

Abstract
Ubiquitin specific protease18 (USP18), previously known as UBP43, is the Interferon-Stimulated Gene 15 (ISG15) deconjugase. USP18 removes ISG15 from substrate proteins. This study reports that USP18 null mice (versus wild-type mice) exhibited lower lipolysis rates, altered fat to body weight ratios and cold sensitivity. USP18 is a regulator of lipid and fatty acid metabolism. Prior work established that USP18 promotes lung tumorigenesis. We sought to learn if this occurs through altered lipid and fatty acid metabolism. Loss of USP18 repressed adipose triglyceride lipase (ATGL) expression; gain of USP18 expression upregulated ATGL in lung cancer cells. The E1-like ubiquitin activating enzyme (UBE1L) promoted ISG15-conjugation of ATGL and destabilization. Immunoprecipitation assays confirmed that ISG15 covalently conjugates to ATGL. Protein expression of thermogenic regulators was examined in brown fat of USP18 null versus wild-type mice. Uncoupling Protein 1 (UCP1) was repressed in USP18 null fat. Gain of USP18 expression augmented UCP1 protein via reduced ubiquitination. Gain of UCP1 expression in lung cancer cell lines enhanced cellular proliferation. UCP1 knock-down inhibited proliferation. Beta-hydroxybutyrate colorimetric assays performed after gain of UCP1 expression revealed increased cellular fatty acid beta-oxidation, augmenting fatty acid beta-oxidation in seahorse assays. Combined USP18, ATGL and UCP1 profiles were interrogated in The Cancer Genome Atlas (TCGA). Intriguingly, lung cancers with increased USP18, ATGL and UCP1 expression had an unfavorable survival. These findings reveal that USP18 is a pharmacologic target that controls fatty acid metabolism.
Keywords: USP18, ATGL, UCP1, lipolysis, fatty acid oxidation and tumorigenesis
Introduction
Metabolic reprogramming is a hallmark of cancer (1–3). Cancer cells have an expanded metabolic capability that can augment tumor survival (4). Warburg and colleagues discovered that cancers favor glycolysis over oxidative phosphorylation (5). These altered metabolic changes contribute to carcinogenesis and affect glucose uptake, glycolysis utilization, glutamine consumption and lipid metabolism. Prior work found that increased lipolysis (6), fatty acid synthesis (7, 8) and fatty acid oxidation (9, 10) can each promote tumorigenesis. Altered lipid metabolism in cancer cells engages fatty acid synthesis, lipolysis, and fatty acid oxidation; these are candidate anti-neoplastic targets (11,12).
Ubiquitin specific peptidase 18 (USP18), previously designated as UBP43, is the Interferon-Stimulated Gene 15 (ISG15) deconjugase that removes this species from substrate proteins. Other components of this pathway include the E1 activating enzyme UBE1L, the E2 conjugating enzyme (UBCH8), and the E3 ligase that together enable ISG15 to conjugate to substrate protein and confer protein destabilization, among other effects (13–15). USP18 is a negative regulator of interferon (IFN) signaling, independent of its ISG15 deconjugase activity (16). We previously reported that the oncogenic proteins PML/RARα, cyclin D1 and Kras are substrates for ISG15 conjugation (17–20). Increased USP18 enzymatic activity can augment lung tumorigenicity (18). Lung tumor numbers in KrasLA2/+/Usp18−/−, are statistically-significantly lower than in KrasLA2/+/Usp18+/+, and KrasLA2/+/Usp18+/− mice (20), implicating USP18 as an antineoplastic target.
This study reports the previously unrecognized finding that increased USP18 activity promotes lipolysis and fatty acid oxidation by stabilizing both adipose triglyceride lipase (ATGL) and Uncoupling Protein 1 (UCP1). Notably, USP18 null mice exhibited in vivo metabolic changes, including altered lipolysis. It was hypothesized that regulators of lipolysis are repressed by loss of UPS18. Consistent with this, expression of ATGL, the enzyme that converts triglycerides to diglycerides, was reduced by loss of USP18 expression. Likewise, immunoprecipitation assays confirmed that ATGL undergoes ISG15-mediated destabilization that is opposed by gain of USP18 expression. Excess ATGL activity augmented cancer cell growth and inhibition of ATGL suppressed this (21). It is reported here that USP18 null mice are exquisitely sensitive to cold exposure. Also, the master thermoregulator, UCP1, was repressed by loss of USP18 expression. In contrast, gain of USP18 expression promoted fatty acid oxidation through increased UCP1 protein expression via reduction of total ubiquitination. This enhanced lung cancer cell growth.
Bioinformatics analysis using The Cancer Genome Atlas (TCGA) revealed that a signature of combined high levels of USP18, ATGL and UCP1 expression conferred an unfavorable survival in lung adenocarcinoma and other human cancers. Taken together, these and other findings that are presented provide evidence for the previously unrecognized findings that USP18 regulates lipid metabolism by affecting ATGL and UCP1 expression. It is hypothesized that the ubiquitin specific peptidase USP18 is an antineoplastic target that influences carcinogenesis through altered lipid metabolism.
Materials and Methods
Generation of USP18 −/− Mice
FVB-Usp18+/− mice were purchased from Jackson Laboratory. Mice were bred to generate FVB-Usp18 knockout mice, as reported (22–24).
Body Composition Analysis
Body composition data for 12-week-old female and male wild-type and USP18 null mice were acquired by whole body metabolic profiling using a Bruker minispec Body Composition Analyzer (Brunker Corp.) performed through the Mouse Metabolic and Phenotyping Core, Baylor College of Medicine (Houston, TX). A total of 10 mice were examined in two replicate experiments conducted with 5 mice each.
Plasma Parameter Analysis
Plasma glucose (Wako), total triglyceride (Thermo Fisher Scientific), total cholesterol (Wako), FFAs (Wako), adiponectin (Invitrogen), leptin (Invitrogen), insulin (Invitrogen), and 3-hydroxybutyrate (Stanbio Laboratory) were measured using enzyme colorimetric assays according to the manufacturer’s methods at the Mouse Metabolic and Phenotyping Core, Baylor College of Medicine (Houston, TX).
In Vivo Lipolysis Assays
For in vivo lipolysis assays, 12-week-old female USP18 null and wild-type mice were injected with 0.1 mg/kg CL316243 (Tocris Bioscience), a 3-adrenergic receptor agonist or saline as a vehicle control. Plasma was collected at 1 hour post-injection. As a measure of lipolysis, free fatty acids (FFAs) (Wako) and glycerol (ZenBio) levels were quantified using enzymatic colorimetric assays according to the manufacturer’s methods at the Mouse Metabolic and Phenotyping Core of Baylor College of Medicine (Houston, TX).
Cold Exposure
Mice housed individually in a cage were transiently placed at 4° C prior to testing. Body temperatures were recorded rectally at the indicated time points. All aspects of this experiment were reviewed and approved by Dartmouth Medical School Institutional Animal Care and Use Committee and also by The University of Texas MD Anderson Cancer Center Institutional Animal Care and Use Committee.
Lipid Metabolite Assay
The lipid metabolite assay was conducted using the AbsoluteIDQ® p400 HR Kit (BIOCRATES Life Sciences AG, Austria). Logarithmically-growing lung cancer cells were harvested by treating them with 0.05% trypsin and resuspending in 100 μl phosphate buffered saline (PBS). Cell lysates were processed by a manual protocol. Samples were analyzed by the Q-Exactive LC/MS system (Thermo Fisher Scientific Inc, Waltham, MA) and data were processed using MetIDQ software (BIOCRATES Life Sciences AG, Austria).
Cell Culture
Murine (ED1, 344SQ and 393P) and human (A549, HOP62 and H226) lung cancer cell lines as well as 393T cells were each cultured in RPMI-1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Thermo Scientific). These cells were cultured at 37°C in a humidified incubator with 5% CO2. Cell lines were obtained from American Type Culture Collection (ATCC) except for the murine ED1 lung cancer cell line that was previously described (25) as were the murine 344P and 393P lung cancer cell lines (26) and murine USP18-null leiomyosarcoma KHC1 and KHC2 cell lines (24). All cell lines were authenticated as reported (24–26) and were each excluded as having mycoplasma contamination using the MycoAlert PLUS Mycoplasma Detection Kit (Lonza).
Proliferation Assays
Logarithmically-growing cells were seeded at optimized densities for each examined lung cancer cell line onto individual wells of 12, 24 or 48 well tissue culture plates (for high-throughput testing) and assays were performed in triplicates. Proliferation was measured using the CellTiter-Glo Luminescent Assay (Promega) after 48–72 hours of transfection. Each assay was performed at least in triplicate independent replicate experiments.
Expression Plasmids and Transfections
Expression plasmids pCMV-HA-ISG15, pSG5-UBE1L, pCMV2-UBCH8, pcDNA4-USP18, were already described (18, 20, 27). Plasmids pReceiver-M12-Ubiquitin and pReceiver-M43-UCP1 were purchased from GeneCopoeia (Rockville, MD). Logarithmically-growing ED-1, LKR13, Hop62, and H522 lung cancer cell lines were each transiently transfected using jetPRIME (Polyplus-transfection, NY).
Reverse Phase Protein Arrays (RPPAs)
RPPAs were performed in the A549 human lung cancer cell line that was engineered by small hairpin (sh) RNA versus control shRNA to confer reduced USP18 expression. RPPAs were done using previously described methods (28).
SiRNA Transfections
RISC-free control siRNA and two siRNAs independently-targeting human or murine USP18 were each purchased (GE Dharmacon), as follows:
human USP18 siRNA1 (5’-CTGCATATCTTCTGGTTTA-3’), human USP18 siRNA2
(5’-GGAAGAAGACAGCAACATG-3’), murine USP18 siRNA1
(5’-CGTTGTTTGTCCAGCACGA-3’), murine USP18 siRNA2
(5’-AGGAACTCGAGGACGGAAA-3’), Human UCP1 siRNA1
(5’-GTACAGAGCTAGTAACATA-3’), human UCP1 siRNA2
(5’-CGTCCAGTGTTATTAGGTA-3’), murine UCP1 siRNA1
(5’-GGACTTATAATGCGTACAG-3’), and murine UCP1 siRNA2
(5’-GACCACGGCTTTCTTCAA-3’). Indicated lung cancer cells were independently transfected with siRNAs using jetPRIME (Polyplus-transfection, NY). The siRNA-mediated silencing of the desired species was confirmed by immunoblot analyses.
Immunoprecipitation, Immunoblot and Antibodies
Cell lysates were placed in RIPA buffer with the Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). Lysates were size-fractionated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. Membranes were independently probed with indicated antibodies and visualized using Clarity Western ECL 10 Substrate (Bio-Rad). Primary antibodies were: ATGL (#2138; Cell Signaling Technology), USP18 (#4813; Cell Signaling Technology), β-actin (#3700; Cell Signaling Technology), HA tag (#3724S, Cell Signaling Technology), UBE1L (#61266, Cell Signaling Technology), Tubulin, UCP1 (#PA1–24894, ThermoFisher Scientific), Myc-tag (#2276S, Cell Signaling Technology), and ISG15 (#2743, Cell Signaling Technology). Secondary antibodies were Goat anti-rabbit IgG (#170–6515; Bio-Rad) or Goat anti-mouse IgG (#170–6516; Bio-Rad).
Statistical Analyses
Differences between analyzed groups were assessed by a Student’s t or Mann-Whitney U test. To control for the overall type I error rate in addressing multiple comparisons, Tukey’s method was used for all pairwise comparisons across cell lines or experimental conditions. Dunnett’s method was applied for comparing the result of different concentrations with the control group. Statistical analyses were with SPSS Statistics software (version 23, SPSS) and GraphPad Prism software (version 8, GraphPad Software). All statistical tests were two-sided and a P value of < 0.05 was considered statistically significant. Other than in in vivo metabolic studies in mice that were conducted in duplicate replicate experiments, all other studies were done in triplicate and in at least three independent replicates.
TCGA Analysis
Kaplan-Meier survivals were assessed by the log-rank test using the ‘survival’ package in the R software. Kaplan-Meier survivals were assessed by the log-rank test using the ‘survival’ package in the R software. Gene expression profiles from RNA-sequencing were downloaded from the TCGA project, and were scored as TPM (transcripts per million) values to compare expression levels. In the survival analyses, high (above median) expression versus low (below median) levels were used for comparisons. When multiple species were evaluated, the ‘high’ group represented the expression profiles of examined species with higher than their median expression levels.
Fatty Acid Oxidation Assays
Logarithmically-growing lung cancer cells were seeded at optimized densities for each examined cell line onto individual 24 or 48 well tissue culture plates and in triplicate. Fatty acid oxidation was measured by β-Hydroxybutyrate (Ketone Body) Colorimetric Assay Kit (Cayman Chemical, MI) using the manufacturer’s methods.
Seahorse Assays
Seahorse assays were conducted using the Seahorse XFe96 Analyzer and the vendor’s procedures. Logarithmically-growing lung cancer cells were plated onto XF96 cell culture microplates (#101085–004, Agilent Technologies) with XFe96 Extracellular Flux Assay Kits (#102416–100, Agilent Technologies) using XF Cell Mito Stress Test Kit (#103015–100, Agilent Technologies). Measurements of fatty acid oxidation was described by the manufacturer: https://www.agilent.com/cs/library/usermanuals/public/XF_Cell_Mito_Stress_Test_Kit_User_Guide.pdf
Results
Loss of USP18 Expression Alters Lipid Metabolism
USP18 is a deubiquitinase (DUB) that stabilizes target proteins, including oncoproteins and other growth-regulatory species as we reported (17–20, 29). Our prior work found that USP18 null mice spontaneously developed leiomyosarcomas in a mouse strain-dependent manner (24). This was observed in the FVB strain, but not in other examined genetic backgrounds (24, 30).
This study comprehensively evaluated lipid metabolism in female USP18 null mice within the murine FVB background. These USP18 null mice exhibited higher fat weight and fat ratios to body weights when compared to wild-type mouse litter mates (n = 10, P < 0.05) (Fig. 1A). In USP18 null mice in vivo lipolysis rates were measured. Findings revealed that these mice exhibited a substantially lower lipolysis rate as compared to USP18 wild-type mice (n = 10, P < 0.05) (Fig. 1B). Other lipid metabolism parameters were measured. It was determined that lower serum adiponectin and HDL levels were detected in USP18 null mice (Fig. 1C). Oxygen consumption and CO2 production rates measured in USP18 null mice did not significantly change between these USP18 null versus wild-type mice (data not shown). To confirm independently that engineered loss of USP18 expression affected metabolic regulators, RPPAs were done and results are displayed in Supplementary Fig. 1.
Figure 1:
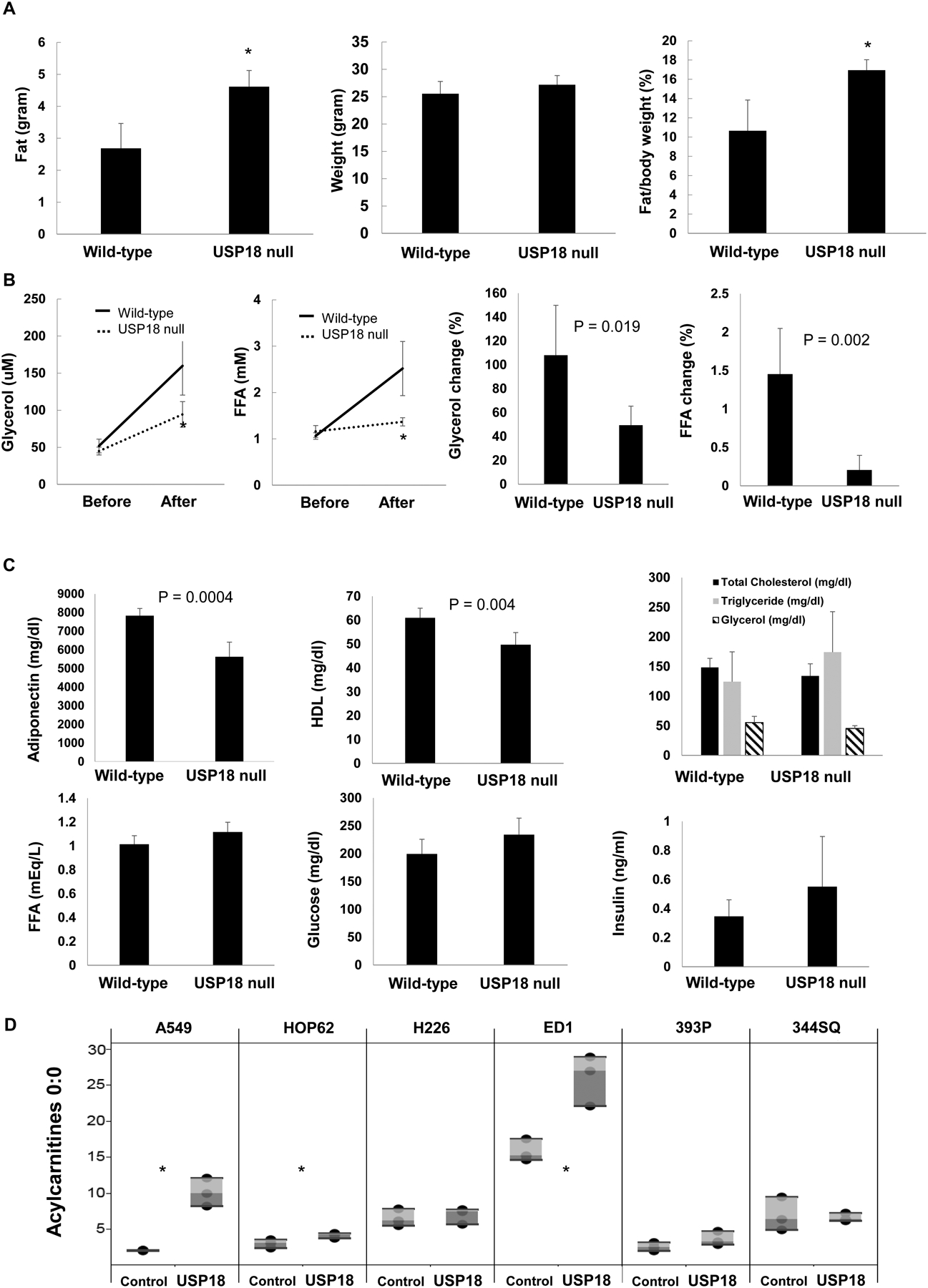
USP18 null mice exhibit reduced lipolysis. (A) USP18 null mice have higher ratios of fat to body weights as assessed by whole body Magnetic Resonance Imaging (MRI), as compared to wild-type mice. (B) USP18 null mice have decreased in vivo rates of lipolysis. Plasma glycerol and free fatty acid (FFA) levels were measured in these mice before and after injection of the selective β3-adrenergic agonist BRL37344. (C) USP18 null mice had lower adiponectin and higher triglyceride plasma levels. (D) Engineered gain of USP18 expression in human and murine lung cancer cell lines increased acylcarnitine 0:0 levels, as shown in this panel. The symbol * refers to P < 0.05.
To extend these in vivo findings, in vitro studies were performed. Ectopic USP18 expression profiles were achieved independently in human and murine lung cancer cells. This conferred changes in lipid metabolism, as displayed in Supplementary Fig. 2. Notably, acylarnitine 0:0 was significantly upregulated in human A549 and HOP62 as well as murine ED1 lung cancer cell lines (P < 0.05) (Fig. 1D).
USP18 Upregulates ATGL via ISG15 Deconjugation
Because USP18 null mice exhibited decreased lipolysis, it was sought to learn whether key regulators of lipolysis were engaged and repressed. Independently engineered loss (Fig. 2A) versus gain (Fig. 2B) of USP18 expression was achieved in cultured lung cancer cell lines and this, respectively, decreased and increased ATGL protein levels relative to controls. In the USP18 null leiomyosarcoma cell line KHC2 (24) USP18 expression was reconstituted and this upregulated ATGL expression in Fig. 2B. To learn whether the ISG15-conjugation pathway was involved, the E1 activating enzyme UBE1L was overexpressed in ED1 lung cancer cells. ATGL protein levels were found to decline as compared to these empty vector-transfected control cells (Fig. 2C). The DUB USP18 removes ISG15 from its conjugated substrates, thus preventing target protein destabilization (31).
Figure 2:
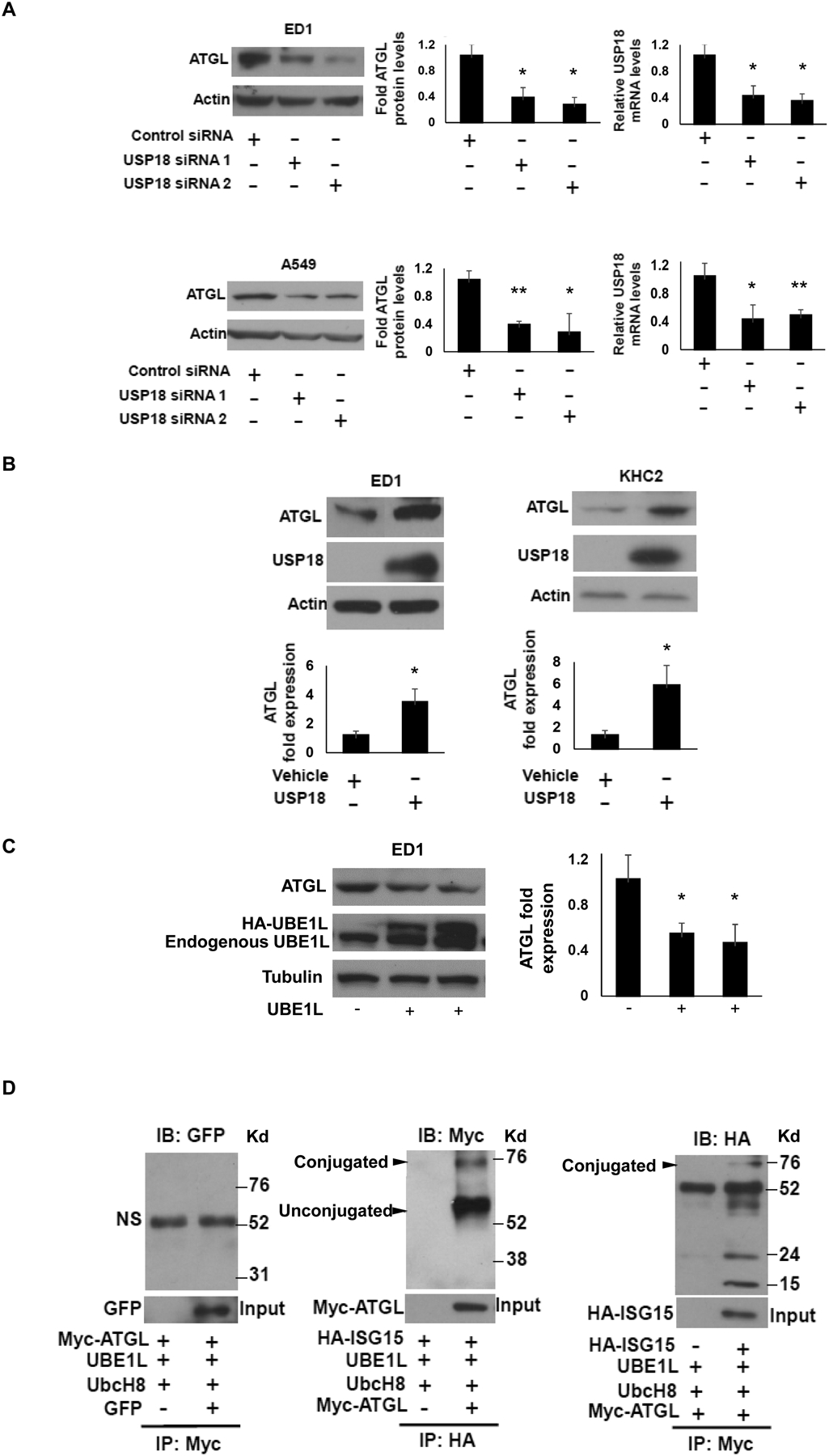
USP18 upregulated ATGL protein by removing the ISG15 conjugate from this substrate. (A) Knock-down of USP18 by individual siRNAs was independently achieved in ED1 and A549 cancer cell lines and this led to decreased ATGL expression, with expression profiles quantified relative to actin expression and normalized to control (inactive) siRNAs independently transfected into these respective lung cancer cell lines. Real-time qPCR assays were done to validate siRNA knockdown of USP18. (B) Gain of USP18 expression was achieved independently in murine ED1 lung cancer and KHC2 leiyomyosarcoma cell lines and this led to increased ATGL protein expression. (C) Gain of expression of the E1-like ubiquitin activating ligase UBE1L conferred reduced ATGL protein expression. Quantification was relative to actin expression as normalized to the empty vector transfected into these respective lung cancer cell lines. (D) Immunoprecipitation assays confirmed that ISG15 and ATGL form conjugated protein, as shown in this panel. The abbreviation NS refers to non-specific and IB indicates immunoblot while IP is immunoprecipitation. The quantification of immunoblots shows the average of three independent experiments. The symbols refer to * P < 0.05 and ** P < 0.01, respectively. Representative immunoblots are displayed.
Since ATGL expression is affected by altering USP18 expression levels, it was hypothesized that ATGL protein was an ISG15ylation target. Given this, immunoprecipitation assays were performed on human lung cancer cells transfected with Myc-tagged ATGL, HA-tagged ISG15, UBE1L and UbcH8. Observed findings revealed that exogenous ATGL and ISG15 can conjugate, as shown in Fig. 2D.
USP18 Null Mice and Cold Sensitivity
It was next sought to learn the extent through which USP18 affected metabolic response in vivo. Intriguingly, USP18 null mice were found to be especially sensitive to cold stimuli. When USP18 null mice were transiently subjected to cold exposure of 4°C, the rate of rectal temperature decrease in USP18 null mice was statistically-significantly more rapid than in wild-type mice (P < 0.01), indicating that USP18 null mice were less able to maintain their body temperature than wild-type littermate mice, as in Fig. 3A.
Figure 3:
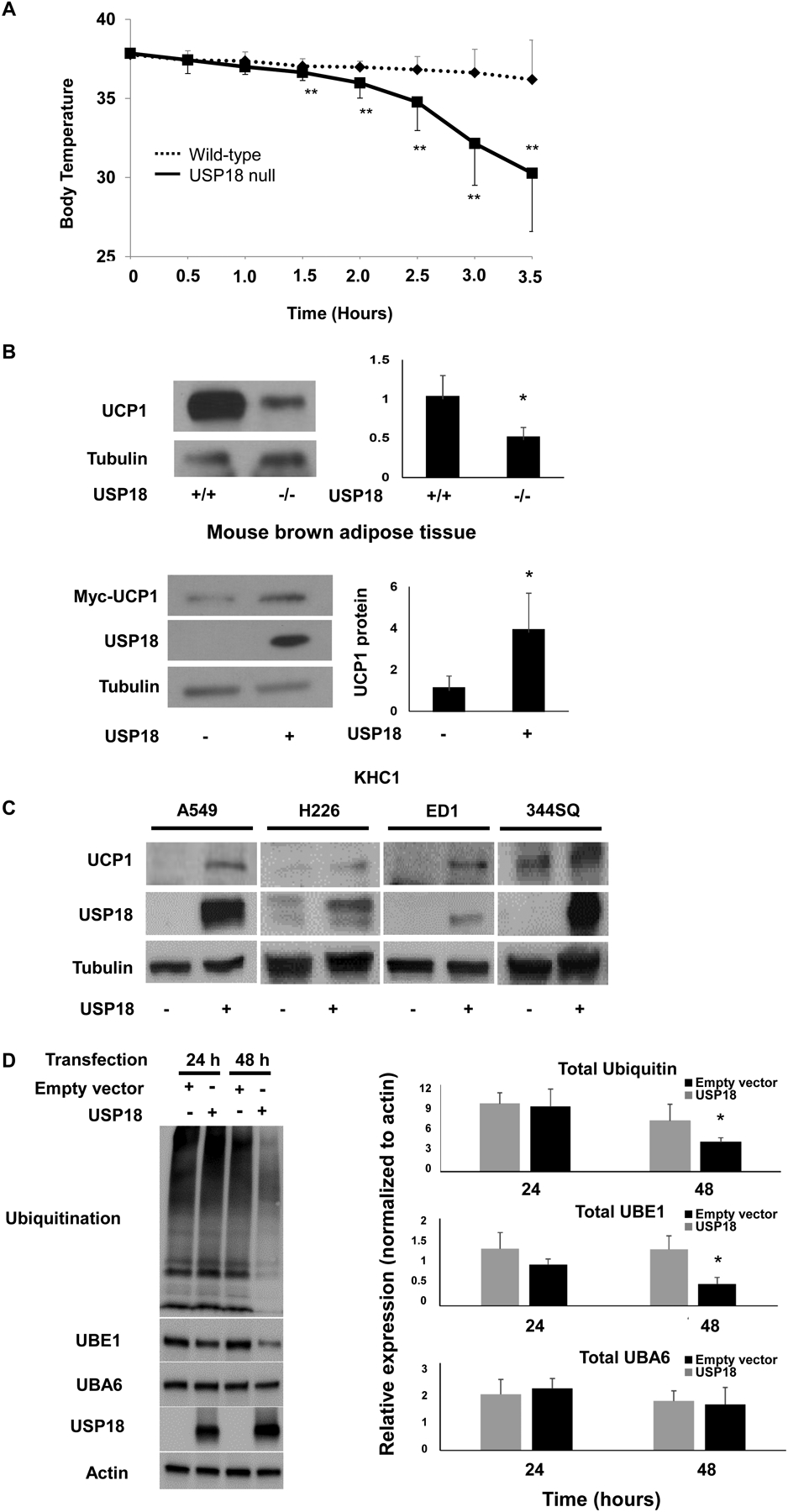
USP18 null mice are markedly sensitive to cold stress and this is associated with reduced UCP1 protein expression. (A) USP18 null mice had impaired ability to maintain their basal temperature after exposure to cold stress. Protein levels were quantified relative to tubulin expression and normalized to examined wild-type murine tissues. (B) UCP1 expression is displayed in brown adipose fat of USP18 null mice versus the murine KHC1 USP18−/− null leiyomyosarcoma cell line. Protein levels were quantified relative to tubulin expression. Protein expression profiles were normalized to empty vector-transfected KHC1 cells. (C) Gain of USP18 expression independently achieved in human and murine lung cancer cell lines conferred enhanced expression of UCP1 protein. (D) Gain of USP18 expression in 293T cells substantially decreased total ubiquitination and UBE1 levels. Protein levels were quantified relative to actin expression and normalized to examined empty-vector transfected cells, as displayed in this panel. The quantification of immunoblots shows the average of three independent experiments. Representative immunoblots are displayed. The symbols refer to * P < 0.05 and ** P < 0.01, respectively.
Specific thermoregulators were hypothesized to be affected by the loss of USP18 expression. To explore this possibility, immunoblot assays were performed in harvested brown adipose tissues isolated respectively from USP18 null mice as compared to wild-type mice. These USP18 null mice exhibited a markedly lower UCP1 expression profile versus wild-type mice, as displayed in Fig. 3B. USP18 expression was next reconstituted in murine KHC1 USP18−/− leiomyosarcoma cells. This conferred a substantial augmentation of UCP1 protein expression as in Fig. 3B. Engineered overexpression of USP18 independently achieved in human and murine lung cancer cells led to enhanced UCP1 protein levels (Fig. 3C).
Prior work found that UCP1 was destabilized through the ubiquitination-proteasome system (32, 33). Given this, it was explored if this was a mechanism engaged by USP18 to affect the stability of UCP1 protein. Immunoprecipitation assays did not find ISG15 conjugated to UCP1 (data not shown). Yet, engineered gain of USP18 expression downregulated total ubiquitination and UBE1 levels (Fig. 3D), providing a mechanistic basis through which the DUB USP18 affects UCP1 expression.
UCP1 Promotes Fatty Acid Oxidation and Lung Cancer Growth
USP18 was known to affect tumorigenicity in lung cancer cells (17, 20) and UCP1 functions as a thermoregulator within brown adipose tissue. It was next examined whether USP18-mediated stabilization of UCP1 protein and if this species played a role in the proliferation of lung cancer cells using β-Hydroxybutyrate (Ketone Body) colorimetric assays. These assays revealed that independently engineered gain of UCP1 expression in both human and murine lung cancer cells markedly promoted fatty acid oxidation as compared to empty vector control transfected lung cancer cells (P < 0.01 and P < 0.001), as shown in Fig. 4A. This result was independently confirmed using the seahorse assay system. It was found that engineered gain of UCP1 expression augmented oxidation of both endogenous and exogenous representative fatty acid (palmitate) oxidation versus empty vector-transfected cellular controls (P < 0.01), as displayed in Fig. 4B. It was next determined if this affected lung cancer cell proliferation. Engineered loss (Fig. 4C) versus gain (Fig. 4D) of UCP1 expression was independently achieved in human and murine lung cancer cells. This led to inhibited and increased, respectively, growth of these transfected lung cancer cells (P < 0.05). To validate that engineered gain of UCP1 expression promoted lung cancer growth, manual cell number counts and CellTiter-Glo assays were done and yielded consistent results, as shown in Supplementary Fig. 3.
Figure 4:
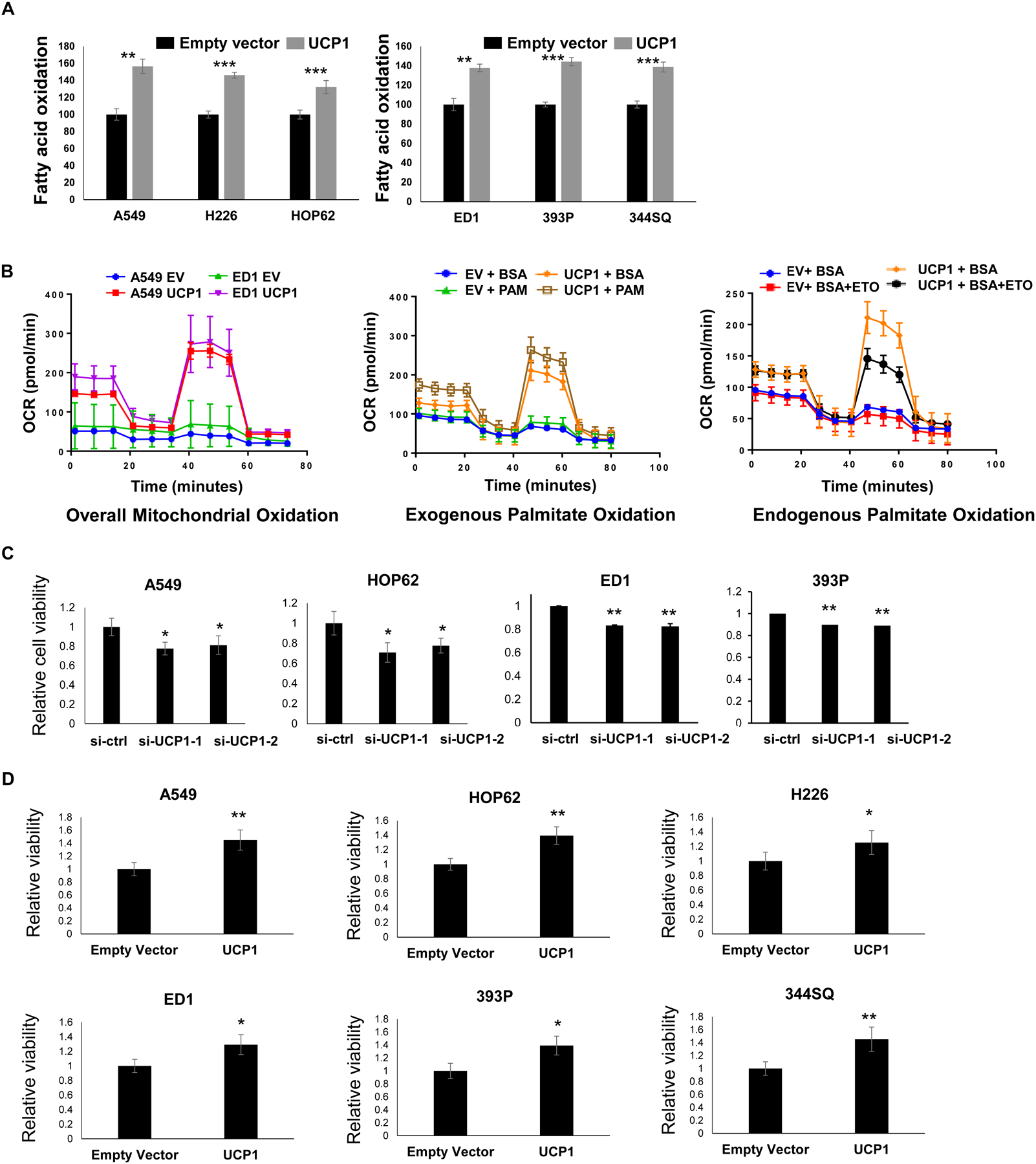
UCP1 expression can promote fatty acid oxidation and proliferation of lung cancer cells. (A) β-Hydroxybutyrate (ketone body) colorimetric assays determined that engineered gain of UCP1 expression substantially enhanced fatty acid beta oxidation in both human and murine lung cancer cells. (B) Engineered UCP1 over-expression increased fatty acid beta oxidation in human A549 and murine ED-1 lung cancer cell lines. The oxidation of representative lipid species (fatty acid and exogenous as well as exogenous palmitate) were measured using Seahorse assays. (C) Knock-down of UCP1 expression reduced human and murine lung cancer cell proliferation, as measured by Celltiter Glo assays that were normalized to control siRNA-transfected lung cancer cells. (D) Gain of UCP1 expression also augmented proliferation in human (A549, HOP62 and H226) and murine (ED1, 393P and 344SQ) lung cancer cell lines, as measured by Celltiter Glo assays that were normalized to empty vector-transfected cells. The symbol * refers to P < 0.05, ** indicates P < 0.01 and *** displays P < 0.001.
Augmented USP18, ATGL and UCP1 Expression and Lung Cancer Outcome
Given that prior work found that augmented ATGL expression promoted cancer cell growth (21) and as reported here augmented UCP1 expression enhanced growth of transfected lung cancer cells, it was examined if increased USP18, ATGL and UCP1 expression profiles detected in human lung cancers affected lung cancer survival. Results appear in Fig. 5A. These mRNA expression levels of these species were also interrogated for other cancer types, as in Supplementary Fig. 4. Interestingly, TCGA data revealed that a signature of high USP18-ATGL-UCP1 expression led to a statistically-significant decline in survival as compared to lung cancers having a lower USP18-ATGL-UCP1 expression profile (P = 0.026), as determined in Fig. 5B.
Figure 5:
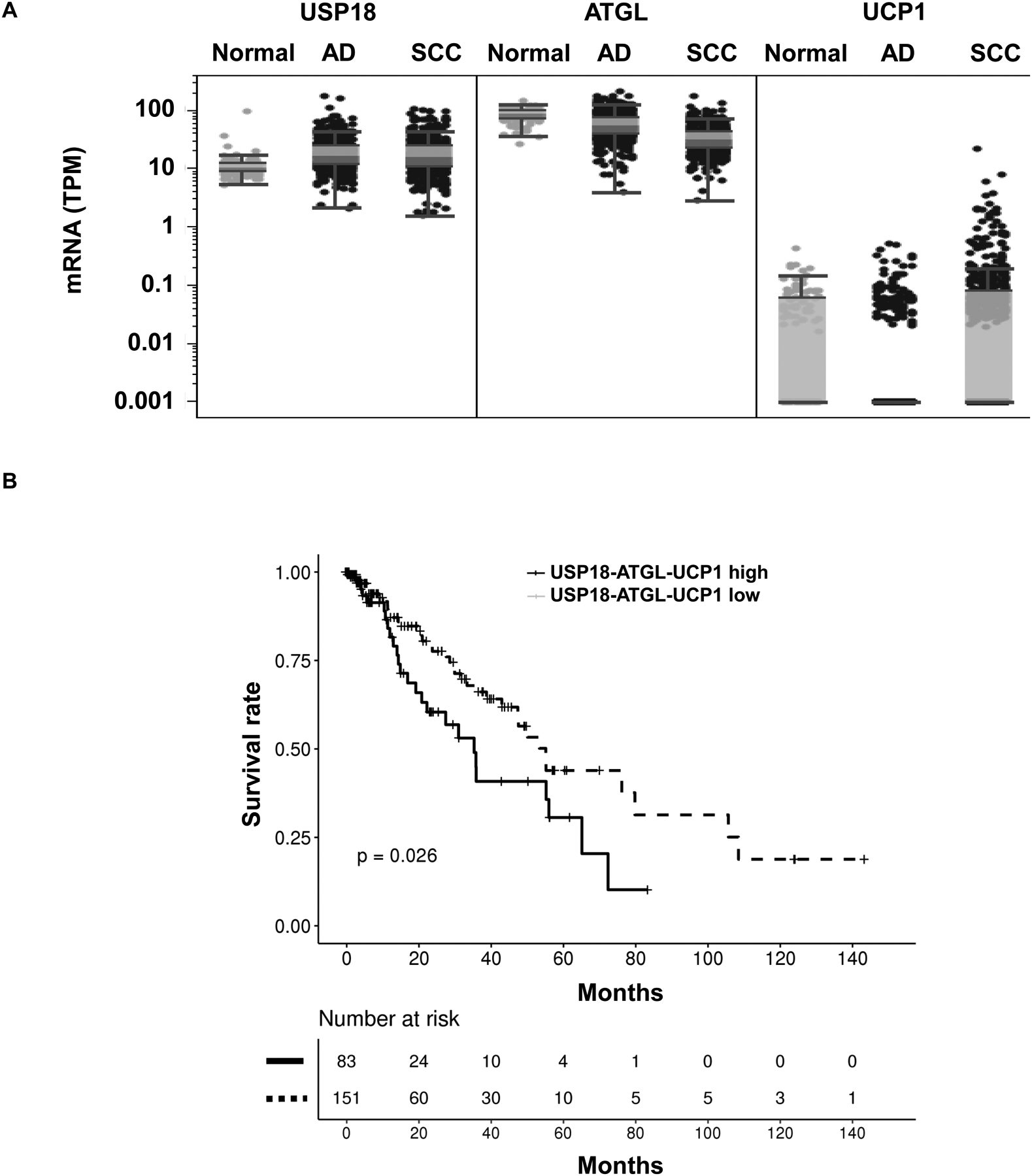
Combined USP18, ATGL and UCP1 expression profiles in human lung cancers were interrogated using TCGA database. (A) Comparisons of USP18, ATGL and UCP1 mRNA levels were between normal and malignant (adenocarcinoma, AD, and squamous cell carcinoma, SCC) lung tissues using the TCGA database. (B) Kaplan-Meier analysis of survival in human lung adenocarcinomas and squamous cell carcinomas are displayed. Cases with higher combined USP18, ATGL and UCP1 expression levels had an unfavorable survival as compared to those cases with lower USP18, ATGL and UCP1 expression profiles, as shown in this panel.
To elucidate if similar survival patterns occurred in other human malignancies, analyses were extended to include assessing the consequences of high versus low levels of expression of these key components of the ISGylation pathway. Notably, a signature of higher USP18-ATGL-UCP1 profiles predicted an unfavorable survival outcome (as compared to all other examined cases in TCGA) for prostatic adenocarcinomas (P=0.024) (Fig. 6D), kidney chromophobes (P=0.03) (Fig. 6E) and low grade gliomas (P=0.00081) as in Fig. 6F. The mRNA expression levels of these species were interrogated (Fig. 6A–C). Other human cancers were assessed for the survival consequences of enhanced expression of these regulators of ISG15ylation. TCGA results appear in Supplementary Fig. 5A–C.
Figure 6:
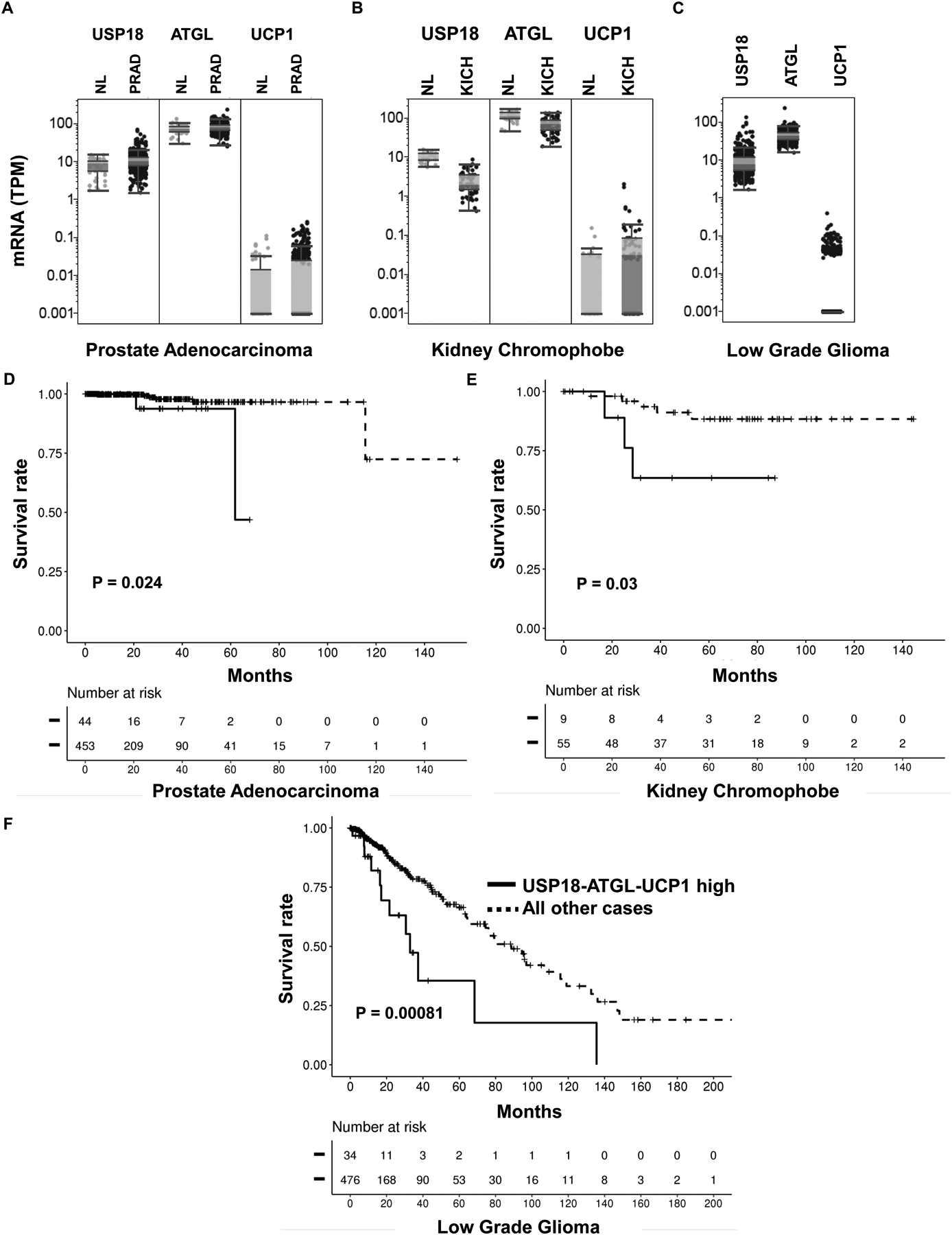
Combined USP18, ATGL and UCP1 expression profiles independently determined in prostate cancers, kidney chromophobes and low grade gliomas. Comparisons of USP18, ATGL and UCP1 mRNA levels were assessed between: (A) normal prostate and malignant prostate (B) normal kidney versus kidney chromophobe and (C) low grade gliomas. Kaplan-Meier analysis of survival was determined in (D) prostate cancer (E) kidney chromophobe and (F) low grade glioma cases comparing high USP18, ATGL and UCP1 expression profiles versus all other cases.
Discussion
Lung cancer is the most common cause of cancer death for men and women in the United State (34). An improved understanding of cancer metabolism could provide new therapeutic insights to combat lung and other cancers. The Warburg effect established that altered glycolysis is a paradigmatic feature of cancers (5). This study advanced prior work by linking the expression levels of the protease USP18 to the regulation of lipid metabolism. This underscores USP18 as an emerging antineoplastic target.
The importance of ISG5ylation and the USP18-mediated pathway is further shown here by reporting a link exists between them and the regulation of lipolysis and thermoregulation, each major components of lipid metabolism. The consequences of this were evident in vivo in examined mice with marked observed changes in basal lipid metabolism in USP18 null versus wild-type mice, as in Fig. 1. This led to marked cold sensitivity of USP18 null mice, as in Fig. 3. Intriguingly, loss of USP18 expression also affected lung cancer cellular lipid metabolism, as shown in Fig. 2.
The net consequence of this is that elevated levels of USP18 promoted lipolysis and increased fatty acid oxidation that augmented lung cancer growth. Intriguingly, enhanced expression of key components of the ISG15ylation pathway coupled with that of the lipolysis regulators ATGL and UCP1 can confer an unfavorable survival when co-expressed with USP18 in lung and other human cancers, as displayed in Fig. 5, Fig. 6, and Supplementary Fig. 5. The implication from these findings is that an inhibitor of USP18 enzymatic activity would interfere with growth of lung and potentially other cancers by antagonizing lipid metabolism in these tumors.
Key components that affect lipolysis are reported to promote cancer growth, including Monoacylglycerol lipase (MAGL) (35) and ATGL (21). This study found that USP18 directly stabilized ATGL protein by removing ISG15 from the conjugated complex (Fig. 2). This finding not only extends an understanding of the role of ISG15ylation in the regulation of lipolysis, but also uncovered a previously unrecognized mechanism through which USP18 promotes tumorigenicity. Future work should learn how USP18 and other protein destabilization pathways precisely regulate lipolysis and by this affect tumorigenicity.
The thermoregulator family members (and in particular UCP1) have key functions in the homeostasis of mammalian body temperature by increasing oxidation in mitochondria of brown adipose fat (33). More attention should be placed on investigating UCP1 actions in other tissue and cell types, including cancer cells. Since this study reported that USP18 null mice exhibited reduced UCP1 levels in fat tissue and altered thermoregulation (Fig. 3A), it was sought to learn the actions of UCP1 in cancer cells.
It was found here that USP18 stabilized UCP1 protein in cancer cells. This promoted both fatty acid oxidation and lung cancer cell growth (Fig. 3 and Fig. 4). This study described a previously unrecognized mechanism through which USP18 stabilized UCP1 protein and by this promoted mitochondrial oxidation in lung cancer cells and enhanced their growth, as shown in Fig 4. It is hypothesized that the relatively modest growth effects observed in the examined UCP1-transfected lung cancer cells were due to the presence of lipid present in FBS-supplemented cultures.
This finding has translational relevance. Subsets of human lung and other cancers do exhibit UCP-1 expression and when combined with increased ATGL and USP18 expression these profiles exert an unfavorable outcome on survival in lung and other cancers types Fig. 5 and Fig. 6. Prior work found that USP18 protein was more abundantly expressed in the malignant than in the adjacent histopathologically normal lung tissues (18). Also, ATGL immunohistochemical expression is detected in a subset of examined human lung cancer cases, as shown in Supplementary Fig. 6. From these observations, it is concluded that deregulation of the thermoregulator UCP-1 has neoplastic actions beyond its known role in lipid metabolism within fat tissue. Future translational research should discern the precise functional role of UCP-1 in cancer biology.
Taken together, this study advances prior work (36) by reporting a novel consequence of regulated USP18 expression. This is enhanced lipolysis, fatty acid oxidation and cancer cell growth. These and other findings provide a strong rationale to develop an inhibitor to USP18. That inhibitor could exhibit anti-neoplastic effects alone or as part of a combined regimen to combat lung and potentially other human cancers.
Supplementary Material
Implications:
USP18 is an antineoplastic target that affects lung cancer fatty acid metabolism.
Acknowledgments:
This work was supported partly by the National Institutes of Health (NIH), National Cancer Institute (NCI) grant R01-CA190722 (XL, SJF and JMK), a Samuel Waxman Cancer Research Foundation Award (ED), an American Cancer Society Professorship (ED), the University of Texas Science and Technology Acquisition and Retention Award (ED), and funding from the NCI Intramural Research Program and Center for Cancer Reward, NCI and NIH contract #75N91019D00024 (ED).
Footnotes
Conflicts of Interest: no conflicts of interest are cited by these authors.
References
- 1.Hanahan D, and Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, and Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 3.Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, et al. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer. 2017;16(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pavlova NN, and Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23(1):27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warburg O, Wind F, and Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8(6):519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, and Smans K. Lipogenesis and lipolysis: The pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res. 2013;52(4):585–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menendez JA, and Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7(10):763–77. [DOI] [PubMed] [Google Scholar]
- 8.Rohrig F, and Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer. 2016;16(11):732–49. [DOI] [PubMed] [Google Scholar]
- 9.Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang XY, et al. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018;435:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carracedo A, Cantley LC, and Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13(4):227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harjes U, Kalucka J, and Carmeliet P. Targeting fatty acid metabolism in cancer and endothelial cells. Crit Rev Oncol Hematol. 2016;97:15–21. [DOI] [PubMed] [Google Scholar]
- 12.Luengo A, Gui DY, and Vander Heiden MG. Targeting metabolism for cancer therapy. Cell Chem Biol. 2017;24(9):1161–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong JJ, Pung YF, Sze NS, and Chin KC. HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc Natl Acad Sci U S A. 2006;103(28):10735–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuan W, and Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 2001;20(3):362–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao C, Beaudenon SL, Kelley ML, Waddell MB, Yuan W, Schulman BA, et al. The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an IFN-alpha/beta-induced ubiquitin-like protein. Proc Natl Acad Sci U S A. 2004;101(20):7578–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malakhova OA, Kim KI, Luo JK, Zou W, Kumar KG, Fuchs SY, et al. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006;25(11):2358–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo Y, Dolinko AV, Chinyengetere F, Stanton B, Bomberger JM, Demidenko E, et al. Blockade of the ubiquitin protease UBP43 destabilizes transcription factor PML/RARalpha and inhibits the growth of acute promyelocytic leukemia. Cancer Res. 2010;70(23):9875–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo Y, Chinyengetere F, Dolinko AV, Lopez-Aguiar A, Lu Y, Galimberti F, et al. Evidence for the ubiquitin protease UBP43 as an antineoplastic target. Mol Cancer Ther. 2012;11(9):1968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah SJ, Blumen S, Pitha-Rowe I, Kitareewan S, Freemantle SJ, Feng Q, et al. UBE1L represses PML/RAR{alpha} by targeting the PML domain for ISG15ylation. Mol Cancer Ther. 2008;7(4):905–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mustachio LM, Lu Y, Tafe LJ, Memoli V, Rodriguez-Canales J, Mino B, et al. Deubiquitinase USP18 loss mislocalizes and destabilizes KRAS in lung cancer. Mol Cancer Res. 2017;15(7):905–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zagani R, El-Assaad W, Gamache I, and Teodoro JG. Inhibition of adipose triglyceride lipase (ATGL) by the putative tumor suppressor G0S2 or a small molecule inhibitor attenuates the growth of cancer cells. Oncotarget. 2015;6(29):28282–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richer E, Prendergast C, Zhang DE, Qureshi ST, Vidal SM, and Malo D. N-ethyl-N-nitrosourea-induced mutation in ubiquitin-specific peptidase 18 causes hyperactivation of IFN-alphass signaling and suppresses STAT4-induced IFN-gamma production, resulting in increased susceptibility to Salmonella typhimurium. J Immunol. 2010;185(6):3593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ritchie KJ, Malakhov MP, Hetherington CJ, Zhou L, Little MT, Malakhova OA, et al. Dysregulation of protein modification by ISG15 results in brain cell injury. Genes Dev. 2002;16(17):2207–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chinyengetere F, Sekula DJ, Lu Y, Giustini AJ, Sanglikar A, Kawakami M, et al. Mice null for the deubiquitinase USP18 spontaneously develop leiomyosarcomas. BMC Cancer. 2015;15:886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Sempere LF, Ouyang H, Memoli VA, Andrew AS, Luo Y, et al. MicroRNA-31 functions as an oncogenic microRNA in mouse and human lung cancer cells by repressing specific tumor suppressors. J Clin Invest. 2010;120(4):1298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ, et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009;23(18):2140–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng Q, Sekula D, Guo Y, Liu X, Black CC, Galimberti F, et al. UBE1L causes lung cancer growth suppression by targeting cyclin D1. Mol Cancer Ther. 2008;7(12):3780–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawakami M, Mustachio LM, Rodriguez-Canales J, Mino B, Roszik J, Tong P, et al. Next-generation CDK2/9 inhibitors and anaphase catastrophe in lung cancer. J Natl Cancer Inst 2017;109(6):djw297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mustachio LM, Kawakami M, Lu Y, Rodriguez-Canales J, Mino B, Behrens C, et al. The ISG15-specific protease USP18 regulates stability of PTEN. Oncotarget. 2017;8(1):3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ketscher L, Hannss R, Morales DJ, Basters A, Guerra S, Goldmann T, et al. Selective inactivation of USP18 isopeptidase activity in vivo enhances ISG15 conjugation and viral resistance. Proc Natl Acad Sci U S A. 2015;112(5):1577–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Honke N, Shaabani N, Zhang DE, Hardt C, and Lang KS. Multiple functions of USP18. Cell Death Dis. 2016;7(11):e2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clarke KJ, Adams AE, Manzke LH, Pearson TW, Borchers CH, and Porter RK. A role for ubiquitinylation and the cytosolic proteasome in turnover of mitochondrial uncoupling protein 1 (UCP1). Biochim Biophys Acta. 2012;1817(10):1759–67. [DOI] [PubMed] [Google Scholar]
- 33.Azzu V, Jastroch M, Divakaruni AS, and Brand MD. The regulation and turnover of mitochondrial uncoupling proteins. Biochim Biophys Acta. 2010;1797(6–7):785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siegel RL, Miller KD, and Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. [DOI] [PubMed] [Google Scholar]
- 35.Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, and Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140(1):49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mustachio LM, Lu Y, Kawakami M, Roszik J, Freemantle SJ, Liu X, Dmitrovsky E. Evidence for the ISG15-specific deubiquitinase USP18 as an antineoplastic target. Cancer Res. 2018; 78(3):587–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.