

SUMMARY
Transcriptional profiling of tumors has revealed a stress-like state among the cancer cells, with the concerted expression of genes such as fos, jun and heat-shock proteins, though this has been controversial given possible dissociation-effects associated with single-cell RNA-Seq. Here, we validate the existence of this state using a combination of zebrafish melanoma modeling, spatial transcriptomics and human samples. We found that the stress-like subpopulation of cancer cells is present from the early stages of tumorigenesis. Comparing with previously reported single-cell RNA-Seq datasets from diverse cancer types, including triple-negative breast cancer, oligodendroglioma and pancreatic adenocarcinoma, indicated the conservation of this state during tumorigenesis. We also provide evidence that this state has higher tumor-seeding capabilities and that its induction leads to increased growth under both MEK and BRAF inhibitors. Collectively, our study supports the stress-like cells as a cancer cell state expressing a coherent set of genes and exhibiting drug-resistance properties.
Keywords: Melanoma, stress-like, single-cell RNA-Seq, spatial transcriptomics, drug-resistant states
Graphical Abstract
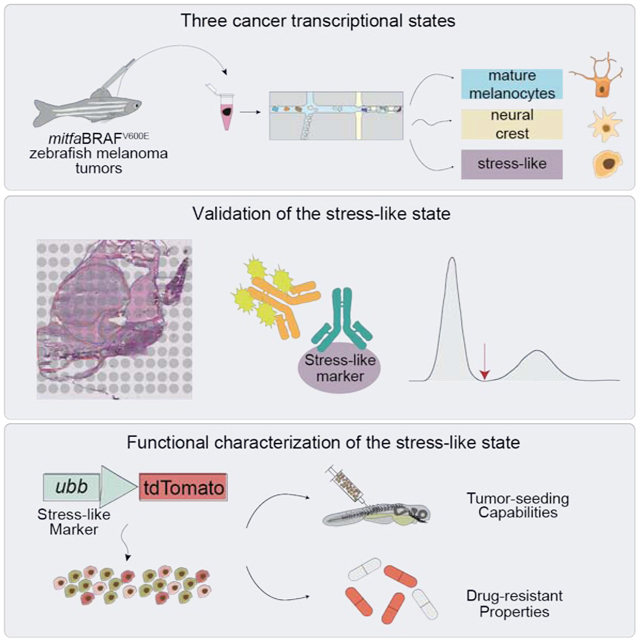
eTOC:
Multiple transcriptional states occur in melanoma cancer cells, one of which is a stress-like state that is conserved across species and other cancer types. The stress-like state has a role in tumor progression and drug response.
INTRODUCTION
A universal feature of cancer is its genetic and phenotypic heterogeneity (Fisher et al., 2013; Lawrence et al., 2013; Meacham and Morrison, 2013; Sharma et al., 2010). Genetically, tumor evolution leads to a recurring set of DNA alterations in genes such as KRAS, BRAF or p53. (Hollstein et al., 1991; Lièvre et al., 2006; Riely et al., 2009; Vogelstein and Kinzler, 2004). In addition to such DNA alterations, transcriptional heterogeneity of cancer cells is increasingly recognized in a diverse array of tumors including melanoma (Jerby-Arnon et al., 2018; Rambow et al., 2018; Tirosh et al., 2016a), glioblastoma (Patel et al., 2014), oligodendroglioma (Tirosh et al., 2016b), breast (Kim et al., 2018) and head and neck cancer (Puram et al., 2017, 2018). In glioblastoma, multiple transcriptional programs of cancer cells co-exist according to classical, proneural, neural, and mesenchymal cell states (Patel et al., 2014). For melanoma, the genetics have been well characterized in terms of recurring genes and pathways across nevus and invasive bulk tumors (Cancer Genome Atlas Network, 2015; Tsao et al., 2012), and recent work using single-cell RNA sequencing (scRNA-Seq) has mapped out multiple transcriptional programs (Jerby-Arnon et al., 2018; Rambow et al., 2018; Tirosh et al., 2016a). However, despite the characterization of this diversity, the functional consequences of cancer cell states are not well understood.
Much like genetic evolution during tumorigenesis, transcriptional evolution can give rise to different cancer cell states over time. Many open questions remain about the nature of these cell states, including how they arise, their physical organization, and their functional consequences (Barkley and Yanai, 2019). In addition, evidence has been presented indicating that cell states are not fixed, which may be an important aspect of tumor cell plasticity (Hoek et al., 2008; Verfaillie et al., 2015; Widmer et al., 2012). Switching between states can be driven by factors from the microenvironment such as WNT5A (O’Connell et al., 2013) and EDN3 (Kim et al., 2017), or from varying levels of MITF (Carreira et al., 2006; Vivas-García et al., 2020), the master transcription factor for melanocyte development. The ability of individual cells to take on these varying transcriptional states also has important consequences for patient prognosis (Sarrió et al., 2008). For example, the acquisition of an AXLhigh/MITFlow state is associated with an invasive, metastatic phenotype (Müller et al., 2014; Tirosh et al., 2016a), and phenotype switching has been associated with drug tolerance (Ahmed and Haass, 2018). More recently, a transcriptional state associated with RXR signaling has been found to be a key factor in resistance to BRAF/MEK inhibitors (Rambow et al., 2018).
Previous scRNA-Seq melanoma studies have revealed multiple cell states in melanoma, including neural crest, pigmented, invasive and starved states (Rambow et al., 2018; Tirosh et al., 2016a). We and others have previously shown that the neural crest transcriptional program, typified by genes such as SOX10, is essential to melanoma initiation because it provides the proper milieu on which DNA mutations can act (Kaufman et al., 2016; Shakhova et al., 2012; Travnickova et al., 2019; White et al., 2011). In contrast, the existence of other cell states such as a “stress-like” state, expressing genes such as fos and jun, has been suggested by prior work but its functional role remains unclear (Tirosh et al., 2016a). Further complicating the matter is the observation that a stress transcriptional program has been shown to arise as an artifact of cell dissociation protocols and flow cytometry sorting (van den Brink et al., 2017). This highlights the challenge of identifying a role for stress signaling in cancer. Collectively, the existence of the stress-like state as a biological property of tumorigenesis remains unclear.
We previously developed a transgenic zebrafish model of melanoma, in which the BRAFV600E oncogene is expressed in melanocytes (Ceol et al., 2011; Patton et al., 2005; White et al., 2011). These fish develop melanomas that resemble the human disease at histological, transcriptomic and genomic level, and have previously been shown to be a powerful model for the study of both metastasis and drug resistance (Heilmann et al., 2015; Patton et al., 2005; Yen et al., 2013). Importantly, the tumors can be repeatedly sampled over time, allowing us to understand when these cell states arise over the course of tumor evolution.
Here, we use the zebrafish model to study subpopulations of cancer cells with scRNA-Seq, identifying the transcriptional programs for mature melanocytes, neural crest, and a population of “stress-like” cells. Using spatial transcriptomics on intact tumors – which does not rely on cell sorting or dissociation – we investigate whether the stress-like transcriptional program is consistently present among cancer cells. We then reanalyzed published scRNA-Seq data to determine whether the stress-like transcriptional program is found across a wide variety of tumor types, including triple-negative breast cancer, oligodendroglioma and pancreatic adenocarcinoma. We also developed a transgenic reporter of this stress-like state, and used it to determine whether stress-like cells have drug-resistant properties.
RESULTS
Cell state mapping reveals conserved cell types between zebrafish and human melanomas
Animal models of cancer allow for both characterization and functional analysis of cell states in cancer. To study this in melanoma, we utilized a transgenic zebrafish model in which human BRAFV600E is expressed in melanocytes through the mitfa promoter. In a p53−/− background, these fish develop melanomas that resemble the human tumors (Patton et al., 2005). To determine if these tumors recapitulate the cellular diversity of human melanoma, we performed scRNA-Seq on 8 tumor biopsies. We processed ~15k cells from the eight samples (Figure 1A), using the inDrop system (Klein et al., 2015; Zilionis et al., 2017). After quality control and filtering (see Methods), we were left with a total 10,012 cells, each with an average of approximately 2,500 transcripts and 1,000 genes detected (Figure S1A). Studying the transcriptomes, we detected eight cell types, each forming a distinct cluster when visualized using tSNE (t-distributed stochastic neighbor embedding, Maaten and Hinton, 2008, Figure 1B). We annotated the cancer cell population by the detection of BRAFV600E transcripts, and annotated the rest of the cell types - keratinocytes, fibroblasts, erythrocytes, natural killer cells, neutrophils, macrophages, other lymphocytes - using known markers (Table S1 and Figure S1B). For example, a cluster was annotated as keratinocytes since it was enriched in the expression of keratin 4 (krt4) and other genes. In humans, keratinocytes are the major cell type that surrounds the melanocytes from which these tumors arise, and contribute important growth factors such as EDN3 that promote tumor growth (Saldana-Caboverde and Kos, 2010). Other identified cell types include immune cells such as T-cells and macrophages, also commonly observed cell types in the human disease. These data confirm that the zebrafish melanomas are composed of multiple cell types that closely resemble the cell types seen in humans.
Figure 1. Single-cell RNA-Seq on zebrafish melanoma.

(A) Eight tumor biopsies were processed from three distinct tumors using scRNA-Seq. (B) tSNE analysis of 7,278 individual cells from tumor 1. Color indicates the inferred cell type. (C) PCA on the cancer cells revealed three transcriptional cell states, indicated by the colored circles. (D) Heatmap showing the Pearson’s correlation coefficients between the three cell states across all eight biopsies. Biopsy samples cluster according to states and not tumor or animal of origin. (E) Serial biopsies were taken from the same tumor (tumor 1), at one week intervals. The tumor at each time point is shown in the micrographs. The stacked bar plot indicates the proportions of the transcriptional cell states detected in panel (C) for each biopsy.
Melanomas exhibit three cell states, including a stress-like cell state
To study transcriptional heterogeneity within the melanoma malignant cells, we applied principal component analysis (PCA) to the cancer cell transcriptomes from a single tumor (tumor 1). This analysis revealed a triangle-shaped arrangement of transcriptomes with a concentration of cells near the vertices (Figure 1C) suggesting that melanoma tumors consist of three cancer cell states. We verified that this shape is not driven by pre-processing methods or any technical aspects such as the number of UMIs detected (Figure S1C-F). Furthermore, analyzing the cells across our eight biopsies, we found that all three transcriptional states are present in each biopsy and tumor (Figure 1D and Figure S2A-C). In order to test whether these three cancer cell states are present in early stages of tumorigenesis, we performed microscopic biopsies on one of the transgenic zebrafish tumors (tumor 1) as soon as they were visible on the skin of the animals followed by scRNA-Seq. We found that all three states were readily identifiable as early as 5 months of age and that their proportion within each biopsy does not change significantly over time (Figure 1E).
To characterize the functional attributes of the three cancer cell states, we studied their underlying transcriptional programs in terms of uniquely expressed genes (Figure 2A). We performed differential gene expression analysis among the groups of cells closest to each PCA vertex in Figure 1C (see Methods and Table S2 for lists of all genes corresponding to each program). We found that transcriptional program 1 (low PC1, low PC2) is enriched for the expression of neural crest genes, such as sox2 and sox10, suggesting co-option of this progenitor program by the cells (Figure S2D). In contrast, transcriptional program 2 (low PC1, high PC2) is enriched with the expression of genes associated with mature melanocytes, such as dct, tyrp1b, and pmela, indicating that these cells co-opt the differentiated melanocyte transcriptional program (Figure S2D). To test for the distinction between program 1 and 2, we compared their expression profiles to a published dataset that measured gene expression changes that accompany human melanocyte differentiation from pluripotent stem cells (Mica et al., 2013). As expected, we found that programs 1 and 2 best correlate with the expression to neural crest cells and mature melanocytes, respectively (Figure 2B). A third transcriptional program was enriched in the expression of genes such as jun, fosb, fosab, ubb, and heat-shock response genes, all associated with a stress-like transcriptional program (Figure 2A and Figure S2D). While this was of interest to us, this class of genes was found to be a potential artifact of scRNA-Seq methods (van den Brink et al., 2017).
Figure 2. Transcriptional program underlying melanoma cancer cell states.

(A) Normalized expression levels of the differentially expressed genes across the cancer cells of tumor 1. Genes are colored by function based on GO annotations indicated at the bottom. Bottom panel - expression of mitfa, egfra, ngfra, ngfrb and crestin, all associated with melanoma cell lines program (B) PCA of bulk melanocyte differentiation from previously reported data (Mica et al., 2013), colors indicate expression levels of SOX2 (purple) and DCT (green). In the heatmap, the Pearson’s correlation levels are shown between the three human melanoma cell type programs and the developmental transcriptomes of stem cells, neural crest, and mature melanocytes. (C,D) Comparison between zebrafish melanoma transcriptional program and (C) human melanoma transcriptional program (Tirosh et al., 2016a) and (D) patient-derived xenografts (Rambow et al., 2018; Tirosh et al., 2016a) (*, P<10−2; ***, P<10−4; ****, P<10−5).
To address whether our identified cancer cell states are unique to the zebrafish system or conserved also in human melanoma, we compared them to the cell states detected in two previously reported human melanoma scRNA-Seq datasets (Rambow et al., 2018; Tirosh et al., 2016a). We first compared to the Tirosh et al. human melanoma scRNA-Seq data. Focusing on the MITF high population of cells, since the zebrafish BRAFV600E transgene is driven by the mitfa promoter, we found that these have a similar pattern of expression to the zebrafish tumor expression (P<10−5, Figure 2C and Figure S2D). We next compared our data to that of Rambow et al. (Rambow et al., 2018), who described four transcriptional states of melanoma cells isolated from patient-derived xenografts that were exposed to RAF/MEK inhibition. We found that the neural crest state, the mature melanocyte, and the stress-like state showed the highest respective gene expression correlation with Rambow et al.’s annotated neural crest stem cells, pigmented cells, and ‘starved’-like melanoma cells respectively (P<10−2, P<10−5 and P<10−4, respectively, Figure 2D). While we did not identify the invasive program in the zebrafish model this perhaps stems from our use of a mitfa promoter in a p53 dominant-negative background, which may constrain some transcriptional heterogeneity.
Spatial transcriptomics supports the presence of stress-like cancer cells
Since the neural crest and melanocyte programs are well described in melanoma, we focused on the poorly characterized stress-like state, whose actual presence has been called into question, as it has been observed that a stress transcriptional program may arise as an artifact of cell dissociation protocols and flow cytometry sorting (van den Brink et al., 2017). Seeking to test for the existence of the stress-like cancer cell state, we found that the associated transcriptional program was not detected in two other non-cancer datasets generated using the same cell-dissociation protocol and scRNA-Seq methods (Figure S2F-H). We also implemented the previously described in silico purification method (van den Brink et al., 2017) to our dataset, and found that the stress-like state in cancer cells remains across a range of thresholds Figure 2SE).
To more directly validate the existence of the stress-like cell state in melanoma, we turned to spatial transcriptomics (Ståhl et al., 2016). This is an in situ RNA-Seq approach that does not depend upon dissociation or flow sorting of cells, which we have previously used to study the architecture of the tumor microenvironment (Moncada et al., 2020). We generated zebrafish melanomas by transplanting a melanoma cell line called ZMEL1 (Heilmann et al., 2015) into transparent casper zebrafish. We prepared two frozen sections of the tumor and surrounding normal tissue, along with a section from non-tumor bearing regions of the fish, and placed these on a spatial transcriptomics slide containing spatially barcoded mRNA probes (see Methods).
Analyzing the spatial transcriptomics data, we sought to ask whether the stress-like state could be evidenced in malignant areas. We thus examined the expression of the 3 melanoma cell states in the tumor and normal areas, as identified by the hematoxylin/eosin stain of the same section (Figure 3A). The expression of genes such as sox2 (neural crest program), pmela (melanocyte program) and fosab (stress-like program) are highly enriched in the tumor areas when compared to the normal surrounding tissue (Figure 3A,B, P<10−7, Mann-Whitney test, Figure S3A,B). Extending this analysis to all genes for each of the 3 cancer cell states, we found that genes associated with the three states are enriched in the tumor area (Figure 3C and Figure S3C) when compared to surrounding normal tissue. As a negative control, we examined a randomly selected gene-set (n=200), and found that these do not show an enrichment in the tumor area (Figure 3C). In addition, we also found that the expression of the genes of each of the cancer cell states were not found in non-tumor bearing sections of animals (Figure S3D).
Figure 3. The transcriptional programs of the cancer cell states are enriched in cancer areas and detected at the protein level across cancer types.

(A) Hematoxylin and eosin stain of a zebrafish transplanted tumor section. Red and blue dotted lines mark cancer and non-cancer areas, respectively. (B) Gene expression profiles of the indicated genes obtained by spatial transcriptomics performed on a section adjacent to the one shown in panel A. (C) Violin plots indicating the enrichment of each gene (Man-Whitney test, −log10 of the P-value) in each of the indicated gene programs. Genes shown in panel B are indicated by arrows in each program. Negative control represents a randomly selected set of 200 genes. (D) FOS protein is localized in the cell nuclei (white arrows) as shown by the DAPI nuclear staining (blue, left), FOS immunofluorescence staining (green, middle) and a merged image (right). (E) PCA on PDAC (Moncada et al., 2020), TNBC (Kim et al., 2018), oligodendroglioma (Tirosh et al., 2016b) and melanoma (Tirosh et al., 2016a) tumor cancer cells. Color indicates normalized expression levels of the stress-like program, significantly enriched in one vertex (*, P<10−2; **, P<10−4).
We further tested for the existence of the stress-like cell state at the protein level by performing immunofluorescence for FOS, a marker of the stress-like program (Figure 2A and Figure S2D) on 100 human biopsy core sections (see Methods), including 62 cases of primary melanoma, 21 metastatic melanoma, and 17 nevus tissues as a control (Figure S3E-H). Consistently, we found that in tumor slices FOS protein expression was localized in the nucleus of cells (Figure 3D).
The stress-like cancer cell state is also observed in other cancer types
Since the program defined for the stress-like cancer state (including fos, jun, and heat shock proteins) does not contain genes specific to melanocyte biology - in contrast to the neural crest and melanocyte states - we hypothesized that it might be conserved in other cancer types. To study this at the transcriptomics level, we reanalyzed four previously published scRNA-Seq tumor datasets: triple negative breast cancer (TNBC, Kim et al., 2018), pancreatic cancer adenocarcinoma (PDAC, Moncada et al., 2020), oligodendroglioma (Tirosh et al., 2016b) and melanoma (Tirosh et al., 2016a). For each cancer type, we analyzed the transcriptomes of the cancer cells in isolation: 388 single cells from a TNBC patient, 462 single cells from a PDAC patient, 692 from an oligodendroglioma patient and 1257 single cells from melanoma patients, all without treatment.
Studying these cancer cells using PCA, we found a triangle-shaped distribution of cells for each cancer type (Figure 3E), reminiscent of that found for zebrafish melanoma (Figure 1C). We further found that in all datasets, one vertex is enriched in the expression of the stress-like gene program (Figure 3E). Using a bootstrapping approach, we found evidence that these enrichments are statistically significant in all four datasets (P<0.01, see Methods). Particularly noteworthy is the fact that the TNBC dataset was uniquely generated using a nuclear scRNA-Seq, which involves snap freezing of the sample followed by nuclei harvesting (Gao et al., 2017; Habib et al., 2017; Kim et al., 2018). Since this approach is not prone to possible cell dissociation induced artifacts (Krishnaswami et al., 2016), the observation of the stress-like state in this sample provides further evidence for the expression of this transcriptional program in cancer cells.
In addition, we again used the FOS protein as a marker for stress-like cells and queried for its expression in various other cancer tissues. IF on pancreatic ductal adenocarcinoma (PDAC) showed similar patterns to melanoma where heterogeneous nuclear staining of FOS is observed across the different cancer cells (Figure S3I). Extending to other tissues, using the human protein atlas dataset (Thul et al., 2017; Uhlén et al., 2015), we further observed that heterogeneous nuclear expression of FOS is present in other cancer types such as breast cancer, glioma, prostate cancer and head and neck cancer (Figure S3J).
A ubb-tdTomato transgenic line as a reporter of the stress-like state
We next sought to functionally assess whether the stress-like cells in our melanomas had unique biological properties that made them particularly pro-tumorigenic. To this end we built a fluorescent transgenic reporter using the ubb gene that allowed us to isolate and characterize the stress-like cancer cells (Figure 4A). The ubb gene encodes for the ubiquitin-B protein, part of the protein degradation system that is commonly activated under periods of diverse types of cell stress (Flick and Kaiser, 2012), including DNA damage, changes in temperature, oxidative damage, hypoxia and starvation. It serves in part to target proteins for degradation by the proteasome, including other stress genes such as fos that we identified in our analysis as part of the stress-like program (Stancovski et al., 1995). The ubiquitin system acts in concert with other stress related genes such as the heat shock proteins, which can serve as chaperones for misfolded proteins (Dai and Sampson, 2016) and together can act as a pivotal mechanism by which the cell can either survive stress or undergo apoptosis if the stress cannot be resolved. ubb itself is transcriptionally induced under stress conditions such as oxidative stress, making it an ideal reporter of the stress-like state (Bianchi et al., 2015). We reasoned that cells with higher levels of ubb transcription would be representative of a more general stress program and could use transcriptional levels of ubb as an indicator of the more general stress state we observed in our melanomas. To test this, we used a previously described ubb transcriptional promoter fragment (Mosimann et al., 2011) to drive tdTomato, and stably inserted this transgene into the ZMEL1-GFP melanoma cell described above.
Figure 4. Zebrafish ubbhigh cells form higher burden tumors and induction of the stress-like state increases drug resistance.
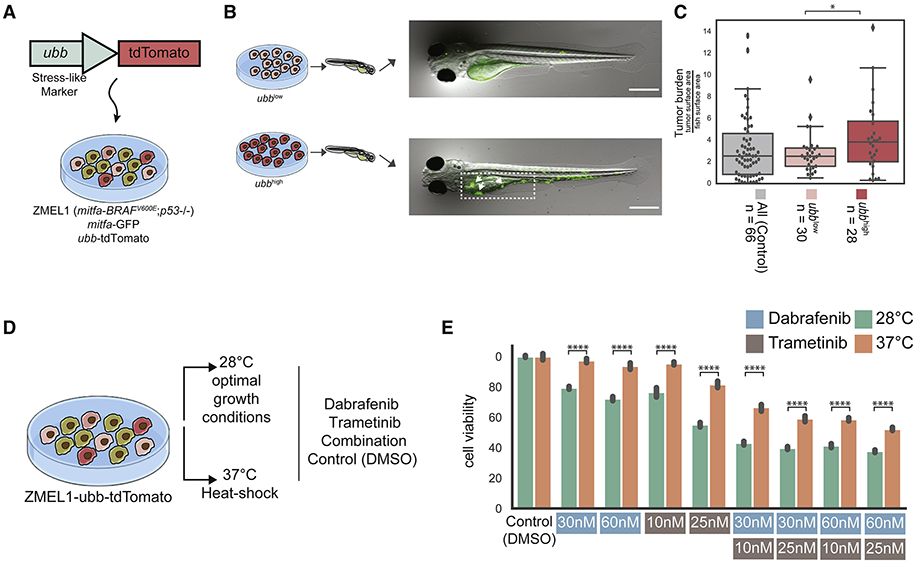
(A) The ZMEL1-GFP;ubb-tdTomato system to track and select for the cells in the stress-like state. (B) ZMEL1-GFP;ubb-tdTomato cells were sorted to high and low levels of tdTomato intensities and injected into zebrafish for tumor initiation assay followed by quantification of GFP intensity (additional representative images can be found in Figure S4J). (C) Boxplot of tumor burden quantified by GFP intensity of the two different levels of tdTomato compared to parental unsorted cells as a control (mix of population with no selection). Tumor sizes were significantly higher when high tdTomato cells were injected (Mann-Whitney test; *, P<0.05). (D) Schematic of ZMEL1-GFP;ubb-tdTomato cells cultured in optimal and heat shock conditions and exposed to different drug concentrations. (E) Bar plot of cell viability across culturing conditions (optimal/heat shock) and drug treatments. Cell viability is significantly higher under drug treatment when cells were cultured in heat shock conditions.
The ubb reporter allowed us to isolate ubbhigh versus ubblow cells from the melanocyte population (Figure S4). We confirmed that the transgene is only at a single copy per cell by measuring genomic ubb levels by qPCR. We validated that high intensity of tdTomato represents the stress-like cells in two ways: (1) Using qPCR, we found that fosab, hsp70.1, junba and ubb are more highly expressed in tdTomato-high cells compared to tdTomato-low cells, providing support for the stress-like identity (Figure S5A). We confirmed that the high fluorescence was only reflected in tdTomato and not GFP intestines to rule out autofluorescence artifacts (Figure S4A-I). (2) The stress-like program includes eight heat shock genes. We induced their expression by culturing the cells in heat shock conditions (37°C overnight, compared to optimum conditions, 28°C). Examining tdTomato intensity as a measure for ubb expression, we detected a significant increase in heat shock conditions compared to the control (P<0.001, effect size = 0.8). This provides evidence for the notion that stress by heat shock leads to enrichment in ubbhigh cells (Figure S5B).
We next inquired into the robustness of ubb expression as a marker for the stress-like program by examining its correlation with the expression of the three programs. For this, we processed cells from the cultured ZMEL1-GFP melanoma cell line for scRNA-Seq using CEL-Seq2 (see Methods). We found that ubb expression across the cells was highly correlated with that of the stress-like program, and anti-correlated with the melanocyte differentiation program (Figure S5C). We did not detect, however, an anti-correlation with the neural crest program. We thus injected the cell line into a zebrafish embryo which resulted in a tumor later in the animal (see Methods). Again sampling the cells using scRNA-Seq, we found that both the melanocyte differentiation and neural crest programs are anti-correlated with ubb expression (Figure S5C). From these experiments, we concluded that high ubb expression is a robust marker for the stress-like program, and that it is mutually exclusive with the induction of the other two programs.
Cells in the stress-like state are more efficient at seeding new tumors
Recent work has pointed to an important role of cell stress in mediating tumor dormancy and the ability to see new tumor sites. For example, an imbalance in the ratio of ERK versus p38/stress signaling can dictate the fate choice between dormancy and proliferation (Harper et al., 2016; Ranganathan et al., 2006), and under the right microenvironment these dormant cells can re-enter the cell cycle and begin to proliferate. Similarly, FBXW7 is a subunit of the SCF ubiquitin ligase complex that regulates HSF1 and stability of the heat shock proteins. Tumors that lose FBXW7 have elevated expression of heat shock proteins and are more efficient at metastatic seeding (Kourtis et al., 2015), supporting the concept that stress signaling can promote tumor progression. We therefore tested whether our stressed population, which occurs endogenously in the transgenic melanomas, might be more efficient at seeding new tumors.
For this we developed a highly stringent assay in which we can test whether a small number of cells can give rise to a tumor after transplantation in the zebrafish. We built upon the logic of blastula transplantation, an assay that is commonly employed in developmental biology (Gansner et al., 2017) in which small numbers of fluorescently labelled “donor” cells are transplanted into an un-labelled “recipient” animal and the fate of those cells is then tracked using in vivo imaging. We adopted this method to use with our ZMEL1-GFP;ubb-tdTomato cells as the donors, and transparent casper animals as recipients. From the ZMEL1-GFP;ubb-tdTomato parental population, we sorted ubbhigh versus ubblow cells using flow cytometry (Figure S4), and then transplanted 5-10 of each of these cells into recipient animals (Figure 4A-B). We performed this assay with three groups of fish: 1. ubbhigh (stress-like) cells, 2. ubblow (non-stress-like cells), and 3. and a mix of the parental cells without prior selection. After 5 days, we then quantified the tumor burden by calculating the percentage of the entire fish that is covered by GFP+ cells intensity in a total of n=124 animals. Because the GFP is driven by the mitfa promoter, it is independent of ubb mediated transcription. This analysis revealed that tumor size (overall tumor burden) in animals seeded by the ubbhigh cells is larger compared to the tumors seeded by the ubblow cells, or from tumors seeded by the parental unsorted cells (Figure 4C, Mann-Whitney test, P< 5×10−2).
Extrinsic induction of the stress-like program induces drug resistance
Finally, we asked whether cancer cells in the stress-like state are intrinsically more drug-resistant, by querying if extrinsic induction of the stress-like state is sufficient to trigger this resistance. Given our finding that heat shock of the melanoma cells was sufficient to trigger increased expression of the ubb reporter (Figure S5B), we reasoned that heat shock of the cells might affect sensitivity to BRAF or MEK inhibitors. To test this, we grew ZMEL1-GFP;ubb-tdTomato cells at either 28°C (the normal zebrafish temperature) or at 37°C (a temperature long known to induce the heat shock response in zebrafish, Figure 4D). Strikingly, this induced resistance to both BRAF as well as MEK inhibitors across a wide range of doses and combinations (Figure 4E and Figure S5D). To exclude the possibility of a zebrafish-specific effect, we repeated this experiment using human A375 melanoma cells, which harbor the same BRAFV600E mutation as the ZMEL1 cells. In this case, we grew the human cells either at 37°C (normal human body temperature) or at 42°C (known to induce heat shock in human cells) and found a similar phenomenon: cells at the higher temperature were more resistant to both BRAF and MEK inhibitors across the doses tested (Figure S5E).
To address whether this response is due to an increase of a specific gene program, such as the stress-like cells or an induction of several cancer cell populations, we queried for the stress-like state in cells undergoing drug treatment. For this, we turned to a dataset in which human melanoma cells (451Lu) were treated with vemurafenib, a BRAF inhibitor (Ho et al., 2018). In that study, cells at confluence were processed for scRNA-Seq with and without the drug treatment, corresponding to parental sensitive (3,295 cells) and resistant cells (1,757 cells). Examining the expression of key genes of the stress-like program – FOS, UBB and the heat shock protein gene HSPH1 – we found higher expression in the resistant cells compared to the parental ones (Figure S5F, P<10−4, Mann-Whitney test, effect sizes of 0.16, 0.53 and 0.68, respectively). Expanding this analysis to the entire set of genes in the stress-like program, we found that it is generally expressed higher in the resistant cells (Figure S5F-I, P<10−4, Mann-Whitney test, effect size of 0.98). The neural-crest program, in contrast, is not induced in the resistant cells (Figure S5G). However, the mature melanocyte program is also significantly induced in the resistant cells, suggesting that the potential drug-resistant properties of this program also require additional research. These results suggest potential drug-resistant properties of the stress-like state. We further explored this association with a zebrafish in vivo experiment (Figure S5J-L), however further work will be required to fully map the drug-tolerant properties across cancer cell states.
DISCUSSION
Here we have studied gene expression programs in zebrafish and human melanomas, detecting 3 recurring cancer cell states: a mature melanocyte, a neural-crest and a stress-like. Other recent findings using scRNA-Seq have also detected transcriptional programs in melanoma cancer cells. Tirosh et al. showed that cancer cells have distinct transcriptional programs that capture the known proliferative and invasive states (Tirosh et al., 2016a), Rambow et al. identified four transcriptional programs in patient-derived xenografts (PDX) formed by BRAF mutant patients that were treated with RAF/MEK inhibition drugs (Rambow et al., 2018) and Tsoi et al. found that bulk melanoma tumors show different states along the trajectory of differentiation (Tsoi et al., 2018). While the frequencies of the states within the cancer cell population varied by studies perhaps, due to different model systems and sampling approaches, similar states were detected overall (Figure 2). Our focused analysis on the stress-like cell state led us to validate it as a significant component of the tumor, and not an artifact of cell dissociation and sorting methods (van den Brink et al., 2017). In this Discussion, we consider the general occurrence of the stress-like state across cancer types, its possible role, qualities and implications for treatment.
Collectively, we have provided 9 lines of evidence that support the existence of the stress-like cancer state: (1) applying a previously introduced method for eliminating stress dissociation biases, confirmed that the cancer state persists (Figure S2), (2) using spatial transcriptomics, a method that avoids biases by immediate sample freezing without a cell dissociation step, we also recovered the stress-like state in cancer regions (Figure 3 and Figure S2), (3) immunofluorescence on 100 tissues indicated the expression of a marker for the state (Figure 3D, Figure S3E-H), (4) the presence of the stress-like cancer state was detected in three additional cancer types (Figure 3E), (5) the stress-like cancer cell state was detected in nuclear single-cell RNA-Seq data (TNBC, Figure 3E), (6) the stress-like subpopulation of cancer cells was present from the early stages of tumorigenesis (Figure 1E), (7) the stress-like cancer state is observed across species, in both zebrafish and human tumors (Figure 2, Figure S2D), (8) cells expressing the stress-like cells state are more efficient at seeding tumors in zebrafish (Figure 4C), and (9) induction of the stress-like state leads to increased growth under both MEK and BRAF inhibitors (Figure 4E). Collectively, this provides strong evidence for the stress-like state as a conserved component of tumorigenesis.
One important clinical implication of this study is that the existence of the stress program can have consequences for the intrinsic responsiveness to therapy. Our study also helps explain prior work in which it has been observed that high levels of the heat shock protein HSP90 can promote BRAF oncogenic activity, since it can bind to and stabilize the mutant BRAFV600E protein (Grbovic et al., 2006). The increased resistance to both BRAF and MEK inhibitors upon application of heat shock may be due to this stabilization of BRAF and increased MAP kinase signaling. Whether other physiologic conditions associated with increased heat, i.e. fever that occurs in cancer patients, affects response to these therapies remains an open question.
One of our main findings is that the stress-like state is a consistent component of the cancer cell population. This raises the question of what adaptive advantages this program might have for tumor initiation. The fos/jun pathway is a critical downstream mediator of MAP kinase signaling (Dunn et al., 2005; O’Donnell et al., 2012), suggesting that activation of BRAFV600E itself may be inducing this state. However, it is likely that other factors such as hypoxia are playing a role as well, during these early stages of tumorigenesis (Webster et al., 1993). The stress population in cancer is likely enacting a set of regulatory mechanisms that balance cell survival/quiescence versus apoptosis, which is critical in the early stages of tumorigenesis. The fact that we see the emergence of this population early in tumorigenesis, even in the absence of drug or other selection factors leads us to speculate that these states could be generated by stable epigenetic mechanisms, rather than genetic mechanisms, since we see it in multiple cancers with very different DNA mutational events (i.e. BRAF, KRAS, CDKN2A). The specific factors that induce the stress state, whether cell intrinsic or relating to the microenvironment, will need further analysis, since they may offer an opportunity for eliminating these intrinsically drug resistant “seeds” even before therapies are applied.
STAR Methods:
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Itai Yanai (Itai.Yanai@nyulangone.org).
Materials Availability
Plasmids generated in this study have been deposited to Addgene (ID 159533).
Data and Code Availability
The complete data that support the findings of this study have been deposited in NCBI GEO database with the accession code GSE115140. The code generated during this study is available in Github: https://github.com/MaayanBaron/sc_melanoma_Baron2020
Experimental Model and Subject Details
Cell lines
Melanoma cell lines:
A375 (human melanoma cell line) was obtained from ATCC. ZMEL1 cells were generated as previously described (Heilmann et al., 2015).
ZMEL1-tdtomato-ubb cell line:
For the generation of ZMEL1 cells expressing tdTomato under the control of the ubb promoter, LR gateway cloning was performed with 5’ubb promoter, middle entry tdTomato-NTR and 3’ SV40 fragment. Following successful cloning, 8 million ZMEL1 cells were electroporated with 15μg of the plasmid using the Neon electroporator. Following electroporation, cells were allowed to recover for 72hr and subsequently grown in 4μg/ml blasticidin containing media for 3 weeks to select for stable integration of the plasmid.
Fish strains and handling
Transgenic minicoopr fish:
minicoopr fish were generated as previously described (Ceol et al., 2011; Iyengar et al., 2012). Fish with the genotype mitfa-BRAFV600E;p53−/−;mitfa−/− were incrossed. 1-cell stage embryos were injected with a plasmid containing mitfa-MITF and mitfa-GFP. Fish were screened at 3 days for melanocyte rescue, visualized as black spots along the skin. The fish were raised to adulthood (4-12 months) and screened for the appearance of GFP-positive tumors. For biopsy experiments, fish with visible tumors were anesthetized with Tricaine (MS222), and a biopsy was taken using a 1mm biopsy punch. The fish were then allowed to recover and were returned to the main aquatics system.
Transplant ZMEL1 fish:
ZMEL1 zebrafish melanoma cells were grown as previously described (Heilmann et al., 2015). Cells were detached from standard tissue culture dishes with trypsin-EDTA and approximately 50 cells in 1nl of 0.9X Dulbecco’s PBS were transplanted intravenously into the Duct of Cuvier of each 2 day-post-fertilization casper embryo as previously described (Heilmann et al., 2015; Kim et al., 2017).
METHOD DETAILS
Single-cell RNA-Seq collection and processing of tumor biopsies.
Tumor biopsy sample collection:
Biopsy samples were obtained from the same location of the tumor and placed in a 1.5mL Eppendorf tube followed by the addition of 500μL 0.25% Trypsin-EDTA for digestion. The digestion was carried out at 37°C in a thermomixer for 15-30 minutes to soften the tissue, with tissue mashings every 5 minutes using a disposable pestle to break up softened tissue. Upon completion of incubation at 37°C, 500μL of DMEM10 were added to deactivate the trypsin. Cells were washed three times by spinning down the sample at 500 rcf for 5 minutes and resuspended in PBS. The sample was then filtered twice using 5mL polystyrene round-bottom tube with 35μm cell-strainer. Viability and single cell consistency were checked prior to encapsulation of the cells with the inDrop system (Klein et al., 2015; Zilionis et al., 2017) for each biopsy taken.
Single-cell encapsulation, processing and bioinformatics pipeline:
inDrop encapsulation of the cells and reverse transcription (RT) reaction was carried out as previously described (Klein et al., 2015; Zilionis et al., 2017). RNA amplification and library preparation was carried out according to this protocol incorporating the changes introduced in Zilionis et al. 2017 based on the CEL-Seq2 protocol (Hashimshony et al., 2016) on batches of 1,500-3,000 cells from each biopsy taken. The number of PCR cycles required for final library amplification ranged from 9-13 cycles. scRNA-Seq library sequencing was carried out using the Illumina NextSeq 500/550 machine. Pair-end sequencing was carried out with read1 (barcodes) for 34bp, index read for 6bp and read2 (transcripts) for 50bp. Raw sequencing data obtained from the inDrop method was processed using a custom-built pipeline, (available at https://github.com/flo-compbio/singlecell). The location of the known “W1” adapter sequence of the inDrop RT primer, was located in the barcode read (read 2). Reads for which the W1 sequence could not be detected were discarded. The start position of the W1 sequence was then used to infer the length of the first part of the inDrop cell barcode in each read, which can range from 8-11bp, as well as the start position of the second part of the inDrop cell barcode, which is 8bp long. Cell barcode sequences were mapped to the known list of 384 barcode sequences for each read. The resulting barcode combination was used to identify the cell from which the fragment originated. Finally, UMI sequence was extracted, and reads with low confidence base calls for the six bases comprising the UMI sequence (minimum PHRED score less than 20) were discarded. The reads containing the mRNA sequence (read 1) were mapped using STAR with parameter “—outSAMmultNmax 1” and default settings otherwise (Dobin et al., 2013). Expression was quantified by counting the number of reads mapped to each gene. The genome and gff file used included the zebrafish genome (Version 10) (Zerbino et al., 2018) and the BRAF human vector.
Quality control and filtering of low-quality cells:
Single-cell transcriptomes with UMIs>750, mitochondrial transcripts < 20% and ribosomal transcripts < 30% were retained for analysis, leaving 7,278 cells. The same approach was applied for the other tumors leaving 1171 and 1563 cells, respectively, yielding a total of 10,012 (out of 15,000 processed for all samples). Expression profiles were smoothed using MAGIC (van Dijk et al., 2018) with parameter k = 7 to reduce noise after optimization using different k values. UMI counts were normalized by the total number of transcripts per cell, and a scale factor equivalent to the median number of transcripts for that cell was applied (transcripts per median, TPM). Expression was transformed using Freeman-Tukey transform (FTT) as described previously (Wagner et al., 2017). In order to avoid processing artifacts and since cell cycle genes were not differentially expressed among the cells we did not perform further processing (Figure S1C-F).
Single cell RNA-Seq analysis.
Cell type clustering:
Clustering was performed by first distinguishing the cancer cells from non-cancer cells by detecting the expression of the human BRAF gene. Next, hierarchical clustering was performed on the non-cancer cells with Ward’s criterion using the most variable genes (defined as Fano factor and mean expression above mean-dependent thresholds). Clustering was performed over correlations computed from the smoothed expression of the selected genes (Z-score of the TPM). This process initially revealed five clusters: cancer, immune, keratinocytes, fibroblasts and erythrocytes. After examining the immune cluster using recently published markers (Carmona et al., 2017) we could separate the immune cluster into three subclusters: macrophages, natural killers and neutrophils. To identify each cluster, we obtained a list of marker genes by examining genes that are differentially expressed (P<10−6, Kolmogorov-Smirnov test; effect size >0.2, Cohen’s d).
Dimensionality Reduction by PCA and tSNE:
Dimensionality reduction methods were performed on TPM transformed data using variable genes (defined as Fano factor and mean expression above mean-dependent thresholds). tSNE was performed using the following parameters: perplexity = 30 and initial dimension = number of principal components explaining >90% of the variance (Maaten and Hinton, 2008).
Cancer cell type analysis:
To identify genes that are uniquely expressed in each cancer cell type we first identified each vertex: 1. low pc2 score, 2. high pc2 score, and 3. high pc1 score. Then we identified the 500 closest cells (Euclidean distance) to create 3 groups of cells. For each gene we then checked if its expression is significantly higher in one group compared to the other two and higher in that program (P < 10−10, Kolmogorov-Smirnov test, effect size > 0.2, Cohen’s d). As a control we also used a different approach which yielded similar results. This included first identifying dynamic genes (defined as Fano factor and mean expression above mean-dependent thresholds) followed by unsupervised clustering to identify 3 clusters. We found a significant overlap between the set of genes differentially expressed genes in these clusters with the ones we had originally identified using the approach described first.
Spatial transcriptomics (ST) of zebrafish tumors.
Tissue preparation, cryosectioning, fixation, staining, and brightfield imaging:
Zebrafish melanoma tumors were obtained by sectioning the entire tumor with its surrounding tissue. Tissue was transferred from 1X-PBS to a dry, sterile 10-cm dish and gently dried prior to equilibration in cold OCT for 2 minutes. The tissue was then transferred to a tissue-mold with OCT and snap-frozen in liquid nitrogen-chilled isopentane. Tissue blocks were stored at −80°C until further use. Prior to cryosectioning, the cryostat was cleaned with 100% ethanol, and equilibrated to an internal temperature of −18°C for 30 minutes. Once equilibrated, OCT embedded tissue blocks were mounted onto the chuck and equilibrated to the cryostat temperature for 15-20 minutes prior to trimming. ST slide was also placed inside cryostat to keep the slide cold and minimize RNase activity. Sections were cut at 10 μm sections, mounted onto the ST arrays, and stored at −80°C until use, maximum of two weeks. Prior to fixation and staining, the ST array was removed from the −80°C and into a RNase free biosafety hood for 5 minutes to bring to room temperature, followed by warming on a 37°C heat block for 1 minute. Tissue was fixed for 10 minutes with 3.6% formaldehyde in 1X PBS, and subsequently rinsed in 1x PBS. Next, the tissue was dehydrated with isopropanol for 1 minute followed by staining with hematoxylin and eosin. Slides were mounted in 65 μl 80% glycerol and brightfield images were taken on a Leica SCN400 F whole-slide scanner at 40X resolution.
Spatial Transcriptomics (ST) barcoded microarray slide information:
Library preparation slides used were purchased from Spatial Transcriptomics (https://www.spatialtranscriptomics.com; lot 10002). Each of the spots printed onto the array is 100 μm in diameter and 200 μm from the center-to-center, covering an area of 6.2 by 6.6 mm. Spots are printed with approximately 2 x 108 oligonucleotides containing an 18-mer spatial barcode, a randomized 7-mer UMI, and a poly-20TVN transcript capture region (Ståhl et al., 2016).
On-slide tissue permeabilization, cDNA synthesis, probe release:
After brightfield imaging, the ST slide was pre-warmed to 42°C and attached to a pre-warmed microarray slide module to form reaction chambers for each tissue section. The sections were pre-permeabilized with 0.2 mg/ml BSA and 200 units of collagenase diluted in 1X HBSS buffer for 20 minutes at 37°C and washed with 100 μl 0.1X SSC buffer twice. Tissue was permeabilized with 0.1% pepsin in HCl for 4 minutes at 42°C and washed with 100 μl 0.1X SSC buffer twice. Reverse transcription (RT) was carried overnight (~18-20h) at 42°C by incubating permeabilized tissue with 75 μl cDNA synthesis mix containing 1X First strand buffer (Invitrogen), 5 mM DTT, 0.5 mM each dNTP, 0.2 μg/μl BSA, 50 ng/μl Actinomycin D, 1% DMSO, 20 U/μl Superscript III (Invitrogen) and 2U/μl RNaseOUT (Invitrogen). Prior to removal of probes, tissue was digested away from the slide by incubating the tissue with 1% 2-mercaptoethanol in RLT buffer (Qiagen) for one hour at 56°C with interval shaking. Tissue was rinsed gently with 100 μl 1X SSC, and further digested with proteinase K (Qiagen) diluted 1:8 in PKD buffer (Qiagen) at 56°C for 1 hour with interval shaking. Slides were rinsed in 2X SSC with 0.1% SDS, then 0.2X SSC, and finally in 0.1X SSC. Probes were released from the slide by incubating arrays with 65 μl cleavage mix (8.75 μM of each dNTP, 0.2 μg/μl BSA, 0.1 U/μl USER enzyme (New England Biolabs) and incubated at 37 °C for 2 hours with interval mixing. After incubation, 65 μl of cleaved probes was transferred to 0.2 ml low binding tubes and kept on ice.
ST library preparation and sequencing:
Libraries were prepared from cleaved probes as previously described, with the following changes. After RNA amplification by in vitro transcription (IVT) and subsequent bead clean-up, the second RT reaction was performed using random hexamers, eliminating the need for a primer ligation step as described previously (Hashimshony et al., 2016).
ST spot selection and image alignment:
Upon removal of probes from ST slide, the slide is kept at 4°C for up to 3 days. The slide was placed into a microarray cassette and incubated with 70 μl of hybridization solution (0.2 μM Cy3-A-probe, 0.2 μM Cy3 Frame probe, in 1X PBS) for 10 minutes at room temperature. The slide was subsequently rinsed in 2X SSC with 0.1 % SDS for 10 minutes at 50°C, followed by one-minute room temperature washes with 0.2X SSC and 0.1X SSC. Fluorescent images were taken on a Hamamatsu NanoZoomer whole-slide fluorescence scanner. Brightfield images of the tissue and fluorescent images were manually aligned with Adobe Photoshop CS6 to identify the array spots beneath the tissue.
ST library sequence alignment and annotation:
The raw paired end sequencing file was processed by custom pipeline CEL-Seq2 (https://github.com/yanailab/celseq2) to generate the UMI-count matrix for 1007 spots. In general, the CEL-Seq2 was adapted to spatial-transcriptomics data in 3 steps: 1) Tagging and demultiplexing. The leftmost 25nt of R1 sequence consist of 18nt for spot-specific barcode and then 7nt for UMI. R2 sequence reads contain the transcript information and its leftmost 35nt were used for mapping. The name of every R2 read is tagged with spot-specific barcode and UMI sequences that are extracted from the paired R1 read. R2 reads are demultiplexed to create the 1007 spot-specific FASTQ files. If the detected spot-specific barcode of a read is not present in the pre-defined barcodes list, the read is excluded from the downstream analysis. 2) Alignment of demultiplexed FASTQ files using Bowtie2 version 2.3.1 (Langmead and Salzberg, 2012). 3) Counting UMI using customized HTSeq (Anders et al., 2015). The reads that are aligned to are collapsed to count only once if they have the same UMI.
Analysis of ST data:
UMI counts in each spot were normalized by the total number of transcripts per spot and then multiplied by a scale factor equivalent to the median number of transcripts per spot (TPM). A pseudocount of 1 was added prior to log10 transformation. To distinguish between cancer and non-cancer areas clustering was performed as described previously (Moncada et al., 2020) and enrichment of each gene in each of the transcription programs was calculated by Wilcoxon rank sum test.
Stress-like cells in vivo and in vitro functional assays.
Heat shock and drug treatments:
ZMEL1 cells were grown at 28°C (optimum temperature) or 37°C (heat shock) for 48hr with 30, 60, 600 and 6000nM of dabrafenib or 10, 25 and 50nM of trametinib, either alone or in combination. For human melanoma cell line A375, cells were grown either at 37°C (optimum temperature) or 42°C (heat shock) and treated with the same drug concentrations as above. Cell viability was measured using the Promega CellTiter-Glo® 2.0 Cell Viability Assay and luminescence was measured using a Biotek plate reader.
Zebrafish blastula transplants and microscopy:
ZMEL1 cells expressing mitf-GFP and ubb-tdTomato were grown as previously described (Heilmann et al., 2015). Cells were trypsinized and resuspended in Dulbecco’s 1X PBS to a concentration of 2x107cells/mL. Approximately 20 cells in 1nL PBS were injected into the blastula of pre-epiboly casper embryos (~2.5-4 hours post-fertilization) using a quartz microneedle. Embryos were grown in E3 for 24 hours before adding drugs. All fish were grown at 28.5°C for the duration of the experiment. For microscopy, fish were anesthetized in Tricaine and placed on a petri dish containing 2% agarose. The fish were imaged using a Zeiss AxioZoom V16 fluorescence stereoscope with a 0.6X lens. Each fish was consecutively imaged with brightfield, GFP and Rhodamine filters. Raw images (CZIs) of each larva were exported for downstream analysis in MATLAB.
Drug treatments:
Compounds used were dabrafenib (working concentration: 1μM; Selleckchem #S2807) and trametinib (working concentration: 25nM; Selleckchem #2673). DmSO in E3 was used as the vehicle for all drugs and for all drug controls. Drugs were added at 24 hours post-fertilization and changed every subsequent 24 hours until imaging on day 5.
Quantification of tumor burden:
Quantifications of tumor area were performed using the MATLAB Image Processing Toolbox and a fully automated custom image analysis pipeline. Brightfield images were automatically segmented to calculate the area of the entire larva. The brightfield segmentation of the whole larva was then applied as a mask to the corresponding images of GFP fluorescence from the same fish to crop the images and reduce background noise. To measure tumor area, tumor images were thresholded and the number of pixels above the threshold within the body of the larva were counted. Images of GFP fluorescence were used to calculate tumor area for all groups, due to consistency in mitf-GFP fluorescence in all tumor cells across all groups. Tumor surface area was normalized to the total fish surface area.
Quantification of ubb cells using flow cytometry:
For each cell group of treatment, all larval fish were disaggregated in trypsin-EDTA by shaking at 37°C for 15 minutes with intermittent gentle agitation with a Disposable Pellet Pestle (Fisher Scientific) every 5 minutes. Cells were pelleted by centrifugation and resuspended in DMEM supplemented with 2% FBS. All samples were filtered through 40μm cell strainers to achieve single cell suspensions prior to FACS. Individual cancer cells, from each treatment group, were selected based on GFP expression using FACS (Fluorescence activated cell sorting) SONY SH800 cell sorter in single cell mode to achieve the highest purity possible. The distribution of tdTomato was recorded and analyzed using FlowJo software v10.6.0 (Tree star, Ashland, OR, USA).
ZMEL1-GFP single cell RNA-Seq.
Embryo Transplantation, sample preparation, dissociation and collection.
ZMEL zebrafish melanoma cells were grown as previously described (Heilmann et al., 2015). Cells were detached from standard tissue culture dishes with trypsin-EDTA and approximately 50 cells in 1nl of 0.9X Dulbecco’s PBS were transplanted intravenously into the Duct of Cuvier of each 2 day-post-fertilization casper embryo as previously described (Heilmann et al., 2015; Kim et al., 2017). Fish with successful transplants were allowed to form widespread metastatic disease until 17 days post-transplant, at which point they were used for scRNA-Seq. Two larval fish with widespread metastases were disaggregated individually in trypsin-EDTA by shaking at 37°C for 20 minutes with intermittent gentle agitation with a Disposable Pellet Pestle (Fisher Scientific). Cells were pelleted by centrifugation and resuspended in DMEM supplemented with 2% FBS. In parallel, ZMEL cells grown in standard tissue culture conditions were trypsinized and resuspended in DMEM with 2% FBS. All samples were filtered through 40μm cell strainers to achieve single cell suspensions prior to FACS. 93 Individual cells, from each cell line and each condition, were sorted based on EGFP expression using a FACS (Fluorescence activated cell sorting) SONY SH800 cell sorter into wells of 384-well plates pre-loaded with the CEL-Seq2 primer mix (Hashimshony et al., 2016). After sorting, the plates were immediately spun down and snapped freeze using liquid nitrogen and kept in −80 for further processing.
Single cell mRNA Sequencing of single cells and analysis.
384 well plates with sorted cells were thawed on ice. Amplification of mRNA and library preparation was performed according to the CEL-Seq2 protocol (Hashimshony et al., 2016). scRNA-Seq library paired-end sequencing was carried out using the Illumina NextSeq 500/550 machine. Fastq files were processed using the custom CEL-Seq2 pipeline (https://github.com/yanailab/celseq2) and UMI counts were normalized by the total number of transcripts per cell and then multiplied by a scale factor equivalent to the median number of transcripts per spot (TPM). The program expression profile was calculated by averaging the TPM expression of all genes associated with each program.
Immunofluorescence of FOS protein on tumor microarray.
Tumor microarrays (TMA) were obtained from US Biomax (ME1004g). The slide was baked for 30 minutes in 60°C and washed three times with Xylene, 100% EtOH and 95% EtOH. After rinsing with DI H20, antigen retrieval was performed for 12 minutes in a boiling TE buffer. Slide was cooled down and rinsed with DI H20 following a wash in TBS+0.05% Tween. The array was stained for FOS (1:1000; Synaptic systems 226 003) for 48 hours at room temperature in a wet chamber. Before applying the secondary antibody, three washes with TBS+0.05% Tween were performed. DAPI and Secondary antibody (1:100) was applied and incubated for 1hr in a wet chamber at room temperature followed by 3 washes in TBS+0.05% Tween. Slide was dried and mounted before scanning for imaging.
Re-analysis of existing datasets.
Re-analysis of Tirosh et al. human scRNA-Seq melanoma dataset:
Normalized scRNA-Seq data was retrieved from the Tirosh et al. publication (Tirosh et al., 2016a) and transformed using the Freeman-Tukey approach (Wagner et al., 2017). We examined the cancer cells as annotated by Tirosh et al., and of those only the MITF high (proliferating) cells.
Re-analysis of Mica et al. human melanocyte differentiation dataset:
Normalized microarray data was retrieved from Mica et al. (Mica et al., 2013) for the wild-type cell line studied using the standard and neural crest optimized protocols. PCA was performed on samples using the most variable genes (defined above). K-means clustering with K=3 yielded the following groupings: samples from day 0 to day 3, samples from day 6 to day 11 and primary and mature melanocytes. To calculate the Pearson correlation of each cancer cell state to each grouping of melanocyte differentiation, in silico bulk expression profiles were created by averaging each cancer cell type from the human scRNA-Seq dataset (Tirosh et al., 2016a). Next, the resulting profiles were scaled to the same range of melanocyte differentiation data set using min/max scaling. Finally, only genes that are differentially expressed among the three groupings were used for the correlation calculation (P < 10−5, Kolmogorov-Smirnov test and expression above a mean dependent threshold).
Re-analysis of Moncada et al. human scRNA-Seq pancreatic adenocarcinoma dataset:
scRNA-Seq data was retrieved from Moncada et al. publication (Moncada et al., 2020), smoothed using the MAGIC method (van Dijk et al., 2018) and transformed using the Freeman-Tukey approach (Wagner et al., 2017). We examined only the cancer cells, 462 in total, using the approach described in the “Dimensionality Reduction (PCA and tSNE)” part. The stress program was defined using only the genes that are enriched for this cancer cell type (see “Cancer cell type analysis” part for details) in the human scRNA-Seq Melanoma dataset.
Re-analysis of Kim et al. human scRNA-Seq triple negative breast cancer dataset:
Normalized scRNA-Seq data was retrieved from Kim et al. publication (Kim et al., 2018), smoothed using the MAGIC method (van Dijk et al., 2018) and transformed using the Freeman-Tukey approach (Wagner et al., 2017). We examined only the cancer cells from donor P_6 before treatment, using the same approach as described in “Re-analysis of Moncada et al. human scRNA-Seq pancreatic adenocarcinoma dataset”.
Re-analysis of Tirosh et al. human scRNA-Seq oligodendroglioma dataset:
Normalized scRNA-Seq data was retrieved from Tirosh et al. publication (Tirosh et al., 2016b), smoothed using the MAGIC method (van Dijk et al., 2018) and transformed using the Freeman-Tukey approach (Wagner et al., 2017) The stress-like transcriptional program was identified using the same approach as described in “Re-analysis of Moncada et al. human scRNA-Seq pancreatic adenocarcinoma dataset”.
Re-analysis of Ho et al. human scRNA-Seq BRAF inhibitor treated melanoma cell line dataset:
scRNA-Seq data was retrieved from Ho et al. publication (Ho et al., 2018), normalized by total transcript per cell and transformed using the Freeman-Tukey approach (Wagner et al., 2017). Each transcriptional program was quantified using the same approach as described in “Re-analysis of Moncada et al. human scRNA-Seq pancreatic adenocarcinoma dataset” for both treated and untreated cells and visualized using violin plots.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of scRNA-seq data, including quality controls is described in the sections “Single-cell RNA-Seq collection and processing of tumor biopsies”,”Single cell RNA-Seq analysis”, “Spatial transcriptomics (ST) of zebrafish tumors” and “ZMEL1-GFP single cell RNA-Seq”. Quantification of tumor burden is described in the section “Stress-like cells in vivo and in vitro functional assays”. All statistical tests outputs (p-value) are described in figures legends, adjusted using Benjamini-Hochberg correction and significance was defined as p-value<0.05.
Graphs and illustrations were performed with Matlab, Python and Illustrator software.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| c-FOS antibody | Synaptic systems | 226 003 |
| Biological Samples | ||
| Melanoma Tumor microarray | US Biomax | ME1004g |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dabrafenib | Selleckchem | #S2807 |
| Trametinib | Selleckchem | #2673 |
| DPBS, no calcium, no magnesium | Themo Fisher Scientific | #14190250 |
| Fetal Bovine Serum | Themo Fisher Scientific | #16000044 |
| Trypsin-EDTA | Themo Fisher Scientific | #25200056 |
| SuperScript III Reverse Transcriptase | Invitrogen | #18080085 |
| PrimeScriptTM Reverse | Takara Clonetech | #2680A |
| Agencourt RNAClean XP magnetic beads | Beckman Coulter | #A63987 |
| Agencourt AMPure XP magnetic beads | Beckman Coulter | #A63881 |
| Critical Commercial Assays | ||
| inDrop scRNA-Seq kit | 1CellBio | 10196 |
| Qubit dsDNA HS Assay Kit | Invitrogen | Q32851 |
| Qubit RNA HS Assay Kit | Invitrogen | Q32852 |
| Bioanalyzer RNA 6000 Pico Kit | Agilent | 5067-1513 |
| Bioanalyzer High Sensitivity DNA Analysis Kit | Agilent | 5067-4626 |
| NEBNext mRNA Second Strand Synthesis Kit | New England Biolabs | E6111S |
| HiScribe T7 High Yield RNA Synthesis kit | New England Biolabs | E2040S |
| NextSeq 500/550 75 cycles High output v2 kit | Illumina | FC-404-2005 |
| Spatial Transcriptomics kits | Spatial Transcriptomics | 10002 |
| CellTiter-Glo Cell Viability Assay | Promega | G9241 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GSE115140 |
| Zebrafish reference genome GRCz10 | Ensemble release 91 | ftp://ftp.ensembl.org/pub/release-91/fasta/danio_rerio/ |
| Zebrafish genome annotation GRCz10 | Ensemble release 91 | ftp://ftp.ensembl.org/pub/release-91/fasta/danio_rerio/ |
| Single cell RNA-Seq human melanoma | Tirosh et al., 2016a | GSE72056 |
| Human melanocyte differentiation microarray | Mica et al., 2013 | GSE45227 |
| Single cell RNA-Seq human pancreatic adenocarcinoma | Moncada et al., 2020 | GSE111672 |
| Single cell RNA-Seq human triple negative breast cancer | Kim et al., 2018 | Navin Lab |
| Single cell RNA-Seq human oligodendroglioma | Tirosh et al., 2016b | GSE70630 |
| Single cell RNA-Seq of human melanoma cell line treated with BRAF inhibitor | Ho et al., 2018 | SRP127328 |
| Experimental Models: Cell Lines | ||
| A375 | ATCC | NA |
| ZMEL1 | White Lab | NA |
| ZMEL1-tdtomato-ubb | This paper | NA |
| Experimental Models: Organisms/Strains | ||
| BRAFV600E Zebrafish | White Lab | NA |
| Oligonucleotides | ||
| inDrop PE2-N6 primer: TCGGCATTCCTGCTGAACCGCTCTTCCGATCTNNNNNN | IDT | N/A |
| inDrop PE1 primer index 1: CAAGCAGAAGACGGCATACGAGATCGTGATCTCTTTCCCTACACGA | IDT | N/A |
| inDrop PE1 primer index 2: CAAGCAGAAGACGGCATACGAGATACATCGCTCTTTCCCTACACGA | IDT | N/A |
| inDrop PE1 primer index 3: CAAGCAGAAGACGGCATACGAGATGCCTAACTCTTTCCCTACACGA | IDT | N/A |
| inDrop PE1 primer index 4: CAAGCAGAAGACGGCATACGAGATTGGTCACTCTTTCCCTACACGA | IDT | N/A |
| inDrop PE1 primer index 5: CAAGCAGAAGACGGCATACGAGATCACTGTCTCTTTCCCTACACGA | IDT | N/A |
| inDrop PE1 primer index 6: CAAGCAGAAGACGGCATACGAGATATTGGCCTCTTTCCCTACACGA | IDT | N/A |
| inDrop PE1 primer index 7: CAAGCAGAAGACGGCATACGAGATGATCTGCTCTTTCCCTACACGA | IDT | N/A |
| inDrop PE1 primer index 8: CAAGCAGAAGACGGCATACGAGATTCAAGTCTCTTTCCCTACACGA | IDT | N/A |
| inDrop Custom Read 1 primer: GGCATTCCTGCTGAACCGCTCTTCCGATCT | IDT | N/A |
| inDrop Custom Index Read primer: AGATCGGAAGAGCGTCGTGTAGGGAAAGAG | IDT | N/A |
| inDrop scRNA-seq: Custom Read 2 primer: CTCTTTCCCTACACGACGCTCTTCCGATCT | IDT | N/A |
| Software and Algorithms | ||
| inDrop pipeline | Yanai Lab | https://github.com/flo-compbio/singlecell |
| Custom codes for scRNA-Seq analysis | This paper | https://github.com/MaayanBaron/sc_melanoma_Baron2020 |
| STAR mapper | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| MATLAB R2017a | Mathworks | https://www.mathworks.com/ |
| MAGIC | van Dijk et al., 2018 | https://github.com/KrishnaswamyLab/MAGIC |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| CEL-Seq2 pipeline | Yanai Lab | https://github.com/yanailab/celseq2 |
| FlowJo v10.6.0 | Tree star | https://www.flowjo.com/ |
| HTSeq | Anders et al., 2015 | https://htseq.readthedocs.io/en/master/ |
Highlights:
Melanomas exhibit 3 cell states: neural-crest, mature melanocyte and stress-like.
The stress-like cancer cell state is conserved across tumor types and species.
Stress-like cancer cells are pro-tumorigenic and efficient at seeding new tumors.
Stress-like cancer cells hold drug-resistant properties induced by heatshock.
ACKNOWLEDGMENTS
We thank Naftalie Senderovich and Anna Yeaton for work on the initial pilot of the project. We thank Matt Maurano, Megan Hogan and the NYU Langone Genome Technology Center for assistance with sequencing. This work is supported by the Leon Lowenstein Foundation, NIH Director’s New Innovator Award (DP2CA186572), NIH Research Program Grant (R01CA229215), Mentored Clinical Scientist Research Career Development Award (K08AR055368), Kirschstein-NRSA Predoctoral Fellowship (F30CA220954), Medical Scientist Training Program Award (T32GM007739), NIH Cancer Center Support Grant to MSKCC (P30CA008748), the Melanoma Research Alliance, and the Melanoma Research Foundation, The Pershing Square Sohn Foundation, The Mark Foundation for Cancer Research, The Alan and Sandra Gerry Metastasis Research Initiative at MSKCC, The Harry J. Lloyd Foundation, Consano, and The Starr Cancer Consortium. This work was also supported by NYU Langone Health start-up funds.
Footnotes
DECLARATION OF INTRESTS
RMW is a paid consultant to N-of-One, Inc., a subsidiary of Qiagen. None of the work described in this manuscript is related to this work. He serves on the Scientific Advisory Board of Consano, a non-profit crowdfunding company and receives no compensation for this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ahmed F, and Haass NK (2018). Microenvironment-Driven Dynamic Heterogeneity and Phenotypic Plasticity as a Mechanism of Melanoma Therapy Resistance. Front. Oncol 8, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkley D, and Yanai I (2019). Plasticity and Clonality of Cancer Cell States. Trends Cancer Res. 5, 655–656. [DOI] [PubMed] [Google Scholar]
- Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, Ryu JH, Wagner BK, Shen-Orr SS, Klein AM, et al. (2016). A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst 3, 346–360.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi M, Giacomini E, Crinelli R, Radici L, Carloni E, and Magnani M (2015). Dynamic transcription of ubiquitin genes under basal and stressful conditions and new insights into the multiple UBC transcript variants. Gene 573, 100–109. [DOI] [PubMed] [Google Scholar]
- van den Brink SC, Sage F, Vértesy Á, Spanjaard B, Peterson-Maduro J, Baron CS, Robin C, and van Oudenaarden A (2017). Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 14, 935–936. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2015). Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona SJ, Teichmann SA, Ferreira L, Macaulay IC, Stubbington MJT, Cvejic A, and Gfeller D (2017). Single-cell transcriptome analysis of fish immune cells provides insight into the evolution of vertebrate immune cell types. Genome Res. 27, 451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira S, Goodall J, Denat L, Rodriguez M, Nuciforo P, Hoek KS, Testori A, Larue L, and Goding CR (2006). Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 20, 3426–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, Fritsch L, Lin WM, Hollmann TJ, Ferré F, et al. (2011). The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 471, 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, and Sampson SB (2016). HSF1: Guardian of Proteostasis in Cancer. Trends Cell Biol. 26, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai S, Liu Z, Yao J, Patel N, Chen J, Wu Y, Ahn EE-Y, Fodstad O, and Tan M (2013). Heat shock factor 1 (HSF1) controls chemoresistance and autophagy through transcriptional regulation of autophagy-related protein 7 (ATG7). J. Biol. Chem 288, 9165–9176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, Burdziak C, Moon KR, Chaffer CL, Pattabiraman D, et al. (2018). Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 174, 716–729.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KL, Espino PS, Drobic B, He S, and Davie JR (2005). The Ras-MAPK signal transduction pathway, cancer and chromatin remodeling. Biochem. Cell Biol 83, 1–14. [DOI] [PubMed] [Google Scholar]
- Eisenhoffer GT, Slattum G, Ruiz OE, Otsuna H, Bryan CD, Lopez J, Wagner DS, Bonkowsky JL, Chien C-B, Dorsky RI, et al. (2017). A toolbox to study epidermal cell types in zebrafish. J. Cell Sci 130, 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmons MF, Faião-Flores F, Sharma R, Thapa R, Messina JL, Becker JC, Schadendorf D, Seto E, Sondak VK, Koomen JM, et al. (2019). HDAC8 Regulates a Stress Response Pathway in Melanoma to Mediate Escape from BRAF Inhibitor Therapy. Cancer Res. 79, 2947–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falletta P, Sanchez-Del-Campo L, Chauhan J, Effern M, Kenyon A, Kershaw CJ, Siddaway R, Lisle R, Freter R, Daniels MJ, et al. (2017). Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes Dev. 31, 18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R, Pusztai L, and Swanton C (2013). Cancer heterogeneity: implications for targeted therapeutics. Br. J. Cancer 108, 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flick K, and Kaiser P (2012). Protein degradation and the stress response. Semin. Cell Dev. Biol 23, 515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gansner JM, Dang M, Ammerman M, and Zon LI (2017). Transplantation in zebrafish. Methods Cell Biol. 138, 629–647. [DOI] [PubMed] [Google Scholar]
- Gao R, Kim C, Sei E, Foukakis T, Crosetto N, Chan L-K, Srinivasan M, Zhang H, Meric-Bernstam F, and Navin N (2017). Nanogrid single-nucleus RNA sequencing reveals phenotypic diversity in breast cancer. Nat. Commun 8, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Jiménez C, and Goding CR (2019). Starvation and Pseudo-Starvation as Drivers of Cancer Metastasis through Translation Reprogramming. Cell Metab. 29, 254–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gistelinck C, Gioia R, Gagliardi A, Tonelli F, Marchese L, Bianchi L, Landi C, Bini L, Huysseune A, Witten PE, et al. (2016). Zebrafish Collagen Type I: Molecular and Biochemical Characterization of the Major Structural Protein in Bone and Skin. Scientific Reports 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grbovic OM, Basso AD, Sawai A, Ye Q, Friedlander P, Solit D, and Rosen N (2006). V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc. Natl. Acad. Sci. U. S. A 103, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, Choudhury SR, Aguet F, Gelfand E, Ardlie K, et al. (2017). Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, Avivar-Valderas A, Nagi C, Girnius N, Davis RJ, et al. (2016). Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 540, 588–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T, Senderovich N, Avital G, Klochendler A, de Leeuw Y, Anavy L, Gennert D, Li S, Livak KJ, Rozenblatt-Rosen O, et al. (2016). CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 17, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy S, Khan P, and Davie JR (2013). Immediate early response genes and cell transformation. Pharmacol. Ther 137, 64–77. [DOI] [PubMed] [Google Scholar]
- Heilmann S, Ratnakumar K, Langdon E, Kansler E, Kim I, Campbell NR, Perry E, McMahon A, Kaufman C, van Rooijen E, et al. (2015). A Quantitative System for Studying Metastasis Using Transparent Zebrafish. Cancer Res. 75, 4272–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y-J, Anaparthy N, Molik D, Mathew G, Aicher T, Patel A, Hicks J, and Hammell MG (2018). Single-cell RNA-seq analysis identifies markers of resistance to targeted BRAF inhibitors in melanoma cell populations. Genome Res. 28, 1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek KS, Eichhoff OM, Schlegel NC, Döbbeling U, Kobert N, Schaerer L, Hemmi S, and Dummer R (2008). In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 68, 650–656. [DOI] [PubMed] [Google Scholar]
- Hollstein M, Sidransky D, Vogelstein B, and Harris CC (1991). p53 mutations in human cancers. Science 253, 49–53. [DOI] [PubMed] [Google Scholar]
- Iyengar S, Houvras Y, and Ceol CJ (2012). Screening for melanoma modifiers using a zebrafish autochthonous tumor model. J. Vis. Exp e50086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, Leeson R, Kanodia A, Mei S, Lin J-R, et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman CK, Mosimann C, Fan ZP, Yang S, Thomas AJ, Ablain J, Tan JL, Fogley RD, van Rooijen E, Hagedorn EJ, et al. (2016). A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 351, aad2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, Crosetto N, Foukakis T, and Navin NE (2018). Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 173, 879–893.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IS, Heilmann S, Kansler ER, Zhang Y, Zimmer M, Ratnakumar K, Bowman RL, Simon-Vermot T, Fennell M, Garippa R, et al. (2017). Microenvironment-derived factors driving metastatic plasticity in melanoma. Nat. Commun 8, 14343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RM, and Higgins PJ (2011). A switch in RND3-RHOA signaling is critical for melanoma cell invasion following mutant-BRAF inhibition. Mol. Cancer 10, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, and Kirschner MW (2015). Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtis N, Moubarak RS, Aranda-Orgilles B, Lui K, Aydin IT, Trimarchi T, Darvishian F, Salvaggio C, Zhong J, Bhatt K, et al. (2015). FBXW7 modulates cellular stress response and metastatic potential through HSF1 post-translational modification. Nat. Cell Biol 17, 322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswami SR, Grindberg RV, Novotny M, Venepally P, Lacar B, Bhutani K, Linker SB, Pham S, Erwin JA, Miller JA, et al. (2016). Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nat. Protoc 11, 499–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkeaw K, and Sugiyama D (2012). Zebrafish erythropoiesis and the utility of fish as models of anemia. Stem Cell Res. Ther 3, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lièvre A, Bachet J-B, Le Corre D, Boige V, Landi B, Emile J-F, Côté J-F, Tomasic G, Penna C, Ducreux M, et al. (2006). KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 66, 3992–3995. [DOI] [PubMed] [Google Scholar]
- van der Maaten L, and Hinton G (2008). Visualizing Data using t-SNE. J. Mach. Learn. Res 9, 2579–2605. [Google Scholar]
- Maruyama S, Nakamura K, Papanicolaou KN, Sano S, Shimizu I, Asaumi Y, van den Hoff MJ, Ouchi N, Recchia FA, and Walsh K (2016). Follistatin-like 1 promotes cardiac fibroblast activation and protects the heart from rupture. EMBO Mol. Med 8, 949–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson D, Bradbury CM, Bisht KS, Curry HA, Spitz DR, and Gius D (2004). Heat shock and the activation of AP-1 and inhibition of NF-kappa B DNA-binding activity: possible role of intracellular redox status. Int. J. Hyperthermia 20, 224–233. [DOI] [PubMed] [Google Scholar]
- Meacham CE, and Morrison SJ (2013). Tumour heterogeneity and cancer cell plasticity. Nature 501, 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L, and Lindquist S (2012). HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150, 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mica Y, Lee G, Chambers SM, Tomishima MJ, and Studer L (2013). Modeling neural crest induction, melanocyte specification, and disease-related pigmentation defects in hESCs and patient-specific iPSCs. Cell Rep. 3, 1140–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada R, Barkley D, Wagner F, Chiodin M, Devlin JC, Baron M, Hajdu CH, Simeone DM, and Yanai I (2020). Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol 38, 333–342. [DOI] [PubMed] [Google Scholar]
- Mosimann C, Kaufman CK, Li P, Pugach EK, Tamplin OJ, and Zon LI (2011). Ubiquitous transgene expression and Cre-based recombination driven by the ubiquitin promoter in zebrafish. Development 138, 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, Kong X, Possik PA, Cornelissen-Steijger PDM, Geukes Foppen MH, et al. (2014). Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun 5, 5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell MP, Marchbank K, Webster MR, Valiga AA, Kaur A, Vultur A, Li L, Herlyn M, Villanueva J, Liu Q, et al. (2013). Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov. 3, 1378–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell A, Odrowaz Z, and Sharrocks AD (2012). Immediate-early gene activation by the MAPK pathways: what do and don’t we know? Biochem. Soc. Trans 40, 58–66. [DOI] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton EE, Widlund HR, Kutok JL, Kopani KR, Amatruda JF, Murphey RD, Berghmans S, Mayhall EA, Traver D, Fletcher CDM, et al. (2005). BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr. Biol 15, 249–254. [DOI] [PubMed] [Google Scholar]
- Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo L, Mroz EA, Emerick KS, et al. (2017). Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 171, 1611–1624.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram SV, Parikh AS, and Tirosh I (2018). Single cell RNA-seq highlights a role for a partial EMT in head and neck cancer. Mol Cell Oncol 5, e1448244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Rogiers A, Marin-Bejar O, Aibar S, Femel J, Dewaele M, Karras P, Brown D, Chang YH, Debiec-Rychter M, et al. (2018). Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 174, 843–855.e19. [DOI] [PubMed] [Google Scholar]
- Ranganathan AC, Adam AP, and Aguirre-Ghiso JA (2006). Opposing roles of mitogenic and stress signaling pathways in the induction of cancer dormancy. Cell Cycle 5, 1799–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riely GJ, Marks J, and Pao W (2009). KRAS Mutations in Non–Small Cell Lung Cancer. Proc. Am. Thorac. Soc 6, 201–205. [DOI] [PubMed] [Google Scholar]
- Saldana-Caboverde A, and Kos L (2010). Roles of endothelin signaling in melanocyte development and melanoma. Pigment Cell Melanoma Res. 23, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santagata S, Xu Y-M, Wijeratne EMK, Kontnik R, Rooney C, Perley CC, Kwon H, Clardy J, Kesari S, Whitesell L, et al. (2012). Using the heat-shock response to discover anticancer compounds that target protein homeostasis. ACS Chem. Biol 7, 340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrió D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, and Palacios J (2008). Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 68, 989–997. [DOI] [PubMed] [Google Scholar]
- Sawai M, Ishikawa Y, Ota A, and Sakurai H (2013). The proto-oncogene JUN is a target of the heat shock transcription factor HSF1. FEBS J. 280, 6672–6680. [DOI] [PubMed] [Google Scholar]
- Shakhova O, Zingg D, Schaefer SM, Hari L, Civenni G, Blunschi J, Claudinot S, Okoniewski M, Beermann F, Mihic-Probst D, et al. (2012). Sox10 promotes the formation and maintenance of giant congenital naevi and melanoma. Nat. Cell Biol 14, 882–890. [DOI] [PubMed] [Google Scholar]
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. (2010). A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss M, et al. (2016). Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82. [DOI] [PubMed] [Google Scholar]
- Stancovski I, Gonen H, Orian A, Schwartz AL, and Ciechanover A (1995). Degradation of the proto-oncogene product c-Fos by the ubiquitin proteolytic system in vivo and in vitro: identification and characterization of the conjugating enzymes. Mol. Cell. Biol 15, 7106–7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei ML, Giannoni E, Raugei G, Scacco S, Sardanelli AM, Papa S, and Chiarugi P (2012). Mitochondrial Oxidative Stress due to Complex I Dysfunction Promotes Fibroblast Activation and Melanoma Cell Invasiveness. J. Signal Transduct 2012, 684592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM, et al. (2017). A subcellular map of the human proteome. Science 356. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016a). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG, et al. (2016b). Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travnickova J, Wojciechowska S, Khamseh A, Gautier P, Brown DV, Lefevre T, Brombin A, Ewing A, Capper A, Spitzer M, et al. (2019). Zebrafish MITF-Low Melanoma Subtype Models Reveal Transcriptional Subclusters and MITF-Independent Residual Disease. Cancer Res. 79, 5769–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao H, Chin L, Garraway LA, and Fisher DE (2012). Melanoma: from mutations to medicine. Genes Dev. 26, 1131–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. (2018). Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 33, 890–904.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G, Christiaens V, Svetlichnyy D, Luciani F, Van den Mooter L, et al. (2015). Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun 6, 6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivas-García Y, Falletta P, Liebing J, Louphrasitthiphol P, Feng Y, Chauhan J, Scott DA, Glodde N, Chocarro-Calvo A, Bonham S, et al. (2020). Lineage-Restricted Regulation of SCD and Fatty Acid Saturation by MITF Controls Melanoma Phenotypic Plasticity. Mol. Cell 77, 120–137.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, and Kinzler KW (2004). Cancer genes and the pathways they control. Nat. Med 10, 789–799. [DOI] [PubMed] [Google Scholar]
- Wagner F, Yan Y, and Yanai I (2017). K-nearest neighbor smoothing for high-throughput single-cell RNA-Seq data. bioRxiv. doi: 10.1101/217737 [DOI] [Google Scholar]
- Webster KA, Discher DJ, and Bishopric NH (1993). Induction and nuclear accumulation of fos and jun proto-oncogenes in hypoxic cardiac myocytes. J. Biol. Chem 268, 16852–16858. [PubMed] [Google Scholar]
- Webster MR, Xu M, Kinzler KA, Kaur A, Appleton J, O’Connell MP, Marchbank K, Valiga A, Dang VM, Perego M, et al. (2015). Wnt5A promotes an adaptive, senescent-like stress response, while continuing to drive invasion in melanoma cells. Pigment Cell Melanoma Res. 28, 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RM, Cech J, Ratanasirintrawoot S, Lin CY, Rahl PB, Burke CJ, Langdon E, Tomlinson ML, Mosher J, Kaufman C, et al. (2011). DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature 471, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, and Lindquist SL (2005). HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5, 761–772. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Santagata S, Mendillo ML, Lin NU, Proia DA, and Lindquist S (2014). HSP90 empowers evolution of resistance to hormonal therapy in human breast cancer models. Proc. Natl. Acad. Sci. U. S. A 111, 18297–18302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmer DS, Cheng PF, Eichhoff OM, Belloni BC, Zipser MC, Schlegel NC, Javelaud D, Mauviel A, Dummer R, and Hoek KS (2012). Systematic classification of melanoma cells by phenotype-specific gene expression mapping. Pigment Cell Melanoma Res. 25, 343–353. [DOI] [PubMed] [Google Scholar]
- Xia B, Yan Y, Baron M, Wagner F, Barkley D, Chiodin M, Kim SY, Keefe DL, Alukal JP, Boeke JD, et al. (2020). Widespread Transcriptional Scanning in the Testis Modulates Gene Evolution Rates. Cell 180, 248–262.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen J, White RM, Wedge DC, Van Loo P, de Ridder J, Capper A, Richardson J, Jones D, Raine K, Watson IR, et al. (2013). The genetic heterogeneity and mutational burden of engineered melanomas in zebrafish models. Genome Biol. 14, R113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Girón CG, et al. (2018). Ensembl 2018. Nucleic Acids Res. 46, D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilionis R, Nainys J, Veres A, Savova V, Zemmour D, Klein AM, and Mazutis L (2017). Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc. 12, 44–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The complete data that support the findings of this study have been deposited in NCBI GEO database with the accession code GSE115140. The code generated during this study is available in Github: https://github.com/MaayanBaron/sc_melanoma_Baron2020