

Abstract
Cigarette smoking is the single most important risk factor for the development of cardiovascular diseases (CVD). However, the role of nicotine, the addictive component of all tobacco products, in the development of CVD is incompletely understood. Although increased public awareness of the harms of cigarette smoking has successfully led to a decline in its prevalence, the use of electronic cigarettes (e-cig) or electronic nicotine delivery system has increased dramatically in recent years due to the perception that these products are safe. This review summarizes our current knowledge of the expression and function of the nicotinic acetylcholine receptors in the cardiovascular system and the impact of nicotine exposure on cardiovascular health, with a focus on nicotine-induced vascular dysfunction. Nicotine alters vasoreactivity through endothelium-dependent and/or endothelium-independent mechanisms, leading to clinical manifestations in both cigarette smokers and e-cig users. In addition, nicotine induces vascular remodeling through its effects on proliferation, migration, and matrix production of both vascular endothelial and vascular smooth muscle cells. The purpose of this review is to identify critical knowledge gaps regarding the effects of nicotine on the vasculature and to stimulate continued nicotine research.
Keywords: nicotine, nAChR, vascular dysfunction, endothelial cells, vascular smooth muscle cells
Introduction
Cigarette smoking is the single most important risk factor for the development of cardiovascular diseases (CVD), and smokers are 2–4 times more likely to develop CVD than non-smokers.1 As summarized in “The Health Consequences of Smoking-50 Years of Progress: A Report of the Surgeon General,” the evidence is sufficient to infer a causal relationship between smoking and coronary heart disease, atherosclerotic aortic aneurysm, cerebrovascular disease, stroke, and all-cause mortality. Nicotine is the addictive component of all tobacco products; however, the role of nicotine in the development of CVD is incompletely understood. Although increased public awareness of the harms of cigarette smoking has successfully led to a decline in its prevalence, the use of electronic cigarettes (e-cig) or electronic nicotine delivery system has increased dramatically in recent years due to the perception that these products are safe. This review summarizes our current knowledge of the expression and function of the nicotinic acetylcholine receptors (nAChR) in the cardiovascular system and the impact of nicotine exposure on cardiovascular health, with a focus on nicotine-induced vascular dysfunction. The purpose of this review is to identify critical knowledge gaps regarding the effects of nicotine on the vasculature and to stimulate continued nicotine research.
Overview of Nicotinic Acetylcholine Receptors
The nAChR belong to the family of ligand-gated cation channels and are ubiquitously expressed in the central nervous system.2 Mature nAChR are pentameric structures of different subunit combinations in defined stoichiometries (humans have 9 genetically distinct ligand-binding α-subunits and 7 modulatory non-α-subunits). The nAChR are divided into three major subtypes (Figure 1A): muscle-type, heteromeric and homomeric nAChR. The muscle-type nAChR consists of (α1)2β1δε (adult) or (α1)2β1δγ (fetal) and is located at the neuromuscular junction, and this receptor is poorly responsive to nicotine. The heteromeric nAChR are composed of five subunits in various combinations of α and β subunits, and this type is predominately expressed on neuronal cells such as (α4)2(β2)3-nAChR implicated in nicotine addiction and (α3)2(β4)3-nAChR involved in neurotransmission in the autonomic nervous system. The homomeric nAChR include the (α7)5 and (α9)5 receptors and are expressed by not only neurons but also many non-neuronal cells, with the α7-nAChR being the most studied subtype.
Figure 1. Nicotinic Acetylcholine Receptors (nAChR).
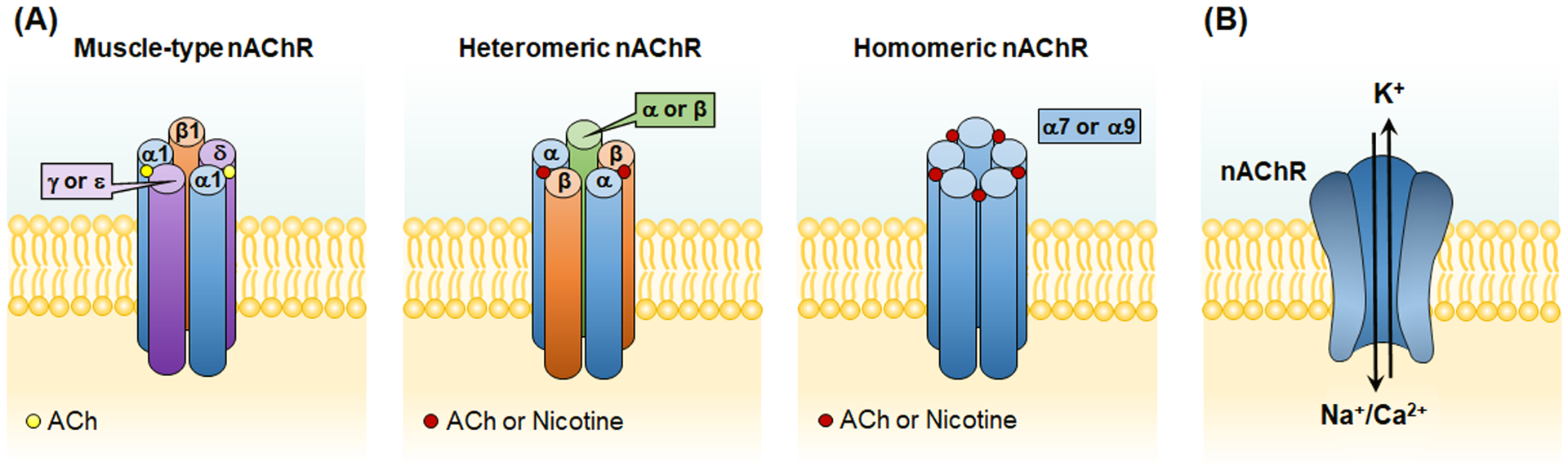
(A) The nAChR are divided into three major subtypes: muscle-type, heteromeric and homomeric nAChR. The muscle-type nAChR consists of (α1)2β1δε (adult) or (α1)2β1δγ (fetal) and is located at the neuromuscular junction, and this receptor is poorly responsive to nicotine. The heteromeric nAChR are composed of five subunits in various combinations of α and β subunits, and the homomeric nAChR are composed of five α subunits (α7 or α9). Ligands (ACh or Nicotine) bind to the α subunits at the subunit interface. (B) The nAChR are ligand-gated cation channels. Upon binding to endogenous ligand ACh or nicotine, the central pore of nAChR opens to allow the flow of cations (Na+ and Ca2+ into and K+ out of the cells). ACh, acetylcholine.
Upon binding to endogenous ligand acetylcholine (ACh) or nicotine, the central pore of nAChR opens to allow the flow of cations (Na+ and Ca2+ into and K+ out of the cells, Figure 1B), resulting in membrane depolarization or activation of intracellular calcium-mediated signaling pathways if calcium permeability is sufficient. Due to its unusual homomeric subunit composition and the presence of five ligand binding sites, α7-nAChR exhibits high permeability to calcium, enough to couple the activity of this receptor to intracellular calcium signaling pathways. Nicotine’s effect on the cardiovascular system is conferred through its ability to bind endogenous nAChR in place of the endogenous agonist ACh.
Overview of Nicotine on Hemodynamics
It is well-recognized and documented that nicotine in tobacco products imposes hemodynamic effects.3 These include acute changes in heart rate (HR) and increases in myocardial contractility and blood pressure (BP).4–7 Treatment with nicotine or a nicotinic agonist induces a brief but pronounced decrease in HR, followed by significant increases in both HR and BP.7,8 The initial parasympathetic bradycardic response has been shown to be mediated by activation of α4β2-nAChR, whereas the subsequent sympathetic tachycardic and pressor responses are mediated by α7-nAChR.8 In addition, inhaled nicotine equivalent to cigarette smoking induces high magnitude fluctuations of BP, irregular pulse BP and cardiac arrhythmia.9 The α7-nAChR also participates in baroreflex regulation that maintains BP homeostasis, as α7-nAChR knockout (KO) mice have impaired sympathetic tachycardic response to sodium nitroprusside (SNP)-induced vasodilation.10
In the medulla, injection of nicotine into the nucleus of the solitary tract (NTS) and area postrema elicits bradycardia and hypotension; in contrast, injection of nicotine into the rostral ventrolateral medulla (RVLM) produces dose-dependent and long-lasting increases in both systolic and diastolic BP.11 Nicotine can activate NTS catecholamine neurons directly through α4β2-nAChR and indirectly through increased glutamate release via α7-nAChR.12 The cardiovascular effects of central α7- and α4β2-nAChR activation have also been shown to involve the release of vasopressin.13 On peripheral postganglionic sympathetic nerve endings including the adrenal medulla, nicotine stimulation of nAChR results in catecholamine release, more potently than ACh.14 Because an increase in cytosolic calcium is a prerequisite for chromaffin cell exocytosis,15 it is likely that α7, α9, and/or α9/α10 nAChR activation would trigger instructive signaling cascades contributing to catecholamine release from these cells.14
The effects of nicotine on long-term BP control is controversial. Although surveys of outpatient BP measurement report that smokers have either similar or slightly lower BP compared to matched nonsmokers,16,17 studies of ambulatory BP monitoring show that long-term cigarette smoking increases average HR and BP throughout the day.18,19 Findings from our laboratory show that chronic nicotine inhalation in mice leads to a transient increase in BP.20 Mice exposed to nicotine (daily 12 h on/12 h off) initially exhibited elevations in both systolic and diastolic BP (weeks 1–3), which then returned to baseline following prolonged exposure (weeks 4–8), indicating development of tolerance and/or activation of compensatory mechanisms. In addition, the BP increase within the first week of nicotine exposure was associated with a lack of systolic BP dipping, which is considered a risk factor for cardiovascular diseases and end organ damage.21,22 Importantly, our study found that 8-week nicotine inhalation exposure leads to elevation in pulmonary BP (pulmonary hypertension) with pulmonary vascular and right ventricular remodeling.20 Interestingly, in RV samples from patients with pulmonary arterial hypertension, α7-nAChR expression was increased and acetylcholinesterase activity was reduced versus controls.23
Nicotine and Vascular Reactivity
Both vascular endothelial cells (EC) and vascular smooth muscle cells (VSMC) express multiple α and β subunits of nAChR,24–28 rendering the vasculature a direct target of nicotine. In terms of vascular reactivity, nicotine exerts primarily vasoconstrictive effects through endothelium-dependent and/or endothelium-independent mechanisms (Figure 2).
Figure 2. Nicotine alters vascular reactivity through endothelium-dependent and endothelium-independent mechanisms.
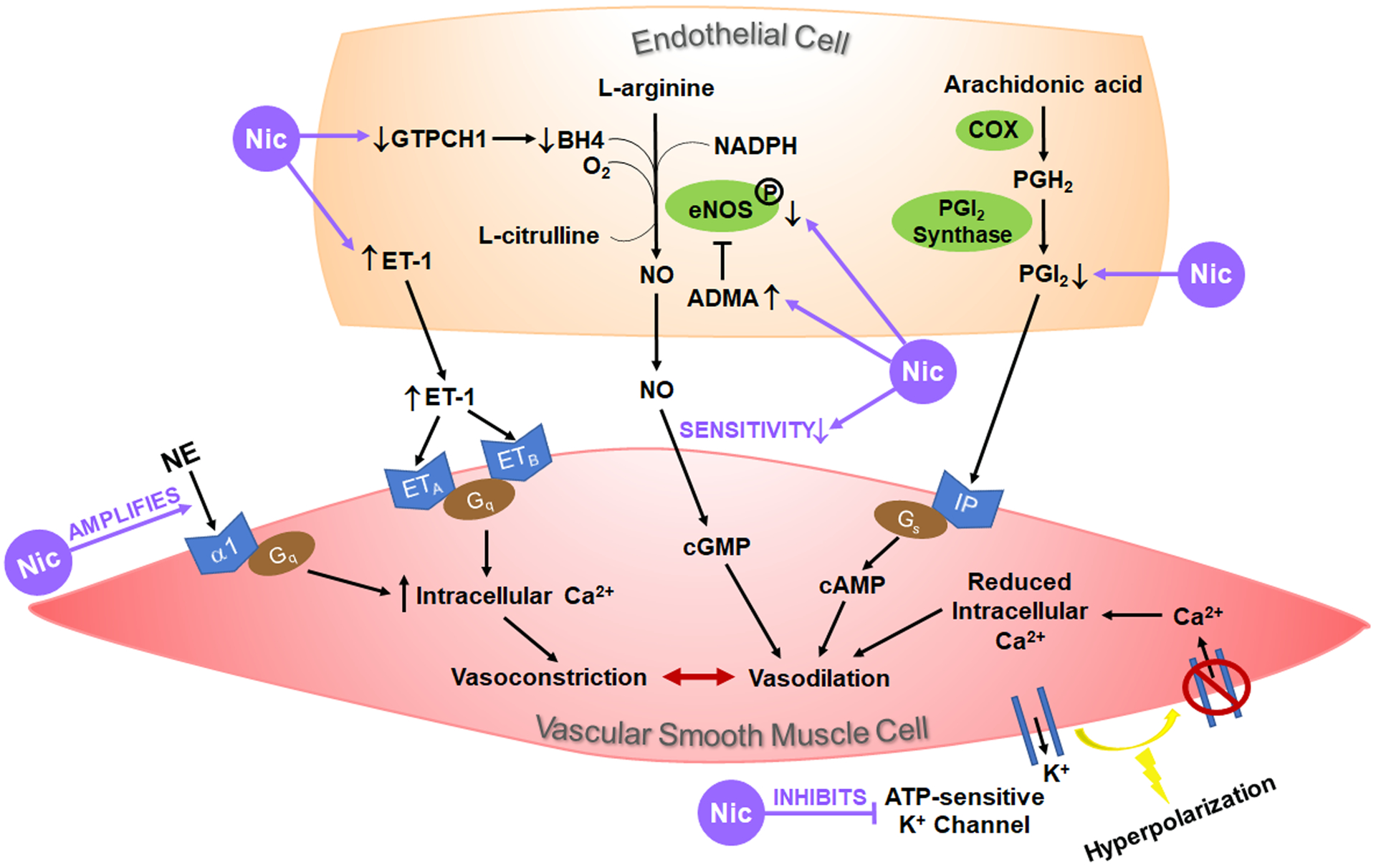
Activation of the endothelium elicits a multitude of pathways with the production of vasoactive substances that travel to the underlying vascular smooth muscle cells (VSMC) to induce vasoconstriction or vasodilation. Nicotine (Nic) has been shown to upregulate the production or release of the vasoconstrictor endothelin-1 (ET-1) and inhibit endothelial production of the vasodilators nitric oxide (NO) and prostacyclin (PGI2). In VSMC, nicotine promotes VSMC contraction by amplifying its response to norepinephrine (NE), by upregulating ET-1 receptor (ETA and/or ETB) expression, and/or by inhibiting ATP-sensitive K+ channels. Up- and down-regulation by nicotine are indicated by ↑ and ↓, respectively. ADMA, asymmetric dimethylarginine (the endogenous eNOS inhibitor); BH4, tetrahydrobiopterin (an essential cofactor for eNOS); COX, cyclooxygenase; eNOS, endothelial nitric oxide synthase; GTPCH1, GTP cyclohydrolase 1 (the rate-limiting enzyme for BH4 production); PGH2, prostaglandin H2.
Nicotine Impairs Endothelium-Dependent Vasodilation
Activation of the endothelium can elicit a multitude of pathways with the production of vasoactive substances that travel to the underlying VSMC to induce vasoconstriction or vasodilation, and these include the endothelin, nitric oxide (NO) and prostacyclin pathways.29 Both human and animal studies have implicated nicotine’s involvement in altering these pathways, hindering blood vessels’ ability to dilate.
Endothelin-1 (ET-1) is a potent vasoconstrictor produced by vascular EC and plays a vital role in maintaining vascular tone. It acts on two G protein-coupled receptors (GPCR), ETA and ETB, located on the underlying VSMC to activate pathways leading to vasoconstriction. To maintain cardiovascular balance, some ET-1 may also bind ETB receptors on the EC to induce vasodilatory effects via formation of prostacyclin or NO.30 Cigarette smoking has been associated with elevated blood ET-1 levels in healthy smokers,31 and pulmonary arteries from smokers and chronic obstructive pulmonary disease patients showed a higher expression of ETA and ETB.32 Cigarette smoke exposure also induces ET-1 and ETA/ETB expression in experimental animals,33–35 and a recent study showed that e-cig exposure for 4 weeks in rats resulted in increased cardiac ET-1 levels.36 In addition, cigarette smoke extract induces ET-1 expression in cultured EC37 and ETA/ETB in cultured VSMC or arterial segments.32,38–40 The expression of ETA/ETB has been shown to be mediated by mitogen-activated protein kinase (MAPK) pathways and downstream transcription factors such as nuclear factor-κB.32,38–40 In contrast, exposure to nicotine alone in culture (24 h) fails to induce the expression of either ET-141 or ETA/ETB.38,39 However, nicotine was shown to increase ET-1 release from cultured human umbilical vein endothelial cells (HUVEC), with a maximal effect observed at 5 minutes post exposure.42 This acute nicotine-induced ET-1 release likely leads to alteration in vasoreactivity. For example, treatment with an ETA receptor antagonist blocked nicotine-induced increase of mean arterial BP in rats, suggesting nicotine’s acute pressor effect is mediated, at least in part, by ET-1 binding of ETA on VSMC.43 Similarly, another study showed that intraportal nicotine infusion in rats decreased hepatic blood flow in a dose-dependent manner through ET-1 and ETA/ETB receptors, while pretreatment with the ganglionic nicotinic receptor blocker hexamethonium attenuated nicotine’s effects.44 In addition, long-term nicotine exposure in animals has been shown to increase ET-1 expression. Nicotine administered orally for 28 days in male rats led to increased ET-1 expression in the aorta.45 Subcutaneous injection of nicotine (0.1 mg/kg per day) for 6 weeks in female ovariectomized rats resulted in increased plasma ET-1 levels, which can be prevented by estrogen replacement therapy, indicating the protective effects of estrogen.46 The role of nicotine in cigarette smoke-induced overexpression of ETA and/or ETB in vivo is not clear.
Another pathway by which endothelium-dependent vasodilation occurs is through the formation of NO from L-arginine catalyzed by endothelial NO synthase (eNOS).47 Cigarette smoking, e-cig use and nicotine administration have all been shown to reduce NO bioavailability.48,49 Nicotine treatment leads to reduced eNOS expression in cultured carotid and uterine arteries,50,51 and in nicotine-treated uterine arteries, the phosphorylation levels of eNOS (Ser1179) are also significantly decreased.50 In a recent study, nicotine exposure in HUVEC was shown to reduce the expression of GTP cyclohydrolase 1 (GTPCH1), the rate-limiting enzyme for the production of tetrahydrobiopterin (BH4), an essential cofactor for eNOS.52 Reduced bioavailability of cofactor BH4 leads to eNOS uncoupling and the generation of superoxide rather than NO.52 Importantly, dietary supplementation of BH4 attenuated nicotine-induced endothelial dysfunction in ApoE−/− mice.52 In addition, oral nicotine administration in experimental animals significantly increased plasma level or EC expression of the endogenous eNOS inhibitor, asymmetric dimethylarginine (ADMA),53,54 and this effect was mediated by nicotine-induced downregulation of dimethylarginine dimethylaminohydrolase (DDAH, a major hydrolase of ADMA) via activation of the α7-nAChR.53 Finally, multiple studies suggest that nicotine-induced production of reactive oxygen species (ROS) contributes to nicotine-induced impairment of NO-mediated endothelium-dependent vasodilation.55–57
Prostacyclin (PGI2) is an effective vasodilator produced by the vascular endothelium from arachidonic acid via pathways involving the enzymes cyclooxygenase and PGI2 synthase. PGI2 then acts on its receptors on VSMC (commonly referred to as IP receptors) to induce VSMC relaxation via the G protein (Gs) linked cAMP pathway.58 Although cigarette smoking has been associated with decreased synthesis of PGI2 in both humans and animal models,59–62 the role of nicotine on the production of PGI2 is less clear. In cultured HUVEC and bovine pulmonary artery EC, nicotine exposure at levels comparable to the plasma levels of human smokers did not affect the levels of PGI2.63,64 However, nicotine perfusion in human umbilical artery in vitro led to a decline in PGI2 production.65 In rabbits, 1-week nicotine infusion in vivo caused a significant reduction of PGI2 in the heart.66 In isolated and perfused rabbit hearts, however, nicotine perfusion alone did not alter PGI2 levels in the myocardium.67 Instead, this study showed that nicotine significantly worsened ischemia-induced decrease in PGI2.67 In rats, nicotine administered in drinking water for 10 days resulted in reduced plasma level of PGI2 in a dose-dependent manner68 and continuous subcutaneous infusion of nicotine over 7 days resulted in reduced PGI2 production in the aorta.69 The above studies indicate that the effects of nicotine on PGI2 may depend on the cell/tissue types as well as in vitro, ex vivo or in vivo conditions. In addition, the mechanisms through which nicotine regulates PGI2 synthesis or metabolism require further investigation.
A standard technique to examine endothelium-dependent vasodilatory response is through stimulation with ACh or methacholine. Both ACh and methacholine bind to muscarinic ACh receptors on vascular EC and induce vasodilation through endothelium-derived NO and/or prostaglandins.70 Aortas isolated from mice exposed to cigarette smoke or e-cig for 8 months exhibited reduced vasodilatory response to methacholine; in contrast, their responses to the NO donor SNP were not affected, indicating intact NO-cGMP signaling in VSMC.71 Importantly, human subjects who chewed nicotine gum also showed reduced vasodilation of the brachial artery in response to methacholine.72
Nicotine Impairs Endothelium-Independent Vasodilation
Most human and animal studies suggest that cigarette smoke, e-cig or nicotine exposure do not alter VSMC’s vasodilatory response to NO as SNP-induced vasodilation is generally preserved.71–73 When subjected to increasing doses of sublingual nitroglycerin, however, smokers exhibited significantly lower dose-response curve to nitroglycerin-induced brachial artery dilatation compared to matched non-smoking controls.74 This study suggests that smokers may suffer reduced sensitivity to NO-induced endothelium-independent vasodilation.
Nicotine may also diminish endothelium-independent vasodilation via interaction with ATP-sensitive K+ channels.75 These channels are ubiquitously expressed on VSMC and upon activation, the outflow of K+ results in hyperpolarization of the membrane and subsequent closure of voltage gated Ca2+ channels, leading to decreased intracellular Ca2+ and VSMC relaxation. In a hamster cheek pouch arteriole reactivity study, both acute and chronic nicotine treatment resulted in significantly reduced vasodilatory response to ATP-sensitive K+ channel activators, and this effect is likely mediated by nicotine-induced superoxide anion production as treatment with superoxide dismutase attenuated the effects of nicotine.75
Nicotine has also been shown to enhance VSMC’s vasoconstrictive response to α1 adrenoceptor agonist. Aortas isolated from mice exposed to cigarette smoke or e-cig for 8 months exhibited enhanced response to phenylephrine, indicating increased α1-adrenergic receptor activation on VSMC.71 In human skin vasculature, acute nicotine treatment has been shown to amplify norepinephrine (NE)-induced vasoconstriction.76 This enhanced constrictor response is likely specific to the NE signaling pathway, as nicotine treatment in rats selectively increased bone vascular constriction to NE, but not arginine vasopressin.77
Nicotine Induces Neurogenic Vascular Relaxation
Nicotine has been shown to induce neurogenic vascular relaxation in multiple vascular beds.78–80 Neurogenic relaxation of cerebral arteries is an important mechanism of maintaining adequate blood flow to the brain, serving as a protective mechanism to meet O2 demand in an acutely stressful situation. Stimulation of nAChR located on perivascular sympathetic nerves by nicotine causes NE release from nerve terminals in cerebral arteries, which subsequently induces NO production in the neighboring cholinergic-nitrergic nerves via β2-adrenergic receptors, leading to nitrergic dilations of cerebral arteries.81,82 The α3β2- and α7-nAChR expressed by perivascular sympathetic nerves contribute to nicotine-induced nitrergic neurogenic vasodilation.81,82 It is important to note, however, that chronic exposure to nicotine promotes oxidative stress and endothelial dysfunction, eventually leading to hypoperfusion of the cerebral arteries.49,83
Nicotine and Vascular Remodeling
In addition to altered vasoreactivity, nicotine has been shown to impact survival, proliferation, migration, as well as matrix production in both EC and VSMC, leading to vascular remodeling (Table 1 and Figure 3).
Table 1.
Summary of Nicotine’s Effects on Vascular Remodeling.
| Vascular Component | Nicotine Exposure | Nicotine’s Effects | |
|---|---|---|---|
| EC | Acute | in vitro (≤ 96 h) | Increased DNA synthesis89 and cell proliferation;85,89,91 increased PDGF BB release;92 increased VEGF* and activation of VEGF receptor 2;84 inhibition of apoptosis;85,93 cytoskeletal reorganization;92 increased cell migration and tube formation*84,85,90,91,95 |
| in vivo (≤ 3 weeks) | Increased vascular growth/capillary density*84,85 | ||
| Chronic | in vitro (> 96 h) | Decreased cell migration and tube formation;95,96 inhibition of apoptosis96 | |
| in vivo (> 3 weeks) | Blunting of nicotine’s acute angiogenic effect95 | ||
| VSMC | Acute | in vitro (≤96 h) | Increased DNA synthesis*99–104 and cell proliferation; *99,101,108 increased PDGF release;102 inhibition of apoptosis;*100 cytoskeletal reorganization and podosome structure alterations;*102,105,106 increased cell migration*105,106,108,109 |
| in vivo (≤ 3 weeks) | None reported | ||
| Chronic | in vitro (> 96 h) | None reported | |
| in vivo (> 3 weeks) | Increased median thickness of the thoracic arteries;108 morphological changes and neointima formation;111 increased mitoses111 | ||
| ECM | Acute | in vitro (≤ 96 h) | Increased collagen production;112,116 increased MMP-2/9113,118 |
| in vivo (≤ 3 weeks) | Increased gelatinase activity113 | ||
| Chronic | in vitro (> 96 h) | Increased collagen production*117 | |
| in vivo (> 3 weeks) | Increased MMP-2/9;*112–114,118 increased gelatinase activity;113 increased elastolytic activity;114 collagen and fibronectin accumulation;112 elastin thinning and fragmentation112,114 | ||
All observations summarized here were at nicotine concentrations ≤10−6 M.
Effect has been shown to be mediated, at least in part, by α7-nAChR.
Figure 3. Nicotine and Vascular Remodeling.
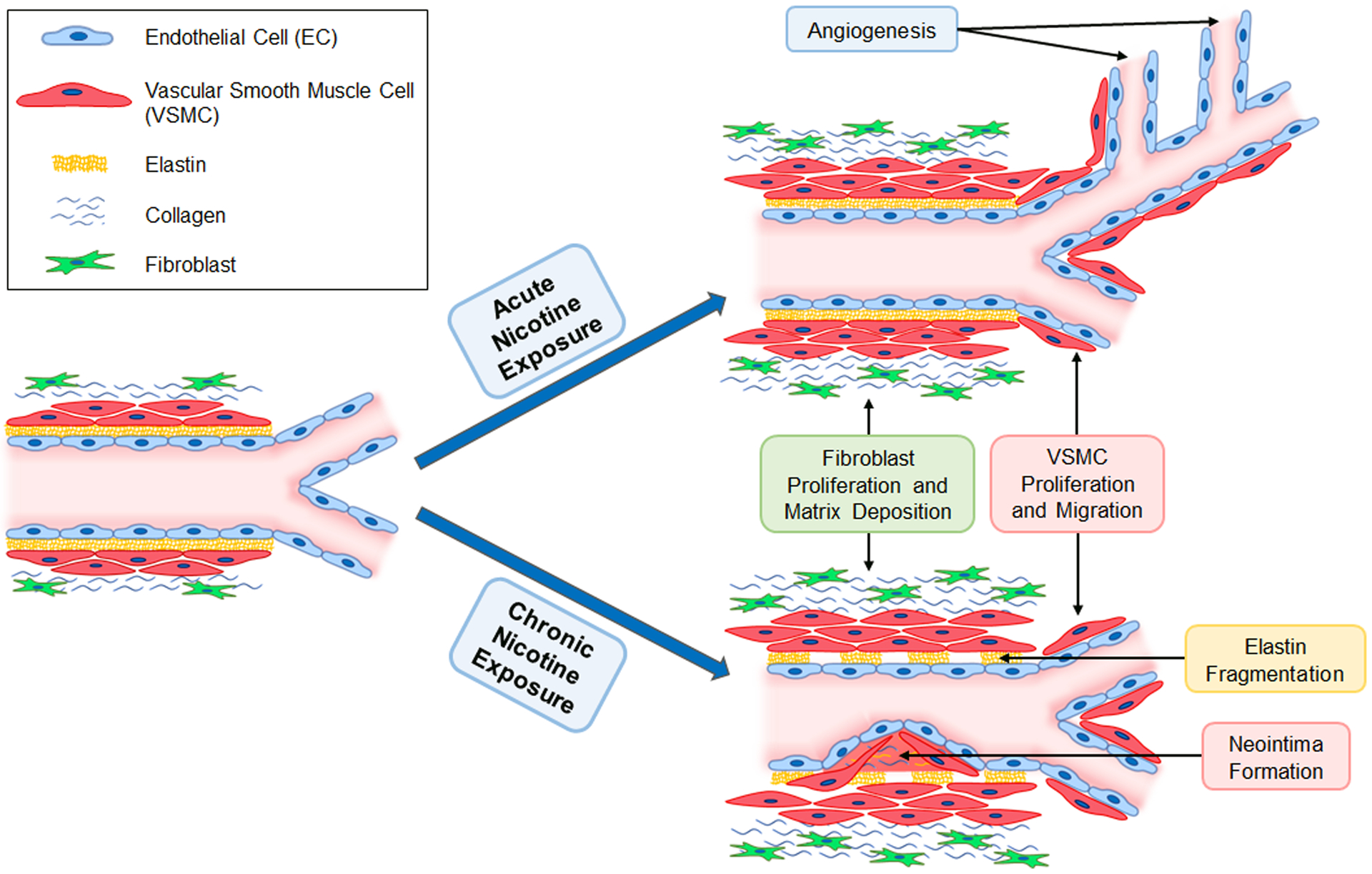
Acute nicotine exposure at concentrations similar to those found in human smokers promotes angiogenesis, whereas chronic exposure to nicotine blunts its proangiogenic response. Both acute and chronic nicotine exposure lead to fibroblast proliferation, extracellular matrix deposition, and vascular smooth muscle cell (VSMC) proliferation and migration. In addition, chronic nicotine exposure is associated with elastin fragmentation and neointima formation.
Nicotine and EC Remodeling
Nicotine at concentrations similar to those found in human plasma after smoking promotes angiogenesis in a variety of models,84,85 and its role in diseases involving pathological angiogenesis has been reviewed in depth.86–88 Nicotine’s proangiogenic effects have been shown to be partly mediated by the α7-nAChR through vascular endothelial growth factor (VEGF), phosphatidylinositol 3-kinase (PI3K) and MAPK signaling pathways.84 Furthermore, the α7-nAChR agonist 3-(2, 4)-dimethoxybenzylidene anabaseine (DMXB) mimics nicotine’s proangiogenic effects.84 Nicotine increases DNA synthesis and cell proliferation in EC in vitro.85,89–91 Acute nicotine exposure also induces EC migration and the formation of capillary-like structures in vitro in a manner similar to that produced by common angiogenic factors,91 and these effects are attenuated by α7-nAChR antagonism.90 Furthermore, calf aortic EC displayed cytoskeletal reorganization of actin filaments and vimentin after stimulation with nicotine, which was shown to be mediated by increased EC release of the homodimer platelet-derived growth factor (PDGF) BB.92 In human coronary artery EC treated with proapoptotic factors, nicotine exposure reduced the number of apoptotic cells as compared to controls, indicating that nicotine has antiapoptotic activity.93 In addition, treatment with the selective α7-nAChR agonist, PNU282987, decreased HUVEC apoptosis in response to radiation exposure.94 Furthermore, no morphological changes consistent with cytotoxicity are observed in EC after exposure to nicotine at concentrations similar to those seen in habitual smokers.91
The aforementioned proangiogenic effects in vitro and in vivo were observed following acute exposures to nicotine. In a murine hindlimb ischemia model, short-term exposure to nicotine (2 weeks in drinking water starting at the time of ischemia surgery) promoted vascular sprouting consistent with the previous findings, however, prior exposure to nicotine for 16 weeks (before the surgery) abolished the effects of nicotine to increase capillary density in the ischemic hindlimb.95 It should be noted that prior exposure to nicotine for 8 weeks did not impact the proangiogenic effect of nicotine, further indicating that angiogenic blunting is due to prolonged exposure. In addition, capillary sprouting was decreased in aortic segments isolated from mice exposed to nicotine in their drinking water for 52 weeks compared to those from vehicle treated control mice.95 In vitro, chronic nicotine exposure for 2 weeks led to decreased cell migration and tube formation.96 Interestingly, chronic nicotine exposure still resulted in decreased EC apoptosis.96 The effects of chronic exposure to nicotine were accompanied by attenuation of nicotine-induced VEGF release.95 Finally, the activation of α9-nAChR has been shown to be anti-proliferative in EC and opposes the action of α7-nAChR.26
The effects of nicotine on EC are likely dose dependent, as EC exposed to nicotine concentrations higher than those seen in habitual smokers (>10−6 M) do not exhibit changes in proliferation and instead display increased release of lactate dehydrogenase and morphological changes consistent with cytotoxicity.91 Similar dose dependency has also been demonstrated in vivo in a murine hindlimb ischemia model.85 Taken together, the effects of nicotine on EC remodeling and angiogenesis vary depending on both the dosage and length of exposure. Thus, further studies are needed to better elucidate the chronic effects of nicotine on EC in habitual users of inhaled nicotine products.
Nicotine and VSMC Remodeling
VSMC reprogramming and dysfunction in response to external stress play important roles in the pathogenesis of vascular diseases.97,98 Acute nicotine exposure at concentrations similar to levels in habitual smokers increases DNA synthesis, promotes proliferation, and protects against apoptosis in VSMC.99–103 Cotinine, an active metabolite of nicotine, is an even greater inducer of DNA synthesis.99 Nicotine’s mitogenic effect on VSMC has been shown to be mediated by basic fibroblast growth factor, transforming growth factor (TGF)-β and autocrine PDGF signaling.101,102,104 In addition, nicotine induces reorganization of cytoskeletal structures in VSMC, including changes in α-actin, vimentin, and β-tubulin.102,105 In response to PKC activation, both nicotine and cigarette smoke extract induce actin cytoskeletal remodeling in the form of podosomes,106 a hallmark of invasive cells.107 The above cytoskeletal changes are important in nicotine-induced VSMC migration, which has been shown to be at least partially mediated by the α7-nAChR;105,106 in addition, myosin light chain kinase,105,108 MAPK activation109,110 and PDGF signaling102 have also been implicated. VSMC proliferation and migration has also been shown to be facilitated by nicotine-induced hypomethylation of microRNA (miR)-200b gene promoter, which results in increased miR-200b expression.108 The increased miR-200b leads to reduced RhoGDIA (Rho specific guanine nucleotide dissociation inhibitor A), increased Rho GTPase activity, and consequently changes in cytoskeletal protein expression involved in VSMC proliferation and migration.108 Finally, aortic VSMC from rats exposed to nicotine for 28 days exhibited elevated expression of thrombospondin-1, TGF-β1 and plasminogen activator inhibitor-1, all of which are makers of neointima formation.111
It should be noted that, similar to nicotine’s effects on EC, nicotine induced VSMC remodeling also exhibits dose dependency. The optimal changes are observed at nicotine concentrations similar to the plasma levels in habitual smokers, whereas higher levels (≥10−4 M) result in reduced VSMC viability.99,101
Nicotine and Vascular Matrix Remodeling
In addition to affecting the major cell types of vessels, nicotine has been shown to elicit changes to the vascular extracellular matrix (ECM).112–114 Aortas harvested from mice exposed to nicotine for 4 weeks via osmotic pumps demonstrated significant collagen and fibronectin accumulation coupled with increased elastin fragmentation.112 Increases in collagen and fibronectin are indicative of fibrogenesis, and nicotine has been shown to induce fibrosis in multiple organ systems.115 Furthermore, both cigarette smoke extract and nicotine increased primary rat cardiac fibroblast (CF) proliferation and collagen synthesis, which was inhibited by mecamylamine (nonselective nAChR antagonist), α-bungarotoxin (selective α7-nAChR antagonist) and knockdown of α7-nAChR with siRNA.116 Similarly, in vitro exposure to nicotine increased production of collagen type I and III in CF isolated from neonatal rats, which was attenuated by α-bungarotoxin.117 Another mechanism by which nicotine can impact the ECM is through increased matrix metalloproteinase (MMP) production by EC and VSMC.112,113,118 The balance between MMP and tissue inhibitors of metalloproteinase is important for vascular integrity, and its disturbance in vascular remodeling has been extensively reviewed.119,120 In human retinal microvascular EC, acute exposure to nicotine (100 nM) significantly increased expression of the gelatinases, MMP-2 and MMP-9, and this effect was mediated by α7-nAChR.118 Increased expression and activity of MMP-2/9 were also observed in VSMC acutely exposed to 150 nM nicotine.113 Similar increases in gelatinase activity were observed in aortic homogenates harvested from rats acutely and chronically exposed to nicotine at concentrations similar to those found in humans after smoking one and three cigarettes.113 In addition to increased gene and protein expression of MMP-2/9, a recent study demonstrated that aortic segments harvested from mice chronically exposed to nicotine exhibited increased elastolytic activity accompanied by elastin thinning and fragmentation, indicative of increased susceptibility to aortic aneurysm.114
The mechanisms through which nicotine elicits changes in vascular matrix are not completely understood. One study investigated the involvement of an oxidized nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylase, sirtuin-1 (SIRT1), as SIRT1 inhibition has been linked to cigarette smoking-induced arterial stiffness.112 In both human VSMC and murine aortas, nicotine exposure reduced the protein level and activity of SIRT1. Importantly, SIRT1 overexpression in mice attenuated nicotine-induced vascular remodeling, leading to reduced accumulation of collagen and fibronectin, reduced elastin fragmentation and MMP-2 expression. This study further showed that the downregulation of SIRT1 by nicotine was due to reduced SIRT1 stability, as nicotine-induced generation of peroxynitrite (ONOO−) irreversibly uncoupled zinc from SIRT1. Furthermore, SIRT1 inactivation by ONOO− activated the YAP (Yes-associated protein)-mediated abnormal ECM remodeling.112
One clinical manifestation of nicotine-induced matrix remodeling is arterial stiffness, which can be measured by pulse wave velocity (PWV), or the rate at which pressure waves move down the vessel. PWV has been established as a highly reliable prognostic parameter for cardiovascular morbidity and mortality in a variety of adult populations including older adults, patients with hypertension, diabetes, and end-stage renal disease. Mice exposed to nicotine via osmotic pumps for 4 weeks exhibited significantly increased PWV and stiffness in both abdominal aorta and carotid artery.112 Long term (8-month) cigarette smoke and e-cig exposure in mice have also been shown to increase PWV.71 Importantly, recent studies in healthy human volunteers showed that vaping of e-liquid containing nicotine, but not e-liquid without nicotine, induced significant increases in PWV and arterial stiffness.121,122
Conclusion and Future Directions
The impact of nicotine exposure on the cardiovascular system is complex, from its modulation of autonomic function to its direct effects on individual cell types in the vasculature. Although the initial presentations of nicotine-induced vascular dysfunction may be insidious (changes in vasoreactivity and vascular remodeling as discussed in this review), these changes contribute to the pathogenesis of serious medical conditions including atherosclerosis, abdominal aortic aneurysm, coronary artery disease and myocardial infarction.123–125
Nicotine has been studied for over 50 years, however, our understanding of the impact of nicotine on cardiovascular function and its associated mechanisms is far from complete. In terms of nicotine-induced vascular dysfunction, most studies have implicated the involvement of α7-nAChR; however, investigation into other nAChR that could play important roles in the vascular effects of nicotine is needed. For example, it has been shown that α1-nAChR mediates nicotine-induced atherogenic response in EC.126 In the setting of nicotine-induced carcinogenesis, α3-containing nAChR have been shown to inhibit programmed cell death and promote cell survival, whereas α9-nAChR regulate cell detachment and migration.127 Identification of the specific nAChR subtypes responsible for the harmful effects of nicotine could help develop targeted therapies for nicotine-associated vascular diseases.
Another important and interesting question remained to be answered is whether and how chronic nicotine exposure alters the expression and/or activity of nAChR in the cardiovascular system. Chronic nicotine exposure has been shown to result in numerous neuroadaptations, including the upregulation of particular nAChR subtypes associated with long-term desensitization of the receptors.128,129 In addition, inactivation of nAChR by ROS130 associated with cigarette smoke or nicotine exposure could lead to upregulation of nAChR as well. Upregulation of ligand binding to nAChRs is observed in the brains of both smokers and animals chronically exposed to nicotine, with functional upregulation of α4β2 and α7 subtypes of nAChR.131 In a recent study, inhalation of e-cig vapor for six months in mice was shown to increase α7-nAChR expression in brain regions involved in nicotine addiction.132 Future research is needed to understand changes in nAChR in the cardiovascular system in smokers and e-cig users and their impact on cardiovascular function.
The e-cig epidemic, especially among young adults and youth, has become a significant public health issue. This review focused specifically on nicotine’s effect on the cardiovascular system; however, e-cig contain additional constituents including humectants (e.g. propylene glycol and vegetable glycerine) and flavorings. While most e-cig additives are “generally recognized as safe” for oral ingestion by the Food and Drug Administration, the safety of direct inhalation of these chemicals is not fully understood. For example, cinnamaldehyde, a compontent of cinnamon flavorings, has been shown to impair respiratory innate immune cell function through alterations of proinflammatory cytokines and the prodcution of ROS.133,134 In addition, carbonyl compounds (formaldehyde, acetaldehyde and acrolein), free radicals and various metals have been detected in e-cig emissions as the result of thermal decomposition of e-cig liquid and the heating of the metal coils in the device.135–137 These harmful chemicals cause oxidative stress and inflammation and may produce additive and synergistic effects with nicotine on the vasculature.138 Finally, newer generation of e-cig devices such as JUUL uses proprietary nicotine salts rather than alkalinized nicotine, producing more efficient nicotine delivery and achieving plasma-nicotine levels as high as or higher than traditional cigarettes.139 The long-term health consequences of e-cig inhalation on the cardiovascular system are still largely unknown and warrant further investigation.
Acknowledgement
We would like to thank Dr. Eric Lazartigues (Professor of Pharmacology at Louisiana State University Health Sciences Center) for insightful discussion during the preparation of this manuscript. This study was supported in part by research grants from the National Institute of Health (HL135635 to X. Yue and COBRE P30GM106392).
Footnotes
Conflict of interests
The authors declare no conflict of interests.
References
- 1.Jamal A, Phillips E, Gentzke AS, et al. Current Cigarette Smoking Among Adults - United States, 2016. MMWR Morb Mortal Wkly Rep. 2018;67(2):53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89(1):73–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oakes JM, Fuchs RM, Gardner JD, Lazartigues E, Yue X. Nicotine and the renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol. 2018;315(5):R895–R906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Najem B, Houssiere A, Pathak A, et al. Acute cardiovascular and sympathetic effects of nicotine replacement therapy. Hypertension. 2006;47(6):1162–1167. [DOI] [PubMed] [Google Scholar]
- 5.Haass M, Kubler W. Nicotine and sympathetic neurotransmission. Cardiovasc Drugs Ther. 1997;10(6):657–665. [DOI] [PubMed] [Google Scholar]
- 6.Houdi AA, Dowell RT, Diana JN. Cardiovascular responses to cigarette smoke exposure in restrained conscious rats. J Pharmacol Exp Ther. 1995;275(2):646–653. [PubMed] [Google Scholar]
- 7.Ren LM, Furukawa Y, Karasawa Y, et al. Effects of tetrodotoxin and imipramine on the cardiac responses to nicotine in isolated, blood-perfused canine heart preparations. J Cardiovasc Pharmacol. 1991;18(1):77–84. [DOI] [PubMed] [Google Scholar]
- 8.Li YF, LaCroix C, Freeling J. Specific subtypes of nicotinic cholinergic receptors involved in sympathetic and parasympathetic cardiovascular responses. Neurosci Lett. 2009;462(1):20–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shao XM, Lopez-Valdes HE, Liang J, Feldman JL. Inhaled nicotine equivalent to cigarette smoking disrupts systemic and uterine hemodynamics and induces cardiac arrhythmia in pregnant rats. Sci Rep. 2017;7(1):16974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franceschini D, Orr-Urtreger A, Yu W, et al. Altered baroreflex responses in alpha7 deficient mice. Behav Brain Res. 2000;113(1–2):3–10. [DOI] [PubMed] [Google Scholar]
- 11.Tseng CJ, Ger LP, Lin HC, Tung CS. The pressor effect of nicotine in the rostral ventrolateral medulla of rats. Chin J Physiol. 1994;37(2):83–87. [PubMed] [Google Scholar]
- 12.Page SJ, Zhu M, Appleyard SM. Effects of acute and chronic nicotine on catecholamine neurons of the nucleus of the solitary tract. Am J Physiol Regul Integr Comp Physiol. 2019;316(1):R38–R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore C, Wang Y, Ramage AG. Cardiovascular effects of activation of central alpha7 and alpha4beta2 nAChRs: a role for vasopressin in anaesthetized rats. Br J Pharmacol. 2008;153(8):1728–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Nardi F, Lefort C, Breard D, Richomme P, Legros C, Guerineau NC. Monitoring the Secretory Behavior of the Rat Adrenal Medulla by High-Performance Liquid Chromatography-Based Catecholamine Assay from Slice Supernatants. Front Endocrinol (Lausanne). 2017;8:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheek TR. Calcium signalling and the triggering of secretion in adrenal chromaffin cells. Pharmacol Ther. 1991;52(2):173–189. [DOI] [PubMed] [Google Scholar]
- 16.Green MS, Jucha E, Luz Y. Blood pressure in smokers and nonsmokers: epidemiologic findings. Am Heart J. 1986;111(5):932–940. [DOI] [PubMed] [Google Scholar]
- 17.Friedman GD, Klatsky AL, Siegelaub AB. Alcohol, tobacco, and hypertension. Hypertension. 1982;4(5 Pt 2):III143–150. [DOI] [PubMed] [Google Scholar]
- 18.Pickering TG, Schwartz JE, James GD. Ambulatory blood pressure monitoring for evaluating the relationships between lifestyle, hypertension and cardiovascular risk. Clin Exp Pharmacol Physiol. 1995;22(3):226–231. [DOI] [PubMed] [Google Scholar]
- 19.Palatini P, Pessina AC, Graniero GR, et al. The relationship between overweight, life style and casual and 24-hour pressures in a population of male subjects with mild hypertension. The results of the HARVEST study. G Ital Cardiol. 1995;25(8):977–989. [PubMed] [Google Scholar]
- 20.Oakes JM, Xu J, Morris TM, et al. Effects of Chronic Nicotine Inhalation on Systemic and Pulmonary Blood Pressure and Right Ventricular Remodeling in Mice. Hypertension. 2020;75(5):1305–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarikaya S, Sahin S, Ozturk S, et al. Detection of atrial electrical and mechanical dysfunction in non-dipper pre-hypertensive subjects. Clin Exp Hypertens. 2014;36(7):465–470. [DOI] [PubMed] [Google Scholar]
- 22.Gorostidi M, Sobrino J, Segura J, et al. Ambulatory blood pressure monitoring in hypertensive patients with high cardiovascular risk: a cross-sectional analysis of a 20,000-patient database in Spain. J Hypertens. 2007;25(5):977–984. [DOI] [PubMed] [Google Scholar]
- 23.da Silva Goncalves Bos D, Van Der Bruggen CEE, Kurakula K, et al. Contribution of Impaired Parasympathetic Activity to Right Ventricular Dysfunction and Pulmonary Vascular Remodeling in Pulmonary Arterial Hypertension. Circulation. 2018;137(9):910–924. [DOI] [PubMed] [Google Scholar]
- 24.Boswijk E, Bauwens M, Mottaghy FM, Wildberger JE, Bucerius J. Potential of alpha7 nicotinic acetylcholine receptor PET imaging in atherosclerosis. Methods. 2017;130:90–104. [DOI] [PubMed] [Google Scholar]
- 25.Vazquez-Padron RI, Mateu D, Rodriguez-Menocal L, Wei Y, Webster KA, Pham SM. Novel role of Egr-1 in nicotine-related neointimal formation. Cardiovasc Res. 2010;88(2):296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu JC, Chruscinski A, De Jesus Perez VA, et al. Cholinergic modulation of angiogenesis: role of the 7 nicotinic acetylcholine receptor. J Cell Biochem. 2009;108(2):433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moccia F, Frost C, Berra-Romani R, Tanzi F, Adams DJ. Expression and function of neuronal nicotinic ACh receptors in rat microvascular endothelial cells. Am J Physiol Heart Circ Physiol. 2004;286(2):H486–491. [DOI] [PubMed] [Google Scholar]
- 28.Bruggmann D, Lips KS, Pfeil U, Haberberger RV, Kummer W. Rat arteries contain multiple nicotinic acetylcholine receptor alpha-subunits. Life Sci. 2003;72(18–19):2095–2099. [DOI] [PubMed] [Google Scholar]
- 29.Durand MJ, Gutterman DD. Diversity in mechanisms of endothelium-dependent vasodilation in health and disease. Microcirculation. 2013;20(3):239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davenport AP, Hyndman KA, Dhaun N, et al. Endothelin. Pharmacol Rev. 2016;68(2):357–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haak T, Jungmann E, Raab C, Usadel KH. Elevated endothelin-1 levels after cigarette smoking. Metabolism. 1994;43(3):267–269. [DOI] [PubMed] [Google Scholar]
- 32.Milara J, Gabarda E, Juan G, et al. Bosentan inhibits cigarette smoke-induced endothelin receptor expression in pulmonary arteries. Eur Respir J. 2012;39(4):927–938. [DOI] [PubMed] [Google Scholar]
- 33.Wright JL, Tai H, Churg A. Cigarette smoke induces persisting increases of vasoactive mediators in pulmonary arteries. Am J Respir Cell Mol Biol. 2004;31(5):501–509. [DOI] [PubMed] [Google Scholar]
- 34.Cao L, Zhang Y, Cao YX, Edvinsson L, Xu CB. Cigarette smoke upregulates rat coronary artery endothelin receptors in vivo. PLoS One. 2012;7(3):e33008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao L, Ping NN, Cao YX, et al. The effects of MEK1/2 inhibition on cigarette smoke exposure-induced ET receptor upregulation in rat cerebral arteries. Toxicol Appl Pharmacol. 2016;304:70–78. [DOI] [PubMed] [Google Scholar]
- 36.Mayyas F, Aldawod H, Alzoubi KH, Khabour O, Shihadeh A, Eissenberg T. Comparison of the cardiac effects of electronic cigarette aerosol exposure with waterpipe and combustible cigarette smoke exposure in rats. Life Sci. 2020;251:117644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee SD, Lee DS, Chun YG, et al. Cigarette smoke extract induces endothelin-1 via protein kinase C in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;281(2):L403–411. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, Zhang W, Edvinsson L, Xu CB. Lipid-soluble Cigarette Smoke Particles Induced Vascular Endothelin Type A Receptor Up-Regulation through Activation of ERK1/2 Signal Pathways. Basic Clin Pharmacol Toxicol. 2017;120(4):327–334. [DOI] [PubMed] [Google Scholar]
- 39.Sandhu H, Xu CB, Edvinsson L. Upregulation of contractile endothelin type B receptors by lipid-soluble cigarette smoking particles in rat cerebral arteries via activation of MAPK. Toxicol Appl Pharmacol. 2010;249(1):25–32. [DOI] [PubMed] [Google Scholar]
- 40.Huang LH, He JY, Yuan BX, Cao YX. Lipid soluble smoke particles upregulate endothelin receptors in rat basilar artery. Toxicol Lett. 2010;197(3):243–255. [DOI] [PubMed] [Google Scholar]
- 41.Zhang S, Day I, Ye S. Nicotine induced changes in gene expression by human coronary artery endothelial cells. Atherosclerosis. 2001;154(2):277–283. [DOI] [PubMed] [Google Scholar]
- 42.Lee WO, Wright SM. Production of endothelin by cultured human endothelial cells following exposure to nicotine or caffeine. Metabolism. 1999;48(7):845–848. [DOI] [PubMed] [Google Scholar]
- 43.Tanus-Santos JE, Sampaio RC, Hyslop S, Franchini KG, Moreno H Jr. Endothelin ET(A) receptor antagonism attenuates the pressor effects of nicotine in rats. Eur J Pharmacol. 2000;396(1):33–37. [DOI] [PubMed] [Google Scholar]
- 44.Hashimoto T, Yoneda M, Shimada T, Kurosawa M, Terano A. Intraportal nicotine infusion in rats decreases hepatic blood flow through endothelin-1 and both endothelin A and endothelin B receptors. Toxicol Appl Pharmacol. 2004;196(1):1–10. [DOI] [PubMed] [Google Scholar]
- 45.Rodella LF, Favero G, Rossini C, Foglio E, Reiter RJ, Rezzani R. Endothelin-1 as a potential marker of melatonin’s therapeutic effects in smoking-induced vasculopathy. Life Sci. 2010;87(17–18):558–564. [DOI] [PubMed] [Google Scholar]
- 46.El-Seweidy MM, Mohamed HE, Asker ME, Atteia HH. Nicotine and vascular endothelial dysfunction in female ovariectomized rats: role of estrogen replacement therapy. J Pharm Pharmacol. 2012;64(1):108–119. [DOI] [PubMed] [Google Scholar]
- 47.Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33(7):829–837, 837a–837d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carnevale R, Sciarretta S, Violi F, et al. Acute Impact of Tobacco vs Electronic Cigarette Smoking on Oxidative Stress and Vascular Function. Chest. 2016;150(3):606–612. [DOI] [PubMed] [Google Scholar]
- 49.Toda N, Toda H. Nitric oxide-mediated blood flow regulation as affected by smoking and nicotine. Eur J Pharmacol. 2010;649(1–3):1–13. [DOI] [PubMed] [Google Scholar]
- 50.Xiao D, Huang X, Yang S, Zhang L. Direct effects of nicotine on contractility of the uterine artery in pregnancy. J Pharmacol Exp Ther. 2007;322(1):180–185. [DOI] [PubMed] [Google Scholar]
- 51.Conklin BS, Surowiec SM, Ren Z, et al. Effects of nicotine and cotinine on porcine arterial endothelial cell function. J Surg Res. 2001;95(1):23–31. [DOI] [PubMed] [Google Scholar]
- 52.Li J, Liu S, Cao G, et al. Nicotine induces endothelial dysfunction and promotes atherosclerosis via GTPCH1. J Cell Mol Med. 2018;22(11):5406–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiang DJ, Jia SJ, Yan J, Zhou Z, Yuan Q, Li YJ. Involvement of DDAH/ADMA/NOS pathway in nicotine-induced endothelial dysfunction. Biochem Biophys Res Commun. 2006;349(2):683–693. [DOI] [PubMed] [Google Scholar]
- 54.Hamasaki H, Sato J, Masuda H, et al. Effect of nicotine on the intimal hyperplasia after endothelial removal of the rabbit carotid artery. Gen Pharmacol. 1997;28(5):653–659. [DOI] [PubMed] [Google Scholar]
- 55.Balakumar P, Sharma R, Singh M. Benfotiamine attenuates nicotine and uric acid-induced vascular endothelial dysfunction in the rat. Pharmacol Res. 2008;58(5–6):356–363. [DOI] [PubMed] [Google Scholar]
- 56.Hanna ST. Nicotine effect on cardiovascular system and ion channels. J Cardiovasc Pharmacol. 2006;47(3):348–358. [DOI] [PubMed] [Google Scholar]
- 57.Mayhan WG, Arrick DM, Sun H, Patel KP. Exercise training restores impaired dilator responses of cerebral arterioles during chronic exposure to nicotine. J Appl Physiol (1985). 2010;109(4):1109–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mitchell JA, Ahmetaj-Shala B, Kirkby NS, et al. Role of prostacyclin in pulmonary hypertension. Glob Cardiol Sci Pract. 2014;2014(4):382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jeremy JY, Mikhailidis DP, Dandona P. Cigarette smoke extracts, but not nicotine, inhibit prostacyclin (PGI2) synthesis in human, rabbit and rat vascular tissue. Prostaglandins Leukot Med. 1985;19(3):261–270. [DOI] [PubMed] [Google Scholar]
- 60.Lubawy WC, Culpepper BT, Valentovic MA. Alterations in prostacyclin and thromboxane formation by chronic cigarette smoke exposure: temporal relationships and whole smoke vs. gas phase. J Appl Toxicol. 1986;6(2):77–80. [DOI] [PubMed] [Google Scholar]
- 61.Mehta P, Mehta J. Effects of smoking on platelets and on plasma thromboxane-prostacyclin balance in man. Prostaglandins Leukot Med. 1982;9(2):141–150. [DOI] [PubMed] [Google Scholar]
- 62.Su YC, Wang DX. Effects of cigarette smoking, hypoxia and vasoactive mediators on the production of PGI2 and TXA2 in cultured pulmonary artery endothelial cells. J Tongji Med Univ. 1991;11(1):6–9. [DOI] [PubMed] [Google Scholar]
- 63.Bull HA, Pittilo RM, Woolf N, Machin SJ. The effect of nicotine on human endothelial cell release of prostaglandins and ultrastructure. Br J Exp Pathol. 1988;69(3):413–421. [PMC free article] [PubMed] [Google Scholar]
- 64.Suzuki N, Ishii Y, Kitamura S. Effects of nicotine on production of endothelin and eicosanoid by bovine pulmonary artery endothelial cells. Prostaglandins Leukot Essent Fatty Acids. 1994;50(4):193–197. [DOI] [PubMed] [Google Scholar]
- 65.Stoel I, vd Giessen WJ, Zwolsman E, et al. Effect of nicotine on production of prostacyclin in human umbilical artery. Br Heart J. 1982;48(5):493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Effeney DJ. Prostacyclin production by the heart: effect of nicotine and carbon monoxide. J Vasc Surg. 1987;5(2):237–247. [PubMed] [Google Scholar]
- 67.Nemr R, Lasserre B, Chahine R. Effects of nicotine on thromboxane/prostacyclin balance in myocardial ischemia. Prostaglandins Leukot Essent Fatty Acids. 2003;68(3):191–195. [DOI] [PubMed] [Google Scholar]
- 68.Hui SC, Wang Z, Zhang H, Ogle CW. Arachidonic acid metabolism in nicotine-treated rats and nicotine-incubated rabbit aortic smooth muscle cells. Clin Exp Pharmacol Physiol. 1992;19(10):689–693. [DOI] [PubMed] [Google Scholar]
- 69.Bull HA, Pittilo RM, Blow DJ, et al. The effects of nicotine on PGI2 production by rat aortic endothelium. Thromb Haemost. 1985;54(2):472–474. [PubMed] [Google Scholar]
- 70.Kellogg DL Jr., Zhao JL, Coey U, Green JV. Acetylcholine-induced vasodilation is mediated by nitric oxide and prostaglandins in human skin. J Appl Physiol (1985). 2005;98(2):629–632. [DOI] [PubMed] [Google Scholar]
- 71.Olfert IM, DeVallance E, Hoskinson H, et al. Chronic exposure to electronic cigarettes results in impaired cardiovascular function in mice. J Appl Physiol (1985). 2018;124(3):573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sarabi M, Lind L. Short-term effects of smoking and nicotine chewing gum on endothelium-dependent vasodilation in young healthy habitual smokers. J Cardiovasc Pharmacol. 2000;35(3):451–456. [DOI] [PubMed] [Google Scholar]
- 73.Chalon S, Moreno H Jr., Benowitz NL, Hoffman BB, Blaschke TF. Nicotine impairs endothelium-dependent dilatation in human veins in vivo. Clin Pharmacol Ther. 2000;67(4):391–397. [DOI] [PubMed] [Google Scholar]
- 74.Lanza GA, Spera FR, Villano A, et al. Effect of smoking on endothelium-independent vasodilatation. Atherosclerosis. 2015;240(2):330–332. [DOI] [PubMed] [Google Scholar]
- 75.Mayhan WG, Sharpe GM. Acute and chronic treatment with nicotine impairs reactivity of arterioles in response to activation of potassium channels. J Cardiovasc Pharmacol. 2002;39(5):695–703. [DOI] [PubMed] [Google Scholar]
- 76.Black CE, Huang N, Neligan PC, et al. Effect of nicotine on vasoconstrictor and vasodilator responses in human skin vasculature. Am J Physiol Regul Integr Comp Physiol. 2001;281(4):R1097–1104. [DOI] [PubMed] [Google Scholar]
- 77.Feitelson JB, Rowell PP, Roberts CS, Fleming JT. Two week nicotine treatment selectively increases bone vascular constriction in response to norepinephrine. J Orthop Res. 2003;21(3):497–502. [DOI] [PubMed] [Google Scholar]
- 78.Lee TJ. Sympathetic modulation of nitrergic neurogenic vasodilation in cerebral arteries. Jpn J Pharmacol. 2002;88(1):26–31. [DOI] [PubMed] [Google Scholar]
- 79.Kawasaki H, Takatori S, Zamami Y, et al. Paracrine control of mesenteric perivascular axo-axonal interaction. Acta Physiol (Oxf). 2011;203(1):3–11. [DOI] [PubMed] [Google Scholar]
- 80.Toda N, Ayajiki K, Okamura T. Neurogenic and endothelial nitric oxide regulates blood circulation in lingual and other oral tissues. J Cardiovasc Pharmacol. 2012;60(1):100–108. [DOI] [PubMed] [Google Scholar]
- 81.Shih CC, Chen PY, Chen MF, Lee TJF. Differential blockade by huperzine A and donepezil of sympathetic nicotinic acetylcholine receptor-mediated nitrergic neurogenic dilations in porcine basilar arteries. Eur J Pharmacol. 2020;868:172851. [DOI] [PubMed] [Google Scholar]
- 82.Si ML, Lee TJ. Alpha7-nicotinic acetylcholine receptors on cerebral perivascular sympathetic nerves mediate choline-induced nitrergic neurogenic vasodilation. Circ Res. 2002;91(1):62–69. [DOI] [PubMed] [Google Scholar]
- 83.Toda N, Okamura T. Cigarette smoking impairs nitric oxide-mediated cerebral blood flow increase: Implications for Alzheimer’s disease. J Pharmacol Sci. 2016;131(4):223–232. [DOI] [PubMed] [Google Scholar]
- 84.Heeschen C, Weis M, Aicher A, Dimmeler S, Cooke JP. A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. J Clin Invest. 2002;110(4):527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Heeschen C, Jang JJ, Weis M, et al. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat Med. 2001;7(7):833–839. [DOI] [PubMed] [Google Scholar]
- 86.Lee J, Cooke JP. Nicotine and pathological angiogenesis. Life Sci. 2012;91(21–22):1058–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Egleton RD, Brown KC, Dasgupta P. Angiogenic activity of nicotinic acetylcholine receptors: implications in tobacco-related vascular diseases. Pharmacol Ther. 2009;121(2):205–223. [DOI] [PubMed] [Google Scholar]
- 88.Cooke JP, Ghebremariam YT. Endothelial nicotinic acetylcholine receptors and angiogenesis. Trends Cardiovasc Med. 2008;18(7):247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Villablanca AC. Nicotine stimulates DNA synthesis and proliferation in vascular endothelial cells in vitro. J Appl Physiol (1985). 1998;84(6):2089–2098. [DOI] [PubMed] [Google Scholar]
- 90.Ng MK, Wu J, Chang E, et al. A central role for nicotinic cholinergic regulation of growth factor-induced endothelial cell migration. Arterioscler Thromb Vasc Biol. 2007;27(1):106–112. [DOI] [PubMed] [Google Scholar]
- 91.Park YJ, Lee T, Ha J, Jung IM, Chung JK, Kim SJ. Effect of nicotine on human umbilical vein endothelial cells (HUVECs) migration and angiogenesis. Vascul Pharmacol. 2008;49(1):32–36. [DOI] [PubMed] [Google Scholar]
- 92.Cucina A, Sapienza P, Borrelli V, et al. Nicotine reorganizes cytoskeleton of vascular endothelial cell through platelet-derived growth factor BB. J Surg Res. 2000;92(2):233–238. [DOI] [PubMed] [Google Scholar]
- 93.Hakki A, Friedman H, Pross S. Nicotine modulation of apoptosis in human coronary artery endothelial cells. Int Immunopharmacol. 2002;2(10):1403–1409. [DOI] [PubMed] [Google Scholar]
- 94.Chen JK, Li ZP, Liu YZ, et al. Activation of alpha 7 nicotinic acetylcholine receptor protects mice from radiation-induced intestinal injury and mortality. Radiat Res. 2014;181(6):666–671. [DOI] [PubMed] [Google Scholar]
- 95.Konishi H, Wu J, Cooke JP. Chronic exposure to nicotine impairs cholinergic angiogenesis. Vasc Med. 2010;15(1):47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Park HS, Cho K, Park YJ, Lee T. Chronic nicotine exposure attenuates proangiogenic activity on human umbilical vein endothelial cells. J Cardiovasc Pharmacol. 2011;57(3):287–293. [DOI] [PubMed] [Google Scholar]
- 97.Li M, Qian M, Kyler K, Xu J. Endothelial-Vascular Smooth Muscle Cells Interactions in Atherosclerosis. Front Cardiovasc Med. 2018;5:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Michel JB, Li Z, Lacolley P. Smooth muscle cells and vascular diseases. Cardiovasc Res. 2012;95(2):135–137. [DOI] [PubMed] [Google Scholar]
- 99.Carty CS, Huribal M, Marsan BU, Ricotta JJ, Dryjski M. Nicotine and its metabolite cotinine are mitogenic for human vascular smooth muscle cells. J Vasc Surg. 1997;25(4):682–688. [DOI] [PubMed] [Google Scholar]
- 100.Cucina A, Fuso A, Coluccia P, Cavallaro A. Nicotine inhibits apoptosis and stimulates proliferation in aortic smooth muscle cells through a functional nicotinic acetylcholine receptor. J Surg Res. 2008;150(2):227–235. [DOI] [PubMed] [Google Scholar]
- 101.Cucina A, Sapienza P, Corvino V, et al. Nicotine-induced smooth muscle cell proliferation is mediated through bFGF and TGF-beta 1. Surgery. 2000;127(3):316–322. [DOI] [PubMed] [Google Scholar]
- 102.Cucina A, Sapienza P, Corvino V, et al. Nicotine induces platelet-derived growth factor release and cytoskeletal alteration in aortic smooth muscle cells. Surgery. 2000;127(1):72–78. [DOI] [PubMed] [Google Scholar]
- 103.Thyberg J Effects of nicotine on phenotypic modulation and initiation of DNA synthesis in cultured arterial smooth muscle cells. Virchows Arch B Cell Pathol Incl Mol Pathol. 1986;52(1):25–32. [DOI] [PubMed] [Google Scholar]
- 104.Pestana IA, Vazquez-Padron RI, Aitouche A, Pham SM. Nicotinic and PDGF-receptor function are essential for nicotine-stimulated mitogenesis in human vascular smooth muscle cells. J Cell Biochem. 2005;96(5):986–995. [DOI] [PubMed] [Google Scholar]
- 105.Li S, Zhao T, Xin H, et al. Nicotinic acetylcholine receptor alpha7 subunit mediates migration of vascular smooth muscle cells toward nicotine. J Pharmacol Sci. 2004;94(3):334–338. [DOI] [PubMed] [Google Scholar]
- 106.Gu Z, Fonseca V, Hai CM. Nicotinic acetylcholine receptor mediates nicotine-induced actin cytoskeletal remodeling and extracellular matrix degradation by vascular smooth muscle cells. Vascul Pharmacol. 2013;58(1–2):87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Linder S, Aepfelbacher M. Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 2003;13(7):376–385. [DOI] [PubMed] [Google Scholar]
- 108.Liang D, Wang Z, Yan Z, et al. Nicotine facilitates VSMC dysfunction through a miR-200b/RhoGDIA/cytoskeleton module. Sci Rep. 2017;7:43798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Di Luozzo G, Pradhan S, Dhadwal AK, Chen A, Ueno H, Sumpio BE. Nicotine induces mitogen-activated protein kinase dependent vascular smooth muscle cell migration. Atherosclerosis. 2005;178(2):271–277. [DOI] [PubMed] [Google Scholar]
- 110.Yoshiyama S, Chen Z, Okagaki T, et al. Nicotine exposure alters human vascular smooth muscle cell phenotype from a contractile to a synthetic type. Atherosclerosis. 2014;237(2):464–470. [DOI] [PubMed] [Google Scholar]
- 111.Rodella LF, Rossini C, Favero G, Foglio E, Loreto C, Rezzani R. Nicotine-induced morphological changes in rat aorta: the protective role of melatonin. Cells Tissues Organs. 2012;195(3):252–259. [DOI] [PubMed] [Google Scholar]
- 112.Ding Y, Han Y, Lu Q, et al. Peroxynitrite-Mediated SIRT (Sirtuin)-1 Inactivation Contributes to Nicotine-Induced Arterial Stiffness in Mice. Arterioscler Thromb Vasc Biol. 2019;39(7):1419–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jacob-Ferreira AL, Palei AC, Cau SB, et al. Evidence for the involvement of matrix metalloproteinases in the cardiovascular effects produced by nicotine. Eur J Pharmacol. 2010;627(1–3):216–222. [DOI] [PubMed] [Google Scholar]
- 114.Wagenhauser MU, Schellinger IN, Yoshino T, et al. Chronic Nicotine Exposure Induces Murine Aortic Remodeling and Stiffness Segmentation-Implications for Abdominal Aortic Aneurysm Susceptibility. Front Physiol. 2018;9:1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jensen K, Nizamutdinov D, Guerrier M, Afroze S, Dostal D, Glaser S. General mechanisms of nicotine-induced fibrogenesis. FASEB J. 2012;26(12):4778–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vang A, Clements RT, Chichger H, et al. Effect of alpha7 nicotinic acetylcholine receptor activation on cardiac fibroblasts: a mechanism underlying RV fibrosis associated with cigarette smoke exposure. Am J Physiol Lung Cell Mol Physiol. 2017;312(5):L748–L759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chuang TD, Ansari A, Yu C, et al. Mechanism underlying increased cardiac extracellular matrix deposition in perinatal nicotine-exposed offspring. Am J Physiol Heart Circ Physiol. 2020;319(3):H651–H660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dom AM, Buckley AW, Brown KC, et al. The alpha7-nicotinic acetylcholine receptor and MMP-2/−9 pathway mediate the proangiogenic effect of nicotine in human retinal endothelial cells. Invest Ophthalmol Vis Sci. 2011;52(7):4428–4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90(3):251–262. [PubMed] [Google Scholar]
- 120.Wang X, Khalil RA. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv Pharmacol. 2018;81:241–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Antoniewicz L, Brynedal A, Hedman L, Lundback M, Bosson JA. Acute Effects of Electronic Cigarette Inhalation on the Vasculature and the Conducting Airways. Cardiovasc Toxicol. 2019;19(5):441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chaumont M, de Becker B, Zaher W, et al. Differential Effects of E-Cigarette on Microvascular Endothelial Function, Arterial Stiffness and Oxidative Stress: A Randomized Crossover Trial. Sci Rep. 2018;8(1):10378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Centner AM, Bhide PG, Salazar G. Nicotine in Senescence and Atherosclerosis. Cells. 2020;9(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li ZZ, Dai QY. Pathogenesis of abdominal aortic aneurysms: role of nicotine and nicotinic acetylcholine receptors. Mediators Inflamm. 2012;2012:103120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gaemperli O, Liga R, Bhamra-Ariza P, Rimoldi O. Nicotine addiction and coronary artery disease: impact of cessation interventions. Curr Pharm Des. 2010;16(23):2586–2597. [DOI] [PubMed] [Google Scholar]
- 126.Ren A, Wu H, Liu L, Guo Z, Cao Q, Dai Q. Nicotine promotes atherosclerosis development in apolipoprotein E-deficient mice through alpha1-nAChR. J Cell Physiol. 2018. [DOI] [PubMed] [Google Scholar]
- 127.Grando SA. Connections of nicotine to cancer. Nat Rev Cancer. 2014;14(6):419–429. [DOI] [PubMed] [Google Scholar]
- 128.Wittenberg RE, Wolfman SL, De Biasi M, Dani JA. Nicotinic acetylcholine receptors and nicotine addiction: A brief introduction. Neuropharmacology. 2020;177:108256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Picciotto MR, Addy NA, Mineur YS, Brunzell DH. It is not “either/or”: activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol. 2008;84(4):329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Krishnaswamy A, Cooper E. Reactive oxygen species inactivate neuronal nicotinic acetylcholine receptors through a highly conserved cysteine near the intracellular mouth of the channel: implications for diseases that involve oxidative stress. J Physiol. 2012;590(1):39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Buisson B, Bertrand D. Nicotine addiction: the possible role of functional upregulation. Trends Pharmacol Sci. 2002;23(3):130–136. [DOI] [PubMed] [Google Scholar]
- 132.Alasmari F, Crotty Alexander LE, Nelson JA, et al. Effects of chronic inhalation of electronic cigarettes containing nicotine on glial glutamate transporters and alpha-7 nicotinic acetylcholine receptor in female CD-1 mice. Prog Neuropsychopharmacol Biol Psychiatry. 2017;77:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Clapp PW, Pawlak EA, Lackey JT, et al. Flavored e-cigarette liquids and cinnamaldehyde impair respiratory innate immune cell function. Am J Physiol Lung Cell Mol Physiol. 2017;313(2):L278–L292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Muthumalage T, Prinz M, Ansah KO, Gerloff J, Sundar IK, Rahman I. Inflammatory and Oxidative Responses Induced by Exposure to Commonly Used e-Cigarette Flavoring Chemicals and Flavored e-Liquids without Nicotine. Front Physiol. 2017;8:1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang P, Chen W, Liao J, et al. A Device-Independent Evaluation of Carbonyl Emissions from Heated Electronic Cigarette Solvents. PLoS One. 2017;12(1):e0169811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Khlystov A, Samburova V. Flavoring Compounds Dominate Toxic Aldehyde Production during E-Cigarette Vaping. Environ Sci Technol. 2016;50(23):13080–13085. [DOI] [PubMed] [Google Scholar]
- 137.Cao DJ, Aldy K, Hsu S, et al. Review of Health Consequences of Electronic Cigarettes and the Outbreak of Electronic Cigarette, or Vaping, Product Use-Associated Lung Injury. J Med Toxicol. 2020;16(3):295–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Buchanan ND, Grimmer JA, Tanwar V, Schwieterman N, Mohler PJ, Wold LE. Cardiovascular risk of electronic cigarettes: a review of preclinical and clinical studies. Cardiovasc Res. 2020;116(1):40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fadus MC, Smith TT, Squeglia LM. The rise of e-cigarettes, pod mod devices, and JUUL among youth: Factors influencing use, health implications, and downstream effects. Drug Alcohol Depend. 2019;201:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]