

Abstract
Type II diabetes (T2D) and specific cancers share many risk factors, however the molecular mechanisms underlying these connections are often not well-understood. BCDIN3D is an RNA modifying enzyme that methylates specific precursor microRNAs and tRNAHis. In addition to breast cancer, BCDIN3D may also be linked to metabolism, as its gene locus is associated with obesity and T2D. In order to uncover metabolic pathways regulated by BCDIN3D in cancer, we performed an unbiased analysis of the metabolome, transcriptome, and proteome of breast cancer cells depleted for BCDIN3D. Intersection of these analyses showed that BCDIN3D-depleted cells have increased levels of Fructose 1,6 Bisphosphate (F1,6-BP), the last six-carbon glycolytic intermediate accompanied by reduced glycolytic capacity. We further show that elevated F1,6-BP is due to downregulation of Aldolase C (ALDOC), an enzyme that cleaves F1,6-BP mainly in the brain, but whose high expression/amplification is associated with poor prognosis in breast cancer. BCDIN3D regulates ALDOC through a non-canonical mechanism involving the crucial let-7 microRNA family and its target site on the 3’UTR of ALDOC. Overall, our results reveal an important connection between BCDIN3D, let-7 and glycolysis that may be relevant to breast cancer, obesity and T2D.
Introduction
RNA end modifications are particularly important for determining the fate of RNA molecules [reviewed in [1]]. We previously uncovered a 5’ end RNA modification that consists of the methylation of the 5’-monophosphate ends [2, 3]. This RNA modification is written by BCDIN3D, a member of the Bin3 family of RNA methyltransferases [4] that are present from S. pombe to humans [3, 5]. BCDIN3D methylates the 5’-monophosphate of mature tRNAHis [2, 6], as well as specific microRNA precursors to inhibit their processing by Dicer [3]. Several reports point to a role of BCDIN3D in cancer, and more specifically breast cancer. Firstly, genome-wide work identified the BCDIN3D messenger RNA as overexpressed in tumorigenic (CD44+; CD24-/low) breast cancer ‘stem’ cells compared to normal breast epithelium, and included it in a 186-gene ‘invasiveness’ gene signature linked to poor prognosis [7]. Secondly, the Curtis et al. gene expression analysis in breast cancer ranked BCDIN3D in the top 1% over-expressed mRNAs in invasive ductal and lobular breast carcinomas [8]. Thirdly, Yao et al. showed that expression of BCDIN3D at the protein level correlates with significantly decreased disease-free survival in invasive ductal breast carcinoma patients, particularly those who also are triple negative [9]. These results are consistent with our results showing that knock-down of BCDIN3D severely impairs anchorage independent growth (an ex vivo model for transformation) and invasion (an ex vivo model for metastasis) in the MDA-MB-231 breast cancer cell line [3], which is a well-established model for the study of triple negative breast cancer of the Mesenchymal Stem Like (MSL) subtype [10]. Interestingly, the BCDIN3D gene locus contains two single nucleotide polymorphisms (SNPs) associated with obesity and type II diabetes [11–13]. In order to address BCDIN3D’s potential role in metabolism in a cancer context, we performed an unbiased analysis of the metabolome, transcriptome, and proteome of MDA-MB-231 breast cancer cells depleted for BCDIN3D. Intersection of these analyses showed that BCDIN3D-depleted cells have increased levels of the last six-carbon intermediate of the glycolysis pathway, Fructose 1,6 Bisphosphate (F1,6-BP), which is due to a decrease in the levels of Aldolase C (ALDOC), one of the enzymes that cleaves F1,6-BP. Consistent with these findings, BCDIN3D-depleted cells showed lower glycolytic function than control cells. We further showed that BCDIN3D regulates ALDOC through a non-canonical mechanism involving the let-7 microRNA and its target site on the 3’UTR of ALDOC, together with other regulators of the let-7 microRNA processing. Overall, our results reveal a connection between BCDIN3D, let-7 and glycolysis that may be relevant to breast cancer, obesity and type II diabetes.
Results
BCDIN3D depletion reduces tumor formation in vivo.
We previously showed that depletion of BCDIN3D slows the growth of MCF-7 cells, but does not affect the growth of MDA-MB-231 cells on a 2D surface [3]. To assess the effect of BCDIN3D on tumor promotion without the potentially confounding effect of decreased cellular proliferation, we focused our work on MDA-MB-231 cells. These cells also have the advantage of being a well-established model for invasive and triple negative breast cancer, which are most relevant to BCDIN3D [7–9]. BCDIN3D knock-down in MDA-MB-231 cells abolishes their growth in soft-agar, an in vitro method for assaying transformation. It also reduces invasion through an extracellular matrix in a Boyden chamber [3], an in vitro read-out for metastatic potential. These results prompted us to test the effect of BCDIN3D knock-down in an in vivo setting. To this end, we used immunodeficient Foxn1nu/nu female mice to xenograft MDA-MB-231-luc-D3H2LN control (shNC) and knocked-down for BCDIN3D (shBCDIN3D) cells (Fig. 1). MDA-MB-231-luc-D3H2LN cells are widely used MDA-MB-231 derivatives displaying long-term stable Luciferase expression in vivo [14]. Using these cells, we performed orthotopic injection into the fourth right mammary pad (Fig. 1a) to assess tumor growth, and tail vein injection to assess metastasis to the lung and bones (Fig. 1b). As seen in Fig. 1a, BCDIN3D-knocked-down cells formed slightly less tumors (8 out of 12) than control cells (10 out of 12), and importantly their final volume 5 weeks after the injection was significantly lower than the controls (p-value of 0.0048 – two-tailed unpaired t test). The majority of the tumors arising from the MDA-MB-231-luc-D3H2LNshBCDIN3D cells became visible late, during the 5th week after orthotopic injection, i.e. two weeks later than the control (Fig. 1a). These late-rising tumors most likely lost the activity of the BCDIN3D small hairpin RNA (shBCDIN3D) in the absence of puromycin selection in vivo, as immunohistochemistry (IHC) with a specific BCDIN3D antibody (Fig. S1a–b) showed BCDIN3D staining of same or higher level as control tumors (Fig. S1c). As shown in Fig. 1b, shBCDIN3D cells did not give rise to any metastasis to the bone, while 3 out of 12 control mice showed tumors on the bone [spine (#940), rib cage (#950) and jaw (#929)] in the tail vein injection experiments 8 weeks post-injection. Overall, these experiments show that BCDIN3D expression is important for tumor growth in vivo.
Figure 1. BCDIN3D depletion reduces tumor formation in vivo.
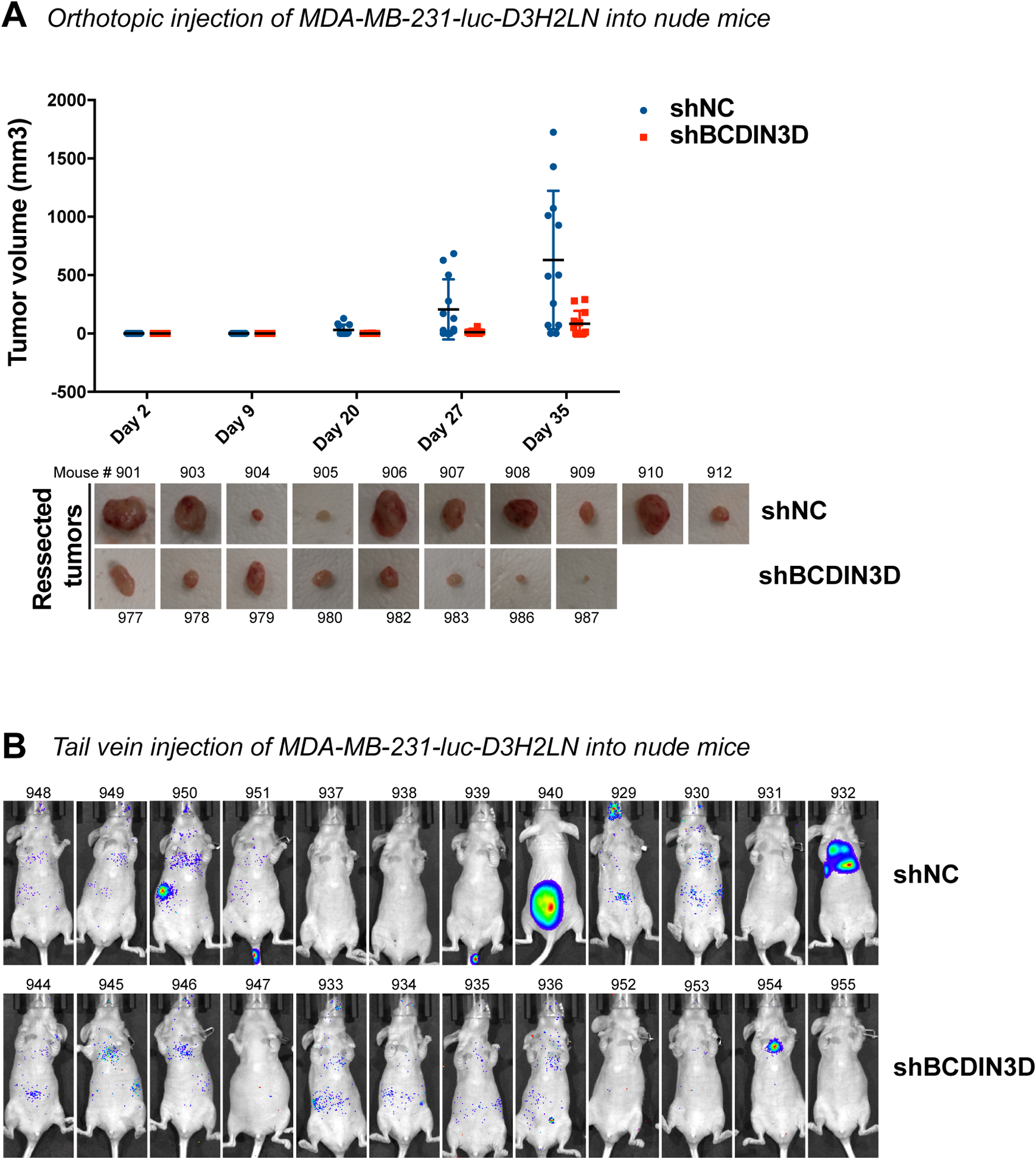
a Foxn1nu/nu mice were orthotopically injected into the #4 mammary fat pad with MDA-MB-231-luc-D3H2LN shNC or shBCDIN3D cells. Tumors were palpated and measured with a caliper over a period of five weeks. The graph shows the tumor volume (mm³) calculated by the formula: volume = (smaller dimension² × larger dimension)/2 as a function of time (scatter plot with mean ± SD, n=10 for shNC, and n=8 for shBCDIN3D, please note that the mice who did not develop tumors are shown on the graph with a dot at 0, but were not taken into account for the calculation of the average size of tumors). The pictures show the resected tumors at day 35 post-injection.
b Shown are IVIS images of mice injected with luciferin intraperitoneally 8 weeks post-tail vein injection of MDA-MB-231-luc-D3H2LN shNC or shBCDIN3D cells. 3 control mice show tumors on the bone [spine (#940), rib cage (#950) and jaw (#929)], 1 control mouse (#932) shows a large tumor in the lungs and 1 mouse injected with shBCDIN3D cells (#954) shows a smaller tumor in the lungs.
BCDIN3D knock-down profoundly affects gene expression of breast cancer cells.
In order to start understanding why cells depleted for BCDIN3D display defects of tumor formation and invasion, we performed combined RNA-Seq, proteomics, and metabolomics of MDA-MB-231 shNC and shBCDIN3D cells (Tables S1–S3). RNA-Seq analysis, which is the most sensitive and robust of the three methods, showed that BCDIN3D depletion deeply affects the transcriptome of MDA-MB-231 cells (Fig. 2a), with 1308 differentially deregulated protein coding genes; 744 being downregulated, and 564 upregulated in shBCDIN3D compared to shNC cells (1.5 fold change, FDR<0.05). Ingenuity pathway analysis (IPA) of this dataset showed a significant enrichment of canonical pathways relevant to cancer (Fig. S2a and Table S4). In particular, 47 protein coding genes belonging to the “Molecular Mechanisms of Cancer” category were differentially regulated in shBCDIN3D compared to shNC cells (Fig. S2b–c), of which many are of clinical relevance for diagnosis and prognosis of cancer (Table S5). Interestingly, 14 of these genes were shared with the mTOR pathway (Fig. 2b and Table S6), which is predicted to be inactivated (z-score=−2; p-value=0.032) by IPA (Table S4). For example, the RPS6KA2 gene - coding p90 RSK1 that indirectly regulates mTOR activation [15] - is significantly downregulated at the mRNA level (Fig. 2c). Consistent with the IPA prediction, shBCDIN3D cell lines show decreased mTOR signaling, as assessed by mTOR phosphorylation at S2448 (see Fig. 2d for mTOR-phospho Ser 2448 quantification in MDA-MB-231 cells, and Fig. S1a for mTOR-phospho Ser 2448 quantification in MDA-MB-231-luc-D3H2NL cells).
Figure 2. BCDIN3D knock-down profoundly affects gene expression in breast cancer cells.
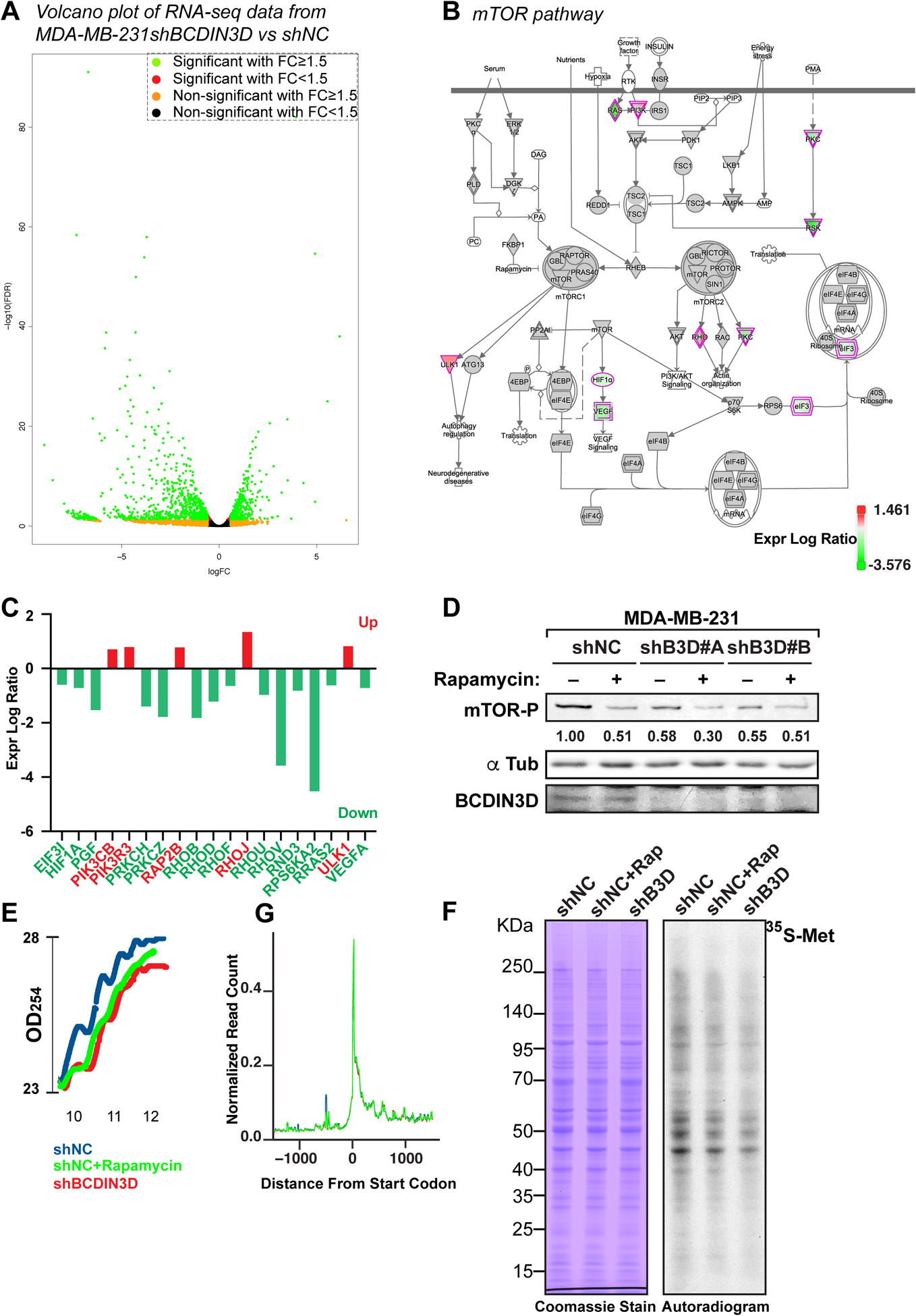
a Volcano plot representing the fold change (FC) of normalized RNA read counts (shBCDIN3D over shNC) on the X-axis (log2) and the False Discovery Rate (FDR) on the Y-axis (log10). Green dots indicate significantly down- or up-regulated genes in shBCDIN3D over shNC with a fold change FC≥1.5 and FDR ≤ 0.05, while the other colors indicate the other combinations of FC and significance, as indicated in the legend in top right of the Volcano plot.
b Ingenuity pathway analysis (IPA) of the RNA-Seq analysis in MDA-MB-231 shNC and shBCDIN3D cells, mapping the position of protein coding genes belonging to the “mTOR pathway” that were differentially regulated in shBCDIN3D compared to shNC cells (green: downregulated; red: upregulated, see legend for exact correspondence between expression log ratio and color).
c Identity and expression log ratio of the mTOR pathway in (B).
d mTOR-phospho Ser 2448 western blot analysis of MDA-MB-231 shNC and shBCDIN3D cells untreated, or treated with 10 nM Rapamycin for 24h. shB3D#A and B represent two independent MDA-MB-231 shBCDIN3D clones. The numbers beneath the anti mTOR-P western blot corresponds to the mTOR-P signal normalized to αTubulin signal from the same western blot, and to shNC.
e Polysome lysates from MDA-MB-231 shNC, shNC+Rapamycin at 10 nM for 24h, and shBCDIN3D cells were fractionated on a 7–50% sucrose gradient. Shown here are the real time recording of OD254, starting at the monosome fraction of a representative experiment.
f Metabolic labeling of MDA-MB-231 shNC, shNC+Rapamycin at 10 nM for 24h, and shBCDIN3D cells for 30 min with Methionine, L-[35S]. The autoradiogram on the left was obtained from exposure on film of the Coomassie-stained gel on the right.
g Meta-analysis of ribosome profiling reads around the translation start site (TSS ± 1kb) of protein coding genes in MDA-MB-231 shNC, shNC+Rapamycin at 10 nM for 24h, and shBCDIN3D cells (See complete analysis in Fig. S3).
BCDIN3D-depleted cells show another hallmark of mTOR pathway inactivation, i.e. a reduction of global translation. This is based on results of three independent types of experiments used to evaluate protein synthesis. Firstly, polysome fractionation experiments showed that shBCDIN3D cells have lower levels of polysomes compared to shNC cells, very similar to cells treated for 24h with 10 nM of Rapamycin, which is an mTORC1 inhibitor [16–18] (Fig. 2e). Secondly, BCDIN3D-depleted cells exhibit lower 35S-Methionine incorporation into proteins after a short 30-min pulse with 35S-Methionine (Fig. 2f). Thirdly, we consistently obtained fewer ribosome-protected footprints in the shBCDIN3D compared to shNC ribosome profiling libraries, in a manner very similar to Rapamycin-treated sample (Fig. S3). As expected for a truly global process, and very similar to Rapamycin-treated cells, no specific defect was detected in shBCDIN3D cells by meta-analysis either at the start codon or internal codons by ribosome profiling, most probably due to normalization at several steps of ribosome profiling: (1) same quantity of library DNA used for each sample at the next generation sequencing step; and (2) normalization to reads from coding genes at the bioinformatic analysis step (Fig. 2g and Fig. S3). Taken together, our results indicate that BCDIN3D depletion inhibits global mRNA translation, either through changing the expression of genes involved in mTOR signaling pathway, or through a yet to be identified mechanism.
Fructose 1,6 Bisphosphate levels are elevated in BCDIN3D-depleted cells.
Given the association of the BCDIN3D locus with obesity and T2D, we hypothesized that BCDIN3D may also regulate metabolism of breast cancer cells. In order to test this hypothesis, we performed untargeted profiling of primary metabolism by Mass Spectrometry in MDA-MB-231 breast cancer cells control or depleted for BCDIN3D (Table S1). We used two different ways to knock-down BCDIN3D, either via siRNA to reveal the effect of short term BCDIN3D depletion, or via shRNA to uncover the effect of long term BCDIN3D knock-down. We performed 8 biological replicates, four using siRNA and four using shRNA, for a total of 16 samples (four of each siBCDIN3D, siNC, shBCDIN3D and shNC samples). As shown on Table S1, the majority of the 405 metabolites that we obtained spectra from, did not change in BCDIN3D knock-down compared to control cell (siBCDIN3D/siNC or shBCDIN3D/shNC ratio of ~1). Some metabolites changed ratio in siRNA vs shRNA condition, but these mostly corresponded to poorly detected metabolites with high sample-to-sample variability (Table S1). Interestingly, our analysis revealed two metabolites, one increased and one decreased in all biological replicates, regardless of the method of BCDIN3D depletion. The metabolite with decreased levels upon BCDIN3D depletion is N-acetylglutamate (Fig. 3a, p-value of 1.25×10−6), while the metabolite with increased levels is Fructose 1,6 Bisphosphate (F1,6-BP) (Fig. 3a, p-value of 0.01). Because of the genetic link between the BCDIN3D locus and T2D, we focused on F1,6-BP, which is an important intermediate of the glycolytic pathway. As seen on Fig. 3a, F1,6-BP is upregulated by ~ 1.6 fold in BCDIN3D-depleted compared to control cells, while other intermediates of the pathway do not significantly change.
Figure 3. BCDIN3D regulates the levels of F1,6-BP and glycolysis through ALDOC.
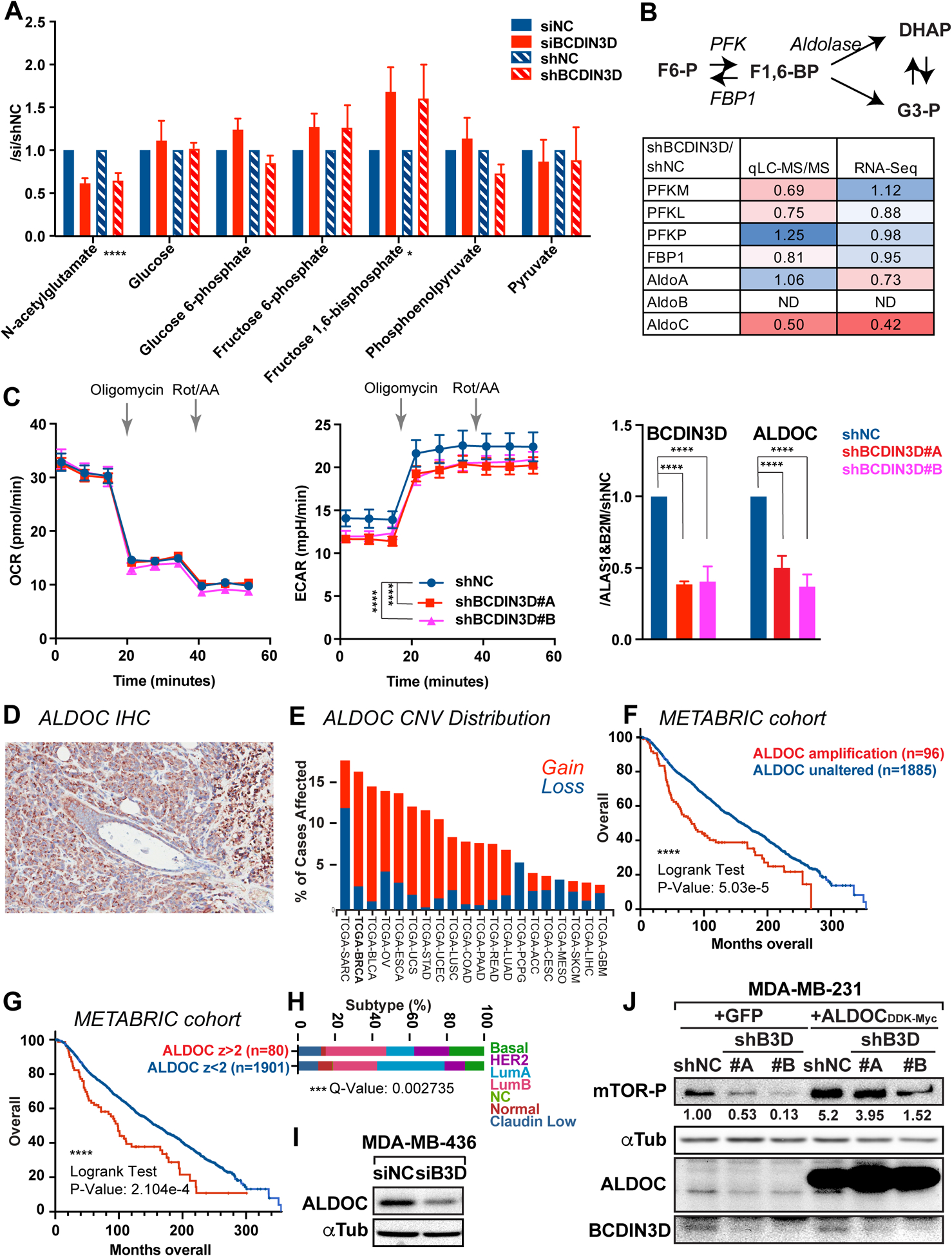
a N-acetylglutamate and glycolysis pathway intermediates results from the untargeted profiling of primary metabolism in MDA-MB-231 cells where BCDIN3D is depleted via siRNA or shRNA. Data is normalized to siNC and shNC controls (mean ± SEM, n=4). The asterisks indicate the significant p-values between the control and BCDIN3D knock-down samples (multiple t-test).
b Results of expression levels of glycolysis pathway enzymes from the proteomic analysis (LC-MS/MS) and RNA-Seq in MDA-MB-231 shBCDIN3D compared to shNC cells. The color scale goes from the deepest shade of red for the lowest value to the deepest shade of blue for the highest value.
c Left and Middle: Kinetic profile of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) measurement of MDA-MB-231 shNC and shBCDIN3D #A and #B cells using a Real Time ATP rate assay kit in a Seahorse XFp apparatus [mean ± SEM, n=4 (i.e. 2×2)]. Injection of oligomycin (1.5 μM final concentration) results in an inhibition of mitochondrial ATP synthesis that results in a decrease in OCR, and an increase of glycolytic ATP production that results in an increase in ECAR. Complete inhibition of mitochondrial respiration with rotenone plus antimycin A (0.5 μM final concentration) enables calculation of mitochondrial-associated acidification, which is negligible in these cells. Right: BCDIN3D and ALDOC gene expression from MDA-MB-231 shNC and shBCDIN3D #A and #B cells. Data is normalized to ALAS1, B2M and shNC (mean ± SEM, n=3). The asterisks indicate significant p-values (2 way Anova with multiple comparisons).
d IHC of ALDOC from a representative xenograft tumor of MDA-MB-231 shNC cells in Foxn1nu/nu mice.
e Copy number variation (Gain in red, Loss in blue) of the ALDOC gene in different cancers from The Cancer Genome Atlas (TCGA), showing that Breast Cancer (BRCA) displays the highest number of copy number gain.
f Overall breast cancer patient survival status as a function of time in weeks (up to 360 weeks) and separated in two groups: having a ALDOC amplification (n=96) or not (n=1806) (METABRIC cohort analyzed on cBioPortal for Cancer Genomics).
g Overall breast cancer patient survival status as a function of time in weeks (up to 360 weeks) and separated in two groups: having high ALDOC mRNA level relative to diploid samples, z-score>2 (n=80) or not (n=1901) (METABRIC cohort analyzed on cBioPortal for Cancer Genomics).
h Distribution of patients defined in g as a function of PAM50 + Claudin-low subtypes (Chi Squared Test, p value = 3.646e-4, q value = 2.735e-4).
i ALDOC expression is also reduced upon siRNA-mediated knock-down of BCDIN3D in MDA-MB-436 basal B breast cancer cells.
j Overexpression of ALDOC increases mTOR signaling in MDA-MB-231 cells. Protein extracts from MDA-MB-231 shNC, shBCDIN3D#A-B cell lines stably expressing GFP or ALDOC were analyzed by western blot with the indicated antibodies. The numbers beneath the anti mTOR-P western blot corresponds to the mTOR-P signal normalized to αTubulin signal from the same western blot, and to shNC+GFP.
BCDIN3D regulates the expression of ALDOC in breast cancer cells.
To establish how BCDIN3D regulates the levels of F1,6-BP, we extracted from our proteomic and transcriptomic data in MDA-MB-231 shNC and shBCDIN3D cells (Tables S2–S3) the information relevant to enzymes involved in the glycolytic pathway (Fig. 3b). This survey clearly pointed to ALDOC as being down-regulated at both the RNA and protein levels in BCDIN3D-depleted MDA-MB-231 cells, providing the most likely explanation for the higher levels of F1,6-BP in these cells. Indeed, ALDOC is one of the three enzymes (Aldolase A, B and C) that cleave the six-carbon F1,6-BP to give 2 three-carbon intermediates, glyceraldehyde-3-phosphate (G3-P) and dihydroxyacetone phosphate (DHAP) (Fig. 3b) in human cells. These lower levels of ALDOC appear to be sufficient to significantly reduce the glycolytic capacity of MDA-MB-231 cells depleted for BCDIN3D, as measured in a real-time ATP rate assay performed in a Seahorse flux analyzer (Fig. 3c).
ALDOC mRNA expression is highest in brain tissues (Fig. S4), however its expression is clearly detected in MDA-MB-231 breast cancer cells grown not only in vitro, but also in vivo (Fig. 3d). Interestingly, analysis of GTEx data showed that ALDOC is also highly expressed in Epstein-Barr virus (EBV) transformed lymphocytes (Fig. S4a), while TCGA data showed that ALDOC is expressed in breast cancer tissue from patients, albeit at a lower level than in the brain, and that breast cancer shows the highest ratio of ALDOC copy number gains (149/1072 cases, 13.4%) (Fig. 3e). Furthermore, analysis of overall survival of breast cancer patients from the METABRIC cohort [19] showed that ALDOC amplification is significantly associated with poorer survival (Fig. 3f, see Table S7 for detailed clinical attributes). Similarly, breast cancer patients with high levels of ALDOC mRNA expression also have poorer survival (Fig. 3g, see Table S8 for detailed clinical attributes). Analysis of ALDOC expression in 17 Triple Negative breast cancer cell lines showed an excellent correlation (R2=0.9045) between ALDOC mRNA and protein levels (Fig. S5), suggesting that these results based on ALDOC mRNA over-expression in breast cancer patients are likely meaningful.
Interestingly, ALDOC overexpression is more prevalent in more aggressive breast cancer subtypes, such as HER2 and Basal, and less frequent in less aggressive breast cancer subtypes, such as Luminal A (Fig. 3h and Table S8). To examine if BCDIN3D also regulates ALDOC in basal cell lines, we depleted BCDIN3D using siRNA in MDA-MB-436, a Basal B breast cancer cell line that expresses BCDIN3D at the protein level (Fig. S6). As shown on Fig. 3i, BCDIN3D knock-down led to a significant decrease of ALDOC protein levels in MDA-MB-436 cells, showing that regulation of ALDOC by BCDIN3D is a prevalent mechanism.
Given that BCDIN3D knock-down reduced mTOR signaling (Fig. 2 and Fig. S1a), we further investigated the part played by ALDOC in this observation. To this end, we performed a complementation experiment, in which we re-expressed ALDOC under CMV promoter control without its native 5’ and 3’UTRs in MDA-MB-231 control (shNC) and two independent BCDIN3D-depleted (shBCDIN3D) cell lines (Fig. 3j). Remarkably, ALDOC overexpression significantly increased mTOR signaling in the MDA-MB-231 shNC cells, showing that ALDOC expression is a limiting factor for mTOR signaling in MDA-MB-231 cells. In BCDIN3D knock-down cells, overexpression of ALDOC increases mTOR-S2448 phosphorylation at a level that is superior to the baseline (shNC+GFP control cells), but still inferior to the ALDOC over-expressing control cells (shNC+ALDOCDDK-MYC). Thus, BCDIN3D knock-down likely affects mTOR signaling in a multitude of ways, including through downregulating ALDOC levels.
BCDIN3D regulates the expression of ALDOC through a non-canonical mechanism involving the let-7 microRNA.
Given the potential importance of ALDOC in BC, we then sought to uncover how BCDIN3D regulates ALDOC expression in MDA-MB-231 cells. Analysis of RNA-Seq reads in MDA-MB-231 shNC and shBCDIN3D cells showed that the number of reads immediately downstream of the transcription start site of the ALDOC gene, corresponding to nascent unspliced mRNAs, are similar in the two conditions (Fig. 4a). This strongly suggests that BCDIN3D affects ALDOC levels post-transcriptionally. Because we previously showed that BCDIN3D can regulate specific miRNA levels, we decided to test whether BCDIN3D regulates ALDOC levels via miRNA-mediated decay. We first used miRanda to examine the 3’UTR of ALDOC for target sites of conserved miRNAs with good mirSVR scores (microRNA.org). This showed that miR-328, miR-153, miR-125a-3p, miR-7, let-7 family, or miR-31, have the potential to target the ALDOC 3’UTR (Fig. 4a). Among these miRNAs, let-7 expression is largely dominant in MDA-MB-231 cells (Fig. 4b) in both the miRNome [20] and our dataset [2], thus being the most likely to have an effect on ALDOC expression. Analysis of our small RNA-Seq dataset [2] with a microRNA-Seq pipeline showed that BCDIN3D depletion does not significantly affect the levels of miR-328, miR-153, miR-125a-3p, miR-7, or miR-31 microRNAs, but downregulates the levels of several let-7 isoforms, of which let-7f shows the highest net decrease. The reduced levels of mature let-7f in shBCDIN3D cells compared to shNC cells were confirmed by northern blot (see image, and its quantitation by ImageJ on Fig. 4d).
Figure 4. BCDIN3D stimulates ALDOC through a non-canonical mechanism involving let-7 microRNAs.
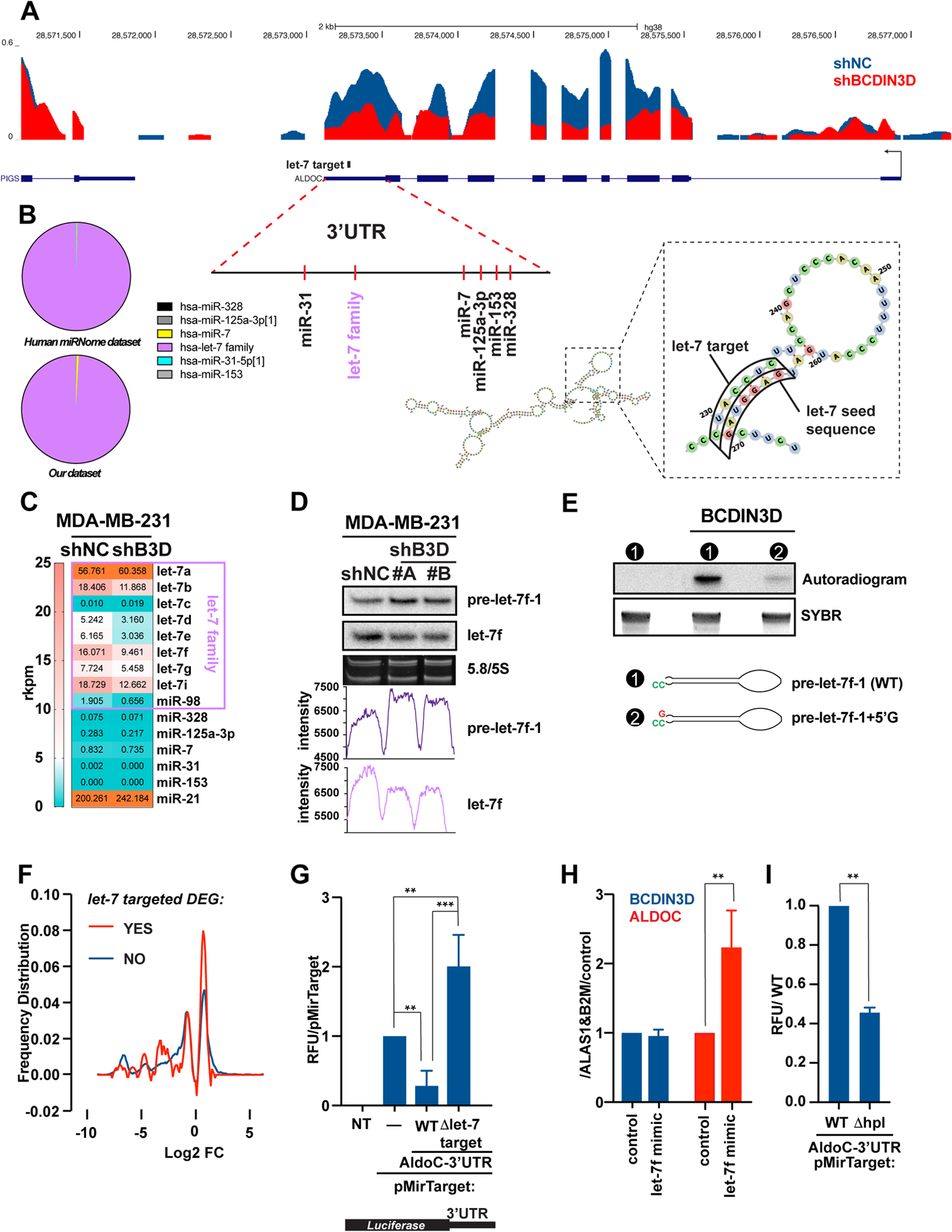
a UCSC genome browser traces of RNA-Seq in MDA-MB-231 shNC and shBCDIN3D cells at the ALDOC locus. The traces are overlaid, with the shNC trace (in blue) in the back position, and the shBCDIN3D trace (in red) in the forward position. The position of predicted miRNA target sites on the ALDOC 3’UTR is also indicated, together with the predicted structure of the ALDOC 3’UTR, overall or focused at the let-7 target site.
b Pie chart showing the relative levels of microRNAs that are predicted to target the ALDOC 3’UTR, in MDA-MB-231 cells from Human miRNome track hub [20] (top) and our dataset [2].
c Levels of microRNAs in reads per kilobase per million (rkpm) that are predicted to target the ALDOC 3’UTR, in MDA-MB-231 shNC and shBCDIN3D cells from our dataset [2] analyzed with a microRNA specific bioinformatic pipeline.
d 1 μg of RNA from MDA-MB-231 shNC and shBCDIN3D cells was analyzed by northern blot probed with the anti-let-7f probe, stripped, and re-probed with the anti-pre-let-7f-1 probe. 5/5.8 S panel shows the SYBR Gold stained gel at the level of the 5/5.8S ribosomal RNA (loading control). The bottom graphs show the raw intensity of the pre-let-7f-1 and let-7f signal quantified with ImageJ.
e in vitro RNA methyltransferase assay with recombinant BCDIN3D using radioactive [3H]-SAM as methyl group donor, and pre-let-7f-1-5’P precursor with either the WT predicted sequence, or containing an extra G nucleotide at the 5’ end. The bottom panel shows the SYBR stained gel that was used for the autoradiography in the top panel. See Fig. S8 for complete analysis.
f Distribution of differentially expressed genes (DEG) in MDA-MB-231 shBCDIN3D compared to shNC cells as a function of their logarithmic fold change, and separated into two groups: having a putative let-7 target site (red) or not (blue) by TargetScan. See Table S9 for complete information.
g MDA-MB-231 cells were transfected with pMirTarget (Origene) containing or not the 3’-UTR of ALDOC, either WT or having a deletion of the let-7 seed target (Δlet-7 target), fused to the Luciferase Reporter gene. 24h after transfection, the cells were lysed and the luciferase activity was measured in a luminometer. Data is normalized to pMirTarget transfected cells (mean ± SD, n=3). The asterisks indicate significant p-values (1 way Anova with multiple comparisons).
h BCDIN3D and ALDOC gene expression from MDA-MB-231 transfected with siNC (control) or let-7f mimic. Data is normalized to ALAS1, B2M and control (mean ± SD, n=2). The asterisks indicate significant p-values (Ratio paired t test).
i MDA-MB-231 cells were transfected with pMirTarget-ALDOC-3’-UTR, either WT or having a deletion of the hairpin loop (Δhpl) targeted by let-7, fused to the Luciferase Reporter gene and analyzed as in (G). The asterisks indicate significant p-values (Ratio paired t test).
The mature let-7 levels are the final product of several consecutive post-transcriptional steps [21]. Firstly, Drosha processes the primary microRNA into a precursor microRNA hairpin with a 5’-monophosphate and a 3’-overhang. Secondly, Dicer processes the primary microRNA into a mature microRNA duplex by cleaving off the loop and generating another 5’-monophosphate end and a 3’-overhang. Finally, the mature microRNA is incorporated into Ago2 of the RNA interference effector complex (RISC), while the passenger microRNA is degraded. In the case of let-7f, there are two primary microRNA loci, pri-let-7f-1 on chromosome 9 and pri-let-7f-2 on chromosome X (Fig. S7). In our RNA-Seq data, neither of these two loci shows reduced RNA levels in shBCDIN3D compared to shNC cells (Fig. S7), strongly suggesting that BCDIN3D depletion affects let-7f levels post-transcriptionally. Interestingly, while the mature let-7f levels decreased, northern blot analysis showed that pre-let-7f-1 levels increased (Fig. 4d), suggesting that processing of pre-let-7f-1 into mature let-7f is less efficient in BCDIN3D depleted cells. This effect may be direct as recombinant BCDIN3D is able to methylate pre-let-7f-1 in vitro with exquisite specificity for the predicted precursor microRNA structure with a 2 nt 3’ overhang (Fig. 4e and Fig. S8–S9).
As shown on Fig. 4f, the lower levels of let-7f have the expected effect on gene expression in MDA-MB-231 shBCDIN3D compared to shNC cells, i.e. differentially expressed genes that have validated or putative let-7 target sites are enriched in upregulated genes compared to genes that are not predicted to be let-7 targets [↓ let-7f ⇒ ↑ let-7 target genes] (See Table S9 for a complete list of these genes). However, in the case of ALDOC, we observed the opposite of what is expected for microRNA-mediated regulation, i.e. both let-7f and its putative target, ALDOC, are down-regulated in shBCDIN3D compared to shNC cells. Three subsequent experiments led us to hypothesize a mechanism by which let-7 may upregulate ALDOC stability, instead of decreasing it (as expected for a microRNA). Firstly, luciferase reporter expression from a pMirTarget vector is highly decreased when the luciferase mRNA contains the ALDOC 3’UTR (Fig. 4g), showing that the ALDOC 3’UTR negatively regulates mRNA stability. When the let-7 seed target sequence is deleted, the ALDOC 3’-UTR loses its inhibitory effect on mRNA stability, as expected for microRNA-mediated regulation (Fig. 4g). However, deletion of the let-7 seed target not only relieves repression, but also stimulates mRNA stability approximately two-fold over the control 3’UTR (Fig. 4g). This result shows that the let-7 seed target sequence constitutes a key element in the ALDOC 3’UTR, whose deletion enhances mRNA stability. Secondly, a similar stabilizing effect is observed when a let-7f mimic is transfected into MDA-MB-231 cells (Fig. 4h), confirming that let-7 can have a stabilizing effect on the ALDOC 3’UTR, as suggested by the observed effect of BCDIN3D depletion on both ALDOC and let-7f levels. An observation that reconciliates both these seemingly contradictory results is that the let-7 seed target sequence on the ALDOC 3’UTR is predicted to form a hairpin by Watson-Crick base-pairing with an adjacent sequence that is identical to the let-7 seed (Fig. 4a). Given that the same effect is obtained with deleting the let-7 seed target sequence, or the let-7f mimic, we hypothesized that the sequence that base-pairs with the let-7 seed target sequence binds to an ALDOC mRNA stabilizing factor. In agreement with this hypothesis, deletion of the entire hairpin reduced reporter luciferase activity even further compared to the WT ALDOC-3’UTR sequence (Fig. 4i), suggesting that the opening of the RNA hairpin by base-pairing with the let-7 microRNA recruits an ALDOC mRNA stabilizing factor.
Other let-7 microRNA processing regulators stimulate the expression of ALDOC.
Our findings also raised the possibility that other let-7 microRNA processing regulators control the levels of ALDOC. We first focused our attention on SRSF3. SRSF3 or SRp20 is a 20 KDa protein of the Serine Arginine SR family of proteins that stimulates the processing of CNNC pri-miRNAs into pre-miRNAs by the Microprocessor complex [22, 23]. Pri-let-7f-1 contains a CNNC motif starting exactly at 17-nt downstream of the Drosha cleavage site, and its processing should be stimulated by SRSF3. Consistent with the predicted role of SRSF3, the levels of mature let-7f were significantly decreased in siSRSF3 depleted cells (Fig. S10a). Remarkably, knock-down of SRSF3 in MDA-MB-231 cells is accompanied by a four-fold decrease of ALDOC mRNA levels (Fig. S10a). This effect is mediated by let-7, as transfection of a let-7f mimic is dominant over siSRSF3, i.e. rescues the levels of ALDOC mRNA (Fig. S10a). Finally, the effect of BCDIN3D down-regulation is epistatic to SRSF3 (Fig. S10b), in accordance with the fact that BCDIN3D most likely regulates let-7 processing downstream of primary microRNA processing (Figure 4d–e and [3]).
We then shifted our attention downstream of SRSF3, on TUT4 and TUT7, which either mono-uridylate let-7 precursors to stimulate their processing through Dicer, or poly-uridylate them after stimulation by LIN28 or defective pre-miRNA structure to lead to their decay [24–28]. In MDA-MB-231 cells, double siRNA depletion of TUT4 and TUT7 (siTUT4/7) decreases ALDOC levels by three to four fold, and this effect is additive with BCDIN3D knock-down (Fig. S10c). Given that MDA-MB-231 cells do not express LIN28A or B, our results showing that mature let-7f levels do not significantly change in siTUT4/7-treated cells are as expected (Fig. S10c) [29]. Moreover, these results do not necessarily mean that TUT4 and TUT7 stimulate the expression of ALDOC in a let-7 independent manner, as the let-7f or other let-7 isoforms produced after TUT4 and TUT7 depletion may not be functional (See Discussion). Taken together, our data clearly shows that BCDIN3D together with other let-7 microRNA pathway regulators tightly control ALDOC levels in breast cancer cells.
Discussion
T2D and specific cancers, including breast cancer, share many risk factors, however the molecular mechanisms underlying this link are not always well understood [30]. Because the BCDIN3D RNA methyltransferase has been linked to both breast cancer [3, 7–9] and T2D [11–13], we decided to use a combination of transcriptomic, proteomic and metabolomic analyses to identify metabolic pathways dependent on BCDIN3D in breast cancer cells. We chose the MDA-MB-231 cell line because it is a well-established model for invasive and triple negative breast cancer in which BCDIN3D depletion does not affect cellular proliferation in vitro. Our analyses showed that BCDIN3D depletion deeply affects both the transcriptome and global protein translation of MDA-MB-231 breast cancer cells (Fig. 2), inducing gene expression changes that are well in line with the observed reduced tumor formation in BCDIN3D knocked-down cells (Fig. 1).
When RNA-Seq and proteomic analyses were intersected with unbiased metabolomic profiling, a defect in the glycolysis pathway emerged in BCDIN3D-depleted cells. More specifically, BCDIN3D knock-down in MDA-MB-231 cells downregulates the expression of ALDOC that cleaves Fructose 1,6 Bisphosphate, the last six-carbon glycolytic intermediate. Interestingly, two aldolases are expressed in MDA-MB-231 cells, ALDOA and ALDOC, but BCDIN3D depletion only affects ALDOC (Fig. 3b), and this seems to be sufficient to upregulate the levels of Fructose 1,6 Bisphosphate (Fig. 3a), and reduce glycolytic capacity (Fig. 3c). ALDOC is normally expressed in the brain, but is also expressed in tumors, including breast cancer. Indeed, TCGA data showed that 13.4 % of breast cancers contain ALDOC copy number gain (Fig. 3e). Furthermore, ALDOC amplification or high mRNA expression is significantly associated with poorer breast cancer patient survival (Fig. 3f–h) and is more prevalent in the more aggressive HER2 and basal cancers (Tables S7–S8). Furthermore, we observed a stimulatory effect of ALDOC overexpression on mTOR signaling (Fig. 3j) in MDA-MB-231 cells. As recently shown by the Lin lab, F1,6BP Aldolases regulate AMPK and mTOR signaling directly through complex Aldolase interactions with the vacuolar H+ v-ATPase and Transient Receptor Potential V (TRPV) channels that are sensitive to F1,6BP levels bound to Aldolase. Aldolase absence mimics a F1,6BP-free Aldolase state, allowing for formation of the AXIN-based lysosomal complex, which leads to activation of AMPK and inactivation of mTOR [31–33]. Thus, it is possible that breast cancers with ALDOC amplification or overexpression may be sensitive to mTOR inhibitors. Altogether, these findings highlight the importance of deciphering the role of ALDOC in breast, as well as other cancers. Indeed, reports about other enzymes of the glycolysis pathway being directly involved in cancer are emerging. For example, the Simon lab recently showed that loss of fructose-1,6-bisphosphatase 2 (FBP2), which functions on the opposite sense of Aldolases to promote glyco- and gluconeogenesis, is a common metabolic feature of soft tissue sarcomas [34].
We focused on F1,6BP and glycolysis because of the genetic link of the BCDIN3D gene locus with T2D, but the significant reduction in the levels of N-acetylglutamate (NAG) observed in shBCDIN3D cells (Fig. 3a) is intriguing. NAG is synthesized from acetyl-CoA and glutamate by N-acetylglutamate Synthase (NAGS) in the mitochondrion to regulate the urea cycle [35]. NAG’s primary role is to allosterically activate Carbamoyl Phosphate Synthetase (CPS I), and provide short term regulation of flux through the urea cycle. The levels of glutamate and N-acetylated amino-acids other than NAG, as well as NAGS, are not changed in BCDIN3D knocked-down cells (Table S1–S3), suggesting that the lower NAG levels are not due to glutamate, acetyl-CoA, or NAGS availability. However, NAG levels may be responding to reduced flux through the urea cycle, which itself could be a direct result of lowered protein turnover to compensate for the global inhibition of protein translation in shBCDIN3D cells (Fig. 2e–g and Fig. S3).
Coming back to ALDOC, its expression in MDA-MB-231 breast cancer cells is controlled via a non-canonical mechanism involving the crucial let-7 microRNA family [36]. Indeed, the ALDOC 3’UTR contains a sequence predicted to form a hairpin loop where both arms exactly correspond to the let-7 seed sequence and its target (Fig. 4a). Deletion of the entire hairpin loop reduced reporter luciferase activity compared to the WT ALDOC-3’UTR sequence (Fig. 4i), suggesting that a sequence within this hairpin loop has an activating role overall. However, deletion of just the let-7 target sequence on one of the hairpin arm sequences, or transfection of a let-7f mimic, both de-repressed ALDOC expression (Fig. 4g–h). A plausible scenario that accounts for all these three results is that opening of the hairpin by binding of the let-7 microRNA may lead to more efficient recruitment of an ALDOC mRNA stabilizing factor. In line with this, both SRSF3 and BCDIN3D, whose knock-downs decrease let-7f levels, also decrease ALDOC expression (Fig. 4c–d and Fig. S10a–b). ALDOC expression additionally depends on two other well-established let-7 microRNA processing regulators, the terminal uridyl transferases TUT4 and TUT7. Their double knock-down decreases ALDOC levels and has an additive effect with BCDIN3D depletion (Fig. S10c). Even though let-7f levels do not decrease after TUT4 and TUT7 depletion, ALDOC regulation by TUT4 and TUT7 is likely to be let-7 dependent. As mentioned above, let-7 microRNAs produced after TUT4 and TUT7 depletion may not be competent for stimulating ALDOC expression. In fact, TUT4 and TUT7 were shown to uridylate mature let-7 miRNAs in vitro, and their depletion in HeLa cells, which like MDA-MB-231 cells, do not express LIN28, causes the selective loss of 3’ mono-uridylation and a concomitant increase in non-templated 3’ mono-adenylation of let-7 microRNAs [29]. These changes in the 3’end sequence of let-7 microRNAs may be sufficient to disrupt interaction with the ALDOC 3’UTR, as previously proposed for other miRNA/mRNA pairs [37, 38] and explain the pronounced effect of the double TUT4/7 knock-down on ALDOC.
We showed that BCDIN3D can methylate the pre-let-7f-1 precursor microRNA in vitro with exquisite specificity for a pre-miRNA structure with a 2 nt overhang (Fig. 4e and Fig. S8–S9) that is the best substrate for Dicer processing [21]. Indeed, this methylation activity is significantly reduced by a single addition of a 5’G residue that could result from the improper Drosha processing of the primary microRNA, or the non-templated addition of G residues - such as in the case of tRNAHis maturation by THG1L [2]. Remarkably, BCDIN3D depletion led to a concomitant increase of pre-let-7f-1 precursor and reduced mature let-7f microRNA levels, suggesting that BCDIN3D is actually required for efficient let-7f Dicer processing in cells. Given that BCDIN3D interacts with Dicer in an RNaseA-dependent manner [3], it is possible that BCDIN3D interacts with properly structured let-7 precursors to facilitate their transition from Drosha to Dicer. The validation of this hypothesis will require the development of efficient pre-miRNA sequencing methods, which to this day remain the holy grail of the microRNA processing field.
Finally, while we discovered that ALDOC expression is controlled by let-7 and several let-7 microRNA processing modulators in breast cancer cells, it will also be important to explore this mechanism in the brain, where ALDOC expression and its impact on glycolysis may be strongest. This mechanism has the potential to at least partially explain the association of the BCDIN3D locus with obesity and type II diabetes. By regulating ALDOC in the brain, let-7, BCDIN3D, and other let-7 microRNA processing regulators may directly modulate glycolysis, and thus impact brain function, and/or body weight and T2D through the brain centered glucoregulatory system [39]. Our findings may have translational relevance, given that BCDIN3D, TUT4 and TUT7 are druggable enzymes [40–42].
Materials and Methods
NGS data are available on GEO (GSE158669). Detailed information about cell lines, xenograft assays, (small)RNA-Seq, western blots, polysome fractionation, metabolic labeling, ribosome profiling, metabolomics, proteomics, immunofluorescence, immunohistochemistry, RNA analysis, Seahorse analysis, meta-analysis of public databases, RNA methyltransferase assays are provided in Supplemental Information.
Supplementary Material
Acknowledgments
BX was supported by NIH (R01-GM127802, DOD-CDMRP-BCRP (W81XWH-16-1-0352), Welch Foundation (F1859), STORM Therapeutics, and start-up funds. CVDB was partly supported by CPRIT (RR160093). This study made use of the MD Anderson Science Park facilities supported by CPRIT Core Facility Support Grants RP120348 and RP170002 and P30 CA16672 DHHS/NCI Cancer Center Support Grant (CCSG).
Footnotes
Conflict of Interest Statement
The authors declare that they have no conflict of interest.
References:
- 1.Shelton SB, Reinsborough C, Xhemalce B. Who Watches the Watchmen: Roles of RNA Modifications in the RNA Interference Pathway. PLoS Genet (Review) 2016; 12: e1006139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reinsborough CW, Ipas H, Abell NS, Nottingham RM, Yao J, Devanathan SK et al. BCDIN3D regulates tRNAHis 3’ fragment processing. PLoS Genet 2019; 15: e1008273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xhemalce B, Robson SC, Kouzarides T. Human RNA methyltransferase BCDIN3D regulates microRNA processing. Cell 2012; 151: 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu W, Hanes SD. Identification of drosophila bicoid-interacting proteins using a custom two-hybrid selection. Gene 2000; 245: 329–339. [DOI] [PubMed] [Google Scholar]
- 5.Zhu L, Liao SE, Ai Y, Fukunaga R. RNA methyltransferase BCDIN3D is crucial for female fertility and miRNA and mRNA profiles in Drosophila ovaries. PLoS One 2019; 14: e0217603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez A, Yamashita S, Nagaike T, Sakaguchi Y, Suzuki T, Tomita K. Human BCDIN3D monomethylates cytoplasmic histidine transfer RNA. Nucleic Acids Res 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med 2007; 356: 217–226. [DOI] [PubMed] [Google Scholar]
- 8.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012; 486: 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yao L, Chi Y, Hu X, Li S, Qiao F, Wu J et al. Elevated expression of RNA methyltransferase BCDIN3D predicts poor prognosis in breast cancer. Oncotarget 2016; 7: 53895–53902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011; 121: 2750–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berndt SI, Gustafsson S, Magi R, Ganna A, Wheeler E, Feitosa MF et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat Genet 2013; 45: 501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thorleifsson G, Walters GB, Gudbjartsson DF, Steinthorsdottir V, Sulem P, Helgadottir A et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet 2009; 41: 18–24. [DOI] [PubMed] [Google Scholar]
- 13.Walley AJ, Asher JE, Froguel P. The genetic contribution to non-syndromic human obesity. Nat Rev Genet 2009; 10: 431–442. [DOI] [PubMed] [Google Scholar]
- 14.Jenkins DE, Hornig YS, Oei Y, Dusich J, Purchio T. Bioluminescent human breast cancer cell lines that permit rapid and sensitive in vivo detection of mammary tumors and multiple metastases in immune deficient mice. Breast Cancer Res 2005; 7: R444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A 2004; 101: 13489–13494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 2004; 6: 1122–1128. [DOI] [PubMed] [Google Scholar]
- 17.Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994; 369: 756–758. [DOI] [PubMed] [Google Scholar]
- 18.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994; 78: 35–43. [DOI] [PubMed] [Google Scholar]
- 19.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 2016; 7:11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCall MN, Kim MS, Adil M, Patil AH, Lu Y, Mitchell CJ et al. Toward the human cellular microRNAome. Genome Res 2017; 27: 1769–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartel DP. Metazoan MicroRNAs. Cell 2018; 173: 20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Auyeung VC, Ulitsky I, McGeary SE, Bartel DP. Beyond secondary structure: primary-sequence determinants license pri-miRNA hairpins for processing. Cell 2013; 152: 844–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim K, Nguyen TD, Li S, Nguyen TA. SRSF3 recruits DROSHA to the basal junction of primary microRNAs. RNA 2018; 24: 892–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faehnle CR, Walleshauser J, Joshua-Tor L. Mechanism of Dis3l2 substrate recognition in the Lin28-let-7 pathway. Nature 2014; 514: 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hagan JP, Piskounova E, Gregory RI. Lin28 recruits the TUTase Zcchc11 to inhibit let-7 maturation in mouse embryonic stem cells. Nat Struct Mol Biol 2009; 16: 1021–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heo I, Ha M, Lim J, Yoon MJ, Park JE, Kwon SC et al. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell 2012; 151: 521–532. [DOI] [PubMed] [Google Scholar]
- 27.Heo I, Joo C, Kim YK, Ha M, Yoon MJ, Cho J et al. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell 2009; 138: 696–708. [DOI] [PubMed] [Google Scholar]
- 28.Kim B, Ha M, Loeff L, Chang H, Simanshu DK, Li S et al. TUT7 controls the fate of precursor microRNAs by using three different uridylation mechanisms. EMBO J (Research Support, Non-U.S. Gov’t) 2015; 34: 1801–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thornton JE, Du P, Jing L, Sjekloca L, Lin S, Grossi E et al. Selective microRNA uridylation by Zcchc6 (TUT7) and Zcchc11 (TUT4). Nucleic Acids Res 2014; 42: 11777–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA et al. Diabetes and cancer: a consensus report. Diabetes Care 2010; 33: 1674–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li M, Zhang CS, Zong Y, Feng JW, Ma T, Hu M et al. Transient Receptor Potential V Channels Are Essential for Glucose Sensing by Aldolase and AMPK. Cell Metab 2019; 30: 508–524.e512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin SC. Aldolase deployed for surveilling glucose. Nat Rev Mol Cell Biol 2020; 21: 714. [DOI] [PubMed] [Google Scholar]
- 33.Zhang CS, Hawley SA, Zong Y, Li M, Wang Z, Gray A et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017; 548: 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huangyang P, Li F, Lee P, Nissim I, Weljie AM, Mancuso A et al. Fructose-1,6-Bisphosphatase 2 Inhibits Sarcoma Progression by Restraining Mitochondrial Biogenesis. Cell Metab 2020; 31: 174–188.e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi D, Allewell NM, Tuchman M. The N-Acetylglutamate Synthase Family: Structures, Function and Mechanisms. Int J Mol Sci 2015; 16: 13004–13022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol 2008; 18: 505–516. [DOI] [PubMed] [Google Scholar]
- 37.Jones MR, Blahna MT, Kozlowski E, Matsuura KY, Ferrari JD, Morris SA et al. Zcchc11 uridylates mature miRNAs to enhance neonatal IGF-1 expression, growth, and survival. PLoS Genet 2012; 8: e1003105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones MR, Quinton LJ, Blahna MT, Neilson JR, Fu S, Ivanov AR et al. Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat Cell Biol 2009; 11: 1157–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwartz MW, Seeley RJ, Tschop MH, Woods SC, Morton GJ, Myers MG et al. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature 2013; 503: 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blazer LL, Li F, Kennedy S, Zheng YG, Arrowsmith CH, Vedadi M. A Suite of Biochemical Assays for Screening RNA Methyltransferase BCDIN3D. SLAS Discov 2017; 22: 32–39. [DOI] [PubMed] [Google Scholar]
- 41.Lin S, Gregory RI. Identification of small molecule inhibitors of Zcchc11 TUTase activity. RNA Biol 2015; 12: 792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schapira M Structural Chemistry of Human RNA Methyltransferases. ACS Chem Biol 2016; 11: 575–582. [DOI] [PubMed] [Google Scholar]
- 43.Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 2012; 7: 1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Popa A, Lebrigand K, Paquet A, Nottet N, Robbe-Sermesant K, Waldmann R et al. RiboProfiling: a Bioconductor package for standard Ribo-seq pipeline processing. 1000Res 2016; 5:1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 2014; 13: 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.