

SUMMARY
Histone crotonylation is a non-acetyl histone lysine modification that is as widespread as acetylation. However, physiological functions associated with histone crotonylation remain almost completely unknown. Here we report that histone crotonylation is crucial for endoderm differentiation. We demonstrate that key crotonyl-coenzyme A (CoA)-producing enzymes are specifically induced in endodermal cells during differentiation of human embryonic stem cells (hESCs) in vitro and in mouse embryos, where they function to increase histone crotonylation and enhance endodermal gene expression. Chemical enhancement of histone crotonylation promotes endoderm differentiation of hESCs, whereas deletion of crotonyl-CoA-producing enzymes reduces histone crotonylation and impairs meso/endoderm differentiation in vitro and in vivo. Our study uncovers a histone crotonylation-mediated mechanism that promotes endodermal commitment of pluripotent stem cells, which may have important implications for therapeutic strategies against a number of human diseases.
Graphical Abstract
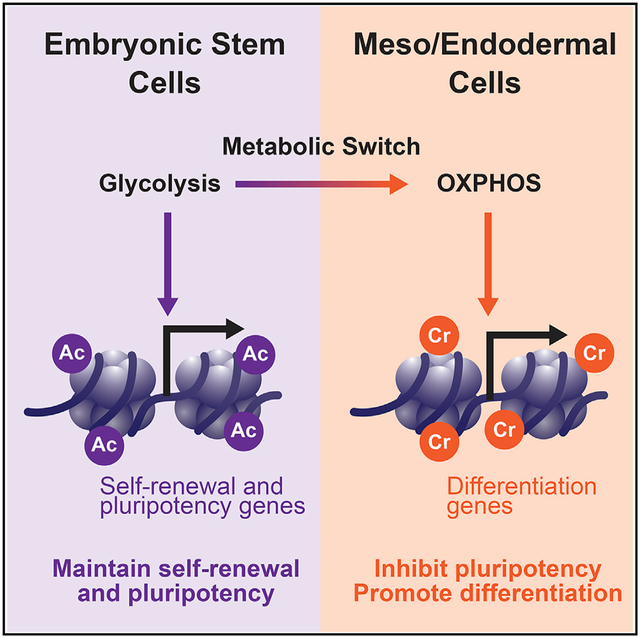
In Brief
Histone crotonylation is as widespread as acetylation. However, its associated physiological outcomes remain largely unknown. Fang et al. discover that histone crotonylation is increased during meso/endoderm differentiation of human embryonic stem cells, which, in turn, enhances expression of meso/endodermal genes and promotes their meso/endoderm commitment in vitro and in vivo.
INTRODUCTION
Emerging evidence suggests that cell-specific metabolic programs are directly linked to their unique epigenetic statuses (Carey et al., 2015; Folmes et al., 2011) because many intermediate metabolites, including acetyl-coenzyme A (CoA), α-ketoglutarate, and S-adenosylmethionine, can function as substrates or cofactors for enzymes that regulate chromatin modification and gene expression (Cai et al., 2011; Shiraki et al., 2014; Shyh-Chang et al., 2013; Wellen et al., 2009; Xu et al., 2011). Pluripotent embryonic stem cells (PSCs) possess a unique set of metabolic programs and epigenetic states to sustain their un-limited proliferation while maintaining their pluripotency (Folmes et al., 2012; Ito and Suda, 2014; Panopoulos et al., 2012; Zhang et al., 2012). For instance, PSCs demand high glycolysis-mediated, glucose-dependent production of acetyl-CoA and histone acetylation for maintenance of pluripotency (Moussaieff et al., 2015). Differentiation of PSCs to mesoderm and endoderm triggers a metabolic switch from glycolysis to oxidative phosphorylation (Cliff et al., 2017). This metabolic switch is believed to be important to release stem cells from pluripotency and promote differentiation because inhibition of glycolytic metabolism has been shown to lead to loss of histone acetylation and the pluripotent state (Gu et al., 2016; Moussaieff et al., 2015). However, whether and how the increase in oxidative phosphorylation during this metabolic remodeling actively affects this early-stage differentiation is still unclear.
Over the past decade, a panel of short-chain and long-chain lysine acylations have been identified by high-sensitivity mass spectrometry as novel histone post-translational modifications (Rousseaux and Khochbin, 2015; Sabari et al., 2017; Tan et al., 2011). Compared with histone acetylation, these lysine acylations are mediated by intermediate metabolites from different metabolic pathways, pose distinct genomic distributions, display diverse affinities to various modifiers (e.g., writers, erasers, and readers), and have a greater ability to activate gene expression (Sabari et al., 2017). For example, histone crotonylation, one of the first discovered non-acetyl histone lysine acylations, is derived from crotonyl-CoA, an intermediate metabolite during mitochondrial or peroxisomal fatty acid oxidation as well as lysine and tryptophan metabolism (Figures 1A and S1A). Experimental, genetic, and environmental perturbations that alter cellular concentrations of crotonyl-CoA, including incubation of nuclear extracts with crotonyl-CoA (Simithy et al., 2017), incubation of cells with crotonate, knocking down the crotonyl-CoA-producing enzyme acyl-CoA synthetase short chain family member 2 (ACSS2) in cultured cells (Sabari et al., 2015), or depletion of short-chain fatty acid-producing gut microbiota in mice (Fellows et al., 2018), notably change levels of histone crotonylation in nuclear extracts, in cells, and in vivo. Biochemically, histone crotonylation can be catalyzed by histone acetyltransferase p300 (Sabari et al., 2015), read by Yaf9, ENL, AF9, Taf14, and Sas5 (YEATS) domain proteins and double plant homeodomain (PHD) fingers (Andrews et al., 2016; Li et al., 2016; Xiong et al., 2016; Zhao et al., 2016), and erased by a number of classically annotated histone deacetylases (Bao et al., 2014; Madsen and Olsen, 2012; Sabari et al., 2017; Wei et al., 2017). However, physiological outcomes associated with histone crotonylation remain largely unknown.
Figure 1. Key crotonyl-CoA-producing enzymes are induced and enriched in endodermal cells.
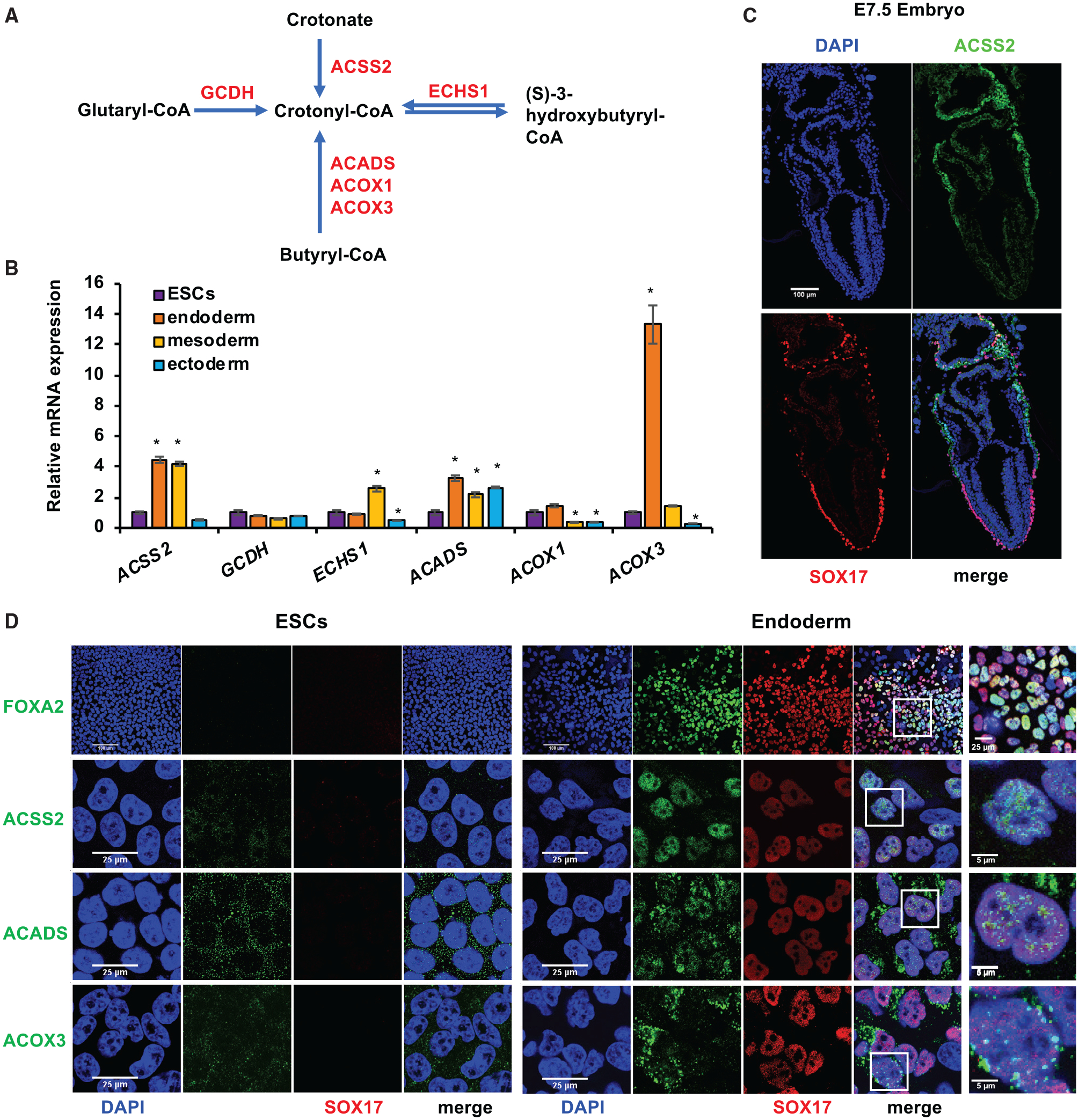
(A) Key metabolic reactions that produce crotonyl-CoA. ACSS2, acyl-CoA synthetase short chain family member 2; ACADS, acyl-CoA dehydrogenase short chain; ACOX1, acyl-CoA oxidase 1; ACOX3, acyl-CoA oxidase 3; ECHS1, enoyl-CoA hydratase, short chain 1; GCDH, glutaryl-CoA dehydrogenase.
(B) ACSS2, ACADS, and ACOX3 are induced during endodermal differentiation of hESCs in vitro. The mRNA levels of the indicated genes in mel1 hESC-differentiated endoderm, mesoderm, or ectoderm were analyzed by qPCR (n = 3 biological replicates/group, *p < 0.05, values are expressed as mean ± SEM).
(C) ACSS2 is enriched in endodermal cells in mouse E7.5 embryos. Expression of ACSS2 was analyzed by immunofluorescence staining in E7.5 mouse embryos. The full embryo images were taken by tiling/stitching multiple fields using ZEN software. Representative images from at least three different embryos are shown. Scale bar, 100 μm.
(D) ACSS2, ACADS, and ACOX3 are induced and translocated into the nucleus during endodermal differentiation of mel1 hESCs in vitro. Mel1 hESCs were induced to differentiate into endoderm for 3 days, and then expression of the indicated proteins was analyzed by immunofluorescence staining. Scale bars for the left 8 panels: 100 μm in the FOXA2 images and 25 μm in the ACSS2, ACADS, and ACOX3 images. Scale bars for the last panel: 25 μm in the FOXA2 image and 5 μm in the ACSS2, ACADS, and ACOX3 images.
See also Figures S1 and S2.
In the present study, we assessed the effect of histone crotonylation on lineage commitment of PSCs and early embryogenesis. Given the observation that crotonyl-CoA is primarily generated from oxidation of fatty acids and amino acids (Figure 1A and S1A), we hypothesized that crotonyl-CoA production, and therefore histone crotonylation, is increased during the metabolic switch in the early differentiation of PSCs and that this increase is important for lineage commitment of these cells.
RESULTS
Key crotonyl-CoA-producing enzymes are induced and enriched in endodermal cells
Crotonyl-CoA is an endogenous intermediate metabolite during fatty acid oxidation and lysine/tryptophan degradation (Figures 1A and S1A). To test our hypothesis that crotonyl-CoA production and histone crotonylation are increased during metabolic remodeling in early differentiation of PSCs, we first analyzed the expression of key crotonyl-CoA-producing enzymes during in vitro differentiation of human embryonic stem cells (hESCs) into three primary germ layers (Figures S1B and S2A). As shown in Figures 1B and S2B, the mRNA levels of ACSS2 as well as two additional enzymes that catalyze conversion of butyryl-CoA to crotonyl-CoA during fatty acid oxidation (a mitochondrial short-chain acyl-CoA dehydrogenase, ACADS, and a peroxisomal acyl-CoA oxidase, ACOX3) were induced significantly upon endoderm and/or mesoderm differentiation but not after ectoderm differentiation of mel1 hESCs in vitro, suggesting that differentiation of endoderm and mesoderm, but not ectoderm, is associated with increased crotonyl-CoA production. This observation is consistent with a recent report that the glycolysis to oxidative phosphorylation metabolic switch occurs only during mesoderm and endoderm differentiation but not during ectoderm differentiation of hESCs (Cliff et al., 2017). Immunofluorescence analysis in embryonic day 7.5 (E7.5) mouse embryos further showed that one crotonyl-CoA-producing enzyme, ACSS2, was specifically enriched in SOX17-positive visceral endodermal cells (Figure 1C), suggesting that endoderm is the major germ layer targeted for possible regulation by crotonylation in mice. As a control, factors involved in acetyl-CoA production and transport (Moussaieff et al., 2015) were not significantly induced during endoderm differentiation of hESCs (Figure S2C), suggesting that endoderm differentiation is specifically associated with increased crotonyl-CoA but not altered acetyl-CoA production.
Further immunofluorescence staining analyses in hESCs and differentiated endodermal cells confirmed that expression of ACSS2, ACADS, and ACOX3 was induced upon endoderm differentiation of mel1 hESCs (Figure 1D, green) and H9 hESCs (Figure S2D, green). Intriguingly, significant fractions of these crotonyl-CoA-producing enzymes were localized to the nucleus, particularly in endodermal cells (Figures 1D, S2D, and S2E), suggesting a nuclear role of these enzymes in endodermal cells. Our findings indicate that endoderm differentiation is specifically associated with increased expression of crotonyl-CoA-producing enzymes in cultured cells and in vivo.
Endoderm differentiation is associated with enhanced histone crotonylation
We next analyzed whether the observed increase in expression of crotonyl-CoA-producing enzymes during endoderm differentiation is accompanied by increased histone crotonylation. Lysine crotonylation (Kcr) was enhanced dramatically in the nucleus of mel1 hESCs after 3-day differentiation into endodermal cells, as detected by a previously used and well-characterized rabbit polyclonal pan-anti-Kcr antibody, PTM-501 (Figure 2A, left panels, Kcr (501), Day 3 endodermal cells [D3 Endo] versus embryonic stem cells [ESCs]; Sabari et al., 2015; Tan et al., 2011; Wei et al., 2017) as well as by a mouse monoclonal pan-anti-Kcr antibody, PTM-502 (Figure 2A, center panels, Kcr (502), D3 Endo versus ESCs). In contrast, lysine butyrylation (Kbu), a structurally similar lysine acylation, displayed comparable levels in ESCs and D3 endodermal cells, as detected by a rabbit polyclonal pan-anti-Kbu antibody (Figure 2A, right panels). These observations suggest that Kcr is specifically induced during endoderm differentiation. We confirmed that all three pan-antibodies have the desired specificity to distinguish Kcr and Kbu modifications in the immunofluorescence staining assay by peptide competition experiments (Figure S3A). Further immunoblot analysis revealed that all three antibodies predominantly detect crotonylation or butyrylation signals on histones (Figures S3B–S3D), and Kcr(502) and Kbu antibodies also have adequate specificity in the immunoblot assay (Figures S3C and S3D).
Figure 2. Endoderm differentiation is associated with enhanced histone crotonylation.
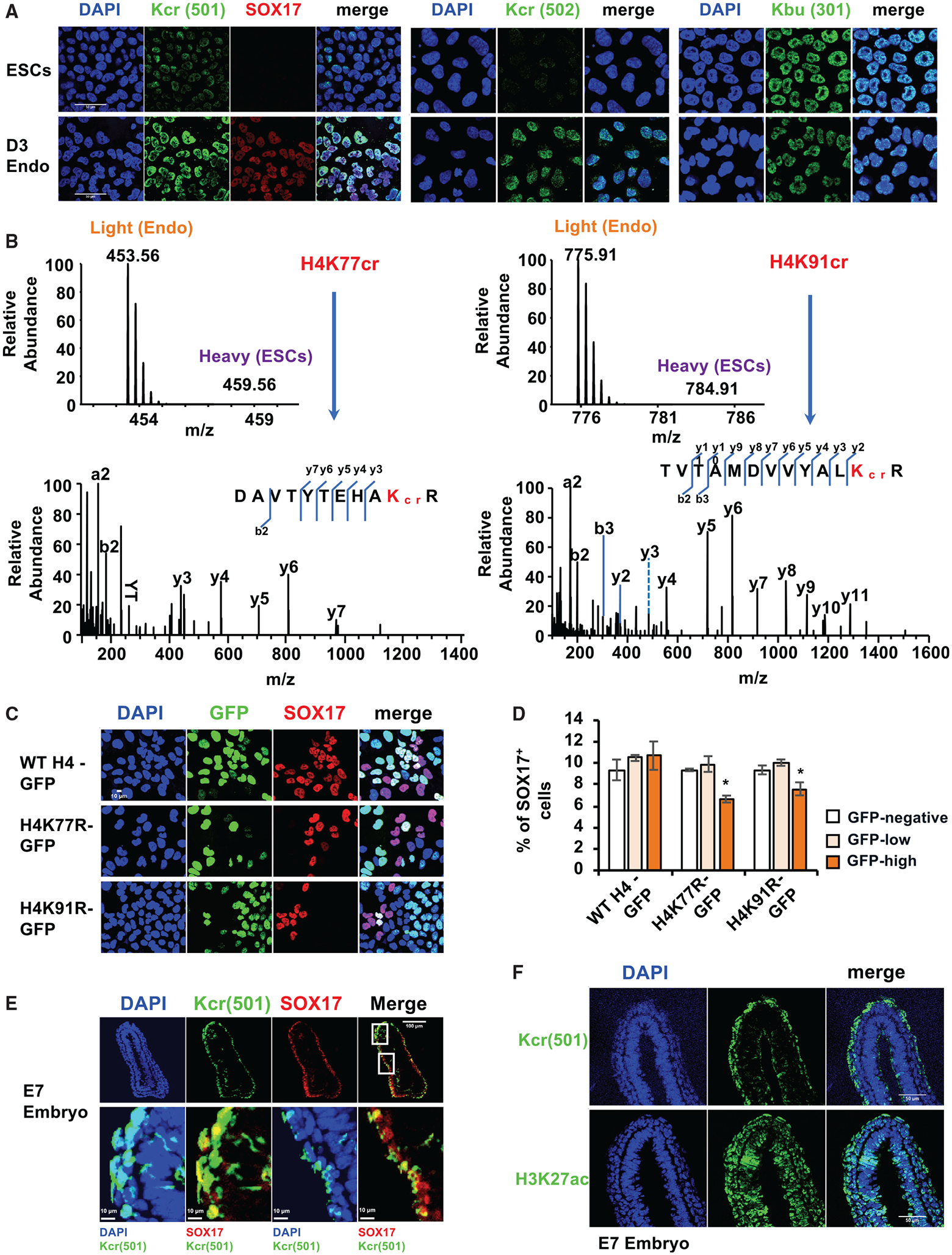
(A) Histone Kcr, but not Kbu, is increased upon endoderm differentiation of hESCs. Mel1 hESCs were induced to differentiate into endoderm for 3 days, and the levels of SOX17, Kcr, and Kbu were analyzed by immunofluorescence staining. Kcr (501), rabbit polyclonal pan-anti-Kcr antibody (PTM-501, 1:10,000 dilution); Kcr (502), mouse monoclonal pan-anti-Kcr antibody (PTM-502, 1:5,000 dilution); Kbu (301), rabbit polyclonal pan-anti-Kbu antibody (PTM-301, 1:5,000 dilution). Scale bars, 50 μm.
(B) H4K77 and H4K91 are two major sites crotonylated upon endoderm differentiation of hESCs. Mel1 hESCs and D3 endodermal cells differentiated from mel1 hESCs were subjected to SILAC analysis as described in Method details. The full mass spectrometry (MS) and MS/MS spectra of H4K77cr and H4K91cr peptides are shown for their identification and quantification. The b and y ions indicate peptide backbone fragment ions containing the N or C terminus, respectively.
(C) K-to-R mutation of H4K77 or H4K91 reduces differentiation of endodermal cells from hESCs. Mel1 hESCs were induced to differentiate into endoderm for 2 days. SOX17 levels in D2 endodermal cells expressing GFP-tagged WT H4, H4K77R, or H4K91R were analyzed by immunofluorescence staining. Scale bar, 10 μm.
(D) K-to-R mutation of H4K77 or H4K91 reduces differentiation of endodermal cells. The fractions of SOX17+ cells were analyzed in D2 endodermal cells expressing GFP-tagged WT H4, H4K77R, or H4K91R by fluorescence-activated cell sorting (FACS) assay as described in Method details (n = 3 biological replicates/group, *p < 0.05, values are expressed as mean ± SEM).
(E) Histone Kcr is enriched in endodermal cells in E7 mouse embryos. The level of Kcr was analyzed by immunofluorescence staining as described in Method details. Representative images from at least three different embryos are shown. Boxed areas are enlarged to show the overlap between Kcr and SOX17 or DAPI. Scale bar in top panels, 100 mm; scale bars in bottom panel, 10 μm.
(F) Histone Kcr, but not H3K27ac, is enriched selectively in endodermal cells in E7 mouse embryos. The distribution of Kcr or H3K27ac was analyzed by immunofluorescence staining in E7 mouse embryos. Representative images from at least three different embryos are shown. Scale bars, 50 μm.
To further confirm that Kcr is increased during endoderm differentiation of hESCs, we quantified histone Kcr levels in endodermal cells differentiated from mel1 hESCs and compared them with those in mel1 hESCs using stable isotope labeling by amino acids in cell culture (SILAC)-based quantitative proteomics analysis (Table S1; Figure 2B). This analysis revealed that 2 of 3 detectable histone Kcr sites, H4K77cr and H4K91cr, were found only in the differentiated endodermal cells compared with hESCs (Figure 2B; Table S1, Kcr). Again, the levels of distinct histone lysine acetylation (Kac) and Kbu sites were differentially altered after endoderm differentiation (Table S1, Kac and Kbu), indicating that histone Kcr is specifically induced upon endoderm differentiation of hESCs.
Notably, SILAC analysis also revealed that Kac and Kbu could be increased on histone tails and cores upon endoderm differentiation, but Kcr was primarily detected and induced on histone globular cores on the nucleosome lateral surface (H4K77) or on the histone-histone interface (H4K91) (Figure 2B; Table S1), suggesting a specific function of Kcr in modulating DNA/nucleosome interaction and nucleosome dynamics and stability (Bowman and Poirier, 2015; Lawrence et al., 2016) during endoderm differentiation. Germline monoallelic missense mutations affecting H4K91 have been reported recently to cause a developmental syndrome in humans (Tessadori et al., 2017). Our further mutagenesis study showed that mutation of K to R, which abolishes the possible crotonylation modification on K77 or K91 of H4, significantly reduced the fraction of SOX17-positive endodermal cells when highly expressed in WT D2 endodermal cells (Figures 2C, 2D, and S3E), indicating that acylation on both sites is important for endoderm differentiation.
In line with these observations in cultured cells, Kcr was also enriched in the SOX17-positive endodermal cells in the mouse E7 embryo (Figure 2E). Intriguingly, compared with a ubiquitous distribution pattern of H3K27ac, an enhancer mark that has been shown previously to prime poised enhancers, increasing the response of endodermal intermediates to inductive signals (Wang et al., 2015), Kcr was enriched selectively in endodermal cells in mouse embryos (Figure 2F), suggesting a specific role of this modification in endoderm.
To better understand this unique role of Kcr in endodermal cells at the genomic scale, we profiled the chromatin landscape of Kcr in comparison with that of H3K27ac in hESCs versus differentiated endodermal cells by chromatin immunoprecipitation sequencing (ChIP-seq) analysis (Figures 3A–3C and S4A; Tables S2 and S3). Gene Ontology (GO) analyses of mapped genomic loci revealed that, in pluripotent hESCs, Kcr was relatively enriched near Transcriptional Start Sites (TSSs) of genes involved in various metabolic processes, whereas the vast majority of H3K27ac was localized on enhancers of genes regulating gastrulation and endoderm formation (Figures S4B and S4C; Table S4). Three-day induction of endoderm differentiation altered the Kcr levels on 77,003 genomic loci (Figure 3A, Kcr Endo versus ESCs), with increased enrichment on enhancer regions (Figure S4D, Kcr Endo/ESCs). In contrast, the H3K27ac levels were changed on 35,010 genomic loci after endoderm differentiation (Figure 3A, H3K27ac Endo versus ESCs), with relatively increased enrichment on promoter regions (Figure S4D, H3K27ac Endo/ESCs). Remarkably, the altered Kcr sites covered 87% (30,465 of 35,010) of the altered H3K27ac sites (Figure 3A, top), and these overlapped loci were enriched near genes involved in gastrulation and endoderm development (Figure 3A, bottom), indicating that histone Kcr and H3K27ac are engaged in activation of endodermal genes upon endoderm differentiation. For example, Kcr and H3K27ac were increased on the SOX17 gene, although enrichment of Kcr was more notable compared with that of H3K27ac (Figure 3B). Further bulk RNA sequencing (RNA-seq) analysis showed that changes in gene expression are correlated with alterations of these two chromatin modifications upon endoderm differentiation. First, Kcr and H3K27ac were highly enriched on TSSs of genes that were regulated during endoderm differentiation (up or down) compared with their signal on TSSs of genes that were not significantly regulated (no change) (Figure 3C). Second, and importantly, the TSSs of highly induced genes (up) displayed enhanced enrichment of Kcr and H3K27ac, whereas the TSSs of repressed genes (down) were depleted of Kcr and H3K27ac (Figures 3C and 3D). ChIP-qPCR results confirmed that levels of Kcr and H3K27ac were increased strongly near the TSSs of endodermal genes but depleted near the TSS of a pluripotent gene upon endoderm differentiation (Figure 3E). Interestingly, GO analysis of 46,538 unique Kcr genomic loci altered during endoderm differentiation (Figure 3A) showed that this modification is also enriched near TSSs of genes involved in mesoderm differentiation, particularly artery and kidney development (Figure S4E), suggesting that, compared with H3K27ac, Kcr may have a special role in modulating mesoderm differentiation. Additionally, further gene set enrichment analysis (GSEA) of the RNA-seq datasets from me1 hESCs and D3 endodermal cells revealed that pathways involved in fatty acid, triacylglycerol, and ketone body metabolism are significantly upregulated in endodermal cells compared with hESCs (Figure S4F), supporting the notion that fatty acid metabolism is induced upon endoderm differentiation. Our findings suggest that endoderm differentiation is associated with enhanced histone Kcr in cultured cells and early mouse embryos.
Figure 3. Histone crotonylation is increased and enriched near endodermal genes upon endoderm differentiation.
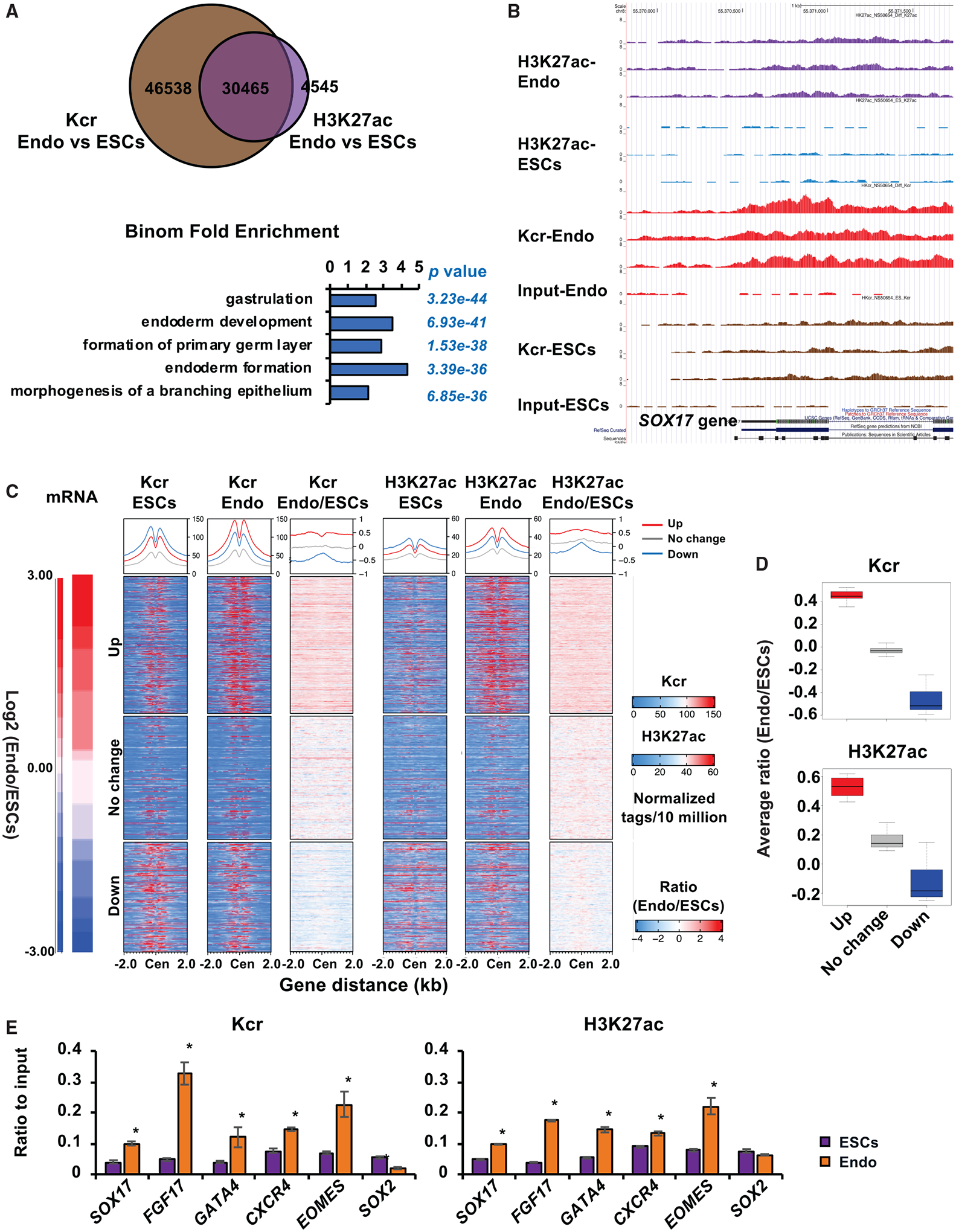
(A) Deposition of histone Kcr and H3K27ac is increased near endodermal genes upon endoderm differentiation of hESCs. Genomic distributions of Kcr and H3K27ac in mel1 hESCs (ESCs) or D3 endodermal cells differentiated in regular differentiation medium (Endo) were analyzed by ChIP-seq analysis as described in Method details. The GO enrichment of biological processes in overlapped 30465 chromatin loci was analyzed by GREAT version 3.0.0.
(B) Abundance of histone Kcr and H3K27ac deposition is induced on the SOX17 gene locus upon endoderm differentiation of hESCs. The deposition of Kcr and H3K27ac on the SOX17 locus in cells described in (A) was visualized in the UCSC Genome Browser.
(C) Histone Kcr and H3K27ac are enriched on the TSSs of induced genes but depleted from the TSS of repressed genes upon endoderm differentiation of hESCs. Transcriptomes (mRNA) of mel1 hESCs and D3 Endo cells were analyzed by bulk RNA-seq. The top 1,864 significantly upregulated genes (up), 1,495 down-regulated genes (down) (cutoff: fold change > 2.0 or < −2.0, q < 0.01), and 1,677 not significantly changed genes (no change) were selected. These genes were then sorted from the largest to the smallest based on their fold changes between endodermal cells and mel1 hESCs (Endo/ESCs). The corresponding ChIP-seq read tags of Kcr and H3K27ac near the TSS of each gene in mel1 hESCs (ESCs) and D3 endodermal cells (Endo) and the ratio of read tags between endodermal cells and hESCs (Endo/ESCs) were graphed.
(D) Boxplot presentation of the average ratio of Kcr and H3K27ac genome coverage on induced (up), not changed (no change), and repressed (down) gene loci upon endodermal differentiation. The gene clusters were selected as in (C), and the average genome coverage of Kcr and H3K27ac on these three gene clusters were plotted. The center line is located at the median; the box length extends from the first quantile (Q1) to the third quantile (Q3), which corresponds to the interquartile range (IQR). The maximum line is located at Q3 + 1.5 3 IQR, whereas the minimum line is located at Q1 −1.5 × IQR. For Kcr and H3K27ac, the difference between three clusters was significant (p < 2.2 × 10−16, Student’s t test between any two clusters).
(E) Endoderm differentiation results in increased deposition of histone Kcr and H3K27ac on endodermal genes. Association of Kcr and H3K27ac on the TSSs of the indicated endodermal and pluripotent genes was analyzed by ChIP-qPCR, and relative enrichment was calculated against the input signal (n = 3 biological replicates/group, *p < 0.05, values are expressed as mean ± SEM).
Crotonate promotes endoderm differentiation of hESCs
To further evaluate the importance of histone crotonylation in endoderm differentiation, we investigated whether crotonate, a crotonyl-CoA precursor, can directly affect endoderm differentiation of hESCs. Acetate, an acetyl-CoA precursor that has been shown to increase intracellular acetyl-CoA levels, thereby maintaining pluripotency and inhibiting endoderm differentiation in H1 hESCs (Moussaieff et al., 2015), was used for comparison. We confirmed that crotonate was able to dramatically increase intracellular levels of crotonyl-CoA and histone crotonylation, as reported (Sabari et al., 2015), in our cell culture systems (Figures 4A and 4B). In contrast, adding acetate did not significantly increase intracellular steady levels of acetyl-CoA (Figure 4A), possibly because the intracellular acetyl-CoA concentration of differentiating mel1 hESCs was much higher than that of H1 hESCs (30 pmol/106 in mel1 cells versus 7 pmol/106 in H1 cells) (Figure 4A; Moussaieff et al., 2015). Our observation is consistent with a previous report where acetate did not change intracellular level of acetyl-CoA in HeLa cells (Sabari et al., 2015) and suggests that the acetyl-CoA level in mel1 hESCs is already saturated. Notably, incubation with crotonate alone was sufficient to dose-dependently activate expression of differentiation markers, endodermal and mesodermal markers in particular, in pluripotent mel1 hESCs maintained in hESC medium (Figure 4C, crotonate). Inducing endoderm differentiation of mel1 hESCs massively increased the levels of endodermal markers (FOXA2, SOX17, and GOOSECOID) while significantly decreasing pluripotency markers (POU5F1 and NANOG) (Figure S2A). Crotonate further dose-dependently enhanced expression of endodermal markers (Figure 4D, crotonate) and key crotonyl-CoA-producing enzymes (Figure 4E) upon endoderm differentiation. In contrast, acetate failed to increase the expression levels of differentiation genes in hESCs (Figure 4C, acetate) or differentiated endodermal cells (Figure 4D, acetate). Similar results were observed in H9 hESCs, where crotonate promoted but acetate repressed endoderm differentiation (Figures S5A and S5B), suggesting that crotonate, but not acetate, promotes endoderm differentiation of hESCs.
Figure 4. Crotonate promotes endoderm differentiation of hESCs.
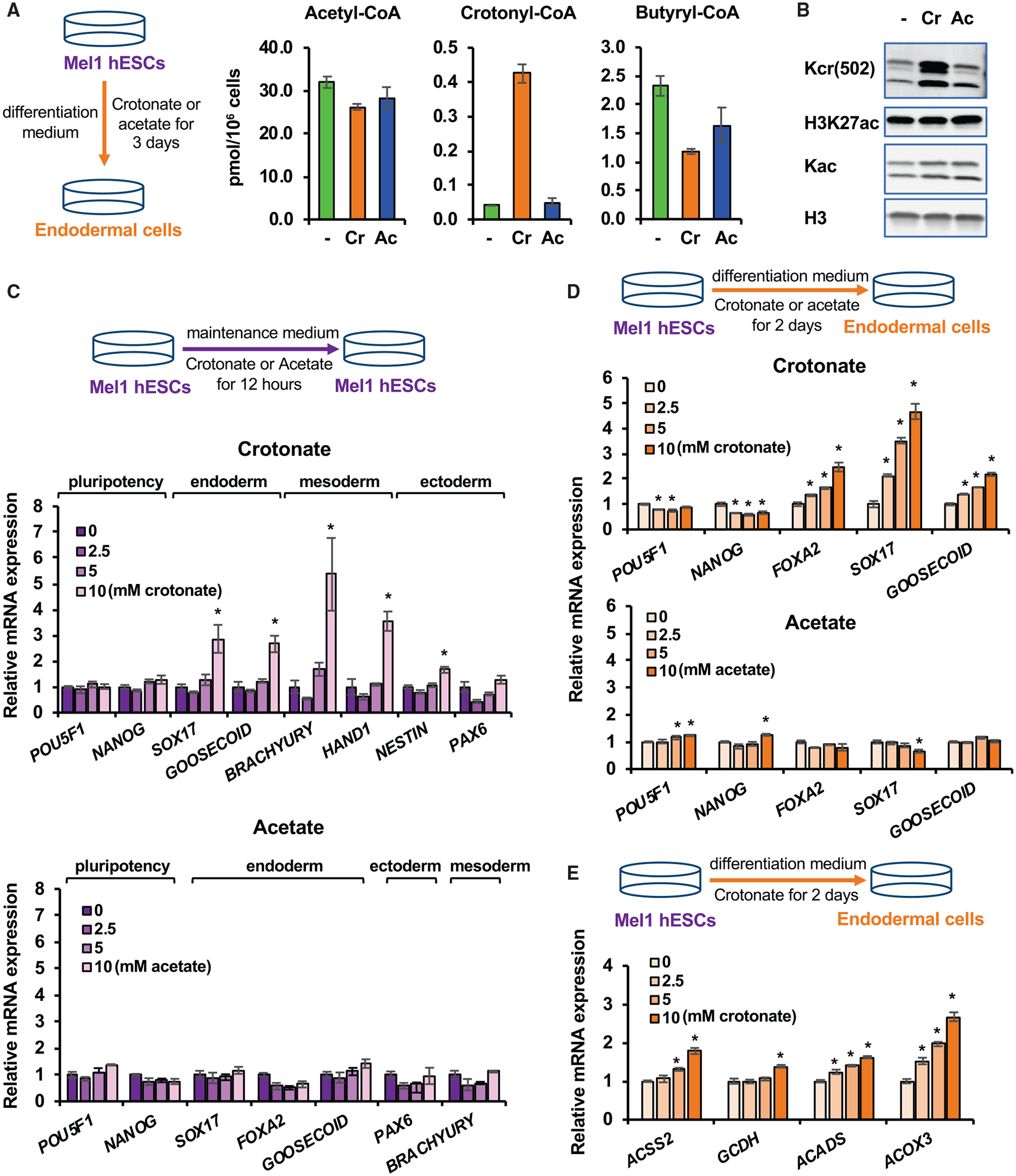
(A) Crotonate increases the cellular level of crotonyl-CoA. The concentrations of acetyl-CoA, crotonyl-CoA, and butyryl-CoA in mel1 hESCs after 3-day differentiation into endoderm in regular differentiation medium (−) or in differentiation medium containing 5 mM crotonate (Cr) or 5 mM acetate (Ac) were analyzed as described in Method details (n = 3 biological replicates/group, values are expressed as mean ± SEM).
(B) Crotonate increases histone crotonylation. Mel1 hESCs were induced for endoderm differentiation with or without 5 mM Cr or 5 mM Ac for 3 days. Whole-cell lysates were immunoblotted with the indicated antibodies.
(C) Crotonate is sufficient to induce expression of endodermal and mesodermal genes in hESCs. Mel1 hESCs were treated with increased doses of Cr or Ac in ESC maintenance medium for 12 h, and the expression of the indicated genes was analyzed by qPCR (n = 3 biological replicates/group, *p < 0.05, values are expressed as mean ± SEM).
(D) Crotonate dose-dependently promotes the expression of endodermal genes upon endoderm differentiation of hESCs. Mel1 hESCs were induced to differentiate into endoderm in differentiation medium containing 0, 2.5, 5, or 10 mM Cr or Ac for the indicated days. The mRNA levels of the indicated genes were analyzed by qPCR (n = 3 biological replicates/group, *p < 0.05, values are expressed as mean ± SEM).
(E) Crotonate dose-dependently enhances the expression of key crotonyl-CoA-producing enzymes upon endoderm differentiation. Mel1 hESCs were differentiated into endoderm in regular differentiation medium containing 0, 2.5, 5, or 10 mM Cr for 2 days. The mRNA levels of the indicated crotonyl-CoA-producing enzymes were analyzed by qPCR (n = 3 biological replicates/group, *p < 0.05, values are expressed as mean ± SEM).
See also Figures S5A and S5B.
Further single-cell RNA-seq analysis (scRNA-seq; Table S5) revealed that mel1 hESCs undergoing in vitro endoderm differentiation can be clustered into three states—the ESC state, intermediate state, and differentiated state—based on the expression patterns of known marker genes and their individual transcriptomes (Figures 5A, center plot, and S5C). Each cell state had a specific list of additional marker genes (Table S6). Particularly, Single-Cell Regulatory Network Inference and Clustering (SCENIC) analysis (Aibar et al., 2017) of candidate transcriptional activities in individual cells (Figure 5B) and Ingenuity Pathway Analysis (IPA) of intermediate marker genes (Figure S5D) showed that the intermediate state has a high activity of the ATF4 transcription network, which mediates an adaptive integrated stress response to amino acid deprivation that has been implicated previously in promoting endoderm differentiation in mouse ESCs (Shan et al., 2013; Xu et al., 2014). This observation underscores the importance of nutrient sensing in regulation of stem cell linage specification.
Figure 5. Crotonate increases the fraction of differentiated endodermal cells upon endoderm differentiation of hESCs.
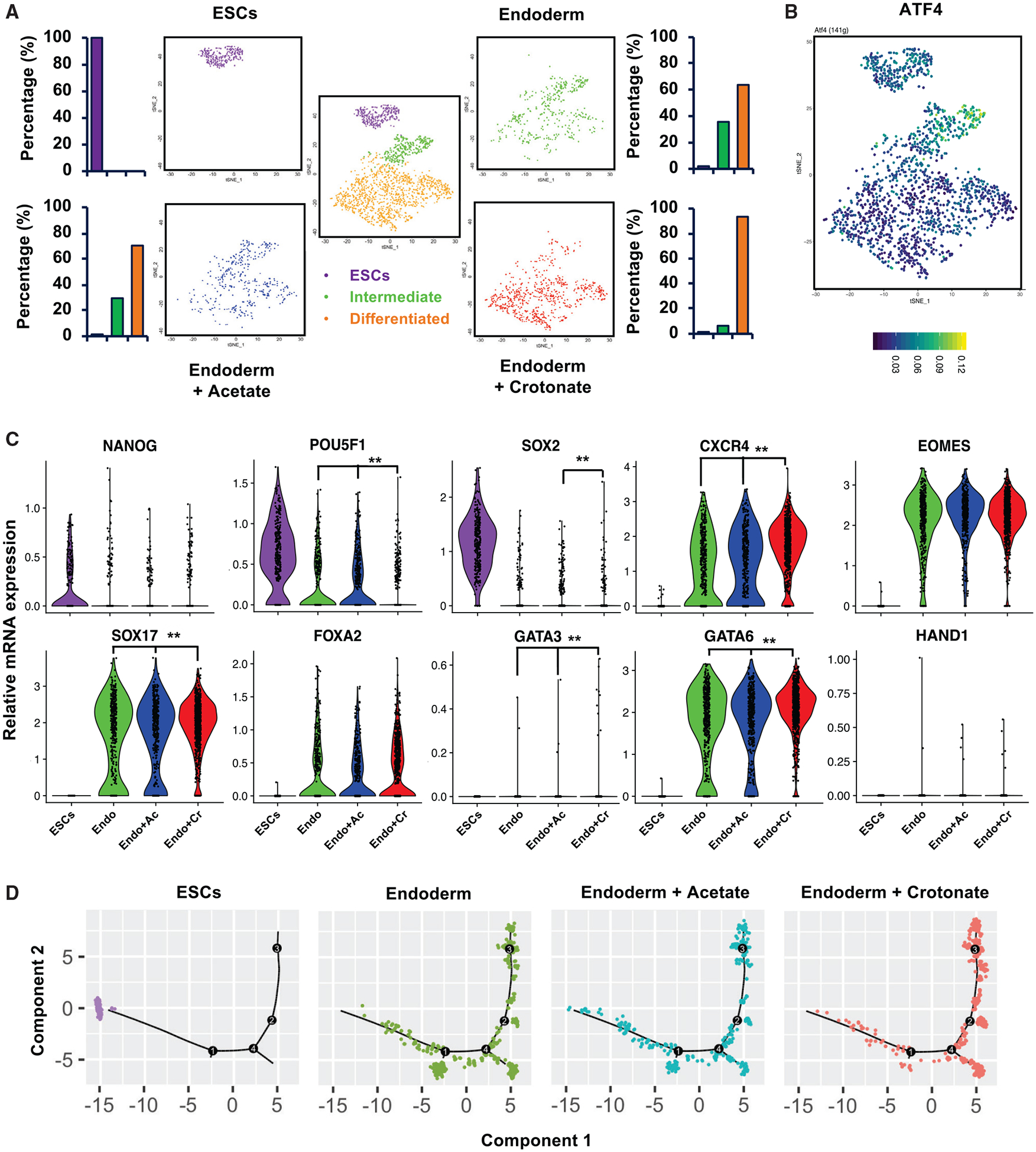
(A) Crotonate, but not acetate, promotes more hESCs to differentiate into endodermal cells when analyzed at the single-cell transcriptome level. Mel1 hESCs and D3 endoderm cells differentiated in regular endodermal differentiation medium (endoderm) or in regular endodermal differentiation medium containing 5 mM acetate (endoderm+Acetate) or 5 mM Crotonate (endoderm+Crotonate) were analyzed by scRNA-seq. A total of 1,863 cells from all four samples were clustered into three clusters (center panel). The tSNE plots at the four corners are for four individual experimental samples, showing their individual clustering patterns and the percentage of cells in each cluster. The percentage of cells within each cluster was quantified by comparing the number of cells in that particular cluster with the total cell number analyzed for that experimental sample (n = 290 cells in ESCs, 428 in endoderm, 397 in endoderm+Acetate, and 748 in endoderm+Crotonate).
(B) ATF4 is enriched in the intermediate cluster during endodermal differentiation of hESCs. Mel1 hESCs and corresponding D3 endodermal cells were analyzed by scRNA-seq. All combined promoters of the genes expressed in a cell were used to infer transcription factor activity by SCENIC, and the activity of the ATF4 transcription factor in different cells was projected onto tSNE space with its activity color coded (AUCell; high, yellow; low, dark blue).
(C) Crotonate, but not acetate, promotes more cells with high endodermal gene expression. The color-coded violin plots display the expression distribution of pluripotent and differentiation markers in four different scRNA-seq samples. Each dot represents a single cell. Gene expression changes between samples were compared, and the significance of change was labeled (**false discovery rate [FDR]-adjusted p < 0.01).
(D) Crotonate, but not acetate, promotes more hESCs to differentiate into endodermal cells, as revealed by the single-cell pseudotemporal analysis. The endodermal differentiation trajectories of the indicated scRNA-seq samples were analyzed by pseudotemporal analysis as described in Method details.
See also Figures S5C and S5D and Tables S5 and S6.
More specifically, our scRNA-seq analysis showed that 3-day endoderm differentiation of mel1 hESCs in regular endoderm differentiation medium shifted 35.7% of cells from the ESC state to the intermediate state and 63.8% further to the differentiated state (Figure 5A, endoderm). Remarkably, crotonate, but not acetate, significantly increased the differentiated cells to 93.5% (Figure 5A, compare endoderm + crotonate with endoderm + acetate). Further individual marker gene analysis showed that, compared with regular differentiation medium and acetate, crotonate significantly represses expression of the pluripotent genes POU5F1 and SOX2 but promotes the endodermal markers CXCR4, SOX17, and GATA6 (Figure 5C). Pseudotemporal analysis of the scRNA-seq data confirmed that crotonate, but not acetate, ordered more cells toward the differentiated state along the endodermal developmental trajectory than regular endoderm differentiation medium alone (Figure 5D). Therefore, in agreement with the elevated expression of crotonyl-CoA-producing enzymes (Figure 1) and enhanced histone crotonylation (Figures 2 and 3) upon endoderm differentiation, an increase in intracellular levels of crotonyl-CoA by crotonate promotes endoderm differentiation of hESCs.
Crotonyl-CoA-producing enzymes modulate histone crotonylation and regulate endoderm differentiation in vitro and in vivo
To directly assess the significance of histone Kcr in mediating endoderm differentiation, we examined whether crotonyl-CoA-producing enzymes are required for endoderm differentiation of hESCs by performing CRISPR-Cas9-mediated knockout of ACADS or ACOX3 in mel1 cells. Fatty acid β-oxidation is a process in which fatty acyl-CoA is broken down gradually via a repeated 4-step cycle to generate acetyl-CoA, with removal of 2 carbons each cycle. Because ACADS is a mitochondrial dehydrogenase catalyzing oxidation of short-chain fatty-acyl CoAs (C4 and C6) but not medium-chain, long-chain, or very-long-chain fatty acyl-CoA, we did not expect its deficiency to affect overall cellular fatty acid metabolism and acetyl-CoA production. As expected, knockout of ACADS (Figure S6A) significantly decreased intracellular levels of crotonyl-CoA without alteration of other tested short-chain fatty acyl-CoAs, including butyryl-CoA, the substrate of crotonyl-CoA, and acetyl-CoA, the end product of fatty acid oxidation (Figure 6A), suggesting that, overall, fatty acid oxidation is not impaired significantly in these deficient cells. ACOX3 has been shown previously to primarily oxidize branched-chain fatty acids as well as C10- and C16-CoA (Ferdinandusse et al., 2018). We detected its oxidase activity toward butyryl-CoA in vitro (Figure S6B) and observed a trend of ~50% reduction of crotonyl-CoA but no changes for other tested short-chain fatty acyl-CoAs in ACOX3 knockout (KO) cells despite a marked variation (Figure 6A).
Figure 6. Crotonyl-CoA-producing enzymes modulate histone crotonylation and regulate endoderm differentiation in vitro.
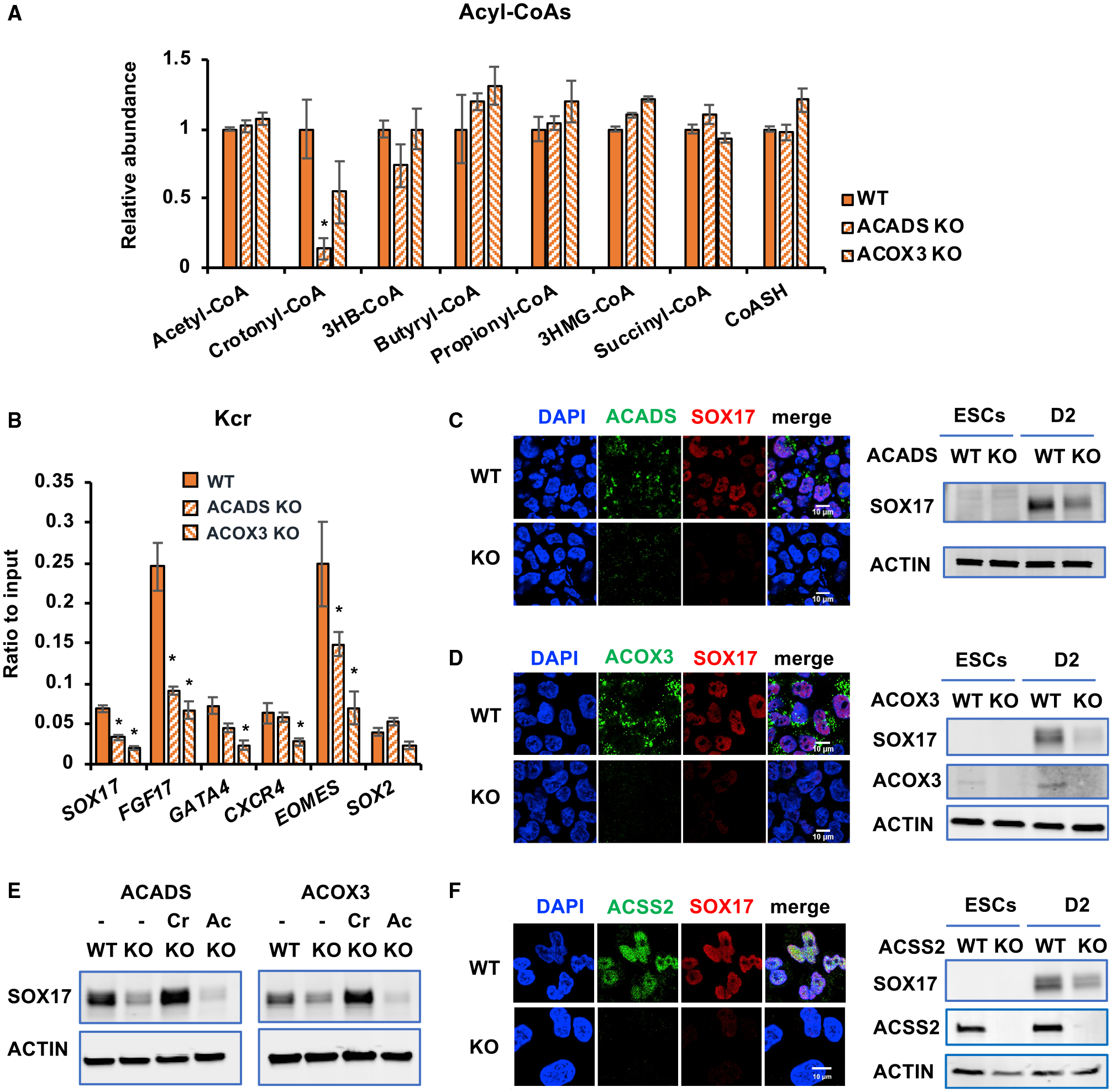
(A) KO of ACADS or ACOX3 reduces crotonyl-CoA levels. The levels of crotonyl-CoA in the indicated mel1-differentiated D3 endodermal cells were measured as described in Method details (n = 3 biological replicates/group, *p < 0.05, values are expressed as mean ± SEM).
(B) Knocking out ACADS or ACOX3 in mel1 hESCs reduces deposition of Kcr at the TSSs of endodermal genes. TSS-associated Kcr levels were analyzed by ChIP-qPCR in the indicated cells, and relative enrichment was calculated against the input signal (n = 3 independent experiments, *p < 0.05, values are expressed as mean ± SEM).
(C) Knocking out ACADS in mel1 hESCs impairs endodermal gene expression. Left: the expression of ACADS and SOX17 was analyzed in WT and ACADS KO D2 endodermal cells by immunofluorescence staining. Right: the expression of SOX17 was analyzed in hESCs and the indicated D2 endodermal cells by immunoblotting. Scale bars, 10 μm.
(D) Knocking out ACOX3 in mel1 hESCs impairs endodermal gene expression. WT and ACOX3 KO D2 endodermal cells were analyzed as in (C). Scale bars, 10 μm
(E) Crotonate, but not acetate, rescues the defective endoderm differentiation induced by ACADS or ACOX3 deficiency. WT and the indicated KO mel1 hESCs were induced into endoderm differentiation in the absence or presence of 5 mM crotonate (Cr) or acetate (Ac) for 2 days.
(F) Deletion of ACSS2 in mel1 hESCs impairs endodermal gene expression. WT and ACSS2 KO D2 endodermal cells were analyzed as in (C). Scale bar, 10 μm.
See also Figure S6.
The reduction of crotonyl-CoA in ACADS KO and ACOX3 KO cells was linked to a significant reduction of total cellular histone Kcr but not Kbu or Kac levels (Figure S6C) as well as a decrease in histone Kcr deposition near the TSSs of endodermal genes (Figure 6B). These defects were further associated with strong repression of SOX17 induction (Figures 6C and 6D). Importantly, SOX17 repression induced by ACADS or ACOX3 deficiency could be rescued by crotonate (Figure 6E, crotonate [Cr]), which induced a robust rise in intracellular levels of crotonyl-CoA (Figure S6D, crotonyl-CoA) and rescued total histone Kcr (Figure S6E, Kcr). However, acetate failed to rescue SOX17 suppression (Figure 6E, Ac). This dichotomy suggests that reduced Kcr is accountable for repressed SOX17 induction in these cells. Consistently, knocking down ACADS by small interfering RNAs (siRNAs) also significantly reduced histone Kcr and repressed induction of SOX17 (Figures S6F–S6H). Additionally, deletion or knockdown of ACSS2 significantly reduced histone Kcr and blunted induction of endodermal markers upon endoderm differentiation (Figures 6F, S6H, and S6I). In contrast to suppressed endoderm differentiation, ACADS KO and ACOX3 KO mel1 hESCs showed enhanced expression of PAX6 and OTX2, two ectodermal markers, when subjected to ectoderm differentiation in vitro (Figure S6J). Therefore, individual deficiency of crotonyl-CoA-producing enzymes reduces histone crotonylation, impairing endoderm differentiation while enhancing ectoderm differentiation of hESCs in vitro.
Finally, to test whether crotonyl-CoA-producing enzymes are required for endoderm differentiation of hESCs in vivo, we injected wild-type (WT), ACADS KO, and ACOX3 KO hESCs into NSG CD34FL mice and analyzed the formation and development of teratomas of these cells. As shown in Figures 7A and S7, ACADS KO and ACOX3 KO teratomas were morphologically immature compared with WT teratomas, containing large cystic portions filled with serous or mucinous fluid (arrowheads). At the histological level, ACADS KO and ACOX3 KO teratomas notably lacked well-defined tissue structures compared with WT teratomas and were particularly blunted in development of gut tube-like endodermal structures (Figure 7B, CDH17 staining) and Actinin α2 (ACTN2)-positive muscle fibers (Figure 7B, ACTN2 staining). Consistent with these observations, the expression levels of many endodermal and mesodermal genes, but not ectodermal genes, were reduced significantly in ACADS-or ACOX3-deficient teratomas compared with WT teratomas (Figure 7C), indicating that ACADS and particularly ACOX3 deficiency specifically suppresses endoderm and mesoderm differentiation in vivo. Collectively, these data support the notion that histone crotonylation is important for meso/endoderm commitment in vitro and in vivo.
Figure 7. Crotonyl-CoA-producing enzyme deficiency impairs endoderm differentiation in vivo.
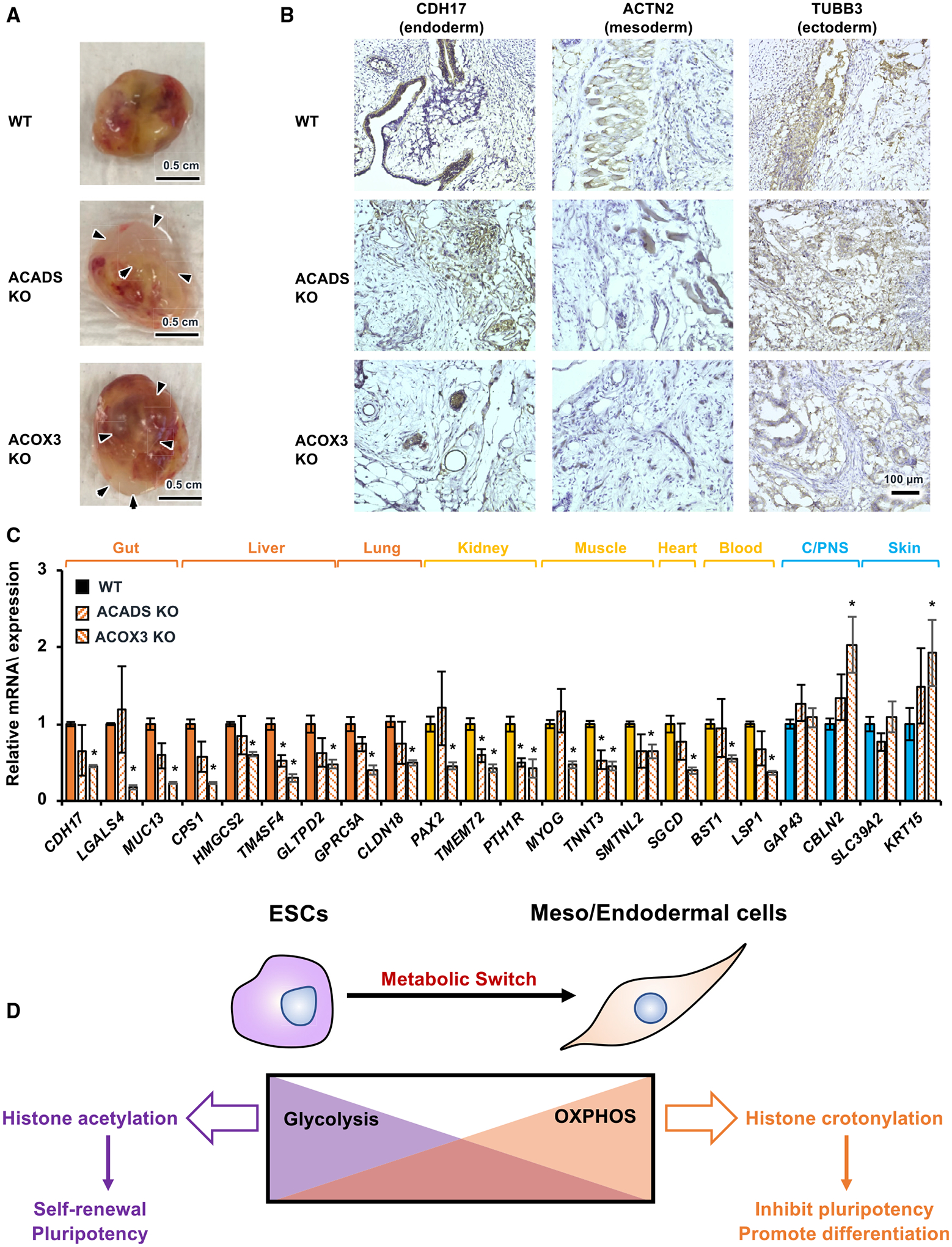
(A) WT, ACADS KO, and ACOX3 KO teratomas. WT, ACADS KO, and ACOX3 KO mel1 hESCs were injected subcutaneously into NSG mice for an in vivo teratoma assay as described in Method details. WT, ACADS KO, and ACOX3 KO teratomas were dissected 9 weeks after injection. Arrowheads, fluid-filled large cysts on ACADS KO and ACOX3 KO teratomas. Representative images are shown (see Figure S7 for all teratomas in this experiment; n = 8 WT teratomas, 8 ACADS KO teratomas, and 10 ACOX3 KO teratomas). Scale bars, 0.5 cm.
(B) ACADS or ACOX3 deficiency in hESCs impairs differentiation of endodermal and mesodermal tissues in vivo. Tissue sections of WT, ACADS KO, and ACOX3 KO teratomas were analyzed by immunohistochemistry (IHC) staining of the indicated endodermal, mesodermal, or ectodermal markers. ACADS KO and ACOX3 KO teratomas display disorganized structures of endodermal and mesodermal tissues. Scale bar, 100 μm.
(C) Deletion of ACADS or ACOX3 in hESCs reduces the expression of endodermal and mesodermal but not ectodermal markers in vivo. Orange, endodermal tissues; yellow, mesodermal tissues; blue, ectodermal tissues. The mRNA levels of the indicated genes were analyzed by qPCR (n = 3 WT, 4 ACADS KO, and 3 ACOX3 KO teratomas, *p < 0.05, values are expressed as mean ± SEM).
(D) Histone crotonylation promotes meso/endodermal commitment of PSCs. Pluripotent ESCs demand high glycolysis-mediated production of acetyl-CoA and histone acetylation for maintenance of pluripotency (left). During differentiation of PSCs to meso/endodermal cells, when a metabolic switch from glycolysis to oxidative phosphorylation occurs, histone crotonylation is enhanced to promote differentiation (right).
See also Figure S7.
DISCUSSION
As one of the first discovered non-acetyl histone lysine acylations, histone crotonylation has been reported to be enriched on genes that are active in post-meiotic male germ cells (Montellier et al., 2012; Sin et al., 2012; Tan et al., 2011) or induced by acute kidney injury (Ruiz-Andres et al., 2016). The status of histone crotonylation has also been shown to affect HIV latency and reactivation (Jiang et al., 2018). However, direct evidence supporting physiological functions of this modification is still lacking. Prior to our present study, ACSS2, which catalyzes addition of CoA to short-chain fatty acids, including crotonate, was the only enzyme shown to enhance the histone Kcr level in vitro (Sabari et al., 2015). We identified two additional fatty acid oxidation enzymes, ACADS and ACOX3, as key crotonyl-CoA producers during endoderm differentiation in this study. Knocking out these enzymes individually specifically reduces intracellular crotonyl-CoA levels without affecting steady levels of many other tested short-chain fatty acyl-CoAs, decreasing histone Kcr and impairing meso/endoderm differentiation in vitro and in vivo (Figures 6 and 7). We further identified two histone Kcr sites, H4K77cr and H4K91cr, that are only detected upon endoderm differentiation and confirmed that crotonylation of these two sites is important for endoderm differentiation (Figure 2). Therefore, our data directly link short-chain fatty acid oxidation to histone crotonylation during meso/endoderm differentiation and demonstrate that Kcr is a metabolic sensitive epigenetic modification that has a role in promoting meso/endodermal gene expression during the glycolysis to oxidative phosphorylation metabolic switch upon differentiation of these two lineages (Figure 7D). This notion also implies that histone crotonylation/acylations might be important in the development and/or functions of tissues with high demand of fatty acid oxidation and/or lysine or tryptophan metabolism. It will be of great interest to test this possibility in future studies.
Kcr-provided transcriptional specificity for meso/endoderm lineage commitment in comparison with Kac and Kbu in hESCs is very intriguing because these three lysine short-chain fatty acylations are structurally similar (Sabari et al., 2017). Moreover, many lysine acylation modifiers display a broad range of specificities. For example, p300 acetyltransferase has been shown to catalyze Kac, Kbu, Kcr, lysine succinylation (Ksucc), and lysine glutarylation (Kglu), whereas SIRT1, SIRT2, and SIRT3 are able to erase Kac, Kbu, and Kcr (Sabari et al., 2017). YEATS domain-containing proteins indeed have been shown to have enhanced binding affinities for Kcr over Kac, but they also have high affinity for lysine propionylation (Kpr) and Kbu compared with Kac (Andrews et al., 2016; Li et al., 2016; Zhao et al., 2016). Therefore, it is unlikely that the specificity of Kcr on meso/endodermal genes is primarily determined by its writers, readers, or erasers. Based on our observations regarding the intracellular concentrations of acetyl-CoA, butyryl-CoA, and crotonyl-CoA in D3 endodermal cells from mel1 hESCs (Figure 4A) as well as our current understanding of the broad range of specificities of lysine acylation modifiers, we propose that Kac/Kbu and Kcr are differentially modulated by two factors involved in the acyl-transfer reaction: acetyltransferases and small molecular acyl-CoAs. On one hand, the intracellular acetyl-CoA pool appears to be maintained in a saturated state in hESCs because the intracellular acetyl-CoA concentration is very high and is not sensitive to acetate or crotonate treatment (crotonate can also be converted to 2 molecules of acetyl-CoA through fatty acid β-oxidation) (Figure 4A, acetyl-CoA). When the substrate reaches saturating levels, acetyltransferases are the limiting factors for the acetyl-transfer reaction. In this case, Kac is sensitive to changes of its enzymatic modifiers and is primarily regulated by the dynamic balance between its writers and erasers. A similar argument could also apply to Kbu. On the other hand, the intracellular crotonyl-CoA concentration is about 600- to 1,000-fold lower than that of acetyl-CoA (and 60- to 100-fold lower than that of butyryl-CoA), crotonyl-CoA is likely to be the limiting factor in the crotonyl transfer reaction. As a result, Kcr is sensitive to any metabolic changes that alter cellular crotonyl-CoA levels and is mainly controlled by the availability of crotonyl-CoA and, hence, by the expression/activity/location/interacting partners of crotonyl-CoA-producing enzymes. Identification of signals and transcription factors that induce expression of crotonyl-CoA-producing enzymes as well as the nuclear interacting partners of these enzymes will be important to further dissect the underlying molecular mechanisms that regulate Kcr during germ layer differentiation of hESCs.
It is worth mentioning that crotonyl-CoA-producing enzymes are involved in fatty acid oxidation. This canonical function could potentially impact global metabolism, affecting meso/endoderm differentiation in a Kcr-independent mechanism. However, we provided evidence that histone crotonylation is a key mechanism mediating the effect of these crotonyl-CoA-producing enzymes on endoderm differentiation. First, deletion of ACADS or ACOX3 primarily reduces intracellular crotonyl-CoA levels with minimal effect on other tested short-chain acyl-CoAs, including acetyl-CoA, the final product of fatty acid oxidation (Figure 6A), suggesting that the effect of these enzymes on overall fatty acid oxidation is minimal in our culture system. Second, crotonate, but not acetate, rescues SOX17 repression induced by ACADS or ACOX3 deficiency (Figure 6E), despite acetate being able to increase the levels of H3K27ac in these deficient cells (Figure S6E). This result implies that restoration of cellular crotonyl-CoA is sufficient to rescue the endoderm differentiation defect despite the potential global metabolic effect induced by deletion of these enzymes. Third, we identified that H4K77 and H4K91 are only crotonylated in endoderm cells (Figure 2; Table S1). H4K77R and H4K91R mutations that abolish crotonylation at these sites also impair endoderm differentiation (Figures 2B–2D), confirming that crotonylation at these sites is important for endoderm differentiation. These observations support our conclusion that histone Kcr is a major mechanism for suppressed endoderm differentiation observed in hESCs deficient in crotonyl-CoA-producing enzymes.
The discovery in our present study has unique clinical relevance and physiological significance. Short-chain acyl-CoA dehydrogenase deficiency (SCADD), an autosomal recessive disorder caused by ACADS dysfunction, is routinely screened for in human newborns. SCADD has a wide range of symptoms, including frequent vomiting, failure to thrive, developmental delay, progressive muscle weakness, hypotonia, and even early death (Bhala et al., 1995; Corydon et al., 2001; Kim et al., 2011). Biochemically, SCADD is characterized by elevation of butyryl-CoA and its by-products in the liver and muscle as well as excessive accumulation of ammonia in the blood and body tissues (van Maldegem et al., 2006; Wolfe et al., 1993). This disorder is traditionally categorized as an energy deficiency disease because of defects in converting short-chain fatty acids into energy. However, the exact etiology remains not well understood. With our discovered roles of ACADS in regulating histone epigenetics, it will be worth examining the potential dysregulation of histone Kcr and other possible short-chain fatty acylations, together with abnormal gene regulation, in SCADD patients.
Notably, our study also has important translational implications. Endoderm germ layer-originating organs, including the liver, lungs, pancreas, and digestive tract, are subject to many life-threatening diseases that affect millions of patients (Yiangou et al., 2018). Because primary cells from endodermal organs are often difficult to grow in vitro, human PSCs hold great promise for generating endoderm cells and their derivatives for therapies against these human diseases. Our discovery that crotonate, a short-chain fatty acid, significantly increases the endoderm differentiation efficiency of hESCs (from 63.8% to 93.5%) (Figure 5A) suggests that crotonate could be developed as a nutritional supplement for current in vitro endoderm differentiation protocols to promote their efficiency. Therefore, this discovery may help to improve PSC-based therapy for human diseases affecting endodermal organs.
Our study uncovers a histone crotonylation-mediated mechanism that promotes endodermal commitment of PSCs, which may have important implications for stem cell therapy.
Limitations of study
Our study has several limitations. First, although we observed induction of crotonyl-CoA-producing enzymes upon endoderm differentiation of hESCs (Figure 1B), we did not directly show the increase of intracellular crotonyl-CoA concentration after endoderm differentiation because of technical limitations. As shown in Figure 4A, the concentration of crotonyl-CoA in endodermal cells is very low (about 1/600 to 1/1,000 of that of acetyl-CoA and 1/50 to 1/60 of that of butyryl-CoA), which is right on the detection limit of the liquid chromatography (LC)-quadrupole/Orbitrap high-resolution mass spectrometry we used for measurement of these metabolites. As a result, sensitivity is limited to detect the concentration of crotonyl-CoA, particularly in hESCs, which prevents us from directly comparing the levels of this metabolite in endodermal cells versus hESCs. On the other hand, based on our observation that a significant fraction of all tested crotonyl-CoA-producing enzymes is located in the nucleus after endoderm differentiation (Figures 1D and S2D), we believe that there is a local surge of crotonyl-CoA in the nucleus upon endoderm differentiation. Development of intra-nuclear crotonyl-CoA biosensors will be important to validate our conclusion. Second, although a previous study has demonstrated that the glycolysis to oxidative phosphorylation metabolic switch occurs only during mesoderm and endoderm differentiation of hESCs but not during ectoderm differentiation (Cliff et al., 2017), and because we showed that three crotonyl-CoA production enzymes (Figure 1B) and pathways involved in fatty acid metabolism (Figure S4F) are significantly induced during endoderm and/or mesoderm differentiation because of the hypersensitivity of hESCs and D3 endodermal cells to glucose deprivation, which is required for measurement of fatty acid oxidation in cells, we were not able to reliably measure fatty acid oxidation rates in these cells. Consequently, we did not provide direct evidence to show that fatty acid oxidation is increased during endoderm differentiation of hESCs in our hands. For the same reason, we also did not directly show that deletion of ACADS or ACOX3 has minimal effects on overall fatty acid oxidation despite our observation that the steady levels of short-chain acyl-CoAs remain consistent after their deletion (Figure 6A). Finally, although we have shown that alterations of histone crotonylation are positively associated with changes in gene expression during endodermal differentiation of hESCs and further demonstrated that histone crotonylation is functionally important for this process, we did not analyze in details how this modification relates to known chromatin states (active versus poised versus heterochromatin) in the present study. Future studies are needed to better understand the correlational and causal relationships between histone crotonylation and overall chromatin remodeling during meso/endoderm differentiation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, Xiaoling Li (lix3@niehs.nih.gov).
Materials availability
Two new deficient cell lines were generated in this study: ACADS KO mel1 hESCs and ACOX3 KO mel1 hESCs. Five new plasmids were generated in this study: LentiCRISPR-ACADS sgRNA, LentiCRISPR-ACOX3 sgRNA, AAVS1_puro_PGK1_H4-GFP, AVS1_puro_PGK1_H4K77R-GFP, and AAVS1_puro_PGK1_H4K91R-GFP. These materials will be made available in accordance with the Guidelines for the Conduct of Research in the Intramural Research Programs at NIH and the NIH Principles for Sharing Biomedical Research Resources, the “Research Tools Guidelines.”
Data and code availability
The bulk RNA-seq (RNA-seq), single-cell RNA-seq (scRNA-seq), and ChIP-seq data have been deposited to Gene Expression Omnibus under the accession number GEO: GSE130345 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130345). Three subseries are linked to GEO: GSE130345:
RNA-seq: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130340
scRNA-seq: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130341
ChIP-seq: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130344
Additional information about ChIP-seq quality control, ChIP-seq peaks, GO lists for ChIP-seq peaks, scRNA-seq quality control, and Marker gene lists in three cell clusters from scRNA-seq is included in Tables S2, S3, S4, S5, and S6.
The SILAC data are available in Table S1.
The source file for unprocessed and uncompressed immuno-blots used in the Figures is available from Mendeley Data: https://doi.org/10.17632/sfpv2m95s6.1.
This study did not generate new code for data analysis.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mammalian cell lines
Mel1 hESCs were obtained with permission from Dr. Andrew Elefanty and Dr. Edouard Stanley at the University of Queensland, Australia (NIH Registration number: 0139), and H9 hESCs were requested with permission from Dr. James A. Thomson at University of Wisconsin-Madison (via WiCell Research Institute, NIH Registration number: 0062; RRID:CVCL_9773). All cells were cultured in plates coated with Matrigel (Corning, #354277) and maintained in TesR™-E8™ medium (Stem Cell Technology Inc.).
Mouse models
NSG CD34FL mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ) were from Jackson Laboratory (#005557). All mice at the NIEHS animal facility are maintained under strict SPF conditions. NSD mice were housed with four mice per cage in micro-isolator static cages (Techni-plast, Exton, PA) in a room with a constant temperature (19–23°C) and humidity (55% ± 10%) and a 12-hour light/dark cycle. They were provided ad libitum with an autoclaved plant-based chow diet (NIH-31, Harlan Laboratories, Madison, WI) and fed with autoclaved acid water. All mice were negative for mouse hepatitis virus, Sendai virus, pneumonia virus of mice, mouse parvovirus 1 and 2, epizootic diarrhea of infant mice, mouse norovirus, Mycoplasma pulmonis, Helicobacter spp., and endo- and ectoparasites, and no pathogens were detected in sentinel mice during our whole study (analyzed weekly). The clinical health status of all experimental mice was checked twice a week during our experiments per our approved animal study protocols.
Mice were randomly assigned to experimental groups after they were allowed to acclimatize for at least one week prior to experiments.
METHOD DETAILS
In vitro differentiation of hESCs
In vitro differentiation of hESCs to endoderm, mesoderm and ectoderm was induced by the STEMDIFF trilineage differentiation kit (Stem Cell Technology, Inc., #05230) according to the kit instruction. Briefly, 3 × 104 mel1 cells/well or 5 × 104 H9 cells/well were seeded to a 12-well plate in the presence of 10 mM Rock inhibitor, 1 mL endoderm differentiation medium was added to each well next day, and medium was changed daily.
To assess the influence of naturally occurred histone acetylation and crotonylation on endoderm differentiation, mel1 or H9 hESCs were induced in endoderm differentiation medium for 3 days. To catch up the early impact of histone crotonylation mutants or crotonyl-CoA producing enzyme deficiency on endoderm differentiation, mel1 hESCs were differentiated for 2 days. To evaluate the impact of acetate or crotonate on endoderm differentiation, endoderm differentiation media containing different concentrations of acetate or crotonate were adjusted to pH7.4, mel1 or H9 hESCs were then incubated in these endoderm differentiation medium for 2–4 days as indicated in figure legends.
Immunofluorescence of cells and mouse embryos
Cells were fixed with 4% paraformaldehyde in PBS for 10 minutes, and permealized with 0.5% triton in PBS for 10 minutes after wash. Cells were then incubated with 10% Fetal Bovine Serum (FBS) for 30 minutes, followed by incubation with primary antibodies at room temperature for 1 hour. After wash, cells were incubated with secondary antibodies at room temperature for 30 minutes.
E7 mouse embryos were isolated and fixed in 4% paraformaldehyde for 2 hours. After incubation with 30% sucrose in PBS overnight, embryos were mounted and sectioned to 10 μm sections with a cryo-stat machine. Samples were permealized with 0.2% Tween in PBS for 20 minutes, blocked with 10% FBS in PBS for 1 hour, and incubated with primary antibodies at room temperature for 2 hours. The samples were incubated with donkey anti-rabbit IgG H&L (Biotin) (Abcam, ab97062) for 30 minutes, followed by incubation with DyLight 488 Streptavidin (Vector laboratories, SA-5488) and/or anti-goat IgG H&L (Alexa Fluor 594) (Abcam, ab150132) for 1 hour. All conducted animal experiments were approved by the NIEHS/NIH Animal Care and Use Committee.
The antibodies used include: rabbit anti-ACSS2 (Cell Signaling, 3658S; RRID:AB_2222710), rabbit anti-ACADS (ThermoFisher, PA5–54580; RRID:AB_2637583), goat anti-human SOX17 (R&D systems, AF1924-SP; RRID:AB_355060), rabbit anti-ACOX3 (ThermoFisher, PA5–57422; RRID:AB_2637608), rabbit anti-FOXA2 (Cell Signaling Technology, 8186T; RRID:AB_10891055), rabbit pan anti-crotonyllysine (PTM Biolabs, PTM-501; RRID:AB_2877694), mouse pan anti-crotonyllysine (PTM Biolabs, PTM-502; RRID:AB_2877695), rabbit anti-H3K27ac (Abcam, ab177178; RRID:AB_2828007), and pan anti-butyryllysine (PTM Biolabs,PTM-301; RRID:AB_2687946). The samples were observed with confocal microscope (ZEISS LSM 780 or ZEISS LSM 710; objective: plan-Apo-chromat 63 ×/1.40 oil DIC M27, or 40 ×/1.2). The full embryo images of ACSS2 and SOX17 staining were obtained by tiling/stitching multiple fields using ZEN software and ZEISS 710 confocal microscope.
SILAC analysis
To quantify the differential abundance of histone Kcr, Kac, and Kbu in hESCs and differentiated endodermal cells, mel1 hESCs were cultured in lysine-free hESCs culture medium with “heavy” lysine (13C6 15N2-Lys) or “light” lysine (12C6 14N2-Lys) (100 mg/L, Cambridge Isotope Laboratories). Cells were grown for more than seven generations before being harvested, to achieve more than 99% labeling efficiency (based on MS analysis). Endodermal cells were differentiated from mel1 hESCs cultured in “light” medium for 3 days. The “heavy” hESCs and “light” endodermal cells were then harvested, and histones were extracted, mixed together at 1:1 ration, digested with trypsin (Promega Corp.), and immunoprecipitated with Pan anti-Kcr, anti-Kac, or anti-Kbu antibody (PTM Biolabs, Chicago, IL, catalog number PTM-501, RRID:AB_2877694; PTM-105, RRID:AB_2877698; and PTM-301, RRID:AB_2687946). The enriched peptides obtained above were then analyzed by HPLC/MS/MS as described (Huang et al., 2018). The acquired MS/MS data was searched with Mascot search engine (Matrix Science) against UniProt Human protein database (88,949 entries). Maximum missing cleavage was set at 4, and mass tolerance was set at 10 ppm for precursor ions and ± 0.05 Da for MS/MS. To ensure that the Kcr, Kac and Kbu level change in endodermal cells versus hESCs is not caused by the protein level change, we also quantified the protein expression levels. Briefly, the proteolytic peptides obtained in previous step were separated on preparative HPLC into 20 fractions, or separated into 12 fractions with SDS-PAGE. These samples were analyzed using the same procedures for the Kcr, Kac and Kbu peptides quantification. Then all the ratios of quantified Kcr, Kac and Kbu peptides were normalized by the ratios of their corresponding protein expression levels.
Immunoblotting
Cells were washed once with PBS, and were then lysed and scraped with 1 x SDS loading buffer without bromophenol blue. Samples were boiled for 10 minutes, and quantified. Equal amount of protein lysates was loaded and resolved on SDS-PAGE gel and transferred onto an PVDF membrane (Millipore). Blots were blocked with 5% BSA for 1 hour, incubated with primary antibodies at 4°C overnight, incubated with secondary antibodies for 2 hours, and detected by Odyssey (LI-Cor inc.). Additional antibodies used include: mouse monoclonal anti-Actin, clone C4 (Millipore, MAB1501; RRID:AB_2223041), rabbit polyclonal anti-H3 (abcam ab1791; RRID:AB_302613), mouse monoclonal anti-Histone H3, clone 6.6.2 (Millipore, 05–499; RRID:AB_309763), rabbit polyclonal anti-acetylated lysine (Cell signaling, 9441s; RRID:AB_331805), rabbit anti-ACSS2 (Cell Signaling, 3658S; RRID:AB_2222710), goat anti-human SOX17 (R&D systems, AF1924-SP; RRID:AB_355060), rabbit anti-ACOX3 (ThermoFisher, PA5–57422; RRID:AB_2637608), rabbit anti-FOXA2 (Cell Signaling Technology, 8186T; RRID:AB_10891055), pan anti-crotonyllysine (PTM Biolabs, PTM-502; RRID:AB_2877695), rabbit anti-H3K27ac (Abcam, ab177178; RRID:AB_2828007), pan anti-butyryllysine (PTM Biolabs,PTM-301; RRID:AB_2687946).
ChIP and ChIP-seq analysis
To profile the chromatin landscape of Kcr and H3K27ac in mel1 hESCs versus differentiated endodermal cells, 1×107 cells from three independent experiments were cross-linked, harvested, and sonicated. The resulting sonicated chromatin was processed for immunoprecipitation with anti-Kcr (PTM Biolabs, PTM-501; RRID:AB_2877694) or anti-H3K27ac (Abcam, ab177178; RRID:AB_2828007) antibodies. The DNA fragments from each sample were then analyzed by the pair-end 75bp protocol using the NextSeq 500 platform (Illumina) per the manufacturer’s protocol.
For each sample, raw reads from different runs were pooled together. Pooled raw reads were filtered based on a mean base quality score of > 20, and adapters were trimmed by “Trim Galore” (version 0.4, https://github.com/FelixKrueger/TrimGalore). Trimmed reads were mapped to hg19 using Bowtie (Langmead et al., 2009) with the parameters “-m1, -v2–best–strata -X 1500.” For each condition/treatment, aligned read files from replicate samples were merged together using function merge from samtools (Version 1.3.1) (Li et al., 2009). For all merged files, duplicates were removed using Picard (Version 1.115, http://broadinstitute.github.io/picard/).
For ChIP-seq quality control assessment, all mapped and deduplicated individual BAM files were analyzed using strand-shift profile (SSP) method (Nakato and Shirahige, 2018) and the normalized strand cross-correlation coefficient (NSC) for each sample was calculated (Table S2). Overall similar NSC values were observed among the biological replicates, with all NSC values from the ChIP-seq samples were higher than input samples. Additionally, the pairwise spearman correlation coefficients between individual BAM files were computed from the read coverages for genomic regions, which were calculated by multiBamSummary, and plotted by plot-Correlation from deepTools (Figure S4A). Again, the overall correlation among biological replicates was high.
The program called findPeaks from Homer (Version 4.8.3) (Heinz et al., 2010) was applied to call ChIP-seq peaks. The basic idea is to identify regions in the genome where we find more sequencing reads than expected by chance. The pooled input sample was used as noise control for peak calling. The findPeaks was run with parameter “-F 2 -L 0 -C 1.5 -fdr 0.001.”
After peak calling was done for each sample, we then obtained the union of peaks called from all paired conditions as the candidate regions for quantitative comparison. For each comparison, union peak list was made by two steps: 1) Two list of peaks were put into one file and sorted; 2) merge overlapping and neighbor peaks within 100nt using mergeBed from bedTools (version 2.25.0) (Quinlan and Hall, 2010). Read count of each union peak for each sample was calculated using coverageBed from bedTools.
The quantification results from coverageBed were imported into the Bioconductor package DESeq2 for pairwise comparisons between ES and differentiated samples. Benjamini and Hochberg method was applied to adjust false discovery rate (FDR). A peak was considered significantly differential bound if the FDR adjusted p value was less than 0.05.
All the significant peaks associated with genes were subjected to GO and pathway analyses using Genomic Regions Enrichment of Annotations Tool (GREAT) (Version 3.0.0) (McLean et al., 2010); and motif analysis by findMotifsGenome.pl from Homer with default parameter.
Heatmap of peaks was analyzed and generated using Deeptools (V2.4.2) (Ramírez et al., 2016).
RNA-seq analysis
Total RNA was extracted from undifferentiated mel1 cells and mel1 cells after 3 days of endoderm differentiation from three independent experiments. All RNA-seq libraries (non-strand-specific) were prepared with the TruSeq RNA Sample Prep kit (Illumina, San Diego, CA). The total RNA samples were subject to poly(A) enrichment as part of the TruSeq protocol. mRNA samples were sequenced using the single-end 75bp protocol using the NextSeq 500 platform (Illumina) per the manufacturer’s protocol.
Raw reads were filtered based on a mean base quality score of > 20, and adapters were trimmed by “Trim Galore” (version 0.4). Trimmed reads were mapped to hg19 using STAR aligner (version 2.6) (Dobin et al., 2013). Subread featureCounts (version 1.4.6) (Liao et al., 2013) was applied for gene quantification based on University of California Santa Cruz (UCSC) hg19 RefSeq gene release (downloaded August 18th, 2017). The quantification results from featureCounts were then analyzed by the Bioconductor package DESeq2 (Love et al., 2014), which fits a negative binomial distribution to estimate technical and biological variability. The comparisons between vehicle and treatment samples were performed to identify the differential expressed genes. Benjamini and Hochberg method was applied to adjust the false discovery rate (FDR). A gene was considered significantly differentially expressed if the FDR adjusted p value for differential expression was less than 0.05. The heatmap was generated using Partek Genomic Suite (Version 6.6, Partek, St. Louis, Missouri).
Single cell analysis
To analyze the impact of crotonate and acetate on endoderm differentiation of hESCs at the single-cell transcriptome level, mel1 hESCs (ESCs), or mel1 hESCs after 3-day differentiation into endoderm in regular differentiation medium (Endoderm), or in differentiation medium containing 5 mM acetate (Endoderm + acetate) or 5 mM crotonate (Endoderm + crotonate) were processed for single cell analysis using a one-touch ddSEQ Single-Cell Isolator. Acetate and crotonate were purchased from Sigma-Aldrich (S2889 and 113018).
Specifically, single-cell suspensions were converted to barcoded scRNA-seq libraries, in which cell capture, lysis, reverse transcription, and cDNA amplification were performed on the ddSEQ Single-Cell Isolator for mRNA-seq on Illumina Bio-Rad Single-Cell Sequencing Solution following the manufacturer’s instructions. Single sample is always processed in a single well of a PCR plate, allowing all cells from a sample to be treated with the same master mix and in the same reaction vessel. All samples were processed in parallel in the same thermal cycler. Libraries were sequenced on an Illumina Next500, and reads were parsed for cell barcode and UMI information, filtering out reads according to the following constraints: no 6nt barcode segment is more than a Hamming distance of 1 from a valid barcode block; all linker segments are within 1nt length of the expected value; the UMI has a length of 8nt; and only 1 mismatch is allowed within the ACG-GAC segments flanking the UMI. Reads were mapped to hg19 using STAR with the UCSC refSeq (Downloaded from UCSC August 18th, 2017) annotation as gene models. Gene assignments were carried out using subread featureCounts version 1.6.3 using this same RefSeq annotation prior to deduplication and count accumulation using UMI-tools (version 0.5.0) (Smith et al., 2017), allowing for a single edit for UMI matching.
Valid cell barcodes were identified using R (https://www.r-project.org/, Version 3.5.0) package DropletUtils (version 1.2.2) (Griffiths et al., 2018).
Single-cell gene expression quantification and determination of the major cell types
Raw gene expression matrices generated per sample were combined and converted to a Seurat object using the Seurat package (version 2.3.4) (Satija et al., 2015). The monocle2 strategy (Qiu et al., 2017) was applied to filter out all the dead cells and doublets. The total number of UMI and genes for each cell were counted. The upper bound was calculated as mean plus two standard deviation (SD) and the lower bound as mean minus two SD for both the total UMI and genes, respectively. Cells with total UMIs or genes outside of the upper and lower bounds were removed. Cells with over 15% UMIs derived from mitochondrial genome were also excluded from further analysis. For the remaining 1863 cells, gene expression matrices were normalized to total cellular read count and to mitochondrial read count using linear regression as implemented in Seurat’s RegressOut function. Variably expressed genes were defined as a normalized expression between 0.125 and 8, and a quantile-normalized variance exceeding 1. To reduce dimensionality of this dataset, the resulting 738 variably expressed genes were summarized by principle component analysis, and the first 18 principle components further summarized using t-Distributed Stochastic Neighbor Embedding (tSNE) dimensionality reduction using the default settings of the RunTSNE function. Cell clusters in the resulting two-dimensional representation were annotated to known biological cell types using canonical marker genes. The cell cycle phase for each cell was examined using cell cycle related marker genes.
Gene regulatory network reconstruction and cell-state identification
To identify transcription factors that are specifically activated in different differentiation stages during 3-day endoderm differentiation of Mel1 hESCs, we used SCENIC (Aibar et al., 2017) to infer activity of transcription factors from scRNA-Seq data according to instructions provided on github (https://github.com/aertslab/SCENIC) using R3.6.2. In short, gene count matrices from Seurat were used to infer a transcriptional state from an underlying gene regulatory network. Sequences 500bp (mm9–500bp-upstream-7species.mc9nr.feather) and 10kb (extended); mm9-tss-centered-10kb-7species.mc9nr.feather) upstream of the TSS from the mm9 annotation were used to infer the transcription factor (TF) activity of these regulons. The TF activity was quantified on a per cell basis using an enrichment score (AUCell) for the regulons and projected onto the tSNE space. Figures contain the number of genes matching genes per transcription factor in parenthesis and are colorized for AUCell (higher score = higher activity) and projected onto two dimensional tSNE space.
Pseudotemporal analysis
To characterize the potential process involved in embryonic stem cells differentiation, Monocle2 (Version 2.10.1) was applied to order cells into trajectories based on their transcriptomic similarity. A total of 1863 cells that passed the filtering criteria were used as input and loaded into Monocle2 to create a CellData Set object with the parameter ‘expressionFamily = negbinomial’ for pseudotime analysis. The function dispersionTable with default parameters was run to identify the top 1970 highly informative genes. The function reduceDimension was applied for dimensions reduction. Cells were ordered by orderCells through the inferred pseudotime to indicate their differentiation progress. The trajectory in the reduced dimensional space was inferred and visualized using plot_cell_trajectory with the default parameters.
Short-chain acyl-CoA measurement
To measure the acetyl-CoA, crotonyl-CoA, butyryl-CoA, and other short-chain fatty acyl-CoA concentrations in mel1 hESCs that were induced for endoderm differentiation for 3 days with different treatments or gene knockouts, 1 × 107 cells were harvested and analyzed for the abundances of acetyl-CoA and crotonyl-CoA using LC-quadrupole/Orbitrap high-resolution mass spectrometry as previously described (Frey et al., 2016). Samples were spiked with Stable isotope labeling by essential nutrients in cell culture (SILEC) acyl-CoA internal standard (Basu and Blair, 2011). Standards for calibration curves were prepared from commercially available acyl-CoAs (Sigma-Aldrich). Standards were spiked with SILEC acyl-CoA internal standard, then treated and extracted in the same manner as the cell samples. Analysts were blinded to sample identity.
CRISPR-mediated gene knockout, siRNA-mediated knockdown
To generate crotonyl-CoA producing enzyme defective hESCs, mel1 cells were transfected with EF1a-cas9-nls together with Edit-R CRISPR-cas9 synthetic tracrRNA and Edit-R Human ACSS2 crRNA (Dharmacon, CM-010396-01-0002), or transfected with lenti-CRISPR v2 vectors containing gRNAs for ACADS, and ACOX3, respectively. After selection with puromycin, single colonies were picked and expanded, and genotyped with genomic DNA sequencing. Protein expression was analyzed by western blot and/or immunofluorescence.
To knockdown ACADS or ACSS2 expression, mel1 cells were transfected with control siRNA, ACSS2 siRNA (S3146, Ambion) or ACADS siRNA (S895, Ambion), respectively. Two days later, cells were induced for endoderm differentiation. Samples were collected on day 2 post differentiation, and knockdown was confirmed by quantitative PCR (qPCR) and/or western blot.
Butyryl-CoA oxidase activity assay for ACOX3
The butyryl-CoA oxidase activity of ACOX3 was measured using butyryl-CoA as substrate as previously described (Small et al., 1985). Briefly, immuno-purified ACOX3-GFP protein was incubated in a reaction buffer (50 mM 2-[N-morpholino] ethanesulfonic acid [pH 8.0], 0.8 mM 4-aminoantipyrine, 11 mM phenol solution, 0.04% Triton X-100, 30 μM butyryl-CoA, 5 μM FAD, and 1 unit peroxidase) at 30°C. During the reaction, H2O2 produced by ACOX3-mediated oxidation of butyryl-CoA was further utilized by peroxidase to convert 4-aminoantipyrine/phenol to quinoneimine dye, leading to absorption at A500 nm. The activity of ACOX3 was calculated by the change of A500 in the first 5 min.
Teratoma assay
WT control, ACADS KO, and ACOX3 KO mel1 cells were resuspended with matrigel diluted by PBS at 1:1 ratio. 2×106 cells/100 μL were injected to NSG CD34FL mice, and mice were euthanized at 9 weeks after injection. Teratomas were cut into halves, with one half fixed for histological analysis, and the other half analyzed for expression of tissue specific markers by qPCR as described (International Stem Cell Initiative, 2018). The antibodies used in immunofluorescence staining of teratomas include: rabbit polyclonal anti-CDH17 (Sigma, HPA026556; RRID:AB_1845897), rabbit monoclonal anti-ACTN2 [EP2529Y] (Abcam, ab68167; RRID:AB_11157538), rabbit polyclonal anti-beta III tubulin (Abcam, ab18207; RRID:AB_444319).
All conducted animal experiments were approved by the NIEHS/NIH Animal Care and Use Committee.
QUANTIFICATION AND STATISTICAL ANALYSIS
Values are expressed as mean ± standard error of mean (SEM) from at least three independent experiments or biological replicates, unless otherwise indicated in the figure legend. Significant differences between the means were analyzed by the two-tailed, unpaired, Student’s t test, and differences were considered significant at *p < 0.05 using Microsoft Office Excel (Version 16.16.27). No methods were used to determine whether the data met assumptions of the statistical approach (e.g., test for normal distribution).
Bioinformatic analyses of ChIP-seq, RNA-seq, and single-cell RNA-seq are detailed in Method details section.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-ACSS2 | Cell Signaling | Cat# 3658S; RRID:AB_2222710 |
| Rabbit polyclonal anti-ACADS | ThermoFisher | Cat# PA5-54580; RRID:AB_2637583 |
| Goat polyclonal anti-SOX17 | R&D systems | Cat# AF1924-SP; RRID:AB_355060 |
| Rabbit polyclonal anti-ACOX3 | ThermoFisher | Cat# PA5-57422; RRID:AB_2637608 |
| Rabbit monoclonal anti-FOXA2 | Cell Signaling | Cat# 8186T; RRID:AB_10891055 |
| Rabbit polyclonal pan anti-crotonyllysine | PTM Biolabs | Cat# PTM-501; RRID:AB_2877694 |
| Mouse monoclonal pan anti-crotonyllysine | PTM Biolabs | Cat# PTM-502; RRID:AB_2877695 |
| Rabbit monoclonal anti-H3K27ac | Abcam | Cat# ab177178; RRID:AB_2828007 |
| Rabbit polyclonal pan anti-butyryllysine | PTM Biolabs | Cat# PTM-301; RRID:AB_2687946 |
| Rabbit polyclonal pan anti-acetyllysine | PTM Biolabs | Cat# PTM-105; RRID:AB_2877698 |
| Mouse monoclonal anti-Actin, clone C4 | Millipore | Cat# MAB1501; RRID:AB_2223041 |
| Rabbit polyclonal anti-H3 | Abcam | Cat# ab1791; RRID:AB_302613 |
| Rabbit polyclonal anti-acetylated lysine | Cell Signaling | Cat# 9441s; RRID: RRID:AB_331805 |
| Rabbit polyclonal anti-CDH17 | Sigma | Cat# HPA026556; RRID:AB_1845897 |
| Rabbit monoclonal anti-ACTN2 [EP2529Y] | Abcam | Cat# ab68167; RRID:AB_11157538 |
| Donkey anti-rabbit IgG H&L (Biotin) | Abcam | Cat# ab97062; RRID:AB_10679678 |
| Mouse monoclonal anti-Histone H3 Antibody, clone 6.6.2 | Millipore | Cat# 05-499; RRID:AB_309763 |
| Rabbit polyclonal anti-beta III tubulin | Abcam | Cat# ab18207; RRID:AB_444319 |
| Bacterial and virus strains | ||
| One shot Stbl3™ chemically competent | Thermo Fisher | Cat# C737303 |
| Chemicals, peptides, and recombinant proteins | ||
| Matrigel hESC-qualified | Corning | Cat# 354277 |
| Y-27632 dihydrochloride (ROCK inhibitor) | TOCRIS | Cat# 1254 |
| Crotonic acid | Sigma | Cat# 113018 |
| Sodium acetate | Sigma | Cat# S2889 |
| L-Arginine-13C6,15N4 hydrochloride | Sigma | Cat# 608033 |
| L-Lysine-13C6,15N2 hydrochloride | Sigma | Cat# 608041 |
| Crotonyllysine peptide library | Dr. Yingming Zhao’s lab | This paper |
| Butyrylysine peptide library | Dr. Yingming Zhao’s lab | This paper |
| Critical commercial assays | ||
| STEMDIFF trilineage differentiation kit | Stem Cell Technology, Inc. | Cat# 05230 |
| Deposited data | ||
| RNaseq data | This paper | GEO: GSE130345 |
| scRNA-seq data | This paper | GEO: GSE130341 |
| ChIP-seq data | This paper | GEO: GSE130344 |
| Unprocessed and uncompressed immuno-blots | This paper | https://doi.org/10.17632/sfpv2m95s6.1 |
| Experimental models: cell lines | ||
| Mel1 hESCs | Dr. Andrew Elefanty and Edouard Stanley at the University of Queensland, Australia | NIH Registration number: 0139 |
| H9 hESCs | Dr. James A. Thomson at University of Wisconsin-Madison (via WiCell Research Institute) | NIH Registration number: 0062; RRID:CVCL_9773 |
| ACADS KO mel1 hESCs | This paper | N/A |
| ACOX3 KO mel1 hESCs | This paper | N/A |
| Experimental models: organisms/strains | ||
| NSG CD34FL (NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ) | Jackson Laboratory | #005557 |
| Oligonucleotides | ||
| Please see Table S7 | N/A | |
| Recombinant DNA | ||
| EF1a-cas9-nls | Stolfi et al., 2014 | Addgene plasmid #59987; RRID:Addgene_59987 |
| LentiCRISPR v2 | Sanjana et al., 2014 | Addgene plasmid #52961; RRID:Addgene_52961 |
| AAVS1_puro_PGK1_3xFLAG_Twin_Strep_EZH2 | Dalvai et al., 2015 | Addgene plasmid #79902; RRID:Addgene_79902 |
| LentiCRISPR-ACADS sgRNA | This paper | N/A |
| LentiCRISPR-ACOX3 sgRNA | This paper | N/A |
| AAVS1_puro_PGK1_H4-GFP | This paper | N/A |
| AAVS1_puro_PGK1_H4K77R-GFP | This paper | N/A |
| AAVS1_puro_PGK1_H4K91R-GFP | This paper | N/A |
| Software and algorithms | ||
| Trim Galore (version 0.4) | https://github.com/FelixKrueger/TrimGalore | |
| Microsoft Office Excel (version 16.16.27) | Microsoft | N/A |
| Bowtie | Langmead et al., 2009 | N/A |
| Samtools (version 1.3.1) | Li et al., 2009 | N/A |
| Picard (version 1.115) | http://broadinstitute.github.io/picard | |
| MultiBamSummary | N/A | |
| FindPeaks from Homer (version 4.8.3) | Heinz et al., 2010 | N/A |
| MergeBed from bedTools (version 2.25.0) | Quinlan and Hall, 2010 | N/A |
| Bioconductor package DESeq2 | Love et al., 2014 | N/A |
| GREAT (version 3.0.0) | Mclean et al., 2010 | N/A |
| Deeptools (version 2.4.2) | Ramírez et al., 2016 | N/A |
| STAR aligner (version 2.6) | Dobin et al., 2013 | N/A |
| FeatureCounts (version 1.4.6) | Liao et al., 2013 | N/A |
| Partek Genomic Suite (version 6.6) | Partek | N/A |
| UMI-tools (version 0.5.0) | Smith et al., 2017 | N/A |
| R (version 3.5.0) | https://www.r-project.org/ | |
| DropletUtils (version 1.2.2) | Griffiths et al., 2018 | N/A |
| Seurat package (version 2.3.4) | Satija et al., 2015 | N/A |
| SCENIC | Aibar et al., 2017 | N/A |
| Github | https://github.com/aertslab/SCENIC | |
| Monocle2 (version 2.10.1) | Qiu et al., 2017 | N/A |
Highlights.
Key crotonyl-CoA-producing enzymes are induced and enriched in endodermal cells
Endoderm differentiation is associated with enhanced histone crotonylation (Kcr)
Chemical enhancement of histone Kcr promotes endoderm differentiation in hESCs
Disrupting histone Kcr impairs meso/endoderm differentiation in vitro and in vivo
ACKNOWLEDGMENTS
We thank Drs. Paul Wade, Guang Hu, G. Greg Wang, Keji Zhao, and members of the Li laboratory for critical reading of the manuscript. We also thank the NIEHS Epigenomics Core Facility for performance of RNA-seq, ChIP-seq, and scRNA-seq experiments and Anna Krause from the NIEHS Photography & Graphics Service Center for the graphical abstract. This research was supported by the Intramural Research Program of the National Institute of Environmental Health Sciences of the NIH (Z01 ES102205 to X.L.), NIH grants DK107868 and GM115961 (to Y.Z.), and NIH grants R03HD092630 and R01GM132261 (to N.W.S.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.stem.2020.12.009.
DECLARATION OF INTERESTS
Y.Z. is on the science advisory board of PTM Biolabs.
REFERENCES
- Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews FH, Shinsky SA, Shanle EK, Bridgers JB, Gest A, Tsun IK, Krajewski K, Shi X, Strahl BD, and Kutateladze TG (2016). The Taf14 YEATS domain is a reader of histone crotonylation. Nat. Chem. Biol 12, 396–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Wang Y, Li X, Li XM, Liu Z, Yang T, Wong CF, Zhang J, Hao Q, and Li XD (2014). Identification of ‘erasers’ for lysine crotonylated histone marks using a chemical proteomics approach. eLife 3, e02999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu SS, and Blair IA (2011). SILEC: a protocol for generating and using isotopically labeled coenzyme A mass spectrometry standards. Nat. Protoc 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhala A, Willi SM, Rinaldo P, Bennett MJ, Schmidt-Sommerfeld E, and Hale DE (1995). Clinical and biochemical characterization of short-chain acyl-coenzyme A dehydrogenase deficiency. J. Pediatr 126, 910–915. [DOI] [PubMed] [Google Scholar]
- Bowman GD, and Poirier MG (2015). Post-translational modifications of histones that influence nucleosome dynamics. Chem. Rev 115, 2274–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Sutter BM, Li B, and Tu BP (2011). Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell 42, 426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Finley LW, Cross JR, Allis CD, and Thompson CB (2015). Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliff TS, Wu T, Boward BR, Yin A, Yin H, Glushka JN, Prestegaard JH, and Dalton S (2017). MYC Controls Human Pluripotent Stem Cell Fate Decisions through Regulation of Metabolic Flux. Cell Stem Cell 21, 502–516.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corydon MJ, Vockley J, Rinaldo P, Rhead WJ, Kjeldsen M, Winter V, Riggs C, Babovic-Vuksanovic D, Smeitink J, De Jong J, et al. (2001). Role of common gene variations in the molecular pathogenesis of short-chain acyl-CoA dehydrogenase deficiency. Pediatr. Res 49, 18–23. [DOI] [PubMed] [Google Scholar]
- Dalvai M, Loehr J, Jacquet K, Huard CC, Roques C, Herst P, Cote J, and Doyon Y (2015). A Scalable Genome-Editing-Based Approach for Mapping Multiprotein Complexes in Human Cells. Cell Rep 13, 621–633. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellows R, Denizot J, Stellato C, Cuomo A, Jain P, Stoyanova E, Balázsi S, Hajnády Z, Liebert A, Kazakevych J, et al. (2018). Microbiota derived short chain fatty acids promote histone crotonylation in the colon through histone deacetylases. Nat. Commun 9, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandusse S, Denis S, van Roermund CWT, Preece MA, Koster J, Ebberink MS, Waterham HR, and Wanders RJA (2018). A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl-CoA oxidase. Biochim. Biophys. Acta Mol. Basis Dis 1864, 952–958. [DOI] [PubMed] [Google Scholar]
- Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, and Terzic A (2011). Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 14, 264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CD, Dzeja PP, Nelson TJ, and Terzic A (2012). Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 11, 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey AJ, Feldman DR, Trefely S, Worth AJ, Basu SS, and Snyder NW (2016). LC-quadrupole/Orbitrap high-resolution mass spectrometry enables stable isotope-resolved simultaneous quantification and 13C-isotopic labeling of acyl-coenzyme A thioesters. Anal. Bioanal. Chem 408, 3651–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths JA, Richard AC, Bach K, Lun ATL, and Marioni JC (2018). Detection and removal of barcode swapping in single-cell RNA-seq data. Nat. Commun 9, 2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Gaeta X, Sahakyan A, Chan AB, Hong CS, Kim R, Braas D, Plath K, Lowry WE, and Christofk HR (2016). Glycolytic Metabolism Plays a Functional Role in Regulating Human Pluripotent Stem Cell State. Cell Stem Cell 19, 476–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Tang S, Ji M, Tang Z, Shimada M, Liu X, Qi S, Locasale JW, Roeder RG, Zhao Y, and Li X (2018). p300-Mediated Lysine 2-Hydroxyisobutyrylation Regulates Glycolysis. Mol. Cell 70, 984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Stem Cell Initiative (2018). Assessment of established techniques to determine developmental and malignant potential of human pluripotent stem cells. Nat. Commun 9, 1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, and Suda T (2014). Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol 15, 243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G, Nguyen D, Archin NM, Yukl SA, Méndez-Lagares G, Tang Y, Elsheikh MM, Thompson GR 3rd, Hartigan-O’Connor DJ, Margolis DM, et al. (2018). HIV latency is reversed by ACSS2-driven histone crotonylation. J. Clin. Invest 128, 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Park HD, Sohn YB, Park SW, Cho SY, Ji S, Kim SJ, Choi EW, Kim CH, Ko AR, et al. (2011). Mutations of ACADS gene associated with short-chain acyl-coenzyme A dehydrogenase deficiency. Ann. Clin. Lab. Sci 41, 84–88. [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Daujat S, and Schneider R (2016). Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 32, 42–56. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Sabari BR, Panchenko T, Wen H, Zhao D, Guan H, Wan L, Huang H, Tang Z, Zhao Y, et al. (2016). Molecular Coupling of Histone Crotonylation and Active Transcription by AF9 YEATS Domain. Mol. Cell 62, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen AS, and Olsen CA (2012). Profiling of substrates for zinc-dependent lysine deacylase enzymes: HDAC3 exhibits decrotonylase activity in vitro. Angew. Chem. Int. Ed. Engl 51, 9083–9087. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montellier E, Rousseaux S, Zhao Y, and Khochbin S (2012). Histone crotonylation specifically marks the haploid male germ cell gene expression program: post-meiotic male-specific gene expression. BioEssays 34, 187–193. [DOI] [PubMed] [Google Scholar]
- Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D, Nemirovski A, Shen-Orr S, Laevsky I, Amit M, et al. (2015). Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 21, 392–402. [DOI] [PubMed] [Google Scholar]
- Nakato R, and Shirahige K (2018). Sensitive and robust assessment of ChIP-seq read distribution using a strand-shift profile. Bioinformatics 34, 2356–2363. [DOI] [PubMed] [Google Scholar]
- Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, Tautenhahn R, Herrerías A, Batchelder EM, Plongthongkum N, Lutz M, et al. (2012). The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 22, 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W1), W160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseaux S, and Khochbin S (2015). Histone Acylation beyond Acetylation: Terra Incognita in Chromatin Biology. Cell J 17, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Andres O, Sanchez-Niño MD, Cannata-Ortiz P, Ruiz-Ortega M, Egido J, Ortiz A, and Sanz AB (2016). Histone lysine crotonylation during acute kidney injury in mice. Dis. Model. Mech 9, 633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Tang Z, Huang H, Yong-Gonzalez V, Molina H, Kong HE, Dai L, Shimada M, Cross JR, Zhao Y, et al. (2015). Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell 58, 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Zhang D, Allis CD, and Zhao Y (2017). Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol 18, 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan J, Hamazaki T, Tang TA, Terada N, and Kilberg MS (2013). Activation of the amino acid response modulates lineage specification during differentiation of murine embryonic stem cells. Am. J. Physiol. Endocrinol. Metab 305, E325–E335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, Aburatani H, Kume K, Endo F, and Kume S (2014). Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 19, 780–794. [DOI] [PubMed] [Google Scholar]
- Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H, et al. (2013). Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 339, 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simithy J, Sidoli S, Yuan ZF, Coradin M, Bhanu NV, Marchione DM, Klein BJ, Bazilevsky GA, McCullough CE, Magin RS, et al. (2017). Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun 8, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin HS, Barski A, Zhang F, Kartashov AV, Nussenzweig A, Chen J, Andreassen PR, and Namekawa SH (2012). RNF8 regulates active epigenetic modifications and escape gene activation from inactive sex chromo-somes in post-meiotic spermatids. Genes Dev. 26, 2737–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small GM, Burdett K, and Connock MJ (1985). A sensitive spectrophoto-metric assay for peroxisomal acyl-CoA oxidase. Biochem. J 227, 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolfi A, Gandhi S, Salek F, and Christiaen L (2014). Tissue-specific genome editing in Ciona embryos by CRISPR/Cas9. Development 141, 4115–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, et al. (2011). Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessadori F, Giltay JC, Hurst JA, Massink MP, Duran K, Vos HR, van Es RM, Scott RH, van Gassen KLI, Bakkers J, and van Haaften G; Deciphering Developmental Disorders Study (2017). Germline mutations affecting the histone H4 core cause a developmental syndrome by altering DNA damage response and cell cycle control. Nat. Genet 49, 1642–1646. [DOI] [PubMed] [Google Scholar]
- van Maldegem BT, Duran M, Wanders RJ, Niezen-Koning KE, Hogeveen M, Ijlst L, Waterham HR, and Wijburg FA (2006). Clinical, biochemical, and genetic heterogeneity in short-chain acyl-coenzyme A dehydrogenase deficiency. JAMA 296, 943–952. [DOI] [PubMed] [Google Scholar]
- Wang A, Yue F, Li Y, Xie R, Harper T, Patel NA, Muth K, Palmer J, Qiu Y, Wang J, et al. (2015). Epigenetic priming of enhancers predicts developmental competence of hESC-derived endodermal lineage intermediates. Cell Stem Cell 16, 386–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Liu X, Chen J, Gao S, Lu L, Zhang H, Ding G, Wang Z, Chen Z, Shi T, et al. (2017). Class I histone deacetylases are major histone decrotonylases: evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 27, 898–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CB (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe L, Jethva R, Oglesbee D, and Vockley J (1993). Short-Chain Acyl-CoA Dehydrogenase Deficiency (revised: 2018). In GeneReviews, Volume R, Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, and Amemiya A, eds. (University of Washington; ). [PubMed] [Google Scholar]
- Xiong X, Panchenko T, Yang S, Zhao S, Yan P, Zhang W, Xie W, Li Y, Zhao Y, Allis CD, and Li H (2016). Selective recognition of histone crotonylation by double PHD fingers of MOZ and DPF2. Nat. Chem. Biol 12, 1111–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. (2011). Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Tsang KS, Wang Y, Chan JC, Xu G, and Gao WQ (2014). Unfolded protein response is required for the definitive endodermal specification of mouse embryonic stem cells via Smad2 and β-catenin signaling. J. Biol. Chem 289, 26290–26301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiangou L, Ross ADB, Goh KJ, and Vallier L (2018). Human Pluripotent Stem Cell-Derived Endoderm for Modeling Development and Clinical Applications. Cell Stem Cell 22, 485–499. [DOI] [PubMed] [Google Scholar]
- Zhang J, Nuebel E, Daley GQ, Koehler CM, and Teitell MA (2012). Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell 11, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Guan H, Zhao S, Mi W, Wen H, Li Y, Zhao Y, Allis CD, Shi X, and Li H (2016). YEATS2 is a selective histone crotonylation reader. Cell Res. 26, 629–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The bulk RNA-seq (RNA-seq), single-cell RNA-seq (scRNA-seq), and ChIP-seq data have been deposited to Gene Expression Omnibus under the accession number GEO: GSE130345 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130345). Three subseries are linked to GEO: GSE130345:
RNA-seq: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130340
scRNA-seq: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130341
ChIP-seq: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130344
Additional information about ChIP-seq quality control, ChIP-seq peaks, GO lists for ChIP-seq peaks, scRNA-seq quality control, and Marker gene lists in three cell clusters from scRNA-seq is included in Tables S2, S3, S4, S5, and S6.
The SILAC data are available in Table S1.
The source file for unprocessed and uncompressed immuno-blots used in the Figures is available from Mendeley Data: https://doi.org/10.17632/sfpv2m95s6.1.
This study did not generate new code for data analysis.