

Abstract
Background:
Opioid abuse poses significant risk to individuals in the United States and epigenetic changes are a leading potential biomarker of opioid abuse. Current evidence, however, is mostly limited to candidate gene analysis in whole blood. To clarify the association between opioid abuse and DNA methylation, we conducted an epigenome-wide analysis of DNA methylation in brain samples of individuals who died from acute opioid intoxication and group-matched controls.
Methods:
Tissue samples were extracted from the dorsolateral prefrontal cortex of 153 deceased individuals (Mage = 35.42; 62% male; 77% European ancestry). The study included 72 opioid samples, 53 psychiatric controls, and 28 normal controls. The epigenome-wide analysis was implemented using the Illumina MethylationEPIC BeadChip; analyses adjusted for sociodemographic characteristics, negative control principal components, ancestry principal components, cellular composition, and surrogate variables. Horvath’s epigenetic age and Levine’s PhenoAge were calculated, and gene set enrichment analyses were performed.
Results:
Although no CpG sites survived false-discovery rate correction for multiple testing, 13 sites surpassed a relaxed significance threshold (p < 1.0 x 10−5). One of these sites was located within Netrin-1, a gene implicated in kappa opioid receptor activity. There was an association between opioid use and accelerated PhenoAge (b = 2.24, se = 1.11, p = .045). Gene set enrichment analyses revealed enrichment of differential methylation in GO and KEGG pathways broadly related to substance use.
Conclusions:
Netrin-1 may be associated with opioid overdose, and future research with larger samples across stages of opioid use will elucidate the complex genomics of opioid abuse.
Keywords: Epigenetic, DNA Methylation, Opioid, Brain, Prefrontal Cortex
1. Introduction
Opioid use continues to pose significant risk to individuals throughout the globe, especially in the United States. Data from population-based epidemiological surveys from 2002-2016 in the United States revealed that approximately 30% of individuals who started heroin use met DSM-IV criteria for opioid dependence within one year of initiation (Santiago Rivera et al., 2018). In addition, the most recent estimates from the Global Burden of Diseases, Injuries, and Risk Factors Study revealed that the global prevalence of opioid dependence was 510 people per population of 100,000, and the United States had the highest prevalence rate among all countries (1,347 persons per 100,000 population) (Degenhardt et al., 2019). These data are alarming given the preponderance of evidence demonstrating robust associations between opioid dependence and harmful physical and psychosocial outcomes such as decreased quality of life, contact with the criminal justice system, increased risk for HIV (among injection drug users), and fatal and non-fatal overdoses (Degenhardt et al., 2019). Given the individual and societal burden of opioid dependence, it is necessary to characterize the biological pathways to dependence. Doing so will aid in identification of specific pathways that can be targeted for treatment and intervention.
Epigenetic changes have emerged as a leading potential biological marker of drug dependence given their implications for transcription regulation and cellular reprogramming (Jaenisch and Bird, 2003). Among these changes, DNA methylation (DNAm) at cytosineguanine (CpG) sites has received the most attention in substance use research (Vanyukov and Tarter, 2019). Studies in animal models provide robust evidence that substance abuse causes changes in gene expression through changes in DNAm (Nestler, 2014). Baker-Andersen et al. (Baker-Andresen et al., 2015) conducted an epigenome-wide study of DNAm in the medial prefrontal cortex (mPFC) of cocaine-using mice, finding that cocaine use was associated with differential methylation in 29 regions of the mPFC and subsequent changes in gene expression. Moreover, enrichment analyses revealed that differentially methylated genes in these regions were associated with known memory and addiction pathways in the brain. Results from human studies yield similar findings (Nielsen et al., 2012). Hagerty et al. (Hagerty et al., 2016) examined epigenome-wide methylation patterns in buccal cells between alcohol use disorder (AUD) cases and controls, finding 561 hypomethylated CpG sites and 485 hypermethylated CpG sites among AUD cases compared to controls; A majority of these sites were located in genes involved in lipid metabolism, immune response, and inflammatory disease pathways. Additionally, in a second cohort of brain tissue samples, 432 of these sites (244 hypomethylated and 188 hypermethylated) were significantly correlated between buccal and brain samples, suggesting some overlap in methylation patterns across tissue type. Altogether, current evidence suggests that DNAm is a potential biomarker of substance use and has implications for broader biological functioning.
To date, epigenetic studies of opioid use have focused on candidate genes, namely OPRM1, which encodes for the μ-opioid receptor (Chorbov et al., 2011; Ebrahimi et al., 2018; Nielsen et al., 2009; Sandoval-Sierra et al., 2020). This receptor plays a role in the tolerance and physical dependence of opiates (Martini and Whistler, 2007). Candidate-epigenetic studies generally find hypermethylation of CpG sites in various regions of OPRM1, including promoter regions (Ebrahimi et al., 2018). Although these studies provide evidence for a potential pathway to opioid dependence in humans, they focus on a single gene, often in small sample sizes, increasing the likelihood of false positive results and limiting the detection of other genomic regions relevant to opioid use.
In the first epigenome-wide association study of DNAm of heroin users, Kozlenkov et al. (Kozlenkov et al., 2017) found three genome-wide significant differentially methylated sites in the brains of individuals who died from heroin intoxication (n = 37) compared to controls (n = 28) in tissue from the medial orbital frontal cortex (mOFC), a subregion of the mPFC that aids in goal-directed behavior and decision-making processes implicated in drug addiction (Gansler et al., 2011). Recently, Montalvo-Ortiz et al. (Montalvo-Ortiz et al., 2019) conducted a genome-wide analysis of DNAm levels in whole-blood samples of opioid-dependent women and matched controls. Results revealed three significantly hypomethylated CpG sites in the PARG, RERE, and CFAP77 genes, which are involved in chromatin remodeling, DNA binding, and cell survival and projection. Notably, PARG and RERE are highly expressed in multiple brain regions, and single nucleotide polymorphisms (SNPs) within RERE have been linked to psychiatric disorders including schizophrenia, attention deficit-hyperactivity disorder, major depressive disorder, and bipolar disorder (Smoller et al., 2013). Although neither of these studies detected differential methylation in the OPRM1 gene, they provide insight into DNAm patterns in separate tissue types (i.e., brain, blood) and elucidate the functional relevance of DNAm.
Although current evidence suggests that opioid use is associated with DNAm, several questions and issues remain. First, the majority of epigenetic studies focused on a single candidate gene, OPRM1, yet there are undoubtedly other potential genes and pathways through which opioid use contributes to dependence. Second, the majority of studies examined DNAm levels in blood or saliva, assuming that DNAm patterns in these tissues mirror levels in the brain. Although overlap in patterns of DNAm across tissues may occur, there is ample evidence suggesting that DNAm levels differ substantially by tissue type and brain region (Davies et al., 2012; Lister et al., 2013). The goal of the present study was to address these issues by assessing epigenome-wide DNAm in postmortem brain tissue from individuals who died of acute opioid intoxication and group-matched controls. The current study builds upon similar work by Kozlenkov et al. by assessing epigenome-wide methylation using the Illumina MethylationEPIC BeadChip, which contains over 400,00 more CpG sites than the Illumina HumanMethylation450 BeadChip, and other improvements such as the inclusion of CpG sites located in enhancers (Pidsley et al., 2016). This study also benefits from a larger sample and examination of a separate brain region, as prior work suggests that patterns of DNAm vary across regions of the brain (Ladd-Acosta et al., 2007). In sum, these epigenome-wide data allow for the detection of numerous CpG sites possibly related to opioid use and dependence, and the examination of brain tissue allows us to test whether methylation patterns in the brain mirror evidence from other tissues, further clarifying the functional relevance of DNAm in regions of the brain implicated in addiction (i.e., PFC).
2. Materials and Methods
2.1. Sample Description
Human postmortem brain samples were donated to the Lieber Institute for Brain Development from the Offices of the Chief Medical Examiner of the State of Maryland (MDH protocol #12-24) and of Western Michigan University Homer Stryker School of Medicine, Department of Pathology (WIRB protocol #1126332), and one brain sample was acquired via material transfer agreement from the NIMH (donated through the Office of the Chief Medical Examiner of the District of Columbia (protocol NIMH#90-M-0142), all with the informed consent of legal next-of-kin at the time of autopsy. The present study used tissue from the dorsolateral prefrontal cortex (dlPFC) of 160 deceased individuals (Mage = 35.15, SD = 9.42 years; 62% male; 78% European ancestry). Tables 1 and 2 provide detailed information on the study samples, which consisted of 73 individuals who died of acute opioid intoxication, 59 group-matched psychiatric controls, and 28 group-matched normal controls.
Table 1.
Sample Demographic Information
| Full Sample (n = 160) | Analytic Sample (n = 153) | |||
|---|---|---|---|---|
| Demographic Categories | n (%) | Mean (SD) | n (%) | Mean (SD) |
| Sex | ||||
| Female | 61 (38%) | - | 58 (38%) | - |
| Male | 99 (62%) | - | 95 (62%) | - |
| Race | ||||
| African American | 35 (21.9%) | - | 35 (23%) | - |
| European American | 124 (77.5%) | - | 118 (77%) | - |
| Multi-Racial | 1 (0.6%) | - | - | - |
| Diagnosis Type | ||||
| Normal Control | 28 (17.5%) | - | 28 (17.5%) | - |
| Opioid User | 73 (45.6%) | - | 72 (45.6%) | - |
| Psychiatric Control | 59 (36.9%) | - | 53 (36.9%) | - |
| Age of Death | - | 35.13 years (9.42) | - | 35.42 years (9.43) |
| Postmortem Interval | - | 27.35 hours (9.65) | - | 27.42 hours (9.60) |
Table 2.
Primary Psychiatric Diagnosis by Study Group (n = 153)
| Psychiatric Diagnosis | Psychiatric Controls | Opioid Users | Normal Controls |
|---|---|---|---|
| Attention-Deficit/Hyperactivity Disorder (ADHD) | 0 | 2 | - |
| Anxiety | 0 | 1 | - |
| Bipolar Disorder | 19 | 17 | - |
| Unspecified Bipolar Disorder | 0 | 1 | - |
| Unspecified Depressive Disorder | 1 | 6 | - |
| Major Depressive Disorder | 33 | 27 | - |
| Neurological Disorder | 0 | 1 | - |
| Obsessive Compulsive Disorder (OCD) | 0 | 2 | - |
| Eating Disorder | 0 | 1 | - |
| Posttraumatic Stress Disorder (PTSD) | 0 | 1 | - |
| Schizophrenia | 0 | 3 | - |
| Substance Use Disorder (SUD) | 0 | 10 | - |
| Total (153) | 53 | 72 | 28 |
Note. Two samples (one opioid user, one psychiatric control) were later removed due to mismatching predicted and observed sex, and five psychiatric controls were removed due to positive opioid tests in toxicology reports.
At the time of donation, a 36-item next-of-kin informant telephone screening was conducted to obtain medical, social, demographic, and psychiatric history. Macroscopic and microscopic neuropathological examinations were conducted on every case by a board-certified neuropathologist to exclude for neurological problems, neuritic pathology, or cerebrovascular accidents. Postmortem interval (PMI) was a calculation of hours between a donor’s time of death and the time of brain freezing. A retrospective clinical diagnostic review was conducted on every brain donor, consisting of the telephone screening, macroscopic and microscopic neuropathological examinations, autopsy and forensic investigative data, forensic toxicology data, extensive psychiatric treatment, substance abuse treatment, and/or medical record reviews, and whenever possible, family informant interviews.
All data were compiled into a comprehensive psychiatric narrative summary that was reviewed by two board-certified psychiatrists in order to arrive at lifetime DSM-5 psychiatric diagnoses (including substance use disorders/intoxication) and medical diagnoses. Non-psychiatric healthy controls were free from psychiatric and substance use diagnoses, and their toxicological data was negative for drugs of abuse. Every brain donor had forensic toxicological analysis, which typically covered ethanol and volatiles, opiates, cocaine/metabolites, amphetamines, and benzodiazepines. Some donors also received supplemental directed toxicological analysis using National Medical Services, Inc., including nicotine/cotinine testing, cannabis testing, and the expanded forensic panel in postmortem blood (or, in rare cases, in postmortem cerebellar tissue) in order to cover any substances not tested. The following substances were considered opioids: codeine, morphine, oxycodone, hydrocodone, oxymorphone, hydromorphone, methadone, fentanyl, 6-monoacetylmorphine, and tramadol. Subsequent epigenomics profiling of DNA derived from human postmortem brain tissue is exempt from institutional ethics committees as research conducted on postmortem tissue is not considered Human Subjects Research by the Department of Health and Human Services.
2.2. DNA Methylation Measurement and Preprocessing
Genomic DNA was extracted from 100mg of dlFPC tissue and isolated with the Qiagen DNeasy kit; bisulfite conversion of 600ng of genomic DNA was then performed with Zymo EZ methylation gold kit at the Johns Hopkins University Center for Inherited Disease Research. Bisulfite-treated DNA was then run on the Illumina Infinium MethylationEPIC BeadChip (Pidsley et al., 2016). Briefly, the EPIC BeadChip contains over 850,000 CpG sites, including >90% of the CpG sites included in the previous 450K BeadChip. Notable improvements in this array include the assessment of an additional ~400,000 CpG sites and inclusion of more than 350,000 CpG sites at regions identified as potential enhancers (Pidsley et al., 2016). Individual-level microarray data are available in the Gene Expression Omnibus (GEO Accession Number: GSE164822).
All quality control and inferential analyses were conducted in R version 3.6.1 (Team, 2018). The minfi package was used to process raw red and green channel intensity files into noob (normal-exponential out-of-band) preprocessed methylation beta values (Aryee et al., 2014). Specifically, we used the ‘preprocessNoob‘ function, which corrects for dye-bias in the raw intensities. Because cell type proportions can confound the association between DNAm and outcomes of interest, we estimated neuronal cell type proportions to control for cellular heterogeneity. Specifically, estimates of the percentage of neurons in each sample were calculated using the ‘estimateCellCounts()‘ function with the ‘compositeCellType = “DLPFC”‘ argument in the minfi R/Bioconductor package (Aryee et al., 2014), which uses the Houseman statistical method (Houseman et al., 2012). Samples were tested for low intensity (n = 0) and inconsistency between predicted and observed sex (n = 2; one opioid user, one psychiatric control) and those failing were removed for quality control. Probes with low methylation intensity also were excluded (n = 1,208) since these probes mostly consisted of background noise rather than true biological signal. Five additional psychiatric controls were dropped due to testing positive for an opioid in the toxicology report. The final analysis consisted of 153 samples and 864,883 probes. Principal components (PCs) from negative control probes were extracted to control for technical variations (Gagnon-Bartsch and Speed, 2012), and surrogate variable analysis (Leek and Storey, 2007) was conducted to account for unknown sources of heterogeneity and remove batch effects in the data.
2.3. Statistical Methods
2.3.1. Epigenome-wide association analysis.
Prior to epigenome-wide analyses, beta values were converted to M-values. Although beta values provide easier biological interpretation since they are on a scale of 0-1, reflecting percentage of methylation at each CpG site, the use of the log-transformed M-value provides a distribution that closer satisfies the assumption of normality in subsequent models, and the use of M-values usually leads to a better detection and true positive rate compared to the beta value (Du et al., 2010). We also collapsed the psychiatric and healthy control samples and created a covariate where individuals were coded as either having been (or not having been) diagnosed with a psychiatric diagnosis other than a substance use disorder (SUD). We then used limma (Ritchie et al., 2015) to run single-site association analyses by linear regression with opioid use status (yes/no) across the epigenome for the 864,883 CpG sites, adjusting for the following covariates: diagnosis of a psychiatric disorder (yes/no), age of death, sex, postmortem interval (PMI), cell composition (i.e., % positive neurons), four negative control PCs, the top four ancestry PCs, and 13 surrogate variables detected using sva in R. The four negative control PCs were reflective of background noise such as batch effects, and the 13 surrogate variables represented unmeasured confounding factors besides known confounders adjusted in the model. Ancestry PCs were estimated from the 59 SNP probes profiled on the EPIC array. Multiple testing correction was applied using the false discovery rate (FDR) procedure (Benjamini and Hochberg, 1995). This method controls the expected proportion of false positives among all statistical tests. It is less conservative than the commonly used Bonferroni correction, which controls for any expected false positive, but benefits from increased statistical power. Statistical significance was determined by an FDR adjusted p < .05 (unadjusted p < 1.0 x 5−8).
2.3.2. Epigenetic and phenotypic age.
In order to understand the cumulative epigenetic consequence of opioid abuse, we calculated Horvath’s epigenetic, or DNAm, age and Levine’s phenotypic age (a.k.a. PhenoAge) using the ENmix package (Xu et al., 2016). Briefly, Horvath’s DNAm age is an estimate of the cumulative effect of an epigenetic maintenance system and is strongly correlated with chronological age (Horvath, 2013). DNA methylation age is calculated using data from 353 CpG sites that are correlated with chronological age, irrespective of tissue or cell type. Similarly, Levine’s PhenoAge (Levine et al., 2018) is an estimate of accelerated aging, but is derived from biomarkers related to clinical phenotypes (e.g., physical functioning, all-cause mortality), and is thought to better reflect epigenetic aging in response to specific environmental exposures (e.g., opioid abuse). PhenoAge is calculated using 513 CpG sites (41 of which overlap with Horvath’s method) that are correlated with chronological age. Although developed using whole blood (as opposed to 51 different tissue and cell types in Horvath’s method), PhenoAge correlates strongly with chronological age across tissue and cell types.
In the present analysis, we first estimated DNAm age and PhenoAge, respectively. We then calculated accelerated DNAm age and PhenoAge by taking the difference between each individual’s DNAm age and PhenoAge and their age of death (i.e., positive values indicated accelerated aging). Since Horvath’s DNAm age is based upon CpG sites spanning across numerous tissue types (including brain tissue), we conducted an independent samples t-test to examine if DNAm age acceleration differed by opioid intoxication status. Although correlated with chronological age across most tissue and cell types, we used a multiple linear regression to examine the association between opioid intoxication status and PhenoAge acceleration, adjusting for cellular composition, since this estimate was originally developed in whole blood only. Significance was determined using p < .05 for both analyses.
2.3.3. Gene set enrichment analyses.
Gene set enrichment analyses (GSEA) were performed on probes with an FDR adjusted p-value < .05; however, if no probes survived FDR correction for multiple testing, GSEA was performed on all probes with an unadjusted p < .05. Analyses were conducted using the “methylgometh” function in the R package methylGSA (Ren and Kuan, 2019). This function calls the “gometh” function in missMethyl (Phipson et al., 2016), which adjusts for the number of CpG sites within each gene. We tested for enrichment of differential methylation across GO, KEGG, and Reactome pathways, selecting gene sets containing at least 25 genes and no more than 500 genes. Significant enrichment was determined using an FDR adjusted p-value < 0.05.
3. Results
3.1. Epigenome-Wide Association Analysis
First, we conducted epigenome-wide analysis to determine if there were significant differences in methylation at individual CpG sites between opioid and control samples. Figure 1 illustrates the quantile-quantile (QQ) plot of p-values for the association between DNAm and opioid intoxication. There was no evidence of inflation (λ = 1.02). As can be seen in Figure 2, no CpG sites survived FDR correction for multiple testing (red line; p < 5.0 x 10−8); however, 13 CpG sites surpassed a relaxedp-value threshold (blue line; p < 1.0 x 10−5). As can be seen in Table 3, which contains these top 13 CpG sites, six of the sites were hypomethylated and the other seven sites were hypermethylated in opioid samples. Among the CpG sites listed in Table 3, only cg24060527, located within Netrin-1 (NTN1), has published evidence demonstrating a potential link to opioid use. Specifically, evidence has linked Netrin-1 activity to stimulation of the kappa opioid receptor (Tsai et al., 2007, 2006), which is implicated in opioid dependence (Laurence Lalanne et al., 2014). Lastly, we examined the effect of the psychiatric diagnosis covariate, as the sample included psychiatric controls and some opioid samples had a primary psychiatric diagnosis other than a substance use disorder. There was no evidence of an association between psychiatric diagnosis and DNAm (all adj. p > .05).
Figure 1.
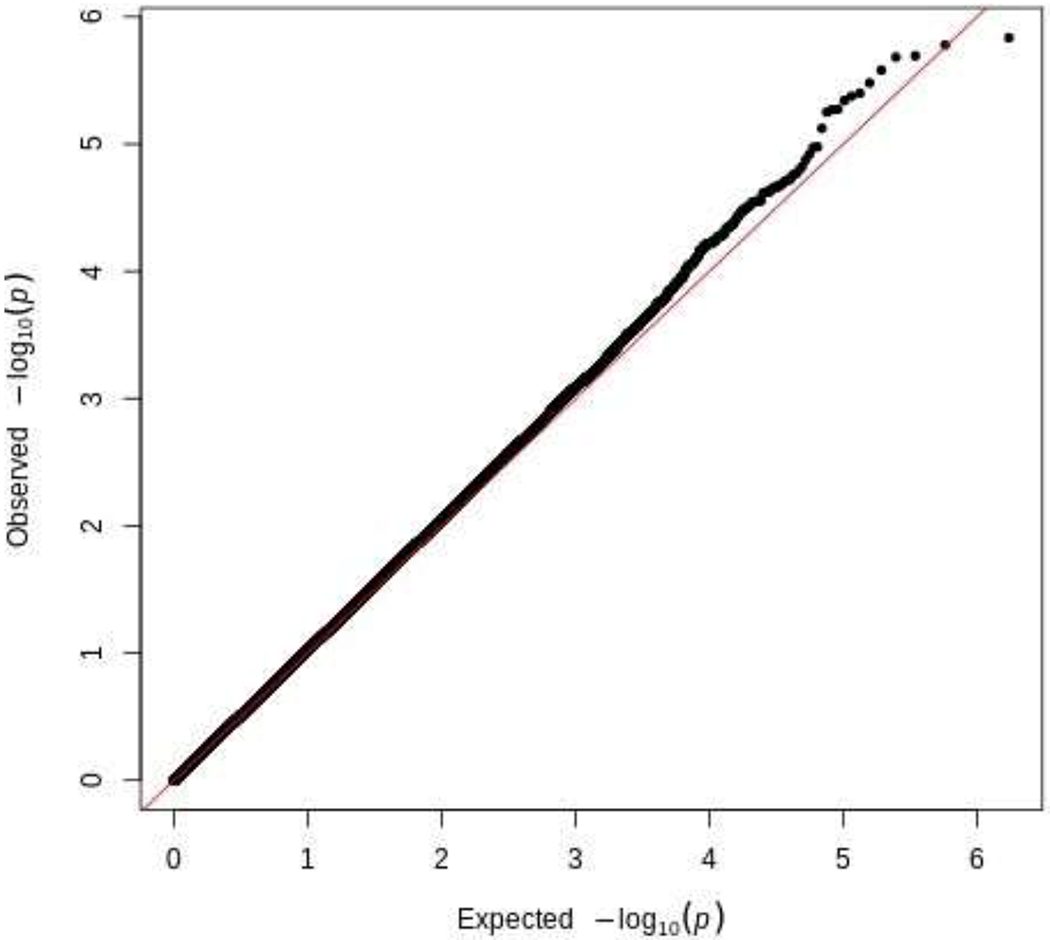
QQ Plot of p-values for the Association Between DNA Methylation and Opioid Use
Figure 2.
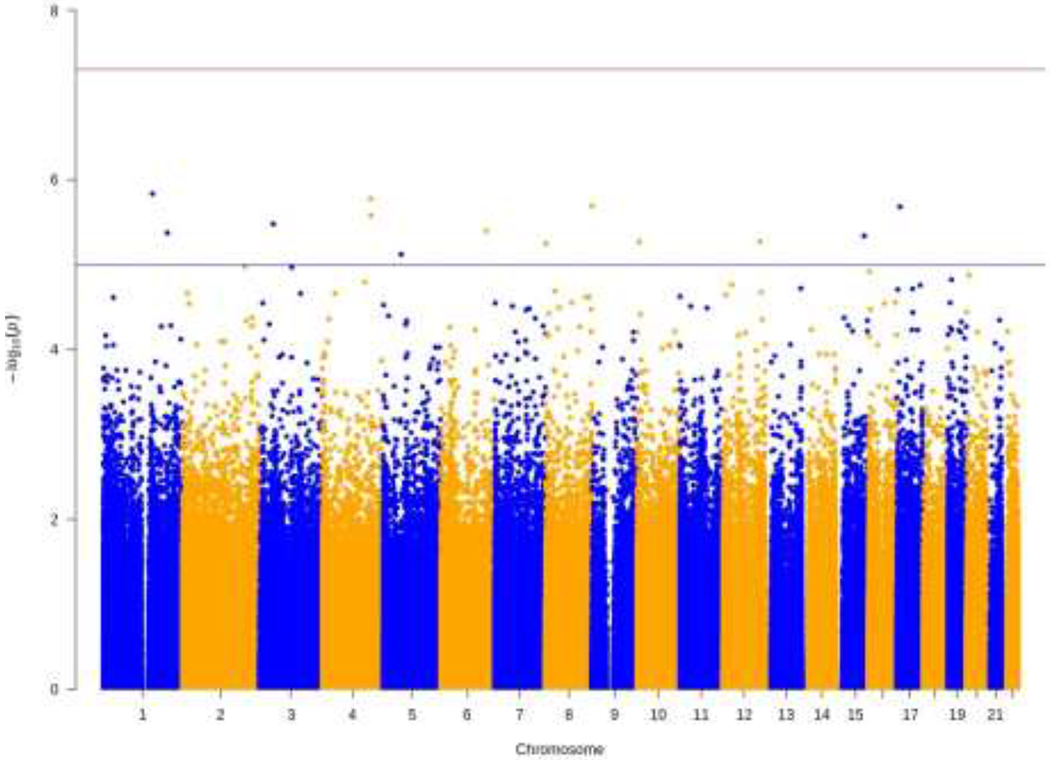
Manhattan Plot for Epigenome-Wide Association Analysis
Note. The red line indicates genome-wide significance (p < 5.0 x 10−8) and the blue line indicates suggestive significance (p < 1.0 x 10−5). Thirteen CpG sites reached suggestive significance.
Table 3.
Top Differentially Methylated CpG Sites
| CpG | UCSC gene symbol | Chr | Position | MMdiff | t-statistic | p-value | Adjusted p-value |
|---|---|---|---|---|---|---|---|
| cg01032200 | RUSC1; RUSC1-AS1 | 1 | 155290641 | −.332 | −5.053 | 1.46E-06 | 4.05E-01 |
| cg16385330 | LRBA; MAB21L2 | 4 | 151503878 | −.216 | −5.023 | 1.66E-06 | 4.05E-01 |
| cg03228209 | KIAA1688 | 8 | 145814162 | .109 | 4.977 | 2.03E-06 | 4.05E-01 |
| cg24060527 | NTN1 | 17 | 9066156 | .293 | 4.972 | 2.07E-06 | 4.05E-01 |
| cg03674718 | GATB | 4 | 152593043 | .140 | 4.917 | 2.63E-06 | 4.05E-01 |
| cg25084741 | -- | 3 | 43115622 | .143 | 4.863 | 3.31E-06 | 4.05E-01 |
| cg21026257 | PHACTR2 | 6 | 143998961 | .225 | 4.820 | 3.98E-06 | 4.05E-01 |
| cg25951285 | LGR6 | 1 | 202182175 | .110 | 4.807 | 4.21E-06 | 4.05E-01 |
| cg15257765 | MRPS11; MRPL46 | 15 | 89010459 | .152 | 4.788 | 4.55E-06 | 4.05E-01 |
| cg09935667 | C12orf49 | 12 | 117164627 | −.121 | −4.751 | 5.33E-06 | 4.05E-01 |
| cg03672997 | TAF3 | 10 | 7859623 | −.454 | −4.749 | 5.37E-06 | 4.05E-01 |
| cg10759972 | -- | 8 | 1971471 | −.202 | −4.738 | 5.61E-06 | 4.05E-01 |
| cg26506680 | -- | 5 | 56820679 | −0.090 | −4.668 | 7.53E-06 | 5.01E-01 |
Note. Chr = Chromosome. MMdiff = difference in mean methylation between cases and controls. Adjusted p-value refers to the Benjamini-Hochberg adjusted p-value.
3.2. Epigenetic and Phenotypic Age
Next, we tested the association between opioid use status and DNAm age and PhenoAge. Tests of bivariate correlations revealed that age of death was associated with DNAm age (r = .90, p < .001) and PhenoAge (r = .69, p < .001) ages. DNAm age and PhenoAge also were correlated (r = .69, p < .001). Results from an independent samples t-test did not reveal evidence of an association between opioid intoxication status and accelerated DNAm age, t(146.45) = .20, p = .84. There was evidence of an association between opioid intoxication status and accelerated PhenoAge (b = 2.24, se = 1.11, p = .045), adjusting for cell composition. Specifically, opioid samples were, on average, two years epigenetically older compared to control samples. To ensure results were not affected by combining psychiatric and health controls, we tested whether psychiatric and healthy controls differed in accelerated DNAm age and PhenoAge. Results did not provide evidence for an association between type of control sample and either accelerated DNAm age [t(67.02) = −1.09, p = .28)] or PhenoAge (b = 1.96, se = 1.56, p = .21).
3.3. Gene Set Enrichment Analyses
In order to test for case-control methylation differences at the biological system level, GSEA were conducted for all probes with unadjusted p-values < .05. This resulted in GSEA using 46,139 CpG sites. Regarding GO terms, 351 terms survived FDR correction for multiple testing. Among the top enriched pathways were “glutamatergic synapse” (cellular component; adj. p = 3.23 x 10−6) and “learning or memory” (biological process; adj. p = 6.99 x 10−3). The complete list of significant GO terms can be found in Supplementary Table 1. Since a large number of GO terms surpasses our significance threshold, we also plotted the results using REVIGO (Supek et al., 2011) to identify themes across the biological process, cellular component, and molecular function ontologies. These results can be found in Supplementary Figures 1–3, but no notable patterns or themes emerged.
Regarding KEGG pathways, 38 pathways survived FDR correction for multiple testing. As can be seen in Supplementary Table 2, “dopaminergic synapse” (adj. p = 3.46 x 10−2) was among the significant pathways, but no other significant pathways were notable. Lastly, we tested for enrichment in differential methylation among Reactome pathways. Twenty-two pathways survived FDR correction for multiple testing; however, no pathways were relevant to opioid use (see Supplementary Table 3).
4. Discussion
The present analysis builds upon emerging literature on epigenome-wide markers of opioid abuse (Kozlenkov et al., 2017; Montalvo-Ortiz et al., 2019) by examining DNAm in the brains of individuals who died of acute opioid intoxication compared to group-matched controls. Specifically, the present findings inform our understanding of potential epigenetic mechanisms through which opioid use may affect pathways within brain regions involved in addiction. We observed no single-locus significant results after FDR correction for multiple testing; however, a CpG site within NTN-1 surpassed a relaxed significance threshold and NTN-1 has been implicated in the stimulation of the kappa opioid receptor, which is linked to addiction and other psychiatric disorders (Laurence Lalanne et al, 2014). Accelerated methylation-age testing via the widely used Horvath clock did not reveal evidence of an association with opioid intoxication status; however, analyses of Levine’s PhenoAge revealed accelerated aging among opioid samples versus controls. Biological pathway analysis yielded several pathways involved in glutamatergic and dopaminergic synapse functioning, but the results were largely nondescript. Moreover, these were very broad pathways with limited specificity for opioid use disorder.
The detection of differential methylation of a CpG within NTN-1 was unexpected, as it has not been identified within prior epigenetic studies of opioid use, nor does it appear to be directly related to opioid use. Moreover, it has not been detected in prior genome-wide association studies (GWAS) of opioid use and dependence. For example, Polimanti et al. (Polimanti et al., 2020) recently conducted a GWAS among ~41,000 individuals that were either diagnosed with opioid dependence, had used opioids at least once in their lifetime, or had reported no lifetime use of opioids. The authors detected a variant, rs9291211, that was associated with lifetime exposure to opioids and regulates transcriptomic profiles of SLC30A9 and BEND4 in brain tissue. Interestingly, no CpG sites within either of these genes was among the top 10,000 probes in our analysis. Similarly, another recent GWAS by Zhou et al. (Zhou et al., 2020) of ~10,000 individuals revealed a functional variants in OPRM1 (rs1799971) associated with opioid use disorder. No CpG sites within OPRM1 were detected among the top 10,000 hits in the present analysis. It is possible that tissue type contributes to some of these disparate findings, although sample size likely plays a role as well. Additional analyses of postmortem brains are needed to validate the current findings.
The association detected between opioid use and older phenotypic age supports evidence that opioid use is a risk factor for age-related negative health outcomes ranging from physical and cognitive ailments, to mortality (Degenhardt et al., 2019). It was unexpected to not detect an association between opioid use status and accelerated Horvath epigenetic age, although Montalvo-Ortiz et al. (Montalvo-Ortiz et al., 2019) also failed to detect this association among a sample of women diagnosed with opioid dependence. Conversely, Kozlenkov et al. (Kozlenkov et al., 2017) found that heroin users were epigenetically younger compared to controls in their sample. Neither study assessed accelerated PhenoAge between opioid users and controls. It is possible that the divergent epigenetic age findings are due to differing samples (e.g., women only vs. men and women), examination of different tissues (whole blood vs. brain), or sampling from different brain regions, in the case of Kozlenkov et al. (Kozlenkov et al., 2017) (mOFC vs. dlPFC). Additional studies examining opioid dependence, and both accelerated epigenetic and phenotypic ages among larger samples in similar brain regions will clarify these findings.
The present study has several strengths. The primary strength of this study is the use of brain tissue in the dlPFC. This allows for testing of epigenetic markers in regions of the brain where genes of interest within addiction pathways are expressed. Second, thoughtful sampling of group-matched controls allowed us to account for potential confounders in the association between opioid use and DNAm. Third, this analysis adds to a limited literature on epigenome-wide associations between opioid use and DNAm. Although these studies have limited sample sizes and sample different tissues, each has detected CpG sites within unique genes that may have functional relevance to opioid dependence. These findings underscore the need to look beyond candidate genes to elucidate the complex etiology of opioid use and dependence.
The primary limitation of this work is the sample size. As with other investigations into the genomics of complex human traits, including substance use disorders, large sample sizes are needed to detect the effect sizes expected for the anticipated architecture of the traits. As noted by Andersen et al. (Andersen et al., 2015), effect sizes for differences in DNAm between opioid users and controls are often fairly small (i.e., < 7% difference in methylation), and none of those studies used an array-based approach assessing thousands of CpG sites simultaneously, which requires larger samples to detect such small effects after correcting for multiple testing. Consequently, we and others are actively working to form consortia to analyze similar postmortem opioid brain tissues samples together. Another limitation of this study is the lack of validation samples. Kozlenkov et al. (Kozlenkov et al., 2017) conducted the only other epigenome-wide analysis of DNAm in the brains of opioid users, examining DNAm in the mOFC. A comparison of the top 100 CpG sites from their analyses did not yield any overlapping sites. Given the dearth of studies looking at epigenome-wide associations among opioid users, it is difficult to interpret the present findings, especially given the sample size and unique population. Lastly, the cross-sectional design does not allow for direct testing of a causal relationship between opioid use and DNAm. Larger, longitudinal studies are needed to clarify the effects of opioid use on DNAm, as well as important confounders such as polysubstance use.
It is important to consider, in light of the limitations, how these results can best be used to inform the field. There are several limitations of work on SUDs in postmortem brain tissue. First is the difficulty in distinguishing the causal role of any differentially methylated regions or pathways discovered in post-mortem tissue when compared to non-SUD control tissue since the tissue is frequently collected after years of chronic drug use. Thus, it is where in the etiologic pathways of the disorder those findings lie – causes of SUD risk or consequences of chronic opioid use disorder (OUD). One method to attempt to overcome this issue would be to follow a cohort of individuals with a drug use disorder [e.g., AIDS Linked to the IntraVenous Experience (ALIVE) Cohort; (Vlahov et al., 1991) and obtain consent to examine brains in the event of an individual’s death.] This is a rather challenging effort but presents one manner in which researchers could go beyond posthumous psychiatric interviews with immediate family to understand patterns of drug use. Second, the utility of such findings is relatively limited with respect to clinical translation. While these findings will help elucidate our understanding of the neurobiology underlying the clinical course of OUDs, inasmuch as these findings do not overlap with findings in peripheral tissue, their potential use as a clinical biomarker is limited. Although there are limitations in the clinical utility of this work, it is important to consider how postmortem brain SUD results can inform the field. As a clearer picture emerges from genetic studies of OUD risk and clinical course, it will be important to integrate the knowledge gained from this work.
4.1. Conclusions
This study adds to a growing literature on genome-wide associations between opioid use and DNAm and is the first to do so using brain tissue from the dlPFC. Although no sites reached significance after correction for multiple testing, opioid users were phenotypically older compared to controls. Future work with larger samples is needed to clarify these associations.
Supplementary Material
Highlights.
Epigenome-wide analysis of DNA methylation in human dorsolateral prefrontal cortex.
Opioid overdose is marginally associated with methylation within the Netrin-1 gene.
Opioid overdose is associated with accelerated phenotypic age.
Acknowledgements
The authors would like to express their gratitude to our colleagues whose efforts have led to the donation of postmortem tissue to advance these studies, including at the Office of the Chief Medical Examiner of the State of Maryland, Baltimore Maryland, and the Office of the Chief Medical Examiner of Kalamazoo County Michigan. We also would like to acknowledge the contributions of Dr. Llewelyn Bigelow for his diagnostic expertise. Finally, we are indebted to the generosity of the families of the decedents, who donated the brain tissue used in these studies.
Role of Funding Source
David W. Sosnowski is supported by the National Institute on Drug Abuse’s Drug Dependence Epidemiology Training Program (T32 DA007292; PI: Brion S. Maher). This research was supported by the National Institute on Drug Abuse (R01 DA039408; PI: Brion S. Maher).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors have no conflicts of interest to declare.
References
- Andersen AM, Dogan MV, Beach SRH, Philibert RA, 2015. Current and future prospects for epigenetic biomarkers of substance use disorders. Genes (Basel). 10.3390/genes6040991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA, 2014. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369. 10.1093/bioinformatics/btu049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Andresen D, Zhao Q, Li X, Jupp B, Chesworth R, Lawrence AJ, Bredy TW, 2015. Persistent variations in neuronal DNA methylation following cocaine self-administration and protracted abstinence in mice. Neuroepigenetics 4, 1–11. 10.1016/j.nepig.2015.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y, 1995. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 57, 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Chorbov VM, Todorov AA, Lynskey MT, Cicero TJ, 2011. Elevated levels of DNA methylation at the OPRM1 promoter in blood and sperm from male opioid addicts. J. Opioid Manag 7, 258–264. 10.5055/jom.2011.0067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S, Coarfa C, Harris RA, Milosavljevic A, Troakes C, Al-Sarraj S, Dobson R, Schalkwyk LC, Mill J, 2012. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 13, R43. 10.1186/gb-2012-13-6-r43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt L, Grebely J, Stone J, Hickman M, Vickerman P, Marshall BDL, Bruneau J, Altice FL, Henderson G, Rahimi-Movaghar A, Larney S, 2019a. Global patterns of opioid use and dependence: harms to populations, interventions, and future action. Lancet. 10.1016/S0140-6736(19)32229-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt L, Grebely J, Stone J, Hickman M, Vickerman P, Marshall BDL, Bruneau J, Altice FL, Henderson G, Rahimi-Movaghar A, Larney S, 2019b. Global patterns of opioid use and dependence: harms to populations, interventions, and future action. Lancet. 10.1016/S0140-6736(19)32229-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM, 2010. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 11, 1–9. 10.1186/1471-2105-11-587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi G, Asadikaram G, Akbari H, Nematollahi MH, Abolhassani M, Shahabinejad G, Khodadadnejad L, Hashemi M, 2018. Elevated levels of DNA methylation at the OPRM1 promoter region in men with opioid use disorder. Am. J. Drug Alcohol Abuse 44, 193–199. 10.1080/00952990.2016.1275659 [DOI] [PubMed] [Google Scholar]
- Gagnon-Bartsch JA, Speed TP, 2012. Using control genes to correct for unwanted variation in microarray data. Biostatistics 13, 539–552. 10.1093/biostatistics/kxr034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gansler DA, Lee AKW, Emerton BC, D’Amato C, Bhadelia R, Jerram M, Fulwiler C, 2011. Prefrontal regional correlates of self-control in male psychiatric patients: Impulsivity facets and aggression. Psychiatry Res. - Neuroimaging 191, 16–23. 10.1016/j.pscychresns.2010.09.003 [DOI] [PubMed] [Google Scholar]
- Hagerty SL, Bidwell LC, Harlaar N, Hutchison KE, 2016. An Exploratory Association Study of Alcohol Use Disorder and DNA Methylation. Alcohol. Clin. Exp. Res 40, 1633–1640. 10.1111/acer.13138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, 2013. DNA Methylation Age of Human Tissues and Cell Types. Genome Biol. 14, R115. 10.1186/gb-2013-14-10-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT, 2012. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13, 1–16. 10.1186/1471-2105-13-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A, 2003. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet 10.1038/ng1089 [DOI] [PubMed] [Google Scholar]
- Kozlenkov A, Jaffe AE, Timashpolsky A, Apontes P, Rudchenko S, Barbu M, Byne W, Hurd YL, Horvath S, Dracheva S, 2017. DNA methylation profiling of human prefrontal cortex neurons in heroin users shows significant difference between genomic contexts of hyper- and hypomethylation and a younger epigenetic age. Genes (Basel). 8, 2–15. 10.3390/genes8060152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladd-Acosta C, Pevsner J, Sabunciyan S, Yolken RH, Webster MJ, Dinkins T, Callinan PA, Fan JB, Potash JB, Feinberg AP, 2007. DNA Methylation Signatures Within the Human Brain. Am. J. Hum. Genet 81, 1304–1315. 10.1086/524110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalanne Laurence, Ayranci Gulebru, Brigitte LK, Lutz PE, 2014. The kappa opioid receptor: From addiction to depression, and back. Front. Psychiatry. 10.3389/fpsyt.2014.00170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Storey JD, 2007. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 3, 1724–1735. 10.1371/journal.pgen.0030161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, Hou L, Baccarelli AA, Stewart JD, Li Y, Whitsel EA, Wilson JG, Reinerl AP, Avivl A, Lohman K, Liu Y, Ferrucci L, Horvath S, 2018. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany. NY). 10, 573–591. 10.18632/aging.101414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, Yu M, Tonti-Filippini J, Heyn H, Hu S, Wu JC, Rao A, Esteller M, He C, Haghighi FG, Sejnowski TJ, Behrens MM, Ecker JR, 2013. Global epigenomic reconfiguration during mammalian brain development. Science (80-.). 341. 10.1126/science.1237905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini L, Whistler JL, 2007. The role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr. Opin. Neurobiol 10.1016/j.conb.2007.10.004 [DOI] [PubMed] [Google Scholar]
- Montalvo-Ortiz JL, Cheng Z, Kranzler HR, Zhang H, Gelenter J, 2019. Genomewide Study of Epigenetic Biomarkers of Opioid Dependence in European-American Women. Sci. Rep 9, 1–9. 10.1038/s41598-019-41110-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, 2014. Epigenetic mechanisms of drug addiction. Neuropharmacology. 10.1016/j.neuropharm.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen DA, Utrankar A, Reyes JA, Simons DD, Kosten TR, 2012. Epigenetics of drug abuse: Predisposition or response. Pharmacogenomics. 10.2217/pgs.12.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen DA, Yuferov V, Hamon S, Jackson C, Ho A, Ott J, Kreek MJ, 2009. Increased OPRM1 DNA methylation in lymphocytes of methadone-maintained former heroin addicts. Neuropsychopharmacology 34, 867–873. 10.1038/npp.2008.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipson B, Maksimovic J, Oshlack A, 2016. MissMethyl: An R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics 32, 286–288. 10.1093/bioinformatics/btv560 [DOI] [PubMed] [Google Scholar]
- Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, Van Djik S, Muhlhausler B, Stirzaker C, Clark SJ, 2016. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 17, 1–17. 10.1186/s13059-016-1066-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polimanti R, Walters RK, Johnson EC, McClintick JN, Adkins AE, Adkins DE, Bacanu SA, Bierut LJ, Bigdeli TB, Brown S, Bucholz KK, Copeland WE, Costello EJ, Degenhardt L, Farrer LA, Foroud TM, Fox L, Goate AM, Grucza R, Hack LM, Hancock DB, Hartz SM, Heath AC, Hewitt JK, Hopfer CJ, Johnson EO, Kendler KS, Kranzler HR, Krauter K, Lai D, Madden PAF, Martin NG, Maes HH, Nelson EC, Peterson RE, Porjesz B, Riley BP, Saccone N, Stallings M, Wall TL, Webb BT, Wetherill L, Edenberg HJ, Agrawal A, Gelernter J, 2020. Leveraging genome-wide data to investigate differences between opioid use vs. opioid dependence in 41,176 individuals from the Psychiatric Genomics Consortium. Mol. Psychiatry 25, 1673–1687. 10.1038/s41380-020-0677-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Kuan PF, 2019. methylGSA: a Bioconductor package and Shiny app for DNA methylation data length bias adjustment in gene set testing. Bioinformatics 35, 1958–1959. 10.1093/bioinformatics/bty892 [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, 2015. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval-Sierra JV, Salgado García FI, Brooks JH, Derefinko KJ, Mozhui K, 2020. Effect of short-term prescription opioids on DNA methylation of the OPRM1 promoter. Clin. Epigenetics 12. 10.1186/s13148-020-00868-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago Rivera OJ, Havens JR, Parker MA, Anthony JC, 2018. Risk of heroin dependence in newly incident heroin users. JAMA Psychiatry 75, 863–864. 10.1001/jamapsychiatry.2018.1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoller JW, Kendler K, Craddock N, Lee PH, Neale BM, Nurnberger JN, Ripke S, Santangelo S, Sullivan PS, Neale BN, Purcell S, Anney R, Buitelaar J, Fanous A, Faraone SF, Hoogendijk W, Lesch KP, Levinson DL, Perlis RP, Rietschel M, Riley B, Sonuga-Barke E, Schachar R, Schulze TS, Thapar A, Kendler KK, Smoller JS, Neale M, Perlis R, Bender P, Cichon S, Daly MD, Kelsoe J, Lehner T, Levinson D, O’Donovan, Mick, Gejman P, Sebat J, Sklar P, Daly M, Devlin B, Sullivan P, O’Donovan, Michael, 2013. Identification of risk loci with shared effects on five major psychiatric disorders: A genome-wide analysis. Lancet 381, 1371–1379. 10.1016/S0140-6736(12)62129-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, Šmuc T, 2011. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS One 6. 10.1371/journal.pone.0021800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team RC, 2018. R: A language and environment for statistical computing. [Google Scholar]
- Tsai NP, Bi J, Loh HH, Wei LN, 2006. Netrin-1 signaling regulates de novo protein synthesis of κ opioid receptor by facilitating polysomal partition of its mRNA. J. Neurosci 26, 9743–9749. 10.1523/JNEUROSCI.3014-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai NP, Bi J, Wei LN, 2007. The adaptor Grb7 links netrin-1 signaling to regulation of mRNA translation. EMBO J. 26, 1522–1531. 10.1038/sj.emboj.7601598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanyukov MM, Tarter RE, 2019. Genetics and Epigenetics of Substance Use, in: Sloboda Z, Petras H, Robertson E,HR (Ed.), Prevention of Substance Use. Advances in Prevention Science. Springer, pp. 57–73. 10.1007/978-3-030-00627-3_4 [DOI] [Google Scholar]
- Vlahov D, Anthony JC, Muñoz A, Margolick J, Nelson KE, Celentano DD, Solomon L, Polk BF, 1991. The Alive Study: A Longitudinal Study of HIV-1 Infection in Intravenous Drug Users: Description of Methods. J. Drug Issues 21, 759–776. 10.1177/002204269102100406 [DOI] [PubMed] [Google Scholar]
- Xu Z, Niu L, Li L, Taylor JA, 2016. ENmix: A novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 44, 20. 10.1093/nar/gkv907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Rentsch CT, Cheng Z, Kember RL, Nunez YZ, Sherva RM, Tate JP, Dao C, Xu K, Polimanti R, Farrer LA, Justice AC, Kranzler HR, Gelernter J, 2020. Association of OPRM1 Functional Coding Variant with Opioid Use Disorder: A Genome-Wide Association Study. JAMA Psychiatry 77, 1072–1080. 10.1001/jamapsychiatry.2020.1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.