

Abstract
Significant rates of atmospheric dihydrogen (H2) consumption have been observed in temperate soils due to the activity of high-affinity enzymes, such as the group 1h [NiFe]-hydrogenase. We designed broadly inclusive primers targeting the large subunit gene (hhyL) of group 1h [NiFe]-hydrogenases for long-read sequencing to explore its taxonomic distribution across soils. This approach revealed a diverse collection of microorganisms harboring hhyL, including previously unknown groups and taxonomically not assignable sequences. Acidobacterial group 1h [NiFe]-hydrogenase genes were abundant and expressed in temperate soils. To support the participation of acidobacteria in H2 consumption, we studied two representative mesophilic soil acidobacteria, which expressed group 1h [NiFe]-hydrogenases and consumed atmospheric H2 during carbon starvation. This is the first time mesophilic acidobacteria, which are abundant in ubiquitous temperate soils, have been shown to oxidize H2 down to below atmospheric concentrations. As this physiology allows bacteria to survive periods of carbon starvation, it could explain the success of soil acidobacteria. With our long-read sequencing approach of group 1h [NiFe]-hydrogenase genes, we show that the ability to oxidize atmospheric levels of H2 is more widely distributed among soil bacteria than previously recognized and could represent a common mechanism enabling bacteria to persist during periods of carbon deprivation.
Subject terms: Soil microbiology, Biodiversity, Bacterial physiology
Introduction
Soil bacteria consume molecular hydrogen (H2) from the Earth’s atmosphere and serve as the main sink in the global biogeochemical H2 cycle as demonstrated in field and laboratory-based investigations [1–6]. Hydrogen oxidation in soil follows biphasic kinetics with both high-affinity (Km < 100 nM) and low-affinity (Km > 1000 nM) enzyme activities [7, 8]. The ability to oxidize H2 stems from the presence of specialized metalloenzymes, called hydrogenases, that catalyze the conversion of H2 to protons and electrons [9]. Bacteria with low-affinity have been known for decades [10] and are believed to grow on high concentrations of H2 produced in microniches, such as N2-fixing root nodules [11]. In contrast, bacteria that consume atmospheric H2 (~0.53 ppmv) [5] remained elusive until recently [12].
While soil microorganisms harbor a range of hydrogenases that catalyze H2 oxidation under oxic conditions [9, 10, 13–16], it is thought that the group 1h [NiFe]-hydrogenases are primarily responsible for (sub-) atmospheric H2 oxidation [13, 14, 17], as seen with several previously isolated actinobacteria [9, 12] that consume atmospheric H2 to conserve energy during persistence [17–19]. Actinobacteria are abundant in soils based on culture-independent studies [9, 14, 20] and were thought to be primarily responsible for atmospheric H2 oxidation [14, 16].
Recent genomic and metagenomic investigations, along with pure culture work, have identified additional bacteria harboring group 1h [NiFe]-hydrogenases within the phyla Acidobacteria, Proteobacteria, Planctomycetes, Chloroflexi and Verrucomicrobia [14, 21–23]. Notably, Acidobacteria are one of the most abundant soil phyla with relative abundances in 16S rRNA libraries ranging from ca. 20 to 40% in temperate soils such as forests, grasslands and pasture soils [24]. They constitute a large and phylogenetically distinct phylum [25, 26] that harbors, diverse physiologies [27]. Two thermophilic acidobacterial strains were previously shown to consume atmospheric levels of H2, due to the presence of high-affinity [NiFe] hydrogenases [21, 28]. A recent large-scale comparative genome analysis of acidobacteria also identified the genes encoding the large and small subunits of the group 1h [NiFe]-hydrogenase (hhyL and hhyS, respectively) in genomes of various mesophilic soil acidobacteria, along with the necessary maturation and accessory genes [29]. Yet, it remained unknown whether mesophilic acidobacteria can scavenge H2, which are highly abundant in temperate soils where atmospheric H2 consumption was previously reported [30–32].
As more and more taxonomic groups have been identified to harbor group 1h [NiFe]-hydrogenases, it was our goal to design broadly inclusive primers for long-read sequencing that allow the investigation of diverse group 1h [NiFe]-hydrogenase communities. Sequencing of almost the complete large subunit gene further enables improved phylogenetic placement and identification of the amplified genes from soil samples. Using this new primer pair, we demonstrate that group 1h [NiFe] hydrogenases are widespread across many phyla, including previously unidentified groups. In addition, we illustrate that mesophilic acido-bacteria are prevalent and active members of the group 1h [NiFe] hydrogenase-harboring community in H2-consuming temperate soils and are capable of atmospheric H2 consumption. This work therefore reveals new mediators in the biogeochemically and ecologically important process of atmospheric H2 oxidation, and supports growing evidence that trace gases might be a universal energy source for bacterial persistence.
Materials and methods
Screening publicly available genomes and MAGs
Publicly available genomes and metagenome-assembled genomes (MAGs) (n = 175509, November 2018) were screened for the presence of hydrogenase large subunit genes using pfam model PF00374.19, as well as models constructed to be more sensitive to [NiFe] lineages 1–4 from HydDB [33]. For lineage-sensitive models, [NiFe] hydrogenases contained in HydDB were separated into the four major lineages [1–4] and models were constructed de novo. Amino acid sequences for each lineage were extracted from HydDB, clustered into centroids using usearch [34] (-sortbylength and –clustersmallmem –id 0.85) and aligned using MAFFT [35]. The resulting alignments were trimmed using trimAl with setting:automated1 [36] and models were constructed using hmmbuild from hmmer3 [37]. [NiFe] hydrogenases were identified in genomes and MAGs using hmmsearch (e-value<0.001). For all hidden Markov model (hmm) hits, the putative genes were back-screened against the Pfam-A database to verify that pfam model PF00374.19 was the best matching Pfam. All putative group 1 [NiFe]-hydrogenase large subunit genes (hhyL) were extracted and further screened using the HydDB online classifier [33]. CheckM was used to estimate completeness, contamination and heterogeneity of the genomes based on lineage-specific markers [38]. The taxonomy of MAGs containing the hhyL gene with a completeness of >50% was determined using the Genome Taxonomy Database (https://gtdb.ecogenomic.org). Duplicate copies of hhyL in a genome were removed and scored as one. The amino acid sequences derived from these hhyL genes were used to explore the phylogeny and confirm the taxonomic assignment of the full-length sequences retrieved in this study. Sequences were aligned with MUSCLE [39] and phylogenetic trees were generated using FastTree JTT + CAT model, along with estimating FastTree confidence [40].
Soil sample collection and nucleic acid extraction
Soil samples were collected from (a) a mature beech forest (Fagus sylvatica L.), ca. 40 km southwest of Vienna, Austria (more details can be found in [41]) collected in summers of 2012, 2013, 2014; (b) a managed grassland from the agricultural research station (AREC) in Raumberg-Gumpenstein, Austria (49°29’37”N, 14°06’10”E; more details can be found in [42]) collected in the summer of 2018; (c) the rhizosphere of Arrhenatherum elatius (tall oat-grass) grown at this aforementioned managed grassland, collected in the summer of 2018; and (d) biological soil crusts (of ~5 mm thickness) from the central Negev Desert, Israel (30°47’N, 34°46’E; more details on the site can be found in [43]) collected in the summer of 2017. DNA and RNA were extracted from ca. 0.4–0.5 g of soil using a modified bead-beating protocol in the presence of a CTAB buffer and phenol as previously described [44]. Samples were purified using OneStep™ PCR Inhibitor Removal Kit (Zymo, Irvine, CA, USA) and quantified using the Qubit dsDNA BR Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). For the generation of cDNA, extracts were purified with the Turbo DNA-free kit, quantified with the Qubit HS RNA Assay and reverse-transcribed using SuperScript IV Reverse Transcriptase, all according to the manufacturers’ protocol. All reagents and kits were purchased at Thermo Fisher Scientific, Waltham, MA, USA.
Sequencing of the group 1h [NiFe]-hydrogenase
Broadly inclusive primers for the large subunit gene (hhyL) of the group 1h [NiFe]-hydrogenase were designed using the program CODEHOP [45] using high-quality, full-length sequences downloaded from the HydDB [33] along with acidobacterial sequences (Acidobacteriaceae bacterium KBS 83 and Acidobacteriaceae bacterium KBS 96, Granulicella mallensis strain MP5ACTX8, Edaphobacter aggregans, “Ca. Solibacter usitatus” Ellin6076) (n = 105 sequences). Primer sequences are listed in Table S1. Reactions were performed in volumes of 25 µl containing the following components: 10 ng to 50 ng of DNA template, 2.5 µl of 10 × DreamTaq Green Buffer, 0.2 mM of each nucleotide dNTP mixture, 0.2 µg µl−1 of BSA, 0.5 µM of each primer, and 1.25 U of DreamTaq Green DNA Polymerase (all from Thermo Fisher Scientific, Waltham, MA, USA). Primers were optimized for annealing temperature and cycle number using soil DNA and genomic DNA of Acidobacteriaceae bacterium KBS 83. The final PCR program for amplification was: 95 °C for 3 min followed by 38 cycles of 95 °C for 30 s, 65 °C for 30 s and 72 °C for 1.5 min, and a single step of final elongation at 72 °C for 10 min.
Primers were adapted with barcodes at the 5’ end for each primer with 16-nt symmetric barcodes to allow multiplexing of samples in a single run [46]. Positive PCR products were purified using the CleanNA NGS magnetic bead-based clean-up system (CleanNA, Waddinxveen, Netherlands) and sequenced on a PacBio Sequel System (Pacific Biosciences, PacBio, Menlo Park, CA, USA) in circular consensus mode at the Vienna BioCenter Core Facilities (https://www.viennabiocenter.org/facilities/). Consensus sequences were generated for each read with more than five passes of circular sequencing and the reads were demultiplexed using pbbioconda package (https://github.com/PacificBiosciences/pbbioconda). Additional filtering, where average quality within a sliding window of 10 bp should be above 70 and no base call should have quality below 10, was performed with Mothur v. 1.34 [47]. Quality-trimmed sequences longer than 1000 bp were clustered at 95% identity using VSEARCH [48]. The 95% OTU cut-off was used based on the comparison between the sequence similarity scores of group 1h [NiFe]-hydrogenase large subunit genes and the 16S rRNA genes [9]; very few sequences had a similarity above 90%, as such we felt that 95% was a conservative cut-off for this functional gene. This cut-off is also consistent with a recent publication where organisms of the same species had an average nucleotide identity of ≥95% [49]. OTU centroids were taxonomically classified based on diamond blastx [50] search against NCBI-Nr database. Specificity of the amplified sequences was assessed with hmm models of [NiFe]-hydrogenase groups generated with data from HydDB [33]. Diversity estimates and β-diversity were assessed using the phyloseq Package in R [51]. Phylogenetic trees were constructed with OTU representatives based on deduced amino acid sequences in FastTree [40] along with a reference database [33] to identify acidobacterial clusters and closest relatives.
Sister libraries using previously published group 1h [NiFe]-hydrogenase primers [NiFe]-244F/568R and [NiFe]-1129F/1640R [18, 52] (Table S1) were generated on the above soil samples; details can be found in Supplemental Results and Discussion S1-1. The raw sequence data were deposited into the NCBI Short Read Archive under BioProject accession number PRJNA649096.
Evaluating group 1h [NiFe]-hydrogenase communities across the amplicon libraries
The near full-length group 1h [NiFe]-hydrogenase sequences stemming from PacBio sequencing were compared to the (i) [NiFe]-244F/568R MiSeq-derived sequences and (ii) [NiFe]-1129F/1640R MiSeq-derived sequences using bidirectional nucleotide blast (Nucleotide-Nucleotide BLAST v2.8.1+). Briefly, the top bit scores were generated using a word size of 7, maximum target sequences of 1 and normalized to the respective MiSeq self-blast ([NiFe]-244F/568R or [NiFe]-1129F/1640R). Data were plotted on a frequency histogram to assess the overlap of the communities.
The classification of the OTU representatives was performed using the Evolutionary Placement Algorithm (EPA) implementation in RAxML [53] using the GTR model of amino acid substitution. Briefly, a hmm model was generated based on a MAFFT [35] alignment (L-INS-I mode) of HydDB [33] sequences using hmmer v3.1b2 (www.hmmer.org) [54]; this model was used to align sequences for the base tree construction and to place sequences using EPA. A base tree of 1,716 amino acid reference sequences (avg. 585 amino acids in length) of the group 1h [NiFe]-hydrogenase was generated in FastTree based on the JTT + CAT model [40] upon aligning the sequences using the hmm model (hmmer v3.1.b2). The OTU representatives from the three amplicon libraries ([NiFe]-244F/568R MiSeq; [NiFe]-1129F/1640R MiSeq; [NiFe]-full-length PacBio) were translated and manually curated upon alignment using the hmm model (hmmer v3.1.b2). Poorly aligned sequences were removed from the final dataset leaving 325 (ca. 100 amino acid in length), 61 (ca. 130 amino acid in length) and 1923 (ca. 486 amino acid in length) sequences from [NiFe]-244F/568R MiSeq, [NiFe]-1129F/1640R MiSeq, and [NiFe]-full-length PacBio amplicon libraries, respectively. Maximum likelihood trees were reconstructed in RAxML.
The average genetic distance was determined for the hmm aligned OTU representatives from [NiFe]-244F/568R MiSeq, [NiFe]-1129F/1640R MiSeq and [NiFe]-full-length PacBio amplicon libraries using MEGA7 [55] by computing the pairwise distances. Briefly, the nearly full-length sequences were trimmed to each respective region of the MiSeq primer pairs and pairwise distances were compared using the Poisson model, with a uniform rate among sites. Gaps and missing data were treated as pairwise deletions.
Bacterial strains and growth conditions
Acidobacterial strains (Acidobacteriaceae bacterium KBS 83 [56] and E. aggregans [57]) were grown using a defined vitamins and salts medium, VSB-6 [56, 58], with 5 mM glucose as the carbon source. The use of a defined medium allows one to vary total carbon (here suppled as glucose) to determine when the final yield (as measured by optical density at 600 nm) reduced to the proportional decrease in glucose concentration. To determine carbon-limiting conditions, strains were grown in the aforementioned defined medium in differing glucose concentrations (5 mM and 10 mM glucose). Carbon-limiting conditions were defined when the cellular density was proportional to the amount of carbon provided. They were incubated on an orbital shaker (ca. 130 RPM) under aerobic conditions at 24 °C. Growth was monitored by measuring the optical density at 600 nm.
Primer design and RT-qPCR
Total RNA was extracted from cultures using a modified standard bead-beating protocol [44] with one-round of bead beating and acidified phenol/choloroform/isoamyl alcohol (pH 4.5). Extracts were purified with the Turbo DNA-free kit according to the manufacturer’s protocol. RNAs were normalized to 3 ng µL−1 and ca. 30 ng were used for cDNA synthesis using SuperScript IV Reverse Transcriptase, following the manufacturer’s protocol.
Quantitative PCR (qPCR) was performed on a C1000 Touch thermocycler equipped with a CFX96 Real Time System in combination with iQ SYBR-Green qPCR Assay (Biorad, Hercules, CA, USA). Primers were designed for the large (hhyL) and small (hhyS) subunits of the group 1h [NiFe]-hydrogenase of E. aggregans and Acidobacteriaceae bacterium KBS 83 targeting the same region of the respective gene, along with the homolog of the hhyS as was done previously [21]. All primers are listed in Table S1. Standard curves were constructed with 10-fold serial dilutions of genomic DNA of each respective strain, typically ranging between 106 to 1 copy. The qPCR assay was performed in 20 µl volume containing the following components: 10 µl of SYBR Green Supermix (Biorad, Hercules, CA, USA), 0.4 ng µl−1 BSA (Thermo Fisher Scientific, Waltham, MA, USA), 0.2 µM (E. aggregans) or 1 µM (Acidobacteriaceae bacterium KBS 83) of each primer and 1–5 µl of cDNA template. The program used was: 95 °C for 3 min, followed by 40 cycles of 95 °C for 15 s, 65 °C (hhyS, hhyL for Acidobacteriaceae bacterium KBS 83 were run at 68 °C and 58 °C, respectively) for 30 s for annealing and 72 °C for 1 min for extension. The expression of the large and small subunit genes of the group 1h [NiFe]-hydrogenase was assessed using these newly designed primer pairs upon normalization to the 16S rRNA gene using the above assay but with an annealing temperature of 68 °C. Melting curves were generated between 65 °C and 95 °C. Data were processed and analyzed using the CFX Manager software (Biorad, Hercules, CA, USA) and data were log-transformed to determine fold-increase. Data on the specificity of the qPCR assay can be found in Fig. S1.
H2 consumption and hydrogenase activity measurement assays for acidobacterial cultures
Briefly, cells were harvested in exponential (OD600nm ranging from 0.25–0.38) and stationary (OD600nm ranging from 0.50–0.53) phase by centrifugation at 10000 RPM for 10 min to allow pelleting of these cells that produce extracellular material. Harvested cells were concentrated 10-fold, resuspended in 10 mL carbon-free VSB medium and transferred to 110 mL serum vials sealed with butyl rubber stoppers. Headspace was flushed with synthetic air (Messer Gas, Bad Soden, Germany) and supplemented with ~20 ppmv H2 (Linde Gas, Dublin, Ireland). Strains were incubated at 24 °C on an orbital shaker (ca. 130 RPM) for 7 days. Headspace samples were periodically sampled and the H2 concentration was determined via a Trace GC Ultra (Thermo Scientific, Austria) with a pulse discharge detector (PDD). This GC has the ability to accurately detect H2 down to concentrations of 0.5 ppmv. A gas chromatograph with a pulsed discharge helium ionization detector (model TGA-6791-W-4U-2, Valco Instruments Company Inc. (VICI, Houston, TX, USA)) was used for sub-atmospheric H2 concentrations (ca. 0.1 to 0.5 ppmv) as previously described [22]. To ensure an oxygenated headspace during these incubations, oxygen concentrations were monitored and were never lower than 18% (v/v) (Fig. S2). Hydrogen consumption controls were run on uninoculated medium, heat-killed Acidobacteriaceae bacterium KBS 83 cells and an acidobacterial strain (Terriglobus roseus KBS 63) that does not contain group 1h [NiFe]-hydrogenase genes [29] over a period of 24 h with starting H2 concentrations of ~80 ppmv to ensure any potential consumption activity would be sufficiently high for detection (Fig. S3).
The enzyme kinetics (Vmax, Km[app]) of the group 1h [NiFe]-hydrogenase of pure cultures were determined using similar methods as described in [17] using gas chromatography. Briefly, cells were harvested as described above, concentrated and resuspended in carbon-free VSB medium with various concentration of H2 (ranging from 0 to 1000 ppmv). Consumption was monitored via gas chromatography over a period of 120 h. H2 uptake rates were determined at each respective concentration and normalized to mg of protein using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Enzyme kinetics for the Michaelis–Menten kinetics, non-linear least squares method was determined in R [59], while the Hanes-Woolf plot was calculated manually.
Soil H2 consumption assays
Soil samples were collected from aforementioned mature beech forest, managed grassland and desert biological soil crusts to explore H2 consumption. Briefly, approximately 1–2 grams of soil (or of natural dry soil crust) was incubated in a sealed 110 mL serum bottle, flushed with synthetic air and supplemented with ~20 ppmv H2. H2 consumption was monitored in the temperate soils using a Trace GC Ultra (Thermo Scientific, Austria) with a PDD-type detector down to a concentration of 0.5 ppmv; sub-atmospheric H2 concentrations (ca. 0.1–0.5 ppmv) were determined on a gas chromatograph with a pulsed discharge helium ionization detector (PDHID) (model TGA-6791-W-4U-2, Valco Instruments Company Inc. (VICI)) as described above. All H2 measurements of biological soil crust samples were performed using the VICI gas chromatograph with a PDHID detector.
To determine the Km[app] of H2 for the beech forest soil, we incubated 1–2 grams of soil in a sealed 110 mL serum bottle. The headspace was flushed with synthetic air and supplemented with H2 ranging from 10 to 1000 ppmv, as higher concentrations are needed to estimate Km[app] and Vmax. The headspace was sampled over 24 h and uptake kinetics were calculated using the Michaelis–Menten kinetics, non-linear least squares method was determined in R [59] and normalized to gram dry soil.
Results
Grassland and forest soils harbor active and diverse communities of atmospheric H2-oxidizing bacteria
We measured bacterial H2 consumption in soils from a beech forest, a managed grassland and a desert biological soil crust. The desert soil crust consumed H2 very slowly over the course of two weeks to sub-atmospheric levels (Fig. 1a). In contrast, the forest and managed grassland soils rapidly consumed H2 to sub-atmospheric levels over a 24-h period (Fig. 1a). Differences between H2 uptake amongst the soils are reflected in the contrasting rate constant (k) values (Fig. 1a). There was no significant difference in the estimated rate constant values between the temperate soils. The apparent Michaelis constant, Km[app], was estimated to be 33 ± 12 nM (Fig. 1b in the beech forest soil); this soil had a significantly (p value < 0.05) higher rate constant value when compared to the biological soil crust.
Fig. 1. Activity and diversity of bacteria mediating atmospheric H2 oxidation in three soil ecosystems.

a Hydrogen consumption by soils collected from a beech forest, managed grassland and desert biological soil crust. Dotted lines represent atmospheric concentrations of H2 (~0.53 ppmv). Data points depict the mean ± standard deviation. b Michaelis–Menten kinetics of H2 oxidation by the beech forest soil. Best-fit curve was determined using a Michaelis–Menten non-linear regression model. c Alpha diversity (Shannon index) and d beta diversity (Bray–Curtis dissimilatory) of the group 1h [NiFe]-hydrogenase large subunit (hhyL) genes in the soils detected by long-read amplicon sequencing. Analysis of variance with a Tukey’s HSD mean separation was performed across the soil types for the Shannon index; similar letters indicate that no significant difference was observed (p value > 0.05). Data were rarefied; unrarefied comparisons can be found in Fig. S4.
To reveal the microorganisms mediating this H2 uptake, we designed degenerate primers to target the group 1h [NiFe]-hydrogenase large subunit genes (hhyL) for long-read amplicon sequencing of the different soil communities. These primers were designed to encompass the diversity of hhyL sequences across ten different phyla. We amplified and sequenced hhyL genes from total community DNA extracted from the managed grassland (bulk and rhizosphere) and forest soils, along with biological soil crusts. Overall, diverse hhyL-encoding communities were observed across the soils, with 2403 OTU95 clusters (95% cut-off) identified in total. The designed primers proved highly specific for the group 1h [NiFe]-hydrogenase subgroup, as no other genes were amplified (Table S2) and rarefaction curves confirmed the sequencing effort was sufficient to capture the diversity of hhyL sequences present (Fig. S4a, Shannon index). Based on the Shannon index, the H2-oxidizing community in the managed grassland soils had a significantly higher diversity as compared to the beech forest soil (avg. p value < 0.02) and biological soil crusts (avg. p value < 0.003), which was observed for rarefied data (Fig. 1c); a similar diversity pattern was observed for the unrarefied data (Fig. S4c). The composition of the hhyL-harboring communities was more similar amongst the managed grassland bulk, managed grassland rhizosphere and forest soils than the biological soil crust based on rarefied (Fig. 1d) and unrarefied data (Fig. S4b).
We subsequently performed taxonomic and phylogenetic analysis of the hhyL sequences derived from the investigated soils (Fig. 2) alongside those that we retrieved from previously published genomes and metagenome-assembled genomes (Figs. 2 and 3). Bacteria within these temperate soils that encoded group 1h [NiFe]-hydrogenase sequences were affiliated with Acidobacteria, Actinobacteria, Chloroflexi, Nitrospirae, Planctomycetes, Proteobacteria (Alpha-, Beta- and Delta-) and Verrucomicrobia (Fig. 2a, b). In contrast, the biological soil crusts harbored group 1h [NiFe]-hydrogenase sequences predominantly affiliated with Actinobacteria and Chloroflexi (Fig. 2a, b).
Fig. 2. Taxonomic and phylogenetic analysis of the group 1h [NiFe]-hydrogenase large subunit (hhyL) genes in three soil ecosystems.
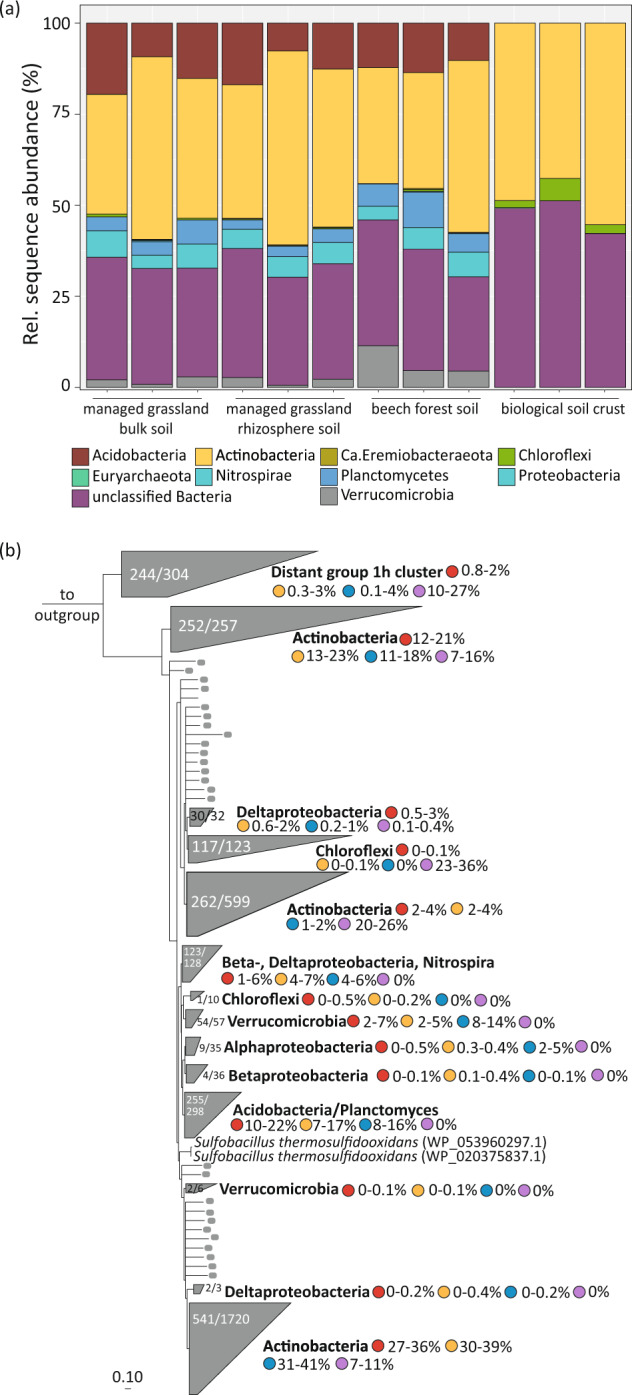
a Predicted taxonomic distribution of amplified hhyL sequences across the investigated soils based on their closest hits to sequences in the NCBI-nr database. Triplicate samples are depicted for each soil. Colors represent different taxonomic groups. b RAxML-EPA tree of amino acid sequences of the group 1h [NiFe]-hydrogenase large subunit (hhyL) from long-read amplicon sequencing and reference sequences. The phylogenetic placements of OTU representatives stemming from nearly full-length sequences are depicted in gray: collapsed clusters containing OTU representative are shaded gray and non-clustered OTU representatives are colored gray. The number of the placed, nearly full-length hhyL sequences and the total number of sequences in each cluster are depicted. The proportions of sequences within each soil are depicted to the right of the clusters managed grassland (red), rhizosphere (orange), beech forest (blue) and biological soil crust (purple). Sequences from group 1g [NiFe]-hydrogenases were used as an outgroup. The scale bar indicates the number of substitutions per site.
Fig. 3. Phylogenetic tree of the group 1h [NiFe]-hydrogenase large subunit (hhyL) sequences stemming from reference genomes and metagenome-assembled genomes (MAGs) based on amino acid sequences (n = 1650 sequences, ca. 570 amino acid positions).
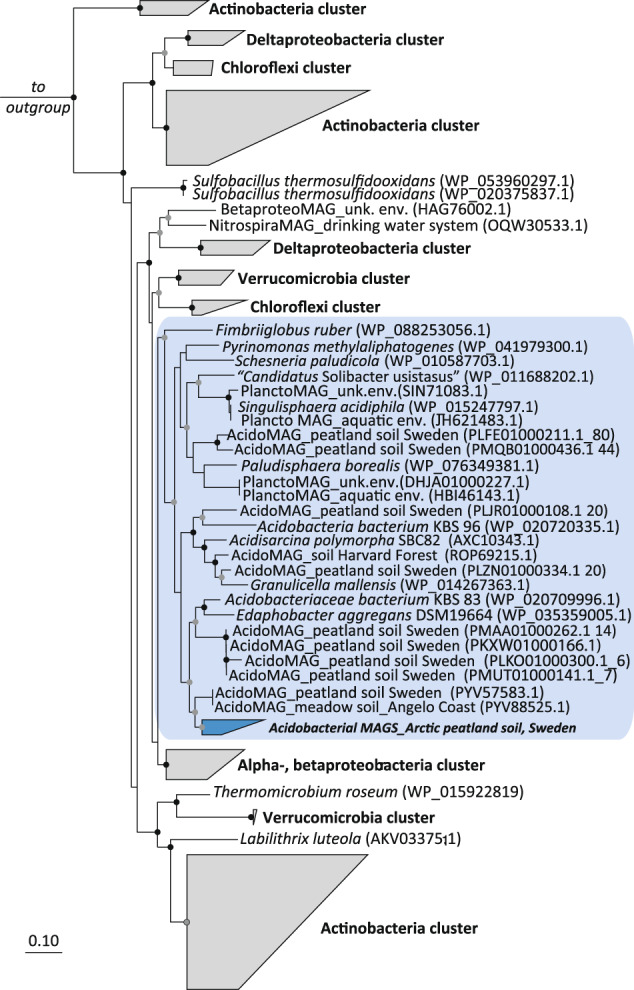
The tree was calculated using FastTree using the JTT + CAT model; FastTree confidence values of >95% (black circles) and >80% (gray circle) are depicted. The scale bar indicates the number of substitutions per site. The acidobacterial cluster is depicted in blue. The environmental source of the sequences (if available) is depicted in the name of each MAG. Additional information on publicly available MAG sequences depicted in the tree can be found in Table S4. Group 1g [NiFe]-hydrogenase sequences were used as an outgroup.
There was an additional deep-branching cluster of sequences containing group 1h [NiFe]-hydrogenases of diverse origin, such as members of the phyla Actinobacteria, Chloroflexi, Bacteriodetes, Acidobacteria, Proteobacteria and Euryarchaeota (Fig. 2b, “Distant group 1h cluster”). Although these sequences were all classified as a group 1h [NiFe]-hydrogenase based on the HydDB [33], they appear to be distantly related to the other, main branch of the tree (Fig. 2b). Approximately 244 OTU representatives of the long-read sequences were placed in this cluster; these sequences were found across all soil samples, but were most prevalent in the biological soil crust samples (ca. 10–27%). In addition, there were various OTU representatives that did not group in clusters that contain reference sequences (Fig. 2b, gray dots).
For an evaluation of the newly designed long-read primer pair, we compared its coverage with the coverage of previously published short-read primer pairs ([NiFe]-244F/568R and [NiFe]-1129F/1640R [18, 52]. These short-read primers led to similar patterns of alpha- and beta-diversity (Fig. S5) compared to the long-read primers (Figs. 1c, d and S4). However, the long-read primer pair captured a breadth of diversity exceeding those attained with the short-read primers (Fig. S6). More specifically, the newly designed long-read primer pair captured an additional group of actinobacterial sequences (Fig. S6c, top-most Actinobacteria cluster), along with putative members in the Deltaproteobacteria, Nitrospira and the deep-branching cluster of group 1h [NiFe]-hydrogenases of diverse origin (Fig. S6c, “Distant group 1h cluster”). A further difference amongst the primer pairs was the increased number of sequences without any reference sequences in the long-read amplicon libraries (Fig. S6c, indicated by gray dots) compared to both short-read primer pair sets (Fig. S6a, b). The increased diversity in the long-read primer pair amplicon libraries was further supported by calculating the average genetic distance in the group 1h [NiFe]-hydrogenase community, which was increased compared to the short-read primer pairs (Supplementary Information SI-1), and via bidirectional nucleotide blast analyses and primer match analyses (Supplementary Information SI-1, Fig. S7, Table S3). Taken together, it appears that the newly designed long-read primers pair targets a wider (and putatively novel) phylogenetic diversity of group 1h [NiFe]-hydrogenases in the investigated soils.
A cluster of acidobacterial high-affinity hydrogenases are abundant and expressed in the grassland and forest soils
The long-read amplicon data further revealed that, after Actinobacteria, Acidobacteria were the second most represented phylum in the hhyL amplicons (Fig. 2). Seven to 22% of amplicon sequences stemming from forest and managed soils were most closely related to hhyL sequences from acidobacterial genomes (Fig. 2b). In contrast, acidobacterial hydrogenases were absent from the biological soil crusts (Fig. 2), where over half of the obtained OTUs were affiliated with three actinobacterial-specific hhyL clades (Fig. 2b). Analysis of publicly available acidobacterial genomes and MAGs confirmed that ~7% (of 745 genomes) harbored the hhyL gene (Fig. S8), including members from subdivisions 1 (genera Acidipila, Edaphobacter and Granulicella), 2 (MAGs only), 3 (genera Bryobacter, ‘Candidatus Solibacter’) and 4 (genus Pyrinomonas). The acidobacterial hhyL sequences stemming from pure cultures [56, 57, 60–63], genomes and MAGs originated from soil (Table S4) and formed a distinct cluster (Fig. 3), as also observed with our derived amplicon sequences (Fig. 2b), together with hhyL sequences from Planctomycetes. Overall the acidobacterial cluster had an average sequence similarity of ca. 83% based on amino acid sequences. Despite the described more limited diversity covered with the short-read primers (Supplementary Information SI-1), amplification of cDNA derived from the soils with these primers revealed that the hhyL genes of various phyla, including the Acidobacteria, were expressed in the temperate soils (Supplementary Information SI-1, Figs. S9–10).
Acidobacteria isolated from temperate forest and grassland soils oxidize atmospheric H2 during carbon limitation
The above culture-independent inferences suggest that members of the Acidobacteria may be important mediators of atmospheric H2 oxidation in temperate soils. As such, we investigated the gene expression and activity of the group 1h [NiFe]-hydrogenase present in two mesophilic acidobacterial isolates, the grassland soil bacterium Acidobacteriaceae bacterium KBS 83 [56] and forest soil bacterium Edaphobacter aggregans [57]. In addition to containing hhyL and hhyS [29], both organisms also possess an additional copy of hhyS ca. 3,600 bp upstream of the structural and maturation genes, as observed in P. methylaliphatogenes [21] (Supplementary Information SI-2).
Transcription of both the large (hhyL) and small (hhyS) subunits of the hydrogenase was upregulated during carbon-limitation (Fig. 4a, d). We used defined media to determine the conditions that induced stationary phase due to carbon-limitation in both strains (Fig. S11). Under stationary phase conditions, the transcription of the hhyL gene (normalized to the 16S rRNA gene) was upregulated by ~125-fold in Acidobacteriaceae bacterium KBS 83 (Fig. 4a) and ~3.5-fold in E. aggregans (Fig. 4d) compared to exponential growth. In both organisms, transcription of the hhyS gene was not detected during exponential phase (< 2 copies per ng cDNA), but was detected during stationary phase (Fig. 4a, d). The apparent differential expression of the hhyS and hhyL genes in exponential phase may be attributed to different promoter regions; potential different promoter regions were computationally identified across the structural genes of Acidobacteriaceae bacterium KBS 83, which presumably also have different transcription factors (Fig. S12). The expression of the hhyS homolog was detected in Acidobacteriaceae bacterium KBS 83, but not in E. aggregans (Supplementary Information SI-2).
Fig. 4. Expression, activity, and kinetics of the enzymes mediating atmospheric H2 oxidation in two acidobacterial strains isolated from temperate soils, Acidobacteriaceae bacterium KBS 83 and Edaphobacter aggregans.

a, d Growth curves of the strains over time (days) (x-axis) and expression levels (inset) of the group 1h [NiFe]-hydrogenase structural subunit genes (large, hhyL; small, hhyS) during exponential and stationary phase. Arrows depict the growth phases in which cells were harvested for gene expression investigations (gray arrow, exponential phase; black arrow, stationary phase). During this experiment, H2 consumption was measured on stationary phase cells (black arrows) and in a parallel experiment on exponential phase cells (Fig. S13), as H2 consumption assays required the entire biomass of such an experiment. b, e H2 consumption of stationary phase stage cells of each respective strain; x-axis depicts the start of measurements for H2 consumption after harvesting cells from growth curves of panels a, d. Dashed lines represent atmospheric H2 concentrations (~0.53 ppmv), whereas red points depict the heat-killed controls for each respective strain. The final sub-atmospheric H2 measurement for each strain was performed on a gas chromatograph with a pulsed discharge helium ionization detector (model TGA-6791-W-4U-2, Valco Instruments Company Inc.); this measurement is indicated by an asterisk. Over the course of the experiment, we observed a slight decrease in H2 for our medium control (8%). Even when this loss is accounted for in the final sub-atmospheric measurement, the concentration is still below atmospheric levels of H2, (0.27 ppmv for Acidobacteriaceae bacterium KBS 83 and 0.39 ppmv for E. aggregans). Additional controls can be found in Fig. S3. c, f Apparent kinetic parameters of H2 oxidation for the strains based on whole-cell assays. Best-fit curves were determined using the Michaelis–Menten non-linear regression model; similar values were observed using Hanes–Woolf plots (Table S5).
We subsequently tested whether these strains consume atmospheric H2 under carbon-limiting conditions. Both strains consumed H2 from levels of ~20 ppmv to a concentration of 0.25 ± 0.03 ppmv (Acidobacteriaceae bacterium KBS 83) and 0.36 ± 0.06 ppmv (E. aggregans) after 172 h (Fig. 4b, e). H2 was only consumed by carbon-limited stationary cells; no H2 oxidation was observed in cells exponentially growing on the defined medium (Fig. S13) or in E. aggregans cultures grown to stationary phase on an undefined medium (presumably due to non-carbon-limiting conditions) (Fig. S14). Likewise, no H2 was consumed in the uninoculated medium, heat-killed controls, or in an acidobacterial strain lacking the group 1h [NiFe]-hydrogenase (Terriglobus roseus) (Fig. S3). The kinetic parameters of the group 1h [NiFe]-hydrogenases were determined on whole cells of strains Acidobacteriaceae bacterium KBS 83 and E. aggregans. The apparent half-saturation constant (Km[app]) measured on cells of Acidobacteriaceae bacterium KBS 83 was 173 ± 63 nM H2 with a saturating rate (Vmax[app]) of 0.27 ± 0.03 µmol H2 mg protein−1 h−1 (Fig. 4c). The Km[app] for H2 uptake by E. aggregans was 95 ± 31 nM with a Vmax[app] of 6.16 ± 1.06 nmol H2 mg protein−1 h−1 (Fig. 4f); similar values were observed using another model (Table S5). These kinetic parameters are consistent with previous estimates for mid- to high-affinity hydrogenases of pure cultures (Fig. 5) and temperate soils, where the Acidobacteria are typically found.
Fig. 5. Comparison of apparent substrate affinity (Km[app], in nM) for H2-oxidizing bacteria and archaea spanning different taxonomic groups.

Figure was amended from [17]; data were extracted from Greening et al. [17] and Islam et al. [22]. Asterisks depict microorganisms harboring a group 1h [NiFe]-hydrogenase. Acidobacteria estimates derived in this study are depicted in blue, and data from the investigated beech forest soil are shown in green.
Discussion
Our genomic surveys and the targeted hhyL amplicon sequencing of soil samples add to the growing evidence that group 1h [NiFe]-hydrogenases appear to be widespread in numerous taxonomic groups (Fig. 2). The genomic survey identified new groups, namely members of the Deltaproteobacteria and Nitrospira, that harbor group 1h [NiFe]-hydrogenase genes, in addition to Acidobacteria, Actinobacteria, Bacteriodetes, Chloroflexi, Planctomycetes, Proteobacteria (Alpha- and Beta-) and Verrucomicrobia (Figs. 2 and S8) in accordance with previous surveys [9, 10, 14, 16]. These phyla are commonly found in soils with varying relative abundances [64]; however, it should be noted that with the exception of the Acidobacteria and Actinobacteria, group 1h [NiFe]-hydrogenases are present in less than 3% of genomes from each of these phyla (Fig. S8). Furthermore, all of the mentioned phyla are amplifiable with our newly designed primers (Fig. 2), thus allowing future investigations to explore the distribution of these groups across environments using long-read amplicon sequencing.
Our newly designed group 1h [NiFe]-hydrogenases primers not only capture the diversity of previously established group 1h [NiFe]-hydrogenases primers [18, 52], but also additional sequence diversity across edaphically different soils (Fig. S6). In comparison to these other primers, phylogenetic analysis revealed the presence of additional clusters as well as OTUs without reference sequences (Fig. S6, in gray), the latter suggesting the presence of putative novel hydrogenases. We suggest that the use of long-read hhyL sequences could allow for improved phylogenetic placement and identification of the amplified sequences from environmental samples, along with the use of the Evolutionary Placement Algorithm implemented in RAxML using a base tree built with not only publicly available hhyL sequences (mostly stemming from isolates) but also with hhyL genes identified in genomes and MAGs of uncultured organisms (Fig. S8). As such, we have added an additional primer pair to the toolbox of exploring the group 1h [NiFe]-hydrogenases in environmental samples. Although it is unclear if it captured the complete diversity of atmospheric H2-oxidizers harboring group 1h [NiFe]-hydrogenases in the investigated soils, it captured some putative novel sequences and previously undetected groups (Fig. 2). These findings will aid future work to attribute which community members and enzyme lineages are responsible for the biogeochemically important process of atmospheric H2 oxidation.
Biological soil crusts exhibited the lowest diversity estimates (Fig. 1c), being dominated by hhyL sequences assigned to Actinobacteria, Chloroflexi and the “Distant group 1h cluster” (Fig. 2b), and the slowest H2 consumption (Fig. 1a, k = 0.0051). Temperate soil libraries had higher diversity estimates, especially in the managed grassland soil (Fig. 1c), and contained sequences related to the Actinobacteria, Acidobacteria, Planctomycetes, Proteobacteria and Verrucomicrobia (Fig. 2). These soils exhibited faster H2 consumption (Fig. 1a; managed grassland k = 0.1752, forest soil k = 0.2495). Follow-up investigations will be necessary to reveal the contributions of these different groups to H2 oxidation (also including hydrogenases other than the group 1h-type), but preliminary sequencing of cDNA shows a diverse community actively transcribing hhyL genes in the temperate soils (Fig. S10).
Many of the hhyL sequences detected by using the new primer pair clustered with the two acidobacterial strains tested here for H2 consumption (Acidobacteriaceae bacterium KBS 83 and E. aggregans), in addition to other mesophilic acidobacteria such as Acidobacteriaceae bacterium KBS 96, G. mallensis, and “Ca. Solibacter usitatus” Ellin6076”. All of these strains are described as aerobic heterotrophs [56, 57, 61, 65], which is consistent with the group 1h [NiFe]-hydrogenase being both oxygen-tolerant [66] and linked to the aerobic respiratory chain [10]. The acidobacterial group 1h [NiFe]-hydrogenase sequences from genomes and MAGs formed a distinct clade (Figs. 2 & 3) together with those from the Planctomycetes. Some of these Planctomycetes-affiliated sequences were generated from MAGs and, as such, this clustering could be a result of poor assemblies, incompleteness or high contamination of MAGs. Yet it is unlikely as this cluster also contains sequences stemming from pure cultures (such as Singulisphaera acidiphila) (Fig. 3).
We demonstrate that mesophilic acidobacteria are capable of scavenging H2 in pure culture (Fig. 4). Although previous work has shown that two thermophilic acidobacterial strains scavenge atmospheric H2 either due to a group 1h or 1f [NiFe]-hydrogenase [21, 28], only very few sequences with an identity of ≥97% could be detected in the NCBI database and of those identified, were primarily from thermophilic environments (Fig. S15). Therefore, investigating the H2-oxidation capability in representative mesophilic strains inhabiting temperate soils was essential. With the data presented in this study on mesophilic acidobacteria, this is first report of a mesophilic bacterium outside the Actinobacteria [9, 20, 67] being capable of atmospheric H2 oxidation via a group 1h [NiFe]-hydrogenase. We were further able to detect expressed acidobacterial hhyL in temperate soils (Fig. S10), illustrating that acidobacteria are active in the soils that consume H2 (Fig. 1a). This is ecologically significant given mesophilic acidobacteria are abundant across temperate soils [68], which together comprise ~20–30% of terrestrial environments [69]. Further investigations are warranted to determine the acidobacterial contribution to H2 consumption in temperate soils, for instance in comparison to the well-studied actinobacteria. The two investigated strains, Acidobacteriaceae bacterium KBS 83 and E. aggregans, exhibit mid to high-affinity H2 uptake kinetics comparable to those measured for other bacteria harboring group 1h [NiFe]-hydrogenases (Fig. 5, asterisks). The whole soil communities within the beech forest soil also exhibited high-affinity H2 uptake kinetics (Fig. 1b), suggesting that H2 is predominantly being oxidized by bacteria expressing high-affinity enzymes (i.e., group 1h [NiFe]-hydrogenases). This is congruent with previous surveys showing these enzymes are the most prevalent hydrogenases in grassland and forest soils [14].
The Km[app] of the investigated mesophilic soil acidobacteria (95 and 172 nM) was higher compared to the thermophilic acidobacterium P. methylaliphatogenes (35 nM), but in line with the higher-end of model high-affinity H2-oxidizing bacteria such as representatives in the Actinobacteria (Fig. 5). Yet the Km[app] of the investigated strains was lower than the Chloroflexi strain Thermomicrobium roseum that consumes H2 from both geological and atmospheric sources [22] (Fig. 5). This further demonstrates that group 1h [NiFe]-hydrogenases spanning both the high- and mid-affinity Km[app] values are capable of using atmospheric concentrations of H2. The Vmax rate for Acidobacteriaceae bacterium KBS 83 were ca. 5-fold higher than that observed for P. methylaliphatogenes [21], but 2 to 6-fold lower than those observed for T. roseum [22] and Mycobacterium smegmatis [17]. In contrast, the Vmax rate of E. aggregans was orders of magnitude lower than the aforementioned strains. This observed variation in Vmax within the strains used in this study and those previously published [17, 21] suggest there is high variability among group 1h [NiFe]-hydrogenases. Furthermore, the relative contributions of group 1h [NiFe]-hydrogenases to atmospheric H2 uptake in-situ remain unclear, as does to what extent duration of carbon starvation or other growth-limiting conditions influences group 1h [NiFe]-hydrogenases expression and potential activity in soil environments.
Our data suggest that mesophilic acidobacteria use atmospheric H2 to adapt to carbon starvation, which is in accordance with other soil strains [16, 17]. The structural genes encoding the hydrogenases were highly upregulated by both species in stationary-phase, carbon-starved cultures compared to exponentially growing, carbon-replete cultures (Fig. 4a, d). Likewise, activity was only observed in carbon-starved cultures (Figs. 4b, e and S14). Exponential bacterial growth is a state rarely found in nature; rather in many ecosystems (such as soil), bacteria enter a non-replicative persistent state [70] for extended periods of time [71]. It was estimated that up to 80% of the soil microorganisms at a given time [72] will be in such a state, often referred to as dormancy. Hydrogen has previously been proposed as a possible universal energy source for survival [73]. It was then proposed more specifically that the use of group 1h [NiFe]-hydrogenases facilitate the ability to scavenge atmospheric H2 to sustain aerobic respiration during periods of starvation in actinobacteria [17] and thermophilic acidobacteria [21, 28]. In this study, we further extend this working hypothesis to encompass mesophilic acidobacteria. It is likely that the oxidation of atmospheric H2, as a ubiquitous, diffusible, high-energy substrate, enables mesophilic acidobacteria to meet maintenance energy needs during persistence [10, 18]. H2-scavenging acidobacteria could have a selective advantage in periods of carbon depletion and therefore an increased likelihood to persist in the soil. Although group 1h [NiFe]-hydrogenase genes could only be detected in ca. 7% of 745 investigated acidobacterial genomes (Fig. S8), this may reflect the lack of adequate representations of Acidobacteria in our public databases relative to the breadth of diversity found in nature. It is also plausible to assume that there are other physiologies that allow persisting periods of carbon starvation, such as atmospheric carbon monoxide oxidation as suggested recently for actinobacteria [74] and chloroflexi [22].
Overall, the finding that mesophilic acidobacteria and likely other diverse microorganisms in soil can oxidize atmospheric H2 has important implications for both atmospheric chemistry and microbial ecology. These bacteria potentially contribute to the major sink of the global H2 cycle. Moreover, atmospheric H2 scavenging is hypothesized to contribute to bacterial persistence, with theoretical estimates predicting that atmospheric H2 provides the necessary maintenance energy for 107 to 108 bacteria per gram of soil [10, 18]. This physiology could, therefore, aid in maintaining populations and genotypes, ultimately sustaining soil microbial biodiversity.
Supplementary information
Acknowledgements
This work was support by an Austrian Science Fund (FWF) project grant [P26392-B20 to DW and SE] and an ERC Starting grant (grant agreement number 636928, to DW) from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program. In addition, CG was supported by an ARC DECRA Fellowship [DE170100310] and NHMRC EL2 Fellowship [APP1178715]. The authors would like to thank Marlies Dietrich, Erich M. Pötsch, Stefanie Imminger, Osnat Gillor, Sean Bay and Florian Strasser for their help with soil sample collection, Katharina Zingerle for her assistance with sample preparation for the primer evaluation, Daniela Trojan for the use of her optimized RNA extraction protocol for acidobacteria, Claus Pelikan & Maximilian Nepel for assisting with pre-processing the MiSeq sequence data, Thanavit Jirapanjawat, Margarete Watzka and Sean Bay for assistance with H2 measurements, and the Division of Computational Systems Biology for providing and maintaining the Life Science Compute Cluster (LiSC) at the University of Vienna. We thank Kristen Deangelis and Grace Pold for allowing using the sequences SIN71083.1 and ROP69215.1 in Fig. 3.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Andrew T. Giguere, Stephanie A. Eichorst
Supplementary information
The online version of this article (10.1038/s41396-020-00750-8) contains supplementary material, which is available to authorized users.
References
- 1.Conrad R. Soil microorganisms as controllers of atmospheric trace gases (H2, CO, CH4, OCS, N2O, and NO) Microbiol Rev. 1996;60:609–40. doi: 10.1128/mr.60.4.609-640.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rhee TS, Brenninkmeijer CAM, Röckmann T. The overwhelming role of soils in the global atmospheric hydrogen cycle. Atmos Chem Phys. 2006;6:1611–25. [Google Scholar]
- 3.Downey NVS, Randerson JT, Eiler JM. Molecular hydrogen uptake by soils in forest, desert, and marsh ecosystems in California. J Geophys Res. 2008;113:G03037. [Google Scholar]
- 4.Schmitt S, Hanselmann A, Wollschläger U, Hammer S, Levin I. Investigation of parameters controlling the soil sink of atmospheric molecular hydrogen. Tellus B Chem Phys Meter. 2009;61:416–23. [Google Scholar]
- 5.Novelli PC, Lang PM, Masarie KA, Hurst DF, Myers R, Elkins JW. Molecular hydrogen in the troposphere: Global distribution and budget. J Geophys Res. 1999;104:30427–44. [Google Scholar]
- 6.Downey NVS, Randerson JT, Eiler JM. Temperature and moisture dependence of soil H2 uptake measured in the laboratory. Geophys Res Lett. 2006;33:1–5. [Google Scholar]
- 7.Häring V, Conrad R. Demonstration of two different H2-oxidizing activities in soil using an H2 consumption and a tritium exchange assay. Biol Fertil Soils. 1994;17:125–8. [Google Scholar]
- 8.Schuler S, Conrad R. Soils contain two different activities for oxidation of hydrogen. FEMS Microbiol Ecol. 1990;73:77–84. [Google Scholar]
- 9.Constant P, Chowdhury SP, Hesse L, Pratscher J, Conrad R. Genome data mining and soil survey for the novel group 5 [NiFe]-hydrogenase to explore the diversity and ecological importance of presumptive high-affinity H2-oxidizing bacteria. Appl Environ Microbiol. 2011;77:6027–35. doi: 10.1128/AEM.00673-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greening C, Constant P, Hards K, Morales SE, Oakeshott JG, Russell RJ, et al. Atmospheric hydrogen scavenging: from enzymes to ecosystems. Appl Environ Microbiol. 2015;81:1190–9. doi: 10.1128/AEM.03364-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maimaiti J, Zhang Y, Yang J, Cen YP, Layzell DB, Peoples M, et al. Isolation and characterization of hydrogen-oxidizing bacteria induced following exposure of soil to hydrogen gas and their impact on plant growth. Environ Microbiol. 2007;9:435–44. doi: 10.1111/j.1462-2920.2006.01155.x. [DOI] [PubMed] [Google Scholar]
- 12.Constant P, Poissant L, Villemur R. Isolation of Streptomyces sp. PCB7, the first microorganism demonstrating high-affinity uptake of tropospheric H2. ISME J. 2008;2:1066–76. doi: 10.1038/ismej.2008.59. [DOI] [PubMed] [Google Scholar]
- 13.Constant P, Hallenbeck PC. Chapter 5 - Hydrogenase. In: Pandey A, Chang JS, Hallenbeck PC, Larroche C, editors. Biohydrogen, 1st edition. Amsterdam: Elsevier; 2013.
- 14.Greening C, Biswas A, Carere CR, Jackson CJ, Taylor MC, Stott MB, et al. Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilized energy source for microbial growth and survival. ISME J. 2016;10:761–77. doi: 10.1038/ismej.2015.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Constant P, Hallenbeck PC. Chapter 3 - Hydrogenase. In . Editors: Pandey A, Mohan SV, Chang JS, Hallenbeck PC, Larroche C, editors. Biohydrogen, 2nd edition. Amsterdam: Elsevier; 2019;49–78.
- 16.Piché-Choquette S, Constant P. Molecular hydrogen, a neglected key driver of soil biogeochemical processes. Appl Environ Microbiol. 2019;85:e02418–18. doi: 10.1128/AEM.02418-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greening C, Berney M, Hards K, Cook GM, Conrad R. A soil actinobacterium scavenges atmospheric H2 using two membrane-associated, oxygen-dependent [NiFe] hydrogenases. Proc Natl Acad Sci USA. 2014;111:4257–61. doi: 10.1073/pnas.1320586111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Constant P, Chowdhury SP, Pratscher J, Conrad R. Streptomycetes contributing to atmospheric molecular hydrogen soil uptake are widespread and encode a putative high‐affinity [NiFe]‐hydrogenase. Environ Microbiol. 2010;12:821–9. doi: 10.1111/j.1462-2920.2009.02130.x. [DOI] [PubMed] [Google Scholar]
- 19.Meredith LK, Rao D, Bosak T, Klepec-Ceraj V, Tada KR, Hansel CM, et al. Consumption of atmospheric hydrogen during the life cycle of soil‐dwelling actinobacteria. Environ Microbiol Rep. 2014;6:226–38. doi: 10.1111/1758-2229.12116. [DOI] [PubMed] [Google Scholar]
- 20.Piché-Choquette S, Khdhiri M, Constant P. Survey of high-affinity H2-oxidizing bacteria in soil reveals their vast diversity yet underrepresentation in genomic databases. Micro Ecol. 2017;74:771–5. doi: 10.1007/s00248-017-1011-1. [DOI] [PubMed] [Google Scholar]
- 21.Greening C, Carere CR, Rushton-Green R, Harold LK, Hards K, Taylor MC, et al. Persistence of the dominant soil phylum Acidobacteria by trace gas scavenging. Proc Natl Acad Sci USA. 2015;112:10497–502. doi: 10.1073/pnas.1508385112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Islam ZF, Cordero PRF, Feng J, Chen YJ, Bay SK, Jirapanjawat T, et al. Two Chloroflexi classes independently evolved the ability to persist on atmospheric hydrogen and carbon monoxide. ISME J. 2019;13:1801–13. doi: 10.1038/s41396-019-0393-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mohammadi S, Pol A, van Alen TA, Jetten MSM, Op den Camp HJM. Methylacidiphilum fumariolicum SolV, a thermoacidophilic ‘Knallgas’ methanotroph with both an oxygen-sensitive and -insensitive hydrogenase. ISME J. 2017;11:945–58. doi: 10.1038/ismej.2016.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janssen PH. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol. 2006;72:1719–28. doi: 10.1128/AEM.72.3.1719-1728.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barns SM, Cain EC, Sommerville L, Kuske CR. Acidobacteria phylum sequences in uranium-contaminated subsurface sediments greatly expand the known diversity within the phylum. Appl Environ Microbiol. 2007;73:3113–6. doi: 10.1128/AEM.02012-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dedysh SN, Yilmaz P. Refining the taxonomic structure of the phylum Acidobacteria. Int J Syst Evol Microbiol. 2018;68:3796–806. doi: 10.1099/ijsem.0.003062. [DOI] [PubMed] [Google Scholar]
- 27.Kielak AM, Barreto CC, Kowalchuk GA, van Veen JA, Kuramae EE. The ecology of acidobacteria: moving beyond genes and genomes. Front Microbiol. 2016;7:744. doi: 10.3389/fmicb.2016.00744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Myers MR, King GM. Isolation and characterization of Acidobacterium ailaaui sp. nov., a novel member of Acidobacteria sub-division I, from a geothermally-heated Hawaiian microbial mat. Int J Syst Evol Microbiol. 2016;66:5328–35. doi: 10.1099/ijsem.0.001516. [DOI] [PubMed] [Google Scholar]
- 29.Eichorst SA, Trojan D, Roux S, Herbold C, Rattei T, Woebken D. Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments. Environ Microbiol. 2018;20:1041–63. doi: 10.1111/1462-2920.14043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gödde R, Meuser K, Conrad R. Hydrogen consumption and carbon monoxide production in soils with different properties. Biol Fertil Soils. 2000;32:129–34. [Google Scholar]
- 31.Meredith LK, Commane R, Keenan TF, Klosterman ST, Munger JW, Templer PH, et al. Ecosystem fluxes of hydrogen in a mid‐latitude forest driven by soil microorganisms and plants. Glob Change Biol. 2017;23:906–19. doi: 10.1111/gcb.13463. [DOI] [PubMed] [Google Scholar]
- 32.Turlapati SA, Minocha R, Bhiravarasa PS, Tisa LS, Thomas WK, Minocha SC. Chronic N-amended soils exhibit an altered bacterial community structure in Harvard Forest, MA, USA. FEMS Microbiol Ecol. 2012;83:478–93. doi: 10.1111/1574-6941.12009. [DOI] [PubMed] [Google Scholar]
- 33.Søndergaard D, Pedersen CNS, Greening C. HydDB: A web tool for hydrogenase classification and analysis. Sci Rep. 2016;6:1–8. doi: 10.1038/srep34212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 35.Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019;20:1160–6. doi: 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inf. 2009;23:205–11. [PubMed] [Google Scholar]
- 38.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641–50. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaiser C, Koranda M, Kitzler B, Fuchslueger L, Schnecker J, Schweiger P, et al. Belowground carbon allocation by trees drives seasonal patterns of extracellular enzyme activities by altering microbial community composition in a beech forest soil. N. Phytol. 2010;187:843–58. doi: 10.1111/j.1469-8137.2010.03321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spohn M, Pötsch EM, Eichorst SA, Woebken D, Wanek W, Richter A. Soil microbial carbon use efficiency and biomass turnover in a long-term fertilization experiment in a temperate grassland. Soil Biol Biochem. 2016;97:168–75. [Google Scholar]
- 43.Šťovíček A, Kim M, Or D, Gillor O. Microbial community response to hydration-desiccation cycles in desert soil. Sci Rep. 2017;7:1–19. doi: 10.1038/srep45735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Angel R. Total nucleic acid extraction from soil. Protocol Exchange. 2012; 10.1038/protex.2012.046.
- 45.Rose TM, Schultz ER, Henikoff JG, Pietrokovski S, McCallum CM, Henikoff S. Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly related sequences. Nucl Acids Res. 1998;26:1628–35. doi: 10.1093/nar/26.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herbold CW, Pelikan C, Kuzyk O, Hausmann B, Angel R, Berry D, et al. A flexible and economical barcoding approach for highly multiplexed amplicon sequencing of diverse target genes. Front Microbiol. 2015;6:731. doi: 10.3389/fmicb.2015.00731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-Source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90 K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9:5114. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 51.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Constant P, Chowdhury SP, Hesse L, Conrad R. Co-localization of atmospheric H2 oxidation activity and high affinity H2-oxidizing bacteria in non-axenic soil and sterile soil amended with Streptomyces sp. PCB7. Soil Biol Biochem. 2011;43:1888–93. [Google Scholar]
- 53.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eichorst SA, Kuske CR, Schmidt TM. Influence of plant polymers on the distribution and cultivation of bacteria in the phylum Acidobacteria. Appl Environ Microbiol. 2011;77:586–96. doi: 10.1128/AEM.01080-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koch IH, Gich F, Dunfield PF, Overmann J. Edaphobacter modestus gen. nov., sp. nov. and Edaphobacter aggregans sp. nov., two novel acidobacteria isolated from alpine and forest soils. Int J Syst Evol Microbiol. 2008;58:1114–22. doi: 10.1099/ijs.0.65303-0. [DOI] [PubMed] [Google Scholar]
- 58.Eichorst SA, Breznak JA, Schmidt TM. Isolation and characterization of soil bacteria that define Terriglobus gen. nov., in the phylum Acidobacteria. Appl Environ Microbiol. 2007;73:2708–17. doi: 10.1128/AEM.02140-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ritz C, Baty F, Streibig JC, Gerhard D. Dose-response analysis using R. PLoS ONE. 2015;10:e0146021. doi: 10.1371/journal.pone.0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Joseph SJ, Hugenholtz P, Sangwan P, Osborne CA, Janssen PH. Laboratory cultivation of widespread and previously uncultured soil bacteria. Appl Environ Microbiol. 2003;69:7210–5. doi: 10.1128/AEM.69.12.7210-7215.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Männistö MK, Rawat S, Starovoytov V, Haggblom MM. Granulicella arctica sp. nov., Granulicella mallensis sp. nov., Granulicella tundricola sp. nov. and Granulicella sapmiensis sp. nov., novel acidobacteria from tundra soil. Int J Syst Evol Microbiol. 2012;62:2097–106. doi: 10.1099/ijs.0.031864-0. [DOI] [PubMed] [Google Scholar]
- 62.Crowe MA, Power JF, Morgan XC, Dunfield PF, Lagutin K, Rijpstra WIC, et al. Pyrinomonas methylaliphatogenes gen. nov., sp. nov., a novel group 4 thermophilic member of the phylum Acidobacteria from geothermal soils. Int J Syst Evol Microbiol. 2014;64:220–7. doi: 10.1099/ijs.0.055079-0. [DOI] [PubMed] [Google Scholar]
- 63.Belova SE, Ravin NV, Pankratov TA, Rakitin AL, Ivanova AA, Beletsky AV, et al. Hydrolytic capabilities as a key to environmental success: chitinolytic and cellulolytic acidobacteria from acidic sub-arctic soils and boreal peatlands. Front Microbiol. 2018;9:1–14. doi: 10.3389/fmicb.2018.02775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fierer N. Embracing the unknown: disentangling the complexities of the soil microbiome. Nat Rev Microbiol. 2017;15:579–90. doi: 10.1038/nrmicro.2017.87. [DOI] [PubMed] [Google Scholar]
- 65.Ward NL, Challacombe JF, Janssen PH, Henrissat B, Coutinho PM, Wu M, et al. Three genomes in the phylum Acidobacteria provide insight into their lifestyles in soils. Appl Environ Microbiol. 2009;75:2046–56. doi: 10.1128/AEM.02294-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schäfer C, Bommer M, Hennig SE, Jeoung JH, Dobbek H, Lenz O. Structure of an actinobacterial-type [NiFe]-hydrogenase reveals insight into O2-tolerant H2 oxidation. Structure. 2016;24:285–92. doi: 10.1016/j.str.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 67.Liot Q, Constant P. Breathing air to save energy–new insights into the ecophysiological role of high-affinity [NiFe]-hydrogenase in Streptomyces avermitilis. Microbiologyopen. 2016;5:47–59. doi: 10.1002/mbo3.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jones RT, Robeson MS, Lauber CL, Hamady M, Knight R, Fierer N. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J. 2009;3:442–53. doi: 10.1038/ismej.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: the unseen majority. Proc Natl Acad Sci USA. 1998;95:6578–83. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kolter R, Siegele DA, Tormo A. The stationary phase of the bacterial life cycle. Ann Rev Microbiol. 1993;47:855–74. doi: 10.1146/annurev.mi.47.100193.004231. [DOI] [PubMed] [Google Scholar]
- 71.Lennon JTJ, Jones SES. Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat Rev. 2011;9:119–30. doi: 10.1038/nrmicro2504. [DOI] [PubMed] [Google Scholar]
- 72.Jones SE, Lennon JT. Dormancy contributes to the maintenance of microbial diversity. Proc Natl Acad Sci USA. 2010;107:5881–6. doi: 10.1073/pnas.0912765107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morita RY. Is H2 the universal energy source for long-term survival? Micro Ecol. 1999;38:307–20. doi: 10.1007/s002489901002. [DOI] [PubMed] [Google Scholar]
- 74.Cordero PRF, Bayly K, Man Leung P, Huang C, Islam ZF, Schittenhelm RB, et al. Atmospheric carbon monoxide oxidation is a widespread mechanism supporting microbial survival. ISME J. 2019;13:2868–81. doi: 10.1038/s41396-019-0479-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.