J Clin Microbiol. 2010;48(10):3575–3581. doi: 10.1128/JCM.00597-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sumikawa Y, Asada H, Hoshino K, et al. Induction of β-defensin 3 in keratinocytes stimulated by bacterial lipopeptides through toll-like receptor 2. Microbes Infect. 2006;8(6):1513–1521. doi: 10.1016/j.micinf.2006.01.008 [DOI] [PubMed] [Google Scholar]
- 137.Nagy I, Pivarcsi A, Kis K, et al. Propionibacterium acnes and lipopolysaccharide induce the expression of antimicrobial peptides and proinflammatory cytokines/chemokines in human sebocytes. Microbes Infect. 2006;8(8):2195–2205. doi: 10.1016/j.micinf.2006.04.001 [DOI] [PubMed] [Google Scholar]
- 138.Chehoud C, Rafail S, Tyldsley AS, Seykora JT, Lambris JD, Grice EA. Complement modulates the cutaneous microbiome and inflammatory milieu. Proc Natl Acad Sci U S A. 2013;110(37):15061–15066. doi: 10.1073/pnas.1307855110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Naik S, Bouladoux N, Wilhelm C, et al. Compartmentalized control of skin immunity by resident commensals. Science (80- ). 2012;337(6098):1115–1119. doi: 10.1126/science.1225152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Belkaid Y, Segre JA. Dialogue between skin microbiota and immunity. Science (80- ). 2014;346(6212):954–959. doi: 10.1126/science.1260144 [DOI] [PubMed] [Google Scholar]
- 141.Cogen AL, Yamasaki K, Muto J, et al. Staphylococcus epidermidis Antimicrobial δ-Toxin (Phenol-Soluble Modulin-γ) Cooperates with Host Antimicrobial Peptides to Kill Group A Streptococcus. DeLeo FR, ed. PLoS One. 2010;5(1):e8557. doi: 10.1371/journal.pone.0008557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Anderson DJ, Marathe J, Pudney J. The Structure of the Human Vaginal Stratum Corneum and its Role in Immune Defense. Am J Reprod Immunol. 2014;71(6):618–623. doi: 10.1111/aji.12230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Boskey ER, Cone RA, Whaley KJ, Moench TR. Origins of vaginal acidity: High D/L lactate ratio is consistent with bacteria being the primary source. Hum Reprod. 2001;16(9):1809–1813. doi: 10.1093/humrep/16.9.1809 [DOI] [PubMed] [Google Scholar]
- 144.Ravel J, Gajer P, Abdo Z, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A. 2011;108(SUPPL. 1):4680–4687. doi: 10.1073/pnas.1002611107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hyman RW, Fukushima M, Jiang H, et al. Diversity of the vaginal microbiome correlates with preterm birth. Reprod Sci. 2014;21(1):32–40. doi: 10.1177/1933719113488838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Witkin SS. The vaginal microbiome, vaginal anti-microbial defence mechanisms and the clinical challenge of reducing infection-related preterm birth. BJOG An Int J Obstet Gynaecol. 2015;122(2):213–218. doi: 10.1111/1471-0528.13115 [DOI] [PubMed] [Google Scholar]
- 147.Onderdonk AB, Delaney ML, Fichorova RN. The human microbiome during bacterial vaginosis. Clin Microbiol Rev. 2016;29(2):223–238. doi: 10.1128/CMR.00075-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Donders GGG, Bosmans E, Dekeersmaecker A, Vereecken A, Van Bulck B, Spitz B. Pathogenesis of abnormal vaginal bacterial flora. Am J Obstet Gynecol. 2000;182(4):872–878. doi: 10.1016/S0002-9378(00)70338-3 [DOI] [PubMed] [Google Scholar]
- 149.Serrano MG, Parikh HI, Brooks JP, et al. Racioethnic diversity in the dynamics of the vaginal microbiome during pregnancy. Nat Med. 2019;25(6):1001–1011. doi: 10.1038/s41591-019-0465-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhou X, Bent SJ, Schneider MG, Davis CC, Islam MR, Forney LJ. Characterization of vaginal microbial communities in adult healthy women using cultivation-independent methods. Microbiology. 2004;150(8):2565–2573. doi: 10.1099/mic.0.26905-0 [DOI] [PubMed] [Google Scholar]
- 151.Zhou X, Brown CJ, Abdo Z, et al. Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. ISME J. 2007;1(2):121–133. doi: 10.1038/ismej.2007.12 [DOI] [PubMed] [Google Scholar]
- 152.Gong Z, Luna Y, Yu P, Fan H. Lactobacilli Inactivate Chlamydia trachomatis through Lactic Acid but Not H2O2. Yang X, ed. PLoS One. 2014;9(9):e107758. doi: 10.1371/journal.pone.0107758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Graver MA, Wade JJ. The role of acidification in the inhibition of Neisseria gonorrhoeae by vaginal lactobacilli during anaerobic growth. Ann Clin Microbiol Antimicrob. 2011;10:8. doi: 10.1186/1476-0711-10-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lai SK, Hida K, Shukair S, et al. Human immunodeficiency virus type 1 is trapped by acidic but not by neutralized human cervicovaginal mucus. J Virol. 2009;83(21):11196–11200. doi: 10.1128/JVI.01899-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Witkin SS, Mendes-Soares H, Linhares IM, Jayaram A, Ledger WJ, Forney LJ. Influence of vaginal bacteria and D- and L-lactic acid isomers on vaginal extracellular matrix metalloproteinase inducer: Implications for protection against upper genital tract infections. MBio. 2013;4(4). doi: 10.1128/mBio.00460-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hearps A, Gugasyan R, Srbinovski D, et al. Lactic Acid, a Vaginal Microbiota Metabolite, Elicits an Anti-inflammatory Response from Vaginal and Cervical Epithelial Cells. AIDS Res Hum Retroviruses. 2014;30(S1):A238–A239. doi: 10.1089/aid.2014.5527.abstract [DOI] [Google Scholar]
- 157.Stoyancheva G, Marzotto M, Dellaglio F, Torriani S. Bacteriocin production and gene sequencing analysis from vaginal Lactobacillus strains. Arch Microbiol. 2014;196(9):645–653. doi: 10.1007/s00203-014-1003-1 [DOI] [PubMed] [Google Scholar]
- 158.Aroutcheva A, Gariti D, Simon M, et al. Defense factors of vaginal lactobacilli. Am J Obstet Gynecol. 2001;185(2):375–379. doi: 10.1067/mob.2001.115867 [DOI] [PubMed] [Google Scholar]
- 159.Ojala T, Kankainen M, Castro J, et al. Comparative genomics of Lactobacillus crispatus suggests novel mechanisms for the competitive exclusion of Gardnerella vaginalis. BMC Genomics. 2014;15(1):1070. doi: 10.1186/1471-2164-15-1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Nunn KL, Forney LJ. Unraveling the dynamics of the human vaginal microbiome. Yale J Biol Med. 2016;89(3):331–337. [PMC free article] [PubMed] [Google Scholar]
- 161.Mendes-Soares H, Suzuki H, Hickey RJ, Forneya LJ. Comparative functional genomics of Lactobacillus spp. reveals possible mechanisms for specialization of vaginal lactobacilli to their environment. J Bacteriol. 2014;196(7):1458–1470. doi: 10.1128/JB.01439-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Miller EA, Beasley DAE, Dunn RR, Archie EA. Lactobacilli dominance and vaginal pH: Why is the human vaginal microbiome unique? Front Microbiol. 2016;7(DEC):1936. doi: 10.3389/fmicb.2016.01936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gajer P, Brotman RM, Bai G, et al. Temporal dynamics of the human vaginal microbiota. Sci Transl Med. 2012;4(132). doi: 10.1126/scitranslmed.3003605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chaban B, Links MG, Jayaprakash TP, et al. Characterization of the vaginal microbiota of healthy Canadian women through the menstrual cycle. Microbiome. 2014;2(1):23. doi: 10.1186/2049-2618-2-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Noyes N, Cho KC, Ravel J, Forney LJ, Abdo Z. Associations between sexual habits, menstrual hygiene practices, demographics and the vaginal microbiome as revealed by Bayesian network analysis. PLoS One. 2018;13(1). doi: 10.1371/journal.pone.0191625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Romero R, Hassan SS, Gajer P, et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome. 2014;2(1):1–19. doi: 10.1186/2049-2618-2-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.DiGiulio DB, Callahan BJ, McMurdie PJ, et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc Natl Acad Sci U S A. 2015;112(35):11060–11065. doi: 10.1073/pnas.1502875112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Frank DN, Feazel LM, Bessesen MT, Price CS, Janoff EN, Pace NR. The Human Nasal Microbiota and Staphylococcus aureus Carriage. Aziz RK, ed. PLoS One. 2010;5(5):e10598. doi: 10.1371/journal.pone.0010598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yan M, Pamp SJ, Fukuyama J, et al. Nasal microenvironments and interspecific interactions influence nasal microbiota complexity and S. aureus carriage. Cell Host Microbe. 2013;14(6):631–640. doi: 10.1016/j.chom.2013.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Bassis CM, Tang AL, Young VB, Pynnonen MA. The nasal cavity microbiota of healthy adults. Microbiome. 2014;2(1):27. doi: 10.1186/2049-2618-2-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fahy J V, Dickey BF. Medical progress: Airway mucus function and dysfunction. N Engl J Med. 2010;363(23):2233–2247. doi: 10.1056/NEJMra0910061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Dickson RP, Erb-Downward JR, Martinez FJ, Huffnagle GB. The Microbiome and the Respiratory Tract. Annu Rev Physiol. 2016;78(1):481–504. doi: 10.1146/annurev-physiol-021115-105238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.West JB. Regional differences in the lung. Chest. 1978;74(4):426–437. doi: 10.1016/S0012-3692(15)37392-X [DOI] [PubMed] [Google Scholar]
- 174.Ingenito EP, Solway J, McFadden ER, et al. Indirect assessment of mucosal surface temperatures in the airways: Theory and tests. J Appl Physiol. 1987;63(5):2075–2083. doi: 10.1152/jappl.1987.63.5.2075 [DOI] [PubMed] [Google Scholar]
- 175.Charlson ES, Bittinger K, Haas AR, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184(8):957–963. doi: 10.1164/rccm.201104-0655OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dickson RP, Erb-Downward JR, Freeman CM, et al. Spatial variation in the healthy human lung microbiome and the adapted island model of lung biogeography. Ann Am Thorac Soc. 2015;12(6):821–830. doi: 10.1513/AnnalsATS.201501-029OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Venkataraman A, Bassis CM, Beck JM, et al. Application of a neutral community model to assess structuring of the human lung microbiome. MBio. 2015;6(1). doi: 10.1128/mBio.02284-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Erb-Downward JR, Thompson DL, Han MK, et al. Analysis of the Lung Microbiome in the “Healthy” Smoker and in COPD. Bereswill S, ed. PLoS One. 2011;6(2):e16384. doi: 10.1371/journal.pone.0016384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Willner D, Haynes MR, Furlan M, et al. Spatial distribution of microbial communities in the cystic fibrosis lung. ISME J. 2012;6(2):471–474. doi: 10.1038/ismej.2011.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Worlitzsch D, Tarran R, Ulrich M, et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest. 2002;109(3):317–325. doi: 10.1172/jci13870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Schmidt A, Belaaouaj A, Bissinger R, et al. Neutrophil elastase-mediated increase in airway temperature during inflammation. J Cyst Fibros. 2014;13(6):623–631. doi: 10.1016/j.jcf.2014.03.004 [DOI] [PubMed] [Google Scholar]