

Abstract
Alzheimer's disease (AD) is characterized by accumulation of β‐amyloid plaques (AP) and neurofibrillary tangles (NFT) in the cortex, together with synaptic loss and amyloid angiopathy. Perturbations in the brain lysosomal system, including the cathepsin family of proteases, have been implicated in AD where they may be involved in proteolytic clearance of misfolded and abnormally aggregated peptides. However, the status of cathepsin D (catD) is unclear in Lewy body dementia, the second most common form of neurodegenerative dementia after AD, and characterized by Lewy bodies (LB) containing aggregated α‐synuclein. Furthermore, earlier reports of catD changes in AD have not been entirely consistent. We measured CatD immunoreactivities in the temporal (Brodmann area BA21) and parietal (BA40) cortices of well characterized AD brains as well as two clinical subtypes of Lewy body dementia, namely Parkinson disease dementia (PDD) and dementia with Lewy bodies (DLB), known to show varying degrees of concomitant AD pathology. Increased catD immunoreactivities in AD were found for both neocortical regions measured, where they also correlated with neuropathological NFT scores and phosphorylated pSer396 tau burden, and appeared to co‐localize at least partly to NFT‐containing neurons. In contrast, catD was increased only in BA40 in DLB and not at all in PDD, did not correlate with LB scores, and did not appreciably co‐localize with α‐synuclein inclusions. Our study suggests that catD upregulation may be an adaptive response to AD‐related processes leading to neurofibrillary degeneration, but may not be directly associated with formation of α‐synuclein inclusions in Lewy body dementia.
Keywords: Alzheimer's disease, cathepsin D, Lewy body dementia, lysosome, neurofibrillary degeneration
Abbreviations
- AD
Alzheimer's disease
- catD
cathepsin D
- CIND
cognitive impaired no dementia
- CNS
central nervous system
- DLB
dementia with Lewy bodies
- NCI
non‐cognitively impaired participants
- NFT
neurofibrillary tangles
- NINCDS‐ADRDA
National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's disease and Related Disorders Association
- PDD
Parkinson's disease dementia
- AP
amyloid plaques
Introduction
Alzheimer disease (AD) is the most common cause of dementia, and represents a substantial, global healthcare burden 3, 33. Although the “amyloid cascade hypothesis” of AD, which attributes pathogenicity to processes favoring the accumulation and aggregation of β‐amyloid (Aβ) peptides into extracellular senile plaques has provided much research impetus 27, 28, doubts remain as to the exact nature of pathogenic Aβ species as well as their precise roles of Aβ species in the disease process 37, 49. Furthermore, therapeutic strategies based on reducing cortical Aβ deposition have, to date, been disappointing 15. This underscores the need to further understand the complex interactions between multiple processes, including amyloidogenesis, tau hyper‐phosphorylation forming neurofibrillary tangles 25, neuroinflammation 16, alternative splicing dysregulation 40, cell cycle dysregulation 51 as well as lysosomal and autophagic dysfunction 44, 52, within which the cathepsin family of proteases are important due to their critical roles in the degradation and turnover of abnormally aggregated or damaged proteins. In particular, cathepsins B and D have been implicated in various cancers as well as in cellular processes which may be relevant in neurodegeneration, including cell cycle regulation, autophagy and neuroinflammation 5, 22, 41, 46, 65. Cathepsin B, a cysteine protease, has been extensively studied in AD for its potential anti‐amyloidogenic and neuroprotective effects 50, 60. In contrast, previous studies on the aspartyl protease cathepsin D (CatD) in AD are less clear. Whilst a number of studies report activation or upregulation in vulnerable neuronal populations 10, 11, 12, there were also reports of decreased monocyte CatD expression in peripheral blood 62 contrasting with unchanged levels in the cortex 38, or increased mRNA in histologically normal neurons but decreased mRNA in degenerating neurons 10. Furthermore, the status of CatD is unclear in the neocortex of Lewy body dementias, whose major clinical subtypes, dementia with Lewy bodies (DLB) and Parkinson's disease dementia (PDD), comprise the second commonest cause of neurodegenerative dementias after AD 1, 2. Whilst DLB and PDD are characterized by cortical Lewy bodies consisting of abnormally aggregated α‐synuclein, both conditions also manifest variable burden of AD pathological hallmarks, including senile plaques and neurofibrillary tangles 24, 32, 34, 57. Because CatD function has been implicated in the clearance of various proteins, including Aβ, tau and α‐synuclein 26, 36, 58, we aimed to measure CatD immunoreactivity in the postmortem neocortex of AD, PDD and DLB and investigate its association with AD and Lewy body dementia neuropathological burden
Methods
Patients, clinical and neuropathological assessments
Tissues for the postmortem study were obtained from subjects recruited into longitudinal studies with postmortem follow up from the University Hospital Stavanger, Newcastle Brain Tissue Resource, the London Neurodegenerative Diseases Brain Bank and the Thomas Willis Brain Collections at Oxford University, the UK sites being part of the Brains for Dementia Research network (http://brainsfordementiaresearch.org.uk). All dementia subjects for this study were selected on the basis of clinicopathological diagnosis. Diagnostic criteria included the Consortium to Establish a Registry for Alzheimer's disease (CERAD) criteria for AD 47, and the Dementia with Lewy bodies Consortium's “one‐year rule” 45 together with the Movement Disorders Society criteria 18 to distinguish between DLB and PDD. Annual cognitive assessments with MMSE 21 were also available, and the average decline per year (MMSE decline) from the time of dementia diagnosis to death was used as an indicator of dementia severity. At death, informed consent was sought from next‐of‐kin before removal of brains, which were divided into hemispheres, with one formalin fixed for neuropathological assessments. Neuropathological diagnoses were based on Thal Aβ phases 61, neurofibrillary tangle Braak stages 7 and CERAD criteria for AD 47 which are all combined in the National Institute on Aging – Azheimer's Association guidelines 48 and the Newcastle/McKeith criteria for Lewy body disease 45. In addition, semi‐quantitative scoring for amyloid plaques (AP, immunostaining with the 4G8 antibody), neurofibrillary tangles/neuropil threads (NFT, immunostaining with the AT8 antibody), and α‐synuclein‐containing inclusions, that is, Lewy bodies/Lewy neurites (LB, α‐synuclein immunostaining) were performed as previously described 32 by neuropathologists blinded to clinical diagnosis on a four point scale: 0 = None, 1 = Sparse, 2 = Moderate and 3 = Abundant. The contralateral hemisphere was coronally sectioned before further dissection to obtain 1 cm3 blocks from selected regions, followed by fresh freezing and storage at −80°C. Brains from controls were neurologically and cognitively normal, had only age‐associated neuropathological changes and no history of psychiatric diseases.
Tissue processing
Frozen blocks of tissues from the middle temporal gyrus (Brodmann area, BA21) and parietal lobe (BA40) were thawed on ice and dissected free of meninges and white matter, then homogenised with an Ultra‐Turrax homogeniser (IKA, Staufen im Breisgau, Germany, on highest setting, 10 s) in ice‐cold buffer (50 mM Tris‐HCl, 120 mM NaCl, 5 mM KCl, pH 7.4) with cOmplete™ protease inhibitor cocktail and PhosSTOP™ phosphatase inhibitor tablets (Roche Life Science, Penzberg, Germany) at the concentration of 50 mg tissue wet weight/mL. Protein content of the homogenates was measured using Pierce Coomassie Plus Reagent (ThermoFisher Scientific, Waltham, MA, USA) before further processing for immunoblotting.
Immunoblotting
Brain homogenates (see above) were added to Laemmli samples buffer (1:1 vol./vol., Bio‐Rad, Hercules, CA, USA) with heating (95°C for 5 min), followed by loading onto 10–12% SDS‐polyacrylamide gels. Proteins were electrophoresed using a Mini‐PROTEAN® system (Bio‐Rad, Hercules, CA, USA) and transferred to nitrocellulose membranes. Membranes were blocked in phosphate‐buffered saline with 0.1% Tween® 20 (PBST) and 5% skimmed milk for 1 h, then incubated with primary antibodies in PBST containing 5% BSA at 4°C overnight. Primary antibodies used were as follows: anti‐cathepsin D (C‐20, goat polyclonal, 1:1000 dilution, Santa Cruz Biotechnology, Dallas, TX, USA); anti‐LAMP‐1 (ab24170, rabbit polyclonal, 1:1000 dilution, Abcam, Cambridge, UK) and anti‐β‐actin (AC‐74, mouse monoclonal, 1:5000 dilution, Sigma Aldrich, St Louis, MO, USA). After primary antibody incubation, membranes were washed three times with PBST (10 min, 25°C), then incubated with appropriate horseradish peroxidase (HRP)‐conjugated secondary antibodies (1:5000 dilution, Jackson ImmunoResearch, West Grove, PA, USA) for 1h at 25°C. Subsequently, membranes were washed three times with PBST (10 min, 25°C), and immunoreactivities were visualized with Luminata™ Crescendo Western HRP substrate (Merck Millipore, Billerica, MA, USA) and quantified with the Alliance 4.7 image analyser (UVItec, Cambridge, UK).
Cortical Aβ and tau measurements
Aβ, total tau, and serine‐396 phosphorylated tau (pSer396 tau) measurements by enzyme‐linked immunosorbent assay (ELISA) kit (Invitrogen, Carlsbad, CA, USA) were performed as previously reported 13, 55. Briefly, tissues from BA21 and BA40 were homogenized in Tris HCl buffer (pH 8.0) with 5M guanidine, and resultant homogenates were assayed in duplicates for the amyloidogenic, neurotoxic 42 amino‐acid species of Aβ (Aβ42) 67, total tau, and pS396 tau according to manufacturer's instructions, and expressed in pg/mg brain protein.
Double labelling immunohistochemistry
Brain tissue was acquired and assessed pathologically as described above (see also Howlett et al. 32). Formalin fixed, paraffin‐embedded blocks of neocortex (BA9 and BA21) were cut into 7μm sections and mounted onto slides, then dewaxed and rehydrated using Histoclear® (ThermoFisher Scientific, Waltham, MA, USA) and alcohol dilutions. Antigen retrieval was carried out by microwaving for 10 min in citrate buffer (pH 6.0) for catD and phosphorylated tau, and by autoclaving for 10 min in EDTA buffer (pH 8.0) for α‐synuclein; all of which were followed by immersion for 15 min in 98% formic acid. Tissue sections were then treated with 0.3% hydrogen peroxide in PBS (30 min) to inhibit endogenous peroxidases prior to addition of primary antibodies: 20G10 anti‐Aβ42 monoclonal antibody, 1:10 000 dilution 31; AT8 phosphorylated (pSer202 and pThr205) tau monoclonal antibody, 1:200 dilution (ThermoFisher Scientific, Waltham, MA, USA); anti‐α‐synuclein monoclonal antibody, 1:100 dilution (Novacastra™ Leico Biosystems, Newcastle, UK); anti‐cathepsin D (C‐20, goat polyclonal, 1:1000 dilution, Santa Cruz Biotechnology, Dallas, TX, USA); with the sections incubated overnight at 4°C. Development of the sections was performed using biotinylated secondary antibodies, ABC reagents and a DAB kit, with NovaRED™ (all from Vector Laboratories, Peterborough, UK) used as a second chromogen for double labelling. In control experiments, the secondary biotinylated antibody was omitted. Sections were imaged on a Leica DMRB microscope equipped with DC420 digital camera. Ten random images were captured from each section and positively labelled cells identified by ImageJ analyses.
Statistical analyses
All analyses were performed using SPSS Statistics (version 23, IBM Inc., Armonk, NY, USA). All differences of demographic and disease variables in Table 1 were compared by one‐way one way‐analyses of variance (ANOVA) followed by post‐hoc Tamhane's T2 tests, except for gender which was compared by Pearson χ 2 tests. On the other hand, differences in western blot immunoreactivities normalised to loading control β‐actin, neuropathological scores as well as levels of Aβ42 and tau species for the postmortem studies were compared by Kruskal‐Wallis ANOVA followed by post‐hoc Dunn's tests for pair‐wise comparisons. Inter‐correlations between variables were performed using Spearman's rank correlation. For all tests, p < 0.05 were considered statistically significant.
Table 1.
Demographic and disease variables of a cohort of patients with postmortem confirmed AD and synucleinopathies
| Variable | Control | PDD | DLB | AD | P Value |
|---|---|---|---|---|---|
| Maximum available n | 18 | 22 | 21 | 16 | NA |
| Age at Death, years | 82.7 ± 1 | 82.4 ± 1 | 83.1 ± 2 | 87.2 ± 2 | 0.086 |
| Female, n (%) | 9 (50.0) | 11 (50.0) | 11 (52.4) | 10 (62.5) | 0.867 |
| Postmortem Interval, h* | 41.5 ± 6 | 37.3 ± 4 | 44.6 ± 6 | 37.1 ± 6 | 0.715 |
| Brain pH | 6.44 ± 0.07 | 6.46 ± 0.07 | 6.23 ± 0.08 | 6.31 ± 0.08 | 0.098 |
| MMSE decline/year† | NA | 2.29 ± 0.3 | 3.00 ± 0.5 | 3.77 ± 1.0 | 0.218 |
| Duration of dementia, years‡ | NA | 3.4 ± 0.6 | 6.6 ± 0.8 | 9.9 ± 0.7 | < 0.01* |
| Duration of PD symptoms, years§ | NA | 12.5 ± 1.2 | 2.9 ± 0.7 | 0.0 ± 0 | < 0.01* |
| Braak stageǁ | |||||
| 0–II | 13 | 15 | 4 | 0 | NA |
| III–IV | 0 | 6 | 8 | 3 | NA |
| V–VI | 0 | 1 | 9 | 12 | NA |
Data are expressed as mean ± SEM, with significant p values of *one‐way analyses of variance (ANOVA) listed in bold type. Abbreviations: AD, Alzheimer’s disease; BA, Brodmann area; DLB, dementia with Lewy bodies; LB, Lewy bodies; MMSE, Mini Mental State Examination; NA, not applicable/not measured; PDD, Parkinson's disease dementia.
Information was not available for one PDD patient.
Mean MMSE decline per year from study commencement to the last interview before death (available for 21 PDD, 15 DLB and 14 AD patients) and used as an indicator of dementia severity.
Years of dementia at death, available for 21 PDD, 15 DLB and 15 AD patients.
Years of PD motor symptoms at death, available for 21 PDD, 14 DLB and 15 AD patients.
Braak staging 7 data not available for five controls and one AD patient.
Results
Disease and demographic characteristics of study cohort
Table 1 shows the available controls as well as community‐based PDD, DLB and AD patients in the cohort were closely matched in terms of age at death, sex distribution, postmortem interval, brain pH (as an indicator of tissue quality 29) and MMSE decline per year (as a measure of dementia severity). For the mean duration of dementia, there were significant differences among the groups, with the longest in AD and shortest in PDD. In contrast, PDD patients had longer mean duration of parkinsonism symptoms than DLB, while no parkinsonism symptoms were apparent in the AD group. Braak staging for extent of AD pathological changes 7 showed all except one PDD with Braak stage 0–II (with the PDD having Braak VI), while all except three AD were stage V–VI (with three AD at stage IV). For DLB, around 57% were stages 0–IV, and 43% were stages V–VI. Taken together, these data suggest a relative lack of AD pathology and short dementia duration in PDD, high burden of AD pathology and long dementia duration in AD, with intermediate levels in DLB. These clinical and neuropathological features are in line with previous observations of AD and Lewy body dementias 17, 24, 32, 64.
Cathepsin D immunoreactivities were increased in AD and DLB neocortex
We first performed catD immunoblotting in two defined areas of the neocortex (BA21 and BA40) known to play important roles in cognition, and are affected in AD and Lewy body dementias 32. Figure 1 shows that catD immunoreactivities were significantly increased in both areas for AD, and only in BA40 for DLB, whilst catD immunoreactivities were not altered in PDD. In AD, DLB and PDD, immunoreactivities of lysosomal associated membrane protein‐1 (LAMP‐1), a major component of lysosomes not directly involved in degradation of protein aggregates, and used commonly to define the lysosome compartment 20, 39, were not significantly altered. Our data therefore suggest that in AD and DLB, increased catD was a specific alteration within the lysosomal compartment rather than a result of changes in lysosomal biogenesis.
Figure 1.
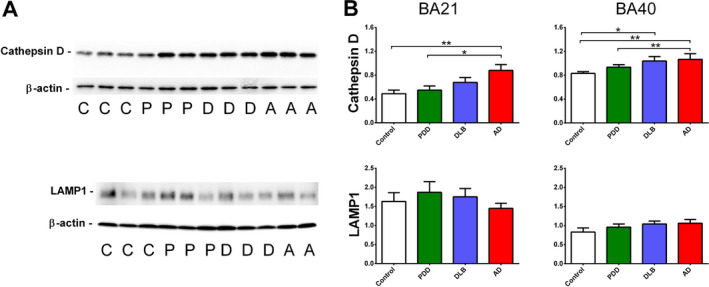
Cathepsin D (catD) immunoreactivities in the postmortem neocortex of Lewy body dementias and AD. A Representative immunoblots and B bar graphs of catD (top) and LAMP‐1 (bottom) immunoreactivities (expressed in mean ± SEM arbitrary units) in Brodmann area 21 (BA21) and BA40 of 18 controls (C), 22 PDD (P), 21 DLB (D) and 16 AD (A) patients, normalised to loading control β‐actin. *p < 0.05 and **p < 0.01; significant pairwise difference using Kruskal‐Wallis ANOVA with Dunn's post‐hoc tests.
Cathepsin D immunoreactivities were associated with neurofibrillary tangles
Because CatD may be involved in the clearance of Aβ, tau and α‐synuclein 26, 36, 58, we compared catD immunoreactivities from immunoblotting across the range of semi‐quantitative neuropathological scores in our cohort for amyloid plaques (AP), neurofibrillary tangles/neuropil threads (NFT) and Lewy bodies/Lewy neurites (LB) to investigate if the observed catD changes may be associated with neuropathological burden. Interestingly, only NFT scores in BA40 associated with catD, with immunoreactivities in the “Abundant” group significantly higher than those in the “Sparse” or “None” groups (Figure 2B). CatD immunoreactivities appeared to trend towards increases with NFT in BA21, and with AP in both regions, but did not reach statistical significance (Figure 2A, B). In contrast, catD did not appear to be associated with LB counts (Figure 2C). To further investigate spatial associations between catD and neuropathological hallmarks, double labelling immunohistochemistry was performed on neocortical sections stained for catD and AP, NFT or LB. Figure 3 shows representative micrographs suggesting that at least some of the catD immunoreactivities were co‐localized with NFT‐containing neurons (as indicated by AT8 phosphorylated tau antibody staining, Figure 3B). In contrast, α‐synuclein immunoractivities (putatively labelling LB) typically showed minimal overlap with those of catD immunoreactivities (Figures C). Similarly, whilst occasional catD immunoreactivity was apparent in Aβ42‐postitive plaques, the preponderance of catD staining was localized to pyramidal neurons (Figure 3A).
Figure 2.
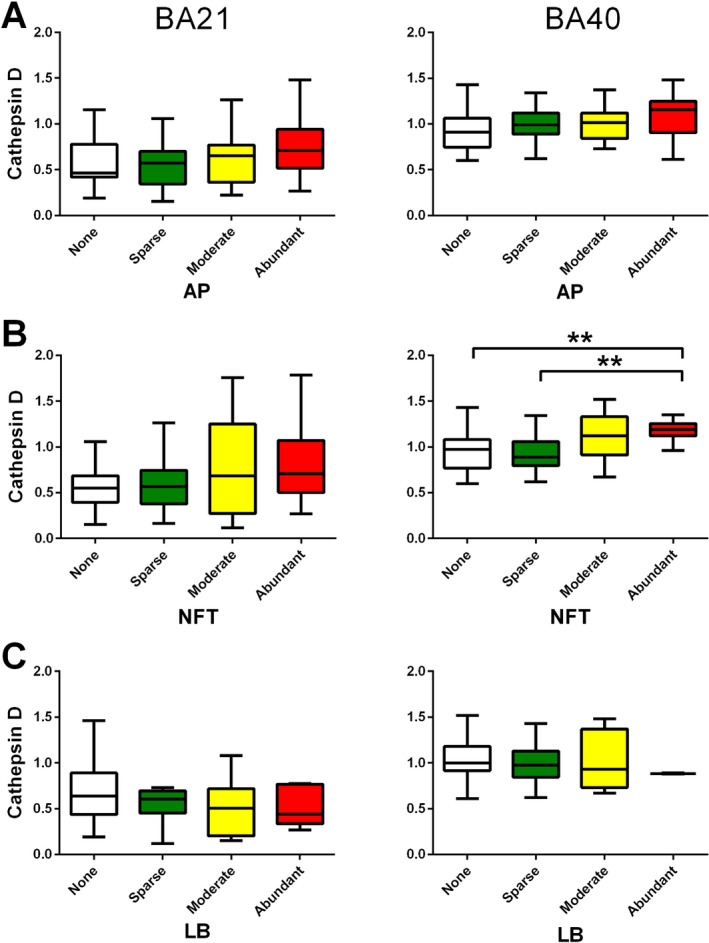
Cathepsin D (catD) immunoreactivities across a range of neuropathological burden. Semi‐quantitative scoring for A amyloid plaques (AP), B neurofibrillary tangles/neuropil threads (NFT) and C α‐synuclein inclusions e.g., Lewy bodies or Lewy neurites (LB) ranged from “None” (score = 0) to “Abundant” (score = 3) were performed according to Howlett et al. 32 (see Methods). Immunoreactivities of catD (expressed in mean ± SEM arbitrary units) in BA21 (left) and BA40 (right) are depicted in box and whiskers plots using Tukey's method. **p < 0.01; significant pairwise difference using Kruskal‐Wallis ANOVA with Dunn's post‐hoc tests.
Figure 3.
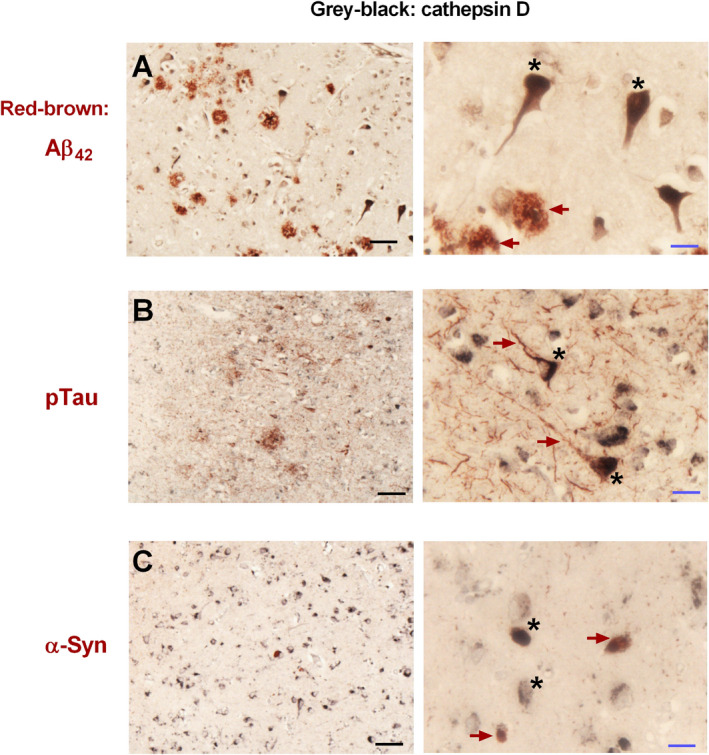
Immunohistochemical staining of neuropathological features and cathepsin D (catD) in postmortem human neocortex. Images of A Aβ42, B phosphorylated Tau reocognized by the AT8 antibody (pTau) and C α‐synuclein (α‐syn) co‐stained with catD (see Methods) in the neocortex of an AD (A, B) and DLB (C) subject at 200x (left) and 630x (right) magnifications. Micrographs were representative images of three AD and three DLB subjects. Scale bars: 50 microns (black), 15 microns (blue). Red‐brown arrows: A Aβ42, B pTau and C α‐syn immunoreactivities. Black asterisks: catD immunoreactivity.
Increased cathepsin D immunoreactivities correlated with phosphorylated (pS396) tau
To corroborate the findings above showing associations between catD and specific AD hallmarks (Figures 2 and 3, we further measured the biochemical substrates of AD, namely guanidine‐treated Aβ42 (previously shown to consist mainly of plaque‐associated Aβ 13, 55) and tau phosphorylated at serine‐396 (pSer396 tau, previously shown to be a specific marker of hyperphosphorylated tau, the major constituent of NFT 4). Similar to previous reports of higher amyloid burden in DLB compared to PDD 11, 17, 24, we found that Aβ42 concentrations in DLB were significantly higher than PDD, and reached levels comparable to AD in both BA21 and BA40 (Figure 4A). However, Aβ42 did not correlate with catD immunoreactivities within the combined dementia group (Figure 4B). In contrast, while pSer396 tau concentrations were higher in DLB compared with PDD, reaching significance in BA21, AD showed the highest pSer396 tau levels, being higher than both DLB and PDD in BA40 (Figure 5A). Changes in pSer396 tau were unlikely to be due to changes in tau expression, as total tau were unchanged among diagnostic groups (Figure 4B). Interestingly, pSer396 tau concentrations positively correlated with catD immunoreactivities in both brain regions (Figure 5C). On the other hand, catD immunoreactivities did not correlate with either pre‐death MMSE scores or with MMSE decline per year (see Supplementary Figure S1). This suggests that catD is a marker of disease processes such as tau hyperphosphorylation rather than directly associated with neuronal function or synaptic plasticity events.
Figure 4.
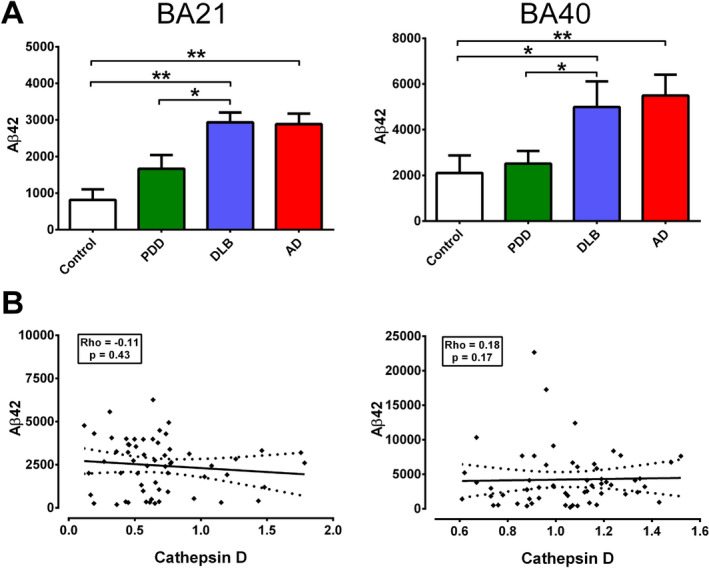
Correlations of cathepsin D (catD) immunoreactivities with Aβ42 concentrations in the postmortem neocortex of Lewy body dementias and AD. A Bar graphs of mean ± SEM Aβ42 concentrations (pg/mg brain protein) in BA21 (left) and BA40 (right). B Scatter plots of Aβ42 concentrations against catD immunoreactivities (arbitrary units) in BA21 (left) and BA40 (right) of the combined dementia cohort (22 PDD, 21 DLB, 16 AD). The solid lines indicate linear regressed best‐fit curves while dashed lines indicate their respective 95% confidence intervals. No significant correlations were found. *p < 0.05 and **p < 0.01; significant pairwise difference using Kruskal‐Wallis ANOVA with Dunn's post‐hoc tests.
Figure 5.
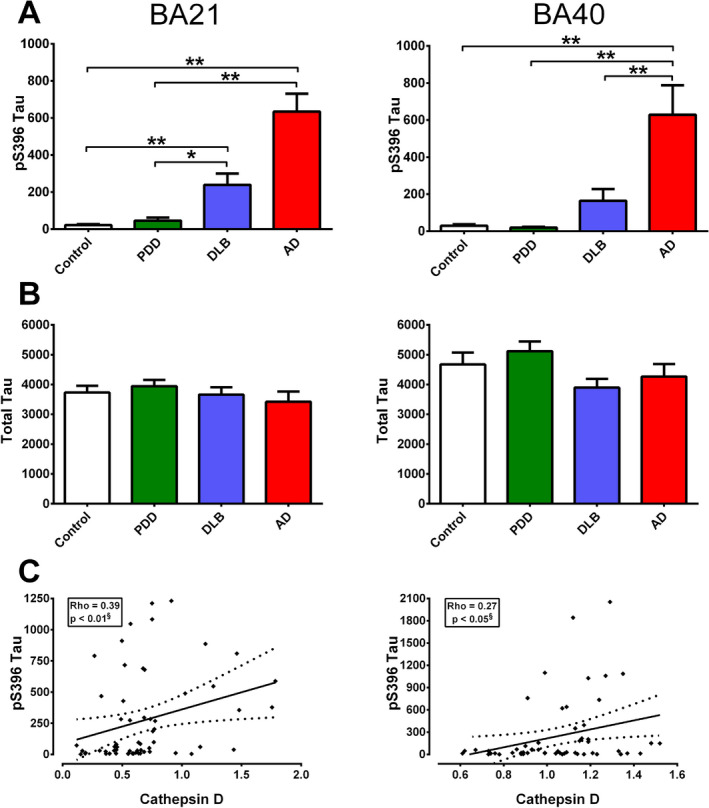
Correlations of cathepsin D (catD) immunoreactivities with pS396 Tau concentrations in the postmortem neocortex of Lewy body dementias and AD. Bar graphs of mean ± SEM A pS396 Tau and B total tau concentrations (pg/mg brain protein) in BA21 (left) and BA40 (right). C Scatter plots of pS396 Tau concentrations against catD immunoreactivities (arbitrary units) in BA21 (left) and BA40 (right) of the combined dementia cohort (22 PDD, 21 DLB, 16 AD). The solid lines indicate linear regressed best‐fit curves while dashed lines indicate their respective 95% confidence intervals. *p < 0.05 and **p < 0.01; significant pairwise difference using Kruskal‐Wallis ANOVA with Dunn's post‐hoc tests. §Significant Spearman correlation.
Discussion
Cathepsin D (catD)'s role in neurodegeneration has been suggested by lysosomal storage disorders such as neuronal ceroid‐lipofuscinosis associated with catD mutations which abolished enzymatic activity, leading inevitably to progressive neurodegeneration and brain atrophy 59. It is therefore not surprising that considerable research have been carried out to elucidate the role of lysosomal constituents, including cathepsins, in neurodegenerative conditions like AD. Previous studies on catD in AD have yielded inconsistent results (see Background). Using postmortem tissues from well‐characterized cohort of AD patients together with Lewy body dementia subgroups with incidental high (DLB) and low (PDD) amyloid pathology 17, 24, 64, but which are otherwise matched in dementia severity (see Table 1, the current study reports robust increases in catD immunoreactivities in AD which correlated with neurofibrillary tangle scores and phosphorylated tau burden, as well as co‐localize at least in part within tangle‐bearing neurons. This is also, as far as we know, the first study to measure catD immunoreactivities in relation to LB pathology in DLB and PDD. In our cohort, catD was unchanged in PDD and increased only in one of the two cortical regions measured in DLB. CatD was also not correlated with LB scores or demonstrated substantial co‐localization with α‐synuclein inclusions.
Taken together, the postmortem data therefore suggest that catD changes may be driven primarily by AD processes, as it is relatively unchanged in PDD characterized by low AD pathological burden, but demonstrated increases in DLB with higher AD pathological burden. In contrast, rodent, nematode or cell culture‐based work have shown protective effects of catD against α‐synuclein aggregation and toxicity 56, suggesting either species differences in how catD interacts with α‐synuclein, or biochemical differences in human LB‐associated α‐synuclein, including higher proportions of phosphorylated forms 63. However, follow up studies are needed to corroborate catD associations with cognitive function as well as investigate underlying mechanisms, the most straightforward of which is the enhanced clearance of abnormally aggregated proteins forming AP and NFT 26, 36. In this context, various studies have previously reported catD association with vulnerable neurons, including those with Aβ deposits 10, 11, 12, although associations were more apparent with NFT than with AP in the current study (Figures 2 to 4. Nevertheless, the available data (including those reported here) suggest the upregulation of catD as a response to abnormal protein aggregation and subsequent formation of AP and NFT, two closely related processes 35, 53.
Besides direct action on abnormally aggregated peptides, catD or its zymogen (pro‐catD) may regulate other cellular responses implicated in AD pathogenesis and progression, including cytokine secretion and initiation of apoptosis 14, 23, 43. Conversely, pro‐inflammatory cytokines like tumor necrosis factor and interferon‐γ, known to be activated in AD by misfolded and aggregated peptides 30, may upregulate pro‐catD and increase catD activities 19, 66. Therefore, follow‐up studies are needed to better characterize the intertwining relationships between protein misfolding, neuroinflammatory responses and catD upregulation in AD.
There are a few limitations in the current studies which requires discussion. First, the cellular basis of catD changes reported in this study are at present unclear, although the current immunohistochemical data suggest localization of catD immunoreactivities to neurons, in agreement with previous work 8, 11. However, catD expression can also be found in astrocytes, microglia and pericytes 6, 8, 42, 54, and further studies are needed to elucidate cell type‐ as well as brain region‐specific catD changes in AD, together with their relative contributions to AD neuropathological and neurochemical features. Additionally, there remains a divergence between in vitro studies showing involvement of catD in α‐synuclein processing (see above), and our postmortem data suggesting no significant associations between catD and α‐synuclein lesions in Lewy body dementias. Findings of relatively unchanged catD in Lewy body dementias are also in need of corroboration from more detailed studies on potential interactions between catD and α‐synuclein, for example, with different α‐synuclein species 63. Furthermore, the regional specifity of associations with catD (both BA21 and 40 for AD versus only BA40 in DLB) may reflect differential vulnerabilities or regional involvement in the neurodegenerative dementias, but further work is needed to validate the regional differences and elucidate the underlying mechanisms. Lastly, the present study adds to growing literature on the involvement of cathepsins in neurological diseases, and gives impetus to further research in this area, both in the role of cathepsin D in other neurodegenerative conditions characterized by tauopathy (eg, progressive supranuclear palsy, corticobasal ganglionic degeneration and primary ageing related tauopathy), and in how other cathepsins shown to be involved in degenerative conditions (eg, cathepsin A‐related arteriopathy with strokes and related leukoencephalopathy, CARASAL) 9 might also be affected in AD and Lewy body dementias.
Conclusion
Using postmortem brain cortical tissues and blood from separate cohorts of aged non‐dementia and dementia patients, the current study reports increased catD immunoreactivities in the neocortex of AD patients which appear to be most strongly associated with neurofibrillary degeneration. In contrast, catD is relatively preserved in the neocortex of Lewy body dementias, and seemed to be associated with concomitant AD pathology rather than with α‐synuclein inclusions. Further studies will help elucidate the mechanisms underlying catD upregulation, as well as its suitability as a prognostic biomarker or therapeutic target for AD.
Funding
Tissue for this study was provided by the Newcastle Brain Tissue Resource, which is funded in part by a grant from the UK Medical Research Council (grant number G0400074) and by Brains for Dementia research, a joint venture between Alzheimer's Society and Alzheimer's Research UK. This study is funded by the National Medical Research Council of Singapore (NMRC/CSA‐SI/007/2016). The funding organizations played no role in the conduct or design of this research.
Authors’ contributions
YLC, PTF, CPC, and MKPL conceived the study and designed the project; YLC, JW, JRC, DH, AH, JHL performed the experiments; JA, DA, CPC, PTF provided postmortem and clinical data; YLC, JW, DH, MKPL analyzed the data; YLC, JW, MKPL wrote the first draft. All authors have read and approved the manuscript.
Ethics approval and consent to participate
For the postmortem study, ethics approval for the collection and study of brain tissues received Institutional Review Board approval in both the UK (08/H1010/4) and Singapore institutions (NUS 12‐062E), and informed consent was obtained from participants’ next‐of‐kin prior to removal of brain. For the clinical study, Institutional Review Board approval from the National Healthcare Group Domain‐Specific Review Board (reference: 2010/00017; study protocol number: DEM4233) and written informed consent had been obtained from participants or their next‐of‐kin before study recruitment and blood collection procedures.
Consent for publication
All authors gave consent for publication.
Competing interests
The authors declare that they have no competing interests.
Supporting information
Fig. S1 Scatter plots of cognitive status (Pre‐death MMSE score and MMSE decline per year; see Methods) against cathepsin D immunoreactivities (arbitrary units) in BA21 (left) and BA40 (right) of the combined dementia cohort. The solid lines indicate linear regressed best‐fit curves while dashed lines indicate their respective 95% confidence intervals. No significance was found using Spearman correlation.
References
- 1. Aarsland D, Andersen K, Larsen JP, Lolk A, Kragh‐Sorensen P (2003) Prevalence and characteristics of dementia in Parkinson disease: an 8‐year prospective study. Arch Neurol 60:387–392. [DOI] [PubMed] [Google Scholar]
- 2. Aarsland D, Rongve A, Nore SP, Skogseth R, Skulstad S, Ehrt U et al (2008) Frequency and case identification of dementia with Lewy bodies using the revised consensus criteria. Dement Geriatr Cogn Dis 26:445–452. [DOI] [PubMed] [Google Scholar]
- 3. ADI (2010) World Alzheimer report 2010: the global economic impact of dementia. Available at:http://www.alz.co.uk/research/world-report.
- 4. Augustinack JC, Schneider A, Mandelkow EM, Hyman BT (2002) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol 103:26–35. [DOI] [PubMed] [Google Scholar]
- 5. Bach AS, Derocq D, Laurent‐Matha V, Montcourrier P, Sebti S, Orsetti B et al (2015) Nuclear cathepsin D enhances TRPS1 transcriptional repressor function to regulate cell cycle progression and transformation in human breast cancer cells. Oncotarget 6:28084–28103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bejarano‐Escobar R, Holguin‐Arevalo MS, Montero JA, Francisco‐Morcillo J, Martin‐Partido G (2011) Macrophage and microglia ontogeny in the mouse visual system can be traced by the expression of Cathepsins B and D. Dev Dyn 240:1841–1855. [DOI] [PubMed] [Google Scholar]
- 7. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 8. Braidy N, Brew BJ, Inestrosa NC, Chung R, Sachdev P, Guillemin GJ (2014) Changes in Cathepsin D and Beclin‐1 mRNA and protein expression by the excitotoxin quinolinic acid in human astrocytes and neurons. Metab Brain Dis 29:873–883. [DOI] [PubMed] [Google Scholar]
- 9. Bugiani M, Kevelam SH, Bakels HS, Waisfisz Q, Ceuterick‐de Groote C, Niessen HW et al (2016) Cathepsin A‐related arteriopathy with strokes and leukoencephalopathy (CARASAL). Neurology 87:1777–1786. [DOI] [PubMed] [Google Scholar]
- 10. Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Bursztajn S et al (1995) Gene expression and cellular content of cathepsin D in Alzheimer's disease brain: evidence for early up‐regulation of the endosomal‐lysosomal system. Neuron 14:671–680. [DOI] [PubMed] [Google Scholar]
- 11. Cataldo AM, Hamilton DJ, Barnett JL, Paskevich PA, Nixon RA (1996) Properties of the endosomal‐lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer's disease. J Neurosci 16:186–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cataldo AM, Nixon RA (1990) Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc Natl Acad Sci USA 87:3861–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chong JR, Chai YL, Lee JH, Howlett D, Attems J, Ballard CG et al (2017) Increased transforming growth factor b2 in the neocortex of Alzheimer's disease and dementia with lewy bodies is correlated with disease severity and soluble Ab42 load. J Alzheimer's Dis 56:157–166. [DOI] [PubMed] [Google Scholar]
- 14. Conus S, Pop C, Snipas SJ, Salvesen GS, Simon HU (2012) Cathepsin D primes caspase‐8 activation by multiple intra‐chain proteolysis. J Biol Chem 287:21142–21151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cummings JL, Morstorf T, Zhong K (2014) Alzheimer's disease drug‐development pipeline: few candidates, frequent failures. Alzheimer's Res Ther 6:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dá Mesquita S, Ferreira AC, Sousa JC, Correia‐Neves M, Sousa N, Marques F (2016) Insights on the pathophysiology of Alzheimer's disease: the crosstalk between amyloid pathology, neuroinflammation and the peripheral immune system. Neurosci Biobehav Rev 68:547–562. [DOI] [PubMed] [Google Scholar]
- 17. Edison P, Rowe CC, Rinne JO, Ng S, Ahmed I, Kemppainen N et al (2008) Amyloid load in Parkinson's disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry 79:1331–1338. [DOI] [PubMed] [Google Scholar]
- 18. Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, Mizuno Y et al (2007) Clinical diagnostic criteria for dementia associated with Parkinson's disease. Movement Disorders 22:1689–1707. [DOI] [PubMed] [Google Scholar]
- 19. Erdmann S, Ricken A, Hummitzsch K, Merkwitz C, Schliebe N, Gaunitz F et al (2008) Inflammatory cytokines increase extracellular procathepsin D in permanent and primary endothelial cell cultures. Eur J Cell Biol 87:311–323. [DOI] [PubMed] [Google Scholar]
- 20. Eskelinen EL (2006) Roles of LAMP‐1 and LAMP‐2 in lysosome biogenesis and autophagy. Mol Aspects Med 27:495–502. [DOI] [PubMed] [Google Scholar]
- 21. Folstein MF, Folstein SE, McHugh PR (1975) “Mini‐mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12:189–198. [DOI] [PubMed] [Google Scholar]
- 22. Fritsch J, Fickers R, Klawitter J, Sarchen V, Zingler P, Adam D et al (2016) TNF induced cleavage of HSP90 by cathepsin D potentiates apoptotic cell death. Oncotarget 7:75774–75789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fusek M, Vetvickova J, Vetvicka V (2007) Secretion of cytokines in breast cancer cells: the molecular mechanism of procathepsin D proliferative effects. J Interferon Cytokine Res 27:191–199. [DOI] [PubMed] [Google Scholar]
- 24. Gomperts SN, Locascio JJ, Marquie M, Santarlasci AL, Rentz DM, Maye J et al (2012) Brain amyloid and cognition in Lewy body diseases. Movement Disorders 27:965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grundke‐Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI (1986) Abnormal phosphorylation of the microtubule‐associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 83:4913–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hamazaki H (1996) Cathepsin D is involved in the clearance of Alzheimer's b‐amyloid protein. FEBS Lett 396:139–142. [DOI] [PubMed] [Google Scholar]
- 27. Hardy JA, Higgins GA (1992) Alzheimer's disease: the amyloid cascade hypothesis. Science 256:184–185. [DOI] [PubMed] [Google Scholar]
- 28. Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356. [DOI] [PubMed] [Google Scholar]
- 29. Hardy JA, Wester P, Winblad B, Gezelius C, Bring G, Eriksson A (1985) The patients dying after long terminal phase have acidotic brains; implications for biochemical measurements on autopsy tissue. J Neural Transm 61:253–264. [DOI] [PubMed] [Google Scholar]
- 30. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL et al (2015) Neuroinflammation in Alzheimer's disease. Lancet Neurol 14:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Howlett DR, Richardson JC, Austin A, Parsons AA, Bate ST, Davies DC, Gonzalez MI (2004) Cognitive correlates of Ab deposition in male and female mice bearing amyloid precursor protein and presenilin‐1 mutant transgenes. Brain Res 1017:130–136. [DOI] [PubMed] [Google Scholar]
- 32. Howlett DR, Whitfield D, Johnson M, Attems J, O'Brien JT, Aarsland D et al. (2015) Regional multiple pathology scores are associated with cognitive decline in Lewy body Dementias. Brain Pathol 25:401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hurd MD, Martorell P, Delavande A, Mullen KJ, Langa KM (2013) Monetary costs of dementia in the United States. N Engl J Med 368:1326–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX et al (2017) Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol 16:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J et al (2010) Dendritic function of tau mediates amyloid‐b toxicity in Alzheimer's disease mouse models. Cell 142:387–397. [DOI] [PubMed] [Google Scholar]
- 36. Kenessey A, Nacharaju P, Ko LW, Yen SH (1997) Degradation of tau by lysosomal enzyme cathepsin D: implication for Alzheimer neurofibrillary degeneration. J Neurochem 69:2026–2038. [DOI] [PubMed] [Google Scholar]
- 37. Kepp KP (2017) Ten challenges of the amyloid hypothesis of Alzheimer's disease. J Alzheimer's Dis 55:447–457. [DOI] [PubMed] [Google Scholar]
- 38. Kohnken RE, Ladror US, Wang GT, Holzman TF, Miller BE, Krafft GA (1995) Cathepsin D from Alzheimer's‐diseased and normal brains. Exp Neurol 133:105–112. [DOI] [PubMed] [Google Scholar]
- 39. Kornfeld S, Mellman I (1989) The biogenesis of lysosomes. Annu Rev Cell Biol 5:483–525. [DOI] [PubMed] [Google Scholar]
- 40. Lee C, Low CY, Francis PT, Attems J, Wong PT, Lai MK, Tan MG (2016) An isoform‐specific role of FynT tyrosine kinase in Alzheimer's disease. J Neurochem 136:637–650. [DOI] [PubMed] [Google Scholar]
- 41. Liu F, Li X, Lu C, Bai A, Bielawski J, Bielawska A et al (2016) Ceramide activates lysosomal cathepsin B and cathepsin D to attenuate autophagy and induces ER stress to suppress myeloid‐derived suppressor cells. Oncotarget 7:83907–83925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu J, Yang L, Tian H, Ma Q (2016) Cathepsin D is involved in the oxygen and glucose deprivation/reperfusion‐induced apoptosis of astrocytes. Int J Mol Med 38:1257–1263. [DOI] [PubMed] [Google Scholar]
- 43. Malik M, Sheikh AM, Wen G, Spivack W, Brown WT, Li X (2011) Expression of inflammatory cytokines, Bcl2 and cathepsin D are altered in lymphoblasts of autistic subjects. Immunobiology 216:80–85. [DOI] [PubMed] [Google Scholar]
- 44. McBrayer M, Nixon RA (2013) Lysosome and calcium dysregulation in Alzheimer's disease: partners in crime. Biochem Soc Trans 41:1495–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 46. Mirkovic B, Markelc B, Butinar M, Mitrovic A, Sosic I, Gobec S et al (2015) Nitroxoline impairs tumor progression in vitro and in vivo by regulating cathepsin B activity. Oncotarget 6:19027–19042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The consortium to establish a registry for Alzheimer's disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41:479–486. [DOI] [PubMed] [Google Scholar]
- 48. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW et al (2012) National institute on aging‐Alzheimer's association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta neuropathol 123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Morris GP, Clark IA, Vissel B (2014) Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer's disease. Acta Neuropathol Commun 2:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mueller‐Steiner S, Zhou Y, Arai H, Roberson ED, Sun B, Chen J et al (2006) Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron 51:703–714. [DOI] [PubMed] [Google Scholar]
- 51. Nagy Z (2007) The dysregulation of the cell cycle and the diagnosis of Alzheimer's disease. BiochimBiophysActa 1772:402–408. [DOI] [PubMed] [Google Scholar]
- 52. Nixon RA, Cataldo AM, Mathews PM (2000) The endosomal‐lysosomal system of neurons in Alzheimer's disease pathogenesis: a review. Neurochem Res 25:1161–1172. [DOI] [PubMed] [Google Scholar]
- 53. Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM (2004) Ab immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43:321–332. [DOI] [PubMed] [Google Scholar]
- 54. Okada R, Wu Z, Zhu A, Ni J, Zhang J, Yoshimine Y et al (2015) Cathepsin D deficiency induces oxidative damage in brain pericytes and impairs the blood‐brain barrier. Mol cell Neurosci 64:51–60. [DOI] [PubMed] [Google Scholar]
- 55. Pham E, Crews L, Ubhi K, Hansen L, Adame A, Cartier A et al (2010) Progressive accumulation of amyloid‐b oligomers in Alzheimer's disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J 277:3051–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Qiao L, Hamamichi S, Caldwell KA, Caldwell GA, Yacoubian TA, Wilson S et al (2008) Lysosomal enzyme cathepsin D protects against alpha‐synuclein aggregation and toxicity. Mol Brain 1:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sabbagh MN, Adler CH, Lahti TJ, Connor DJ, Vedders L, Peterson LK et al (2009) Parkinson disease with dementia: comparing patients with and without Alzheimer pathology. Alzheimer Dis Assoc Dis 23:295–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sevlever D, Jiang P, Yen SH (2008) Cathepsin D is the main lysosomal enzyme involved in the degradation of a‐synuclein and generation of its carboxy‐terminally truncated species. Biochemistry 47:9678–9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Siintola E, Partanen S, Strömme P, Haapanen A, Haltia M, Maehlen J et al (2006) Cathepsin D deficiency underlies congenital human neuronal ceroid‐lipofuscinosis. Brain 129:1438–1445. [DOI] [PubMed] [Google Scholar]
- 60. Sun B, Zhou Y, Halabisky B, Lo I, Cho SH, Mueller‐Steiner S et al (2008) Cystatin C‐cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer's disease. Neuron 60:247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Thal DR, Rüb U, Orantes M, Braak H (2002) Phases of Ab‐deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 62. Tian L, Zhang K, Tian ZY, Wang T, Shang DS, Li B et al (2014) Decreased expression of cathepsin D in monocytes is related to the defective degradation of amyloid‐beta in Alzheimer's disease. J Alzheimer's Dis 42:511–520. [DOI] [PubMed] [Google Scholar]
- 63. Walker DG, Lue LF, Adler CH, Shill HA, Caviness JN, Sabbagh MN et al (2013) Changes in properties of serine 129 phosphorylated a‐synuclein with progression of Lewy‐type histopathology in human brains. Exp Neurol 240:190–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Walker L, McAleese KE, Thomas AJ, Johnson M, Martin‐Ruiz C, Parker C et al (2015) Neuropathologically mixed Alzheimer's and Lewy body disease: burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol 129:729–748. [DOI] [PubMed] [Google Scholar]
- 65. Wang L, Chen Y, Li X, Zhang Y, Gulbins E, Zhang Y (2016) Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B‐mediated Nlrp3 inflammasome activation. Oncotarget 7:73229–73241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Whitaker JN, Herman PK, Sparacio SM, Zhou SR, Benveniste EN (1991) Changes induced in astrocyte cathepsin D by cytokines and leupeptin. J Neurochem 57:406–414. [DOI] [PubMed] [Google Scholar]
- 67. Yoshiike Y, Chui D‐H, Akagi T, Tanaka N, Takashima A (2003) Specific compositions of amyloid‐β peptides as the determinant of toxic β‐aggregation. J Biol Chem 278:23648–23655. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Scatter plots of cognitive status (Pre‐death MMSE score and MMSE decline per year; see Methods) against cathepsin D immunoreactivities (arbitrary units) in BA21 (left) and BA40 (right) of the combined dementia cohort. The solid lines indicate linear regressed best‐fit curves while dashed lines indicate their respective 95% confidence intervals. No significance was found using Spearman correlation.