

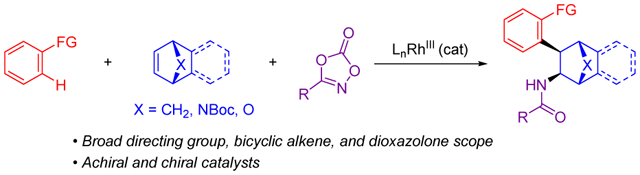
Abstract
A three-component method is described for the preparation of syn-1,2-disubstituted bridged bicyclic compounds. The reaction was demonstrated for readily available aromatic and heteroaromatic C–H bond substrates with tertiary and secondary amide, lactam, pyrazole and triazole directing groups, and a variety of bridged bicyclic alkenes, including norbornene, benzonorbornadiene, oxygen and nitrogen-bridged analogs, and an unsaturated tropinone. Broad dioxazolone scope was also observed. The use of a chiral Cp-derived RhIII catalyst enables asymmetric synthesis of products.
Graphical Abstract
Bridged bicyclic systems are an important chemical motif due to their prevalence in approved drugs and drug candidates. For example, [2.2.1]-bridged bicycles are present in molecules such as Ifetroban, which is currently in Phase II Clinical Trials for Duchenne muscular dystrophy.1 Similarly, the [3.2.1] tropane framework is present in important drugs such as the asthma and COPD medication Spiriva and the HIV drug Selzentry.1 Development of new methodologies for the elaboration of these structures thus has value in the discovery of pharmaceutical agents.
Bridged bicyclic alkenes have also served as versatile reactants for transition-metal catalysis. For example, Lautens carried out seminal work on RhI catalyzed asymmetric ring opening reactions [Scheme 1, (eq 1)],2 and related approaches for transition-metal catalyzed C–H bond addition and ring-opening have subsequently been reported.3 Of the C–H functionalization methods that leave the strained bicycle intact, most proceed with monofunctionalization,4 with relatively few bifunctionalizations having been reported.5 The carboamidation of bridged bicyclic alkenes using N-aryloxy aliphatic amides as internal redox C–H bond substrates is a notable example of bifunctionalization as recently disclosed by the Zhao and Cramer groups to provide ortho-substituted phenolic products (eq 2).6 In 2019, our lab reported a modular, functional group compatible three-component reaction for the synthesis of α-branched amines (eq 3).7,8 Because three different components were used, C–H bond substrates with various directing groups and different amidating reagents could be employed to generate different classes of 1,1-disubstituted α-branched amine derivatives.
Scheme 1.
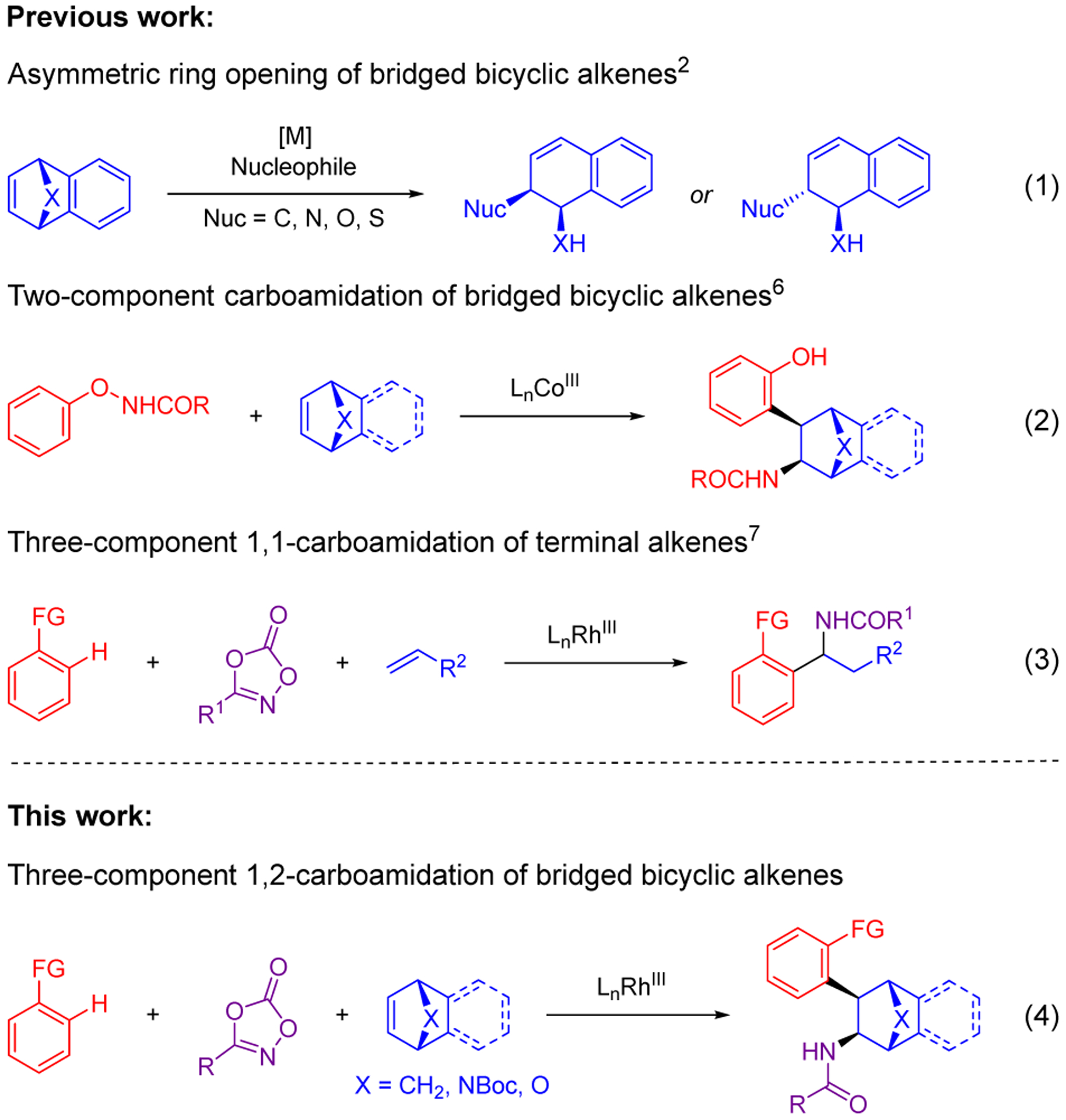
Background for Three-Component 1,2-Carboamidation of Bridged Bicyclic Alkenes
Herein, we describe a modular three-component reaction to access 1,2-disubstituted [2.2.1]-bridged bicycles via RhIII-catalyzed 1,2-addition of C–H bonds and amidating reagents to bridged bicyclic alkenes (eq 4). Different types of C–H bond substrates, both aliphatic and aromatic amidating reagents, and a variety of bridged bicyclic alkenes, including norbornene, benzonorbornadiene, oxygen and nitrogen-bridged analogs, and an unsaturated tropinone, led to a broad range of 1,2-disubstituted products. Additionally, asymmetric catalysis was achieved using a chiral RhIII catalyst employing a chiral ligand designed by Cramer.9,10 A reaction mechanism is proposed that provides a rationale for exclusive 1,2-disubstitution for bridged bicyclic alkenes in contrast to the 1,1-disubstitution that had been observed for three-component carboamidation of terminal alkenes.7
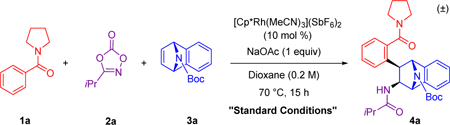
Extensive reaction optimization was conducted for the three-component coupling of pyrrolidine benzamide 1a, isopropyl dioxazolone 2a,11 and azabenzonorbornadiene 3a (Table 1). The cationic RhIII catalyst [Cp*Rh(MeCN)3](SbF6)2 was critical to the reaction (entry 2), and the combination of the [Cp*RhCl2]2 dimer and AgSbF6, which forms the active catalyst in situ, was found to be a comparable system to the chosen catalyst (entry 3). Other d6 metal catalyst systems such as [Cp*Co(CO)I2], [Cp*IrCl2]2, and [RuCl2(p-cymene)]2, paired with AgSbF6, did not provide any product (entries 4–6). The addition of sodium acetate as an additive was found to be important in facilitating the initial C–H activation step, leading to enhanced conversion to product (entry 7). In contrast, acidic additives such as HOAc led primarily to two-component alkylation products. No reaction occurred with Cp*Rh(OAc)2 in the absence of NaOAc, but when AgSbF6 was included in the reaction mixture, 86% conversion to product was observed (entries 8–9). The optimal solvent for this transformation was 1,4-dioxane, although 1,2-dichloroethane (DCE) resulted in only a modest reduction in yield (entry 10). The reaction concentration could be increased from 0.2 to 0.5 M with minimal effect on the reaction yield (entry 11). In addition, lowering the temperature from 70 to 50 °C gave a modest drop to 85% yield (entry 12). For some substrates, lowering the temperature to 50 °C and/or reducing the reaction time to 2 h was necessary to avoid the formation of overalkylation products (vide infra). Finally, when the alkene was employed as the limiting reagent, product was obtained, but with a reduction in the yield (entry 13).
Table 1.
Reaction Condition Deoptimization
 | ||
|---|---|---|
| Entrya | Variation from the standard conditions | Yield 4ab |
| 1 | None | 97%c |
| 2 | No [Cp*Rh(MeCN)3](SbF6)2 | 0% |
| 3 | [Cp*RhCl2]2 (5%) / AgSbF6 (20%) | 91% |
| 4 | [Cp*IrCl2]2 (5%) / AgSbF6 (20%) | 0% |
| 5 | [Cp*Co(CO)I2] (10%) / AgSbF6 (20%) | 0% |
| 6 | [RuCl2(p-cymene)]2 (5%) / AgSbF6 (20%) | 0% |
| 7 | No NaOAc | 87% |
| 8 | Cp*Rh(OAc)2 (10%)d | 0% |
| 9 | Cp*Rh(OAc)2 (10%) / AgSbF6 (20%)d | 86% |
| 10 | DCE as solvent | 77% |
| 11 | 0.5 M | 91% |
| 12 | 50 °C | 85% |
| 13 | 2:3:1 ratio 1a:2a:3a | 63% |
Conditions: 0.10 mmol of 1a, 0.30 mmol of 2a, 0.20 mmol of 3a
Yields determined by 1H NMR relative to trimethylphenylsilane as an external standard.
Isolated yield of the pure material was 86%.
No NaOAc was added.
After determining optimal reaction conditions, we explored the scope of the bridged bicyclic alkene (Scheme 2). We evaluated a variety of [2.2.1]-bridged bicyclic systems with nitrogen, oxygen and carbon at the bridge position. The reaction proceeded in good yields for bicyclic systems containing nitrogen (4a-4b) and oxygen bridges (4c), even though these bridges might have been expected to act as leaving groups through a strain-relieving elimination. It is notable that the transformation was effective for a [3.2.1]-nitrogen-bridged bicyclic alkene (4b), thereby providing a promising new method for the elaboration of the tropinone core, which is present in a wide variety of natural products, approved drugs and drug candidates, including Selzentry and Spiriva.1 When the alkene contained a methylene bridge, the reaction also proceeded in good to excellent yields for commercially available norbornene (4d) and the more strain-activated benzonorbornadiene (4e), respectively. With norbornadiene no conversion of starting material was observed, likely due to non-productive complexation to the catalyst. The scalability of this method was demonstrated by the preparation of 4e in 71% yield on the 1.0 mmol scale with a catalyst loading of 5 mol %.
Scheme 2. Alkene and C–H Bond Scopea.
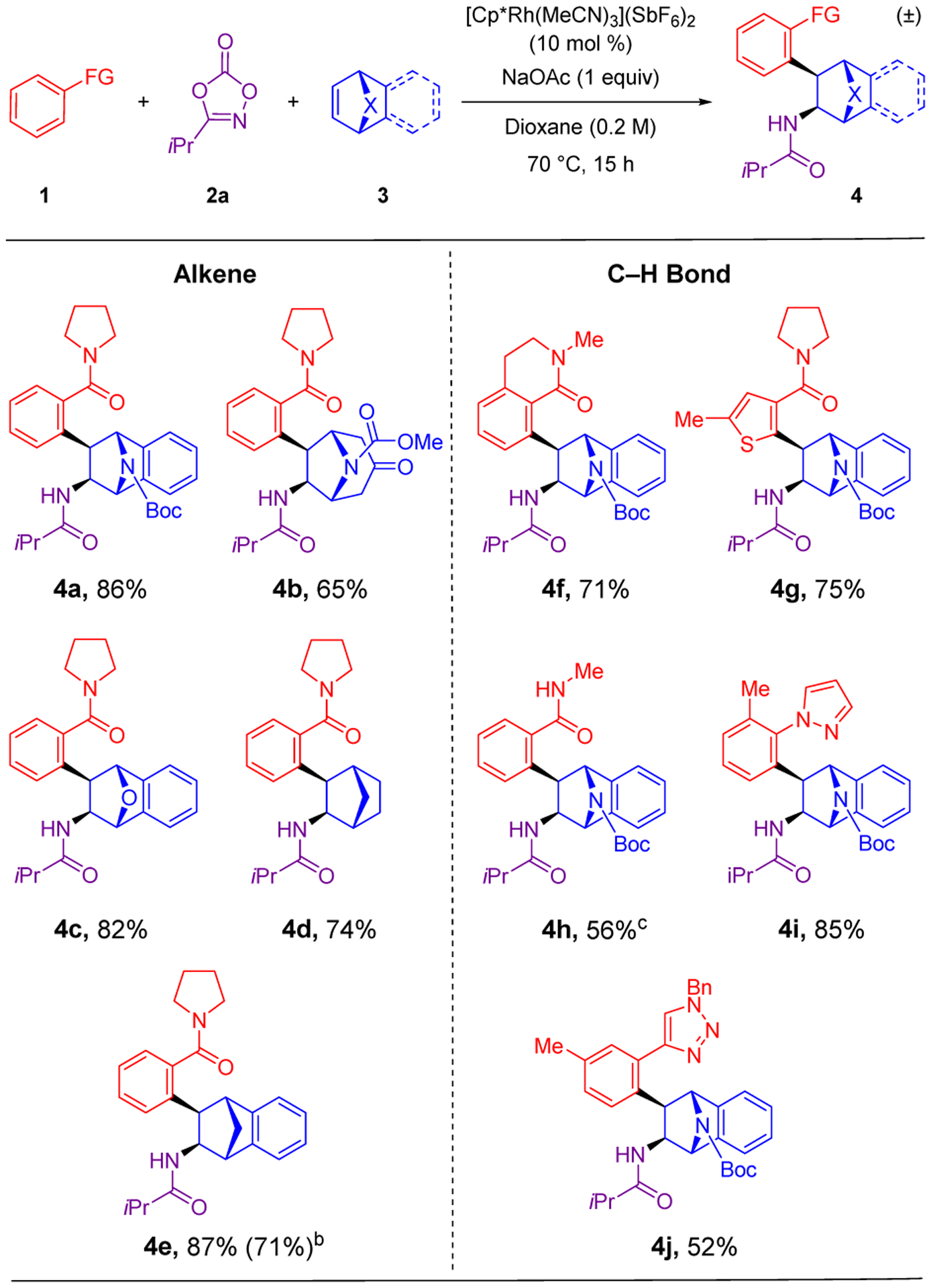
aConditions: 0.20 mmol of 1, 0.60 mmol of 2a, 0.40 mmol of 3. Isolated yields of products after purification by chromatography are reported. bReaction performed on a 1.0 mmol scale in the C–H bond substrate with 5 mol % catalyst. cReaction performed at 70 °C for 2 h.
A variety of C–H bond substrates were successfully employed (Scheme 2). Commonly encountered motifs such as tertiary (4a, 4f-4g) and secondary (4h) amides, pyrazoles (4i), and triazoles (4j) were effective directing groups for this transformation. Product 4f is of interest due to the presence of a fused bicyclic lactam motif. Product 4g is also of interest, as it requires activation of a heteroaromatic C–H bond. For the sterically unencumbered secondary amide in product 4h, the reaction time was reduced to 2 h in order to prevent the addition of a second alkene.
This reaction demonstrated a wide scope for dioxazolone amidating reagents (Scheme 3), with both aliphatic (4a, 4k-4m), aromatic (4n-4r) and heteroaraomatic (4s) dioxazolones coupling in moderate to high yields. Aliphatic dioxazolones were particularly effective, including for both an unhindered acetamide (4k) and a sterically encumbered pivalamide (4l). For some of the aromatic amide products, 4o-4r, it was necessary to reduce the reaction time to 2 h and the temperature to 50 °C to prevent overalkylation by C–H functionalization at the ortho position of the newly formed aromatic or heteroaromatic amide in the product.
Scheme 3. Dioxazolone Scopea.
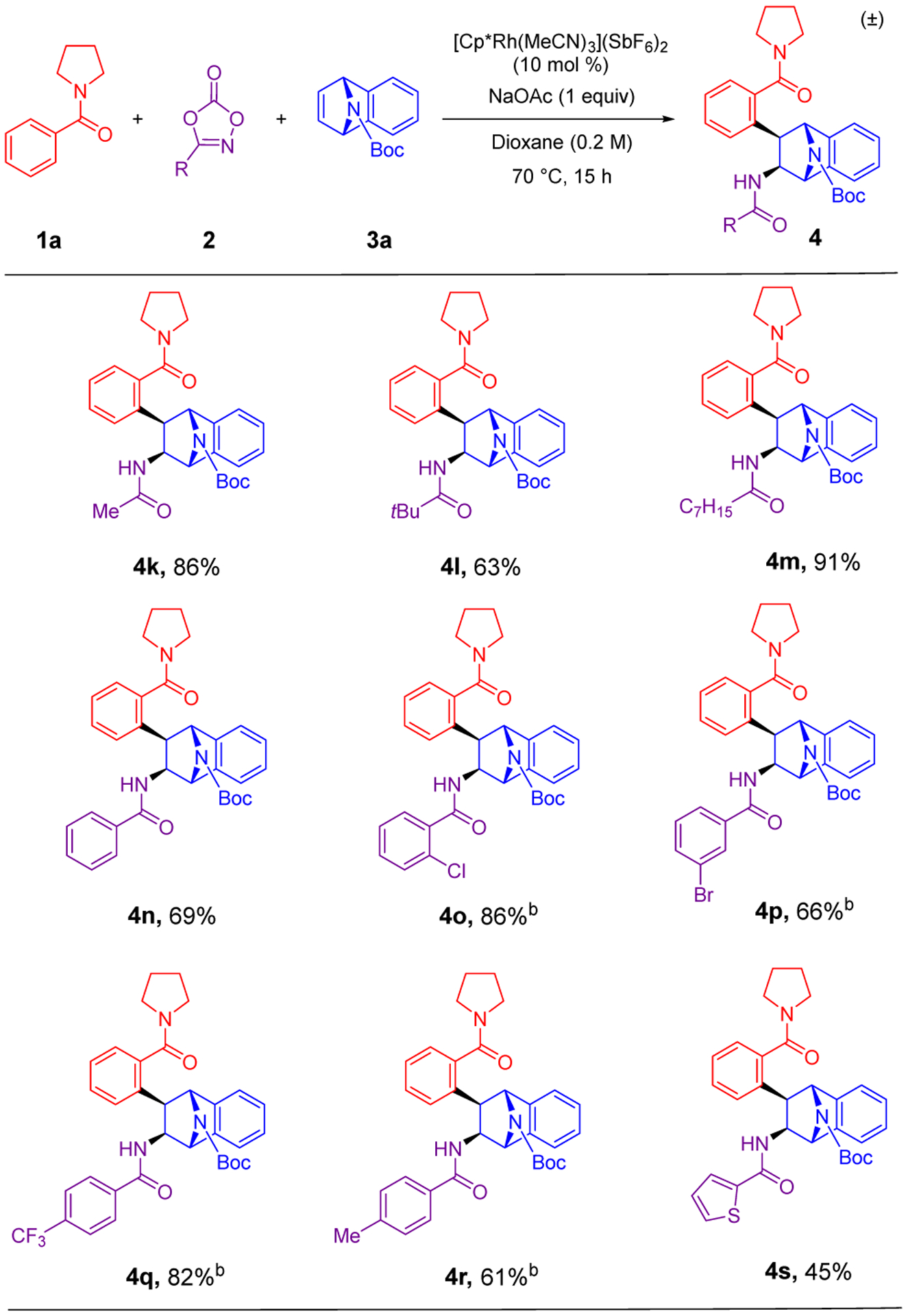
aConditions: 0.20 mmol of 1a, 0.60 mmol of 2, 0.40 mmol of 3a. Isolated yields of products after purification by chromatography are reported. bReactions performed at 50 °C for 2 h.
We were interested in exploring asymmetric catalysis for this system, given the recent impressive advances in asymmetric RhIII-catalyzed C–H functionalization methodology.9,10 The Rh complex 6 employing Cramer’s chiral ligand led to promising enantioselectivity (Scheme 4).9c Optimization of the asymmetric reaction showed that increasing the AgSbF6 loading beyond what was necessary to abstract the Rh-bound iodides increased the yield.9e In addition, for products 5a and 5b, use of DCE instead of dioxane led to higher yields while maintaining similar enantioselectivities. The scope of this enantioselective reaction was constrained to norbornene and benzonorbornadiene as the bridged bicyclic alkene substrate. Oxygen and nitrogen-bridged alkenes did not provide the desired products, possibly due to elimination of the heteroatomic bridge and non-productive anionic coordination to the metal center.3 Both pyrrolidine carboxamide (5a) and pyrazole (5b-5e) were effective directing groups. In addition, 3,5-dimethylphenyl (5a, 5b, 5d) and phenyl (5c, 5e) dioxazolone coupling partners provided comparable yields and enantioselectivity. The absolute configuration of 5d was rigorously determined by amide hydrolysis and X-ray structural characterization of the resulting amine 7 (see Supporting Information). The sense of induction for these enantioenriched products is opposite that of previously reported additions to N-tosyl-azabenzonorbornadienes using Cramer’s chiral ligand, presumably due to the decreased steric bulk of the methylene bridge in our bridged bicyclic alkenes.3j,k
Scheme 4. Asymmetric Synthesis with Chiral Catalysta.
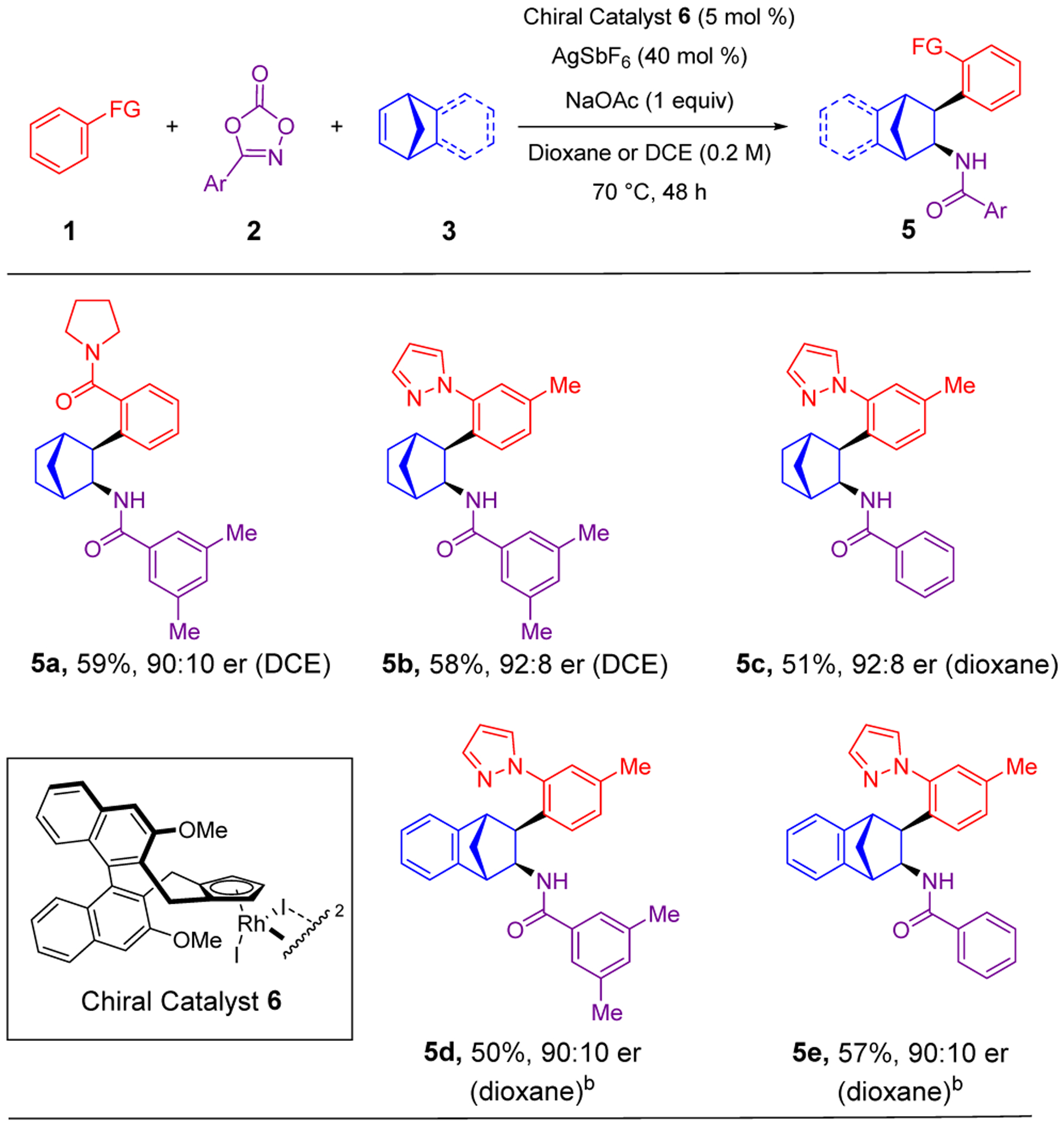
aConditions: 0.05 mmol of 1, 0.15 mmol of 2, 0.20 mmol of 3. Isolated yields of products after purification by chromatography are reported. bReaction performed with 0.05 mmol of 1-[3-methylphenyl]pyrazole, 0.15 mmol of aryl dioxazolone, 0.10 mmol of benzonorbornadiene.
The three-component addition to the bicyclic alkenes reported here proceeds exclusively with syn-1,2-carboamidation, while the previously reported three-component addition to terminal alkenes proceeded solely with 1,1-addition (see eq 3, Scheme 1).7 As supported by a number of mechanistic experiments,7 the reaction first proceeds by concerted metallation deprotonation to generate rhodacycle I (Scheme 5). Then, migratory insertion of the bridged bicyclic alkene into the Rh–C bond of rhodacycle I forms the 7-membered rhodacycle II. In contrast to the previously reported work with terminal alkenes, syn-β-hydride elimination and reinsertion is not possible due to the lack of an appropriately oriented syn hydride. Additionally, the bridgehead hydrogens cannot undergo elimination because this would result in the formation of a high-energy anti-Bredt alkene. Nitrene insertion of the 7-membered rhodacycle II with retention of configuration then forms the amidated species III, which upon protodemetallation produces the observed 1,2-disubstituted product IV. The AgSbF6 additive was found to be necessary in order to enable formation of product when using Cp*Rh(OAc)2 (see entries 8–9, Table 1). The AgSbF6 might facilitate anion exchange with Cp*Rh(OAc)2 to provide the cationic Cp*Rh(OAc)+ which is necessary for coordination to the C–H bond substrate. Alternatively, based upon literature reports, it could facilitate breakdown of the 7-membered rhodacycle III to release the product IV.12
Scheme 5.
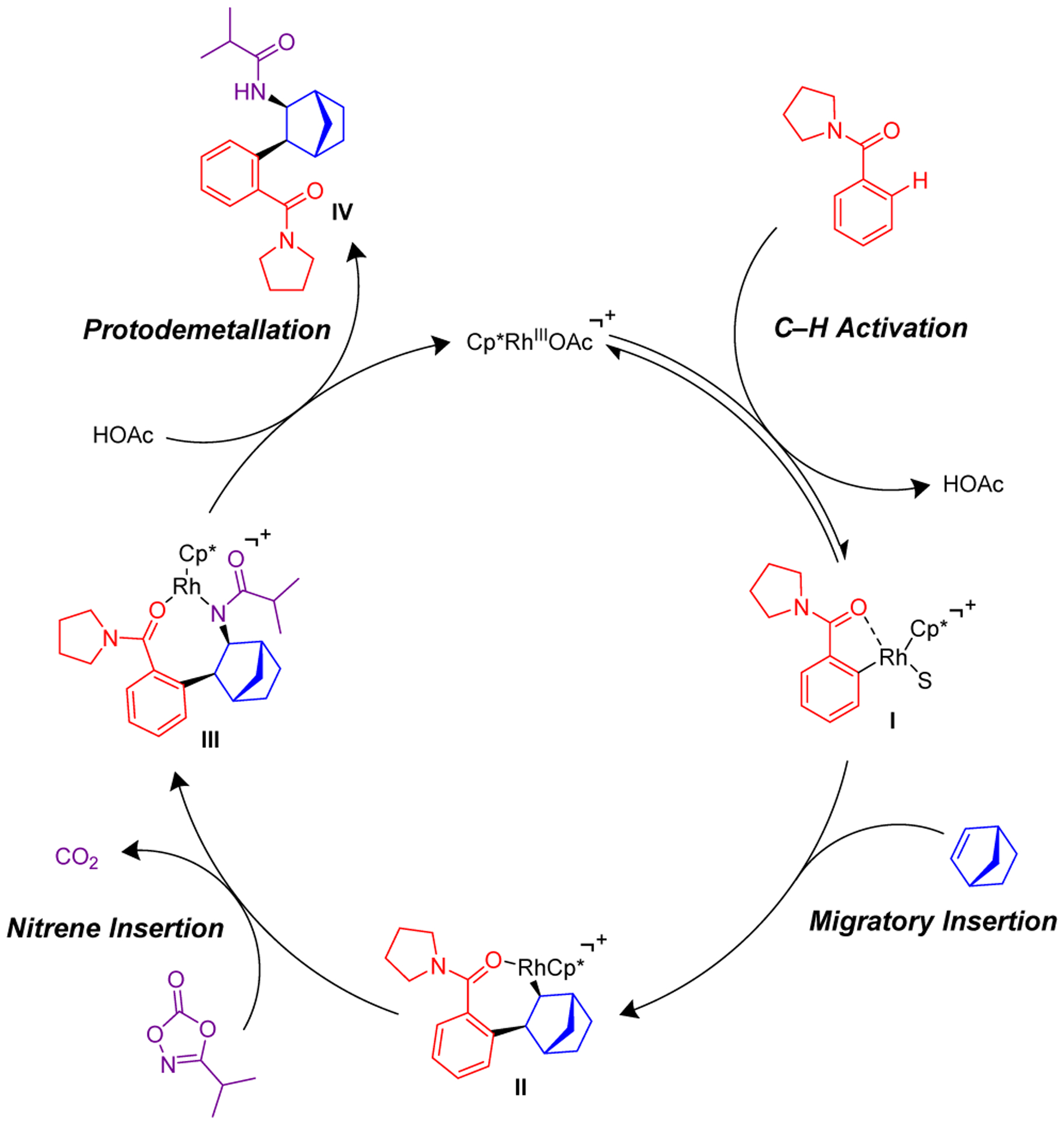
Proposed Mechanism for the Three-Component Transformation
In summary, we have developed a modular, three-component RhIII-catalyzed 1,2-carboamidation of bridged bicyclic alkenes. This methodology shows potential for elaborating the common [2.2.1]- and [3.2.1]-bridged bicyclic frameworks found in a variety of approved drugs and drug candidates. In addition, we showed that the use of a chiral RhIII catalyst enables the asymmetric synthesis of the three-component products.
Supplementary Material
ACKNOWLEDGMENT
The NIH (R35GM122473) is gratefully acknowledged for supporting this work. AS is also grateful for a fellowship from the Spanish Ministry of Education for funding a short stay at Yale University.
Footnotes
SUPPORTING INFORMATION.
The Supporting Information is available free of charge via the Internet at http://pubs.acs.org.
Procedure details and NMR spectra (PDF)
Crystallographic data for 7 (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).Typing the names of these drugs and drug candidates into PubChem provides the compound structure, bioactivity, full list of literature, and access to ongoing clinical trials, applications, and usage. For additional drugs and drug candidates that may be derivatized by this strategy, see:; Stockdale TP; Williams CM Pharmaceuticals that Contain Polycyclic Hydrocarbon Scaffolds. Chem. Soc. Rev 2015, 44, 7737–7763. [DOI] [PubMed] [Google Scholar]
- (2).For leading references, see:; (a) Lautens M; Fagnou K; Rovis T Rhodium-Catalyzed Asymmetric Alcoholysis and Aminolysis of Oxabenzonorbornadiene: A New Enantioselective Carbon–Heteroatom Bond Forming Process. J. Am. Chem. Soc 2000, 122, 5650–5651. [Google Scholar]; (b) Lautens M; Fagnou K; Taylor M; Rovis T Rhodium-Catalysed Asymmetric Ring Opening of Oxabicyclic Alkenes with Heteroatom Nucleophiles. J. Organomet. Chem 2001, 624, 259–270. [Google Scholar]; (c) Lautens M; Fagnou K; Yang D Rhodium-catalyzed Asymmetric Ring Opening Reactions of Oxabicyclic Alkenes: Application of Halide Effects in the Development of a General Process. J. Am. Chem. Soc 2003, 125, 14884–14892. [DOI] [PubMed] [Google Scholar]; (d) Zhang L; Le CM; Lautens M The Use of Silyl Ketene Acetals and Enol Ethers in the Catalytic Enantioselective Alkylative Ring Opening of Oxa/Aza Bicyclic Alkenes. Angew. Chem. Int. Ed 2014, 53, 5951–5954. [DOI] [PubMed] [Google Scholar]; (e) Loh CCJ; Schmid M; Peters B; Fang X; Lautens M Exploiting Distal Reactivity of Coumarins: A Rhodium-Catalyzed Vinylogous Asymmetric Ring-Opening Reaction. Angew. Chem. Int. Ed 2016, 55, 4600–4604. [DOI] [PubMed] [Google Scholar]; (f) Yen A; Choo K-L; Yazdi SK; Franke PT; Webster R; Franzoni I; Loh CCJ; Poblador-Bahamonde AI; Lautens M Rhodium-Catalyzed Enantioselective Isomerization of meso-Oxabenzonorbornadienes to 1,2-Naphthalene Oxides. Angew. Chem. Int. Ed 2017, 56, 6307–6311. [DOI] [PubMed] [Google Scholar]
- (3).For leading references on ring opening reactions, see:; (a) Villeneuve K; Tam W Ruthenium-Catalyzed Isomerization of Oxa/Azabicyclic Alkenes: An Expedient Route for the Synthesis of 1,2-Naphthalene Oxides and Imines. J. Am. Chem. Soc 2006, 128, 3514–3515. [DOI] [PubMed] [Google Scholar]; (b) Qi Z; Li X Rhodium(III)-Catalyzed Coupling of Arenes with 7-Oxa/Azabenzonorbornadienes by C–H Activation. Angew. Chem. Int. Ed 2013, 52, 8995–9000. [DOI] [PubMed] [Google Scholar]; (c) Yang T; Zhang T; Yang S; Chen S; Li X Rhodium(III)-Catalyzed Coupling of N-Sulfonyl 2-Aminobenzaldehydes with Oxygenated Allylic Olefins Through C–H Activation. Org. Biomol. Chem 2014, 12, 4290–4294. [DOI] [PubMed] [Google Scholar]; (d) Zhang Y; Wu Q; Cui S Rh(III)-Catalyzed C–H Activation–Desymmetrization of Diazabicycles with Arenes: Facile Synthesis of Functionalized Cyclopentenes. Chem. Sci 2014, 5, 297–302. [Google Scholar]; (e) Unoh Y; Satoh T; Hirano K; Miura M Rhodium(III)-Catalyzed Direct Coupling of Arylphosphine Derivatives with Heterobicyclic Alkenes: A Concise Route to Biarylphosphines and Dibenzophosphole Derivatives. ACS Catal. 2015, 5, 6634–6639. [Google Scholar]; (f) Zeng C; Yang F; Chen J; Wang J; Fan B Iridium/Copper-Catalyzed Asymmetric Ring Opening Reaction of Azabenzonorbornadienes with Amines. Org. Biomol. Chem 2015, 13, 8425–8428. [DOI] [PubMed] [Google Scholar]; (g) Li DY; Jiang LL; Chen S; Huang ZL; Dang L; Wu XY; Liu PN Cascade Reaction of Alkynols and 7-Oxabenzonorbornadienes Involving Transient Hemiketal Group Directed C–H Activation and Synergistic RhIII/ScIII Catalysis. Org. Lett 2016, 18, 5134–5137. [DOI] [PubMed] [Google Scholar]; (h) Kong L; Yu S; Tang G; Wang H; Zhou X; Li X Cobalt (III)-Catalyzed C-C Coupling of Arenes with 7-Oxabenzonorbornadiene and 2-Vinyloxirane via C–H Activation. Org. Lett 2016, 18, 3802–3805. [DOI] [PubMed] [Google Scholar]; (i) Wang X; Li Y; Knecht T; Daniliuc CG; Houk KN; Glorius F Combination of Cp*RhIII-Catalyzed C–H Activation and a Wagner-Meerwein-Type Rearrangement. Angew. Chem. Int. Ed 2017, 56, 1381–1384. [DOI] [PubMed] [Google Scholar]; (j) Yang X; Zheng G; Li X Rhodium(III)-Catalyzed Enantioselective Coupling of Indoles and 7-Azabenzonorbornadienes by C–H Activation/Desymmetrization. Angew. Chem. Int. Ed 2019, 58, 322–326. [DOI] [PubMed] [Google Scholar]; (k) Mi R; Zheng G; Qi Z; Li X Rhodium-Catalyzed Enantioselective Oxidative [3+2] Annulation of Arenes and Azabicyclic Olefins through Twofold C−H Activation. Angew. Chem. Int. Ed 2019, 58, 17666–17670. [DOI] [PubMed] [Google Scholar]; (l) Kumar SV; Banerjee S; Punniyamurthy T Rh-Catalyzed C–C/C–N Bond Formation via C–H Activation: Synthesis of 2H-indazol-2-yl-benzo[a]carbazoles. Org. Chem. Front 2019, 6, 3885–3890. [Google Scholar]
- (4).For leading references on monofunctionalization reactions, see:; (a) Dong W; Parthasarathy K; Cheng Y; Pan F; Bolm C Hydroarylations of Heterobicyclic Alkenes through Rhodium-Catalyzed Directed C–H Functionalizations of S-Aryl Sulfoximines. Chem. Eur. J 2014, 20, 15732–15736. [DOI] [PubMed] [Google Scholar]; (b) Cheng H; Dong W; Dannenberg CA; Dong S; Guo Q; Bolm C Ruthenium-Catalyzed Hydroarylations of Oxa- and Azabicyclic Alkenes. ACS Catal. 2015, 5, 2770–2773. [Google Scholar]; (c) Shibata K; Natsui S; Tobisu M; Fukumoto Y; Chatani N An Unusual endo-Selective C–H Hydroarylation of Norbornene by the Rh(I)-Catalyzed Reaction of Benzamides. Nat. Commun 2017, 8, Article number: 1448. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li D-Y; Huang Z-L; Liu P-N Heterobicyclic Core Retained Hydroarylations through C–H Activation: Synthesis of Epibatidine Analogues. Org. Lett 2018, 20, 2028–2032. [DOI] [PubMed] [Google Scholar]
- (5).For leading references on bifunctionalization, see:; (a) Liu S; Jin Z; Teo YC; Xia Y Efficient Synthesis of Rigid Ladder Polymers via Palladium Catalyzed Annulation. J. Am. Chem. Soc 2014, 136, 17434–17437. [DOI] [PubMed] [Google Scholar]; (b) Gandeepan P; Rajamalli P; Cheng C-H Diastereoselective [3+2] Annulation of Aromatic/Vinylic Amides with Bicyclic Alkenes through Cobalt-Catalyzed C–H Activation and Intramolecular Nucleophilic Addition. Angew. Chem. Int. Ed 2016, 55, 4308–4311. [DOI] [PubMed] [Google Scholar]; (c) Skhiri A; Chatani N Nickel-Catalyzed Reaction of Benzamides with Bicylic Alkenes: Cleavage of C–H and C–N Bonds. Org. Lett 2019, 21, 1774–1778. [DOI] [PubMed] [Google Scholar]; (d) Wang P; Xu Y; Sun J; Li X Rhodium(III)-Catalyzed Chemo-divergent Couplings of Sulfoxonium Ylides with Oxa/azabicyclic Olefins. Org. Lett 2019, 21, 8459–8463. [DOI] [PubMed] [Google Scholar]; (e) Annamalai P; Hsiao H-C; Raju S; Fu Y-S; Chen P-L; Horng J-C; Liu Y-H; Chuang S-C Synthesis, Isolation, and Characterization of Mono- and Bis-norbornene-Annulated Biarylamines through Pseudo-Catellani Intermediates. Org. Lett 2019, 21, 1182–1186. [DOI] [PubMed] [Google Scholar]; (f) Jeong S; Kim E; Kim M; Hwang YJ; Padhi B; Choi J; Lee Y; Joo JM Divergent Strategies for the π-Extension of Heteroaryl Halides Using Norbornadiene as an Acetylene Synthon. Org. Lett 2020, 22, 9670–9676. [DOI] [PubMed] [Google Scholar]; (g) Padhi B; Kang G; Kim E; Ha J; Kim HT; Lim J; Joo JM Pd-Catalyzed C–H Annulation of Five-Membered Heteroaryl Halides with Norbornene Derivatives. ACS Catal. 2020, 10, 1792–1798. [Google Scholar]
- (6).(a) Zhu Y; Chen F; Zhao X; Yan D; Yong W; Zhao J Cobalt(III)-Catalyzed Intermolecular Carboamination of Propiolates and Bicyclic Alkenes via Non-Annulative Redox-Neutral Coupling. Org. Lett 2019, 21, 5884–5888. [DOI] [PubMed] [Google Scholar]; (b) Ozols K; Onodera S; Woźniak Ł; Cramer N Cobalt(III)-Catalyzed Enantioselective Intermolecular Carboaminations Via C–H Functionalization. Angew. Chem. Int. Ed 2021, 60, 655–659. [DOI] [PubMed] [Google Scholar]
- (7).Maity S; Potter TJ; Ellman JA α-Branched Amines by Catalytic 1,1-Addition of C–H Bonds and Aminating Agents to Terminal Alkenes. Nat. Catal 2019, 2, 756–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).For related carboamidation of alkenes, see:; (a) Pinkert T; Wegner T; Mondal S; Glorius F Intermolecular 1,4-Carboamination of Conjugated Dienes Enabled by Cp*RhIII-Catalyzed C–H Activation. Angew. Chem. Int. Ed 2019, 58, 15041–15045. [DOI] [PubMed] [Google Scholar]; (b) Chen C; Shi C; Yang Y; Zhou B Rh(III)-Catalyzed Tandem Annulative Redox-Neutral Arylation/Amidation of Aromatic Tethered Alkenes. Chem. Sci 2020, 11, 12124–12129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For leading references on asymmetric C–H functionalization with Group IX metal catalysts, see:; (a) Ye B; Cramer N Chiral Cyclopentadienyl Ligands as Stereocontrolling Element in Asymmetric C–H Functionalization. Science 2012, 338, 504–506. [DOI] [PubMed] [Google Scholar]; (b) Hyster TK; Knörr L; Ward TR; Rovis T Biotinylated Rh(III) Complexes in Engineered Streptavidin for Accelerated Asymmetric C–H Activation. Science 2012, 338, 500–503. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ye B; Cramer N A Tunable Class of Chiral Cp Ligands for Enantioselective Rhodium(III)-Catalyzed C–H Al-lylations of Benzamides. J. Am. Chem. Soc 2013, 135, 636–639. [DOI] [PubMed] [Google Scholar]; (d) Jia Z; Merten C; Gontla R; Daniliuc CG; Antonchick AP; Waldmann H General Enantioselective C−H Activation with Efficiently Tunable Cyclopentadienyl Ligands. Angew. Chem. Int. Ed 2017, 56, 2429–2434. [DOI] [PubMed] [Google Scholar]; (e) Potter TJ; Kamber DN; Mercado BQ; Ellman JA Rh(III)-Catalyzed Aryl and Alkenyl C–H Bond Addition to Diverse Nitroalkenes. ACS Catal. 2017, 7, 150–153. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Satake S; Kurihara T; Nishikawa K; Mochizuki T; Hatano M; Ishihara K; Yoshino T; Matsunaga S Pentamethylcyclopentadienyl Rhodium(III)-Chiral Disulfonate Hybrid Catalysis for Enantioselective C–H Bond Functionalization. Nat. Catal 2018, 1, 585–591. [Google Scholar]; (g) Trifonova EA; Ankudinov NM; Mikhaylov AA; Chusov DA; Nelyubina YV; Perekalin DS A Planar-Chiral Rhodium(III) Catalyst with a Sterically Demanding Cyclopentadienyl Ligand and its Application in the Enantioselective Synthesis of Dihydroisoquinolones. Angew. Chem. Int. Ed 2018, 57, 7714–7718. [DOI] [PubMed] [Google Scholar]; (h) Wang S-G; Cramer N An Enantioselective CpX-Rh(III)-Catalyzed C–H Functionalization/Ring-Opening Route to Chiral Cyclopentenylamines. Angew. Chem. Int. Ed 2019, 58, 2514–2518. [DOI] [PubMed] [Google Scholar]
- (10).For recent reviews on asymmetric Rh(III) catalysis, see:; (a) Newton CG; Kossler D; Cramer N Asymmetric Catalysis Powered by Chiral Cyclopentadienyl Ligands. J. Am. Chem. Soc 2016, 138, 3935–3941. [DOI] [PubMed] [Google Scholar]; (b) Newton CG; Wang S-G; Oliveira CC; Cramer N Catalytic Enantioselective Transformations Involving C–H Bond Cleavage by Transition-Metal Complexes. Chem. Rev 2017, 117, 8908–8976. [DOI] [PubMed] [Google Scholar]; (c) Yoshino T; Satake S; Matsunaga S Diverse Approaches for Enantioselective C−H Functionalization Reactions Using Group 9 CpXMIII Catalysts. Chem. Eur. J 2020, 26, 7346–7357. [DOI] [PubMed] [Google Scholar]; (d) Mas-Rosello J; Herraiz AG; Audic B; Laverny A; Cramer N Chiral Cyclopentadienyl Ligands: Design, Syntheses, and Applications in Asymmetric Catalysis. Angew. Chem. Int. Ed 2020, 59, 2–29. [DOI] [PubMed] [Google Scholar]
- (11).For leading references on C–H amination and amidation, see:; Park Y; Kim Y; Chang S Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev 2017, 117, 9247–9301. [DOI] [PubMed] [Google Scholar]
- (12).Neely JM; Rovis T Rh(III)-Catalyzed Decarboxylative Coupling of Acrylic Acids with Unsaturated Oxime Esters: Carboxylic Acids Serve as Traceless Activators. J. Am. Chem. Soc 2014, 136, 2735–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.