

Abstract
Defects in DNA repair and the protection of stalled DNA replication forks are thought to underlie the chemosensitivity of tumors deficient in the hereditary breast cancer genes BRCA1 and BRCA2 (BRCA). Challenging this assumption are recent findings that indicate chemotherapies such as cisplatin used to treat BRCA-deficient tumors do not initially cause DNA double-strand-breaks (DSB). Here we show that single-stranded DNA (ssDNA) replication gaps underlie the hypersensitivity of BRCA-deficient cancer and that defects in homologous recombination (HR) or fork protection (FP) do not. In BRCA-deficient cells, ssDNA gaps developed because replication was not effectively restrained in response to stress. Gap suppression by either restoration of fork restraint or gap filling conferred therapy resistance in tissue culture and BRCA patient tumors. In contrast, restored FP and HR could be uncoupled from therapy resistance when gaps were present. Moreover, DSB were not detected after therapy when apoptosis was inhibited, supporting a framework in which DSB are not directly induced by genotoxic agents, but rather are induced from cell death nucleases and are not fundamental to the mechanism of action of genotoxic agents. Together, these data indicate that ssDNA replication gaps underlie the BRCA cancer phenotype, “BRCAness,” and we propose they are fundamental to the mechanism-of-action of genotoxic chemotherapies.
INTRODUCTION
Mutations in the hereditary breast cancer genes, BRCA1 and BRCA2, first demonstrated that cancer is a genetic disease in which susceptibility to cancer could be inherited (1). In addition to breast cancer, mutated BRCA1 or BRCA2 cause a predisposition to other cancer types, including ovarian, pancreatic, and colorectal cancers. Importantly, cancers with mutated BRCA genes are hypersensitive to cisplatin, a first-line anti-cancer chemotherapy that has been the standard of care for ovarian cancer for over 40 years (2). BRCA-deficient cancers are thought to be hypersensitive to cisplatin due to their inability to repair cisplatin-induced DNA double strand breaks (DSBs) by homologous recombination (HR) (3). Accordingly, it is proposed that the DSBs are created when replication forks collide with the cisplatin-DNA crosslinks, causing the fork to collapse into DSBs (4). This broken-fork-model was further supported by reports that mutations in the BRCA genes also lead to defective fork protection (FP), which is thought to render forks vulnerable to fork collapse and subsequent double strand break induction (5–7). Correspondingly, chemoresistance in BRCA cancer is proposed to occur when either HR or FP is restored, with the latter largely preventing DSBs and therefore eliminating the requirement for HR. Importantly, this hypersensitivity phenotype is known as BRCAness and is thought to arise in a range of cancers via mutations in genes that function similar to BRCA1 and BRCA2 in DSB repair.
However, recent findings challenge the fundamental premise that DSBs are the critical lesion for cisplatin sensitivity. Notably, DNA crosslinks do not appear to initially cause replication forks to collapse and can be bypassed (8, 9). Moreover, in the majority of genetic models currently reported, restored FP fails to restore cisplatin resistance, suggesting the cisplatin lesions do not collapse forks, and therefore calls into question how cisplatin crosslinks could be converted into DSBs (4, 10). Most saliently, indicating that the fundamental sensitizing lesion may in fact not be a DSB, reports indicate even HR proficient cells can nevertheless display hypersensitivity to cisplatin and other genotoxic agents (11–13). Moreover, in addition to cisplatin, BRCA-deficient cells and patient tumors have recently been found to be hypersensitive to a wide range of genotoxic agents that were previously thought to be mechanistically distinct, including doxorubicin, Poly(ADP-ribose) polymerase 1 inhibition (PARPi), and other first-line agents, even including the platinum analog oxaliplatin, which is not thought to generate DSBs (14). Moreover, recent reports indicate that cisplatin toxicity in triple negative breast cancer is unrelated to loss of DNA repair factors (15). Taken together, these findings indicate an opportunity to revise the current framework for both BRCAness as well as the mechanism-of-action of first-line genotoxic chemotherapies.
Here, we propose a model for genotoxic chemotherapy in which hypersensitivity derives from single stranded DNA (ssDNA) formation, and not from the failure to repair or prevent the induction of DSBs due to defects in HR or FP. Specifically, we observed in hypersensitive BRCA-deficient cells that ssDNA gaps develop because DNA replication is not effectively restrained in response to genotoxic stress. Moreover, we observed ssDNA gaps could be suppressed by either restored fork restraint or by gap filling, both of which conferred resistance to genotoxic therapy in tissue culture and BRCA patient tumors. In contrast, we observed that cells with proficient HR and FP are nevertheless hypersensitive to chemotherapy if ssDNA gaps remain. Finally, we find that when apoptosis is inhibited, DSBs are no longer detectable after therapy, suggesting that DSBs are instead created by the programmed cell death nucleolytic machinery and that ssDNA gaps are the critical lesion that determines therapy response. Accordingly, we propose that ssDNA replication gaps underlie the BRCA cancer phenotype, “BRCAness,” and are fundamental to the mechanism-of-action of genotoxic chemotherapies.
MATERIALS AND METHODS
Cell Culture
PEO1, C4-2, VC-8, and MDA-MB-436 cell lines were cultured in DMEM + 10% FBS + 1% P/S. HCC1937 Deficient and HCC1937 + WT BRCA1 were cultured in RPMI1640 + L-Glutamine + 10% FBS + 1% P/S. The Fanconi Anemia RAD51 T131P cells were cultured in DMEM + 15% FBS + Glutamax supplemented with non-essential amino acids. All cells were confirmed mycoplasma free with the MycoALERT kit according to the manufacturer’s instructions (Lonza), with the most recent test in September 2020. PEO1 and C4–2 cells were obtained from the Toshi Taniguchi Lab in September of 2014; VC-8 cells were obtained from the Maria Jasin Lab in September of 2014; HCC1937 cells were obtained from the Lee Zhou Lab in October of 2017; and the RAD51 T131P cells were obtained from the Agata Smogorzewska Lab in January of 2019. The MDA-MB-436 were obtained from ATCC and validated by STR profiling. Cells were validated by western blot and/or Cell Titer Glo toxicity assays as described in the manuscript. Cells were briefly expanded to frozen stocks and used in experiments within ten passages.
DNA Fiber Assays
DNA fiber assays were performed as previously described. Briefly, cells were plated at 106 cells per 10cm dish and allowed to adhere for 36h. Subsequently, DNA was labeled for 30 minutes with 50uM IdU and washed with PBS, and treated with 50uM CldU and replication stress depending on the assay. For fork restraint assays, cells were exposed to 50uM CldU with 0.5mM HU for 2h. For fork restraint with continued stress, cells were exposed to 50uM CldU with 0.5mM HU for 2h, followed by 4mM HU for 2–3h. For fork degradation assays, cells were labeled with 50uM CldU alone, followed by 4mM HU for 3–5h. After labeling, cells were collected with trypsin, washed with PBS, and resuspended in PBS at 250,000 cells/ml. 2ul of cell solution was placed on a positively charged slide, followed by lysis for 8 minutes with 12.5ul of spreading buffer (0.5% SDS, 200mM Tris-HCl, pH 7.4, 50mM EDTA). Slides were tilted to a 45 degree angle to allow fibers to spread, allowed to dry for 20 minutes, fixed in 3:1 Methanol:Acetic Acid for 3 minutes, rehydrated in PBS for 5 minutes, denatured with 2.5mM HCl for 30 minutes, blocked with PBS + 0.1% TritonX-100 + 3% BSA for 1h, and treated with primary (2.5h, 1:100) and secondary antibodies (1h, 1:200) in PBS + 0.1% TritonX-100 + 3% BSA. Slides were washed with PBS and mounted with ProLong Gold antifade. Track lengths were measured in Fiji (16). The antibody used to detect IdU was anti-BrdU (Becton Dickinson 347580, detects both BrdU and IdU); the antibody used to detect CldU was anti-BrdU (Abcam ab6326, detects both BrdU and CldU). The secondary antibodies used were Alexa 488 anti-mouse (detects the primary IdU antibody) and Alexa 594 anti-rat (detects the primary CldU antibody).
Non-Denaturing ssDNA Fiber Assay
The nondenaturing fiber assay to detect ssDNA was performed using the DNA Fiber Assay protocol above with the following modifications: first, all acid steps were removed (both acetic acid from the fixation step, and the HCl denaturing step), and EDTA was removed from the lysis buffer (EDTA impairs Click Chemistry). In addition, IdU was replaced with EdU and detected by ClickIT EdU Alexa 488 Imaging Kit (Thermo Scientific) to label analog in non-denatured DNA per the manufacturer’s instructions. After Click Chemistry, ssDNA was detected by incubating DNA with the primary anti-BrdU antibody (Abcam ab6326, detects both BrdU and CldU) and the secondary antibody Alexa 594 anti-rat as described above. Images were analyzed in Fiju. We classified ssDNA-positive forks based on their line graph; specifically, if ssDNA signal was found adjacent to the EdU labeled regions, the fork was classified as ssDNA positive. In contrast, if there were no regions of ssDNA signal adjacent to the EdU, the fork was classified as ssDNA negative.
S1 Nuclease Fiber Assay
As described previously, cells were exposed to 50uM IdU to label replication forks, followed by 50uM CldU with 0.5mM HU for 2h. Subsequently, cells were permeabilized with CSK buffer (100 mM NaCl, 10 mM MOPS, 3 mM MgCl2 pH 7.2, 300 mM sucrose, 0.5% Triton X-100) at room temperature for 8 minutes, followed by S1 nuclease (20U/ml) in S1 buffer (30 mM Sodium Acetate pH 4.6, 10 mM Zinc Acetate, 5% Glycerol, 50 mM NaCl) for 30 minutes at 37C. Finally, cells were collected by scraping, pelleted, resuspended in 100–500ul PBS; 2ul of cell suspension was spotted on a positively charged slide and lysed and processed as described in the DNA fiber assay section above.
PDX Methods
PNX0204 was derived at Fox Chase Cancer Center under IRB and IACUC approved protocols. PDX tumors were grown in NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice. Cisplatin resistant PDX tumors were obtained from mice after tumors progressed on serial treatments of 6 mg/kg cisplatin. The tumors were harvested at approximately 500 mm3 and dissociated in 0.2% collagenase, 0.33 mg/ml dispase solution for 3h at 37°C. The dissociated cells were maintained at 37°C in RPMI1640 + 10% FBS and used for DNA fiber assays within 24h of tumor extraction. DNA fiber and S1 nuclease fiber assays were performed as described above.
RESULTS
To analyze the mechanism underlying the hypersensitivity of BRCA-deficient cancers to chemotherapy, we monitored the immediate response of DNA replication forks to replication stress with DNA fiber assays. Following the incorporation of nucleotide analogs into nascent DNA as the cells replicate in the presence or absence of stress, the progression of replication forks was detected by immunofluorescence. Specifically, we measured the lengths of the labeled DNA when the cells were exposed to 0.5mM hydroxyurea (HU), a dose that induces replication stress without fully depleting nucleotide pools (17). The condition yields high quality DNA fibers and has been used as a model to study fork responses to genotoxic therapy such as cisplatin, which yields lower quality fibers because cisplatin covalently damages DNA (17, 18). We compared the parental PEO1 cancer cell line, which expresses a truncated BRCA2 protein and is hypersensitive to cisplatin, to the BRCA2 proficient PEO1 reversion cell line, C4–2, which expresses a full-length BRCA2 protein and is resistant to cisplatin (19) (Figure 1A). Both cell lines were incubated with the DNA analog 5-Iodo-2′-deoxyuridine (IdU) for thirty minutes as an internal control to label regions of active replication, followed by the DNA analog 5-chloro-2′-deoxyuridine (CldU) for two hours in the presence of 0.5mM HU in order to monitor the immediate response of DNA replication to genotoxic stress. An additional set of cells were exposed to CldU without HU to serve as untreated controls.
Figure 1: BRCA2 deficient cancer cells fail to restrain replication in the presence of stress, generating regions of ssDNA gaps that are destroyed after continued exposure.
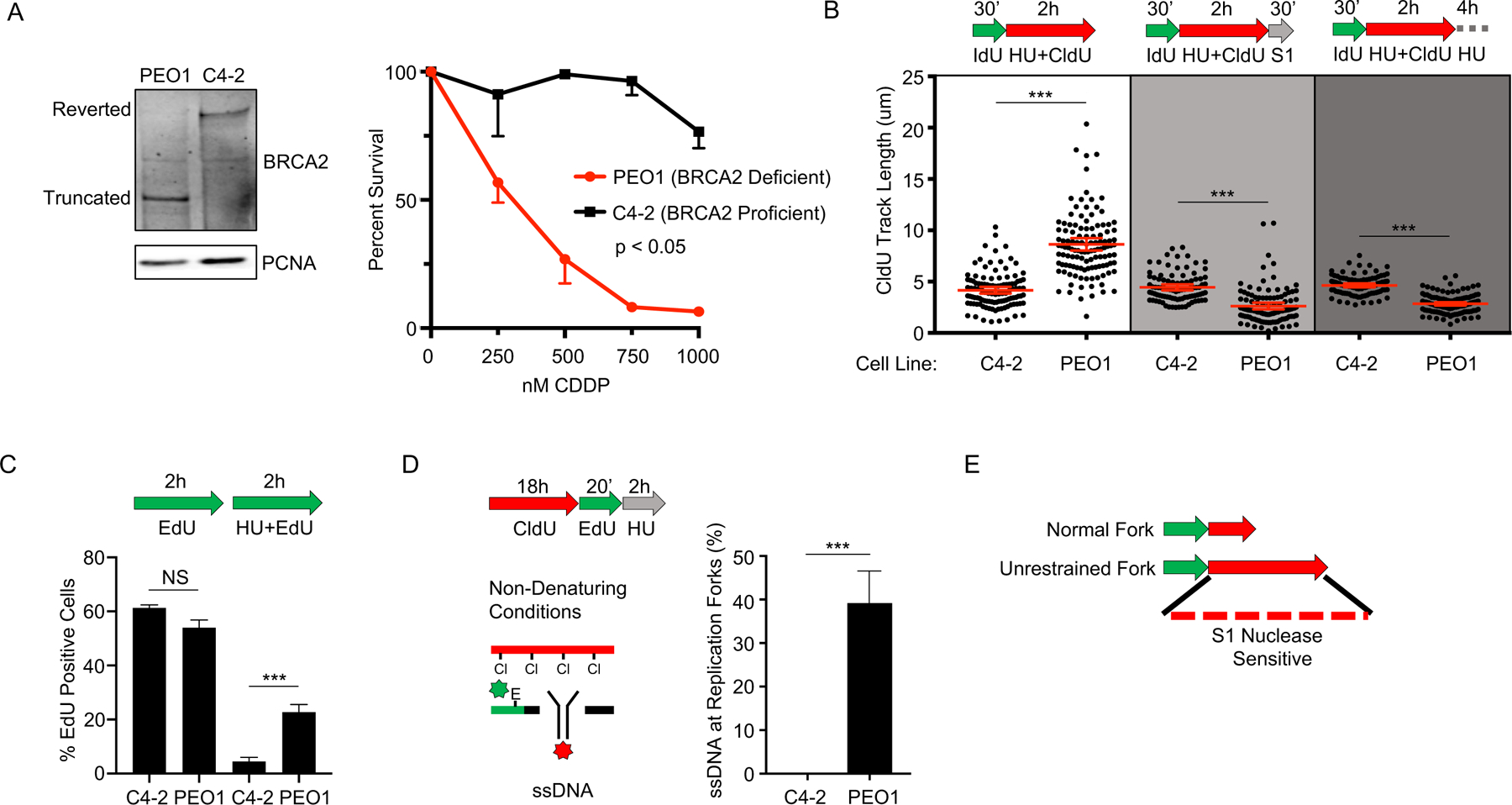
A) Left, Western blot detects truncated BRCA2 protein in BRCA2 deficient PEO1 cells and detects full-length BRCA2 protein in BRCA2 proficient C4–2 cells that are derived from PEO1 cells. Right, cell survival assay confirms PEO1 cells are hypersensitive to cisplatin compared to C4–2 cells. B) Schematic and quantification of CldU track length shows (white panel) that PEO1 cells fail to arrest replication in the presence of stress. These regions are degraded by S1 nuclease (light grey panel), and are also destroyed after continued exposure to replication stress (dark grey panel). Each dot represents one fiber. Experiments were performed in biological triplicate with at least 100 fibers per replicate. Statistical analysis according to two-tailed Mann-Whitney test; *** p< 0.001. Mean and 95% confidence intervals are shown. C) Schematic and quantification of nuclear imaging identifies a greater percentage of EdU positive cells in PEO1 as compared to C4–2. p< 0.05 (*) as determined by t-test of biological triplicate experiments. D) Nondenaturing fiber assay identifies exposed ssDNA adjacent to newly replicating regions after stress in PEO1, but not C4–2 cells. Regions of active replication were detected with EdU-Click chemistry; p < 0.01 (***) as determined by t-test of biological triplicate experiments. E) Model of fiber assay interpretation.
We observed that the BRCA2-deficient PEO1 cells failed to fully restrain replication in response to HU when compared to the BRCA2-proficient C4–2 cells, as indicated by the longer CldU tracks observed in PEO1 compared to C4–2 (Figure 1B). As expected, both untreated controls displayed substantially longer CldU tracks than either of the HU treated cells (Figure S1A, B), therefore indicating that replication is restrained after stress, and that this restraint is less effective in BRCA2 deficient cells. Moreover, we observed similar replication-restraint defects in other BRCA-deficient cells that are hypersensitive to cisplatin, including the BRCA2 deficient Chinese hamster cell line VC-8 (6), BRCA2 depleted C4–2 cells, and BRCA1 deficient breast cancer lines (HCC1937 and MDA-MB-436) (Figure S1C–H). We also observed that the replication restraint defects were not exclusive to HU, but also detected following cisplatin (Figure S1I). In agreement with the DNA fiber assays, analysis of global cellular DNA replication based on incorporation of the analog 5-Ethynyl-2´-deoxyuridine (EdU) similarly indicated that BRCA2 deficient cells failed to properly restrain DNA replication during stress (Figure 1C).
We hypothesized that failure to fully restrain replication during stress in BRCA-deficient cells would result in poorly replicated regions that contain ssDNA. To test this hypothesis, we performed the DNA fiber assay followed by incubation with S1 nuclease. S1 cuts at ssDNA regions and secondary DNA structures, but does not cut dsDNA (20). Indeed, labelled nascent DNA tracks were S1 sensitive in BRCA2-deficient PEO1 cells, but not in the BRCA2-proficient C4–2 cells (Figure 1B). These S1 sensitive nascent DNA regions were also degraded after continued exposure to replication stress, indicating that nascent DNA in regions behind the fork are degraded under continued stress (Figure 1B). Similar to BRCA2, BRCA1 deficient cancer cells (HCC1937 and MDA-MB-436) also displayed DNA replication tracks that were sensitive to S1 nuclease after treatment with HU (Figure S1J). In addition, we employed a non-denaturing DNA fiber assay that detects ssDNA in regions of active DNA replication and confirmed that following HU, ssDNA (detectable by the CldU antibody only in exposed ssDNA regions) was present adjacent to newly replicating regions (detected as EdU signal) in the BRCA2 deficient PEO1 cells, but not in the BRCA2 proficient C4–2 cells (Figure 1D). In contrast, ssDNA was not detected in the untreated cells (Figure S1K). Thus, BRCA-deficient cancer cells fail to fully restrain replication in the presence of stress, creating ssDNA regions (Figure 1E) that are degraded after additional exposure to stress.
We hypothesized that ssDNA gaps confer chemosensitivity in BRCA cancer, and that mechanisms of chemoresistance would suppress these gaps. Indeed, we previously found that depletion of the chromatin remodeling enzyme CHD4 confers cisplatin resistance in BRCA2 deficient PEO1 cells (Figure 2A) (21). Therefore, we tested if CHD4 depletion would reduce ssDNA gaps in PEO1 cells in the S1 fiber assay. When CHD4 was depleted, we observed protection from S1 nuclease after HU compared to the PEO1 non-silencing control, which was degraded to a length below even the arrested forks found in BRCA2 proficient C4–2 cells, therefore indicating ssDNA gaps were reduced in the resistant cells after HU treatment (Figure 2B, Figure S2A–D). Moreover, when CHD4 was depleted, we found nascent DNA tracks were not degraded after continued exposure to HU (Figure 2B). Collectively, these findings indicate that CHD4-depletion in BRCA2 deficient cells reduced ssDNA gaps during replication stress. Notably, however, replication restraint in response to stress was not observed upon CHD4 depletion. Instead, the replication tracks during HU appeared to be longer in CHD4-depleted PEO1 cells compared to PEO1 control cells (Figure 2B, Figure S2B, C, D). Moreover, in agreement with the fiber assays, analysis of global cellular replication by EdU incorporation demonstrated that CHD4-depleted PEO1 cells increased replication after HU treatment as compared to PEO1 or C4–2 control cells (Figure 2C). In addition, we also observed a significant reduction in ssDNA adjacent to regions of active replication in the non-denaturing DNA fiber assay (Figure 2D, Figure S2E). Thus, ssDNA gap formation was suppressed in chemoresistant BRCA2 deficient cells with CHD4-depletion, but fork restraint was not restored (Figure 2E). Taken together, these data indicate that chemoresistant cells display either restored fork restraint, as observed in the BRCA2 reversion cell line C4–2, or continuous replication without ssDNA gap formation, as in the CHD4-depleted PEO1 cells (Figure 2E).
Figure 2: CHD4 depletion suppresses ssDNA gaps but does not restore fork restraint.
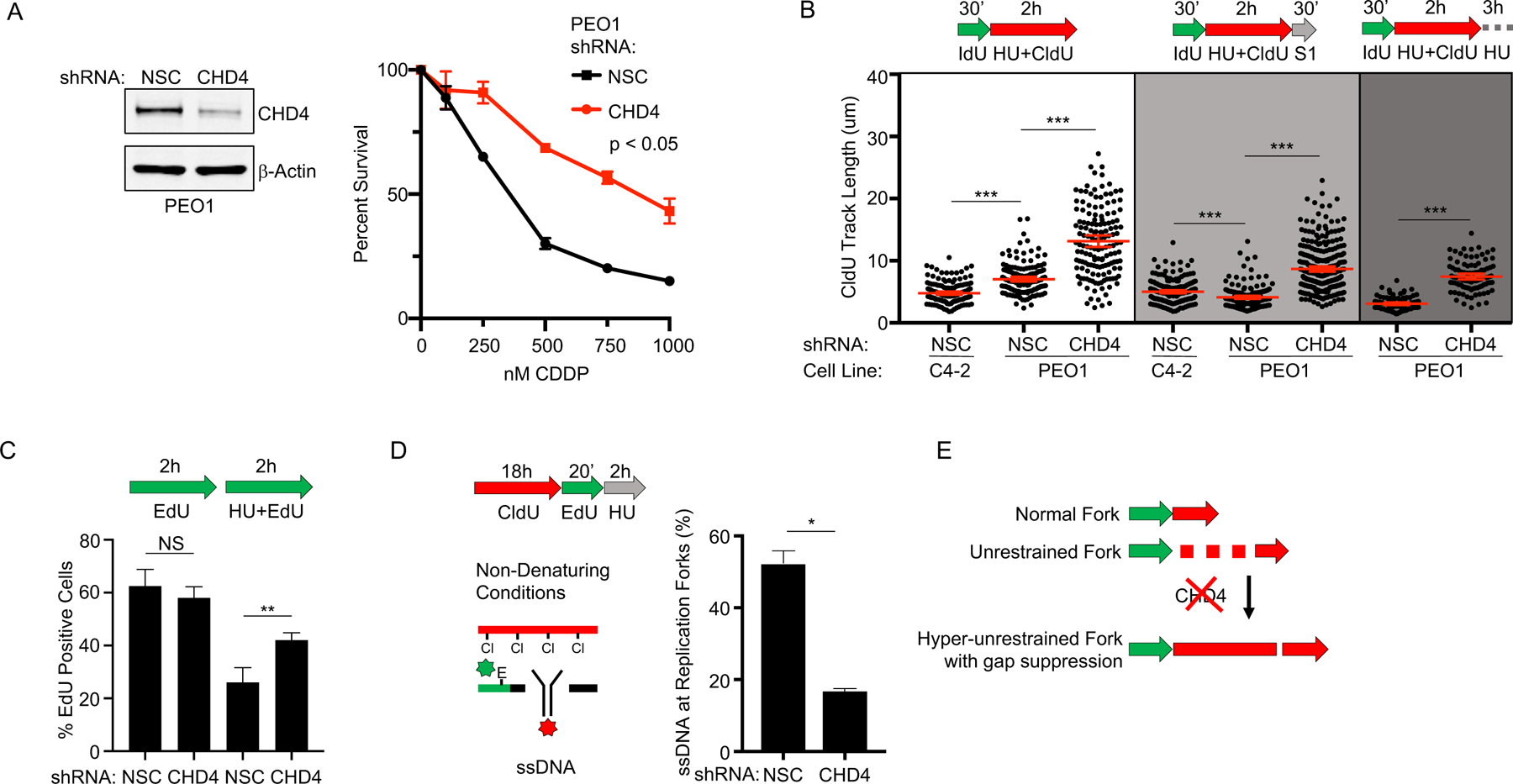
A) Left, Western blot confirms CHD4 is depleted by shRNA compared to non-silencing control (NSC) in BRCA2 deficient PEO1. Right, cell survival assay confirms PEO1 with depleted CHD4 are resistant to cisplatin compared to PEO1 NSC. B) Schematic and quantification of CldU track length shows that PEO1 with depleted CHD4 increase replication in the presence of stress (white panel). These regions are protected from S1 nuclease (light grey panel), and are also protected after continued exposure to replication stress (dark grey panel). Each dot represents one fiber. Experiments were performed in biological triplicate with at least 100 fibers per replicate. Statistical analysis according to two-tailed Mann-Whitney test; p < 0.001 (***). Mean and 95% confidence intervals shown. C) Schematic and quantification of nuclear imaging identifies a greater percentage of EdU positive cells in CHD4 depleted PEO1 as compared to NSC. p < 0.01 (**) as determined by t-test of biological triplicate experiments. D) Nondenaturing fiber assay identifies that ssDNA adjacent to newly replicating regions after stress is reduced when CHD4 is depleted in PEO1 cells. Regions of active replication were detected with EdU-Click chemistry; p < 0.05 (*) as determined by t-test of biological duplicate experiments. E) Model of fiber assay interpretation.
Our data indicate that suppression of ssDNA replication gaps in BRCA-deficient cancer could confer chemoresistance. To address this possibility, we sought to identify additional genes similar to CHD4 that confer chemoresistance when depleted in BRCA2 deficient cells, and subsequently determine if gaps were suppressed. Therefore, we performed quantitative mass spectrometry proteomics to compare the CHD4-interactome in BRCA2 deficient and BRCA2 proficient cells after cisplatin treatment (Figure 3A). Indeed, in addition to known CHD4-interactors (22), we also observed that CHD4 interacted with two proteins associated with chemoresistance in BRCA2 deficient cells: EZH2, which confers chemoresistance when inhibited, and FEN1, which confers chemoresistance when depleted, but is synthetic lethal when knocked out (Figure 3B) (21, 23–25). In BRCA2 deficient cells, we also found enrichment of the known CHD4-interacting protein ZFHX3 (26) and that ZFHX3 depletion enhanced cisplatin resistance in PEO1 cells (Figure 3C). Furthermore, analysis of TCGA patients revealed that low ZFHX3 mRNA levels predicted poor tumor-free survival in ovarian cancer patients with germline BRCA2 deficiency (Figure 3D) as previously found for CHD4, EZH2, and FEN1 (21, 23, 24). Strikingly, as found for CHD4-depletion, we observed that depletion of ZFHX3 or FEN1, or inhibition of EZH2, increased replication in BRCA2 deficient cells in the presence of HU, and as shown in the S1 nuclease assay, ssDNA gaps were suppressed (Figure 3E, S2F). Together, these findings suggest that loss of CHD4, EZH2, FEN1, and ZFHX3 suppress ssDNA gaps during stress to confer chemoresistance.
Figure 3: Suppression of ssDNA gaps accurately predicts poor therapy response in both cell culture and patient xenografts.
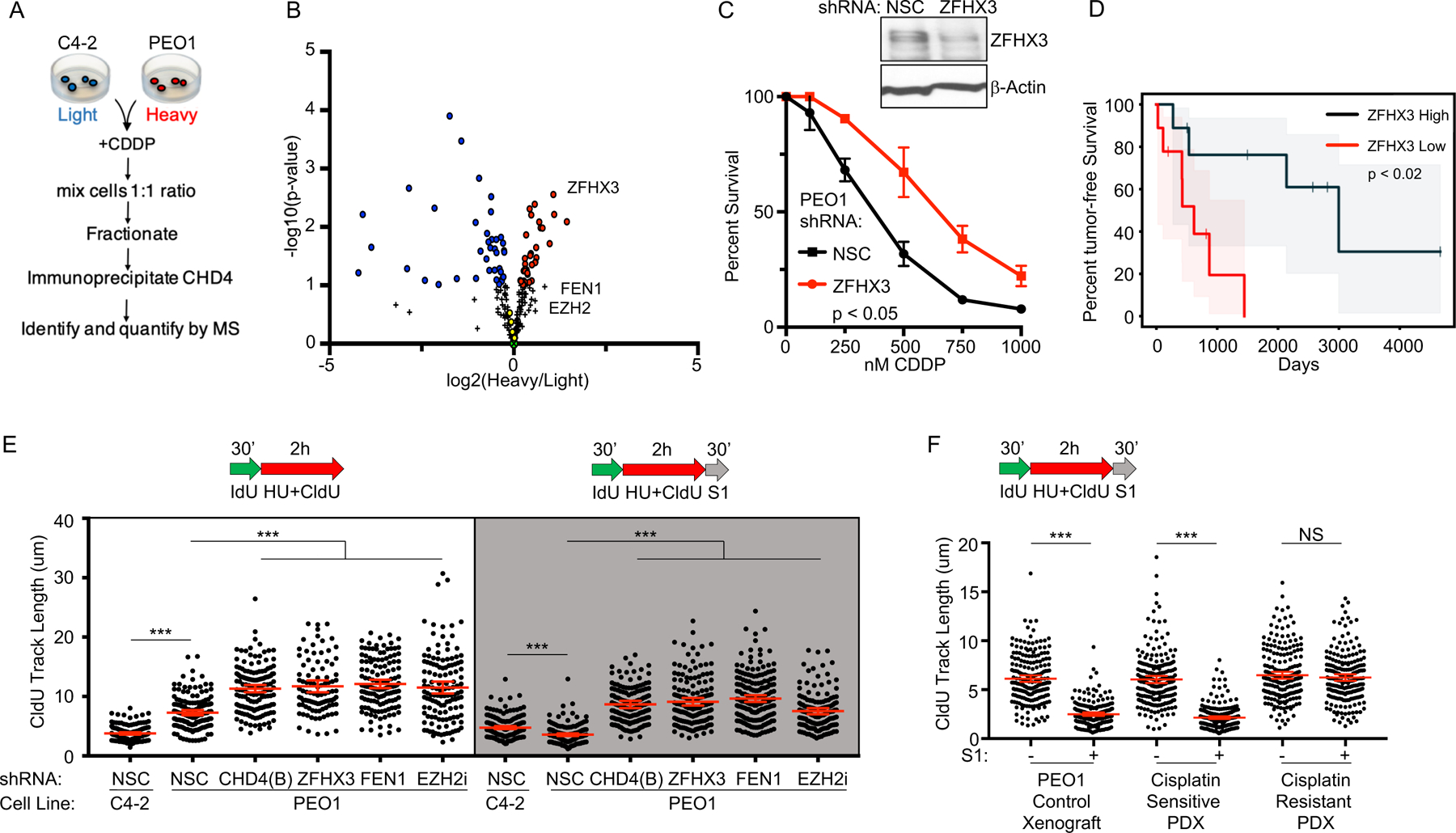
A) Overview of the SILAC CHD4 immunoprecipitation experiment. B) SILAC immunoprecipitation reveals that CHD4 interacts with ZFHX3, FEN1, and EZH2 after cisplatin treatment. Red and blue circles are proteins significantly enriched in the CHD4 network of either PEO1 or C4–2 cells. Green (X) represents CHD4. Yellow circles are known CHD4 interacting partners from the NurD complex, including MTA1, HDAC1, MTA2, and HDAC2 (22); ZFHX4 was also identified and is a known CHD4 interacting partner (26). Black plus signs represent proteins not significantly enriched in the CHD4 network of either PEO1 or C4–2. Three biological replicates were performed; see methods section for statistical analysis. C) Western blot confirms ZFHX3 is depleted by shRNA in PEO1 as compared to NSC. Cell survival assay confirms PEO1 with depleted ZFHX3 are resistant to cisplatin compared to PEO1 NSC. D) Reduced ZFHX3 mRNA levels predict poor patient response to therapy (progression free survival) for ovarian cancer patients with germline BRCA2 deficiency from the TCGA database (p < 0.02). Shaded area represents the 95% confidence interval. E) Schematic and quantification of CldU track length shows that depletion of CHD4 (shRNA(B)), ZFHX3 or FEN1, or inhibition of EZH2, increase replication in the presence of stress (white panel) and protect nascent DNA from S1 nuclease (gray panel). F) Schematic and quantification of CldU track length shows S1 fiber sensitivity is suppressed in BRCA1 deficient patient derived xenografts that have acquired chemoresistance. Each dot represents one fiber. Experiments were performed in biological triplicate with at least 100 fibers per replicate; the xenograft fiber assay was performed in duplicate. Statistical analysis according to two-tailed Mann-Whitney test; p < 0.001 (***). Mean and 95% confidence intervals are shown.
Next, we tested if ssDNA gaps could predict chemosensitivity and resistance in BRCA patient tumor samples. Specifically, we utilized a triple-negative breast cancer patient-derived xenograft (PDX), PNX0204, from a patient with a hemizygous germline BRCA1 mutation (1105insTC); the wild type BRCA1 allele was lost in the tumor, following a Loss of Heterozygosity model (Figure S2G). PNX0204 tumors were originally hypersensitive to cisplatin treatment. After several rounds of cisplatin treatment and serial passage in mice, resistant tumors developed. The sensitive and resistant tumors were then tested for S1 sensitivity, with PEO1 (Figure 3F) and MDA-MB-436 (Figure S2H) xenografts serving as controls. After HU, we observed that the DNA fibers of cisplatin-sensitive PDX cells were degraded by S1 nuclease, but the fibers of cisplatin-resistant PDX cells were not, indicating ssDNA gaps had been suppressed in the resistant patient samples (Figure 3F). Notably, in resistant PDX, ssDNA gaps were suppressed either by continuous replication (Figure 3F), or by restored fork slowing (Figure S2I), indicating that loss of ssDNA gaps had occurred in BRCA patient tumors de novo and accurately predicted acquired cisplatin resistance.
These findings present the idea that ssDNA gaps underlie chemosensitivity, and that loss of FP or HR do not. If so, when gaps are present, it should be possible to uncouple FP and HR from therapy response. To test this prediction, we first restored FP by inhibition of MRE11 or depletion of SMARCAL1 in BRCA2-deficient PEO1 cells (6, 27, 28). Nevertheless, even though FP was restored, cisplatin resistance was not conferred and, as predicted by our model, ssDNA gaps remained as demonstrated by S1 nuclease degradation (Figure 4A,B and S3A–F). Moreover, neither SMARCAL1, nor MRE11 or other reported FP factors, were predictive of BRCA2 cancer patient response based on mRNA levels in the TCGA database (Figure 4C and S3G), suggesting that ssDNA gaps, but not FP, determines therapy response.
Figure 4: ssDNA replication gaps, and not FP or HR, determine patient response to chemotherapy.
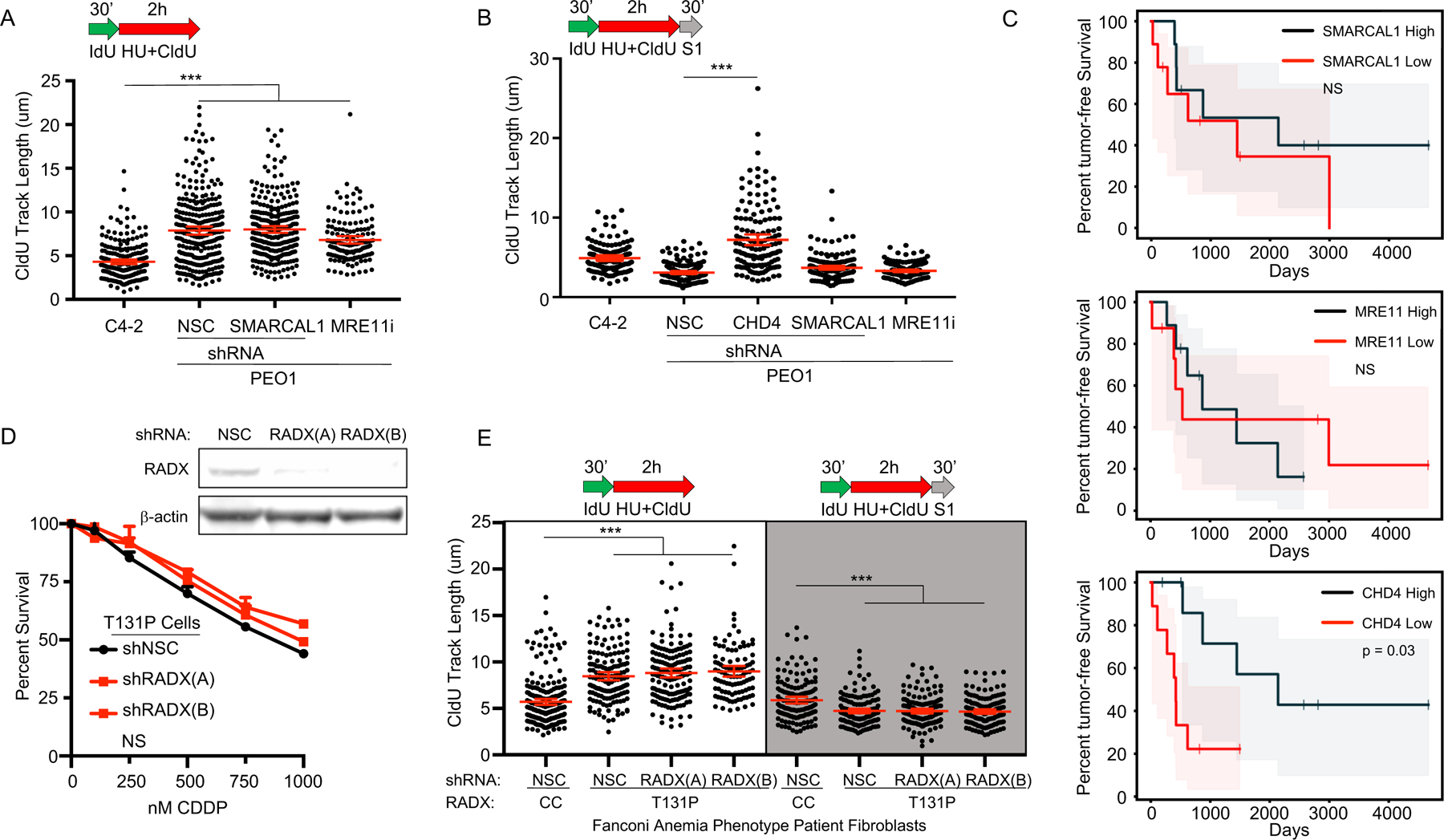
A) Schematic and quantification of CldU track length in PEO1 cells shows that depleted SMARCAL1 or inhibited MRE11 does not increase replication in the presence of stress, and B) does not protect from S1 nuclease, unlike CHD4 depletion. C) Neither SMARCAL1 nor MRE11 mRNA levels predict response of ovarian cancer patients with germline BRCA2 deficiency in TCGA dataset (p > 0.8 and p > 0.5, respectively). In contrast, CHD4 mRNA levels do predict response in these patients (p = 0.03). Shaded area represents the 95% confidence interval. D) Top, Western blot confirms RADX is depleted by two shRNA reagents in T131P cells compared to non-silencing-control (NSC). Bottom, cell survival assay confirms RAD51 T131P cells remain hypersensitive to cisplatin even when RADX is depleted. E) Schematic and quantification of CldU track length shows (white panel) that fibroblasts from a Fanconi Anemia-like patient with a mutant allele of RAD51 (T131P; HR proficient cells, cisplatin hypersensitive) fail to arrest replication in the presence of stress even when RADX is depleted, and these regions are degraded by S1 nuclease (light grey panel). WT FA cells are corrected by CRISPR to delete the dominant-negative T131P RAD51 allele. Each dot represents one fiber. Experiments were performed in biological triplicate with at least 100 fibers per replicate. Statistical analysis according to two-tailed Mann-Whitney test; p < 0.001 (***). Mean and 95% confidence intervals are shown.
Additionally, we tested if ssDNA gaps were distinct from fork degradation. Specifically, we analyzed gaps in VC-8 cells that express either wild-type BRCA2 or a BRCA2 mutant version (S3291A) that is deficient for FP yet resistant to chemotherapy (6). We did not detect ssDNA gaps in the S3291A cells, thereby confirming that fork degradation can occur without the accumulation of ssDNA gaps (Figure S3H,I) and that BRCA function in ssDNA gap suppression is distinct from FP.
We also considered the possibility that our ssDNA gap model could explain a discrepancy in the literature in which cells from a patient with Fanconi Anemia (FA) were sensitive to cisplatin and other genotoxic agents as expected, but were surprisingly found to be proficient in HR (12). Indeed, we found wide-spread ssDNA gap induction in the S1 assay in these FA patient cells; specifically, we observed S1 sensitivity in the FA patient fibroblasts that maintain a RAD51 mutant (T131P) allele as compared to isogenic RAD51 wild type fibroblasts (CRISPR corrected after isolation from the patient) (Figure S4A). Importantly, the T131P cells are deficient for FP, but FP can be restored by depletion of the RAD51 negative regulator RADX in T131P (29). However, despite both proficient HR and FP, even the T131P cells with depleted RADX remained cisplatin hypersensitive, and we observed ssDNA gaps remained by S1 assay; importantly, these gaps were eliminated in the wild type (CRISPR corrected) fibroblast control (Figure 4D,E, S4B). Together, these results suggest that the ssDNA gap model has superior predictive power compared to either the FP or HR models of therapy response and suggests that ssDNA replication gaps are fundamental to the mechanism-of-action of first-line genotoxic chemotherapies.
We next tested a surprising prediction of the ssDNA gap model, namely that DSBs are not fundamental to the mechanism-of-action of genotoxic chemotherapies, but rather a byproduct of the programmed cell death nucleolytic machinery (Figure 5A). To address this possibility, we first confirmed that genotoxic therapy induces programmed cell death via apoptosis. We treated BRCA2 deficient PEO1 with an approximate IC50 dose of cisplatin (0.5uM), and we measured apoptosis with Annexin V and cell death with propidium iodide (PI) in a flow cytometry time course experiment. We observed early apoptosis beginning 24h after treatment, with a minority of cells staining Annexin V positive and PI negative (Figure 5B and Figure S5A). By 120h after cisplatin treatment, we observed approximately fifty percent of cells were in late apoptosis with Annexin V and PI co-staining, as expected for the IC50 dose (Figure S5A). As controls, we confirmed that the BRCA2 proficient C4–2 cells displayed reduced PI and Annexin V signal at all time points following cisplatin as expected (Figure S5B). Moreover, in response to high dose camptothecin, a topoisomerase inhibitor that is reported to induce DSBs (30), we confirmed that PEO1 cells were hypersensitive as compared to C4–2 cells, and underwent apoptosis that was suppressed by the pan-caspase inhibitor Z-VAD-FMK (Figure 5B, S5C–F) (31). In addition, as a control, we confirmed that treatment with Z-VAD-FMK did not alter cell cycle progression (Figure S5G). Taken together, these results indicate that BRCA2 deficient cells undergo programmed cell death via apoptosis after genotoxic treatment.
Figure 5: DNA Double Strand Breaks are not Detected when Apoptosis is Inhibited.
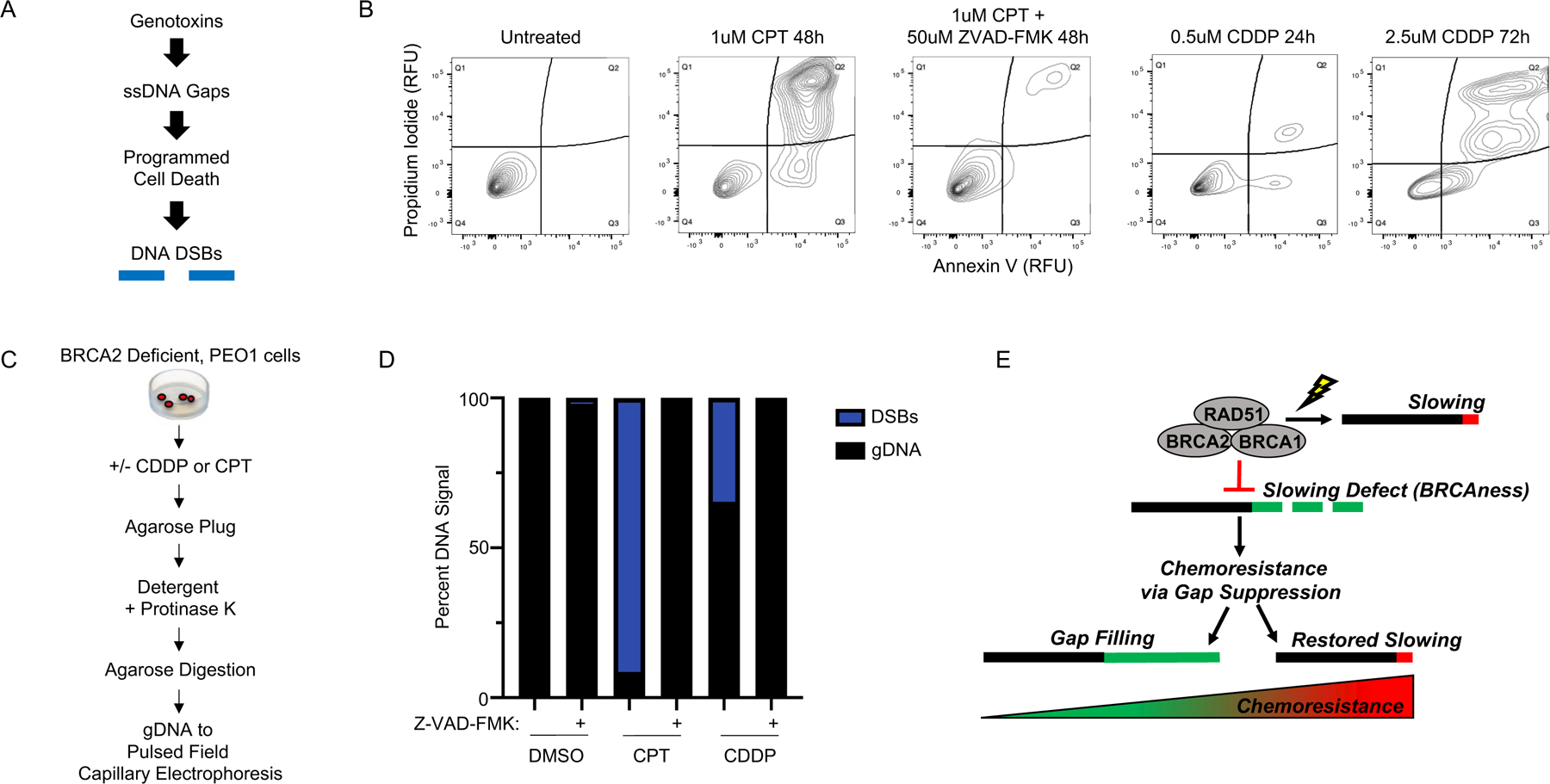
A) Overview of model: therapy induces ssDNA gaps that trigger programmed cell death, and the nucleolytic machinery creates DNA DSBs. B) Left, flow cytometry with propidium iodide and annexin V shows that apoptosis is eliminated by 50uM Z-VAD-FMK in BRCA2 deficient PEO1 cells treated with 1uM CPT for 48h. Right, flow cytometry detects apoptosis in BRCA2 deficient PEO1 cells treated with 0.5uM cisplatin for 24h (see Figure S5A for matched untreated control) or 2.5uM cisplatin for 72h (see Figure S5F for matched untreated control). C) Overview of isolation procedure that maintains high molecular weight (megabase-scale) genomic DNA for pulsed field capillary electrophoresis (PFCE). D) PFCE of PEO1 genomic DNA reveals 50uM Z-VAD-FMK eliminates all detectable DNA DSBs for both 1uM CPT 48h and 2.5uM CDDP 24h. E) Model of BRCAness and chemoresponse. During stress, BRCA-deficient cells fail to effectively restrain replication, leading to ssDNA gaps that determine chemosensitivity: BRCAness. These cells acquire chemoresistance by eliminating the ssDNA gaps, either by gap filling, or by restoring fork slowing.
Finally, we tested if we could detect DSBs following cisplatin or camptothecin. Following approximately the IC90 dose of camptothecin or cisplatin, we isolated intact genomic DNA (gDNA) in agarose plugs, which were subsequently analyzed by pulsed field capillary electrophoresis (Figure 5C). As expected, we observed extensive DNA fragmentation by DSBs in PEO1 cells following 48h treatment with 1uM camptothecin, and to a lesser extent with 24h 2.5uM cisplatin, as indicated by the reduced DNA capillary retention time after treatment that corresponds to sub-megabase sized DNA standards (Figure 5D). In contrast, when apoptosis was inhibited with Z-VAD-FMK, we were unable to detect DSBs after either agent, with the capillary retention time corresponding to megabase sized gDNA and indistinguishable from the retention time observed in the untreated controls (Figure 5D). Moreover, we found that a second pan-caspase inhibitor, Emricasan, similarly eliminated apoptosis by flow cytometry as well as all detectable DSBs after genotoxin treatment (Figure S5H). Taken together, these results support a framework where genotoxic agents create ssDNA gaps, which induce programmed cell death signaling via cleaved caspases to activate the DNA nucleolytic machinery, which ultimately creates DSBs.
DISCUSSION
Although ssDNA gaps are a common indicator of genotoxicity and result from loss of the BRCA-RAD51 pathway, they have been overlooked as the determinant of toxicity in favor of defects in HR and FP (6, 12, 28, 30, 32–38). However, there are several genetic systems in which the DSB model does not appear to accurately predict therapy response, and therefore presents an opportunity to revise the underlying framework. Indeed, in light of our findings in different genetic backgrounds, including both BRCA1 and BRCA2 deficient cancers (Figure S6), we propose that replication gaps underlie the mechanism-of-action of genotoxic chemotherapies, and it is the failure to suppress gaps, and not defects in HR or FP, that underlies the hypersensitivity of BRCA-deficient cancer to treatment. In support of this concept, when gaps persist, we demonstrate that HR or FP proficient cells can nevertheless be hypersensitive to genotoxins. Moreover, when gaps are suppressed by loss of CHD4, FEN1, EZH2, or ZFHX3, BRCA2 mutant cells are resistant to genotoxins without restoring HR (21, 23, 24). Similarly, without HR, FP is proposed to mediate cisplatin resistance (18), however we find restored FP in BRCA2 deficient cells achieved by MRE11 inhibition or SMARCAL1 depletion does not enhance cisplatin resistance. We also find that other fork protection factors fail to accurately predict therapy response in the TCGA.
In addition, the emerging evidence indicates that gaps are distinct lesions arising from replication defects, are suppressed by the BRCA-RAD51 pathway, and are located behind the fork at sites distinct from stalled or broken replication forks (28, 37, 39–42). When replication fails to be fully restrained due to loss of the BRCA-RAD51 pathway, we predict that replication gaps derive from replication dysfunction rather than overactive nuclease activity (28, 43). While nucleases could extend nicks or gaps, we found S1 nuclease digestion was unaffected by MRE11 inhibition or depletion of the fork remodeler SMARCAL1, which generates the replication fork structure degraded by MRE11 in BRCA2 deficient cells (27, 28, 44). Thus, gaps likely form in newly replicated DNA prior to remodeling or degradation of replication forks. We find that gaps are suppressed by at least two mechanisms: gap filling when replication proceeds during exposure to genotoxins, or by restored fork restraint as achieved by BRCA reversion mutation that provides a more robust gap suppression and in turn greater chemoresistance (Figure 5E).
Importantly, our findings do not exclude the possibility that ssDNA gaps are in fact converted, albeit at undetectable levels, into DSBs that drive hypersensitivity. However, it is unclear how low levels of DSBs would lead to hypersensitivity, especially considering that BRCA-deficient cells employ backup DSB repair mechanisms, such as end joining pathways. Although the resulting genomic instability introduced by end joining pathways could conceivably trigger hypersensitivity in BRCA cancer, this model does not appear to fit the observed data. Specifically, the FP deficient VC-8 cells with the BRCA2 S3291A mutant display substantial genomic instability, yet simultaneously display cisplatin resistance that is indistinguishable from the WT BRCA2 control (6) (Figure S3H, I). Similarly, if ssDNA gaps are ultimately converted into DSBs, then cells proficient for HR would be expected to successfully repair these DSBs and therefore be resistant; however, the Fanconi Anemia RAD51 T131P cells are HR proficient, yet are nevertheless hypersensitive to chemotherapy (12) even when FP is restored (Figure 4D, E, S4). Indeed, these hypersensitive T131P cells also conflict more generally with models where DSBs are proposed to be the sensitizing lesion, even if the DSBs are assumed to be generated at levels that are undetectable by PFCE/PFGE; why would DSBs cause hypersensitivity in cells that efficiently repair DSBs with HR? In addition, hypersensitivity with proficient HR has also been observed in other genetic systems (45), suggesting this is not an aberrant observation, and further reduces confidence in DSB models of BRCAness.
Instead, as we report here and as previously shown (46, 47) genotoxin-induced DSBs appear to be created by the programmed cell death process rather than by the genotoxins themselves. Indeed, the observed DSBs from cisplatin and other genotoxic agents result in initial DNA fragments approximately 500–100kb in size (48), which match the early DNA fragments generated by the ordered nucleolytic degradation process carried out by the programmed cell death machinery (49). Accordingly, we also considered that programmed cell death could be the source of the DSBs that cause hypersensitivity; however, we found this model also did not appear to agree with experiment for reasons identical to those described above. In particular, cell death induced DSBs would not be expected to confer hypersensitivity in the HR proficient T131P cells because the DSBs would be effectively resolved by HR repair.
Similarly, we also considered that BRCA deficient cells could instead be uniquely “primed” for programmed cell death, leading to increased cell death nuclease activity that creates higher levels of DSBs to overwhelm even intact HR machinery. However, this model is inconsistent with reports that disruption of programmed cell death nucleases eliminates observable DSBs, but does not eliminate programmed cell death or hypersensitivity (50). This observation also indicates that ssDNA gaps likely can induce cell death by a variety of different mechanisms within the programmed cell death repertoire. Therefore, we propose DSBs are generated either as an unrelated byproduct or as a minority lesion that does not substantially contribute to hypersensitivity, whereas ssDNA gap induced cell killing is the basis for the toxicity of genotoxic agents and BRCAness.
Lastly, we also propose that it will be critical to design experiments to further test both models. Specifically, it will be important to determine if there are latent and unappreciated DSB repair defects in HR proficient cells that are hypersensitive to genotoxins. Likewise, it will be important to determine if persistent ssDNA gaps that occur during active replication under genotoxins can be identified in resistant cells, or if such gaps are found to be absent in hypersensitive cells. Furthermore, it will be important to assess if the cellular introduction of ssDNA or DSB substrates differentially induce programmed cell death as previously described (51, 52); exploring this concept further by gene editing techniques will overcome the limitations of cell transfection and help elucidate the link between ssDNA gaps, DSBs, and genomic instability. It will also be critical to identify gap filling mechanisms that can be targeted to restore hypersensitivity; one possible target is translesion synthesis (TLS). Indeed, CHD4 depletion elevates TLS that suppresses replication gaps (21, 39, 53). Not surprisingly, TLS confers chemotherapy resistance, is a cancer adaptation, and is actively being targeted for cancer therapy (53, 54). Moreover, we find that replication gaps due to BRCA deficiency is the basis for synthetic lethality to PARP inhibitors (55). Understanding how gap suppression functions align with other BRCA roles in genome preservation, cell viability, and tumor suppression will also be critical future questions.
In summary, this study supports a new model that predicts cancer cells with the BRCAness phenotype will be effectively treated by therapies that exacerbate replication gaps. Similarly, preventing gap suppression pathways will improve the effectiveness of therapy as well potentially re-sensitize chemoresistant disease to therapy. Based on our findings, we also propose that ssDNA gaps could serve as biomarkers for BRCAness, and that gap induction is fundamental to the mechanism-of-action of chemotherapies that dysregulate replication.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
This study suggests that ssDNA replication gaps are fundamental to the toxicity of genotoxic agents and underlie the BRCA-cancer phenotype “BRCAness,” yielding promising biomarkers, targets, and opportunities to re-sensitize refractory disease.
ACKNOWLEDGEMENTS
We thank the members of the Cantor laboratory for helpful discussions. We thank Dr. Agata Smogorzewska for providing the FA patient cells, Toshi Taniguchi for the PEO1 and C4–2 cells, Maria Jasin for the VC-8 cells, and Lee Zhou for the HCC1937 cells. This work was supported by R01 grants CA247232, CA225018, and CA254037 (Cantor) and R01 CA214799 and OC130212 (Johnson) as well as charitable contributions from Mr. and Mrs. Edward T. Vitone, Jr. and the Lipp Family Foundation. The Vermont Genetics Network Proteomics Facility is supported through NIH grant P20GM103449 from the INBRE Program of the National Institute of General Medical Science. We thank Drs. Igor Astutarov and Vladimir Khazak for establishing and sharing PNX0204.
Footnotes
CONFLICT OF INTERESTS
The authors declare no conflict of interests.
REFERENCES:
- 1.King MC et al. , Genetic analysis of human breast cancer: literature review and description of family data in workshop. Genet Epidemiol Suppl 1, 3–13 (1986). [PubMed] [Google Scholar]
- 2.Munnell EW, The changing prognosis and treatment in cancer of the ovary. A report of 235 patients with primary ovarian carcinoma 1952–1961. Am J Obstet Gynecol 100, 790–805 (1968). [DOI] [PubMed] [Google Scholar]
- 3.Li X, Heyer WD, Homologous recombination in DNA repair and DNA damage tolerance. Cell Res 18, 99–113 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng W, Jasin M, Homologous Recombination and Replication Fork Protection: BRCA2 and More! Cold Spring Harb Symp Quant Biol 82, 329–338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lomonosov M, Anand S, Sangrithi M, Davies R, Venkitaraman AR, Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev 17, 3017–3022 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schlacher K et al. , Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schlacher K, Wu H, Jasin M, A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer cell 22, 106–116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang J et al. , The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Molecular cell 52, 434–446 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mutreja K et al. , ATR-Mediated Global Fork Slowing and Reversal Assist Fork Traverse and Prevent Chromosomal Breakage at DNA Interstrand Cross-Links. Cell Rep 24, 2629–2642 e2625 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cantor SB, Calvo JA, Fork Protection and Therapy Resistance in Hereditary Breast Cancer. Cold Spring Harb Symp Quant Biol, (2018). [DOI] [PMC free article] [PubMed]
- 11.Bunting SF et al. , BRCA1 Functions Independently of Homologous Recombination in DNA Interstrand Crosslink Repair. Molecular cell 46, 125–135 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang AT et al. , A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol Cell 59, 478–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barazas M et al. , Radiosensitivity Is an Acquired Vulnerability of PARPi-Resistant BRCA1-Deficient Tumors. Cancer Res 79, 452–460 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bruno PM et al. , A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat Med 23, 461–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heijink AM et al. , Modeling of Cisplatin-Induced Signaling Dynamics in Triple-Negative Breast Cancer Cells Reveals Mediators of Sensitivity. Cell Rep 28, 2345–2357 e2345 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schindelin J et al. , Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koc A, Wheeler LJ, Mathews CK, Merrill GF, Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J Biol Chem 279, 223–230 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Chaudhuri AR et al. , Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakai W et al. , Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res 69, 6381–6386 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quinet A, Carvajal-Maldonado D, Lemacon D, Vindigni A, DNA Fiber Analysis: Mind the Gap! Methods Enzymol 591, 55–82 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Guillemette S et al. , Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev 29, 489–494 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Shaughnessy A, Hendrich B, CHD4 in the DNA-damage response and cell cycle progression: not so NuRDy now. Biochemical Society transactions 41, 777–782 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rondinelli B et al. , EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol 19, 1371–1378 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Meghani K et al. , Multifaceted Impact of MicroRNA 493–5p on Genome-Stabilizing Pathways Induces Platinum and PARP Inhibitor Resistance in BRCA2-Mutated Carcinomas. Cell Rep 23, 100–111 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mengwasser KE et al. , Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol Cell 73, 885–899 e886 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chudnovsky Y et al. , ZFHX4 interacts with the NuRD core member CHD4 and regulates the glioblastoma tumor-initiating cell state. Cell Rep 6, 313–324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taglialatela A et al. , Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol Cell 68, 414–430 e418 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolinjivadi AM et al. , Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell 67, 867–881 e867 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhat KP et al. , RADX Modulates RAD51 Activity to Control Replication Fork Protection. Cell Rep 24, 538–545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zellweger R et al. , Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208, 563–579 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Noorden CJ, The history of Z-VAD-FMK, a tool for understanding the significance of caspase inhibition. Acta Histochem 103, 241–251 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Henry-Mowatt J et al. , XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Mol Cell 11, 1109–1117 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Sugimura K, Takebayashi S, Taguchi H, Takeda S, Okumura K, PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J Cell Biol 183, 1203–1212 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su X, Bernal JA, Venkitaraman AR, Cell-cycle coordination between DNA replication and recombination revealed by a vertebrate N-end rule degron-Rad51. Nat Struct Mol Biol 15, 1049–1058 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V, Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17, 1305–1311 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luke-Glaser S, Luke B, Grossi S, Constantinou A, FANCM regulates DNA chain elongation and is stabilized by S-phase checkpoint signalling. EMBO J 29, 795–805 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vallerga MB, Mansilla SF, Federico MB, Bertolin AP, Gottifredi V, Rad51 recombinase prevents Mre11 nuclease-dependent degradation and excessive PrimPol-mediated elongation of nascent DNA after UV irradiation. Proc Natl Acad Sci U S A 112, E6624–6633 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kolinjivadi AM et al. , Moonlighting at replication forks - a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett 591, 1083–1100 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Wong RP, Garcia-Rodriguez N, Zilio N, Hanulova M, Ulrich HD, Processing of DNA Polymerase-Blocking Lesions during Genome Replication Is Spatially and Temporally Segregated from Replication Forks. Mol Cell, (2019). [DOI] [PubMed]
- 40.Elvers I, Johansson F, Groth P, Erixon K, Helleday T, UV stalled replication forks restart by re-priming in human fibroblasts. Nucleic Acids Res 39, 7049–7057 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mouron S et al. , Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 20, 1383–1389 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Bianchi J et al. , PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol Cell 52, 566–573 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu X et al. , BRCA1 represses DNA replication initiation through antagonizing estrogen signaling and maintains genome stability in parallel with WEE1-MCM2 signaling during pregnancy. Hum Mol Genet, (2018). [DOI] [PMC free article] [PubMed]
- 44.Mijic S et al. , Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nature communications 8, 859 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zimmermann M et al. , CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559, 285–289 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Faivre S, Chan D, Salinas R, Woynarowska B, Woynarowski JM, DNA strand breaks and apoptosis induced by oxaliplatin in cancer cells. Biochem Pharmacol 66, 225–237 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Sane AT, Bertrand R, Distinct steps in DNA fragmentation pathway during camptothecin-induced apoptosis involved caspase-, benzyloxycarbonyl- and N-tosyl-L-phenylalanylchloromethyl ketone-sensitive activities. Cancer Res 58, 3066–3072 (1998). [PubMed] [Google Scholar]
- 48.Nowosielska A, Marinus MG, Cisplatin induces DNA double-strand break formation in Escherichia coli dam mutants. DNA repair 4, 773–781 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Kawane K, Motani K, Nagata S, DNA degradation and its defects. Cold Spring Harb Perspect Biol 6, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sakahira H, Enari M, Nagata S, Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 391, 96–99 (1998). [DOI] [PubMed] [Google Scholar]
- 51.Huang LC, Clarkin KC, Wahl GM, Sensitivity and selectivity of the DNA damage sensor responsible for activating p53-dependent G1 arrest. Proc Natl Acad Sci U S A 93, 4827–4832 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tidd DM et al. , Oligodeoxynucleotide 5mers containing a 5’-CpG induce apoptosis through a mitochondrial mechanism in T lymphocytic leukaemia cells. Nucleic Acids Res 28, 2242–2250 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nayak S et al. , Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci Adv 6, eaaz7808 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tonzi P, Huang TT, Role of Y-family translesion DNA polymerases in replication stress: Implications for new cancer therapeutic targets. DNA Repair (Amst) 78, 20–26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cong K et al. , PARPi synthetic lethality derives from replication-associated single-stranded DNA gaps. bioRxiv, 781989 (2019).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.