

Abstract
The drivers of ductal carcinoma in situ (DCIS) to invasive ductal carcinoma (IDC) transition are poorly understood. Here, we conducted an integrated genomic, transcriptomic, and whole-slide image analysis to evaluate changes in copy number profiles, mutational profiles, expression, neoantigen load and topology in 6 cases of matched pure DCIS and recurrent IDC. We demonstrate through combined copy number and mutational analysis that recurrent IDC can be genetically related to its pure DCIS despite long latency periods and therapeutic interventions. Immune “hot” and “cold” tumors can arise as early as DCIS and are subtype-specific. Topologic analysis showed a similar degree of pan-leukocyte-tumor mixing in both DCIS and IDC but differ when assessing specific immune subpopulations such as CD4 T-cells and CD68 macrophages. Tumor-specific copy number aberrations in MHC-I presentation machinery and losses in 3p,4q, and 5p are associated with differences in immune signaling in estrogen receptor (ER) negative IDC. Common oncogenic hotspot mutations in genes including TP53 and PIK3CA are predicted to be neoantigens yet are paradoxically conserved during the DCIS-to-IDC transition, and are associated with differences in immune signaling. We highlight both tumor and immune-specific changes in the transition of pure DCIS to IDC, including genetic changes in tumor cells that may have a role in modulating immune function and assist in immune escape, driving the transition to IDC.
Keywords: evolution, ductal carcinoma in situ, immune escape, neoantigen, breast cancer
INTRODUCTION
Ductal carcinoma in situ (DCIS) of the breast is a pre-invasive cancer characterized by abnormal proliferation of neoplastic epithelial cells confined within the mammary ductal-lobular unit and separated from the stroma by an intact myoepithelial cell layer and basement membrane (1). Whilst over 50,000 cases of DCIS are diagnosed each year in the US, only one third of these will progress to invasive ductal carcinoma (IDC) within 30 years, presenting a natural evolutionary bottleneck (1). Accurate prediction of the likelihood of progression would alleviate over-treatment of the disease. However, achieving this is hampered by our relatively limited knowledge of the molecular processes underlying the DCIS-to-IDC transition.
Characterizing determinants of progression remains challenging due to difficulties with acquiring matched DCIS-IDC samples from treatment-naïve patients and long latency periods after the initial diagnosis of pure DCIS. It is estimated that 10% of patients will relapse with invasive disease after 10 years, with ipsilateral recurrences at least twice as common as contralateral recurrence (2). This trend of local recurrence suggests that the new lesion is genetically related to the prior lesion, and studies of matched pure DCIS and IDC may be informative in uncovering driver events.
Genetic studies of DCIS and IDC cases have shown limited stage-specific differences that could predict progression. Whole-exome sequencing (WES) of DCIS and IDC has shown high similarity in copy number profiles between the two histologic entities, with notable copy number aberrations (CNAs) including 1q, 8q, 17q, 20q amplification and 8p, 11q, 16q, 17p loss (3–6). Activation of oncogenes including PIK3CA, GATA3, ERBB2, and loss of tumor-suppressors TP53 and CDKN2A, are thought to be early breast cancer driver events due to their similar frequencies in DCIS and IDC (5,7). Coordinate selection of multiple genes in chromosome scale aberrations that provide low level fitness advantages has also been implicated as a survival mechanism (8). High genetic similarity has been noted in studies of matched synchronous DCIS and IDC (4,9–12), however, these are reflective of a timepoint beyond the evolutionary bottleneck and the full repertoire of mediators of the successful transition from DCIS to IDC cannot be established from these studies.
In contrast to tumor epithelial cells and genetic changes, multiple cell types composing the tumor microenvironment show tumor progression stage-specific gene expression and epigenetic differences (13,14). Genes involved in extracellular matrix (ECM) organization including collagens, matrix metalloproteinases (MMPs) and cell adhesion molecules are upregulated in IDC compared to DCIS (15,16). Differences in immune cells including T, B, and natural killer (NK) cells have also been noted between DCIS and IDC (16,17), and interleukin (IL4, IL12, IL23) signaling is differentially expressed in DCIS compared to normal breast tissue with changes maintained in IDC (17). These studies suggest that the tumor microenvironment and immune system play major roles in the DCIS to IDC transition.
Immunosurveillance exerts constant selective pressure against tumor cells, which may lead to intratumor heterogeneity observed in DCIS (18). We have previously demonstrated that in HER2+ and triple-negative (TN) DCIS there is an activated immune environment characterized by higher frequency of cytotoxic CD8+GZMB+ T-cells compared to IDC (19). In contrast, the microenvironment of TN IDC is often immunosuppressive with higher PD-L1 expression in tumor epithelial cells and higher expression of T-regulatory cell (Treg) markers (19). Although PD-L1 expression by DCIS tumor epithelial cells is rare (19,20), high proportions of PD-L1+ tumor infiltrating lymphocytes (TILs), Tregs, and CD68+ macrophages have been observed in high-grade DCIS (20,21), implying immune-editing (22).
We hypothesize that the DCIS to IDC transition is mediated by concurrent tumor-specific and immune microenvironmental changes. To explore this, here we describe, for the first time, integrated genetic, gene expression and topologic analysis of a small cohort of matched pure DCIS and recurrent IDC, focusing on immune-related alterations including changes in predicted neoantigens.
MATERIALS AND METHODS
Patient cohort
Patient material was collected from Yonsei University and Seoul National University, Korea between 2000 and 2014. All cases were reviewed by expert pathologists (S.Y.P, J.J) and scored for tumor purity, lymphocyte and stromal infiltration as well as grade, estrogen receptor (ER), progesterone receptor (PR), and HER2 status. Written informed consent was obtained from all patients, and all protocols used were approved by the ethical and institutional review boards, in concordance with the declaration of Helsinki. The patient cohort (recurrence cohort) consisted of three ER+, two HER2+, and one TN tumors with variable treatment regimens (Supplementary Fig. S1A, Supplementary Table S1). Of note, Case 5 was a HER2+ DCIS that recurred as a contralateral ER+ IDC, and Case 1 had a lymph node recurrence. Due to the small cohort size, we used genomic data from two larger DCIS cohorts (Abba (NER-=12, NER+=17) and Lesurf (Nluminal=15, Nbasal=7, NHER2+=7, NNormal=17) cohorts) (5,16) to validate our findings (Supplementary Methods).
Exome and RNA Sequencing
Patient samples were macro-dissected into stromal and epithelial fractions from FFPE (formalin fixed paraffin embedded) tissue-slides, with examples of segmented regions highlighted in Supplementary Fig. S1B. DNA and RNA were extracted from epithelial fractions from matched DCIS and IDC cases and corresponding normal breast using AllPrep DNA/RNA FFPE Kit (Qiagen, Cat. No. 80234). Exome and RNA libraries were prepared by the Genomic Platform at the Broad Institute using Nextera Rapid Capture Exome Kit (Illumina, Cat. No. FC-140–1000) and TruSeq RNA Access Library Prep kit (Illumina, Cat.No. 20020189) and sequenced on HiSeq4000 and Hiseq2000 flow cells respectively. Reads were aligned to the hg37 genome using bwa (RRID:SCR_010910) for exome-data (23) and STAR (RRID:SCR_015899) (24) for RNA-seq data. Further details are described in Supplementary Methods and exome coverage is summarized in Supplementary Fig. S2.
Histology and cyclic immunofluorescence (cycIF)
Whole tissue mounts were sectioned to a thickness of 4 μm and stained with hematoxylin and eosin. The cyclicIF method was applied to cases 8 and 9 to create highly multiplexed images as previously described (25), and further details including antibodies are described in Supplemental Methods and Supplemental Table 2. 10 cycles of immunofluorescence using 4 antibodies of interest and DAPI, imaging and fluorophore bleaching was performed. Collected images are then registered based on DAPI staining, and image segmentation and feature extraction were performed. Intensities within a tissue were scaled to a [0, 1] distribution and phonograph (RRID:SCR_016919) (26) was used to classify cells into clusters.
Data Analysis
Code to reproduce figures and statistical calculations in this study is available at https://github.com/polyak-lab/matchedDCISIDC and is described in Supplementary Methods.
WES:
The Getz Lab CGA WES Characterization pipeline at the Broad Institute (https://docs.google.com/document/d/1VO2kX_fgfUd0x3mBS9NjLUWGZu794WbTepBel3cBg08/edit) has been optimized for analysis of both frozen tissue and archival FFPE. Segmented contiguous chromosomal regions were called using GATK (RRID:SCR_001876) CNV (27). A log fold change of ±0.3 was used to define gains and losses from array CGH data from Lesurf et al (16). Variant discovery was performed using a combination of MuTect1 (27), MuTect2 (27) and Strelka (RRID:SCR_005109) (28) and was annotated using Oncotator (RRID:SCR_005183) (29). Ploidy and variants in normal exome samples were called using GATK GermlineCNVCaller and HaplotypeCaller respectively(27) and annotated for pathogenicity using ClinVar (RRID:SCR_006169) (30).
Topology:
H&E images were digitally segmented using Qupath (RRID:SCR_018257) (31) and cells were classified into epithelial, stromal, and immune cells. Spatial-pattern analysis was performed using k-nearest neighbor distances and Morisita-Horn index for spatial overlap (32).
Gene expression:
RNA sequencing of Cases 1, 5, 2DCIS, 3DCIS were excluded from analysis due to poor quality. Differential gene expression analysis was performed using DESeq2 (RRID:SCR_000154) (33), and reported fold changes used in gene set enrichment analysis was performed using HTSAnalyzeR (34) using the c2 compendium (35,36). Immune signature analysis was performed using single-sample GSEA (ssGSEA; RRID:SCR_003199) using signatures manually curated from the literature (19,37). Proportions of different immune cell populations was inferred using a combination of TIMER (RRID:SCR_018737) (38), CIBERSORT (RRID:SCR_016955) (39), xCELL (40) and EPIC (41). For inferred fractions, statistical differences were assessed using a beta regression model whereas enrichment scores were assessed using Wilcoxon-rank sum test.
Neoantigen prediction:
All non-synonymous mutations and frame shift mutations were in silico translated using SIFT (RRID:SCR_012813) (42) into peptides of length 8–11 amino acids. HLA-type was inferred from exome data using Polysolver (43). Neoantigens were predicted using NetMHCPan4 (RRID:SCR_018182) (44), and estimated binding affinity of <50nM (strong) and <200nM (weak) were used to predict potential binders. Neoantigens were validated as having at least 2 RNA-seq reads supporting the mutation.
BCR/TCR repertoire:
CDR3 recombined regions in B-cells and T-cells were inferred from RNA using MixCR (RRID:SCR_018725) (45). CDR3 reads were filtered to those that have at least 2 supporting reads and have an amino acid sequence length of at least 6. These were searched against the IEDB database (46) of known CDR3 sequences.
RESULTS
Copy number aberrations are early events in the DCIS-to-IDC transition
We performed WES to identify differences in CNAs and mutational profiles between matched pure DCIS and recurrent IDC (Supplementary Fig. S1, S2A–B and Supplementary Table 1). Amongst all DCIS and IDC cases within our recurrence cohort, frequently observed changes included 1q, 8q, 16p, 17q amplification and 11q and 16q loss, consistent with previous observations in other DCIS datasets including the Abba and Lesurf cohorts (5,16) (Fig. 1A). For most DCIS-IDC pairs, copy number profiles of the IDC closely resembled that of the DCIS, where on average 84% of bases had similar calls and Case 8 showed high correlation in copy number ratios (Supplementary Fig. S2C–E). In contrast to the other cases, 2IDC exhibited aberrations in large contiguous chromosomal regions.
Figure 1. Copy number alterations and somatic mutations in the DCIS-to-IDC transition.
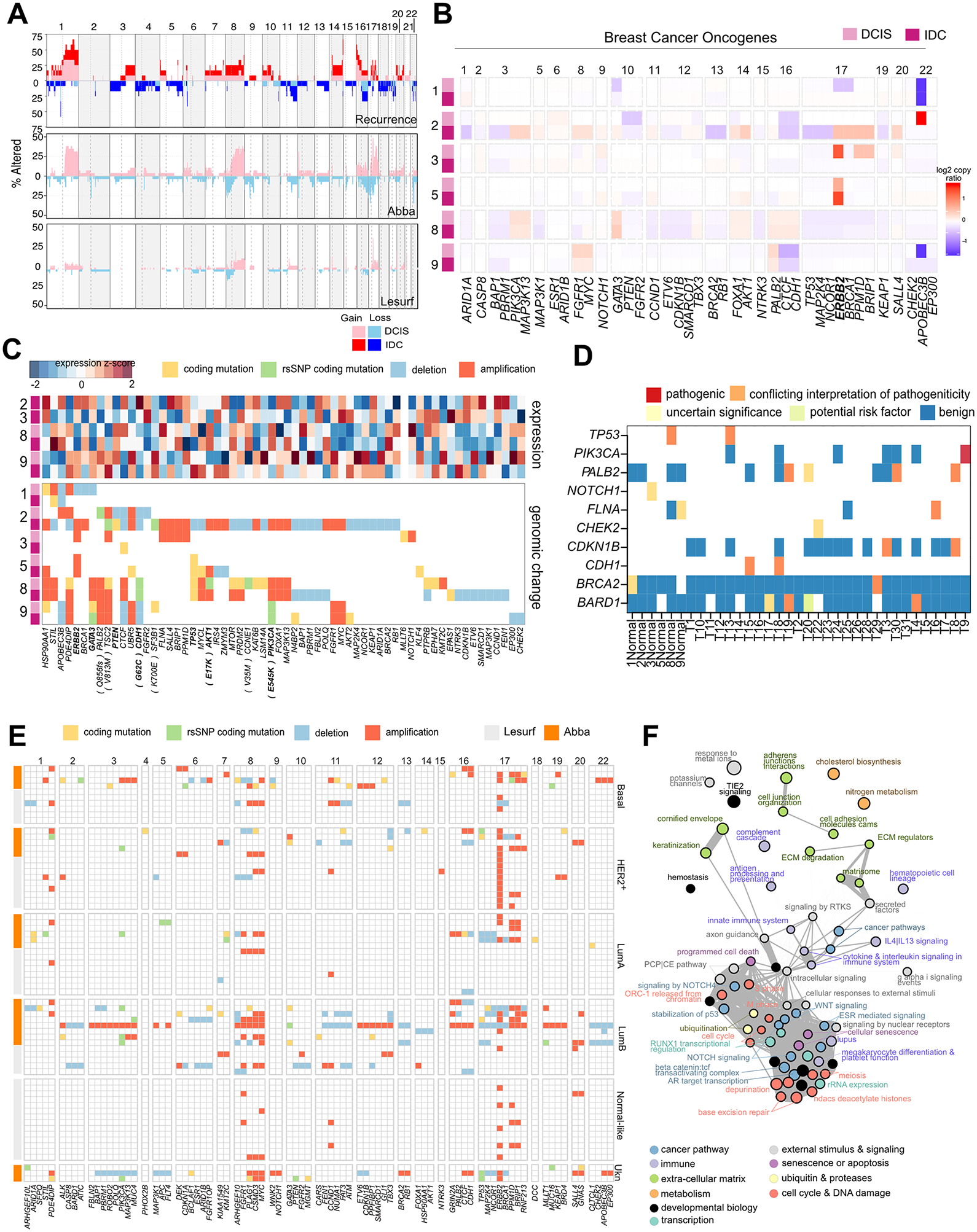
A, Genome-wide summary of proportion of patients with observed CNAs in the Recurrence (our data), Abba (5), and Lesurf (16) cohorts. A z-score of 2 in GATK CNV was set to call gains and losses in the recurrence and Abba cohorts (exome-seq), and a threshold of ±0.3 in the Lesurf set (aCGH). Both DCIS and IDC samples are shown in the recurrence cohort. B, Summary of CNAs in breast cancer-associated oncogenes and tumor suppressors in the recurrence cohort. C, Summary of cancer-related CNAs and coding mutations in the recurrence cohort, and corresponding gene expression profiles. D, variants found in adjacent normal mammary tissue. E, Summary of cancer-related CNAs and mutations in the Abba and Lesurf cohorts grouped by PAM50 subtype. F, Pathways implicated by genetic changes in all three cohorts and by RNA-expression in the Abba cohort comparing DCIS to normal. Circle size reflective of the average enrichment score and line width reflective of the number of common genes in two pathways.
Focusing specifically on breast cancer-related oncogenes, the most commonly observed gain across all cohorts was at the ERBB2 (17q21) locus (Fig. 1B–C). 17q gain in 2IDC may drive weak HER2+ expression in the IDC. In contrast, 5IDC retains this amplicon despite being characterized as ER+HER2−, and 3IDC did not show 17q21 amplification despite being characterized as HER2+ by immunohistochemistry and showed very weak RNA expression. ERBB2 amplifications in PAM50 HER2− cases was observed in other cohorts – the overall proportion of cases with ERBB2 amplification was 48% (14/29) and 40% (17/42) in the Abba and Lesurf cohorts. Other common gains included MYC (8q24) and FGFR1 (8p11). Loss of heterozygosity was observed at 16q22 (CDH1, CTCF) in all cohorts; however, gains were also noted in the Abba cohort. Losses were also noted in genes associated with DNA repair (BRCA2, BRCA1, TP53) and cell cycle regulation (RB1).
Overall, the high genetic similarity between DCIS and IDC shown in our data combined with consistency in copy number profiles with other DCIS cohorts (3,4,16) supports the early acquisition of copy number changes (47).
Genetic overlap between pure DCIS and recurrent IDC
We assessed mutational burden in coding regions and found very little overlap both across patients and even within the same patient (Supplementary Table S3, Supplementary Fig. 3A–B). The total and coding mutational burden was similar in both DCIS and IDC, although a decrease was noted in Cases 8 & 3IDC and an increase in 5IDC (Supplementary Fig. S3C). However, variant allele frequencies less than 0.08 cannot be reliably detected given the sample coverage (Supplementary Fig. S3D), and sampling bias, clonal expansion of a dominant clone, neutral drift or bottleneck selection could explain the differences in these profiles.
Mutations in breast cancer-related genes shared between DCIS and IDC occurred in driver events including TP53 (Case5 & 8), PIK3CA (Case9, E545K activation), and CDH1 (Case8) and RAD50 (Case8) (Fig. 1C). The Abba cohort also showed frequent mutations in the known driver genes TP53 (17%), PIK3CA (20%), and GATA3 (14%) (Fig. 1E). Other conserved coding mutations lie in genes associated with ubiquitin (DCAF16, UCHL5, USP11) and metabolism and mitochondrial activity (GGT7, GUSB, HADHB, SLC25A37) (Supplementary Fig. S3B).
Oncogenic mutations have previously been identified in normal human tissues (48). Thus, we sought to determine if such events occur in our cohort by profiling adjacent matched mammary normal tissue (Fig. 1D). Most samples harbored benign rsSNP variants for common oncogenes including BRCA1/2 and KMT2C. However, we also found some variants which have been proposed to be risk factors or have uncertain clinical effect in TP53, BARD1, CDH1, and PALB2. One patient sample (T9) displayed a pathogenic mutation PIK3CAE545K in the normal tissue and maintained in the DCIS (Supplemental Fig. S3E), indicating the possibility of a very early driver event.
Similarity in CNAs and the presence of conserved mutations points to a genetic relationship between pure DCIS and recurrent IDC despite long latency periods in Case 8 and 9. Case 5 had many shared mutations, suggesting contralateral seeding at the time of the DCIS. Cases 1 and 3 do not display mutational overlap and have quiet copy number profiles. It is unclear whether they are directly genetically related. However, an ipsilateral recurrence only after 3 years in Case 3 suggests the recurrence is likely to have stemmed from residual primary disease.
Immune-related changes between DCIS and IDC: differences in cellular composition and topology
Pathway enrichment analysis of genetic alterations appearing in all cohorts included those associated with cell cycle, DNA damage, apoptosis, and cancer-related pathways including RTK, ESR1, NOTCH, WNT signaling, which was also supported by RNA-seq data comparing normal breast tissue with DCIS in the Abba cohort (Fig. 1F, Supplementary Table S4). Moreover, also implicated were pathways pertaining to the extracellular matrix (ECM) and the immune system with cytokine signaling (the IL13/IL4 pathway supporting Th2 differentiation), innate immunity, and antigen processing and presentation. Both genetic and RNA profiling implied that the immune system has a major impact on the DCIS-to-IDC transition. Thus, we investigated immune-related changes in further detail.
We quantified TILs in whole slide H&E images (Fig. 2A, Supplementary Fig. S1C) and assessed the spatial co-localization of tumor and immune cells using three metrics: (1) The interacting fraction, i.e., the proportion of TILs within 10μm of a tumor epithelial cell and capable of direct interaction (Fig. 2B) (2) the average distance of the 3 closest neighbors to each TIL (kNN, k=3) of cell type x and assessed the proportion within 50 μm (Fig. 2C) and (3) the Morisita-Horn (MH) index of spatial correlation of two cells types (32), which provides a global metric for mixing independent of composition. To take into account differences in tumor architecture between DCIS and IDC, we omitted regions containing only tumor cells when computing MH indices (Fig. 2D, Supplementary Methods).
Figure 2. Immune composition and spatial distribution in DCIS and IDC.
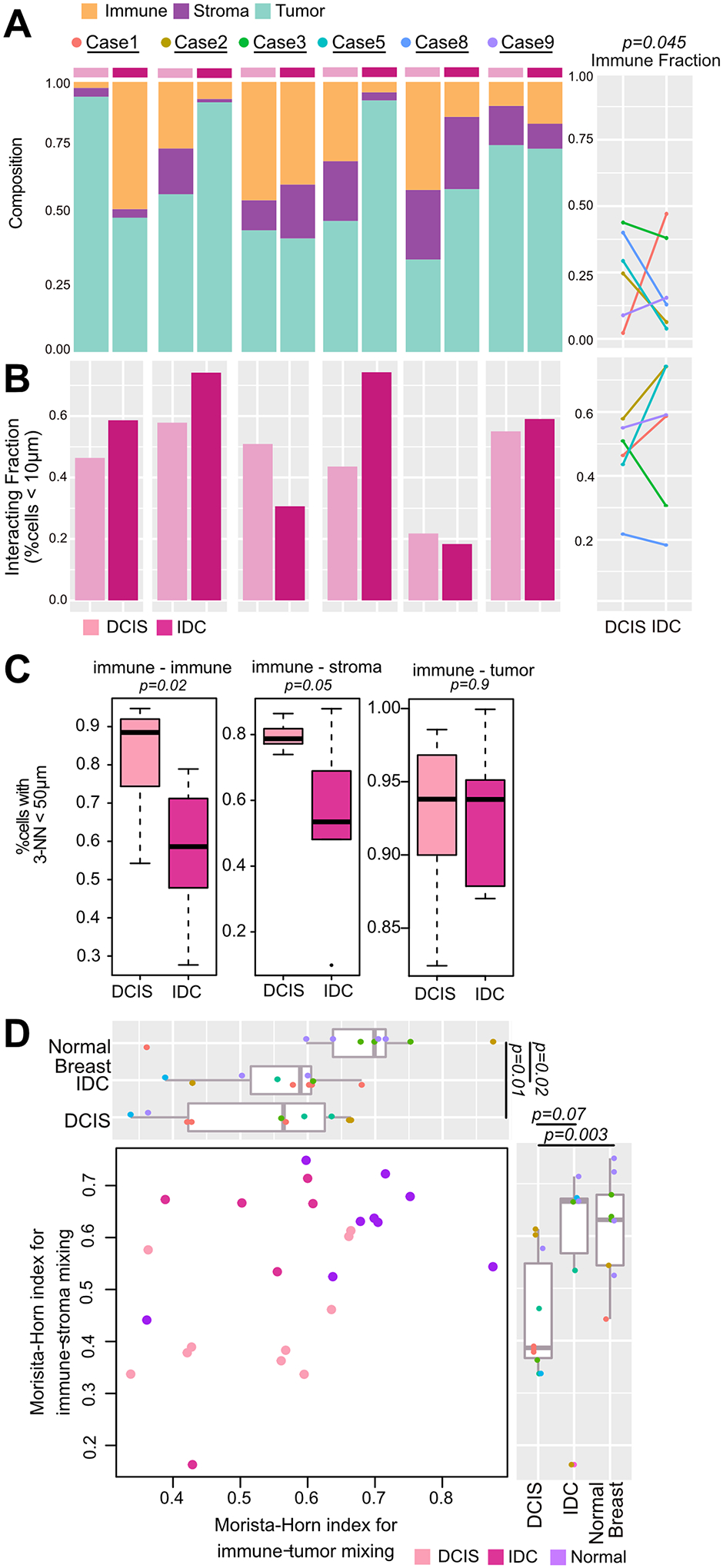
A-C, Compositional and spatial features in the recurrence set based on whole slide H&E images. A, Cellular composition. Significance computed using a beta-regression for bounded fractions (P=0.009) and by paired one-tailed t-test (P=0.045). B, Proportion of immune cells within 10μm of an epithelial cell within digitally macrodissected DCIS, IDC or normal regions. C, Proportion of cells with k-Nearest neighbor (k=3) distances less than 50μm. Significance computed using Wilcoxon rank sum test and beta-regression for bounded fractions (PImmune-Immune=0.003, PImmune-Stroma=0.02). D, Morisita-Horn index of tumor-lymphocyte and stroma-lymphocyte mixing in digitally macrodissected DCIS, IDC or normal regions. Significance between groups computed using Wilcoxon rank sum test.
IDC had either lower or the same fraction of immune cells compared to DCIS with the exception of Case1 which was a lymph node recurrence (paired one-tailed t.test P=0.045). No difference was observed for stromal or tumor cells (Supplementary Fig. S4A). Using the interacting fraction and kNN analysis, we did not detect a difference between DCIS and IDC (Fig. 2B–C), suggesting that there is a similar capacity for immune cells to interact with tumor cells in both DCIS and IDC. However, our kNN analysis highlighted that immune cells tended to co-occur with both immune cells and stromal cells in DCIS compared to IDC (Fig. 2C), which is evident in tertiary lymphoid structures found in Cases 2 and 8 (Supplementary Fig. S1B). This difference in spatial patterns could be attributed to lower TIL frequencies in IDC, which can be taken into account using the MH-index. The MH-index for tumor-lymphocyte mixing was lower in all neoplastic lesions compared to adjacent normal breast tissue, indicating that some degree of compartmentalization and reduced immune-epithelial interaction occurs during tumorigenesis (Fig. 2D). Furthermore, once normalized for TIL content, there was less immune-stromal mixing in DCIS compared to normal tissue and IDC.
The analyses show that in matched pure DCIS and IDC the immune fraction in DCIS is comparable or higher. Furthermore, whilst compartmentalization occurs and there is less interaction between the three different cell types in neoplastic lesions, TIL-tumor spatial distances are comparable in DCIS and IDC.
Immune hot and cold tumors arise in DCIS
To understand differences in DCIS compared to IDC, we performed GSEA across all DCIS and IDC samples and conducted paired analysis for Cases 8 and 9 (Fig. 3A, Supplementary Table S5). Common pathways enriched in DCIS include E-cadherin stabilization & ATM pathways whereas WNT signaling was enriched in IDC, consistent with our genomic analysis. Sample 8 was enriched for cell cycle markers in IDC, indicating higher proliferation. Interrogation of immune-related terms showed downregulation of BCR signaling and upregulation of its inhibitor CD22 specifically in Case 8. In contrast, Case 9 showed an upregulation of terms associated with infection and immunodeficiency in IDC, including FCGR and FCER1 mediated signaling.
Figure 3. Immune expression signature analysis in DCIS and IDC.
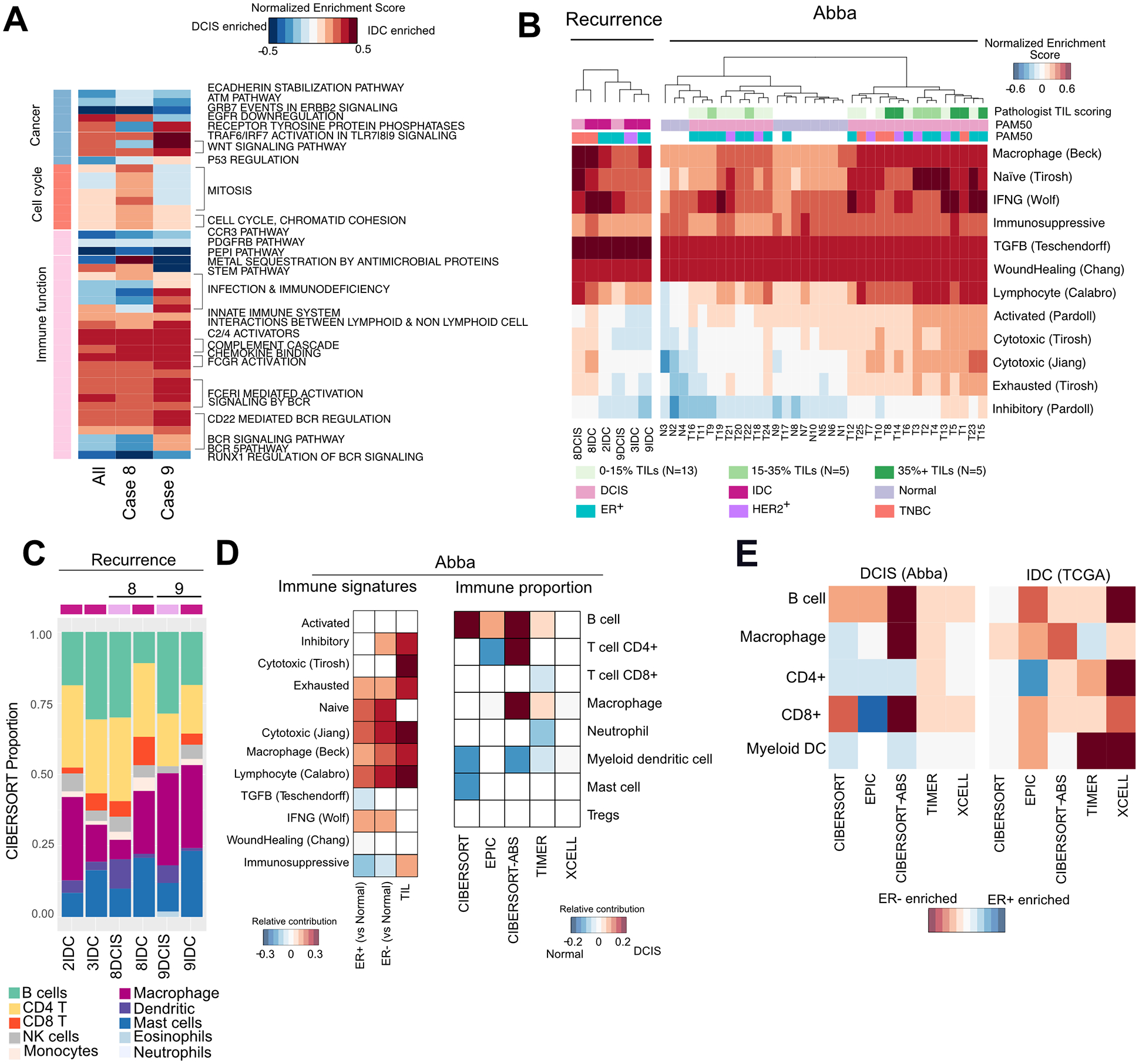
A, Enriched pathways in DCIS compared to IDC across all cases, case 8 and case 9 (FDR<0.1). B, Heatmap of single-sample GSEA scores for published immune signatures in the recurrence and Abba cohort. C, Immune composition of tumors inferred by CIBERSORT in the recurrence cohort D, Heatmap showing relative contribution of ER status and TILs to immune signatures, and the enrichment of immune cell types in normal compared to DCIS using several deconvolution methods. Only significant contributors are shown (P<0.05, Wilcoxon rank sum test for enrichment scores, beta-regression for proportion data). E, Comparison of enriched immune subsets in ER+ and ER− DCIS and IDC. Only significant contributors are shown (P<0.05, Wilcoxon rank sum test for enrichment scores, beta-regression for proportion data).
To further investigate immune related differences, we calculated single-sample GSEA for curated immune-related gene signatures (Fig. 3B, Supplementary Methods). Enrichment scores for immune signatures were variable even in DCIS, as seen in the Abba cohort. 8DCIS was “immune hot” and was enriched for macrophage and lymphocytic infiltration signatures, which persisted in IDC despite reduced TILs by H&E. 8IDC showed a higher enrichment for the cytotoxic IFNγ signature and reduced enrichment for naïve immune response, suggesting an inefficient adaptive immune response in the DCIS. In contrast Case 9 had a “colder” immune microenvironment which was maintained in IDC. Immune cell deconvolution based on RNA-seq data using a number of methods suggested a reduction of B-cells, dendritic cells and CD4+ T-cells in DCIS to IDC (Fig. 3C, Supplementary Fig. S4B).
Similar compositional analysis in the Abba cohort showed an enrichment for dendritic cells normal breast tissue, and B-cells in DCIS (Fig. 3D). Immune signature analysis presented significant upregulation of macrophage, naïve, lymphocytic and cytolytic signatures in both ER+ and ER− DCIS compared to normal. Contrasting ER+ and ER− tumors, we found an enrichment of B-cells in ER− DCIS whereas ER− IDC was enriched for most immune cell types (Fig. 3E). These results suggest that increased immune infiltration in DCIS may exert both anti-tumor and immunosuppressive responses.
To obtain a more comprehensive view of immune composition, we performed cyclic immunofluorescence (cycIF), contrasting immune “hot” Case 8 with immune “cold” Case 9 (Fig. 4A, Supplementary Fig. S4C–D). In Case 8, the total proportion of immune cells relative to luminal epithelial cells decreased in the IDC (Fig. 4C). Amongst the different immune cell types, a large drop in CD20+ B-cells and CD4+ T-cells was observed, whilst similar frequencies of CD68+ macrophages were present in DCIS and IDC, consistent with RNA-based inference (Fig. 4A, Supplementary Fig. S4B). In contrast to our RNA-data, we observed a decrease in CD8+ accompanied by an increase in immunosuppressive Foxp3+ Tregs. GZMB+CD8+ T-cells was a rare population which increased from 1.8% of CD8+Tcells in 8DCIS to 8.2% in 8IDC. One population observed specifically in Case 8 was the exhausted Ki67+PD1+CD4+ T-cell which increased in IDC.
Fig. 4. Cyclic immunofluorescence in DCIS-to-IDC.
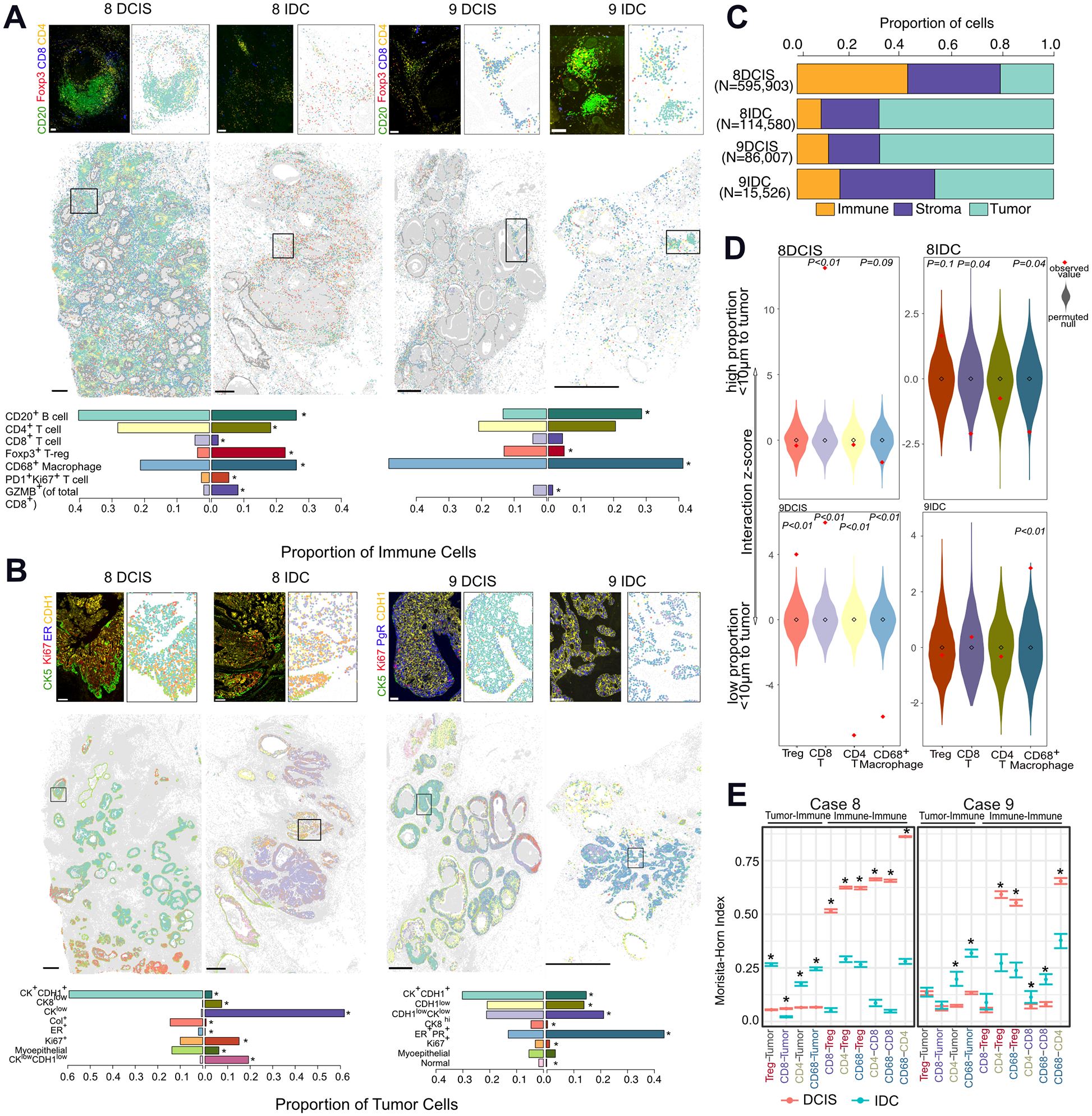
A, Whole slide image of classified immune cells in cases 8 and 9, representative image, and relative proportions of immune cells in each sample. Scale bar whole slide image: 1mm, insert: 100μm. Differences were computed using proportionality test, *P <0.05. B, Whole slide image of classified tumor cells in cases 8 and 9, representative image, and relative proportions of tumor cells in each sample. Scale bar whole slide image: 1mm, insert: 100μm. C, Cellular composition in each sample. D, Z-score of the interacting fraction of immune cells to tumor cells. Null distribution was calculated from 1000 permutations of immune cell labels E, Morisita-Horn index of spatial correlation of two interacting-cell populations, *P <0.05.
Spatial pattern analysis showed co-localization of the different T-cell subtypes in DCIS, often within tertiary lymphoid structures, rather than interaction with the tumor cells as indicated by low MH-indices and the low interacting z-scores (Fig. 4D–E). CD8+ T-cells had a higher interacting score than expected with a random spatial distribution with the tumor in 8DCIS which was reversed in 8IDC where a higher proportion of Tregs were in close contact with the tumor (Fig. 4D). This observation held true beyond an interacting distance of 10μm, where CD8+T-cells were closer to and Tregs further from tumor cells in DCIS whereas the reverse was observed in IDC (Supplementary Fig. S4E).
In contrast, Case 9 had a low proportion of immune cells, which slightly increased within the profiled IDC region (Fig. 4C). The main immune population present in both DCIS and IDC were CD68+ macrophages, which had greater interaction with tumor cells in IDC (Fig. 4A,D–E). An increase in B-cells and decrease in Tregs was observed in IDC, and the proportion of CD4+ and CD8+ (including GZMB+) T-cells remained similar within this transition (Fig. 4A), consistent with RNA-seq data. In DCIS, both CD8+ T-cells and Tregs were in close proximity to the tumor (Fig. 4D) and whilst not significant by permutation testing at a 10μm distance, Tregs continued to be closest to tumor cells in IDC (Supplementary Fig. S4F). The low MH-index index between CD8+ T-cells with any other cell type suggests spatial exclusivity, which in conjunction with its low frequency may result in difficulty in coordinating any cytotoxic response (Fig. 4E).
In both cases, greater intermixing between CD4+ and CD68+ cells with tumor cells was observed in IDC. Co-localization was observed between Tregs with both CD8+ and CD4+ cells; and CD68+ cells with CD4+ cells in DCIS (Fig. 4E) consistent with our H&E analysis, which could be a reflection of a coordinated immune response in DCIS which is diminished in IDC.
Our data shows that the composition and spatial organization of immune cells can be highly variable in DCIS and IDC. One specific “immune hot” TN DCIS becomes more immunosuppressive in IDC through the recruitment of Tregs that intermix with tumor cells. In contrast, an “immune cold” ER+ DCIS shows a population dominated by macrophages and Tregs close to the tumor and has similar expression and compositional properties to IDC. These immune-related differences could be due to differences in tumor subtype and indicate that these differences are already present in the pre-invasive stage.
Spatial heterogeneity of tumor cells in the DCIS-to-IDC transition
To assess heterogeneity within tumor cells, we identified 8 major populations based on cycIF (Fig. 4B, Supplementary Fig. S4D). The most dominant cluster had high expression of cytokeratins (CK7,8,17,19) and CDH1, however, subpopulations with decreased cytokeratin expression, particularly of cytokeratin 8, were also detected, particularly in the IDC (Fig. 4B). These CKlow populations often displayed heterogeneous expression of CDH1, consistent with observed genomic loss or mutation in this gene.
Most ducts in 8DCIS were surrounded by an intact CK5+CK14+ myoepithelial cell layer, which was lost in IDC. In contrast, 9DCIS displayed showed thinning of this heterogeneous SMA+ or CK5+CK14+ myoepithelial layer, a common feature of DCIS (Supplementary Fig. S4F) (49). Case 8 was more proliferative than Case 9 based on Ki67 expression (10% vs <5% positivity), consistent with gene-expression data. A minor ER+ population was observed in 8DCIS (2%) but not the IDC. In contrast, Case 9 had a large PR+ER+ population that was primarily located in the outer regions of tumor-filled ducts. However, despite treatment with tamoxifen, the IDC saw a marked increase in this population (12% to 45%) and intermingling with CK+CDH1+ cells. These observations are consistent with reports of high intratumor heterogeneity even in DCIS (4,18). Some of these observed differences could also be due to tumor subtype, since ER+ tumors (Case 9) are more architecturally organized than TN (Case 8) with tumor cells located in immune excluding nests primarily in ER+ tumors (50).
Genetic aberrations associated with immune changes
Given the changes observed in the immune microenvironment, we investigated whether genomic alterations, specifically in MHC-I presentation or immune suppression (51–53), could contribute to this phenotype. Subclonal losses in PSME3, B2M, TAP1/2 and ERAP1/2 were observed in Cases 1, 8, and 9 (Fig. 5A) and in 7/29 and 2/42 patients in the Abba and Lesurf cohorts respectively (Supplementary Fig. S5A), which may abrogate MHC-I presentation and lead to immune evasion. Correlation analysis showed an association between copy number and RNA-expression in proteasomal proteins involved in MHC-I presentation but not in immune checkpoint proteins, suggesting that MHC-I loss by CNAs could be one mechanism of immune evasion (Fig. 5B).
Figure 5. CNAs associated with immune changes.
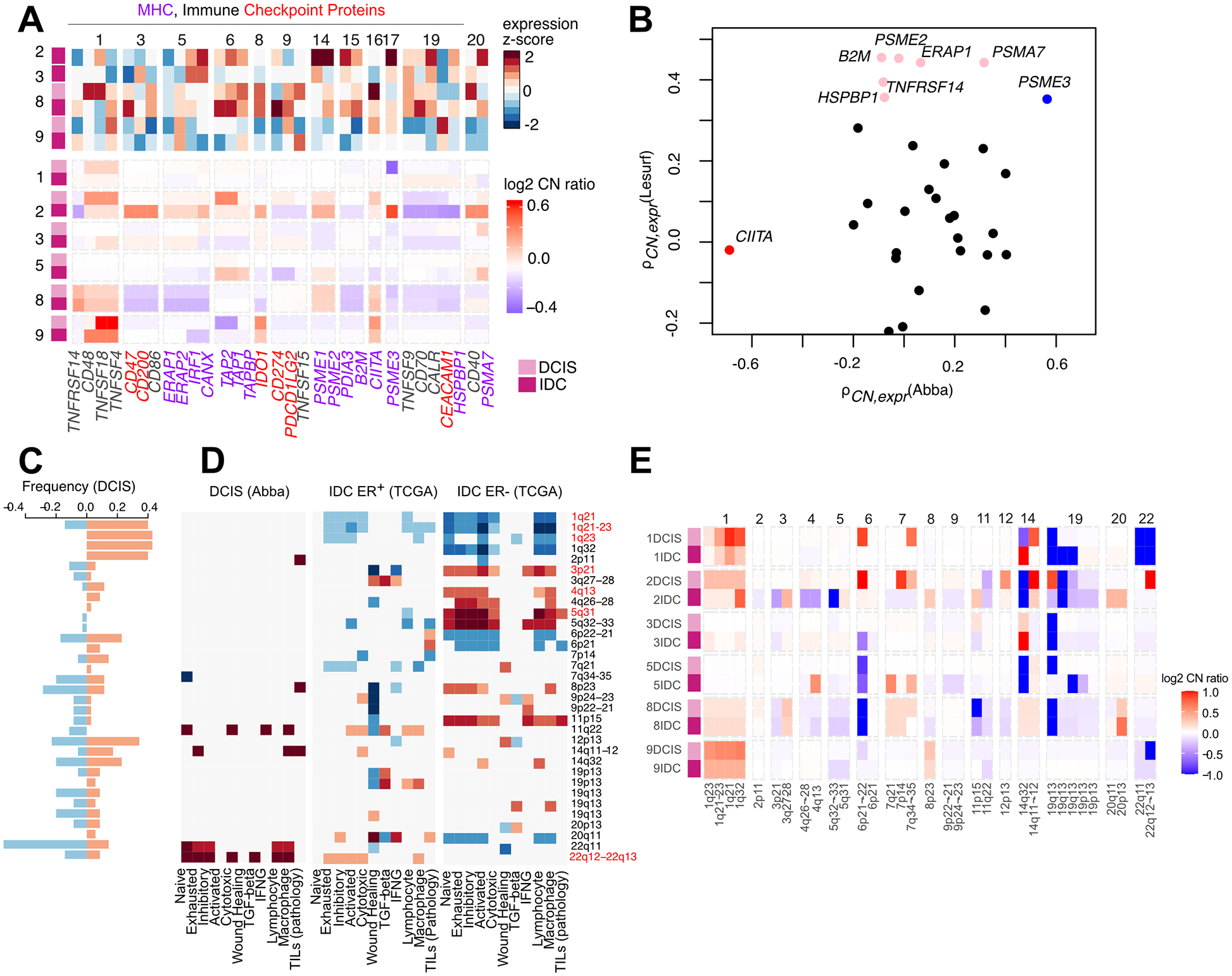
A, CNAs and RNA-expression of in MHC-I presentation and immune checkpoint proteins. In purple are genes involved in MHC-I presentation and in red are immune checkpoint proteins. B, Association between copy-number and gene-expression in the Abba and Lesurf cohorts of the genes shown in (A). Colored genes show a spearman correlation P < 0.1, blue indicates significance in both sets. C, Frequency of CNAs at immune-enriched loci in the Abba cohort. Significant enrichment defined by hypergeometric testing FDR < 0.1. D, Heatmaps of associations between immune signatures and copy number at loci shown in (C) in DCIS (Abba cohort), ER+ IDC (TCGA) and ER− IDC (TCGA) determined by generalized linear models. (Significantly associated beta-scores are shown, P < 0.05). E, CNAs of the regions highlighted in C in the recurrence cohort.
PTEN loss is known to have some correlations with immune hot/cold tumors (54). Interestingly, case 2 DCIS has a loss in PTEN (Fig. 1B) and the IDC has frame-shift insertion of PTEN (VAF: 0.67, Fig. 1C) suggesting biallelic loss, and the IDC is more immune cold than the DCIS.
We have previously identified subtype-specific focal amplifications including gain of 17q12 chemokine cluster and STAT2-CD274 at 9p24 as potential mechanisms of immune escape (19). To determine other loci associated with differences in immune microenvironment, we divided the genome into 5-MB long regions and identified 33 that were enriched for immune-related genes (hypergeometric test, FDR < 0.1, Fig. 5C, Supplementary Fig. S5B, Supplementary Table S6, Supplementary Methods). We then used a combined DCIS cohort (Abba and Recurrence cohorts) and The Cancer Genome Atlas (TCGA) to test for associations between copy number ratios and immune signature scores (37). Amplification of 1q was negatively associated with immune signaling in both ER+ and ER− IDC but not in DCIS, and contains genes associated with inflammation including the PYHIN, S100A, and IL10 family of genes, and the CD1 family involved in MHC-like presentation of lipids and glycoproteins (Fig. 5D). Notably, 11q22 (encoding for the caspase family of proteins) and 22q12–13 (encoding for the APOBEC family of proteins and IL2RB) are two regions which are positively associated with immune scores in both DCIS and ER+ IDC, and losses in these regions are observed at a frequency of 20% in DCIS. Focusing specifically on ER− IDC, we observed a positive association between macrophage and lymphocyte signatures with amplification of the CCR, CXCL, and IL loci at 3p21, 4q13 and 5q31. Losses in these regions are more frequently observed in ER− breast cancers (55), which was indeed observed in TNBC 8IDC (Fig. 5E). Thus, chromosomal gains and losses could contribute to differences in the immune microenvironment between ER+ and ER− tumors.
Hotspot mutations are predicted to be neoantigens
Cells presenting mutant peptides on MHC-class I can be detected and eliminated by the immune system. For each patient, we determined the HLA-types (43) and predicted which oncogenic mutations may present as neoantigens (Supplementary Fig. S5C). As expected, higher mutational burden correlated with higher neoantigen load (ρ = 0.92, P << 0.001) and neoantigen load correlated with TIL counts in DCIS (ρ = 0.5, P = 0.006, Recurrence and Abba cohorts).
RNA-expressed neoantigens included CDH1, PIK3CA, which were the same hotspot driver mutations preserved in the DCIS-to-IDC transition, alongside other reported driver events including PTEN, AKT1 (Fig. 6A). Other expressed neoantigens included SF3B1, RAD50, and AKT2. In the Abba cohort we identified PIK3CA (E545K and H1047R), TP53 (R116W), and GATA3 (P95S) mutations as potential neoantigens supported by RNA-seq (Fig. 6B).
Figure 6. Neoantigen prediction in DCIS and IDC.
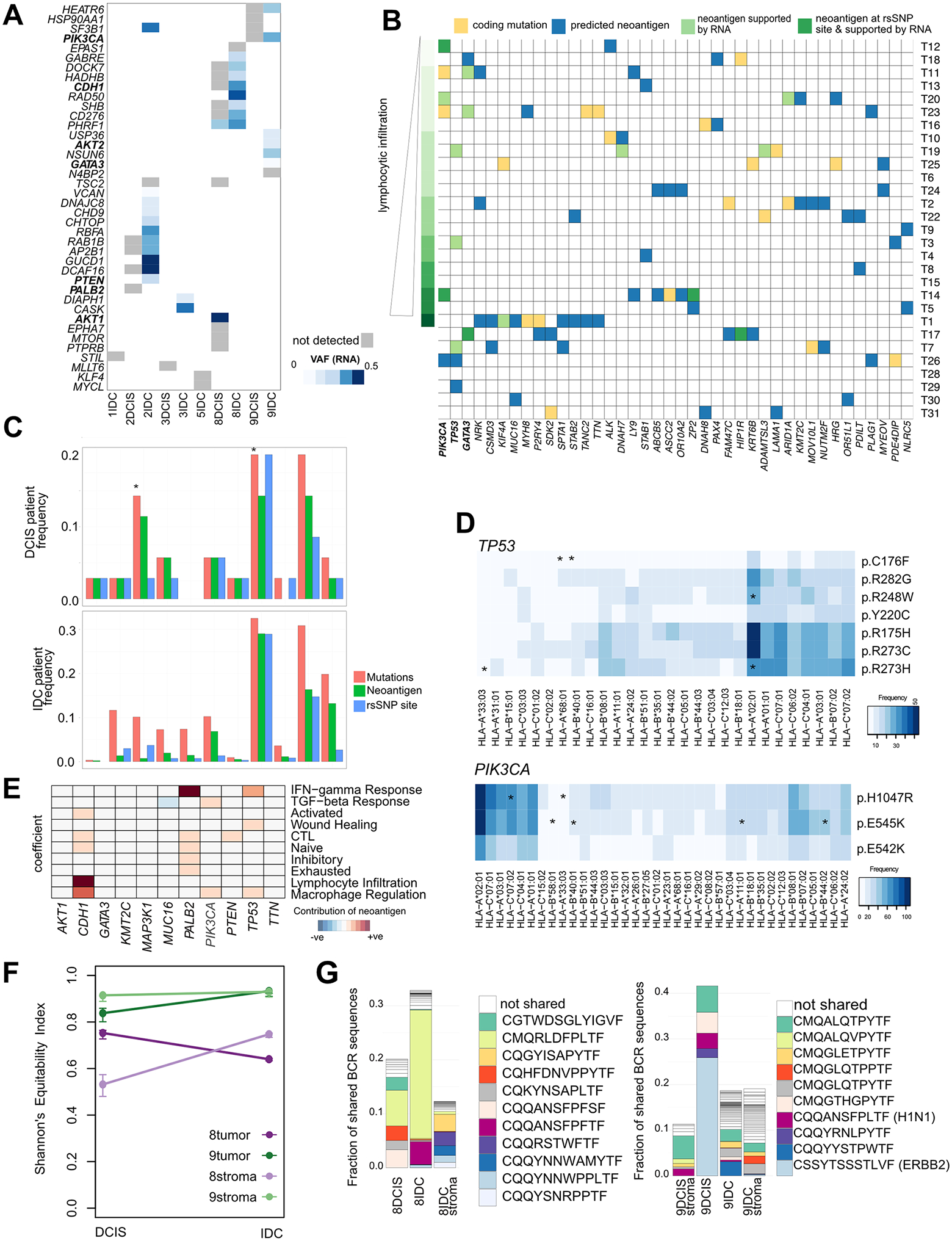
(A-B), Predicted neoantigens supported by expression of the mutation in RNA-seq data in (A) Recurrence cohort and (B) Abba cohort. C, Frequency of mutation, neoantigen and rsSNP sites in the most commonly mutated genes in breast cancer in DCIS (Abba and recurrence cohorts) compared to IDC (TCGA cohort). Differences computed using proportion test *P<0.05. D, HLAs predicted to bind to the most common TP53 and PIK3CA mutant peptides in the TCGA cohort. Asterisked are HLAs predicted to recognize these peptides in the Recurrence/Abba cohort. E, Heatmap showing relative contribution of specific neoantigen to immune signaling pathways in TCGA ER+ patients using a generalized linear model (P< 0.1 shown). F, Changes in BCR repertoire diversity in DCIS and IDC in case 8 and 9. G, Changes in BCR repertoire. Only clonotypes appearing at frequency > 1% are shown, and colored clonotypes are shared between samples.
Given the high frequency of PIK3CA and TP53 mutations predicted as neoantigens in DCIS (14% of patients), we investigated its frequency in the IDC TCGA cohort to determine whether these neoantigens can progress through the DCIS-IDC bottleneck (37) (Fig. 6C, Supplementary Table S7). The proportions of patients with neoantigens between DCIS and IDC were similar, except for GATA3 which saw a reduction, and PIK3CA which saw an increase in frequency (proportion test, p <0.05). Mutations in PIK3CA occurred in 30% of patients and 95% of all PIK3CA mutations were predicted to be neoantigens, predominantly at E545K and H1047R (Supplementary Fig. S5D) Similarly, 60% of TP53 mutations were predicted to be neoantigens, which was much higher than the proportion in other commonly mutated genes including CDH1, GATA3, PTEN, MAP3K1, and KMT2C. Despite TTN and MUC16 being predicted as neoantigens in a high proportion of patients, the mutation sites are variable and do not frequently occur at hotspot or rsSNP sites. PIK3CA and TP53 neoantigens were predicted to bind multiple HLAs common across the population, including HLA-A:01:01 and HLA-A:02:01 (Fig. 6D, Supplementary Fig. S5E), supporting potential presentation as a neoantigen.
We hypothesized the persistence rather than elimination of neoantigens in the DCIS-to-IDC transition could be due to immune-modulatory effects by tumor cells bearing the mutation. We compared immune signatures of tumors with wild-type vs. mutant genes predicted to be neoantigens, accounting for subtype and stage. ER+ patients harboring neoantigenic mutations in PIK3CA, TP53, and CDH1 are associated with increased macrophage signature (Fig. 6E). PIK3CA mutation was also associated with higher TGFβ signaling, and TP53 mutation was associated with IFNγ signaling and wound healing response. Neoantigenic PALB2 was also associated with immune signaling, including IFNγ response.
Our data show that hotspot mutations in common oncogenes including PIK3CA and TP53 can be predicted as neoantigens but are not eliminated in the DCIS-to-IDC transition, and may be associated with immune response. PIK3CA and GATA3 mutations are more common in immune cold luminal breast tumors (50), whereas TP53 mutations are more common in TIL-rich ER− tumors. Thus, breast tumor subtype-specific differences in the immune environment might also influence these results.
B cell receptor (BCR) repertoire in DCIS and IDC
We assessed the evolutionary trajectory of the microenvironment through profiling of the BCR and TCR repertoire, which can serve as molecular barcodes to monitor changes in subpopulations of T or B-cells. We focused on changes in the BCR repertoire due to its higher richness than TCR (Supplementary Fig. S5F), as well as the fact that B-cells play an active role in anti-tumor immune responses both by regulating T-cells and also by direct antibody production. Using the Shannon Equitibility Index as a metric of BCR diversity, we found a reduction in 8IDC compared to the DCIS suggesting a clonal expansion, whereas the reverse was observed in case 9 (Fig. 6F). Indeed, focusing on clones which comprise at least 1% of the BCR repertoire, we found an expansion of the clonotype “CMQRLDFPLTF” in 8IDC which was also detected at low frequency in the IDC stroma (Fig. 6G).
In Case 9, the dominant clonotypes were present in all samples (with the exception of 9DCIS which had lower overall read depth) suggesting little change in the immune microenvironment between DCIS-to-IDC. One dominant clone (“CSSYTSSSTLVF”) detected in 9DCIS was lost in 9IDC, although it was detected at low frequency in the IDC-adjacent tissue. This CDR3 chain can recognize the extracellular domain of HER2-precursor peptides based on the IEDB database (46). This patient is strongly ER+ and scored as HER2 1+ by immunohistochemistry.
DISCUSSION
In this study, we investigated the genomic profiles of matched pure DCIS and recurrent IDC from an immunologic perspective, which provided an invaluable snapshot of how both the tumor and immune system can collectively influence tumor evolution.
It remains unknown whether recurrent IDC arising many years after diagnosis of pure DCIS is genetically related to the initial lesion. Our study demonstrates this is the case in four of our six cases. Our data suggests that CNAs are early clonal events which are common to both DCIS and IDC, consistent with other studies supporting a linear evolutionary trajectory (3,4,12,56). However, our mutational spectra were vastly different between DCIS and IDC, with many coding mutations lost during this transition. Nevertheless, the presence of conserved mutations in genes including PIK3CA, TP53, CDH1 in matched samples of both DCIS-IDC and normal-DCIS suggest a fitness benefit of these mutations. Studies of matched synchronous cases have also shown DCIS or IDC-specific mutations in select cases and the expansion of a minor subclone in the progression from DCIS to IDC (9,57), suggesting the possibility of selection but also consistent with models of clonal expansion of a dominant clone under neutral drift.
We have observed a spectrum of immune hot and cold tumors as early as in DCIS, consistent with other immunofluorescence based studies pure DCIS (19,20,58,59). The increase of TILs in DCIS was shown to be associated with both activated and immunosuppressive gene signatures, suggesting that whilst the immune system may be recruited to the site of the lesion immunosuppressive pathways may be upregulated to avoid detection. Spatial analysis demonstrated similar metrics for TIL-immune mixing even after accounting for global tissue architecture and composition, highlighting that in our cohort TILs have similar capacities to interact with the tumor front in both DCIS and IDC but higher immune-immune mixing is characteristic of DCIS.
We have noted subtype specific differences in immune composition, specifically ER− tumors are more immune hot compared to ER+ tumors, in line with previous observations (11,58,60,61), and these differences could be attributed to subtype-specific genetic properties. Not only do TNBC and HER2+ have a higher average mutational burden compared to their ER+ counterparts (62), but are also enriched for CNAs in immune-related regions including the PD-L1 locus at 9p21 in a subset of TNBC (19) and chemokine cluster at 17q21 in HER2+ tumors (19,63). We additionally show an association between chromosomal loss in chemokine and interleukin loci with reduced immune signaling in ER− tumors (55). TP53 mutation, genomic instability and telomere crisis are more common in ER− DCIS and have been associated with higher TILs which could be recruited following activation of the cGAS-STING pathway mediated by cytosolic double-stranded DNA (47,60,61). We found that TP53 neoantigen presenting tumors were associated with both activated interferon and inflammatory wound healing macrophage signatures. Indeed, TP53 mutation has been associated with increased CD3+, CD4+, and Foxp3+ cells (61), suggesting that immunosuppressive pathways are also upregulated to counteract this anti-tumor response. Hence copy number aberrations at immune-rich loci may be a mechanism of immune evasion specifically in ER− tumors.
We have found that some well-characterized oncogenic mutations in genes including CDH1, and PIK3CA are present in both DCIS and IDC despite being predicted to present as neoantigens. In fact, the PIK3CA E545K mutation, alongside KRAS G12D and BRAF V600E, is one of the most commonly predicted neoantigens in invasive cancer independent of disease type, and studies are underway to determine the potential of these as public neoantigens (37). These data suggest that cells harboring these mutations may upregulate immune-modulatory mechanisms to avoid detection and elimination. Whilst secondary immune effects have not been historically studied, a number of recent studies have begun to dissect the immunological effect of specific oncogenic transformations. For example, TP53 loss through genome instability has been associated with higher TILs in breast cancer (47,60,61). PIK3CA E545K activation in combination with CDH1 loss results in an immunocompromised phenotype with increased macrophage and Treg infiltration in a mouse model of lobular breast cancer (64). Furthermore, PIK3CA mutation is associated with a mesenchymal, secretory phenotype (65), and can create an inflammatory environment through the secretion of cytokines. These studies have highlighted that mutations in driver genes including PIK3CA may directly modulate immune activity in order to evade immune detection and contribute to the immune cold phenotype common in ER+ cancers. Whilst functional studies are warranted to validate the immunological consequences of genetic events, these results present a starting point in evaluating the emergence of subtype specific differences from an immunogenomics perspective.
The profiling of DCIS has been particularly challenging due to small patient cohorts attributed to long latency periods between DCIS and IDC, low recurrence rates, as well technical challenges in molecular characterization due to low patient material and artifacts from FFPE preservation. Our cohort is particularly limited in size due to the added challenges in collecting matched cases of pure DCIS, and we have complete data for 2 cases. Nonetheless, we have showcased an integrative approach to profile this transition using complementary technologies and datasets that have yielded consistent molecular insights. Given that genetic drivers of this transition still remain largely unknown and there are strong relationships between subtype, genomic profiles and immune microenvironment, integrating data from different cohorts which may have been profiled with different technologies will be essential in uncovering the mechanisms of this transition.
In summary, we profiled matched pure DCIS and recurrent IDC samples to provide an invaluable snapshot of the tumor trajectory in the immune context. This concordant interrogation of both types of information provides a more comprehensive overview into the dynamic interplay between tumor cells and the microenvironment, thereby advancing the search for potential drivers of tumor progression and mechanisms of immune escape.
Supplementary Material
Implications.
We demonstrate that the in situ to invasive breast carcinoma evolutionary bottleneck is shaped by both tumor and immune cells.
ACKNOWLEDGEMENTS
We thank Drs. Deborah Dillon, Paul Spellman, Doris Tabassum, Michalina Janiszewska, and members of the Polyak lab for their critical reading of our manuscript. We thank Drs. Thomas O. McDonald, Hua-Jun Wu, and Daniel Temko for their useful suggestions with computational analyses. This work was supported by the National Cancer Institute R35CA197623 (K.P.), U01CA195469 (K.P. and J.G.), NIH-U24CA224331 (C.J.W), R50RCA211482 (S.A.S.), (U54CA209988) (K.P. and J.G.), DFCI Helen Gurley Brown Presidential Initiative Award (A.T.), and the Breast Cancer Research Foundation (K.P.).
Footnotes
Conflicts of Interest Statement: C.J.W. is a cofounder of Neon Therapeutics, and a member of its scientific advisory board. K.P. is a member of the scientific advisory board of Acrivon Therapeutics and Scorpion Therapeutics, has equity positions in Scorpion Therapeutics, and is a consultant to twoXAR Pharmaceuticals. S.A.S. had previously served as a paid consultant for Neon Therapeutics and owns equity in Agenus Inc., Agios Pharmaceuticals, Breakbio Corp., Bristol-Myers Squibb, and NewLink Genetics. J.W.G. has licensed technologies to Abbott Diagnostics and Danaher and has equity positions in PDX Pharmaceuticals and Convergent Genomics. J.W.G. serves as an advisor to New Leaf Ventures and KromaTid. J.W.G receives research funding or other support from Zeiss, ThermoFisher (FEI), Danaher (Cepheid), Micron Technology, Inc, Miltenyi Biotec, PDX Pharmaceuticals, and Quantitative Imaging (Qi).
ACCESSION CODES
RNA-Seq datasets have been deposited to GEO with accession number GSE144020.
REFERENCES
- 1.Cowell CF, Weigelt B, Sakr RA, Ng CK, Hicks J, King TA, et al. Progression from ductal carcinoma in situ to invasive breast cancer: revisited. Molecular oncology 2013;7:859–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elshof LE, Schaapveld M, Schmidt MK, Rutgers EJ, van Leeuwen FE, Wesseling J. Subsequent risk of ipsilateral and contralateral invasive breast cancer after treatment for ductal carcinoma in situ: incidence and the effect of radiotherapy in a population-based cohort of 10,090 women. Breast Cancer Res Treat 2016;159:553–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorringe KL, Hunter SM, Pang J-M, Opeskin K, Hill P, Rowley SM, et al. Copy number analysis of ductal carcinoma in situ with and without recurrence. Modern Pathology 2015;28:1174–84. [DOI] [PubMed] [Google Scholar]
- 4.Casasent AK, Schalck A, Gao R, Sei E, Long A, Pangburn W, et al. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell 2018;172:205–17 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abba MC, Gong T, Lu Y, Lee J, Zhong Y, Lacunza E, et al. A Molecular Portrait of High-Grade Ductal Carcinoma In Situ. Cancer Res 2015;75:3980–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vincent-Salomon A, Lucchesi C, Gruel N, Raynal V, Pierron G, Goudefroye R, et al. Integrated genomic and transcriptomic analysis of ductal carcinoma in situ of the breast. Clin Cancer Res 2008;14:1956–65. [DOI] [PubMed] [Google Scholar]
- 7.Pang JB, Savas P, Fellowes AP, Mir Arnau G, Kader T, Vedururu R, et al. Breast ductal carcinoma in situ carry mutational driver events representative of invasive breast cancer. Mod Pathol 2017;30:952–63. [DOI] [PubMed] [Google Scholar]
- 8.Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 2006;10:529–41. [DOI] [PubMed] [Google Scholar]
- 9.Pareja F, Brown DN, Lee JY, Da Cruz Paula A, Selenica P, Bi R, et al. Whole-Exome Sequencing Analysis of the Progression from Non-Low-Grade Ductal Carcinoma In Situ to Invasive Ductal Carcinoma. Clin Cancer Res 2020;26:3682–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hernandez L, Wilkerson PM, Lambros MB, Campion-Flora A, Rodrigues DN, Gauthier A, et al. Genomic and mutational profiling of ductal carcinomas in situ and matched adjacent invasive breast cancers reveals intra-tumour genetic heterogeneity and clonal selection. J Pathol 2012;227:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim SY, Jung SH, Kim MS, Baek IP, Lee SH, Kim TM, et al. Genomic differences between pure ductal carcinoma in situ and synchronous ductal carcinoma in situ with invasive breast cancer. Oncotarget 2015;6:7597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kroigard AB, Larsen MJ, Laenkholm AV, Knoop AS, Jensen JD, Bak M, et al. Clonal expansion and linear genome evolution through breast cancer progression from pre-invasive stages to asynchronous metastasis. Oncotarget 2015;6:5634–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 2004;6:17–32. [DOI] [PubMed] [Google Scholar]
- 14.Hu M, Yao J, Cai L, Bachman KE, van den Brule F, Velculescu V, et al. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet 2005;37:899–905. [DOI] [PubMed] [Google Scholar]
- 15.Vargas AC, Reed AEM, Waddell N, Lane A, Reid LE, Smart CE, et al. Gene expression profiling of tumour epithelial and stromal compartments during breast cancer progression. Breast Cancer Research and Treatment 2012;135:153–65. [DOI] [PubMed] [Google Scholar]
- 16.Lesurf R, Aure MR, Mork HH, Vitelli V, Oslo Breast Cancer Research C, Lundgren S, et al. Molecular Features of Subtype-Specific Progression from Ductal Carcinoma In Situ to Invasive Breast Cancer. Cell Rep 2016;16:1166–79. [DOI] [PubMed] [Google Scholar]
- 17.Kristensen VN, Vaske CJ, Ursini-Siegel J, Van Loo P, Nordgard SH, Sachidanandam R, et al. Integrated molecular profiles of invasive breast tumors and ductal carcinoma in situ (DCIS) reveal differential vascular and interleukin signaling. Proc Natl Acad Sci U S A 2012;109:2802–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerdes MJ, Gokmen-Polar Y, Sui Y, Pang AS, LaPlante N, Harris AL, et al. Single-cell heterogeneity in ductal carcinoma in situ of breast. Mod Pathol 2018;31:406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gil Del Alcazar CR, Huh SJ, Ekram MB, Trinh A, Liu LL, Beca F, et al. Immune Escape in Breast Cancer During In Situ to Invasive Carcinoma Transition. Cancer Discovery 2017;7:1098–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson E, Taube JM, Elwood H, Sharma R, Meeker A, Warzecha HN, et al. The immune microenvironment of breast ductal carcinoma in situ. Mod Pathol 2016;29:249–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campbell MJ, Baehner F, O’Meara T, Ojukwu E, Han B, Mukhtar R, et al. Characterizing the immune microenvironment in high-risk ductal carcinoma in situ of the breast. Breast Cancer Res Treat 2017;161:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gil Del Alcazar CR, Aleckovic M, Polyak K. Immune Escape during Breast Tumor Progression. Cancer Immunol Res 2020;8:422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eng J, Thibault G, Luoh SW, Gray JW, Chang YH, Chin K. Cyclic Multiplexed-Immunofluorescence (cmIF), a Highly Multiplexed Method for Single-Cell Analysis. Methods Mol Biol 2020;2055:521–62. [DOI] [PubMed] [Google Scholar]
- 26.Levine JH, Simonds EF, Bendall SC, Davis KL, Amir el AD, Tadmor MD, et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 2015;162:184–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012;28:1811–7. [DOI] [PubMed] [Google Scholar]
- 29.Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, et al. Oncotator: cancer variant annotation tool. Hum Mutat 2015;36:E2423–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D862–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep 2017;7:16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horn H Measurement of “Overlap” in Comparative Ecological Studies The American Naturalist 1966;100:419–24. [Google Scholar]
- 33.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Terfve C, Rose JC, Markowetz F. HTSanalyzeR: an R/Bioconductor package for integrated network analysis of high-throughput screens. Bioinformatics 2011;27:879–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011;27:1739–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity 2018;48:812–30 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol 2016;17:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Racle J, de Jonge K, Baumgaertner P, Speiser DE, Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 2012;40:W452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol 2015;33:1152–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J Immunol 2017;199:3360–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods 2015;12:380–1. [DOI] [PubMed] [Google Scholar]
- 46.Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, et al. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res 2019;47:D339–D43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chin K, de Solorzano CO, Knowles D, Jones A, Chou W, Rodriguez EG, et al. In situ analyses of genome instability in breast cancer. Nat Genet 2004;36:984–8. [DOI] [PubMed] [Google Scholar]
- 48.Yizhak K, Aguet F, Kim J, Hess JM, Kubler K, Grimsby J, et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 2019;364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hilson JB, Schnitt SJ, Collins LC. Phenotypic alterations in ductal carcinoma in situ-associated myoepithelial cells: biologic and diagnostic implications. Am J Surg Pathol 2009;33:227–32. [DOI] [PubMed] [Google Scholar]
- 50.Nagarajan D, McArdle SEB. Immune Landscape of Breast Cancers. Biomedicines 2018;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019;567:479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marin-Acevedo JA, Soyano AE, Dholaria B, Knutson KL, Lou Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J Hematol Oncol 2018;11:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cetintas VB, Batada NN. Is there a causal link between PTEN deficient tumors and immunosuppressive tumor microenvironment? J Transl Med 2020;18:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Loo LW, Wang Y, Flynn EM, Lund MJ, Bowles EJ, Buist DS, et al. Genome-wide copy number alterations in subtypes of invasive breast cancers in young white and African American women. Breast Cancer Res Treat 2011;127:297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martelotto LG, Baslan T, Kendall J, Geyer FC, Burke KA, Spraggon L, et al. Whole-genome single-cell copy number profiling from formalin-fixed paraffin-embedded samples. Nat Med 2017;23:376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sakr RA, Weigelt B, Chandarlapaty S, Andrade VP, Guerini-Rocco E, Giri D, et al. PI3K pathway activation in high-grade ductal carcinoma in situ--implications for progression to invasive breast carcinoma. Clin Cancer Res 2014;20:2326–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hussein MR, Hassan HI. Analysis of the mononuclear inflammatory cell infiltrate in the normal breast, benign proliferative breast disease, in situ and infiltrating ductal breast carcinomas: preliminary observations. J Clin Pathol 2006;59:972–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim M, Chung YR, Kim HJ, Woo JW, Ahn S, Park SY. Immune microenvironment in ductal carcinoma in situ: a comparison with invasive carcinoma of the breast. Breast Cancer Res 2020;22:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hendry S, Pang JB, Byrne DJ, Lakhani SR, Cummings MC, Campbell IG, et al. Relationship of the Breast Ductal Carcinoma In Situ Immune Microenvironment with Clinicopathological and Genetic Features. Clin Cancer Res 2017;23:5210–7. [DOI] [PubMed] [Google Scholar]
- 61.Toss MS, Abidi A, Lesche D, Joseph C, Mahale S, Saunders H, et al. The prognostic significance of immune microenvironment in breast ductal carcinoma in situ. Br J Cancer 2020;122:1496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.TCGA. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Safonov A, Jiang T, Bianchini G, Gyorffy B, Karn T, Hatzis C, et al. Immune Gene Expression Is Associated with Genomic Aberrations in Breast Cancer. Cancer Res 2017;77:3317–24. [DOI] [PubMed] [Google Scholar]
- 64.An Y, Adams JR, Hollern DP, Zhao A, Chang SG, Gams MS, et al. Cdh1 and Pik3ca Mutations Cooperate to Induce Immune-Related Invasive Lobular Carcinoma of the Breast. Cell Rep 2018;25:702–14 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chakrabarty A, Surendran S, Bhola NE, Mishra VS, Wani TH, Baghel KS, et al. The H1047R PIK3CA oncogene induces a senescence-like state, pleiotropy and acute HSP90 dependency in HER2+ mammary epithelial cells. Carcinogenesis 2019;40:1179–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.