

Abstract
Long-read and strand-specific sequencing technologies together facilitate the de novo assembly of high-quality haplotype-resolved human genomes without parent–child trio data. We present 64 assembled haplotypes from 32 diverse human genomes. These highly contiguous haplotype assemblies (average contig N50: 26 Mbp) integrate all forms of genetic variation even across complex loci. We identify 107,590 structural variants (SVs), of which 68% are not discovered by short-read sequencing, and 278 SV hotspots (spanning megabases of gene-rich sequence). We characterize 130 of the most active mobile element source elements and find that 63% of all SVs arise by homology-mediated mechanisms. This resource enables reliable graph-based genotyping from short reads of up to 50,340 SVs, resulting in the identification of 1,526 expression quantitative trait loci as well as SV candidates for adaptive selection within the human population.
One Sentence Summary:
Structural variation from diverse human genome haplotype assemblies facilitates genotyping and new associations.
INTRODUCTION
Advances in long-read sequencing, coupled with orthogonal genome-wide mapping technologies, have made it possible to fully resolve and assemble both haplotypes of a human genome (1–3). While such phased human genome assemblies generally improve variant discovery compared to Illumina or “squashed” long-read genome assemblies (4), the largest gains in sensitivity have been among structural variants (SVs)—inversions, deletions, duplications, and insertions ≥50 bp in length. Typical Illumina-based discovery approaches identify only 5,000–10,000 SVs (1, 5, 6) in contrast to long-read genome analyses that now routinely detect >20,000 SVs (1, 3, 4, 7). Among the different classes of SVs, the greatest gains in sensitivity have been noted specifically for insertions where >85% of the variation has been reported as novel (1). In addition, repeat-mediated alterations within SV classes, such as variable number of tandem repeats (VNTRs) and short tandem repeats (STRs), have been challenging to delineate from short-read sequencing technologies and are underrepresented in the reference genome and often collapsed in unphased genome assemblies (8). The integration of long-read sequencing with new technologies such as single-cell template strand sequencing (Strand-seq) has further catalyzed the unambiguous confirmation of both heterozygous- and homozygous-inverted configurations in a genome (1, 9). Long-read phased genome assemblies (1) also better resolve larger full-length mobile element insertions (MEIs), providing an opportunity to systematically investigate their origins, distribution, and the mutational processes underlying their mobilization within more complex regions of the genome, including transductions (10, 11).
The Human Genome Structural Variation Consortium (HGSVC) recently developed a method for phased genome assembly that combines long-read PacBio whole-genome sequencing (WGS) and Strand-seq data to produce fully phased diploid genome assemblies without dependency on parent–child trio data (Fig. 1A) (3). These phased assemblies enable a more complete sequence-resolved representation of variation in human genomes.
Fig. 1. Trio-free phased diploid genome assembly using Strand-seq (PGAS).
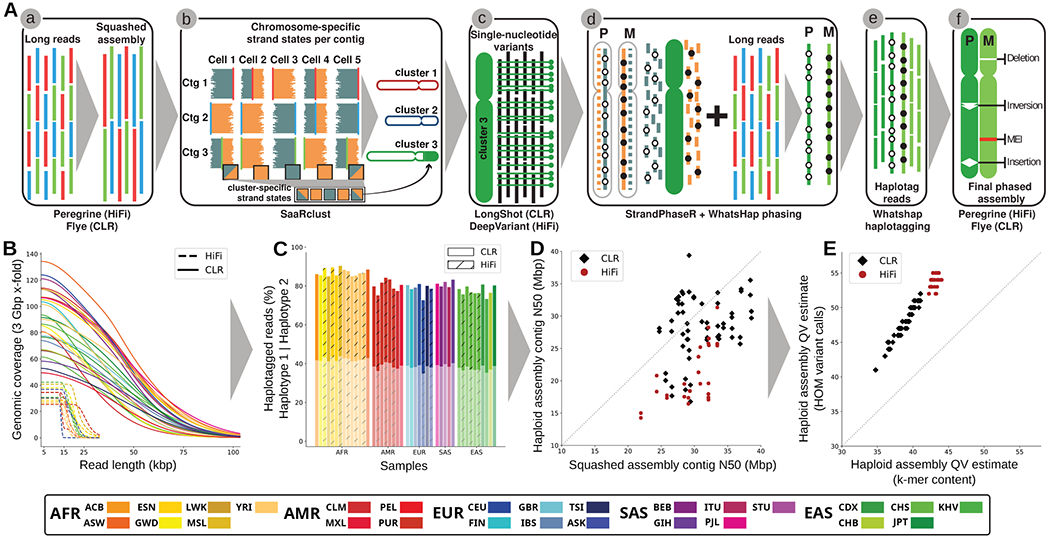
(A) A schematic of the PGAS pipeline (3): (a) generation of a non-haplotype-resolved (“squashed”) long-read assembly; (b) clustering of assembled contigs into “chromosome” clusters based on Strand-seq Watson/Crick signal; (c) calling of single-nucleotide variants (SNVs) relative to the clustered squashed assembly; (d) integrative phasing combines local (SNV) and global (Strand-seq) haplotype information for chromosome-wide phasing; (e) tagging of input long reads by haplotype; (f) phased genome assembly based on haplotagged long reads and subsequent variant calling (18). (B) Genomic coverage (y-axis) as a function of the long-read length (x-axis). (C) Fraction of reads that can be assigned (“haplotagged”) to either haplotype 1 (semitransparent) or haplotype 2 for HiFi (hatched) and CLR (solid) datasets. (D) Contig-level N50 values for squashed (x-axis) and haploid assemblies (y-axis) for CLR (black diamonds) and HiFi (red circles) samples. (E) Haploid assembly QV estimates computed from unique and shared k-mers (x-axis) based on homozygous Illumina variant calls (y-axis). Samples colored according to the 1000GP population color scheme (15) with exception of the added Ashkenazim individual NA24385/HG002 (Coriell family ID 3140) (ASK/dark blue).
Here, we present a resource consisting of phased genome assemblies, corresponding to 70 haplotypes (64 unrelated and 6 children) from a diverse panel of human genomes. We focus specifically on the discovery of novel SVs performing extensive orthogonal validation using supporting technologies with the goal of comprehensively understanding SV complexity, including in regions that cannot yet be resolved by long-read sequencing (fig. S1). Further, we genotype these newly defined SVs using a pangenome graph framework (12–14) into a diversity panel of human genomes now deeply sequenced (>30-fold) with short-read data from the 1000 Genomes Project (1000GP) (15, 16). These findings allow us to establish their population frequency, identify ancestral haplotypes, and discover new associations with respect to gene expression, splicing, and candidate disease loci. The work provides fundamental new insights into the structure, variation, and mutation of the human genome providing a framework for more systematic analyses of thousands of human genomes going forward.
RESULTS
Sequencing and phased assembly of human genomes.
We initially selected 34 unrelated individual genomes for de novo sequencing, with the goal of at least one representative from each of the 26 1000GP populations, of which 30 samples passed initial QC (tables S1 and S2). We additionally sequenced three previously studied child samples completing three parent–child trios, and we included for analysis publicly available sequencing data for two samples, NA12878 and HG002/NA24385, generated as part of the Genome in a Bottle effort (17). The complete set of 35 genomes includes 19 females and 16 males of African (AFR, n=11), Admixed American (AMR, n=5), East Asian (EAS, n=7), European (EUR, n=7) and South Asian (SAS, n=5; table S1) descent. All genomes were sequenced (Methods) using continuous long-read (CLR) sequencing (n=30) to an excess of 40-fold coverage or high-fidelity (HiFi) sequencing (n=12) to an excess of 20-fold coverage (Fig. 1B, table S1, (18)).
As a control for phasing and platform differences, we sequenced nine overlapping samples with both CLR as well as HiFi sequence data corresponding to the three parent–child trios (tables S1, S2) that had been studied for SVs previously by the HGSVC (1). For the purpose of phasing, we generated corresponding Strand-seq data (74-183 cells, fig. S2) for each of the samples. We used these data to successfully produce 70 (64 unrelated) phased and assembled human haplotypes (5.7 to 6.1 Gbp in length for the diploid sequence, table S1) using a reference-free assembly approach (Fig. 1A) (3), which works in the absence of parent–child trio information.
We find that the phased genomes are accurate at the base-pair level (QV > 40) and highly contiguous (contig N50 > 25 Mbp, Fig. 1C–E, table S1) with low switch error rates (median 0.12%, table S3) providing a diversity panel of physically resolved and fully phased single-nucleotide variant (SNV) and indel (insertion/deletion) haplotypes flanking sequence-resolved SVs (table S4). Using two different metrics from variant calling and k-mer content methods, respectively (Fig. 1E), we find that sequence accuracy is higher for human genome assemblies generated by HiFi (median QV = 54 [hom. var.] / 43 [k-mer], Fig. 1E) when compared to CLR (median QV = 48 [hom. var.] / 39 [k-mer], Fig. 1E) sequencing. Considering only accessible regions of the genome (18), the MAPQ60 contig coverage of HiFi and CLR genomes are similar (95.43% and 95.12%, table S5). CLR assemblies, however, are more contiguous (HiFi median contig N50 was 19.5 vs. 28.6 Mbp for CLR; p-value <10e-9, t-test). Fifteen of our assembled haplotypes exceed a contig N50 of 32 Mbp, all of which were based on CLR sequencing where insert libraries are much larger and sequence coverage is higher with half the number of single-molecule, real-time (SMRT) cells (Fig. 1D, fig. S3, table S6).
Comparing Strand-seq phasing accuracy for six samples where parent–child trio data are available (table S3, figs. S4, S5; see Methods in (3)), we estimate on average 99.86% of all 1 Mbp segments are correctly phased from telomere-to-telomere (average switch error rate of 0.18% and Hamming distance of 0.21%, table S3). Predictably (3), remaining assembly gaps are enriched (18) in regions of segmental duplications (SDs) and acrocentric and centromeric regions of human chromosomes (figs. S6, S7, table S7). As a final QC of assembly quality, we analyzed Bionano Genomics optical mapping data for 32 genomes and found a median concordance of >97% between the optical map and the phased genome assemblies (figs. S8, S9, table S8).
Phased variant discovery.
Unlike previous population surveys of structural variation (1, 4, 19–21), which mapped reads or unphased contigs to the human reference genome, we developed the Phased Assembly Variant (PAV) caller (88) to discover genetic variants on the basis of a direct comparison between the two sequence-assembled haplotypes and the human reference genome, GRCh38 (18). In the end, each human genome is rendered into two haplotype-resolved assemblies (each 2.9 Gbp) where all variants are physically linked (table S4). We classify variants as SNVs, indels (1-49 bp), and SVs (≥50 bp), which includes copy number variants (CNVs) and balanced inversion polymorphisms. After filtering (18), our nonredundant callset of unrelated samples contains 107,590 insertion/deletion SVs, 316 inversions, 2.3 million indels, and 15.8 million SNVs.
We observe a 2 bp periodicity for indels (dinucleotide repeats) and modes at 300 bp and 6 kbp for Alu and L1 MEIs, respectively (Fig. 2A), with only a small fraction intersecting functional elements (22) (Fig. 2B). PAV readily flags all reference-based artefacts or minor alleles by pinpointing regions where the 64 phased human genomes consistently differ from GRCh38 (1,573 SVs, 18,630 indels, and 91,537 SNVs, “shared variants”) (Fig. 2C, (18)). The greater haplotype diversity allows us to reclassify 50% of previously annotated shared SVs (4) as minor alleles and correct the coding sequence annotation of five genes with tandem repeats (RRBP1, ZNF676, MUC2, STOX1) or extreme GC content (SAMD1) (table S9). We estimate a false discovery rate (FDR) of 5–7% for SVs on the basis of support from sequence-read-based callers, as well as an independent alignment method (18). A comparison against SVs called from the benchmark Genome in a Bottle sample (HG002), including orthogonal datasets, suggests an FDR of ~4% although this estimate is restricted to a subset of the genome where events could be more reliably called (18).
Fig. 2. Variant discovery and distribution.
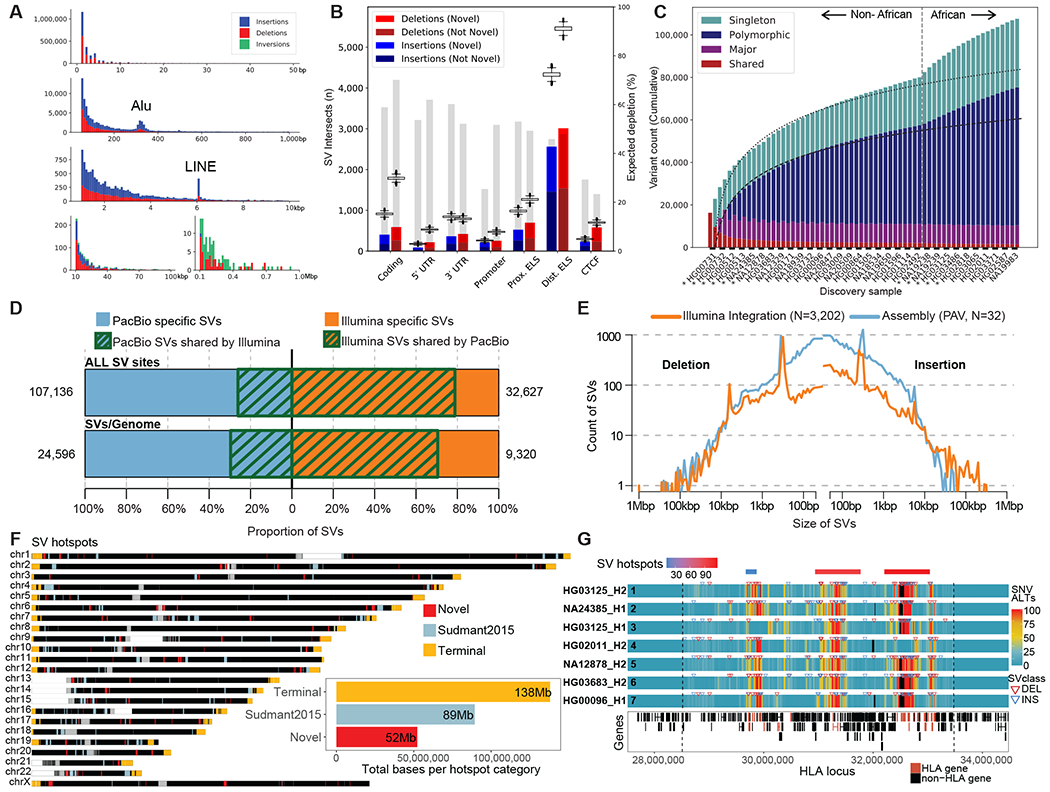
(A) Size distribution of indels and SVs from 64 unrelated reference genomes shows a 2 bp periodicity for indels, 300 bp peak for Alu insertions (second row), and 6 kbp peak for L1 MEIs. (B) The number of SVs intersecting functional elements (horizontal axis) compared to randomly permuting SV locations (box plots). Gray bars depict percent depletion (right axis scale). ELS: Enhancer-like signature. CTCF: CCCTC-binding factor. (C) Cumulative number of unique SVs when adding samples one-by-one, from left to right. The rate of SV discovery slows with each new haplotype (regression lines); however, the addition of haplotypes of African origin (dashed line) increases SV yield. Colors indicate SVs shared among all haplotypes and not present in GRCh38 (red), major allele variants (AF≥50%, purple), polymorphisms (≥2 haplotypes, blue) and singletons (teal). Asterisks indicate samples sequenced using PacBio HiFi. (D) Overlap between SVs detected by PacBio long-read assemblies and Illumina short-read alignments on 31 matched samples (NA24835, HG00514, HG00733 and NA19240 excluded). Top bar shows overall SV sites across 31 samples, while the bottom bar displays the average count of SVs per sample, with green stripes representing concordant SV calls between technologies. (E) Length distribution of SVs detected by PacBio long-read assemblies and Illumina short-read alignments across all 31 matched samples. (F) Genome-wide distribution of SV hotspots divided in three categories: last 5 Mbp of chromosomes (yellow), overlapping (light blue), and novel (red) when compared to short-read SV analysis of 1000GP (23). The total sequence length is represented by each hotspot category (inset). (G) Heatmap of seven selected SV haplotypes for 4 Mbp MHC region (chr6:28,510,120-33,480,577 dashed lines) comparing regions of high SNV (red) and low diversity (blue) regions based on the number of alternate SNVs compared to the reference (GRCh38; alignment bin size 10 kbp, step 1 kbp). Phased SV insertions (blue arrows) and deletions (red arrows) are mapped above each haplotype. The most diverse regions correspond to SV hotspots (red/blue bars top row) and cluster with HLA genes (red bottom track).
Similarly, we estimate a 6% FDR for indels and 4% for SNVs based on an assessment of Mendelian transmission error from the HiFi and CLR parent–child trios (table S10, (18)). We find that 42% of the SVs are novel when compared to recent long-read surveys of human genomes (1, 4, 19–21) (fig. S10). The addition of African samples more than doubles the rate of new variant discovery when compared to non-Africans for all classes of variation (2.21× SVs (809 vs. 366), 3.70× indels (11,514 vs. 3,109), and 2.97× SNVs (160,232 vs. 54,006) for the 64th haplotype (Fig. 2C, table S11, (18)). On average, we detect 24,653 SVs, 794,406 indels, and 3,895,274 SNVs per diploid human genome (table S4).
Structural variant discovery from short-read alignments.
To enable comparison of the PAV calls with genetic variants discovered by WGS, we performed Illumina-based short-read sequencing for 3,202 samples from the 1000GP (34.5-fold coverage) (18) and discovered SVs using three analytic pipelines: GATK-SV (5), SVTools (6) and Absinthe (88). When focusing on the 31 unrelated samples with matching PacBio long-read sequences and callsets included in this study (NA24385, HG00514, HG00733 and NA19240 excluded), we observed 9,320 SVs per genome at 1.8% FDR by comparison to 24,596 SVs per genome from long-read assembly (Fig 2D; Fig S11). On average 77.4% of SVs detected by short-read pipelines were concordant with long-read assemblies, but only 29.6% of long-read SVs were observed in the short-read WGS callset (Fig. 2D). The greatest gains in sensitivity from long-read assemblies were observed among smaller SVs, where ~83.3% of events (<250 bp) were novel (Fig. 2E), while the short-read SV pipelines displayed greater sensitivity among large SVs > 5 kbp (Fig. 2E, figs. S11, S12, tables S12, S13).
Structural variant distribution and mechanisms.
SVs are known to be clustered (4, 15) and we identify 278 SV hotspots on the basis of our PAV callset (Fig. 2F, fig. S13, table S14, (18)) spanning ~279 Mbp of the genome (Fig. 2F inset). We find that 30.6% (32,222/105,327) of SVs on autosomes and chromosome X map within the last 5 Mbp of chromosome arms, corresponding to a ~4-fold enrichment (p=0.001, z-score=301.3, permutation test), with few notable exceptions—the long arm of the X chromosome and the short arms of chromosomes 3 and 20 (Fig. 2F, fig. S14A). Focusing on SVs >5 Mbp from chromosome ends (73,105), we identify 221 hotspots (fig. S14B). Of these, 49% (109/221) have not been previously identified by short-read analyses of the 1000GP data (23). These interstitial hotspots are enriched 6.6-fold (p=0.001, z-score=26.6, permutation test) for SDs consistent with homologous recombination and frequently correspond to gene-rich regions of exceptional diversity among human populations. For example, we identify three distinct hotspots mapping to the major histocompatibility complex (MHC) region that distinguish seven selected structural haplotypes (Fig. 2G, fig. S15, table S15). Our analysis indicates that a majority (98.85%) of this 4 Mbp region has been sequence resolved at the base-pair level (29 of the assemblies are a single assembled contig and 18 have a single gap; 17/19 individual HLA genes are fully sequence resolved in all assemblies; tables S15, S16).
A detailed analysis of the SVs with unambiguous breakpoint locations provided an opportunity to examine mechanisms of SV formation. Excluding MEIs and SVs with ambiguous breakpoints, we assessed 52,974 insertions and 30,467 deletions (table S17). We find 58% of insertions and 70% of deletions, including SVs in VNTRs, are flanked by at least 50 bp of homologous sequence suggesting formation by homology-directed repair (HDR) processes or non-allelic homologous recombination (NAHR). Amongst those, 15% of insertions and 25% of deletions showed >200 bp flanking homology and are more likely mediated by NAHR. VNTRs with short repeat units (<50 bp) account for a smaller number of events (1.6% insertions and 0.4% deletions) and suggest replication slippage-mediated expansion and contraction. Additionally, 40% of insertions and 29% of deletions show blunt-ended breakpoints or microhomology (<50 bp flanking sequence identity), consistent with nonhomologous end joining, microhomology-mediated end joining, or microhomology-mediated break-induced replication (24). Homology-associated SVs are twofold more frequent than expected from reports using short reads (25–27), and when considering Illumina sequencing-based SV calls from the same samples, only 2% of insertions and 19% of deletions appear to be NAHR-mediated SVs with ≥200 bp flanking homology (p-value <2.2e-16; Fisher’s exact test; table S17).
SVs and their breakpoints are generally more depleted within protein-coding sequences and other functional elements; with the exception of specific gene families where variability in the length of amino acid sequences relates to the function of the molecule (lipoprotein (e.g., LPA), mucins (MUC1, MUC3A, MUC4, MUC12, MUC20, MUC21), zinc finger genes (ZNF99, ZNF285, ZNF280), among others; table S18). We identify 9.4% of all SV breakpoints that intersect functional elements, such as exons (n=993), untranslated regions (UTRs; n=1,097), promoters (n=466), and enhancer-like elements (n=6,796) (Fig. 2B, table S19).
When we consider structural polymorphisms that arise from perfect triplet repeats, expansions outnumber contractions 3 to 1 (271 expansions, 88 contractions) consistent with such regions being systematically underrepresented in the original reference (8, 28). Over the 64 haplotypes, there are six such SVs per haplotype and we identify a total of 106 nonredundant loci (tables S20, S21). Of note, 5/7 of the largest insertions of uninterrupted CTG or CGG repeat insertions mapping within exons correspond to genes already associated with triplet repeat instability diseases or fragile sites. For example, we identify a 21-copy CTG repeat expansion in ATXN3 (Machado-Joseph disease), a 17-copy gain of CAG in HTT (Huntington’s disease), a 21-copy gain of a CGG repeat in ZNF713 (Fragile site 4A), and a 36-copy CGG gain in DIP2B (Fragile site 12A) (18). The discovery of these perfect repeat insertion alleles with respect to the human reference provides an important reference for future investigations of triplet repeat instability.
Mobile element insertions.
On the basis of the phased genome assemblies, we identified a collection (n=9,453) of fully sequence-resolved non-reference MEIs, including 7,738 Alus, 1,175 L1Hs, and 540 SVAs (18) and used sequence content of the elements and their flanking sequences to provide insight into their origin and mechanisms of retrotransposition. Retroelement insertions typically display the classic hallmarks of integration via target-site primed reverse transcription. These include endonuclease cleavage motifs at insertion breakpoints, polyadenylate tracts at their 3ʹ end, target site duplications ranging from 3 to 52 bp (mode = 14 bp), in addition to frequent inversion and truncation for L1 elements (fig. S16). Full-length L1 (FL-L1) elements are an especially relevant source of genetic variation since they can mutagenize germline and somatic cells and can lead to gene disruptions that cause human disease (29, 30). While a minority of non-reference L1s are full length (fig. S16, table S22), we find that 78% of FL-L1s possess two intact open reading frames (ORF1 and ORF2), encoding the proteins that drive L1, Alu, SVA, and processed pseudogene mobilization. Indeed, 23% of these sequences show evidence of activity as they are part of a database of 198 FL-L1s known to be active in vitro (31, 32), in human populations (33), and in cancers (34–36). Most active copies (72%; 142/198) are either in our callset or present in the reference genome and are now fully sequence resolved (table S23). We note that 19% of the active FL-L1s have at least one ORF disrupted, which includes a hot element at 9q32 reported to be highly active in diverse tumors (34).
Using L1 Pan troglodytes as an outgroup, we construct a phylogeny (85) of active human L1s and estimate their age in million years (Myr) (Fig. 3A, fig. S17). As expected, copies of the Ta-1 subfamily are the youngest (mean = 1.00 [95% CI: 0.88-1.13]), followed by Ta-0 (mean = 1.63 [95% CI: 1.49-1.77]) and pre-Ta (mean = 2.15 [95% CI: 1.91-2.40]) (fig. S18). Notably, the evolutionary age correlates with L1 features such as subfamily, level of activity, and allele frequency (Fig. 3B, fig. S19)—with the youngest FL-L1s typically corresponding to highly polymorphic and active Ta-1 sequences. Indeed, three out of the four youngest active FL-L1s, namely 2q24.1, 6p24.1 and 6p22.1-2, are Ta-1 copies reported to be extremely active in cancer genomes (34). In contrast, 1p12 is a fixed Pre-Ta insertion that despite integrating into the human genome approximately 1.8 Myr ago remains highly active both in the germline (33) and somatically associated with tumors (34–36). This indicates that a small set of pre-Ta representatives possibly remain very active in the human genome.
Fig. 3. Mobile element insertions.
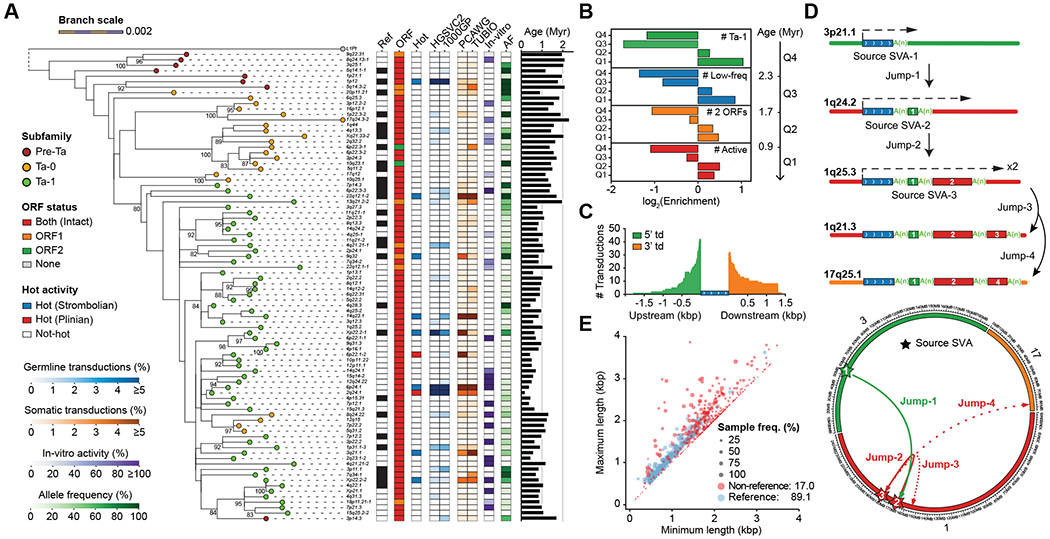
(A) Maximum-likelihood phylogenetic tree (85) for highly active sequence-resolved FL-L1s annotated by subfamily designation, presence/absence on the reference, ORF content, and hot activity profile (34–36) (bootstrap values ≥80% shown). Tree branch lengths are scaled according to the average number of substitutions per base position. Dashed lines map each L1 cytoband identifier to its corresponding branch on the tree. Pan troglodytes (L1Pt) is included as an outgroup. Heatmaps represent allele frequency (AF) based on the assembly discovery set, activity estimates based on in vitro assays (31, 32) and the number of transduction events detected in human populations (33) or cancer studies (34–36). (B) Enrichment and depletion in the number of FL-L1s belonging to the Ta-1 subfamily at age quartiles (Q1-Q4) compared with a random distribution. Same applies for the other features, including the number of FL-L1s with low allele frequency (MAF<5%), with two intact ORFs, or with evidence of activity. (C) Size distribution and number of 5′ and 3′ SVA-mediated transductions (td) based on the analysis of flanking sequences. (D) Schematic and circos representation for serial SVA-mediated transduction events. Dashed arrows indicate SVA transcription initiation and end. Transduced sequences are shown as colored boxes with their length proportional to transduction size. (E) Distributions of VNTR length (x-axis: the minimum, y-axis: the maximum) of reference and non-reference SVA elements. Reference SVAs are shown as blue dots and non-reference SVAs as red dots. The dot size represents the sample frequency of SVAs among discovery samples in the HGSVC.
SVA source elements are able to produce 5′ and 3′ transductions through alternative transcription start sites or bypassing of normal poly(A) sites during retrotransposition (10, 11). We detected 77 transduced non-repetitive DNA sequences at SVA insertion ends (table S24). Interestingly, 5′ transductions are more abundant (58%, 45/77) than 3′ transductions (Fig. 3C), as opposed to L1s, which primarily mediate 3′ transduction events (95%, 89/94). We used these unique transduced sequences to trace the origin of all 77 SVAs to 56 source SVA elements (fig. S20, table S25). A majority of source loci (84%) belong to the youngest human-specific SVA-E and SVA-F subfamilies (37), and only 11 source elements generate 38% of the offspring insertions.
SVA transductions can occasionally shuffle coding sequences as illustrated by the mobilization of a complete exon of HGSNAT by an intronic SVA in antisense orientation (fig. S21). In addition, one SVA source element appears to have caused three sequential mobilization events as indicated by nested transductions flanked by poly(A) tails (Fig. 3D, fig. S22). Finally, SVA elements harbor CpG-rich VNTRs in their interior regions that can expand and contract; we find that non-reference SVAs show significantly greater variability in VNTR copy number compared to those present in the reference (p-value < 10e-5, student’s t-test, two-sided, Fig. 3E).
Inversions.
Copy number neutral inversions are among the most difficult SVs to detect and validate (1). We applied multiple approaches integrating Strand-seq, Bionano optical mapping, and PAV-based variant discovery to generate a comprehensive and orthogonally validated set of inversions. PAV specifically increases inversion detection sensitivity for smaller events (fig. S23) by including a novel k-mer density assessment to resolve inner and outer breakpoints of flanking repeats, which does not rely on alignment breaks to identify inversion sites (18). PAV identifies an additional 43 inversions, on average, increasing sensitivity >2-fold compared to previous phased assembly callsets (2). In total, we discover on average 117 inversions per sample (316 nonredundant calls across samples) (fig. S23). As expected, inversions flanked by SDs tend to be larger than those in unique regions of the genome (38) (Wilcoxon rank sum test (one-sided, greater), p-value: 3.2x10−13, fig. S24). We focus on one complex region mapping to chromosome 16p12 where we observed a large number of polymorphic inversions flanked by SDs (9) (fig. S25A). The region harbors 11 different inversions (red and gray arrows) distinguishing 22 different structural configurations that span a ~2.5 Mbp gene-rich region of chromosome 16p (up to 13 protein-coding genes are flipped in orientation depending on human haplotypes) (Fig. 4A, (18)). These configurations are distributed among human populations, but do not correspond to unique haplotypes (Fig. 4A). For example, an analysis of the flanking sequence shows that at least five of the inversions occur in multiple haplotype backgrounds, indicative of recurrent inversion toggling (38, 39) between a direct and inverted state (fig. S26, (18)). Although Strand-seq data allow us to unambiguously identify the inversion status of the unique regions, most of the breakpoints themselves are not yet fully sequence resolved due to the presence of large repeats (Fig. 4A, fig. S25B, (3)).
Fig. 4. Complex patterns of structural variation.
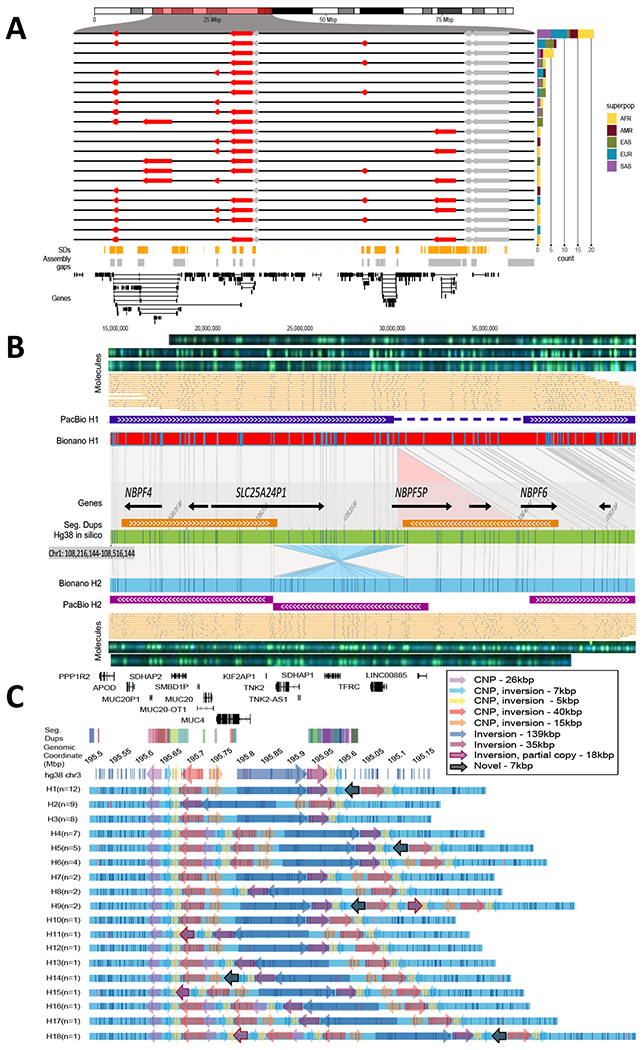
(A) An inversion hotspot mapping to a 2.5 Mbp gene-rich region of chromosome 16p12 (highlighted portion of ideogram). Haplotype structure of inversions (red arrows) are compared to the GRCh38 reference orientation (black lines) as well as additional inversions (gray), which could not be haplotype integrated because of uninformative markers. A barplot (right panel) enumerates the frequency of each distinct inversion configuration (n=22) by superpopulation for the 64 phased genomes. Bottom panels: Shows distribution of SDs (orange), assembly gaps (gray), and genes (black) in a given region. (B) A partially resolved complex SV locus (HG00733 at chr1:108,216,144-108,516,144). Optical maps generated by DLE1 digestion predict a deletion (red bar, Bionano H1) and an inversion (blue bar, Bionano H2) when compared to GRCh38 (green bar). Haplotype structures are strongly supported by extracted single molecules (beige) and raw images (green dots). Phased assembly correctly resolves the hap1 deletion (purple top) and Strand-seq detects the inversion (blue) but misses the flanking SD, which is a gap in the H2 assembly (gap). (C) Haplotype structural complexity at chromosome 3q29. Optical mapping of a 410 kbp gene-rich region (chr3:195,607,154-196,027,006) predicts 18 distinct structural haplotypes (H1-H8) that vary in abundance (n=1 to 12) and differ by at least nine copy number SDs and associated inversion polymorphisms (see colored arrows). This hotspot leads to changes in gene copy and order (GENCODE v34 top panel): 26 haplotypes are fully resolved by phased assembly (21 CLR, 5 HiFi) and the median MAP60 contig coverage of the region is 96.1%.
Complex structural variation.
We investigated the remaining gaps in our assemblies that map near or within centromeres, acrocentric regions, and SDs (figs. S6, S7, table S7). Because such repetitive regions have long been known to be enriched in complex variation (40) and refractory to sequence assembly even with long-read data (1), we re-examined the genome-wide optical maps to assess additional regions of structural variation. In 30 samples, we find that 72% of the large insertions and deletions (≥5 kbp) discovered by optical mapping are completely sequence resolved and concordant with the assembly (table S26), but the remainder show additional complexity. As an example, our analysis of the Puerto Rican phased genome assembly (HG00733) originally identified a 75 kbp deletion between the two haplotypes at chromosome 1p13.3, but a comparison with Bionano Genomics data shows a more complex pattern than a single deletion event: An inversion of 75 kbp is found in the alternate allele flanked by inverted SDs of 100 kbp involving NBPF genes (Fig. 4B). Interestingly, such discrepant regions appear to cluster in the genome.
A comparison between the phased assemblies and Bionano Genomics optical maps revealed 1,175 nonredundant SV clusters not detected in the phased assemblies and an additional 482 SV clusters with support in a different individual (table S27). Among the 1,175 Bionano SV clusters not detected in the PacBio phased assemblies, 71 overlapped unresolved sequence (“N” gaps), and 69.3% (765/1104) of the remaining SV clusters were detected from the Illumina short-read alignment pipelines (table S28). We manually inspected the 339 Bionano SV clusters that could not be detected in any of the short-read or assembly-based analyses and found read-depth evidence supporting 13.9% (47/339).
We estimate that there are still ~35 unresolved regions per phased assembly that are >50 kbp in length where there are five or more distinct SV haplotypes in the human population. On chromosome 3q29, for example (Fig. 4C), we identify 18 distinct structural haplotypes involving at least nine copy number and inversion polymorphisms affecting hundreds of kilobases of gene-rich sequence (min. 375 kbp, max. 690 kbp) (Fig. 4C). This pattern of structural diversity maps to the proximal breakpoint of the chromosome 3q29 microdeletion and microduplication syndrome rearrangement (chr3:195,999,954-197,617,802) associated with developmental delay and adult neuropsychiatric disease (41).
Genotyping.
We applied PanGenie (42), a method designed to leverage a panel of assembly-based reference haplotypes threaded through a graph representation of genetic variation that takes advantage of the linkage disequilibrium inherent in the phased genomes. We initially performed this genotyping step using a reference set of 15.5M SNVs, 1.03M indels (1-49 bp), and 96.1k SVs (where there was <20% allelic dropout; fig. S1, table S29) and genotyped these variants into the 1000GP WGS dataset (18) observing expected patterns of diversity (15) (Fig. 5A, figs. S27, S28).
Fig. 5. SV genotyping and eQTL analysis.
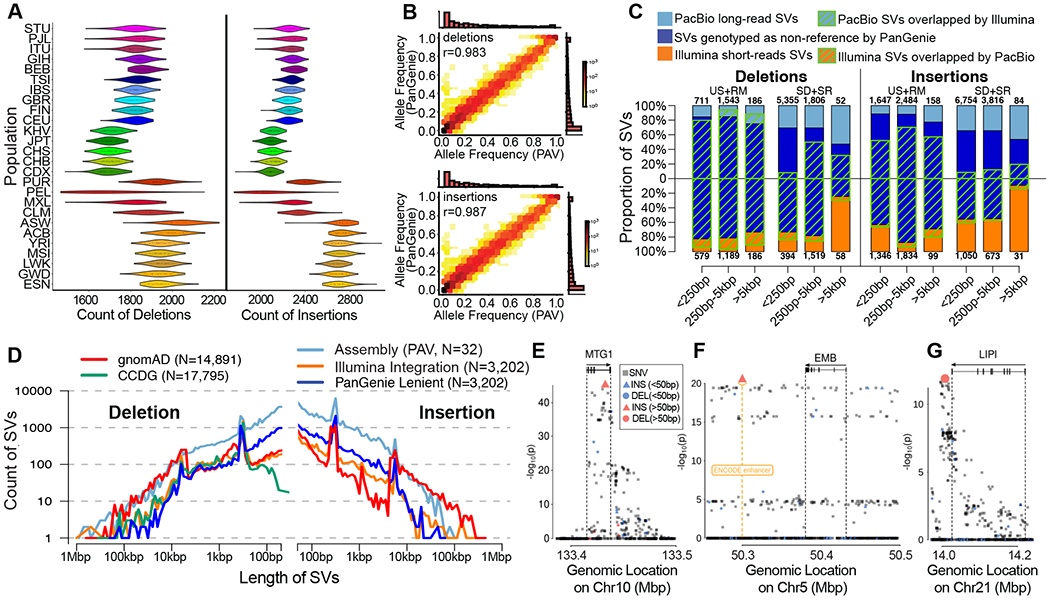
(A) Distribution of heterozygous SV counts per diploid genome broken down by population, based on PanGenie genotypes passing strict filters. (B) Concordance of allele frequency (AF) estimates from the assembly-based PAV discovery callset and AF estimates from genotyping unrelated Illumina genomes (n=2,504) with PanGenie (strict genotype set of 24,107 SVs); marginal histograms are in linear scale. (C) Count of short- and long-read SVs across variant class, size distribution, and genomic sequence localization. Blue bars represent the proportion of SVs genotyped by PanGenie with AF>0 and green stripes represent concordant SVs between technologies. SD: segmental duplications; SR: simple repeats; RM: repeat masked (not SD or SR); US: unique sequence. (D) Length distribution of common SVs sites (AF>5%) represented in assembly-based callset, including variants genotyped using PanGenie and all common variants from population-scale studies from the Genome Aggregation Database (gnomAD-SV) and the Centers for Common Disease Genetics (CCDG; insertions from CCDG omitted due to lack of data). Length distributions for all variants (not restricted to common) are provided in fig. S23. (E-G) Examples of lead SV-eQTLs (large symbols) in context of their respective genes, overlapping regulatory annotation, and other variants (small symbols). (E) An 89 bp insertion (chr10-133415975-INS-89) is linked to decreased expression of MTGI (q-value = 4.10e-11, Beta = −0.55 [−0.51 — −0.59]). (F) A 186 bp insertion (chr5-50299995-INS-186), overlapping an ENCODE enhancer mark (orange), is the lead variant associated with decreased expression of EMB (q-value = 2.92e-06, Beta = −0.44 [−0.39 — −0.49]). (G) A 1,069 bp deletion (chr21-14088468-DEL-1069) downstream of LIPI is linked to increased expression of LIPI (q-value = 0.0022, Beta = 0.44 [0.38 — 0.50]).
As one measure of genotyping quality, we compare the allele frequencies derived from assembly-based PAV calls across the 64 reference haplotypes to short-read-based allele frequencies obtained from PanGenie for the 2,504 unrelated individuals. From the raw output of PanGenie, we observe an allele frequency correlation (Pearson’s) of 0.98 for SNVs, 0.95 for indels, and 0.85 for SVs. To further improve SV genotyping, we filter the variants by assessing Mendelian consistency, the ability to detect the non-reference allele, genotype qualities, and concordance to assembly-based calls in a leave-out-one experiment into account (18). Using these criteria, we define a subset of strict and lenient SVs for genotyping containing 24,107 SVs (25%) and 50,340 SVs (52%), respectively, with excellent allele frequency correlation of 0.99 (strict, Fig. 5B) and 0.95 (lenient, fig. S29). Performance metrics for deletions and insertions are comparable (strict set: SV deletions, r=0.98; SV insertions, r=0.99; Fig. 5B), highlighting the value of sequence-resolved insertion alleles being part of our reference panel, as well as the algorithm’s ability to leverage it (fig. S30). Beyond SVs, 12,283,650 SNVs (79%) and 705,893 indels (68%) met strict filter criteria (note: given this larger fraction, we did not define a lenient set for these variant classes).
Added value from graph-based genotyping into short read WGS data.
To determine the value added by PanGenie genotyping, we next focused on an integrated comparison of long-read SV discovery (PAV), state-of-the-art short-read SV discovery, and the set of genotypable SVs by PanGenie. Consistent with our previous analyses (43), we observed that most SVs specific to long-read discovery localized to highly repetitive sequences, which collectively harbored 95.8% of long-read-specific deletions, and 85.7% of long-read-specific insertions (table S30). We also discovered variation that was uniquely detected (although not sequence-resolved) and genotyped by sequencing read-depth from short reads. On average, there are 167 large CNVs (>5 kbp) per sample – 88.2% of which are not captured by long-read assemblies (Fig 5C, figs. S11, S31). A large fraction of these calls maps to large repetitive regions such as segmental duplications that are not fully sequence-resolved. Remarkably, we find that 42.5% (strict) and 59.9% (lenient) of PanGenie-genotypable SVs are absent from the short-read callset. We examined the distribution of common long-read SVs genotyped at >5% AF across all the 3,202 Illumina genomes against the short-read SVs from large population studies, including the Centers for Common Disease Genomics (CCDG, (6)) and Genome Aggregation Database (gnomAD, (5)) (Fig. 5D, fig. S12). The ability to genotype variation typically not detected in Illumina callsets is reflected in increased numbers of common SVs (AF>5%), particularly deletions below 250 bp and insertions under 1 kbp, genotyped by PanGenie but not seen in CCDG and gnomAD-SV, while also emphasizing the overall value of large-scale short-read datasets to capture rare variation and large CNVs in the population (fig. S31).
QTL analyses.
We applied PanGenie genotypes (strict set) to systematically discover quantitative trait loci (eQTL) associated with structural variation. First, we performed deep RNA-seq (>200M fragments) of the corresponding 34 lymphoblastoid cell lines and integrated these data with 397 transcriptomes of 1000GP samples from GEUVADIS (44). We pursued cis expression quantitative trait loci (eQTL) and cis splicing quantitative trait loci (sQTL) mapping across the merged set of 427 donors, using a window of 1 Mbp centered around the gene or splice cluster, respectively, testing all variants with a minor allele frequency of ≥1% and at Hardy-Weinberg equilibrium (HWE exact test p-value ≥ 0.0001). We considered 23,953 expressed genes (15,504 of which were protein-coding) and 36,100 splicing clusters (linked to 11,278 genes).
Using this design, we identify 58,152 indel-eQTLs (linked to 6,748 unique genes) and 2,109 SV-eQTLs (linked to 1,526 unique genes; table S31) at an FDR of 5%. The set includes 819 lead indel-eQTLs and 38 lead SV-eQTLs at distinct genes, respectively (table S31). In the sQTL analysis we identified 3,382 SV-sQTLs (FDR 5%, linked to 758 unique genes; table S32) of which 65 SV-sQTLs at distinct genes were the lead association at the locus (18). In line with prior studies (23, 45), the lead variants are enriched for SVs (Fisher’s exact eQTL p-value = 1.0e-6, OR = 1.2; sQTL p-value = 1.6e-4, OR = 1.2) as well as smaller indels (Fisher’s exact eQTL: p-value = 8.8e-113, OR = 1.2; sQTL: p-value = 3.5e-72, OR = 1.2), whereas they are depleted for SNVs (Fisher’s exact eQTL p-value = 1.8-e118, OR = 0.84; sQTL: p-value = 1.2e-75, OR = 0.84). Among SVs, deletions show the greatest effect when compared to insertion events (table S33, (18)).
We overlapped lead SV-eQTLs with our Illumina-based discovery callset (18) and a recent large-scale SV study of 17,795 genomes (6) and find that 42% (16 out of 38 SVs) of the lead eQTL associations reported here are novel. Of these previously inaccessible SVs, 12 (75%) correspond to insertions (2 Alu MEIs, 3 tandem duplications, and 7 repeat expansions)—SV classes typically under-ascertained in short-read datasets (1). For example, one of our top novel lead SVs is an 89 bp VNTR insertion in the terminal intron of the mitochondrial ribosome-associated GTPase 1 gene (MTG1; Fig. 5E) and is seen in conjunction with decreased expression. Similarly, we identify a 186 bp insertion in an ENCODE enhancer for B-cell lymphomas, which is associated with reduced expression of the immunoglobulin superfamily gene embigin (EMB; Fig. 5F). In contrast, we sequence resolve a 1,069 bp deletion located in an SD region downstream of the Lipase I gene (LIPI; Fig. 5G) and find that it is associated with increased gene expression of LIPI. Single-nucleotide polymorphisms at this locus have been linked to heart rate in patients with heart failure with reduced ejection fraction in a previous genome-wide association study (GWAS, p-value 9.0e-06 reported in (46)).
Ancestry and population genetic analyses.
The availability of haplotype-phased assemblies provides an opportunity to explore the ancestry and population genetic properties of the genomes and SVs at multiple levels. We applied a machine-learning method (47) and developed a hidden Markov model to identify ancestry-informative SNVs and to assign ancestral segments per block based on population genetic data from the Simons Genome Diversity Project (SGDP, (48)) (18). The two methods, as well as the different sequencing platforms, produce highly concordant results (>90%, fig. S32). At the family level, we can accurately assign paternal and maternal haplotypes and distinguish recombination crossover events in the child compared to parental haplotypes (Fig. 6A).
Fig. 6. Ancestry and population differentiation inferences using haplotype-phased diploid assemblies.

(A) Inferred local ancestries (18) for maternal (upper) and paternal (bottom) haplotypes of HG00733 are compared to parental haplotypes (maternal: HG00732, paternal: HG00731). Ancestral segments are colored (African: yellow, Native American: red, and European: blue) and are consistent with the recent demographic history of the island (18). HG0733 SVs (≥50 bp; insertion: green, deletion: purple), inferred recombination breakpoints (triangles), and transmission of recombinant parental haplotypes (dashed lines) are shown. (B) Length distribution (log10) of ancestry tracts among the 64 genomes assigned to five superpopulations shows evidence of recent (Admixed American) and more ancient (South Asian) admixture. (C) Top population-specific Fst variants (dark color) and top superpopulation-specific Fst variants (light color). The number of stratified SVs differs by orders of magnitude depending on population. (D) Top SV PBS (population branch statistic) values within 5 kbp of genes identify SV candidates for selection and disease. A high PBS statistic suggests AF differences among populations are a result of selection.
At the population level, on average 87.2% of the assembled sequence can be assigned ancestry. 1000GP samples originating from the African continent show the largest tracts of uniform ancestry (mean length = 23.6 cM, Fig. 6B, fig. S33) in contrast to North and South American populations (mean length=2.65 cM, Fig. 6B, fig. S33) and South Asians (mean length=4.38 cM, Fig. 6B), consistent with recent and more ancient admixture. For example, the African American, African Caribbean, and Admixed American 1000GP samples show the greatest diversity of ancestral segments (Fig. 6B, figs. S33, S34) most likely as a result of the transatlantic slave trade and colonial era migration (49).
Focusing on our more comprehensive genotyping of SVs into WGS data, we searched for population-stratified variants since these are potential candidates for local adaptation (50, 51) that could not have been characterized in the original study of 1000GP populations (15). Using Fst as a metric, we find that the number of such population-stratified variants varies widely among different groups likely as a consequence of ancestral diversity (Africans), population bottlenecks (East Asians), and admixture (South Asians) (Fig. 6C). Restricting our analysis to SVs located within 5 kbp of genes and applying population branch statistics (PBS) (51), we identify 117 stratified SVs (PBS >3 s.d., tables S34, S35) and further characterize these by the number of base pairs deleted or inserted per locus (Fig. 6D). The greatest outlier is a 4.0 kbp insertion within the first intron of LCT (lactase gene) originally reported based on fosmid sequencing from European samples (52). We determine that the corresponding insertion is ancestral (i.e., the human reference genome carries the derived deleted allele), the insertion harbors 11 predicted transcription factor binding sites, and the deletion likely occurred as a result of an Alu-mediated NAHR event ~520,000 years ago (fig. S35).
LCT variation is one of the most well-known genes under adaptive evolution among Europeans. Notably, the reported causal, derived allele of lactase persistence in Europeans (−13910*T; rs4988235) is in complete linkage disequilibrium (D′=1) with the reference allele of this SV, and it will be interesting to determine the functional roles of these two mutations in lactase persistence (53). In other cases, the population-stratified variants are nested among known regulatory elements or intersect them directly, such as a 76 bp tandem repeat expansion in a PLEC intron, a cytoskeleton component, seen only in Africans (AF=0.82) and Admixed Americans (AF=0.06). Similarly, we identify a 2.8 kbp insertion mapping near potential repressor-binding sites in a CLEC16A intron, a gene associated with type 1 diabetes when disrupted (54). This variant shows a high frequency in American populations (AF=0.28), with the highest PBS signal among Peruvians (AF=0.39), but is rarely observed in other populations (AF≤0.04). Further studies are needed to confirm functional effect; however, it is interesting to note that type 1 diabetes in Peruvians is among the highest in the world (55).
DISCUSSION
We have generated a diversity panel of phased long-read human genome assemblies that has significantly improved SV discovery and will serve as the basis to construct new population-specific references. Previous large-scale efforts have largely been inferential and biased when it comes to the detection of SVs. Here, we develop a method to discover all forms of genetic variation (PAV) directly by comparison of assembled human genomes. In contrast, SV discovery from the 1000GP was indirect and limited given the frequent proximity of SVs to repeat sequences inaccessible to short reads (15, 23). The 1000GP, for example, reported 69,000 SVs based on the analysis of 2,504 short-read sequenced genomes. In contrast, our analysis of 32 genomes (64 unrelated haplotypes) recovers 107,136 SVs, more than tripling the rate of discovery when compared to short-read Illumina SV analyses on the same samples (Fig. 2D). Recent large-scale short-read sequencing studies (5, 6), interrogating tens of thousands of samples, show even lower SV sensitivity reporting 5,000 to 10,000 SVs per sample, when compared to our phased-assembly approach, which identifies 23,000 to 28,000 SVs per sample. This lack of sensitivity for SV discovery from short reads also affects common variation (AF>5%) and we increase the amount of common SVs by 2.6-fold. The predominant source of this increase in sensitivity was among small SVs (<250 bp) localized to SDs and simple repeat sequences, where we observed a dramatic 8.4-fold increase in variant discovery (12,109 SVs per genome from long-read assembly, 1,444 per genome from Illumina short-read alignment; Fig. 5C). Notably, all discovered genetic variation is physically phased and therefore SVs are fully integrated with their flanking SNVs.
Compared to previous reports based on short-read sequencing (25–27), a surprising finding has been the larger fraction of SVs (63%) now assigned to homology-based (>50 bp) mutation mechanisms, including HDR, NAHR and VNTR. Breakpoint characterization with short-read data apparently biased early reports toward relatively unique regions concluding that <30% of SVs were driven by homology-based mutational mechanisms (25–27). Since a majority of unresolved structural variation still maps to large repeats, including centromeres and SDs subject to NAHR, we conclude that homology-based mutational mechanisms will contribute even further and are, therefore, the most predominant mode shaping the SV germline mutational landscape. Notwithstanding, access to fully assembled retrotransposons and their flanking sequence provides the largest collection of annotated source elements for both L1 and SVA mobile elements. We find that 14% of SVA insertions are associated with transductions compared to 8% of L1s—a difference driven in part by the proclivity of SVAs to transduce sequences at their 5′ and 3′ ends. We find a surprisingly large number of L1 source elements (19%) with defective ORFs suggesting either trans-complementation (56) or polymorphisms leading to the recent demise of these active source elements. Of note, some of the youngest L1 copies (e.g., 6p22.1-1 and 2q24.1) have been reported to be rare polymorphisms able to mediate massive bursts of somatic retrotransposition in cancer genomes (57). This suggests that recently acquired hot L1s, which have not yet reached an equilibrium with our species, contribute disproportionately to disease-causing variation (58).
Genome-wide QTL scans can bridge the gap between molecular and clinical phenotypes and serve as a proxy for functional effects mediated by genetic variant classes (23, 44, 59). Taking advantage of the fully phased sequence-resolved genetic variation, we demonstrate this by applying PanGenie, a new pangenome-based genotyping method, to 3,202 1000GP genomes, resulting in reliable genotype calls for 705,893 indels and up to 50,340 SVs (lenient genotype set). Of these, 59.9% are presently missed in multi-algorithm short-read discovery callsets and the majority (68.2%) of these novel SVs are insertions. Our work, thus, provides a framework for the discovery of eQTLs and disease-associated variants with the potential to discriminate among SNVs, indels, and SVs as the most likely causal variants (lead variants) associated with human genetic traits. The fact that 31.9% of SV-eQTLs and 48% of lead SV-eQTLs are rendered accessible to short reads only through the availability of our panel of haplotype-resolved assemblies testifies to the importance of this resource for future GWAS. Once again, among the lead SV-eQTLs, 75% are insertions, although there are also promising deletion eQTLs. For example, we identify a 1,069 bp deletion eQTL near LIPI, a GWAS disease locus for cardiac failure (46). Indeed, Summary-data-based Mendelian Randomization analysis (SMR, (60)) suggests that this SV-eQTLs of LIPI may be driving this association (SMR p-value adj.: 5.6e-4).
Haplotype-resolved SVs with accurate genotypes will also facilitate evolutionary and population genetic studies of SVs, including estimations of the rates of recurrent mutation, population stratification, and selective sweeps. As part of this analysis, we identify 117 loci associated with genes where allele frequencies differ radically between populations and are candidates for local adaptation (50, 51). Ancestral reconstructions of haplotype-resolved SVs can be further extended to identify introgressed SVs from Neanderthals and Denisovans (61). While archaic SNV haplotypes have been identified in modern-day humans, little is known regarding SV content given the degraded nature of ancient DNA. Combined with coalescent estimates of evolutionary age, it should now be possible to systematically identify associated introgressed SVs and assess them for signatures of adaptive evolution as was recently demonstrated (62). Even though we estimate that 96% of SVs with an allele frequency above 2% have been theoretically discovered (63), a greater diversity of human genomes are required to adequately account for population differences, effects of selection, as well as archaic introgression. Our findings clearly indicate that genomes of African ancestry represent the deepest reservoir of untapped structural variation. Ongoing efforts from the HGSVC, All of Us, and the Human Pangenome Reference Consortium (HPRC, https://humanpangenome.org) exploring the normal pattern of structural variation using long-read sequences over the next few years will be critical to better understand human genetic variation.
Currently, our understanding of the full spectrum of structural variation is not yet complete, despite the advances presented here. There are two important limitations. First, comparison with optical mapping data identifies hundreds of gene-rich regions near and within SDs harboring more complex forms of SVs that are still not fully resolved by long-read assembly. The remaining gaps in human genomes cluster and a subset represent complex SV differences between human haplotypes. Second, only ~50% of our long-read discovery set of SVs can, at present, be reliably genotyped in short-read data using PanGenie. Expanding the number of assembly-based haplotypes available as pangenomic reference will likely mitigate this, but multiallelic VNTRs/STRs as well as SVs embedded in larger repeats such as SDs and centromeres are particularly problematic and novel methods are needed to characterize these. Recent advances coupling both HiFi and ultra-long-read Oxford Nanopore data show promise in resolving the sequence of these more complex regions from both haploid (64) and diploid human genome assemblies (65). Once a larger number of such complex regions are haplotype resolved across diversity panels of human genomes—and algorithms continue to evolve to exploit this information—we expect larger portions (fig. S36) of the human genome to become amenable to genotyping and association with human traits.
METHODS (short)
Libraries were prepared from high-molecular-weight DNA from lymphoblast lines (Coriell Institute). Long-read CLR and HiFi sequencing data (25-50X) were generated on the Sequel II platform (Pacific Biosciences) using 15-hour (CLR) or 30-hour (HiFi) movie times. Strand-seq data were produced from the same samples and used to identify and phase heterozygous SNVs (LongShot (66) and DeepVariant (67)) from the squashed genome assemblies (Peregrine (68) or Flye (69)). StrandphaseR (70), SaaRclust (71) and WhatsHap (72, 73) partitioned long reads into haplotypes to generate phased genome assemblies (PGAS (3)). MAPQ60 phased assembly contig coverage is estimated for autosomes (chr 1-22) and the X chromosome to balance male and female comparisons, excluding regions of heterochromatin (Giemsa pos./var. staining) and unresolved reference sequence (N-gaps). We generated optical maps for 30 of the 32 samples based on DLE1 digestion (Bionano Genomics).
PAV was used to characterize SNVs, indels, and SVs compared to the human reference GRCh38. Inversions were detected using Strand-seq (1, 9, 38), optical mapping data (Bionano Solve v3.5) and PAV (88), which detects inversion signatures using a novel k-mer density approach to identify inner and outer breakpoints of flanking repeats without relying on alignment truncation. The diploid callset is created by merging two independent haploid callsets. We removed variants in collapses by SDA (74) and misaligned contig clusters, then merged variants from all samples to create a nonredundant callset that was subsequently filtered by additional support (18). SVs required support from at least one of seven other sources, including read-based callers (MELT, PBSV, PALMER) (33, 75), optical mapping data, breakpoint k-mer analysis, and PAV replication with LRA (76). Indels required support from at least two of four sources and SNVs required support from at least two of five sources. MEIs were primarily discovered using PAV which were then annotated using MEIGA-PAV (89). In addition, Illumina and PacBio alignments were processed using MELT and PALMER, respectively, in order to increase sensitivity for MEI discovery. Finally, MEI calls across different platforms were merged into an integrated callset.
We estimated functional element depletion for SVs by simulation permuting SVs within their 1 Mbp bin 100,000 times and recording functional element hits for insertions and deletions for each functional category (CDS, 5′ UTR, 3′ UTR, promoter, proximal enhancer, distal enhancer, CTCF, and intron). SV hotspots were defined by searching for regions of increased SV density using kernel density estimation implemented with the ‘hotspotter’ function from the primatR package (38, 77). Illumina WGS short reads (250 bp paired end) were generated (34.5-fold) (18) from 1000GP samples (2,504 unrelated individuals and additional samples from children to form 602 trios). SVs were called from an ensemble of three methods: GATK-SV (5), SVTools (6) and Absinthe (88) and detailed comparisons between long- and short-read data were performed for the 31 matched samples (18).
We genotyped all 3,202 genomes using PanGenie (42), which determines k-mer abundances from an input set of unaligned short reads and infers the genotypes of this short-read sample at all loci represented in the reference set. The method exploits both the linkage disequilibrium structure inherent to the reference haplotypes and the sequence resolution they provide and, hence, makes full use of the haplotype resource provided. RNA-seq data QC was conducted with Trim Galore! (78) and mapped to the reference genome using STAR (79), followed by gene-level quantification using FeatureCounts (80) and quantification of splice events using leafCutter (81). We mapped the effect of genetic variation on both expression levels and splicing ratios using a QTL mapping pipeline based on a linear mixed model implemented in LIMIX (82–84). We combined our QTL statistics with published GWAS results to assess the link among genetic variation, GWAS traits, and either gene expression or splicing ratios using SMR (60). To identify population-stratified SVs in the 26 populations, we computed the FST-based PBS (18). For each focal population, we constructed population triplets by choosing sister- and out-groups inside and outside the continent where the focal population resides, respectively. For each focal population, we selected the maximum PBS per gene for all possible PBS triplets and selected the subset that are at least three standard deviations (Z transformation) beyond the PBS mean as potential targets of selection. Detailed descriptions of materials and methods are available in the supplementary materials (18).
Supplementary Material
Acknowledgements:
We thank T. Brown for assistance in editing this manuscript and K. Hoekzema and C. Baker for the preparation of cell line DNA. We also recognize the computational support (P.H. Rehs and C. Siebert) and infrastructure provided by the Centre for Information and Media Technology (ZIM) at the University of Düsseldorf, the EMBL IT Services, and additional computational analyses (C. Alkan, F. Hormozdiari, D.S. Gordon and S. Murali). We thank M. Paulsen from the EMBL Flow Cytometry Core Facility, as well as J. Zimmermann and V. Benes from the EMBL Genomics Core Facility for assisting in Strand-seq sample preparation and sequencing. We thank the Human Pangenome Reference Consortium (HPRC) for use of the publicly available GIAB sequence data for the Ashkenazim benchmark sample HG002/NA24385. We are grateful to the people who generously contributed samples as part of the 1000 Genomes Project (1000GP). We thank the Pan-UKB project and UK Biobank for making the GWAS results available.
Funding:
Funding for this research project by the Human Genome Structural Variation Consortium (HGSVC) came from the following grants: National Institutes of Health (NIH) U24HG007497 (to C.L., E.E.E., J.O.K., T.M., M.E.T., A.B., M.B.G., S.E.D., I.H., S.A.M., R.E.M., M.J.P.C., and K.C.J.S.), NIH R01HG002898 (to S.E.D.), NIH R01HD081256 (to M.E.T.), NIH 1R01HG007068-01A1 (to R.E.M.), NIH R01HG002385 (to E.E.E.), R01MH115957 (to M.E.T.), NIH R15HG009565 (to X.S.), NIH 1U01HG010973 (to M.J.P.C., T.M., and E.E.E.), NIH 1R35GM138212 and a subaward from 1OT3HL147154 (to Z.C.), NIH/NHGRI Pathway to Independence Award K99HG011041 (to PH.H.), the German Research Foundation (391137747 and 395192176 to T.M.), the European Research Council (Consolidator grant 773026 to J.O.K. and Starting Grant 716290 to J.M.C.T.), the German Federal Ministry for Research and Education (BMBF 031L0184 to J.O.K. and T.M. and BMBF 031L0181A to J.O.K), the Spanish Ministry of Economy, Industry and Competitiveness (SAF2015-66368-P to J.M.C.T.), the Wellcome Trust grants WT085532 and WT104947/Z/14/Z and the European Molecular Biology Laboratory (to S.F., L.C., E.L., H.Z.-B., P.F., J.O.K.), National Science Foundation of China (32070663 to K.Y., 61702406 to X.Y.), National Key R&D Program of China (2017YFC0907500 to K.Y., 2018YFC0910400 to K.Y., 2018ZX10302205 to X.Y.). This work was supported by the BMBF-funded de.NBI Cloud within the German Network for Bioinformatics Infrastructure (de.NBI) (031A537B, 031A533A, 031A538A, 031A533B, 031A535A, 031A537C, 031A534A and 031A532B). E.E.E. is an investigator of the Howard Hughes Medical Institute. J.O.K. and J.M.C.T. are European Research Council (ERC) investigators. C.L. was a distinguished Ewha Womans University Professor supported, in part, by an Ewha Womans University research grant for 2019–2020. Also, this study was supported, in part, by funds from The First Affiliated Hospital of Xi’an Jiaotong University (to C.L.). A.C., W.E.C., and M.C.Z. were supported in part by a Centers for Common Disease Genomics (CCDG) grant from the National Human Genome Research Institute (UM1HG008901). M.S.G. is supported by a PhD fellowship from Xunta de Galicia (Spain). Illumina sequencing data from the 1000GP samples were generated at the New York Genome Center with funds provided by NHGRI Grants 3UM1HG008901-03S1 and 3UM1HG008901-04S2.
Footnotes
Competing interests: A.R.H. and J.L. are employees and shareholders of Bionano Genomics. A.M.W. is an employee and shareholder of Pacific Biosciences. M.C.Z. is a shareholder of Merck & Co. and Thermo Fisher Scientific Inc. P.F. is a member of the Scientific Advisory Boards of Fabric Genomics, Inc., and Eagle Genomics, Ltd. A.D.S., J.O.K., T.M., M.G., and D.P. have a pending patent application relevant to the subject matter (method relevant to Strand-seq).
Data and materials availability: All data generated are available via the official HGSVC2 data portal at www.internationalgenome.org/data-portal/data-collection/hgsvc2. Primary data are available at INSDC under the following accessions and project IDs: Illumina high-coverage genomic sequence (PRJEB37677), HiC and RNA-seq (ERP123231), Bionano Genomics (ERP124807), PacBio (PRJEB36100, ERP125611 and PRJNA698480), and Strand-seq (PRJEB39750). Alignments used for L1 phylogenetic tree construction (Fig. 3), merged PAV callsets, full eQTL results and project code are available via Zenodo (85–89). The following cell lines/DNA samples were obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research: [NA06984, NA06985, NA06986, NA06989, NA06991, NA06993, NA06994, NA06995, NA06997, NA07000, NA07014, NA07019, NA07022, NA07029, NA07031, NA07034, NA07037, NA07045, NA07048, NA07051, NA07055, NA07056, NA07340, NA07345, NA07346, NA07347, NA07348, NA07349, NA07357, NA07435, NA10830, NA10831, NA10835, NA10836, NA10837, NA10838, NA10839, NA10840, NA10842, NA10843, NA10845, NA10846, NA10847, NA10850, NA10851, NA10852, NA10853, NA10854, NA10855, NA10856, NA10857, NA10859, NA10860, NA10861, NA10863, NA10864, NA10865, NA11829, NA11830, NA11831, NA11832, NA11839, NA11840, NA11843, NA11881, NA11882, NA11891, NA11892, NA11893, NA11894, NA11917, NA11918, NA11919, NA11920, NA11930, NA11931, NA11932, NA11933, NA11992, NA11993, NA11994, NA11995, NA12003, NA12004, NA12005, NA12006, NA12043, NA12044, NA12045, NA12046, NA12056, NA12057, NA12058, NA12144, NA12145, NA12146, NA12154, NA12155, NA12156, NA12234, NA12239, NA12248, NA12249, NA12264, NA12272, NA12273, NA12274, NA12275, NA12282, NA12283, NA12286, NA12287, NA12329, NA12335, NA12336, NA12340, NA12341, NA12342, NA12343, NA12344, NA12347, NA12348, NA12375, NA12376, NA12383, NA12386, NA12399, NA12400, NA12413, NA12414, NA12485, NA12489, NA12546, NA12707, NA12708, NA12716, NA12717, NA12718, NA12739, NA12740, NA12748, NA12749, NA12750, NA12751, NA12752, NA12753, NA12760, NA12761, NA12762, NA12763, NA12766, NA12767, NA12775, NA12776, NA12777, NA12778, NA12801, NA12802, NA12812, NA12813, NA12814, NA12815, NA12817, NA12818, NA12827, NA12828, NA12829, NA12830, NA12832, NA12842, NA12843, NA12864, NA12865, NA12872, NA12873, NA12874, NA12875, NA12877, NA12878, NA12889, NA12890, NA12891, NA12892].
References and Notes
- 1.Chaisson MJP et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun 10, 1784 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garg S et al. Chromosome-scale, haplotype-resolved assembly of human genomes. Nat. Biotechnol (2020), doi: 10.1038/s41587-020-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porubsky D et al. Fully phased human genome assembly without parental data using single-cell strand sequencing and long reads. Nat. Biotechnol (2020), doi: 10.1038/s41587-020-0719-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Audano PA et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell. 176, 663–675.e19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collins RL et al. A structural variation reference for medical and population genetics. Nature. 581, 444–451 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abel HJ et al. Mapping and characterization of structural variation in 17,795 human genomes. Nature. 583, 83–89 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wenger AM et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol 37, 1155–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sulovari A et al. Human-specific tandem repeat expansion and differential gene expression during primate evolution. Proc. Natl. Acad. Sci. U. S. A 116, 23243–23253 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanders AD et al. Characterizing polymorphic inversions in human genomes by single-cell sequencing. Genome Res. 26, 1575–1587 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xing J et al. Emergence of primate genes by retrotransposon-mediated sequence transduction. Proc. Natl. Acad. Sci. U. S. A 103, 17608–17613 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damert A et al. 5’-Transducing SVA retrotransposon groups spread efficiently throughout the human genome. Genome Res. 19, 1992–2008 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Computational Pan-Genomics Consortium, Computational pan-genomics: status, promises and challenges. Brief. Bioinform 19, 118–135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paten B, Novak AM, Eizenga JM, Garrison E, Genome graphs and the evolution of genome inference. Genome Res. 27, 665–676 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eizenga JM et al. Pangenome Graphs. Annu. Rev. Genomics Hum. Genet (2020), doi: 10.1146/annurev-genom-120219-080406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature. 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zody MC, 3,202 Illumina cohort dummy. bioRxiv (February 5., 2021), doi: 10.1101/2021.02.05.000000. [DOI] [Google Scholar]
- 17.Zook JM et al. An open resource for accurately benchmarking small variant and reference calls. Nat. Biotechnol 37, 561–566 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Materials and methods are available as supplementary materials.
- 19.Huddleston J et al. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 27, 677–685 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi L et al. Long-read sequencing and de novo assembly of a Chinese genome. Nat. Commun 7, 12065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seo J-S et al. De novo assembly and phasing of a Korean human genome. Nature. 538, 243–247 (2016). [DOI] [PubMed] [Google Scholar]
- 22.ENCODE Project Consortium et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 583, 699–710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sudmant PH et al. An integrated map of structural variation in 2,504 human genomes. Nature. 526, 75–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carvalho CMB, Lupski JR, Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet 17, 224–238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conrad DF et al. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat. Genet 42, 385–391 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lam HYK et al. Nucleotide-resolution analysis of structural variants using BreakSeq and a breakpoint library. Nat. Biotechnol 28, 47–55 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mills RE et al. Mapping copy number variation by population-scale genome sequencing. Nature. 470, 59–65 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaisson MJP et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature. 517, 608–611 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hancks DC, Kazazian HH Jr, Roles for retrotransposon insertions in human disease. Mob. DNA. 7, 9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott EC, Devine SE, The Role of Somatic L1 Retrotransposition in Human Cancers. Viruses. 9 (2017), doi: 10.3390/v9060131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brouha B et al. Hot L1s account for the bulk of retrotransposition in the human population. Proc. Natl. Acad. Sci. U. S. A 100, 5280–5285 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beck CR et al. LINE-1 retrotransposition activity in human genomes. Cell. 141, 1159–1170 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gardner EJ et al. The Mobile Element Locator Tool (MELT): population-scale mobile element discovery and biology. Genome Res. 27, 1916–1929 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodriguez-Martin B et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat. Genet 52, 306–319 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung H, Choi JK, Lee EA, Immune signatures correlate with L1 retrotransposition in gastrointestinal cancers. Genome Res. 28, 1136–1146 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tubio JMC et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 345, 1251343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang H et al. SVA elements: a hominid-specific retroposon family. J. Mol. Biol 354, 994–1007 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Porubsky D et al. Recurrent inversion toggling and great ape genome evolution. Nat. Genet 52, 849–858 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zody MC et al. Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat. Genet 40, 1076–1083 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Locke DP et al. Large-scale variation among human and great ape genomes determined by array comparative genomic hybridization. Genome Res. 13, 347–357 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ballif BC et al. Expanding the clinical phenotype of the 3q29 microdeletion syndrome and characterization of the reciprocal microduplication. Mol. Cytogenet 1, 8 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ebler J et al. Pangenome-based genome inference. Cold Spring Harbor Laboratory; (2020), p. 2020.11.11.378133. [Google Scholar]
- 43.Zhao X et al. Expectations and blind spots for structural variation detection from short-read alignment and long-read assembly. Cold Spring Harbor Laboratory; (2020), p. 2020.07.03.168831. [Google Scholar]
- 44.Lappalainen T et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 501, 506–511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiang C et al. The impact of structural variation on human gene expression. Nat. Genet 49, 692–699 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evans KL et al. Genetics of heart rate in heart failure patients (GenHRate). Hum. Genomics. 13, 22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maples BK, Gravel S, Kenny EE, Bustamante CD, RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am. J. Hum. Genet 93, 278–288 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mallick S et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature. 538, 201–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mathias RA et al. A continuum of admixture in the Western Hemisphere revealed by the African Diaspora genome. Nat. Commun 7, 12522 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nielsen R et al. Darwinian and demographic forces affecting human protein coding genes. Genome Res. 19, 838–849 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yi X et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. 329, 75–78 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kidd JM et al. Characterization of missing human genome sequences and copy-number polymorphic insertions. Nat. Methods. 7, 365–371 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bersaglieri T et al. Genetic signatures of strong recent positive selection at the lactase gene. Am. J. Hum. Genet 74, 1111–1120 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soleimanpour SA et al. The diabetes susceptibility gene Clec16a regulates mitophagy. Cell. 157, 1577–1590 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seclen SN, Rosas ME, Arias AJ, Medina CA, Elevated incidence rates of diabetes in Peru: report from PERUDIAB, a national urban population-based longitudinal study. BMJ Open Diabetes Res Care. 5, e000401 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wei W et al. Human L1 retrotransposition: cis preference versus trans complementation. Mol. Cell. Biol 21, 1429–1439 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium, Pan-cancer analysis of whole genomes. Nature. 578, 82–93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cordaux R, Batzer MA, The impact of retrotransposons on human genome evolution. Nat. Rev. Genet 10, 691–703 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Consortium GTEx, The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 369, 1318–1330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu Z et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet 48, 481–487 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Sankararaman S et al. The genomic landscape of Neanderthal ancestry in present-day humans. Nature. 507, 354–357 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hsieh P et al. Adaptive archaic introgression of copy number variants and the discovery of previously unknown human genes. Science. 366 (2019), doi: 10.1126/science.aax2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eberle MA, Kruglyak L, An analysis of strategies for discovery of single-nucleotide polymorphisms. Genet. Epidemiol 19 Suppl 1, S29–35 (2000). [DOI] [PubMed] [Google Scholar]
- 64.Miga KH et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature. 585, 79–84 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Logsdon GA et al. The structure, function, and evolution of a complete human chromosome 8. Cold Spring Harbor Laboratory; (2020), p. 2020.09.08.285395. [Google Scholar]
- 66.Edge P, Bansal V, Longshot enables accurate variant calling in diploid genomes from single-molecule long read sequencing. Nat. Commun 10, 333 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poplin R et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol 36, 983–987 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Chin C-S, Khalak A, Human Genome Assembly in 100 Minutes. Cold Spring Harbor Laboratory; (2019), p. 705616. [Google Scholar]
- 69.Kolmogorov M, Yuan J, Lin Y, Pevzner PA, Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol 37, 540–546 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Porubsky D et al. Dense and accurate whole-chromosome haplotyping of individual genomes. Nat. Commun 8, 1293 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ghareghani M et al. Strand-seq enables reliable separation of long reads by chromosome via expectation maximization. Bioinformatics. 34, i115–i123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Patterson M et al. WhatsHap: Weighted Haplotype Assembly for Future-Generation Sequencing Reads. J. Comput. Biol 22, 498–509 (2015). [DOI] [PubMed] [Google Scholar]
- 73.Martin M et al. WhatsHap: fast and accurate read-based phasing. Cold Spring Harbor Laboratory; (2016), p. 085050. [Google Scholar]
- 74.Vollger MR et al. Long-read sequence and assembly of segmental duplications. Nat. Methods. 16, 88–94 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou W et al. Identification and characterization of occult human-specific LINE-1 insertions using long-read sequencing technology. Nucleic Acids Res. 48, 1146–1163 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ren J, Chaisson MJP, lra: the Long Read Aligner for Sequences and Contigs. Cold Spring Harbor Laboratory; (2020), p. 2020.11.15.383273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bakker B et al. Single-cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol. 17, 115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krueger F, Trim Galore: a wrapper tool around Cutadapt and FastQC. Trim Galore! (2012), (available at http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). [Google Scholar]
- 79.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liao Y, Smyth GK, Shi W, The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li YI et al. Annotation-free quantification of RNA splicing using LeafCutter. Nat. Genet 50, 151–158 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Casale FP, Rakitsch B, Lippert C, Stegle O, Efficient set tests for the genetic analysis of correlated traits. Nat. Methods. 12, 755–758 (2015). [DOI] [PubMed] [Google Scholar]
- 83.Mirauta BA et al. Population-scale proteome variation in human induced pluripotent stem cells. Elife. 9 (2020), doi: 10.7554/eLife.57390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bonder MJ et al. Systematic assessment of regulatory effects of human disease variants in pluripotent cells. Cold Spring Harbor Laboratory; (2019), p. 784967. [Google Scholar]
- 85.García MS, Multiple sequence alignments of full-length L1 elements with evidence of retrotransposition activity (2021), , doi: 10.5281/zenodo.4475905. [DOI] [Google Scholar]
- 86.Audano PA, HGSVC Key Callset Resources (2020), , doi: 10.5281/zenodo.4268828. [DOI] [Google Scholar]
- 87.Bonder MJ, HGSVC2 full eQTL results (2020), , doi: 10.5281/zenodo.4271574. [DOI] [Google Scholar]
- 88.Ebert P, HGSVC2 project code contributions (2021), , doi: 10.5281/zenodo.4482026. [DOI] [Google Scholar]
- 89.Martín BR, MEIGA-tk/MEIGA-PAV: MEIGA-PAV (2021), , doi: 10.5281/zenodo.4487121. [DOI] [Google Scholar]
- 90.Zook JM et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Scientific Data. 3, 1–26 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fairley S, Lowy-Gallego E, Perry E, Flicek P, The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 48, D941–D947 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gong L, Wong C-H, Idol J, Ngan CY, Wei C-L, Ultra-long Read Sequencing for Whole Genomic DNA Analysis. J. Vis. Exp (2019), doi: 10.3791/58954. [DOI] [PubMed] [Google Scholar]
- 93.Sanders AD, Falconer E, Hills M, Spierings DCJ, Lansdorp PM, Single-cell template strand sequencing by Strand-seq enables the characterization of individual homologs. Nat. Protoc 12, 1151–1176 (2017). [DOI] [PubMed] [Google Scholar]
- 94.Falconer E et al. DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat. Methods. 9, 1107–1112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sanders AD et al. Single-cell analysis of structural variations and complex rearrangements with tri-channel processing. Nat. Biotechnol 38, 343–354 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Quinlan AR, Hall IM, BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kent WJ, Zweig AS, Barber G, Hinrichs AS, Karolchik D, BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics. 26, 2204–2207 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Holley G, Melsted P, Bifrost – Highly parallel construction and indexing of colored and compacted de Bruijn graphs. Cold Spring Harbor Laboratory; (2019), p. 695338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Song L, Florea L, Langmead B, Lighter: fast and memory-efficient sequencing error correction without counting. Genome Biol. 15, 509 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zerbino DR, Wilder SP, Johnson N, Juettemann T, Flicek PR, The ensembl regulatory build. Genome Biol. 16, 56 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kent WJ et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Karolchik D et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 32, D493–6 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Haeussler M et al. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 47, D853–D858 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schneider VA et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.1000 Genomes Project Consortium et al. A map of human genome variation from population-scale sequencing. Nature. 467, 1061–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A, Versatile genome assembly evaluation with QUAST-LG. Bioinformatics. 34, i142–i150 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Seppey M, Manni M, Zdobnov EM, in Gene Prediction: Methods and Protocols, Kollmar M, Ed. (Springer New York, New York, NY, 2019), pp. 227–245. [Google Scholar]
- 108.Frankish A et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sheffield NC, Bock C, LOLA: enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics. 32, 587–589 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vollger MR et al. Improved assembly and variant detection of a haploid human genome using single-molecule, high-fidelity long reads. bioRxiv, 635037 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li H et al. A synthetic-diploid benchmark for accurate variant-calling evaluation. Nat. Methods. 15, 595–597 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Heller D, Vingron M, SVIM-asm: Structural variant detection from haploid and diploid genome assemblies. Bioinformatics (2020), doi: 10.1093/bioinformatics/btaa1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nurk S et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30, 1291–1305 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cheng H, Concepcion GT, Feng X, Zhang H, Li H, Haplotype-resolved de novo assembly with phased assembly graphs. arXiv [q-bio.GN] (2020), (available at http://arxiv.org/abs/2008.01237). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Porubsky D, Ebert P, Audano PA, Vollger MR, A fully phased accurate assembly of an individual human genome. bioRxiv (2019) (available at 10.1101/855049v1.abstract). [DOI] [Google Scholar]
- 116.Miller DE et al. Targeted long-read sequencing resolves complex structural variants and identifies missing disease-causing variants. Cold Spring Harbor Laboratory; (2020), p. 2020.11.03.365395. [Google Scholar]
- 117.Hiatt SM et al. Long-read genome sequencing for the diagnosis of neurodevelopmental disorders. Cold Spring Harbor Laboratory; (2020), p. 2020.07.02.185447. [Google Scholar]
- 118.Li H, A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 27, 2987–2993 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li H, Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ruan J, Li H, Fast and accurate long-read assembly with wtdbg2. Nat. Methods. 17, 155–158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Regier AA et al. Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects. Nat Commun. 9 (2018), doi: 10.1101/269316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.McKenna A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Poplin R et al. Scaling accurate genetic variant discovery to tens of thousands of samples. Cold Spring Harbor Laboratory; (2017), p. 201178. [Google Scholar]
- 124.Delaneau O, Marchini J, Zagury J-F, A linear complexity phasing method for thousands of genomes. Nat. Methods. 9, 179–181 (2011). [DOI] [PubMed] [Google Scholar]
- 125.O’Connell J et al. A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet. 10, e1004234 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Loh P-R et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet 48, 1443–1448 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen X et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 32, 1220–1222 (2016). [DOI] [PubMed] [Google Scholar]
- 128.Kronenberg ZN et al. Wham: Identifying Structural Variants of Biological Consequence. PLoS Comput. Biol 11, e1004572 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Layer RM, Chiang C, Quinlan AR, Hall IM, LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 15, R84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Abyzov A, Urban AE, Snyder M, Gerstein M, CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rausch T et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 28, i333–i339 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Becker T et al. FusorSV: an algorithm for optimally combining data from multiple structural variation detection methods. Genome Biol. 19, 38 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ke G et al. in Advances in Neural Information Processing Systems 30, Guyon I, Luxburg UV, Bengio S, Wallach H, Fergus R, Vishwanathan S, Garnett R, Eds. (Curran Associates, Inc., 2017), pp. 3146–3154. [Google Scholar]
- 134.Collins RL et al. An open resource of structural variation for medical and population genetics. bioRxiv (2019), p. 578674. [Google Scholar]
- 135.Klambauer G et al. cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 40, e69 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.R Core Team, R: A Language and Environment for Statistical Computing (2020), (available at http://www.R-project.org/). [Google Scholar]
- 137.Babadi M et al. Abstract 2287: Precise common and rare germline CNV calling with GATK. Cancer Res. 78, 2287–2287 (2018). [Google Scholar]
- 138.Zhao X, Weber AM, Mills RE, A recurrence-based approach for validating structural variation using long-read sequencing technology. Gigascience. 6, 1–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chen S et al. Paragraph: a graph-based structural variant genotyper for short-read sequence data. Genome Biol. 20, 291 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Katoh K, Standley DM, MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Larson DE et al. svtools: population-scale analysis of structural variation. Bioinformatics. 35, 4782–4787 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Anantharaman TS, Mysore V, Mishra B, Fast and cheap genome wide haplotype construction via optical mapping. Pac. Symp. Biocomput, 385–396 (2005). [DOI] [PubMed] [Google Scholar]
- 143.Porubsky D et al. breakpointR: an R/Bioconductor package to localize strand state changes in Strand-seq data. Bioinformatics. 36, 1260–1261 (2020). [DOI] [PubMed] [Google Scholar]
- 144.Lander ES et al. Initial sequencing and analysis of the human genome. Nature. 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 145.Smit AF, Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr. Opin. Genet. Dev 9, 657–663 (1999). [DOI] [PubMed] [Google Scholar]
- 146.Hancks DC, Kazazian HH Jr, Active human retrotransposons: variation and disease. Curr. Opin. Genet. Dev 22, 191–203 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Muotri AR et al. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 435, 903–910 (2005). [DOI] [PubMed] [Google Scholar]
- 148.Batzer MA, Deininger PL, Alu repeats and human genomic diversity. Nat. Rev. Genet 3, 370–379 (2002). [DOI] [PubMed] [Google Scholar]
- 149.Zook JM et al. A robust benchmark for detection of germline large deletions and insertions. Nat. Biotechnol (2020), doi: 10.1038/s41587-020-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Koren S et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. (2017), doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Li H, Durbin R, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Boissinot S, Chevret P, Furano AV, L1 (LINE-1) retrotransposon evolution and amplification in recent human history. Mol. Biol. Evol 17, 915–928 (2000). [DOI] [PubMed] [Google Scholar]
- 153.Kearse MG, Wilusz JE, Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev. 31, 1717–1731 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jukes TH, Osawa S, Evolutionary changes in the genetic code. Comp. Biochem. Physiol. B. 106, 489–494 (1993). [DOI] [PubMed] [Google Scholar]
- 155.Osawa S, Jukes TH, Watanabe K, Muto A, Recent evidence for evolution of the genetic code. Microbiol. Rev 56, 229–264 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Skowronski J, Fanning TG, Singer MF, Unit-length line-1 transcripts in human teratocarcinoma cells. Mol. Cell. Biol 8, 1385–1397 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.The UniProt Consortium, UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Edgar RC, MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ, Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 25, 1189–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Paradis E, Claude J, Strimmer K, APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics. 20, 289–290 (2004). [DOI] [PubMed] [Google Scholar]
- 161.Schliep KP, phangorn: phylogenetic analysis in R. Bioinformatics. 27, 592–593 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ, IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol 32, 268–274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Marchani EE, Xing J, Witherspoon DJ, Jorde LB, Rogers AR, Estimating the age of retrotransposon subfamilies using maximum likelihood. Genomics. 94, 78–82 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Salem AH et al. LINE-1 preTa elements in the human genome. J. Mol. Biol 326, 1127–1146 (2003). [DOI] [PubMed] [Google Scholar]
- 165.Hancks DC, Kazazian HH Jr, SVA retrotransposons: Evolution and genetic instability. Semin. Cancer Biol 20, 234–245 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ostertag EM, Goodier JL, Zhang Y, Kazazian HH Jr, SVA elements are nonautonomous retrotransposons that cause disease in humans. Am. J. Hum. Genet 73, 1444–1451 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mills RE, Bennett EA, Iskow RC, Devine SE, Which transposable elements are active in the human genome? Trends Genet. 23, 183–191 (2007). [DOI] [PubMed] [Google Scholar]
- 168.Roy-Engel AM et al. Active Alu element “A-tails”: size does matter. Genome Res. 12, 1333–1344 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bennett EA et al. Active Alu retrotransposons in the human genome. Genome Res. 18, 1875–1883 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Flasch DA et al. Genome-wide de novo L1 Retrotransposition Connects Endonuclease Activity with Replication. Cell. 177, 837–851.e28 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Jurka J, Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc. Natl. Acad. Sci. U. S. A 94, 1872–1877 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Feng Q, Moran JV, Kazazian HH Jr, Boeke JD, Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 87, 905–916 (1996). [DOI] [PubMed] [Google Scholar]
- 173.Chaisson MJP et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. bioRxiv (2017), p. 193144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lu T-Y, The Human Genome Structural Variation Consortium, Chaisson M, Profiling variable-number tandem repeat variation across populations using repeat-pangenome graphs. Cold Spring Harbor Laboratory; (2020), p. 2020.08.13.249839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Benson G, Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Marçais G et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol 14, e1005944 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Khelik K, Lagesen K, Sandve GK, Rognes T, Nederbragt AJ, NucDiff: in-depth characterization and annotation of differences between two sets of DNA sequences. BMC Bioinformatics. 18, 338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chikhi R, Limasset A, Medvedev P, Compacting de Bruijn graphs from sequencing data quickly and in low memory. Bioinformatics. 32, i201–i208 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rausch T, Fritz MH-Y, Untergasser A, Benes V, Tracy: basecalling, alignment, assembly and deconvolution of sanger chromatogram trace files. BMC Genomics. 21, 1–9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Andrews S, Others, FastQC: a quality control tool for high throughput sequence data (2010). [Google Scholar]
- 182.Martin M, Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 17, 10–12 (2011). [Google Scholar]
- 183.Robinson MD, McCarthy DJ, Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jun G et al. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am. J. Hum. Genet 91, 839–848 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Cotto KC et al. RegTools: Integrated analysis of genomic and transcriptomic data for the discovery of splicing variants in cancer. Cold Spring Harbor Laboratory; (2021), p. 436634. [Google Scholar]
- 186.Purcell S et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ongen H, Buil A, Brown AA, Dermitzakis ET, Delaneau O, Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics. 32, 1479–1485 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Storey JD, Tibshirani R, Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U. S. A 100, 9440–9445 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Gymrek M et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet 48 (2016), doi: 10.1038/ng.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gusev A et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am. J. Hum. Genet 95 (2014), doi: 10.1016/j.ajhg.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Yang J, Lee SH, Goddard ME, Visscher PM, GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet 88 (2011), doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Buniello A et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kamat MA et al. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. 35, 4851–4853 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Staley JR et al. PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics. 32, 3207–3209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Smigielski EM, Sirotkin K, Ward M, Sherry ST, dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 28, 352–355 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Benjamini Y, Hochberg Y, Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Series B Stat. Methodol 57, 289–300 (1995). [Google Scholar]
- 197.Alexander DH, Novembre J, Lange K, Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Martin AR et al. Human Demographic History Impacts Genetic Risk Prediction across Diverse Populations. Am. J. Hum. Genet 100, 635–649 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Speidel L, Forest M, Shi S, Myers SR, A method for genome-wide genealogy estimation for thousands of samples. Nat. Genet 51, 1321–1329 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Gordon D et al. Long-read sequence assembly of the gorilla genome. Science. 352, aae0344–aae0344 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kronenberg ZN et al. High-resolution comparative analysis of great ape genomes. Science. 360 (2018), doi: 10.1126/science.aar6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Speidel L, Forest M, Shi S, Myers SR, A method for genome-wide genealogy estimation for thousands of samples. Nat. Genet 51, 1321–1329 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Human Genome Structural Variation Working Group et al. Completing the map of human genetic variation. Nature. 447, 161–165 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Regier AA et al. Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects. Nat. Commun 9, 4038 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Eichler EE, in Proceedings of the sixth annual international conference on Computational biology (Association for Computing Machinery, New York, NY, USA, 2002), RECOMB ’02, p. 155. [Google Scholar]
- 206.Sudmant PH et al. Global diversity, population stratification, and selection of human copy number variation. Science. 349, aab3761 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.