

SUMMARY
The adipose tissue stroma is a rich source of molecularly distinct stem and progenitor cell populations with diverse functions in metabolic regulation, adipogenesis, and inflammation. The ontology of these populations and the mechanisms that govern their behaviors in response to stimuli such as overfeeding, however, are unclear. Here we show that the developmental fates and functional properties of adipose PDGFRβ+ progenitor subpopulations are tightly regulated by mitochondrial metabolism. Reducing the mitochondrial β-oxidative capacity of PDGFRβ+ cells via inducible expression of MitoNEET drives a pro-inflammatory phenotype in adipose progenitors and alters lineage commitment. Furthermore, disrupting mitochondrial function in PDGFRβ+ cells rapidly induces alterations in immune cell composition in lean mice and impacts expansion of adipose tissue in diet-induced obesity. The adverse effects on adipose tissue remodeling can be reversed by restoring mitochondrial activity in progenitors, suggesting therapeutic potential for targeting energy metabolism in these cells.
Keywords: Mitochondria, Metabolism, Stem cells, Adipocyte
eTOC
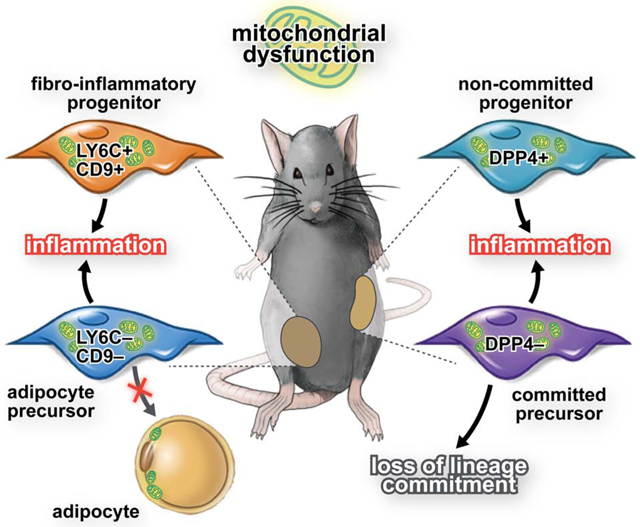
Factors controlling the degree of lineage commitment of PDGFRβ+ subpopulations are poorly defined. Here, Joffin et al. demonstrate that mitochondrial activity determines the cellular identity of adipocyte precursors and lineage commitment and pinpoint PdgfRβ+ cells as key regulators of inflammation and adipose tissue remodeling during caloric excess.
Graphical Abstract
Introduction
White adipose tissue (WAT) represents the principal site for energy storage in mammals. The absence of WAT (i.e. lipodystrophy) or dysfunctional WAT leads to deleterious ectopic lipid accumulation in the liver and other tissues, frequently resulting in cardiometabolic disease. As such, understanding the mechanisms governing the establishment and maintenance of the adipose tissue lineage remain a high priority.
In the setting of caloric excess, WAT expands its mass due to the enlargement of adipocyte size (hypertrophy) or by the recruitment of new adipocytes (hyperplasia) (Hirsch and Han, 1969). Hyperplastic expansion requires the recruitment of adipocyte precursor cells (APCs), a process referred to as adipogenesis.
APCs that contribute to the hyperplastic expansion of WAT in obese mice express the perivascular cell marker, platelet-derived growth factor receptor b (Pdgfrb), and are enriched in the expression of Pparg, Zfp423, and other pro-adipogenic factors (Gupta et al., 2012). The ability of these cells to undergo adipogenesis in the setting of HFD-induced obesity is critical for healthy hyperplastic expansion of WAT (Vishvanath et al., 2016).. Hepler et al. identified functionally distinct subpopulations of perivascular cells in eWAT of adult mice (Hepler et al., 2018). The LY6C- CD9- subpopulation of PDGFRβ+ cells in eWAT represent APCs, whereas LY6C+ PDGFRβ+ cells represent a distinct subpopulation of cells termed, “fibro-inflammatory progenitors” (“FIPs”). FIPs lack adipogenic capacity and instead have the ability to activate a pro-inflammatory gene expression program triggered by HFD feeding (Shan et al., 2020). In the accompanying manuscript, Shao et al. describe the functional heterogeneity of PDGFRβ+ cells in the inguinal WAT (iWAT) depot. in line with, previous studies (Burl et al., 2018; Merrick et al., 2019). Importantly, how the adipogenic vs. pro-inflammatory phenotypes of PDGFRβ+ cells are regulated is currently unknown. Moreover, factors controlling the degree of lineage commitment of PDGFRβ+ subpopulations are poorly defined. The data presented here sheds light on the mechanisms that determine the relative abundance of these subpopulations.
Mitochondrial activity and energy metabolism at large play a critical role in cell fate identity and plasticity in various cell types, such as immune cells and macrophages. Adipogenesis imposes a high demand on cellular energy levels. Thus, differentiating cells enhance the oxidative phosphorylation system (OXPHOS) (Choi et al., 2016; Michalek et al., 2011; Pearce et al., 2009). The “stemness” of cells is conventionally characterized by low mitochondrial activity and a relatively underdeveloped mitochondrial network (Facucho-Oliveira and St John, 2009; Lonergan et al., 2007). Therefore, a shift from generally glycolytic state to more oxidative metabolism is crucial during adipocyte differentiation from precursor cells (Chen et al., 2015).
In this study, we set out to explore the metabolic properties of functionally discrete adipose tissue progenitor subpopulations in distinct WAT depots. Using an inducible genetic mouse model of altered mitochondrial activity specifically in PDGFRβ+ cells, we demonstrate that the developmental fate and cellular function of adipose progenitors can be determined by manipulating their mitochondrial function. Moreover, mitochondrial dysfunction of PDGFRβ+ cells in vivo has rapid effects on the degree of inflammation and expansion of adipose tissue. These deleterious effects on adipose tissue remodeling can be reversed by restoration of mitochondrial function in progenitors, highlighting the significant therapeutic potential for targeting energy metabolism in these cells.
Results
eWAT FIPs and APCs are metabolically distinct stromal cell subpopulations
Our initial question was whether there is a fundamental difference between FIPs and APCs with respect to their metabolic preferences. FIPs and APCs can be separated on the basis of CD9 and LY6C expression (Fig. S1A). Gene expression analysis of freshly isolated cells validated our cell purification approach. In comparison to APCs, FIPs were enriched in the expression of Ly6c1 and numerous transcripts encoding inflammatory genes (e.g. Il-6, Ccl2) and fibrogenic genes (Col1a1, Col3a1, Col5a1, Fn1, Ctgf) (Fig. S1B). Isolated APCs were enriched in the expression of genes associated with adipogenesis (e.g. Zfp423, Pparg2, Cebpa, Cd36) (Fig. S1B). These results are consistent with prior results indicating that APCs have intrinsic adipogenic potential whereas FIPs represent a fibro-inflammatory subpopulation of PDGFRβ+ cells in WAT (Hepler et al., 2018). We observed that the total content of mitochondrial DNA (mtDNA) was the same in both populations (Fig. S1C); however, mitochondrial and metabolic gene expression was notably different. mRNA levels of Tfam were higher in APCs than in FIPs (Fig. S1D). Interestingly, APCs have higher mRNA expression of ATP6 (Fig. S1E) but have lower mRNA expression for Cpt1b and Vhl, which are involved in β-oxidation (Fig. S1F). Glut1, Glut4, Pk, Pdk1 and Ldha are higher in APCs suggesting a glycolytic preference compared to FIPs (Fig. S1G).
To investigate further, we compared mitochondrial respiratory and glycolytic rates between FIPs and APCs. We seeded both subpopulations at confluency (80,000 cells). This was valided by confirming similar protein content between FIPs and APCs in the wells of the assay plates (Fig. S2A). Notably, FIPs have a higher mitochondrial respiration rate than APCs (Fig. S2B–C). This metabolic difference is likely due to the function of mitochondria and not due to mitochondria number, as there was no differences in mtDNA content between the two subpopulations (Fig. S1C). The glycolytic rate was comparable between the two (Fig. S2D); however, during a glycolytic stress test when pyruvate is lacking, FIPs have higher glycolytic capacity than APCs (Fig. S2E–F). The two subpopulations respond differently to a metabolic challenge that mimics a high-fat diet (HFD) by exposing the cells to a fatty acid overload in vitro. Both FIPs and APCs displayed enhanced oxidative and glycolytic rates in response to BSA-palmitate pretreatment; however, metabolic rates remained lower in APCs compared to that observed in FIPs (Fig. S2G–H, Fig. S2I–J). We further evaluated the cellular metabolic responses of FIPs and APCs to short-term HFD feeding in vivo. FIPs and APCs were sorted by FACS from the eWAT of wild type mice fed either control chow diet (CD) or HFD for 3 days. FIPs exhibited higher oxidative and glycolytic rates, similar to what was observed in in vitro following BSA-palmitate treatment (Fig. S2K–L, Fig. S2M–N). We observed a slight increase in oxidative metabolism in APCs, albeit these differences did not reach statistical significance. On the other hand, APCs responded to HFD feeding by robustly increasing glycolytic metabolism (Fig. S2M–N). Together, these data indicate that FIPs and APCs within adult eWAT represent metabolically distinct stromal cell subpopulations.
Genetic reduction of mitochondrial activity impacts the fate and function of eWAT FIPs and APCs in vitro.
The notable differences in metabolic activity between FIPs and APCs raises the question of how the mitochondrial activity of these stromal subpopulations affects their functional properties. To address this, we derived an inducible transgenic model in which the mitochondrial activity of PDGFRβ+ cells can be suppressed in a doxycycline-inducible manner through the expression of MitoNEET. MitoNEET is an iron-containing outer mitochondrial membrane protein that regulates oxidative capacity (Kusminski et al., 2012; Kusminski et al., 2014).This bi-transgenic “Tet-On” model (herein, MitoNEETTG mice) consists of the previously described PdgfrbrtTA allele (Vishvanath et al., 2016) and the TRE-MitoNEET allele which expresses murine Cisd1 (MitoNEET) (Kusminski et al., 2016; Kusminski et al., 2012; Kusminski et al., 2014) under the control of the tet-responsive element (Fig. S3A). The addition of doxycycline results in increased mRNA levels of Cisd1 in the stromal vascular fraction, but not in mature adipocytes, from WAT depots (Fig. S3B).
The addition of doxycycline to cultures of FIPs and APCs for 24 hours led to a comparable degree of Cisd1 overexpression and an increase in protein levels (Fig. S3C–E). Neither doxycycline nor MitoNEET overexpression is cytotoxic for FIPs or APCs (Fig. S3F–G). Using this validated model, we measured in vitro metabolism in both PDGFRβ+ subpopulations. We treated FIPs and APCs at confluency for 24 hours with doxycycline and controlled for the same protein content per well (Fig. S3H–I). We then measured mitochondrial respiration and glycolysis. Consistent with the previously reported function of MitoNEET (Kusminski et al., 2016; Kusminski et al., 2012; Kusminski et al., 2014), we observed a dramatic decrease in the mitochondrial respiration rate in FIPs and APCs upon transgene induction in these cells (Fig. 1A–D). This decrease is independent of mitochondria number as mtDNA content remained unchanged in FIPs and APCs (Fig. 1E–F). The decrease in mitochondrial respiration is mainly due to the reduction of electron transport chain complexes in MitoNEET-expressing cells, particularly complexes enriched in iron-sulfur (Fe-S) clusters, such as complex I, complex II and complex III (Fig.1G–H). Interestingly, MitoNEET-expressing FIPs but not APCs compensate for the decrease in mitochondrial oxidative metabolism by increasing glycolytic rate (Fig. 1I–J). It is possible that the shift to increased anaerobic glycolysis is the result of increased cytosolic pyruvate due to diminished mitochondrial function and enhanced LDH acitivity via mass action; however, no differences in maximal lactate dehydrogenase (LDH) activity were observed in isolated cells (Fig. 1K). These data suggest that both subpopulations are sensitive to the disruption of mitochondrial function, but only FIPs have the capacity to activate an alternative energy-generating pathway. Interestingly, the MitoNEET-induced switch from oxidative to glycolytic metabolism does not alter FIP proliferation compared to control FIPs (Fig 1L). However, MitoNEET-expressing FIPs had higher levels of pro-inflammatory and fibrogenic genes compared to control FIPs. In particular, levels of Il-6, Tnfa, Il-18 and Ccl2, were robustly elevated in response to MitoNEET overexpression in FIPs (Fig. 1M). Lowering oxidative metabolism in APCs with MitoNEET adversely affected the ability of the cells to proliferate long-term in vitro (Fig. 1N). Moreover, their ability to undergo spontaneous differentiation into mature adipocytes was also significantly diminished (Fig. 1O–Q). Interestingly, the mitochondrial dysfunction in APCs led to increased expression of inflammation-related genes, including Il-6, Ccl2, Tnfa, Tgfb1, and Tgfb2 (Fig. 1R). Collectively, these data highlight the differential response of FIPs and APCs to perturbation of mitochondrial function, and reveal the importance of proper mitochondrial metabolism in controlling the unique functional properties of these stromal subpopulations.
Figure 1. Inducible mitochondrial dysfunction in visceral adipose tissue progenitors (FIPs and APCs) promotes inflammation while decreasing adipogenic capacity in vitro.
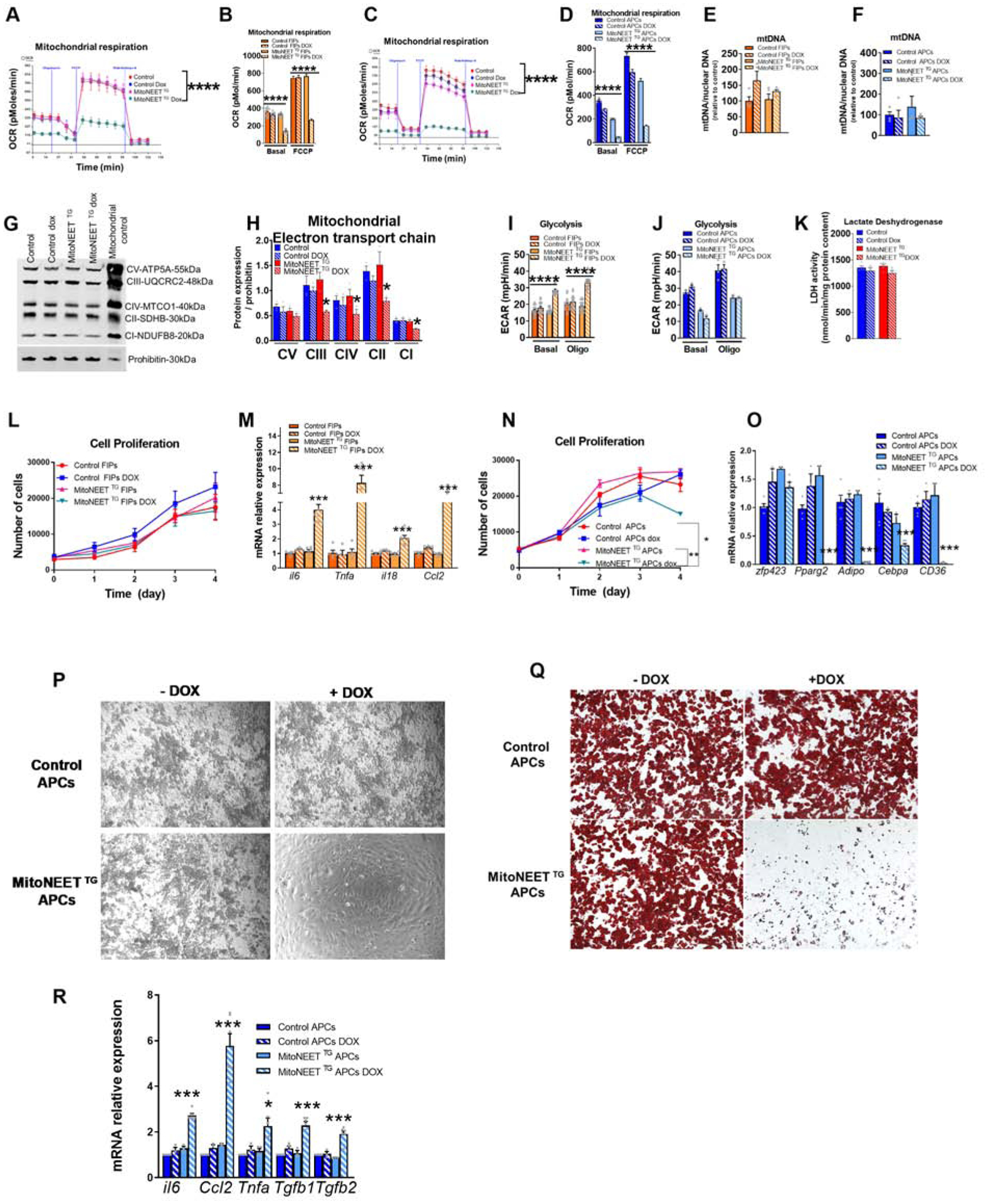
Determination of mitochondrial function in FIPs and APCs using PdgfRβ-rtTA mice (controls) and PdgfRβ-rtTA × TRE-MitoNEET mice (MitoNEET TG). FIPs and APCs were sorted from control or MitoNEET TG (eWAT) (A-K,M,O,P,Q,R) FIPs and APCs were wuth 4.4 μM doxycycline for 24h. (A-B) Oxygen consumption rate of FIPs (n=5–15) or APCs cells (n=3) in pmol/min. (C,D). (E,F) mtDNA content of FIPs (n=4–5) (E) and APCs (F) (n=4–5) ratio between mtDNA and nuclear DNA relative to control. (G-H) Western blot representation and quantification of mitochondrial electron transport chain complexes of PDGFRβ+ cells (n=3). (I) Glycolysis rate of FIPs (n=18) (J) and APCs cells (n=3) (K) Lactate dehydrogenase activity of PDGFRβ + cells (n=9) in nmol/min/mg protein content. (L) Cell proliferation of FIPs treated with or without 4.4 uM doxycycline (n=4). (M) Gene expression profile of control or MitoNEET TG FIPs (n=6). (N) Cell proliferation of APCs treated with or without 4.4 uM doxycycline (n=3) (O) Gene expression profiles of adipocyte markers after 6 days of differentiation of APCs (n=3–6). (P) Microscopic images of APCs from control or MitoNEET TG fat pads differentiated for 6 days doxycycline pretreatment (Q) Oil Red O staining of APCs differentiated for 6 days. (R) Gene expression of inflammatory markers of APCs differentiated into adipocyte for 6 days (n=3–6).Significance in (A,C,L,N) was calcultated using a 2-way Anova with Tukey’s post-test for multiple comparisons. Significance in (B,D,E,F,H,I,J,K,M,O,R) was calculated using a Kruskal-Wallis (One-Way Anova) test with Dunn’s post-test for multiple comparisons. Error bars represent mean ± S.E.M. * (P<0.05), ** (p<0.01), *** (p<0.0001), **** (p<0.00001).
Alteration of mitochondrial activity in PDGFRβ+ cells leads to reversible macrophage activation in eWAT
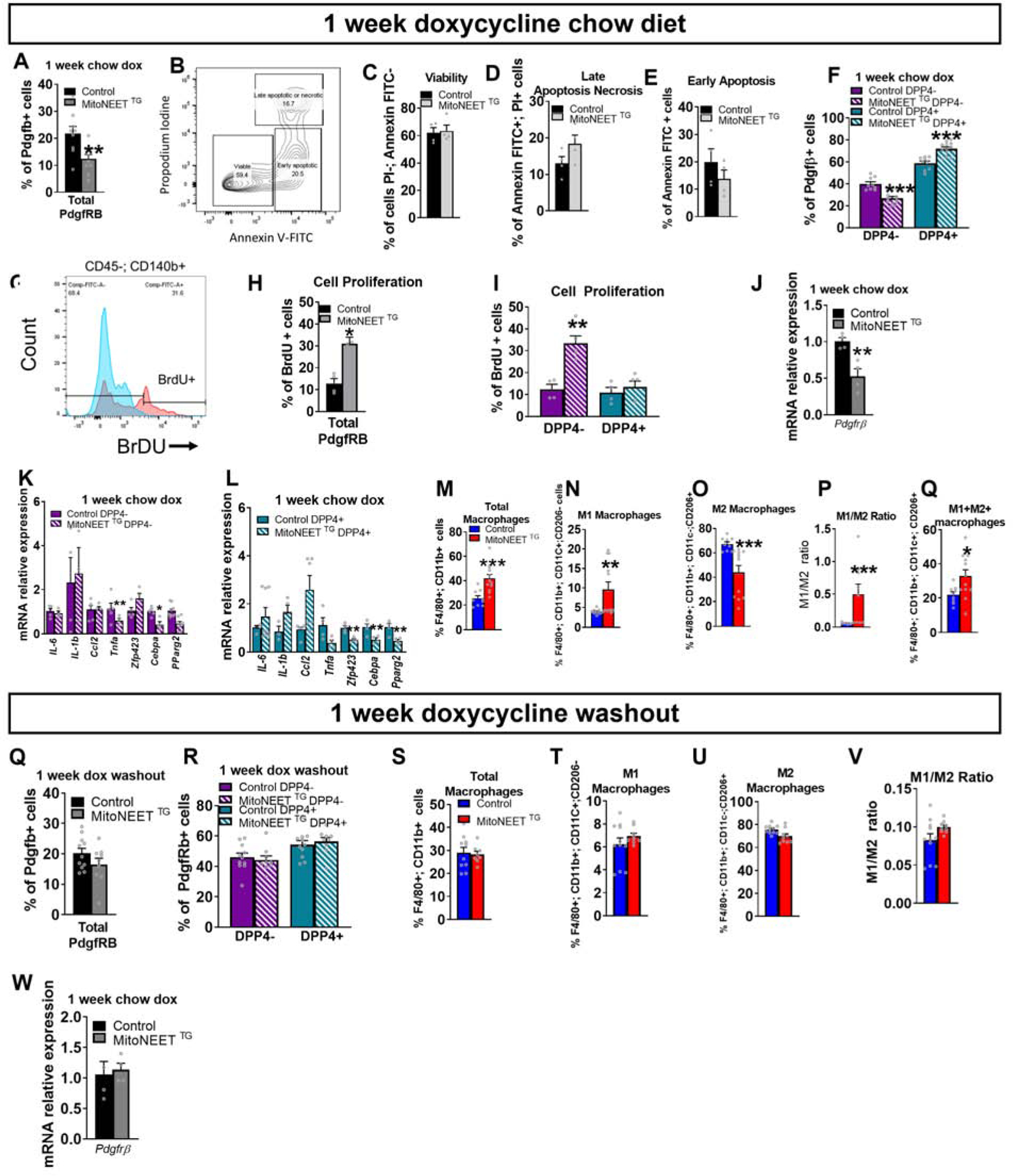
These results prompted us to assess whether this phenomenon occurs in vivo, and whether this impacts the activity of adipose tissue macrophages (ATMs). Control (PdgfrbrtTA or TRE-MitoNEET single transgenics) and MitoNEETTG mice were maintained for 1 week on chow diet containing doxycycline in order to acutely induce MitoNEET mRNA and protein expression in PDGFRβ+ cells (Fig. S3J–L). We then analyzed the frequency of the PDGFRβ+ subpopulations in eWAT by flow cytometry. Total PDGFRβ+ population is decreased upon MitoNEET overexpression (Fig. 2A). This decrease is independent of cell death as PDGFRβ+ cell viability were not affected by mitochondrial dysfunction (Fig. 2B–E). In control animals, ~20% of gated eWAT PDGFRβ+ cells can be clearly defined as APCs (LY6C− CD9−), whereas ~70% of cells in this pool represent FIPs (Hepler et al., 2018). In MitoNEETTG mice, we observed a relative shift in the frequencies of these two subpopulations in eWAT (Fig. 2F). PDGFRβ+ cells overexpressing MitoNEET displayed a similar BrdU incorporation compared to controls (Figure 2G–I). Thus, mitochondrial dysfunction does not impact proliferation of either APCs or FIPs (Fig. 2I). We investigated further to explain the decrease in total PDGFRβ+ cells upon MitoNEET overexpression. MitoNEET overexpression significantly decreased expression of Pdgfrβ gene (Fig. 2J). This result suggests that mural cells lose their identity in response to mitochondrial dysfunction. In MitoNEETTG mice, the reduction of mitochondrial function in FIPs is accompanied by an increase in expression of pro-inflammatory genes, such as Il-6, Il1-β, and Il-18 (Fig. 2K), as seen in vitro. Moreover, mRNA levels of Il-6 are induced by nearly 1,000-fold in FIPs of MitoNEETTG mice (Fig. 2K). These results show that in vivo, a reduction in mitochondrial function exacerbates inflammation in FIPs by increasing the number of these cells, as well as by promoting their inflammatory properties. In APCs fron MitoNEETTG mice, mitochondrial dysfunction led to a significant suppression of pro-adipogenic genes, including Zfp423, Pparg2, and Cebpa, while the expression Tnfa and Ccl2 are markedly elevated (Fig. 2L). These data are in line with the in vitro studies indicating that MitoNEET overexpression in APCs leads to a loss of adipogenic capacity and the activation of a pro-inflammatory gene program.
Figure 2: Mitochondrial dysregulation of FIPs and APCs in vivo increases FIPs and promotes inflammatory gene expression in visceral adipose tissue.
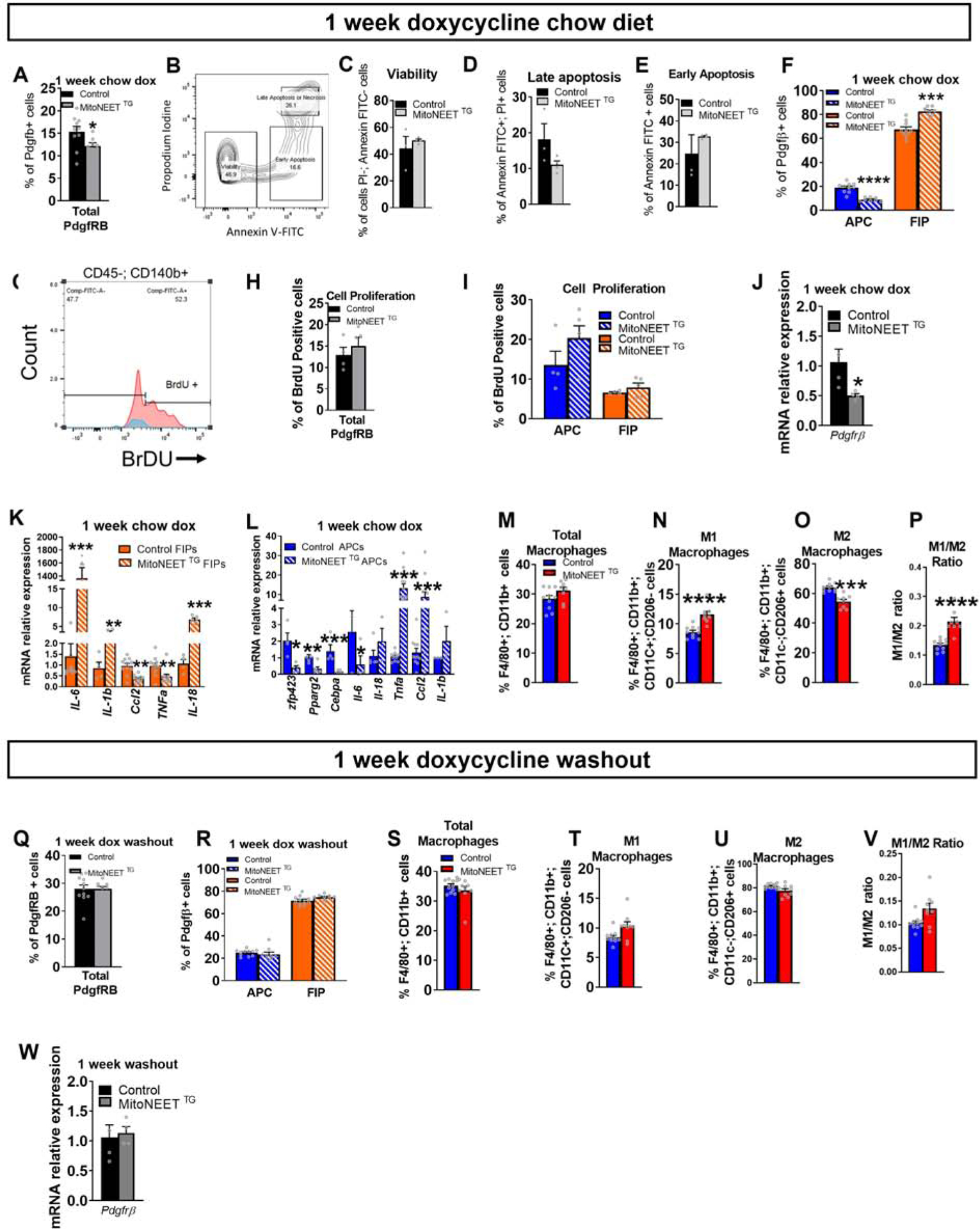
In vivo disruption of mitochondrial function in FIPs and APCs using control or PdgfRβ-rtTA × TRE-MitoNEET (MitoNEETTG) mice fed 1 week with chow diet containing 600 mg/kg of doxycycline. (A) Frequency of PdgfRβ+ cells in control or MitoNEET TG eWAT (n=8–10). (B) Gating strategy of Annexin-V and propodium iodine. (C) Viability of all PdgfRβ+ cells (n=3–4) (D) Late apoptosis or necrosis (n=3–4) (E) Early apoptosis of all PdgfRβ+ cells (n=3–4) (F) Frequency of FIPs and APCs from PdgfRβ+ cells (n=8–10) (G) Gating for BrdU-positive PdgfRβ+ cells (H) Quantification of BrdU incorporation into PdgfRβ+ cells (n=4–5) (I) Quantification of BrdU incorporation into FIPs or APCs (n=4–5). (J) Gene expression of Pdgfrb marker in PdgfRβ+ cells (n=4) (K) Gene expression of inflammatory markers in FIPs (n=4–11). (L) Gene expression of adipogenic and inflammatory markers in APCs (n=3–6). (M-P) eWAT macrophage frequencies (n=8–10). (M) Total macrophages. (N) M1 macrophages. (O) M2 macrophages (P) Ratio between M1 and M2 macrophages. (Q-W) Control mice or MitoNEET TG mice were fed 1 week with chow diet containing 600 mg/kg of doxycycline and fed one more week with chow diet without doxycycline (“doxycycline washout”). (Q) Frequency of the total PdgfRβ+ cells. (R) Frequency of FIPs and APCs from total PdgfRβ+ cells (S-V) eWAT macrophage frequencies. (S) Total macrophages (T) M1 macrophages. (U) M2 macrophages (V) Ratio of M1 vs M2 macrophages (W) Gene expression of Pdgfrb marker in PdgfRβ+ cells. Data are presented in mRNA relative to control cells. Significance between control and MitoNEET TG was calculated using a two-tailed student’s t-test. Error bars represent mean ± S.E.M. * (P<0.05), ** (p<0.01), *** (p<0.0001), **** (p<0.00001).
The potent increase in expression of pro-inflammatory cytokines in PDGFRβ+ cells of MitoNEETTG mice raises the question as to whether these changes have an impact on the activation of adipose-tissue macrophages (ATMs) in this depot. One week of MitoNEET overexpression in chow-fed mice did not change the total macrophage content in eWAT (Fig. 2M). However, it led to a change in the relative proportion of classically activated vs. alternatively activated macrophages (Fig. 2N–P). eWAT depots of MitoNEETTG mice contained more proinflammatory ATMs than control mice, with a corresponding reduction in the numbers of anti-inflammatory ATMs.
The data above suggests a model in which altered mitochondrial metabolism activates the pro-inflammatory phenotype of eWAT PDGFRβ+ stromal cells. This in turn impacts local adipose tissue macrophage activation. To test this further, we asked whether these phenotypes were reversible, upon cessation of transgene expression. Control and MitoNEETTG mice were maintained for 1 week on chow dox diet to induce transgene expression and the associated phenotype. Subsequently, animals were maintained on standard chow diet (without dox) for an additional week prior to analysis. Remarkably, one week after turning off MitoNEET expression, the frequency of total PDGFRβ+ cells, FIPs and APCs in eWAT of MitoNEETTG mice returned to levels comparable to control Fig. 2Q–R). Moreover, the frequency of both M1 and M2 macrophages in MitoNEETTG mice also returned to levels found in control (Fig. 2S–V). Interestingly, Pdgfrβ mRNA levels have been restored upon cessation of MitoNEET overexpression (Fig. 3W). Taken together, these data highlight the sensitivity of ATMs to the frequency and pro-inflammatory state of PDGFRβ+ stromal cells.
Metabolic properties of iWAT APC subpopulations
In the accompanying manuscript, Shao et al. define two molecularly distinct subpopulations of PDGFRβ+ cells within the iWAT depots of adult mice that can be isolated by FACS on the basis of DPP4 expression. These two subpopulations of PDGFRβ+ cells largely overlap with the DPP4+ and DPP4− progenitor populations described by Merrick et al. demonstrated that DPP4+ cells, but not DPP4− cells, have the capacity to differentiate into multiple mesenchymal lineages in vitro (Merrick et al., 2019). Given that DPP4− PDGFRβ+ cells are enriched in the expression of Pparg2 and other markers of lineage commitment, these cells likely represent committed APCs, or “preadipocytes”. On the other hand, DPP4+ PDGFRβ+ cells represent the less committed APC population.
We explored the metabolic properties of these iWAT stromal cell subpopulations that reflect different stages of lineage commitment. We isolated DPP4+ PDGFRβ+ cells and DPP4− PDGFRβ+ cells from iWAT of wild type mice using FACS (Fig. S4A), and compared their gene expression pattern to one another. We confirmed that DPP4+ cells are enriched in Dpp4 mRNA when compared to DPP4− cells (Fig. S4B). DPP4+ cells were also enriched in the expression of pro-inflammatory genes, including Ccl2, Cxcl2, and Il-6 (Fig. S4B). DPP4− APCs were enriched in the expression of Pparg2, Cebpα, and Cd36, consistent with their commitment to the adipocyte lineage (Fig. S4B). Further analysis revealed that DPP4− express lower levels of the mitochondrial marker Nrf1 than DPP4+ cells; however, mtDNA content was comparable in both populations (Fig. S4C,D). Interestingly, DPP4− express reduced levels of mRNA for Cox1, Cox3, Cox4, Cox5b, Cad, and Dhohd, in comparison to DPP4+ (Fig. S4E). Moreover, DPP4− APCs also express reduced levels of Acadvl, Vhl, Pk and Pdk1, but higher levels of Glut4 than DPP4+cells (Fig. S4F–G). We tested whether these gene expression patterns reflect functional differences in rates of oxidative metabolism and glycolytic rates between the two APC subpopulations. We seeded both population at confluency as valided by similar protein content between subpopulations (Fig. S5A). We observed that both mitochondrial respiration and the glycolytic rates in presence of pyruvate are comparable between the two subpopulations at baseline (Fig. S5B–D) and mitochondrial content was similar (Fig. S4D). However, DPP4+ cells show a higher glycolytic rate without pyruvate compared to DPP4− cells during exposure to a glycolytic stress (Fig. S5E–F).
In response to a 4h pre-treatment with BSA-palmitate, mitochondrial respiration was unchanged in DPP4+ and DPP4− cells (Fig. S5G–H). However, it blunted the glycolytic rate in DPP4− cells, whereas no effect was observed in DPP4+ cells (Fig. S5I–J). Interestingly, glycolysis was increased in DPP4+ compared to DPP4− independently of treatment (Fig. S5I).
Short-term HFD-feeding (3 days) had a different effect than palmitate alone on the metabolic responses of DPP4+ and DPP4− APCs. Seahorse analysis was performed on isolated cell populations from control or HFD mice mice, and revealed that DPP4+ APCs showed a significant increase in maximal mitochondrial respiration (FCCP), but not in glycolysis (Fig. S5K–N). Acute HFD feeding also increases maximal mitochondrial respiration (FCCP) in DPP4− APCs, and the glycolytic rate is increased upon exposing the cells to oligomycin (Fig. S5M–N). Interestingly, DPP4+ have a higher glycolytic rate compared to DPP4− after a control diet (Fig.S5M). Altogether, these data highlight the shared and distinct metabolic properties of iWAT APC subpopulations in vitro and in vivo.
Genetic reduction of mitochondrial activity impacts lineage commitment of iWAT APC subpopulations in vitro.
We examined the functional consequences of altered mitochondrial activity of iWAT DPP4+ and DPP4− APCs, using the same approach used to study eWAT FIPs and APCs. In vitro, 24 hours of doxycyline treatment of cultured DPP4+ and DPP4− APCs lead to overexpression of Cisd1 (Fig. S3M–N). No differences in the viability of DPP4+ and DPP4− from either control mice or MitoNEET TG mice were observed (Fig. S3O–P). We seeded cells at confluency, yielding the same amount of protein in each well (Fig S3Q–R). As expected, MitoNEET overexpression led to a decrease in mitochondrial respiration in both subpopulations (Fig. 3A–D). As previously shown for FIPs and APCs, the reduction in mitochondrial function was not due to a decrease in mitochondrial number as mitochondrial content was similar between groups (Fig. 3E,F). However, the decreases in complexes III, II, and I suggest a direct dysfunction of the electron transport chain (Fig. 3G–H). Interestingly, the decrease in mitochondrial respiration was less pronounced than what we observed in FIPs or APCs (Fig 1 A and B). While the mitochondrial respiration rates in FIPs are decreased by ~60% and by more than 70% in APCs, mitochondrial respiration rates in DPP4+ cells are only decreased by ~30% and by ~40% in DPP4− cells. Importantly, DPP4+ APCs effectively compensate the lack of mitochondrial function with an increase in glycolysis, while DPP4− APCs are unable to compensate (Fig. 3I–J). As previously shown for eWAT, LDH activity was similar in iWAT mural cells (Fig. 3K). Importantly, the decrease in mitochondrial respiration and the shift towards glycolytic metabolism did not affect the cell proliferation of either DPP4+ or DPP4− (Fig. 3L–M).
Figure 3: Inducible mitochondrial dysfunction in vitro in subcutaneous progenitors promotes inflammatory function while decreasing adipogenic capacity.
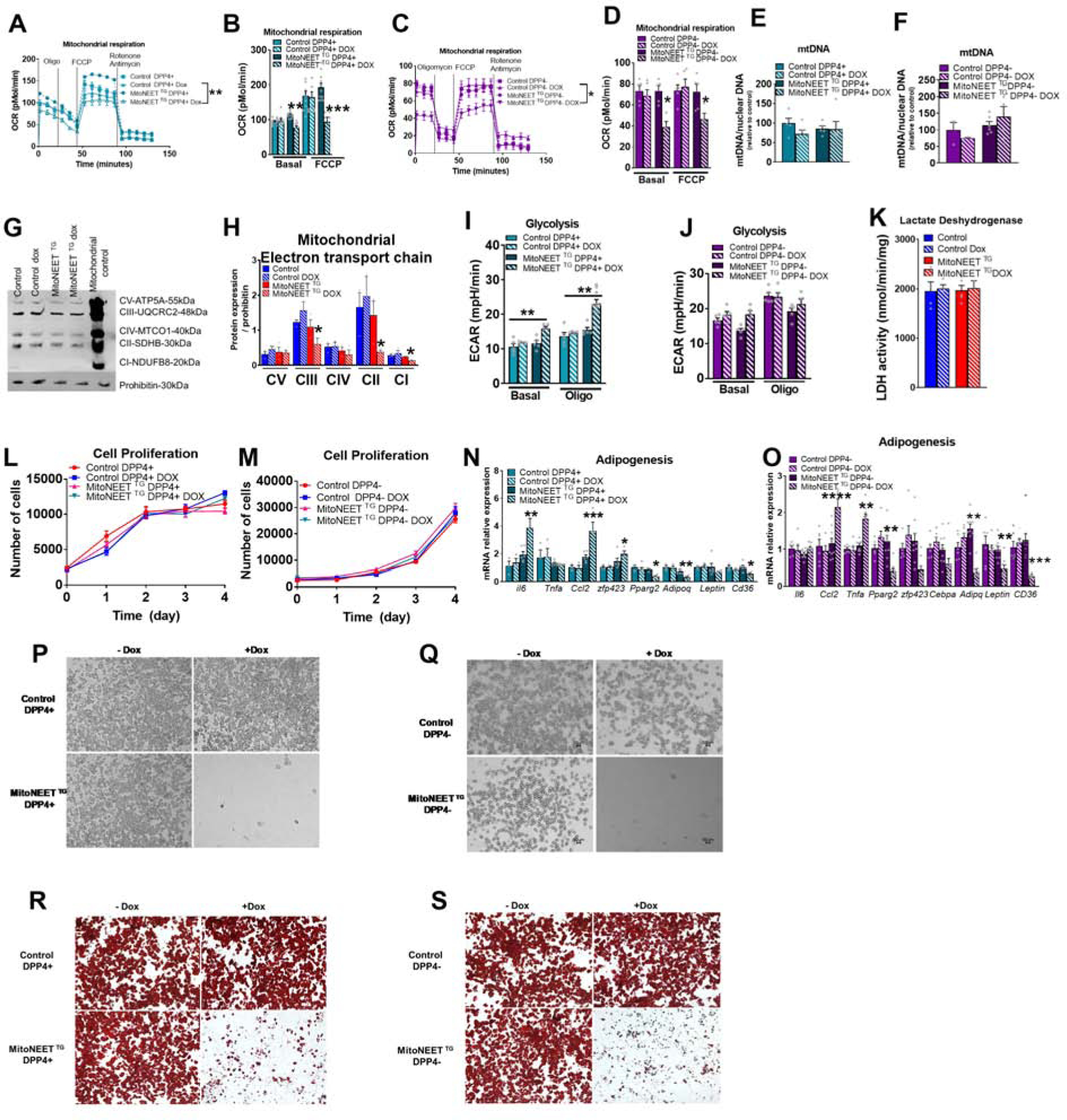
Disruption of mitochondrial function in DPP4+ and DPP4− using PdgfRβ-rtTA (control) and PdgfRβ-rtTA TRE-MitoNEET fat pads (MitoNEETTG). DPP4+ and DPP4− cells were sorted from control or MitoNEETTG fat pads. (A-K) DPP4+ and DPP4− were with 4.4μM doxycycline for 24h. (A-B) Oxygen consumption rate of control or MitoNEETTG DPP4+ cells (n=10–12) (C-D) Oxygen consumption rate control or MitoNEET TG DPP4− cells (n=5). (E) mtDNA content of DPP4+ cells (n=5). (F) mtDNA content of DPP4− cells (n=3–6). (G) Representative and quantification Western blot of electron transport chain (n=3). (I) Glycolysis rate of control or MitoNEETTG DPP4+ (n=5–10) (J) and DPP4− cells (n=5) (K) Lactate dehydrogenase activity of PDGFRβ+ cells (n=9) (L) Cell proliferation of DPP4+ cells treated with 4.4uM doxycycline (n=6). (M) Cell proliferation of DPP4− cells treated with 4.4uM doxycycline (n=6) (N-O) Gene expression of control or MitoNEETTG DPP4+ (N) or DPP4− cells (O) cells pretreated with doxyxycline for 24h and differentiated for 8 days (n=3–6) (n=6–9). (P) Microscopic images of differentiated DPP4+ cells pretreated with 4.4 uM doxyxycline for 24h. (Q) Microscopic images of day 8 differentiated DPP4− cells from control or MitoNEET TG fat pads pretreated with 4.4 uM doxyxycline for 24h and differentiated for 8 days. (R) Oil Red O staining of DPP4+ and (S) DPP4− cells after 8 days of differentiation. Significance in (A,C,L,M) was calculated using a 2-way Anova with Tukey’s post-test for multiple comparisons. Significance in (B,D,E,F,H,I,J,K,N,O) was calculated using a Kruskal-Wallis (One-Way Anova) test with Dunn’s post-test for multiple comparisons. Error bars represent mean ± S.E.M. * (p<0.05), ** (p<0.01), *** (p<0.0001), **** (p<0.00001).
DPP4+ and DPP4− APC populations are both highly adipogenic in vitro. DPP4+, but not DPP4− APCs, exhibit multilineage potential. Additionally, DPP4+ cells can differentiate into osteoblasts in culture (Merrick et al., 2019). In vitro adipocyte differentiation assays revealed that both DPP4+ and DPP4− APCs lose their ability to undergo adipogenesis when MitoNEET is transiently overexpressed prior to the induction of differentiation (Fig. 3N–S). In contrast, the metabolic shift induced by MitoNEET in DPP4+ APCs lead to an increase in osteogenic differentiation potential, reflected by the increased expression of osteoblast markers, such as Cbfa1, Bglap2, Alpl, iBSP and Osx, and calcium deposit following the induction of osteogenesis (Fig. S6A–B). Moreover, the decrease in mitochondrial respiration in DPP4− APCs rendered the cells competent to undergo osteoblast differentiation (Fig. S6C–D). These results are very important, because they indicate that mitochondrial activity greatly influences mesenchymal lineage determination: Reduced mitochondrial oxidative capacity reprograms DPP4− APCs to cell less committed towards adipogenesis.
Altered mitochondrial activity in PDGFRβ+ cells impacts macrophage homeostasis in iWAT
Given the potential pro-inflammatory properties of iWAT PDGFRβ+ cells (particularly the DPP4+ APCs), we wondered whether lowering mitochondrial function in PDGFRβ+ cells can elicit similar effects in vivo. One week of chow dox treatment in MitoNEETTG mice led to a similar level of Cisd1 overexpression and a two-fold increase in MitoNEET protein expression in DPP4+ and DPP4− APCs (Fig. S3S,T). Similar to what was observed in eWAT, MitoNEET overexpression in PDGFRβ+ cells led a decrease in the frequency of PDGFRβ+ cells (Fig. 4A). The viability and the level of early and late apoptosis/necrosis were comparable between groups (Fig. 4B–E). Interestingly, the relative proportion of DPP4+ APCs is increased by MitoNEET overexpression, with a corresponding decrease in the frequency of the more adipocyte committed DPP4− APC precursors (Fig. 4F). These in vivo observations are in line with our in vitro data showing that a decrease in mitochondrial respiration in DPP4− cells reprograms the cells to less committed progenitors. Therefore, it is tempting to speculate that DPP4− cells switch back to DPP4+ cells that represent a less committed stage of differentiation. iWAT PDGFRβ+ cells overexpressing MitoNEET displayed a higher BrdU incorporation compared to controls (Figure 4G–H). A reduction in mitochondrial function increased BrdU incorporation into DPP4− cells (Figure 4I). This result differs from our in vitro data showing a similar proliferation rate between control or MitoNEET TG cells (Fig. 2L–M). These observations reflect that DPP4+ and DPP4− respond differentially in vivo vs. in vitro in response to a reduction in oxidative phosphorylation. As shown for PDGFRβ+ in eWAT, Pdgfrβ mRNA levels were decreased in iWAT mural cells upon overexpression of MitoNEET (Fig. 4J). This highlights that eWAT and iWAT mural cells both tend to lose their identity in response to mitochondrial dysfunction. Consistent with the in vitro studies, overexpression of MitoNEET led to a decrease in pro-adipogenic gene expression in both DPP4+ and DPP4− APC subpopulations in vivo (Fig. 4K–L). Levels of pro-inflammatory transcripts were only modestly impacted by MitoNEET overexpression (Fig. 4K–L). However, the overall increase in pro-inflammatory DPP4+ APCs was associated with an increased total number of macrophages as well as an increase in classically activated macrophages (Fig. 4M–Q). Moreover, the numbers of protective alternatively-activated macrophages in the iWAT of MitoNEETTG mice was decreased (Fig. 4O). As a consequence, the M1/M2 ratio was increased (Fig. 4P). We also observed an increase in the number of macrophages that simultaneously express both classically- and alternatively-activated macrophages, perhaps reflecting macrophage populations in transit from one subtype to another (Fig. 4Q). Similar to what we observed in eWAT, all of these effects of MitoNEET overexpression are reversible. Within 1 week of cessation of transgene expression, the relative frequency of DPP4+ and DPP4− subpopulations and pro- and anti-inflammatory macrophages return to levels found in control animals (Fig. 4Q–R). Pdgfrβ mRNA levels also returned to the same level between groups after the doxycycline washout (Fig. 4W). It is important to note that there is little, if any, active adipogenesis that occurs over a 1-week period in adult chow-fed mice. As such, these results from both iWAT and eWAT illustrate the essential roles of the diverse resident precursor populations in the regulation of WAT homeostasis, beyond their impact on adipogenesis per se. The rapid response of these cell populations to mitochondrial dysfunction, and the reversibility of these phenotypes, highlight a surprising plasticity of these cells in response to mitochondrial activity.
Figure 4. In vivo mitochondrial dysregulation of DPP4+ and DPP4− cells compromises adipogenesis while promoting inflammation in subcutaneous adipose tissue.
In vivo disruption of mitochondrial function in DPP4+ and DPP4− using control or PdgfRβ-rtTA × TRE-MitoNEET (MitoNEETTG) mice fed for 1 week with chow diet containing 600 mg/kg of doxycycline. (A) Frequency of the total PdgfRβ+ cells in control or MitoNEET TG sWAT. (B) Gating strategy of annexin-V and propodium iodine of total PdgfRβ+ cells (C) Viability of all PdgfRβ+ cells (n=4) (D) Late apoptosis or necrosis of PdgfRβ+ cells (E) Early apoptosis of all PdgfRβ+ cells: (n=4) (F) Frequency of DPP4+ and DPP4− cells in control and MitoNEET TG sWAT. (G) Gating for BrdU-positive PdgfRβ+ cells. (H) Quantification of BrdU incorporation into PdgfRβ+ cells (n=4–5) (I) Quantification of BrdU incorporation (n=4–5). (J) Gene expression of Pdgfrb marker in PdgfRβ+ cells (n=4) (K) Gene expression of inflammatory markers in DPP4+ cells (n=4–8) (L) Gene expression of adipogenic and inflammatory markers in DPP4− cells (n=4–10). (M-Q) sWAT macrophage abundance was measured in control and MitoNEETTG (n=9–12) (M) Total macrophages (N) M1 macrophages (O) M2 macrophages (P) Ratio M1 and M2 macrophages (Q) Macrophages expressing both M1 and M2 markers (Q-W) Control and MitoNEETTG mice were fed for 1 week with chow diet containing 600 mg/kg doxycycline and fed another week with chow diet (doxycycline washout) (Q) Frequency of the total PdgfRβ+ cells (n=9–11). (R) Frequency of DPP4+ and DPP4− cells in control and MitoNEETTG mice (n=9–11). Data are presented in percent of PdgfRB+ cells. (S-V) sWAT macrophages abundance was measured in control and MitoNEETTG (n=8–10). (S) Total macrophages (T) M1 macrophages (U) M2 anti-inflammatory macrophages (V) Ratio M1/ M2 (W) Gene expression of Pdgfrb marker in PdgfRβ+ cells (n=4). Significance between control and MitoNEET TG was calculated using a two-tailed student’s t-test. Error bars represent mean ± S.E.M. * (P<0.05), ** (p<0.01), *** (p<0.0001), **** (p<0.00001).
Disruption of mitochondrial metabolism in PDGFRβ+ cells leads to a reversible partial lipodystrophy upon HFD feeding.
Diet-induced obesity is associated with mitochondrial dysfunction and a chronic state of subclinical inflammation. This phenomenon may negatively impact WAT function and expandability. We therefore wondered whether reduced mitochondrial function in PDGFRβ+ cells could impact WAT health in the setting of caloric excess (i.e. HFD feeding). Based on our results obtained above on chow diet, we hypothesized that mitochondrial dysfunction in PDGFRβ+ stromal subpopulations would reduce de novo adipogenesis and exacerbate the pro-inflammatory response of WAT depots in HFD-fed animals. To address this question, we exposed male control and MitoNEETTG mice to doxycycline-containing HFD (HFD dox) for up to 20 weeks. Average body weights of the two groups diverged as early as 5 weeks after the onset of HFD feeding, with MitoNEETTG mice gaining less weight than control animals (Fig. 5A). Body composition analysis after 6 weeks of HFD dox-feeding largely driven by differences in fat mass rather than lean mass (Fig. 5B), with a decrease in iWAT mass, while eWAT mass remains comparable between groups (Fig. 5C). Notably, post-prandial clearance of triglycerides is impaired in MitoNEETTG. This suggests a dysfunctional uptake and esterification of lipids in the hypertrophic adipocytes, which predominate at early stages of HFD feeding (Fig. 5D). Histological analysis at 6 weeks post-HFD dox reveals the presence of smaller adipocytes in the iWAT depot, but not the eWAT depot (Fig. 5E). Histological analysis of livers reveals a striking increase in hepatic lipid content (Fig. 5E), suggesting ectopic lipid accumulation in the liver.
Figure 5. Disruption of mitochondrial metabolism in PDGFRβ+ cells leads to a reversible partial lipodystrophy upon HFD feeding.
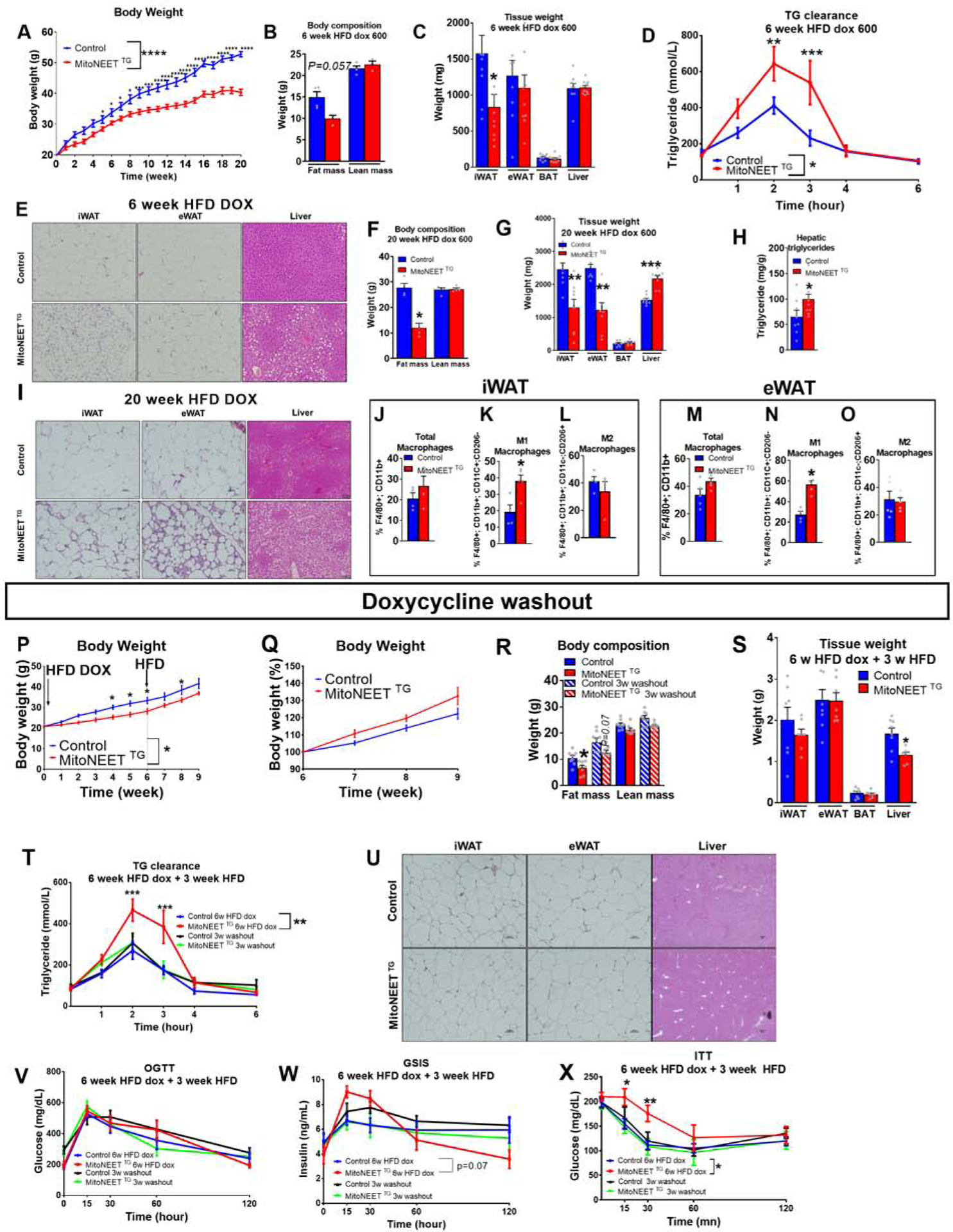
(A) Body weights (g) of PdgfRβ-rtTA (Control) or PdgfRβ-rtTA TRE-MitoNEET (MitoNEETTG) mice fed HFD 600 mg/kg doxycycline (HFD dox) during 20 weeks (n=10–14). (B-E) Control and MitoNEET TG mice fed HFD dox for 6 weeks. (B) Body composition (g) (n=3–4). (C) Tissue mass (mg) (n=9–10). (D) Triglyceride clearance (n=8–9). (E) Histology of sWAT, eWAT and liver. (F-O) Control and MitoNEETTG mice fed HFD doxycycline for 20 weeks. (F) Body composition (g) (n=3–4). (G) Tissue weights (mg) (n=8–9). (H) Hepatic triglyceride (n=8–10) (I) Histology of sWAT, eWAT and livers. (J-L) Macrophage frequency in sWAT (n=4).(J) Total macrophages (K) M1 macrophages (L) M2 macrophages (M-O) Macrophage frequency in eWAT (n=5). (M) Total macrophages . (N) M1 macrophages. (O) M2 macrophages. (P-X) Control and MitoNEETTG mice fed with HFD doxycycline for 6 weeks and 3 weeks with HFD (“doxycycline washout”) (n=9). (P) Body weights (g) (n=9). (Q) Body weights as a percentage of the body weight after 6 weeks under HFD doxycycline (n=9). (R) Body composition (g) (n=8). (S) Tissue weights (in grams) (n=8). (T) Triglyceride clearance (n=7–9). (U) Histology of sWAT, eWAT and liver. (V) Glucose level during oral Glucose Tolerance Test (OGTT) (n=7–10). (W) Insulin level during OGTT (n=7–10) (X) Glucose level during Insulin Tolerance Test (ITT) (n=7–10). Significance between Control and MitoNEET TG in (A,D,P,Q,R,T,V,W,X) was calculated using a 2-way Anova with Tukey’s post-test for multiple comparisons. Significance between control and MitoNEET TG in (B,C,F,G,H,J,K,L,M,N,O,S) was calculated using a two-tailed student’s t-test. Error bars represent mean ± S.E.M. * (P<0.05), ** (p<0.01), *** (p<0.0001), **** (p<0.00001)
By 20 weeks of HFD dox feeding, MitoNEETTG mice exhibited a robust phenotype reminiscent of a partial lipodystrophy. Total fat mass, but not lean mass, was substantially lower than observed in control animals (Fig. 5F). Both iWAT and eWAT in transgenic animals were much smaller. Livers of HFD-dox-fed MitoNEETTG mice were significantly larger than controls (Fig. 5G), likely due to a substantial increase in hepatic triglyceride content (Fig. 5H). Histological analysis confirmed a striking increase in hepatic steatosis in transgenic mice, along with a pathologic remodeling of WAT depots (Fig. 5I). In particular, both the iWAT and eWAT depots displayed numerous crown-like structures, indicative of interstitial inflammation.Both depots of transgenic mice contain significantly more pro-inflammatory macrophages (Fig. 5J–O). These data are in line with our previous data showing an increase of pro-inflammatory macrophages in both iWAT and eWAT depots of MitoNEETTG mice maintained on chow diet.
Remarkably, the early stages of this partial lipodystrophy can be reversed by simply terminating expression of the MitoNEET transgene in PDGFRβ+ cells. After 6 weeks of HFD-dox-feeding, we switched control and MitoNEETTG mice to the equivalent HFD feed lacking doxcycyline for 3 weeks (Fig. 5P). Within 3 weeks of terminating transgene expression, body weights of MitoNEETTG mice rebounded to levels statistically indistinguishable from control mice (Fig. 5P,Q). Body composition analysis revealed that overall fat mass and tissue weights of the iWAT and eWAT depots returned to levels found in controls (Fig. 5R,S). The ability of MitoNEETTG mice to clear triglycerides is also restored, with MitoNEETTG (Fig. 5T). Histological analysis confirmed a decrease in hepatic steatosis in transgenic mice, and a similar overall appearance of WAT depots (Fig. 5U). Transgenic animals tended to be glucose intolerant, as seen by an increase insulin production during a glucose tolerance test at 6 week HFD dox that is restored after 3 weeks of a doxycycline washout (Fig. 5V,W). Moreover, insulin sensitivity of transgenic animals was significantly reduced under HFD-dox and returned to the level of control mice after washout (Fig. 5X). To decipher whether the reversibility of the detrimental phenotype is due to reduced macrophage recruitment signals from FIPs or a consequence of the restoration of de novo adipogenesis, we evaluated the reversibility earlier, after one week of dox wash out. Triglyceride clearance was already restored after one week (Fig. 6A). The frequency of total PDGFRβ+ cells from eWAT was now comparable between control and transgenic groups (Fig. 6B). While FIP frequency returned to control levels, APCs remained lower in MitoNEET TG compared to controls (Fig. 6C). The frequency of total PDGFRβ+ cells from sWAT was now similar between groups (Fig. 6D). Both DPP4− and DPP4+ subpopulations were at the same frequency in both groups. Recruitment of eWAT and sWAT macrophages were also comparable, and the level of activated macrophages M1 did not differ between groups (Fig. 6F–K). These results highlight the rapid response of PDGFRβ+ cells to mitochondrial regulation and the involvement of inflammation in the deterioration of adipose tissue function. As previsouly described in chow diet experiments, a reduction of inflammation from precursors is able to reduce macrophage recruitment in both depots under HFD. Moreover, the reduction of inflammation seems to be the first response to restore adipocyte function, as APCs are still at a lower level in transgenic animals. We also demonstrated that triglyceride clearance was impaired at 1 week of chow dox diet and restored after 1 week doxycycline washout (Fig. 6L). This result argues that inflammation elicited by the precusor cells has a strong detrimental effect on the ability of the adipocytes to store lipids. We have determined that the reduction of inflammation from mural cells is a key parameter to restore adipocyte function. Altogether, these observations highlight the critical role that the metabolic activity of stromal cells plays for the adaptation of WAT to excess caloric intake. Additionally, the reversibility of these phenotypes demonstrates the exquisite sensitivity of adipose tissue to mitochondrial perturbations occurring within PDGFRβ+ subpopulation of cells in the tissue.
Figure 6: Reversibility of partial lipodystrophy upon disruption of mitochondrial metabolism in HFD feeding is due to a rapid restoration of inflammation level in PDGFRβ+ cells after 1 week cessation of doxycycline.
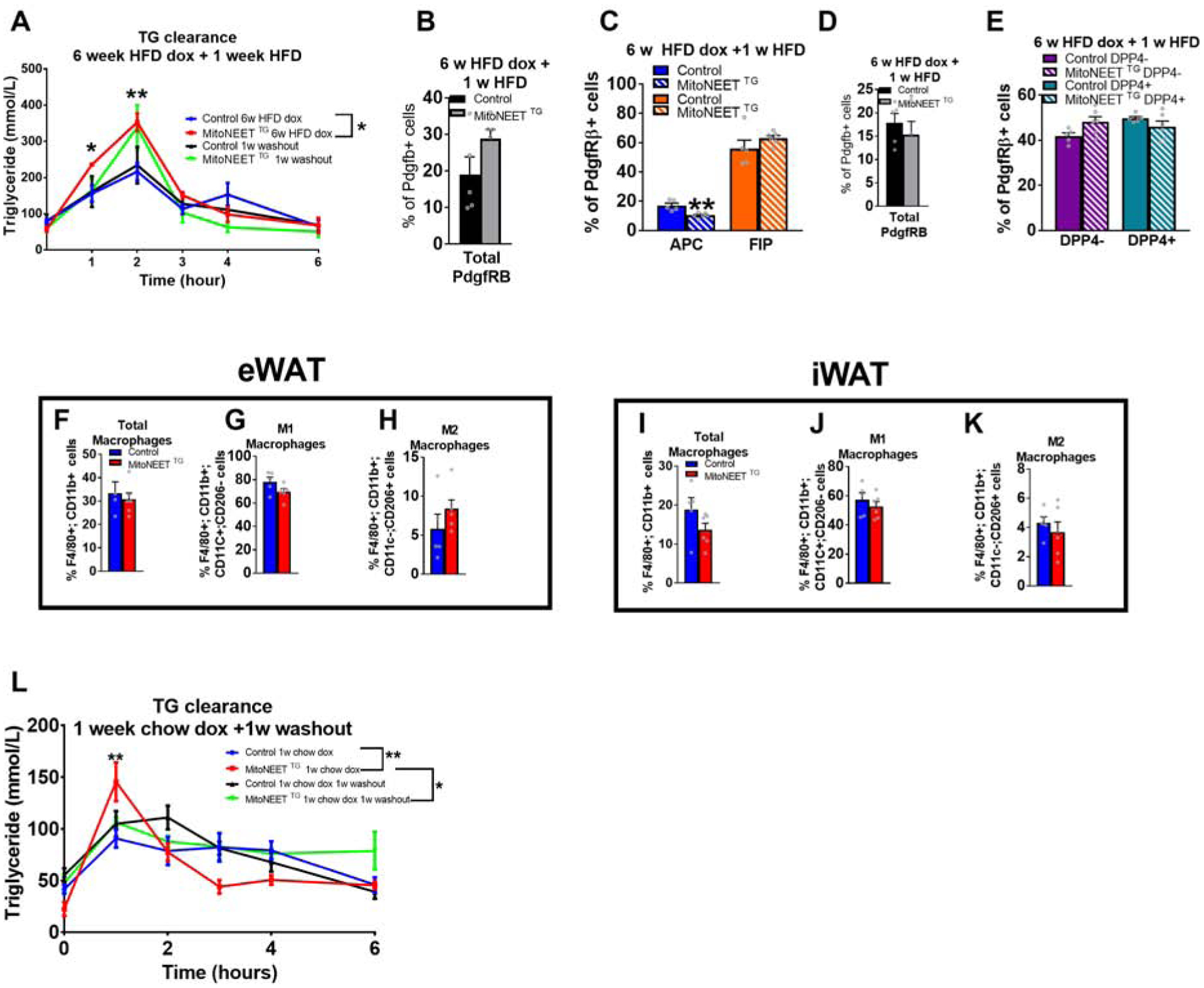
Control and MitoNEET TG mice fed with HFD doxycycline for 6 weeks and switched for 1 week to HFD (“doxycycline washout”) (n=5–6). (A) Triglyceride clearance. (B-E) Frequency of mural cells and mural subpopulations from both eWAT and sWAT doxycycline washout (n=5–6). (B) Frequency of PdgfRβ+ cells in eWAT. (C) Frequency of APCs and FIPs. (D) Frequency of PdgfRβ+ cells in sWAT (n=5–6). (E) Frequency of DPP4− and DPP4+ cells. (F-H) eWAT macrophage frequencies (G) M1 macrophages. (H) M2 macrophages. (I-K) sWAT macrophage frequencies (n=5–6). (I) Total macrophages (J) M1 macrophages. (K) M2 macrophages (L) Triglycerides clearance after doxycycline washout. Significance in (A,L) was calculated using a 2-way Anova with Tukey’s post-test for multiple comparisons. Significance in (B-E; F-K) was calculated using a two-tailed student’s t-test. Error bars represent mean ± S.E.M. * (P<0.05), ** (p<0.01), *** (p<0.0001), **** (p<0.00001).
Discussion
Fine-tuning of mitochondrial function is a key regulator of progenitor fate and function
Recent single-cell sequencing efforts have highlighted the molecular and functional heterogeneity of adipose tissue stromal cells in distinct depots (Burl et al., 2018; Hepler et al., 2018; Merrick et al., 2019). Our results presented here, along with the data described by Shao et al. (see accompanying manuscript), collectively highlight the depot differences in the functional and metabolic properties of depot-resident progenitor populations. In particular, FIPs of the eWAT depot are non-adipogenic PDGFRβ+ cells that have the capacity to exert a strong pro-inflammatory phenotype. APCs within this depot are relatively refractory to pro-inflammatory stimuli and instead are highly committed to undergo adipogenesis. Here, we reveal that the unique functional properties of these cells are controlled by their respective mitochondrial activity. In eWAT, both APCs and FIPs have comparable mitochondrial content; however, APCs are less metabolically active than FIPs and respond differently to mitochondrial dysfunction. MitoNEET-driven alterations in mitochondrial metabolism render these cells unable to undergo adipogenesis. FIPs enhance their glycolytic rate when prompted with mitochondrial dysfunction. The shift from mitochondrial respiration to glycolysis in FIPs is associated with an increase in pro-inflammatory gene expression. Notably, this phenomenon is well described in the literature for immune cells. Enhanced glycolysis occurs in lipopolysaccharide (LPS)-activated macrophages and dendritic cells (Krawczyk et al., 2010; Rodriguez-Prados et al., 2010), in activated natural killer cells (Donnelly et al., 2014), in activated effector T cells (Michalek et al., 2011) and in activated B cells (Doughty et al., 2006). Effector T cell subsets all show an increase in glycolysis following activation, most notably T helper 17 (TH17) cells (Shi et al., 2011), TH1 and TH2 cells (Michalek et al., 2011) and activated effector CD8+ T cells. It is important to note that short-term HFD feeding, which elevates mitochondrial metabolism, as well as a genetic intervention that leads to a decrease in mitochondrial function, both result in an upregulation of inflammatory properties of FIPs. As such, this suggests a “U-shaped” dose response to mitochondrial function which is optimized with both increases and decreases in mitochondrial function, resulting in enhanced inflammation.
Cellular metabolism also plays a critical role in the regulation of stem cell biology and lineage commitment. Our analysis of DPP4+ and DPP4− APCs from iWAT highlight that APCs at different levels of commitment have distinct metabolic responses to mitochondrial perturbation. MitoNEET overexpression significantly diminished the capacity of these two APC populations to undergo adipogenesis. DPP4− APCs, as committed preadipocytes, lack potential to undergo osteoblast differentiation. MitoNEET-driven alterations in energy metabolism renders these cells developmentally competent to undergo differentiation to this alternative lineage. The maintenance of mural identity requires proper mitochondrial function. We show that mitochondrial dysfunction leads to a loss of mural cell identity. Mitochondrial activity impacts progenitor identities and their fates into different cell types, such as neural stem cells, hematopoietic stem cells or mesenchymal stem cells (Atashi et al., 2015; Jang and Sharkis, 2007; Khacho et al., 2016). Collectively, our results highlight the impact of mitochondrial perturbations on adipocyte lineage determination and differentiation.
Alterations in mitochondrial activity of PDGFRβ+ stromal cells impact WAT remodeling
Much attention has been placed on the role of mature adipocytes and classical immune cells in regulating WAT inflammation. Our data highlights the sensitivity of adipose tissue macrophages in lean and obese mice to the metabolic activity of PDGFRβ+ stromal cells. In eWAT, MitoNEET-driven alterations in the mitochondrial activity of APCs and FIPs result in a heightened pro-inflammatory phenotype of these cells, and a shift in the number of FIPs vs. APCs in vivo. Likewise, in iWAT, mitochondrial dysfunction in PDGFRβ+ cells led to an increase in the number of DPP4+ APCs, which themselves exhibit a greater pro-inflammatory expression profile than DPP4+ APCs. Within one week of activating the MitoNEET transgene in PDGFRβ+ cells in lean mice, adipose tissue macrophages become activated in both eWAT and iWAT. Remarkably, these phenotypes are fully reversible within 1 week of terminating transgene expression. It is important to note that PDGFRβ+ cells in eWAT represent only ~20% of the stromal-vascular fraction, with FIPs representing ~15% and APCs about ~5%. For the iWAT depot, DPP4+ and DPP4− cells represent around 10% each of the total SVF fraction. Despite their abundance, mitochondrial dysregulation of these cells leads to a rapid and reversible impact on macrophage activity in lean mice. These findings strongly implicate PDGFRβ+ cells as key regulators of inflammatory balance in adipose tissue.
Disruption of mitochondrial activity in PDGFRβ+ cells at the onset of HFD feeding has an appreciable impact on WAT expansion. The iWAT depot of MitoNEETTG mice fails to expand in the early stages of HFD feeding. It is important to note that iWAT of male HFD-fed C57BL/6 mice expands almost exclusively through cellular hypertrophy rather than through de novo adipogenesis. As such, the inability of the iWAT depot to expand in these transgenic mice is likely due to aberrant activity of DPP4+ and DPP4− APCs, rather than their adipogenic potential. These data highlight the contribution of these cells to WAT remodeling, beyond adipogenesis. A failure to expand eWAT in MitoNEETTG mice was apparent after longer periods of HFD feeding. This depot expands through both adipocyte hyperplasia and adipocyte hypertrophy. Mitochondrial dysfunction in eWAT APCs and the loss of their adipogenic potential is likely contributing to the phenotype. Moreover, the hyperinflammatory responses of FIPs and APCs themselves are likely contributing to increased macrophage infiltration/activation in eWAT. It is notable that the ensuing partial lipodystrophy in MitoNEETTG mice is also reversible. This demonstrates that the restoration of mitochondrial function in progenitors can rescue adipose tissue dysfunction, highlighting significant therapeutic potential for this area of intervention. Altogether, we provide evidence that adipogenic progenitors in both eWAT and iWAT play a key role in the regulation of inflammation and adipogenesis in adipose tissue. Their fate and function are critically dependent on finely-tuned mitochondrial activity.
Going forward, it will be important to establish the precise mechanisms by which altered mitochondrial function impacts the lineage commitment and function of these diverse stromal cell subpopulations. Of note, several recent studies from Olefsky and colleagues highlight the impact of altered mitochondrial activity in mature adipocytes on the pro-inflammatory and pro-fibrogenic gene programs (Ejarque et al., 2019; Seo et al., 2019). Moreover, in the accompanying manuscript, Shao et al. highlight the function of HIF-1α signaling in controlling the fate and function of PDGFRβ+ stromal cell subpopulations. Ultimately, a deeper understanding of how mitochondrial activity in adipose tissue progenitors is fine-tuned may lead to innovative strategies to improve adipose tissue function and metabolic health in obesity.
Limitations of the study
A limitation of our current study is that we are unable to distinguish the effects of mitochondrial dysfunction on specific precursor subpopulations independent of other subpopulations. It would be interesting in the future to develop mouse models in which we can manipulate selectively only a subset of the precursor cell population. Furthermore, the present study does not identify specific metabolites that are altered in response to mitochondrial dysfunction and their relative contributions on adipose tissue inflammation and expansion during caloric excess.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Philipp Scherer (Philipp.Scherer@utsouthwestern.edu).
Materials Availability
Unique materials and reagents generated in this study are available upon request from the Lead Contact with a completed Material Transfer Agreement.
Data and Code Availability
This study did not generate any code or large dataset.
EXPERIEMENTAL MODEL AND SUBJECT DETAILS
Mouse Models -
All animal experimental protocols were approved by the Institutional Animal Care and Use Committee of University of Texas Southwestern (UTSW) Medical Center at Dallas, TX (APN# 2015–101207). We generated a mouse model with doxycycline-inducible, mural cell-specific mitoNEET overexpression by crossing the TRE-mitoNEET transgenic mice with Pdgfrβ promoter driven-rtTA transgenic mice (Pdgfrβ-rtTA, C57BL/6-Tg(Pdgfrb-rtTA)58Gpta/J; JAX028570). These two models have been described previously (Kusminski et al., 2012; Vishvanath et al., 2016). All animals used in this study were littermate-controlled male mice and on a pure C57BL/6 background. Mice were maintained on a 12 h light/dark cycle in a temperature-controlled environment (22°C) and had free access to food and water. Water and cages were autoclaved. Cages were changed every other week, and the health status of the mice was monitoring using the Allentown sentinel filter,which started in the second quarter of 2017. The mouse genotype did not cause visible changes in initial weight, health or immune status. In all experiments, only male mice were used, as female mice are resistance to obesity and type 2 diabetes. The age and number of the mice used for the experiments are indicated for each experiments in the figure legends. In order to ensure the reproducibility, two separate cohorts of mice were used. Mo inclusion or exclusion criteria were used. The experiments were not randomized. No statistical method was used to predetermine the sample size for the animals. All of our experimental animals were kept under barrier conditions under constant veterinary supervision and did not display any signs of distress or pathological changes that warranted veterinary intervention. Mice were fed a standard rodent chow diet or 600 mg kg−1 doxycycline-containing chow or HFD diet containing or not 600mg-kg-1 doxycycline. Chow and HFD diet containing doxycycline was initiated at 6 weeks of age. In all cases where doxycycline was required to induce gene expression, it was supplied in the mouse diet. All mice received doxycycline diet, including controls (lacking the inducible transgenes (TRE-mitoNEET). 5-Bromo-2′-deoxyuridine (BrDU) was administrated in the drinking water at 0.8mg/mL for 1 week. BrdU water was replaced every 2 days.
METHOD DETAILS
Body Composition Analysis -
Body fat mass and lean mass were measured in conscious mice using EchoMRI-100 (UTSW Metabolic Phenotyping Core).
Hepatic Triglyceride Measurements -
For liver triglyceride measurements, 100 mg of liver tissue was homogenized in phosphate-buffered saline (PBS) and mixed sufficiently with 1.6 ml of CHCL3-CH3OH (2:1, v/v). After centrifugation at 3,000 r.p.m. for 10 min at room temperature, the lower organic phase was transferred and air dried completely in a chemical hood. Samples were re-suspended using 1% Triton X-100 in absolute ethanol and triglycerides were measured using Infinity Triglycerides kit based on the method of Wako and the modifications by McGowan et al and Fossati et al (McGowan MW, et al. Clin Chem 1983;29:538; Fossati P, Prencipe L. Clin Chem 1982;28:2077–80.)
Systemic Tests -
Triglyceride clearance tests:
Mice were fasted for 16 h prior to administration of 15 μl g−1 body-weight of 20% intralipid by gastric gavage. Blood was collected from the tail vein at timed intervals (0, 1, 2, 3, 4 and 6 hours), then assayed for triglyceride levels using Infinity Triglycerides kit as mentioned above.
Oral glucose tolerance test (OGTT):
Mice were fasted for 4 h prior to administration of glucose (2.5 g kg−1 body-weight by gastric gavage). Blood was collected from the tail vein at each time point indicated (0, 15min, 30min, 60 min and 120min) and then the blood samples were centrifuged at 6000 rpm for 5 min and stored for further measurements. Glucose concentrations were measured using Bayer Contour glucometers and insulin was measured using Ultra Sensitive Mouse Insulin ELISA kit (Crystal Chem, 90080).
Insulin tolerance test (ITT):
Mice were fasted for 4 h prior to administration of insulin (0.75U kg−1 body-weight by intraperitoneal injection). Blood was collected from the tail vein at time 0, 15min, 30min, 60 min and 120min. Glucose concentrations were measured using Bayer Contour glucometers.
RNA isolation, cDNA Reverse Transcription and Quantitative RT-PCR –
Cells were harvested with lysis buffer solution and snap-frozen before RNA extraction. Then RNA was isolated using an RNAqueous®-micro kit. The RNAqueous®-Micro Kit is used for phenol-free total RNA isolation from micro-sized samples (e.g., from <500,000 cultured cells, from few as 10 laser capture microdissected cells, or from ≤10 mg of tissue) using a guanidinium-based lysis/denaturant and glass fiber filter separation technology. Cell pellets were resuspend in 100 μL Lysis Solution and briefly vortexed. 50 uL of 100% ethanol was added and samples were vortex briefly. The mixture was loaded onto a Micro Filter Cartrige Assembly. Centrifuge 10 secondes at 13 000g at room temperature. 180 uL of Wash Solution 1 (working solution mixed with ethanol) was added to the filter. Centrifuge for 10 secondes at 13,000g at room temperature. 180 uL of Wash Solution 2/3 (working solution mixed with ethanol) was added to the filter. Centrifuge 10 seconds at 13,000g at room temperature. Repeat a second 180 uL aliquot of Wash Solution 2/3. Centrifuge for 10 seconds at 13,000g at room temperature. Open the Micro Filter Cartridge assembly, remove the filter cartridge from the collection tube, and pour out the flow-through. Replace the Micro Filter Cartridge into the same collection tube, close the cap, and centrifuge at maximum speed for 1min to remove residual fluid and dry the filter. Transfer the Micro Filter Cartridge into 1.5mL tube. Apply 5–10 uL or Elution Solution, preheated to 75°C, to the center of the filter. Close the cap and store the assembly for 1 min at room temperature. Centrifuge the assembly for 30 seconds to elute RNA from the filter. Repeat with a second 5–10 uL elution solution, collecting the eluate in the 1.5 mL tube. Then, RNA was quantified by spectrophotometric assay (Nanodrop). cDNA was prepared by reverse transcription with an iScript cDNA Synthesis Kit. Up to 1000ng RNA were reversed transcribed in 20 uL volume containing 4 uL of 5X iScript Reaction Mix and 1 uL iScript Reverse Transcriptase. Samples were then incubated in a thermal cycler using the following protocol: Priming: 5min at 25°C, Reverse transcription: 20 min at 46°C, Reverse Transcription inactivation 1 min at 95°C, hold at 4°C. Samples of cDNA were diluted 1:20, used for RT-qPCR measurements using SYBR Green. RT-qPCR was performed in a QuantStudio 6 Flex. Results were calculated using the threshold cycle method, with B2M gene for normalization. Quantification of mRNA was carried out by comparing the number of cycles required to reach reference and target threshold values (2^-(δ−δ Ct) method) as described previously (Livak and Schmittgen, 2001). Sequences of the mouse sense and antisense primers have listed in the Table S1.
Histology -
Tissues were excised and fixed in 10% PBS-buffered formalin for 24h and keep in 50% ethanol. Following paraffin embedding and sectioning (5 μm), tissues were stained with H&E.
Stromal Vascular Fraction Isolation –
Stroma Vascular Fraction isolation was adapted from Peics, J et al. (Peics et al., 2020). White adipose tissues were minced with scissors in Hank’s balanced salt solution containing 1.5% BSA, and 1mg/mL collagenase D. For both epididymal and subcutanesous adipose tissue, 2 pads were combined together. Minced adipose tissues were placed at 37°C in 100rpm shaking water bath for 45 minutes (epididymal) or 1 hour and 20 minutes (subcutaneous). Samples were mixed by pippeting up and down with a 10mL pipette every 20 minutes during incubation to allow the tissue to digest smoothly. Digested tissues were then filtered through a 100 μm cell strainer and 30mL of cold 2%FBS/PBS was added through the filter to wash it and dilute digestion buffer. Then, cells were centrifuged for 5 minutes at 600g at 4°C to pellet the stroma vascular cells. Supernatant was discarded and cells re-suspend in 500μL red blood cell lysis to eliminate red blood cells. Then, 6mL of cold 2%FBS/PBS was added to stop the reaction and cell suspension was filtered through a 40μm cell strainer in a 15 mL falcon tube containing 6mL of cold 2% FBS/PBS. Cells were centrifuge 5min at 4°C at 600×g to pellet the stroma vascular cells. Stroma vascular cells were then used for cell culture or flow cytometry experiments as described below in Flow Cytometry and Fluorescent Activated Cell Sorting.
Flow Cytometry and Fluorescent Activated Cell Sorting -
Flow cytometry experiments were carried out on a BD Bioscience LSRFortessa SORP flow cytometer (Children’s Medical Center Research Institute Flow Core at UT Southwestern). Cells were sorted on a BD Bioscience FACS Aria Fusion (Children’s Medical Center Research institute flow Core at UT Southwestern). Antibodies used for flow cytometry can be found in the Key Ressour Table.
Key Resource Table
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies for Flow Cytometry | ||
| Purified Rat Anti-mouse CD16/CD32 FC Block clone 2.4G2 | BD biosciences | Cat#: 553142 |
| FITC CD45 clone 30-F11 | BD biosciences | Cat#: 553080 |
| FITC CD31 clone 390 | BD biosciences | Cat#: 558738 |
| PE anti-CD140b clone APB5 | Biolegend | Cat#: 136006 |
| PE-CF594 anti-F4/80 clone T45–2342 | BD biosciences | Cat#: 565613 |
| BB700 anti-CD11b clone M1/70 | BD biosciences | Cat#: 566417 |
| BV421 anti-CD11c clone | BD biosciences | Cat#: 565452 |
| Alexa Fluor 647 anti-CD206 clone MR5D3 | BD biosciences | Cat#: 565250 |
| PerCPcy5.5 anti-CD45 clone 30–711 | Biolegend | Cat#: 103132 |
| PerCPcy5.5 anti-CD31 clone 390 | Biolegend | Cat#: 102420 |
| FITC anti-CD9 clone MZ3 | Biolegend | Cat#: 124808 |
| APC anti-Lyc6 clone HK1.4 | Biolegend | Cat#: 128016 |
| Antibodies for Western blots | ||
| Rabbit polyclonal anti-mouse MitoNEET | Homemade | Kusminski et al 2012 |
| Total OPHOS Rodent antibody cocktail | Abcam | Cat#: ab110413 |
| Anti-Prohibitin antibody - Mitochondrial Marker | Abcam | Cat#: ab28172 |
| Monoclonal Anti-β-Actin antibody | Sigma-Aldrich | Cat#: A5441 |
| IR-Dye 800CW Donkey antibody against rabbit | Li-COR | Cat#: 92632213 |
| IR-Dye 800CW Goat antibody against Mouse | Li-COR | Cat#: 92632213 |
| IR-Dye 680RD Donkey antibody against Mouse | Li-COR | Cat:# 92668072 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 3-isobutyl-1-methylxanthine | Sigma-Aldrich | Cat#: I7018 |
| 5-Bromo-2′-deoxyuridine | Sigma-Aldrich | Cat#: B5002 |
| 5mL polystyrene round-bottom tube with cell-strainer cap | Corning | Cat#: 352235 |
| 20% intralipid | Sigma-Aldrich | Cat#: I141–100mL |
| 96-well Clear Round Bottom Polystyrene Not Treated Microplate | Corning | Cat#: 3795 |
| BDCytofix | BDBioscience | Cat#: 554655 |
| BioCoat™ Collagen I 24-well Clear Flat Bottom TC-treated Multiwell Plate, with Lid | Corning | Cat#: 356408 |
| BSA (Fatty acid free, low endotoxin) | Sigma-Aldrich | Cat#: A8806 |
| Collagenase D | Roche | Cat#: 11088882001 |
| Corning® BioCoat™ Collagen I 24-well Clear Flat Bottom TC-treated Multiwell Plate, with Lid | Corning | Cat#: 356408 |
| Corning® BioCoat™ Collagen I 48-well Clear Flat Bottom TC-treated Multiwell Plate, with Lid, | Corning | Cat#: 354505 |
| Dexamethasone | Sigma-Aldrich | Cat#: D4902 |
| DMEM low glucose glucose | Corning | Cat#: 10–014-CV |
| DMEM F/12+Glutamax | Gibco | Cat#: 10565–018 |
| D-Glucose | Fischer scientific | Cat#: D16–1 |
| Fetal Bovine serum | Fischer Scientific | Cat#: 03-600-511 Lot 140E |
| Gentamicin | Gibco | Cat#: 15750–060 |
| Glycolysis Stress Test Kit | Agilent | Cat#: 103017–100 |
| Glutamax | Gibco | Cat#: 35050 |
| Hank’s balanced salt solution | Sigma-Aldrich | Cat#: H8264–1L |
| HEPES | gibco | Cat#: 15630–080 |
| Insulin-transferrin-Selenium | Gibco | Cat#: 41400–045 |
| L-ascorbic acid 2-phosphate | Sigma-Aldrich | Cat#: A8960–5G |
| mFGF basic | R&D System | Cat#: 3139-FB |
| MCDB201 | Sigma Aldrich | Cat#: M6770 |
| Penicillin-Streptomycin Solution (5,000 U/mL) | Corning | Cat#: 30–001-CI |
| Protease Inhibitor | ThermoFisher | Cat#: A32983 |
| Red Blood Cell Lysis Buffer | Roche | Cat#: 30020500 |
| RIPA buffer | Sigma-Aldrich | Cat#: R0278–500ML |
| TRIzol reagent | Fisher Scientific | Cat#: 12034977 |
| Trypsin-EDTA solution | Sigma-Aldrich | Cat#: 4049–100mL |
| Seahorse XF Calibrant Solution | Agilent | Cat#: 100840–000 |
| Seahorse XF assay medium | Agilent | Cat#: 102365–100 |
| Seahorse XFe24 sensor mini-flux packs | Agilent | Cat#: 102342–100 |
| Sodium Pyruvate | Sigma-Aldrich | Cat#: S8636 |
| SYBR Green PCR Master Mix Power-up | Applied Biosystems | Cat#: A25742 |
| Critical Commercial Assays | ||
| CyQUANT cell Proliferation assay | ThermoFisher | Cat#: C7026 |
| FITC BrdU Flow kit | BD Pharmingen | Cat#: 557891 |
| Infinity Triglycerides | Thermofisher | Cat#: TR22421 |
| iScript cDNA Synthesis Kit | BIORAD | Cat#: 1708890 |
| Mito Stress Test Kit | Agilent | Cat#: 103010–100 |
| Pierce™ BCA Protein Assay Kit | ThermoFisher | Cat#: 23225 |
| RNAqueous-micro kit | Invitrogen | Cat#: AM1931 |
| QIAamp DNA Micro Kit | Qiagen | Cat#: 56304 |
| Experimental Models: Organism/Strains | ||
| C57BL/6-Tg(Pdgfrb-rtTA)58Gpta/J | Jackson laboratory | 028570 |
| TRE-mitoNEET | Kusminski et al 2012 | |
| Chow Diet | LabDiet | Cat#: 5058 |
| Mouse Diet, High Fat Fat Calories (60%), Paste | Bio-Serv | Cat#: S1850 |
| 600mg kg-1 doxycycline-chow diet | Bio-Serv | Cat#: S4107 |
| Mouse Diet, 600mg-kg-1 doxycycline High Fat Fat Calories (60%), Paste | Bio-Serv | Cat#: S5867 |
| Oligonucleotides | ||
| See Table S1related to RNA isolation Star Methods | ||
| Software and Algorithms | ||
| Word | Microsoft | |
| Excel | Microsoft | |
| Fiji (Image J) | Fiji | https://fiji.sc/ |
| Flowjo | Flowjo | https://www.flowjo.com/ |
| Image J | NIH | https://imagej.nih.gov/ij/ |
| Prism | GraphPad Software | Graphpad Software |
| Other | ||
| EchoMRI | N/A | |
| FACS Aria Fusion | BDBioscience | N/A |
| LSRFortessa SORP | BDBioscience | N/A |
| Zeiss LSM8800 Airyscan Confocal Microscope | Zeiss | N/A |
| Keyence BZ-X700 Fluorescence Microscope | Keyence | N/A |
| QuantStudio 6 Flex | Applied Biosystem | N/A |
| Seahorse XFe24 Extracellular Flux Analyzer | Agilent | N/A |
| SynergyMx BioTek | BioTeK | N/A |
| Trans-Blot® Turbo™ transfer system | BioRad | Cat#: 1704150 |
Samples of SVF cells from either inguinal or epididymal WAT depots (isolated as described in Isolation of adipose SVF cells section) were first incubated at 4°C for 10 min in 400 μL of 2% FBS/PBS containing anti-mouse CD16/CD32 Fc Block (clone 2.4G2) (1:200). Cells were then incubated at 4°C for 20 minutes with primary antibodies. For epididymal fat pads, the SVF fraction was incubated with PerCP-cy5.5 anti-CD45 (1:400), PerCP-cy5.5 anti CD31 (1:400), PE anti-CD140b (1:80), APC anti-Ly6c (1:400) and FITC anti-CD9 (1:400). For subcutaneous fat pads, the SVF fraction was incubated with FITC anti-CD45 PE anti-CD140b (1:400), FITC anti-CD31 (1:400), PE anti-CD140b (1:80). BV421 anti-CD26 (DPP4) (1:400).
For macrophages:
SVF fraction from either subcutaneous or epididymal adipose tissue was incubated with FITC CD45 clone 30-F11 (1:200), PE-CF594 anti F4/80 clone T45–2342 (1:200), BB700 anti CD11b clone M1/70 (1:200), Alexa Fluor 647 anti CD206 clone MR5D3 (1:200), BV421 anti CD11c clone N418 (1:200).
Stained SVF fractions were then washed once with 400 ul of cold 2% FBS/PBS and centrifuged 5 min at 4°C at 600g. For flow cytometry analyzes only, cells were then fixed during 20 min at 4°C with 100ul of BD Cytofix (BD Bioscience, 554655). Cells were centrifuged for 5 min at 4°C at 600g and re-suspended in 300μL of cold 2% FBS/PBS. Flow cytometry analyses were done with the Flowjo software.
For fluorescence-activated single cell sorting (FACS), stained cells were washed with cold 2% FBS-PBS and centrifuged 5min at 4°C at 600g and resuspend cold 2% FBS-PBS. Cells were filtered using a 5mL polystyrene round-bottom tube with cell-strainer cap. Cells were sorted into 100% FBS and then either cultured (see mural cell culture and adipocyte differentiation assays section) or resuspended after centrifugation in lysis solution for total RNA extraction using the RNAqueous-micro kit described aboved under the section RNA isolation, cDNA Reverse Transcription and Quantitative RT-PCR.
Flow Cytometry Gating Strategy -
Live cells have been selected on a forward scatter (FSC) versus side scatter plot (SSC) and gated to exclude the debris and dead cells, which are found in the left lower corner. Live cells were then gated on both SSC and FSC Width singlets, ensuring that individual cells were analyzed. Unstained SVF cells isolated from wild-type mice were used as a negative control to determine background fluorescence levels. Cell subpopulations can be distinguished on the basis of different cell markers. For macrophages, anti-CD45 FITC antibody was used to select the hematopoietic cells, anti-F4/80 PE-CF594 and anti-CD11b BB700 antibodies were used to gate the macrophage population (F4/80+;CD11b+, Mac) and distinguish the non-macrophage population (F4/80−;CD11b−, No Mac). From the F4/80+;CD11b+ population, M1 pro-inflammatory macrophages and M2 anti-inflammatory macrophages were distinguished by specific M1 markers anti-CD11c BV421 and specific M2 antibody anti-CD206 Alexa fluor 647.
For FIPs and APCs, cells were selected from the singlets populations as described in (Hepler et al., 2018; Peics et al., 2020). Percp Cy5.5 Anti-CD31, Percp Cy5.5 anti-CD45 and PE anti-CD140b antibodies have been used to gate the mural cell population (CD31−, CD45, CD140b+, PdgfRβ+ cells). From PdgfRβ± cells, APCs and FIPs were distinguished using FITC anti-CD9 and APC anti-Lyc6 antibodies (Lyc6+, CD9+, FIP) and double-negative for APC see gating in Fig. 1SA.
For DPP4± and DPP4−, cells were selected from the singlet populations. FITC anti-CD31, FITC anti-CD45 and PE anti-CD140b antibodies have been used to gate the mural cell population (CD31−, CD45−, CD140b+, PdgfRβ+ cells). From this population, DPP4+ and DPP4− were distinguished using BV421 anti-CD26 antibody see gating in Fig. S3A.
Mural cell culture and adipocyte differentiation assays -
Freshly isolated mural cell fractions were obtained by FACS as described in Flow cytometry section from the SVF of male mice isolated as described in Stroma vascular isolation section. Mural cells from epididymal adipose tissue, FIPs and APCs were plated onto Corning® BioCoat™ Collagen plates and cultured in grown at 10% CO2 in growth media (60% pH7–7.4 low glucose DMEM, 40% pH 7.25 MCDB20, 2% FBS supplemented with 1% ITS premix (Insulin-Transferrin-Selenium), 0.1 mM L-ascorbic acid-2–2phosphate, 10ng/mL FGF basic, 0.5% penicillin-streptomycin, and 0.2% gentamicin (ITS media). For adipocyte differentiation assays, APC cells were plated directly onto Corning® BioCoat™ Collagen I 48-well Clear Flat Bottom TC-treated Multiwell Plate, with Lid and grown to confluence at 10% CO2 in ITS media described above. Once confluent, cells were allowed to maintain in ITS media changed every 2 days for up to 6–8 days to observe the spontaneous adipocyte differentiation.
For mural cells from subcutaneous adipose tissue, DPP4+ and DPP4− were plated onto Corning® BioCoat™ Collagen I 24-well Clear Flat Bottom TC-treated Multiwell Plate, with Lid and grown to confluence at 10% CO2 in growth media containing DMEM F12, 10% FBS, 1% penicillin-streptomycin, 0.1% gentamycin (sWAT media).
For adipocyte differentiation assays, subcutaneous cells DPP4+ and DPP4− subpopulations were plated directly onto Corning® BioCoat™ Collagen I 24-well Clear Flat Bottom TC-treated Multiwell Plate, with Lid and grown until confluence in sWAT media described above. At confluency, differentiation was induced by sWAT media supplemented with 0.5 mM 3-isobutyl-1-methylxanthine, 1μM dexamethasone, 5μg ml−1 Insulin for 48 hours. Then, media was replaced with sWAT media containing 5μg ml−1 Insulin and changed every 2 days for up to 6–8 days to observe adipocyte differentiation.
For osteogenic differentiation, cells from subcutaneous fat pad DPP4+ or DPP4− were incubated with complete MesenCult™ Osteogenic Medium from Stem Cell Technologies and replaced every 3 days for 12 days (Merrick et al., 2019). Osteogenic differentiation may be detected by qPCR analysis of bone-specific transcripts and stained for Alizarin Red S sed for checking the calcium deposits in osteogenic culture(Lysdahl et al., 2013).
BSA-Palmitate Treatment - For Seahorse experiments,
Cells were seeded at confluency with 80,000 cells per well of XFe/XF24 cell culture plate (102342–100, Agilent). Cells were cultured overnight in sWAT media or ITS media. The next morning, the media was removed and cells were rinsed with PBS. Then, media was replaced with sWAT media or ITS containing either 7uM BSA or 0.2 mM palmitate conjugated with 7 uM BSA and pretreated for 4 hours before performing Seahorse experiment to evaluation metabolism.
Cell Viability test -
For viability testx, cells were cultured in 24-well plates. At confluency, cells were treated with 4.4 uM doxycycline for 24h. Viability staining of cells was carried out using ReadyProbes® Cell Viability Imaging Kit from Thermo Fisher Scientific according to manufacturer’s protocol. Briefly, 2 drops of live cell, NucBlue® Live reagent reagent (Hoechst 3342) and NucGreen® dead reagent DNA stain were added per mL of cell culture medium needed and then incubated with cultures for 15 minutes at 37 °C. Cell viability was assessed via the incorporation of DNA stain into the nucleus and imaged using the Keyence BZ-X700 Fluorescence Microscope. DNA Stain only incorporates into cells with a compromised cell membrane, while Hoechst 33342 can permeate the membranes of all cells and bind to the DNA in the nucleus.
Cell proliferation tests -
Cell proliferation was measured using the CyQUANT Cell Proliferation Assay using DNA quantification. On Day 0, cells were plated at 1,000 cells per well in a 96-well plate and grown as previously described in the section ‘Mural cell culture and adipocyte differentiation assays’. A cell dilution series ranging from 0 to 40,000 cells was created for each cell type. One microplate was prepared for each harvest day. Microplates of cells were harvested on days 0, 1, 2, 3, and 4. After harvest, each microplate was kept at −80°C until all microplates had been harvested. Measurement of cell number was done the same day for all experiments. A solution of CyQUANT GR dye in cell lysis buffer was made just prior measurement by diluting the CyQUANT GR dye stock solution 400-fold into cell lysis buffer. Cells were thawed at room temperature and 200uL of CyQUANT GR dye/lysis buffer. The samples were then incubated for 5 min at room temperature in the dark. The fluorescence was measured with a SynergyMx BioTek fluorescence microplate reader with 485 nm (+/− 10 nm) excitation and 530 nm (+/− 12.5 nm) emission filters. For each cell type, a standard calibration curve was generated by plotting measured fluorescence versus cell number (0 to 40 000 cells). Growth curves were plotted as cell number versus time.
Protein content-
Cells were plated at confluency and then treated with 4.4 uM doxycycline for 24h. Cells were then harvested in RIPA buffer and protein content was measured with Pierce™ BCA Protein Assay Kit from ThermoFisher.
Lactate dehydrogenase assay -
Cells were resuspended in 10 mM MOPS, 1 mM EDTA, pH 7.4 (0.4 to 0.8 mg/mL) and lysed by sonication. Lactate dehydrogenase activity was assayed as the rate of NADH oxidation (340 nm, ε = 6200 M-1•cm-1) upon addition of 2.5 mM pyruvate and 0.1 mM NADH to sonicated cells diluted (4 to 8 μg/mL protein) in 25 mM MOPS, pH 7.4 containing 0.05% Triton X-100. Activity was measured in the linear range of protein concentration and was fully suppressed upon addition of the lactate dehydrogenase inhibitor sodium oxamate (10 mM). NADH consumption by the mitochondrial electron transport chain was inhibited by the presence of 0.05% Triton X-100 in the assay buffer.
Seahorse in vitro set up –
According to Agilent Seahorse protocol, to effectively examine metabolic and bioenergetic function using Agilent Seahorse XFe24 Extracellular Flux Analyzer, it is essential to first characterize optimal cell seeding number typically between 1 × 104 and 8 × 104 cells per well (0.32 cm2). Generally, densities resulting in 50–90% confluency generate metabolic rates in the desirable/dynamic range of the instrument. In this study, 80,000 cells were used and represend 100% confluency for all cell types and validated by viability test, microscopy morphology, mtDNA content and protein content for all cell types (FIPs, APCs, DPP4+, DPP4−). On day 0, cells were counted and plated at 100% confluency with 80,000 cells per well overnight in an Agilent Seahorse XF24 cell culture microplate at 37°C under 10% CO2 and culture in 300 μL of sWAT or ITS media (See mural cell culture and adipocyte differentiation assays section). Wells A1, B4, C3, and D6 are used for background temperature correction: add 300 μl of media (no cells). Day 1, cells were treated for 24h with or without 4.4 μM doxycycline or 4h with BSA or BSA palmitate to mimick lipid overload before evulating metabolism (see details in BSA palmitate experimental section). In parallel, an Agilent Seahorse XFe24 extracellular flux assay plate with 1ml Seahorse XF Calibrant Solution was equilibrated overnight at 37°C. Day 2, Mito Stress Test or Glycolysis Stress Tests were performed as described in In vitro Seahorse XF Cell Mito Stress Test Kit and in vitro Seahorse XF Cell Glycolysis Stress Test Kit sections.
In vitro Seahorse XF Cell Mito Stress Test Kit:
Day 2, cells were first rinsed with 7.4 pH Seahorse media containing 1mM Sodium Pyruvate, 2mM Glutamax, 7mM Glucose and incubated in 625uL of Seahorse media for 1h at 37°C in a non-CO2 incubator with 7.4 pH Seahorse media. This 1-hour incubation is necessary for de-gassing the plate, allowing for CO2 diffusion from the cells, medium, and plate. In parallel, each modulator Olygomycin (inhibition of ATP synthase complex V), Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) (uncoupling agent that collapses the proton gradient, disrupts mitochondrial membrane potential, resulting maximum oxygen comsumption by complex IV, maximal OCR), rotenone and antimycin-A (inhibitor of complex I and inhibitor of complex III respectively) are prepared individually at the indicated concentration in 3mL of Seahorse media: for oligomycin 3μL of 10mM oligomycin (final concentration 10uM), for FCCP 9μL of 10mM FCCP (final concentration 30 μM), for Rotenone 3μL of 1mM rotenone is combined with 3μL (final concentration 1uM) of 1mM Antimycin-A (final concentration 1μM). Agilent Seahorse XFe24 extracellular flux assay plate containing all modulators is placed into the Seahorse Agilent instrument for calibration. The Sensor plate will calibrate for ~25 mins. Cell plate is then placed in the instrument. OCR and ECAR values were determined using the XF24 Extracellular Flux Analyzer (Seahorse Bioscience) following the manufacturers’ protocols. For electron-flow (EF) measurements, Oxygen Consumption Rate (OCR) and extracellular acidification rate (ECAR) data were obtained first at the basal level according to basal reading: 3 times: Mix 3 min, wait 0 min, measure 3 min. Then, OCR and ECAR were measured following sequential additions of modulators of respiration into cell during assay using the built-in injection ports on XF sensor cartridge. After basal measurement, 75uL of oligomycin from port A (final concentration in the well 1uM is injected and mixed into each well of the cell plate and OCR and ECAR are measured 3 times: Mix 3 min, wait 0 min, measure 3 min (concentration in the well: 1.20μM oligomycin) (inhibition of Complex V). Then, 83μL of 30μM FCCP is injected and mixed to each well of the cell plate (concentration in the well: 4μM FCCP) and OCR and ECAR are measured 5 times: Mix 3 min, wait 0 min, measure 3 min (Maximal respiration rate). Finally, 93μL of the mix 1 μM rotenone / antimycin-A is injected to each well of the cell plate (concentration in the well: 0.15 μM rotenone and 0.15 μM antimycin-A) 3 times: Mix 3 min, wait 0 min, measure 3 min (non-mitochondrial oxygen).
In vitro Seahorse XF Cell Glycolysis Stress Test Kit:
Day 2, cells were first rinsed with 7.4 pH Seahorse media containing 2mM Glutamax without glucose or pyruvate (glycolysis stress media). Cells are incubated in 625uL of (glycolysis stress media) for 1 hour in t 37°C in a non-CO2 incubator. In parallel, 30mL of Glucose (20mM), 3mL Oligomycin (1μM) and 3mL 2-deoxyglucose (100mM) are prepared. 80 uL of Glucose is placed in each port A, 80 uL of Oligomycin in each port B and 80 uL of 2DG in each port C of the Agilent Seahorse XFe24 extracellular flux assay plate. The glycolysis stress test starts with a baseline measurement of the extracellular acidification rate ECAR in the previously starved cells for 1h in 625uL of Seahorse media containing 2mM Glutamax without glucose or pyruvate. For glycolysis measurements, ECAR data were obtained following sequential additions of glucose (20mM), Oligomycin (1 μM), and 2-deoxyglucose (100mM). Agilent Seahorse XFe24 extracellular flux assay plate containing all modulators is placed into the Seahorse Agilent instrument for calibration. The Sensor plate will calibrate for ~25 mins. Cell plate is then placed in the instrument. OCR and ECAR values were determined using the XF24 Extracellular Flux Analyzer (Seahorse Bioscience) following the manufacturers’ protocols for glycolysis stress test. First, basal acidification of media is measured in starved cells as following 3 times: Mix 3 min, wait 0 min, measure 3 min. Then 80 uL of 20 mM glucose is injected and mixed to each well (2.6mM final concentration in the well). ECAR is measured as following 3 times: Mix 3 min, wait 0 min, measure 3 min. Then, 80 uL of 1uM Oligomycin is injected and mixed to each well ( inal concentration of Oligomycin in the well: 0.12 μM). ECAR is measured as following 3 times: Mix 3 min, wait 0 min, measure 3 min and correspond to the maximal glycolytic rate relying on the glycogen stock of the cells. Finally, 80 uL of 2DG is injected and mixed in each well (final concentration of 2DG in the well: 12.80 mM). ECAR is measured as following 3 times: Mix 3 min, wait 0 min, measure 3 min and corresponds to the non-glycolotic residual acidification.
In vivo Seahorse on cells isolated from HFD or CD fed mice -
Cell were sorted from 3 day control diet or 3 day HFD fed wild-type mice. Cells from each mouse and conditions were seeded directly in the Seahorse plate after sorting. Seahorse experiments were performed the following morning. In these particular in vivo experiments, number of cells sorted might vary regarding mouse and diet. Moreover, seeding cells after sorting increased the risk of cell death that can vary between conditions and cell types. In effort to limit any variabilities from cell death or number, seahorse data are normalized by protein content. Mito Stress Test or Glycolysis Stress Test were performed as described in In vitro Seahorse XF Cell Mito Stress Test Kit section.
Mitochondrial DNA Content -
Mitochondria DNA Content was assessed by absolute quantification using real time PCR. Primers for mouse MtDNA were adopted from Malik et al. Fwd: 5’CTAGAAACCCCGAAACCAAA3’ and Rev: 5’ CCAGCTATCACCAAGCTCGT3 and mouse B2M primers Fwd: 5’ ATGGGAAGCCGAACATACTG3’ and Rev: 5’ CAGTCTCAGTGGGGGTGAA3’ were used to amplify mouse genomic DNA as the nuclear DNA control (Malik et al., 2016). DNA was isolated from cell samples using QIAamp DNA Micro Kit (Qiagen). PCR products were purified and dilutions standards were prepared with a range of 102–108 copies per 2μl for absolute quantification. MtDNA and nucDNA copy number per cell were determined from template DNA by qPCR using a Quant Studio 6 Flox (Applied Biosystems) and PowerUP SYBR Master Mix (Applied Biosystems) using the following protocol: preincubation at 95°C for 20s; denaturation at 95°C for 1s; annealing and extension at 60°C for 20s (repeated denaturation and extension for 40 cycles). Specificity of the primers were confirmed by a single melt peak after melt curve analysis.
Western Blots –
Protein was extracted from cells by homogenization in RIPA buffer supplemented with protease inhibitor. Protein content were measured using Pierce™ BCA Protein Assay Kit. Proteins were resolved on 4–12% bis-Tris gels and transferred 30min to nitrocellulose membrane using BioRad Trans-Blot® Turbo™ transfer system. The blots were then incubated overnight at 4°C with primary antibody. Rabbit polyclonal antibody against mouse MitoNEET was utilized at 1:1000 as previous described (Kusminski et al., 2012). Total OXPHOS Rodent antibody cocktail was used at 1:1000 and 10ug of Rat Heart Tissue Lysate- Mitochondria extract was used as positive control in OXPHOS Western blotting. Rabbit Anti-Prohibitin antibody - Mitochondrial Marker was used at 1:1000 dilution. Monoclonal Anti-β-Actin antibody was used at 1:10 000 dilution. Primary antibodies were detected using secondary immunoglobulin Gs labeled with infrared dyes emitting at 700 nm or 800 nm. Membrane was incubated with secondary antibody for 1h at room temperature. Secondary IR-Dye 800CW Donkey antibody against rabbit was used at 1:5000. Secondary IR-Dye 680RD Donkey antibody against Mouse was used at 1:5000. Secondary IR-Dye 800CW Goat antibody against Mouse was used at 1:5000. Western blots were then visualized on a Li-Cor Odyssey infrared scanner (Li-Cor Bioscience). The scanned data were analyzed using ImageJ software
QUANTIFICATION AND STATISTICAL ANALYSIS
No statistical method was used to predetermine sample size. The experiments were not blind. Data are presented as mean ± standard error of the mean (SEM). All statiscal analyses were perfomed using Prism GraphPad Prism Software. Differences between multiple conditions and two groups over time were evaluated using two-way ANOVA with Tukey’s post-test for multiple comparisons. For comparisons between several different groups a Kruskal-Wallis (One-Way Anova) test with Dunn’s post-test for multiple comparisons. For comparisons between two independent groups, a Student’s t-test was utilized. *p<0.05, ** p<0.01, ***p<0.001, ****p<0.0001. For primary cell culture experiments, the n value corresponds to a cell preparation from separate mice. For all mouse studies, the n value corresponds to individual mice of a given treatment.
Supplementary Material
Highlights.
Adipogenic and inflammatory precursors have distinct metabolic properties.
Mitochondrial activity is crucial for precursors to differentiate into mature adipocytes.
Mitochondrial dysfunction promotes inflammatory characteristics of mural cells.
Maintenance of mural identity requires proper mitochondrial function.
Acknowledgements
We thank the following UT Southwestern Core Units: Histology Core for assistance in embedding and processing of tissue samples, the Metabolic Core for hepatic lipid content measurements. We also thank The Flow Cytometry Core at the Children’s Medical Center Research Institute at UT Southwestern. We would like to acknowledge the help of Dr. Jan-Bernd Funcke for the graphical abstract. This study was supported by US National Institutes of Health (NIH) grants R01-DK55758, P01-DK088761, R01-DK099110 and P01-AG051459 (P.E.S.), F32-DK113704 (C.C), F31-DK113696 (C.H)., R01-DK104789 and R01 DK119163 (R.K.G), RC2-DK118620 (P.E.S. & R.K.G.). N.J. was supported by a post-doctoral fellowship from the Lipedema Foundation (LFA #18).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Atashi F, Modarressi A, and Pepper MS (2015). The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: a review. Stem Cells Dev 24, 1150–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burl RB, Ramseyer VD, Rondini EA, Pique-Regi R, Lee YH, and Granneman JG (2018). Deconstructing Adipogenesis Induced by beta3-Adrenergic Receptor Activation with Single-Cell Expression Profiling. Cell Metab 28, 300–309 e304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Wu YT, and Wei YH (2015). Depletion of mitoferrins leads to mitochondrial dysfunction and impairment of adipogenic differentiation in 3T3-L1 preadipocytes. Free Radic Res 49, 1285–1295. [DOI] [PubMed] [Google Scholar]
- Choi JW, Jo A, Kim M, Park HS, Chung SS, Kang S, and Park KS (2016). BNIP3 is essential for mitochondrial bioenergetics during adipocyte remodelling in mice. Diabetologia 59, 571–581. [DOI] [PubMed] [Google Scholar]
- Donnelly RP, Loftus RM, Keating SE, Liou KT, Biron CA, Gardiner CM, and Finlay DK (2014). mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J Immunol 193, 4477–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doughty CA, Bleiman BF, Wagner DJ, Dufort FJ, Mataraza JM, Roberts MF, and Chiles TC (2006). Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood 107, 4458–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejarque M, Ceperuelo-Mallafre V, Serena C, Maymo-Masip E, Duran X, Diaz-Ramos A, Millan-Scheiding M, Nunez-Alvarez Y, Nunez-Roa C, Gama P, et al. (2019). Adipose tissue mitochondrial dysfunction in human obesity is linked to a specific DNA methylation signature in adipose-derived stem cells. Int J Obes (Lond) 43, 1256–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facucho-Oliveira JM, and St John JC (2009). The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev Rep 5, 140–158. [DOI] [PubMed] [Google Scholar]
- Gupta RK, Mepani RJ, Kleiner S, Lo JC, Khandekar MJ, Cohen P, Frontini A, Bhowmick DC, Ye L, Cinti S, et al. (2012). Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab 15, 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler C, Shan B, Zhang Q, Henry GH, Shao M, Vishvanath L, Ghaben AL, Mobley AB, Strand D, Hon GC, et al. (2018). Identification of functionally distinct fibro-inflammatory and adipogenic stromal subpopulations in visceral adipose tissue of adult mice. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch J, and Han PW (1969). Cellularity of rat adipose tissue: effects of growth, starvation, and obesity. J Lipid Res 10, 77–82. [PubMed] [Google Scholar]
- Jang YY, and Sharkis SJ (2007). A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 110, 3056–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khacho M, Clark A, Svoboda DS, Azzi J, MacLaurin JG, Meghaizel C, Sesaki H, Lagace DC, Germain M, Harper ME, et al. (2016). Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 19, 232–247. [DOI] [PubMed] [Google Scholar]
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, et al. (2010). Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusminski CM, Chen S, Ye R, Sun K, Wang QA, Spurgin SB, Sanders PE, Brozinick JT, Geldenhuys WJ, Li WH, et al. (2016). MitoNEET-Parkin Effects in Pancreatic alpha- and beta-Cells, Cellular Survival, and Intrainsular Cross Talk. Diabetes 65, 1534–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusminski CM, Holland WL, Sun K, Park J, Spurgin SB, Lin Y, Askew GR, Simcox JA, McClain DA, Li C, et al. (2012). MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat Med 18, 1539–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusminski CM, Park J, and Scherer PE (2014). MitoNEET-mediated effects on browning of white adipose tissue. Nat Commun 5, 3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Lonergan T, Bavister B, and Brenner C (2007). Mitochondria in stem cells. Mitochondrion 7, 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysdahl H, Baatrup A, Nielsen AB, Foldager CB, and Bunger C (2013). Phenol red inhibits chondrogenic differentiation and affects osteogenic differentiation of human mesenchymal stem cells in vitro. Stem Cell Rev Rep 9, 132–139. [DOI] [PubMed] [Google Scholar]
- Malik AN, Czajka A, and Cunningham P (2016). Accurate quantification of mouse mitochondrial DNA without co-amplification of nuclear mitochondrial insertion sequences. Mitochondrion 29, 59–64. [DOI] [PubMed] [Google Scholar]
- Merrick D, Sakers A, Irgebay Z, Okada C, Calvert C, Morley MP, Percec I, and Seale P (2019). Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Science 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, and Rathmell JC (2011). Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 186, 3299–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, and Choi Y (2009). Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peics J, Vishvanath L, Zhang Q, Shan B, Pedersen TA, and Gupta RK (2020). Isolation of Adipogenic and Fibro-Inflammatory Stromal Cell Subpopulations from Murine Intra-Abdominal Adipose Depots. J Vis Exp. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, Cascante M, and Bosca L (2010). Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 185, 605–614. [DOI] [PubMed] [Google Scholar]
- Seo JB, Riopel M, Cabrales P, Huh JY, Bandyopadhyay GK, Andreyev AY, Murphy AN, Beeman SC, Smith GI, Klein S, et al. (2019). Knockdown of Ant2 Reduces Adipocyte Hypoxia And Improves Insulin Resistance in Obesity. Nat Metab 1, 86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B, Shao M, Zhang Q, Hepler C, Paschoal VA, Barnes SD, Vishvanath L, An YA, Jia L, Malladi VS, et al. (2020). Perivascular mesenchymal cells control adipose-tissue macrophage accrual in obesity. Nat Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, and Chi H (2011). HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 208, 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishvanath L, MacPherson KA, Hepler C, Wang QA, Shao M, Spurgin SB, Wang MY, Kusminski CM, Morley TS, and Gupta RK (2016). Pdgfrbeta+ Mural Preadipocytes Contribute to Adipocyte Hyperplasia Induced by High-Fat-Diet Feeding and Prolonged Cold Exposure in Adult Mice. Cell Metab 23, 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any code or large dataset.