

Abstract
Kainic acid, an analogue of the excitatory neurotransmitter glutamate, can trigger seizures and neurotoxicity in the hippocampus and other limbic structures in a manner that mirrors the neuropathology of human temporal lobe epilepsy (TLE). However, the underlying mechanisms associated with the neurotoxicity remain unclear. Since amyloid‐β (Aβ) peptides, which are critical in the development of Alzheimer's disease, can mediate toxicity by activating glutamatergic NMDA receptors, it is likely that the enhanced glutamatergic transmission that renders hippocampal neurons vulnerable to kainic acid treatment may involve Aβ peptides. Thus, we seek to establish what role Aβ plays in kainic acid‐induced toxicity using in vivo and in vitro paradigms. Our results show that systemic injection of kainic acid to adult rats triggers seizures, gliosis and loss of hippocampal neurons, along with increased levels/processing of amyloid precursor protein (APP), resulting in the enhanced production of Aβ‐related peptides. The changes in APP levels/processing were evident primarily in activated astrocytes, implying a role for astrocytic Aβ in kainic acid‐induced toxicity. Accordingly, we showed that treating rat primary cultured astrocytes with kainic acid can lead to increased Aβ production/secretion without any compromise in cell viability. Additionally, we revealed that kainic acid reduces neuronal viability more in neuronal/astrocyte co‐cultures than in pure neuronal culture, and this is attenuated by precluding Aβ production. Collectively, these results indicate that increased production/secretion of Aβ‐related peptides from activated astrocytes can contribute to neurotoxicity in kainic acid‐treated rats. Since kainic acid administration can lead to neuropathological changes resembling TLE, it is likely that APP/Aβ peptides derived from astrocytes may have a role in TLE pathogenesis.
Keywords: amyloid precursor protein, β‐amyloid peptide, β‐secretase, epilepsy, γ‐secretase, neurodegeneration, reactive astrocytes
Abbreviations
- Aβ
amyloid β
- AD
Alzheimer's disease
- APH1
anterior pharynx defective 1
- APP
amyloid precursor protein
- BACE1
β‐site APP cleaving enzyme
- BCA
bicinchoninic acid
- CTF
C‐terminal fragment
- ECL
enhanced chemiluminescence
- ERK1/2
extracellular signal‐regulated kinases1/2
- GFAP
glial fibrillary acidic protein
- HRP
horseradish peroxidase
- MAP kinase
mitogen activated protein kinase
- MTT
3‐(4,5‐dimethylthiozolyl)‐2,5‐diphenyltetrazolium bromide
- PBS
phosphate‐buffered saline
- PEN2
presenilin enhancer 2
- PFA
paraformaldehyde
- PS1/2
presenilins 1/2
- TLE
temporal lobe epilepsy
- SBDP
αII‐spectrin breakdown products
Introduction
Kainic acid, an analogue of the excitatory neurotransmitter glutamate, can induce seizures in adult rats originating from the CA3 region of the hippocampus and then spreading to other limbic structures. The epileptogenic effects of kainic acid are preeminently caused by the activation of ionotropic kainate receptors located on CA3 mossy fiber synapses. This triggers Ca+2 influxes into the cells, generating synchronized, network‐driven glutamatergic currents which subsequently induce seizures and facilitate neurotoxicity 10, 44, 80. The consequences of kainic acid administration include neuronal loss in CA1‐CA3 regions, the reorganization of mossy fibers and gliosis; neuropathologies which resemble human temporal lobe epilepsy (TLE) 45, 89. Glutamate, derived from both synaptic and non‐synaptic (ie, astrocytes) sources, plays a critical role in kainic acid‐induced epilepsy, as its administration can induce seizures and loss of neurons in animals, which can be precluded by glutamate receptor antagonists 19, 22, 29. Nevertheless, the cellular mechanisms by which kainic acid triggers cell death remain unclear as apoptotic, necrotic and autophagic markers are found to be altered in affected neurons 81, 82. Thus, defining the signaling pathways that underlie neurotoxicity following kainic acid‐induced seizure may provide a better understanding of the pathology associated with TLE.
Numerous studies have shown that amyloid β (Aβ) peptides, produced constitutively in the normal brain, play a critical role in reducing neuronal viability and subsequent development of Alzheimer's disease (AD), the most common type of senile dementia affecting the elderly 21, 48. These peptides are derived from the amyloid precursor protein (APP), which can be proteolytically processed either by non‐amyloidogenic α‐secretase or amyloidogenic β‐secretase pathways. The α‐secretase (eg, ADAM10) cleaves APP within the Aβ domain, yielding soluble N‐terminal APPα (sAPPα) and a C‐terminal fragment (α‐CTF) that can be further processed by γ‐secretase to generate Aβ17–40/Aβ17–42 fragments. The β‐secretase cleaves APP to generate soluble APPβ (sAPPβ) and an Aβ‐containing C‐terminal fragment (β‐CTF), which is processed via γ‐secretase to yield full‐length Aβ1–40/Aβ1–42 peptides. While β‐secretase is an aspartyl protease called β‐site APP cleaving enzyme (BACE1), γ‐secretase comprises the aspartyl protease presenilin 1/2 (PS1/PS2) and three cofactors, that is, nicastrin, presenilin enhancer 2 (PEN2) and anterior pharynx defective 1 (APH1) 3, 28.
There is now considerable evidence suggesting that toxicity induced by Aβ‐related peptides is partly mediated by glutamate, as transient inactivation and/or blockade of the NMDA receptor by antagonists can protect neurons against Aβ toxicity 1, 40, 75, 78, 85. These results raise the possibility that enhanced glutamatergic activity that renders hippocampal neurons vulnerable to excitotoxicity in animal models of TLE 50, 67, 82 may involve Aβ peptides. This is supported by three lines of evidence (i) prolonged activation of glutamatergic NMDA receptors can enhance amyloidogenic processing of APP 13, 43, 64, (ii) neuronal vulnerability to excitotoxicity is enhanced in the presence of Aβ 53 and (iii) transgenic mice overexpressing Aβ exhibit spontaneous seizures or decreased threshold to seizures compared to wild‐type mice 20, 58, 87. Nevertheless, little is known regarding the involvement of Aβ peptides in the loss of neurons following kainic acid‐induced toxicity. To address this issue, we evaluated alterations in the levels/distribution and processing of APP in the hippocampus of kainic acid‐treated rats. In parallel, using rat hippocampal neuronal and mixed hippocampal neuronal/astrocyte cultures, we determined the significance of Aβ peptides in kainic acid‐mediated neurotoxicity. Collectively, our results reveal that increased levels of astrocyte‐derived Aβ peptides have a role in reducing neuronal viability in the kainic acid model of TLE.
Materials and Methods
Materials
Polyacrylamide electrophoresis gels (4%–20%), cell culture reagents such as Hank's balanced salt solution, fetal bovine serum, Neurobasal media, B27 and N2 supplements, and ELISA kits for the detection of rat Aβ1–40 and Aβ1–42 were purchased from Invitrogen (Burlington, ON, Canada), whereas the enhanced chemiluminescence kit (ECL) and the bicinchoninic acid (BCA) protein assay kit were from Thermo Fisher Scientific (Montreal, QC, Canada). Kainic acid, γ‐secretase inhibitors (DAPT and L‐685,458) and 3‐(4,5‐dimethylthiozolyl)‐2,5‐diphenyltetrazolium bromide (MTT) were obtained from Sigma‐Aldrich (Oakville, ON, Canada). Human and rat Aβ1–42 and the reverse sequence Aβ42‐1 were purchased from American Peptides (Sunnyvale, CA, USA) and the Live/Dead cell viability assay kit was from Molecular Probes (Eugene, OR, USA). Fluoro‐Jade C was from Chemicon Int., (Temecula, CA, USA), whereas β‐secretase BACE1 activity assay kit was from Abcam (Cambridge, United Kingdom). Sources of all primary antibodies used in the study are listed in Table 1. All horseradish peroxidase (HRP)‐conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Paso Robles, CA, USA). Vivaspin filtration columns were from GE Healthcare Ltd. (Mississauga, ON, Canada), whereas protease inhibitor cocktail, BACE1 inhibitor BIV and fluorogenic γ‐secretase substrate were from Calbiochem (San Diego, CA, USA). All other chemicals were from either Sigma‐Aldrich or Thermo Fisher Scientific.
Table 1.
Primary antibodies used.
| Antibody | Type | IHC/IF dilution | WB dilution | Source |
|---|---|---|---|---|
| α‐Fodrin | Monoclonal | n/a | 1:1000 | Enzo Life Sciences, Inc. |
| β‐actin | Monoclonal | n/a | 1:5000 | Sigma‐Aldrich, Inc. |
| μ‐Calpain | Polyclonal | n/a | 1:1000 | Sigma‐Aldrich, Inc. |
| APH1 | Polyclonal | 1:200 | 1:1000 | Kind gift from Dr. G. Thinakaran, The University of Chicago, US |
| APP (clone Y188) | Monoclonal | n/a | 1:5000 | Abcam Inc. |
| APP (clone 369) | Polyclonal | n/a | 1:1000 | Kind gift from Dr. G. Thinakaran, The University of Chicago, US |
| APP (clone CTM1) | Polyclonal | 1:2000 | n/a | Kind gift from Dr. G. Thinakaran, The University of Chicago, US |
| BACE1 | Polyclonal | 1:200 | 1:1000 | EMD Millipore Co. |
| CD68 (clone ED1) | Monoclonal | 1:50 | n/a | AbD Serotec |
| Cleaved Caspase‐3 | Monoclonal | 1:200 | n/a | Cell Signaling Technology |
| GFAP | Polyclonal and monoclonal | 1:500 | n/a | Dako |
| Nicastrin (N‐19) | Polyclonal | 1:200 | 1:500 | Santa Cruz Biotechnology, Inc. |
| p44/42 MAPK (ERK1/2) | Monoclonal | n/a | 1:1000 | Cell Signaling Technology, Inc. |
| Phospho‐p44/42 MAPK (ERK1/2) | Polyclonal | n/a | 1:1000 | Cell Signaling Technology, Inc. |
| Phospho‐CREB (Ser133) | Monoclonal | n/a | 1:1000 | Cell Signaling Technology, Inc. |
| PEN2 | Polyclonal | 1:200 | 1:1000 | Kind gift from Dr. G. Thinakaran, The University of Chicago, US |
| PS1 | Polyclonal | 1:500 | 1:3000 | Kind gift from Dr. G. Thinakaran, The University of Chicago, US |
| sAPPα (2B3) | Monoclonal | n/a | 1:1000 | IBL Co., Ltd |
| sAPPβ (Wild‐type) | Monoclonal | n/a | 1:1000 | IBL Co., Ltd |
Abbreviations: IHC/IF = Immunohistochemistry/Immunofluorescence; WB = western blotting; n/a = not used in specified application.
Systemic administration of kainic acid
Adult male Sprague‐Dawley rats obtained from Charles River (St Constant, QC, Canada) were housed with access to food and water ad libitum. The experiments were performed in accordance with Institutional and Canadian Council on Animal Care guidelines. Adult rats were injected intraperitoneally with either kainic acid dissolved in normal saline (12 mg/kg) or equal volume (0.2–0.25 mL) normal saline as described previously 8, 79. Following treatment, the control and kainic acid‐treated rats were euthanized at 12 h, 2, and 12 days by decapitation; their hippocampi were dissected out of the brain and frozen immediately in dry‐ice for biochemical assays. For histological studies, control and kainic acid‐treated rats were anesthetized with 8% chloral hydrate and then perfused with phosphate‐buffered saline (PBS), followed by 4% paraformaldehyde (PFA). Brains were sectioned (20 or 40 µm) on a cryostat and collected in a free‐floating manner for further processing.
Fluoro‐Jade C Staining
Fluoro‐Jade C labels degenerating neurons, dendrites and axons 68. Sections from control and kainic acid‐treated rat brains from various time periods were stained with 0.0001% Fluoro‐Jade C in 0.1% acetic acid as described earlier 38.
Immunostaining
Brain sections from control and kainic acid‐treated rats (3–5 animals/group) were processed as described earlier 38. For the enzyme‐linked procedure, 40 μm sections were incubated overnight at 4°C with anti‐APP (CTM1), anti‐BACE1, anti‐PS1, anti‐nicastrin, anti‐PEN2 or anti‐APH1 antiserum at dilutions listed in Table 1. Sections were then exposed to HRP‐conjugated secondary antibodies for 1 h and developed using the glucose‐oxidase‐nickel enhancement method. For double immunofluorescence staining, brain sections (20 μm) from control and kainic acid‐treated rats were sequentially labeled with anti‐APP (CTM1), anti‐BACE1, anti‐PS1, anti‐nicastrin, anti‐PEN2 or anti‐APH1 antisera, followed by anti‐ED1, anti‐glial fibrillary acidic protein (GFAP) or anti‐cleaved caspase‐3 antisera at dilutions listed in Table 1. The primary antibodies were labeled with either Texas Red‐ or FITC‐conjugated secondary antibodies (1:200), washed and then mounted with Vectashield medium. Immunostained sections were examined and photographed using a Zeiss Axioskop‐2 fluorescence microscope 49.
Immunoblotting
Hippocampal tissues from control and kainic acid‐treated rats from different time periods (6–8 animals/group) were homogenized in ice‐cold radioimmunoprecipitation assay‐lysis buffer and protein content was determined using a BCA kit. The proteins were separated by electrophoresis on 4%–20% polyacrylamide gels and then transferred to nitrocellulose membranes. The membranes were blocked with 5% non‐fat milk and incubated overnight at 4°C with anti‐APP (369), anti‐BACE1, anti‐PS1, anti‐nicastrin, anti‐PEN2, anti‐APH1, anti‐sAPPα, anti‐sAPPβ, anti‐Y1888, anti‐extracellular signal‐regulated kinases 1/2 (ERK1/2), anti‐phospho‐cAMP response element‐binding protein (phospho‐CREB), anti‐α‐fodrin, anti‐µ‐calpain and anti‐cleaved caspase‐3 antibodies at dilutions listed in Table 1. Membranes were then incubated with the HRP‐conjugated secondary antibodies and developed using an ECL detection kit. Membranes were subsequently reprobed with either anti‐β‐actin or anti‐total ERK1/2 antibody and immunoreactive proteins were quantified using MCID image analysis system 37.
β‐ and γ‐secretase activity assays
The β‐secretase activity in the hippocampus of control and kainic acid‐treated rats (4–6 animals/group) was measured using the fluorometric β‐secretase activity assay kit according to the manufacturer's instructions as described earlier 83. The fluorescence was measured at excitation wavelength of 355 nm and emission wavelength of 495 nm and specific activity of the enzyme was determined by incubating parallel samples with a β‐secretase inhibitor BIV. The γ‐secretase activity assay was performed (4–6 animals/group) as described previously 49. The fluorescence was measured at an excitation wavelength of 355 nm and emission wavelength of 440 nm at 37°C and the specific activity was determined by incubating a parallel set of samples with 100 μM γ‐secretase inhibitor L‐658,458. All samples were assayed in duplicate and data were obtained from three independent experiments.
Rat Aβ1–40 and Aβ1–42 levels
Aβ peptides from hippocampus of control and kainic acid‐treated rats (4–6 animals/group) were solubilized in 5M guanidine buffer 16, centrifuged and then assayed for rat Aβ1–40 or Aβ1–42 levels using ELISA kits as described earlier 49. In a separate series of experiments, cellular and secretory levels of Aβ1–40 and Aβ1–42 were measured in cultured astrocytes treated with or without kainic acid in a similar manner. All samples were assayed in duplicate and the data were obtained from three independent experiments.
Primary hippocampal neuronal, astrocyte and astrocyte/neuronal cultures
Primary rat hippocampal neuronal cultures were prepared from 17/18‐day‐old embryos of time‐pregnant Sprague‐Dawley rats as described previously 86, 96. In brief, the hippocampal region was dissected in Hanks balanced salt solution supplemented with 100 µM HEPES, 10 mM Na‐pyruvate, 50 µM L‐glutamine, 10 U/mL penicillin and 10 mg/mL streptomycin and digested with 0.25% trypsin/EDTA. The cell suspension was filtered through a cell strainer and then plated on either 96‐well plate or 12 mm glass coverslips. The cultures were grown at 37°C in a 5% CO2‐humidified atmosphere in Neurobasal medium supplemented with B27, 50 µM L‐glutamine, 100 µM HEPES, 10 mM Na‐pyruvate, 10 U/mL penicillin and 10 mg/mL streptomycin, and 1% FBS. The medium was replaced 1 day later without FBS and all experiments were performed on day 6 after plating. Primary rat hippocampal astrocytes (Sciencell, CA, USA) were seeded on Poly‐D‐lysine‐coated plates and grown in the associated astrocyte media at 37°C as per the manufacturer's instructions. Cells were grown to confluency and passaged using TrypLE Express. To prepare astrocyte‐neuronal co‐cultures, hippocampal neurons were prepared as described above, and then seeded directly on a layer of astrocytes grown to confluency. Media conditions for co‐cultures follow the protocol for neuronal culture and all experiments were performed 6 days following cell plating.
Treatment of cultured neurons, astrocytes and mixed neurons/astrocytes
The oligomeric forms of rat and human Aβ1–42 used in all experiments related to toxicity were prepared as described earlier 75 and added to neurons to the final concentrations (0.01–20 µM) for different periods of time (ie, 12, 24, 48 or 72 h). Control and Aβ‐treated cultures were then processed for MTT cell viability and live/dead cell toxicity assays 75. In a separate set of experiments, cultured rat hippocampal astrocytes were treated with or without 20 µM γ‐secretase inhibitor DAPT for 24 h and then exposed to 100 µM kainic acid for an additional 24 h. Subsequently, the cellular and secretory levels of rat Aβ1–40/Aβ1–42 as well as cell viability were measured. In parallel, cultured neurons and mixed neurons/astrocytes were exposed to 100 µM kainic acid with or without 20 µM DAPT for 24 or 48 h and their cell viability was assessed 49.
Cell viability assays
Viability of neurons or astrocytes was assessed using the cell proliferation colorimetric MTT assay 83. Control and treated culture plates were replaced with new medium containing 0.25% MTT and then incubated for 2 h at 37°C. The reaction was terminated and measured spectrophotometrically at 570 nm. Neuronal viability following treatment with Aβ peptide was also assessed using the Live/Dead assay kit containing calcein AM and ethidium homodimer (EthD‐1) as the fluorescent probes. Control and treated cultured neurons were incubated with medium containing 2 µM calcein AM and 4 µM EthD‐1 for 30 min at 37°C, fixed in 4% PFA and then visualized under a Zeiss Axioskop‐2 fluorescent microscope 75.
Statistical analysis
Data are expressed as means ± SEM. Statistical significance of differences was determined by one‐way ANOVA followed by Newman‐Keuls post hoc analysis for multiple comparisons or unpaired two‐tailed Student's t test for single comparison with a significance threshold of P < 0.05. All analyses were performed using GraphPad Prism Software.
Results
Loss of neurons and proliferation of glial cells in kainic acid‐treated rats
In keeping with earlier studies 8, 10, 45, 80, we observed that systemic administration of kainic acid evoked seizures characterized by rearing and falling, facial and forelimb clonus, and “wet‐dog” shakes that lasted about 8–10 h. After a seizure‐free period lasting a few days, a high proportion of animals surviving such an attack exhibited a chronic phase of spontaneous recurrent limbic seizures with no remission over 12 days of the experimental paradigm 18, 89. These behavioral changes are accompanied by a loss of neurons, as apparent by Fluoro‐Jade C and cleaved caspase‐3‐labeling, as well as glial hypertrophy in the hippocampus of the rat brain (Figure 1A–K). As expected, reduced neuronal viability progressed with time primarily in CA1‐CA3 pyramidal cells and the hilar region of the dentate gyrus, whereas the granule cell layer was relatively spared 8, 80, 84. The hypertrophy of astrocytes and microglia (data not shown) was apparent to some extent at 12 h and then gradually increased over 12 days post‐treatment period 19, 35, 80 (Figure 1A–I). The progressive loss of neurons and activation of glial cells were seen in the hippocampi of both hemispheres in kainic acid‐treated rats.
Figure 1.
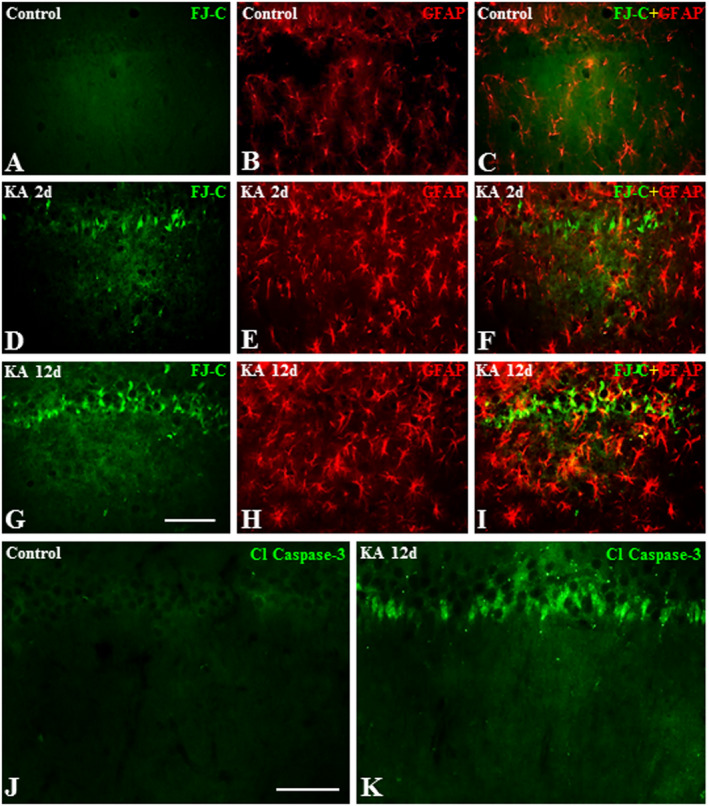
A–I. Photomicrographs depicting Fluoro‐Jade C labeled neurons (A, D, G), GFAP‐labeled astrocytes (B, E, H) and their spatial co‐distribution (C, F, I) in the hippocampus of control (A–C), 2day (D–F) and 12day (G–I) kainic acid‐treated rats. Note the lack of Fluoro‐Jade C labeling and proliferation of astrocytes in control rat hippocampus (A–C), whereas the loss of neurons and proliferation of astrocytes increased gradually in the hippocampus of kainic acid‐treated rats over 12day post‐treatment period. J and K, Photomicrographs depicting cleaved‐caspase‐3 labeling in the hippocampus of control (J) and 12day kainic acid‐treated (K) rats. Note the cleaved‐caspase‐3 labeled neurons following administration of kainic acid but not in control rat. Scale bar, 100 µM.
Earlier studies have indicated that kainic acid induces neurotoxicity by increasing Ca+2 influx, the production of reactive oxygen species and the activation of multiple downstream signaling cascades including mitogen activated protein (MAP) kinase and the cysteine protease calpain 4, 10, 11, 34, 80, 82, 93. While calpain exists in two different isoforms (µ‐ and m‐calpains), the family of MAP kinase is composed of various enzymes such as stress‐activated protein kinases and ERK1/2 (34, 80, 93). A significant induction of phospho‐ERK1/2 and its downstream substrate phospho‐CREB was evident following kainic acid administration at 12 h and 2 days, respectively, and remained elevated over the 12 day post‐treatment period (Figure 2A). Concurrently, we also observed a protracted increase in calpain levels in the hippocampus of kainic acid‐treated rats. To validate the activation of calpain, we measured the breakdown products of non‐erythroid αII‐spectrin (ie, fodrin)—a signature downstream substrate which yields unique proteolytic fragments following cleavage by caspase and calpain 4, 11, 82. While calpain‐specific 150/145kD αII‐spectrin breakdown products (SBDP) were increased at all time points following administration of kainic acid, with maximal changes apparent at 2 day post‐treatment, caspase‐3 specific 120 kD SBDP showed alterations mostly at 2 day post‐treatment over the 12 days experimental paradigm (Figure 2B). Caspase activation is further supported by our Western blot (Figure 2B) and the detection of immunoreactive cleaved‐caspase‐3‐positive neurons (Figure 1J,K) in kainic acid‐treated rat hippocampus compared to control rats.
Figure 2.
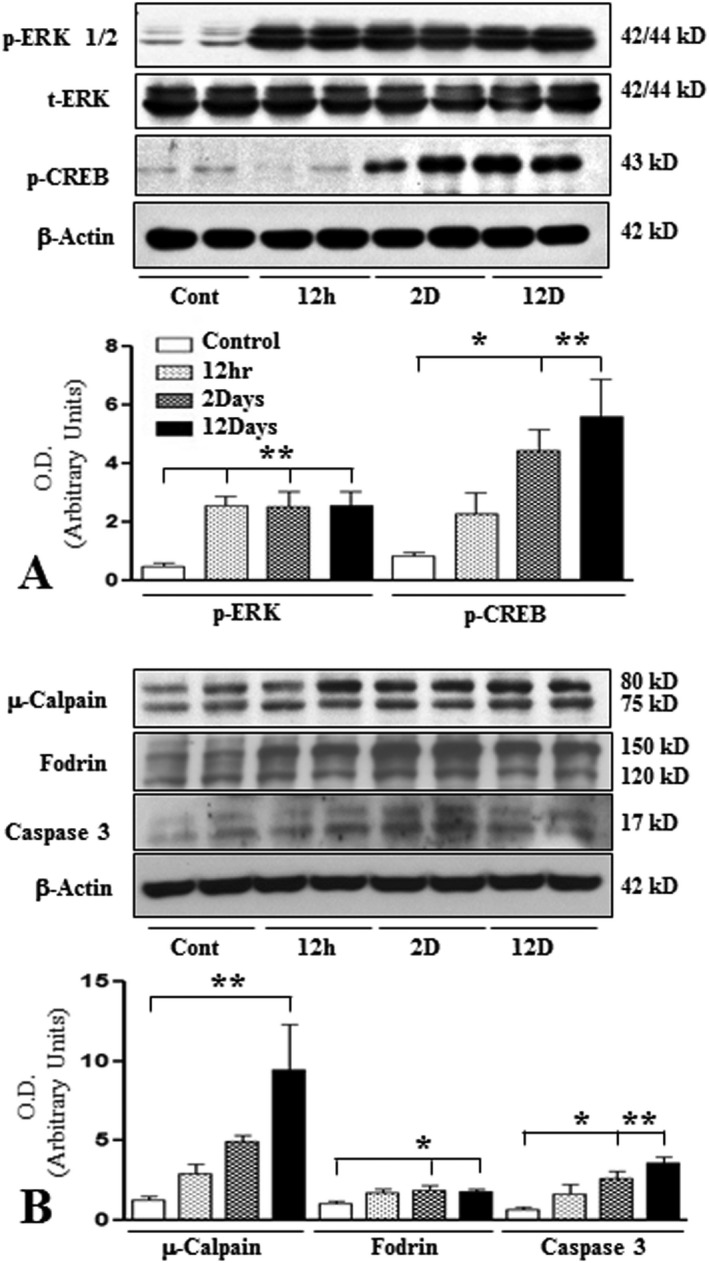
A. Histogram and representative immunoblots showing the levels of phospho‐ERK1/2, total ERK, phospho‐CREB in the hippocampus of control and kainic acid‐treated rats. Note the relative increase in the levels of these markers following treatment with kainic acid compared to saline‐treated control rats. B. Histogram and representative immunoblots showing the levels of µ‐calpain, fodrin, caspase 3 in the hippocampus of control and kainic acid‐treated rats. Note the relative increase in the levels of these markers following treatment with kainic acid compared to saline‐treated control rats. All blots were re‐probed with anti‐β‐actin to monitor protein loading. Values represent means±SEM from 6–8 rats. *P < 0.05, **P < 0.01.
Increased levels and expression of APP and its processing enzymes in kainic acid‐treated rats
APP levels were markedly increased at 2 and 12 days, but not at 12 h, following kainic acid administration compared to control rats (Figure 3A). At the cellular level, APP immunoreactivity in the control hippocampus was evident mostly in CA1‐CA3 pyramidal neurons and granule cells of the dentate gyrus but not in glial cells (Figure 4A,B; Supporting Information Figure S1A–D). Occasionally, APP‐immunoreactive neurons were also apparent in the hilus region of the dentate gyrus (Figure 4A). While the overall expression of APP decreased with time in the degenerating hippocampal neurons, a number of glial cells were found to express APP from 2 days onwards after kainic acid treatment (Figure 4C–F). Our double labeling showed that immunoreactive APP is evident in a subset of GFAP‐labeled reactive astrocytes, but not ED1‐labeled activated microglia, following kainic acid treatment (Figure 4G–J).
Figure 3.
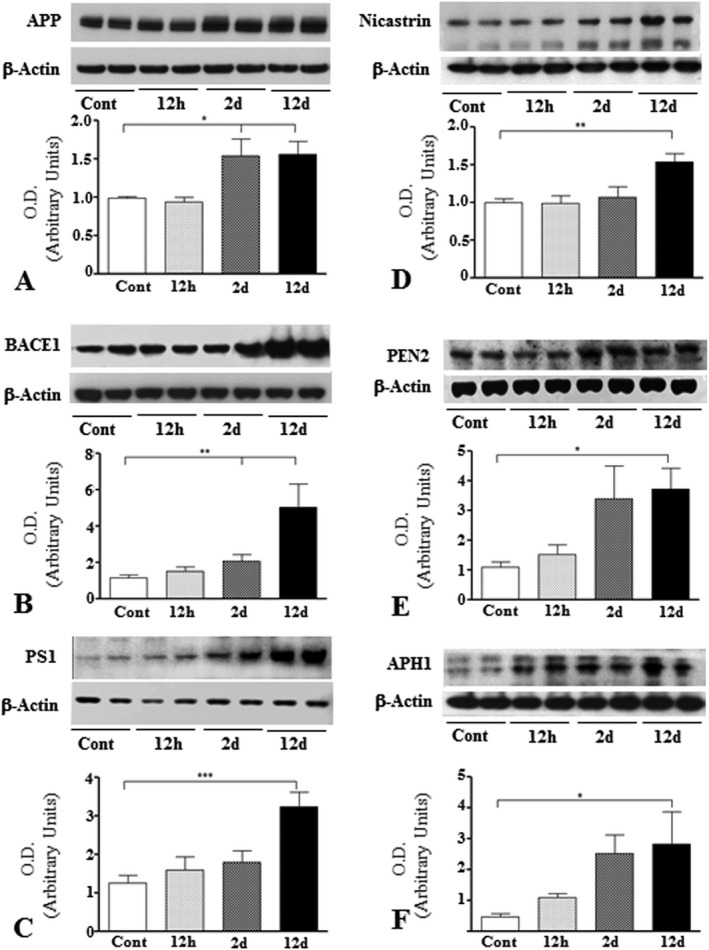
Histograms and representative immunoblots showing the levels of APP (A), BACE1 (B), PS1 (C), nicastrin (D), PEN2 (E) and APH1 (F) in the hippocampus of control and kainic acid‐treated rats. Note the time‐dependent increase in the levels of APP, BACE1 and four components of the γ‐secretase complex following treatment with kainic acid compared to saline‐treated control rats. All blots were reprobed with anti‐β‐actin to monitor protein loading. Values represent means±SEM from 6–8 rats. *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 4.
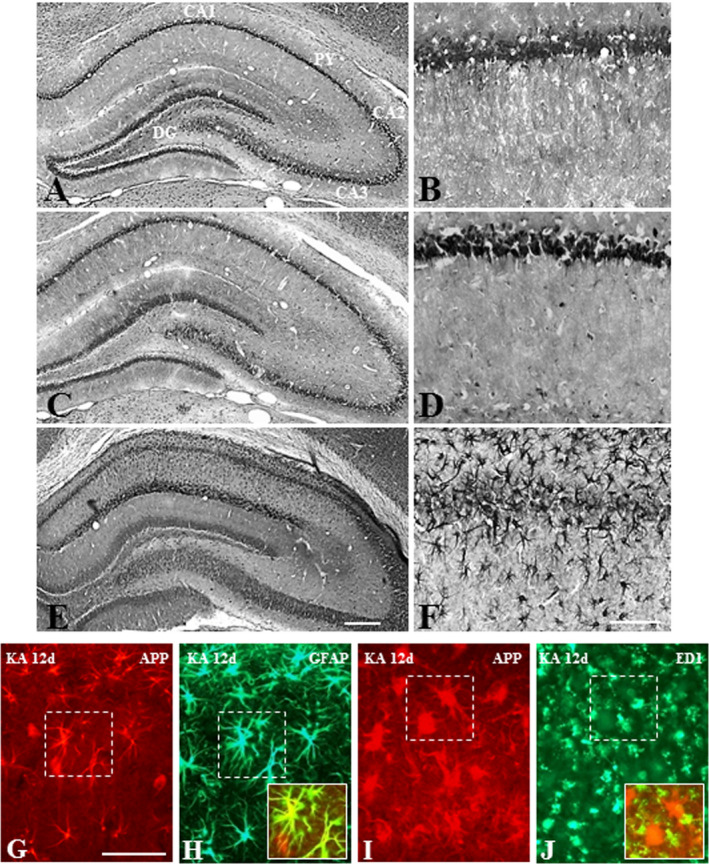
A‐F; Photomicrographs depicting cellular distribution of APP in the hippocampus of saline‐treated control (A, B) and following 12 h (C, D) and 12 day (E, F) kainic acid‐treated rats at lower (A, C, E) and higher (B, D, F) magnifications. Note the expression of APP in pyramidal and granule cell layers in control rats, whereas after 12 day kanic acid treatment APP is evident mostly in glial cells of the hippocampus. G–J. Double immunofluorescence photomicrographs showing a subset of GFAP‐labeled astrocytes (G, H), but not ED1‐labeled microglia (I, J), exhibit immunoreactive APP in 12 day kainic acid‐treated rat. Py, pyramidal cell layer; DG, dentate gyrus. Scale bar, 100 µM.
To determine if enhanced APP levels in kainic acid‐treated rats were accompanied by a parallel increase in APP processing via the β‐secretase pathway, we measured the levels/expression of BACE1 and four components of the γ‐secretase complex (Figures 3B, Figures 5–6; Supporting Information Figures S1–S5) in the hippocampus of treated and control rats. We observed a significant increase in BACE1 levels at 2 and 12 days following kainic acid treatment compared to control rats (Figure 3B). The components of the γ‐secretase complex, PS1 (Figure 3C), nicastrin (Figure 3D), PEN2 (Figure 3E) and APH1 (Figure 3F) were likewise found to be enhanced in kainic acid‐treated rats. Consistent with earlier studies 37, 71, immunoreactive BACE1 and four components of γ‐secretase complex were evident mostly in neurons of the normal brain. In the hippocampal region, intense immunoreactivity for BACE1 (Figure. 5A,B; Supporting Information Figure S1E–H) and components of γ‐secretase complex, that is, PS1 (Figure 6A,B; Supporting Information Figure S1I–L), nicastrin (Supporting Information Figures S2A,B and S5A–D), PEN2 (Supporting Information Figures S3A,B and S5E–H) and APH1 (Supporting Information Figures 4A,B and S5I–L) was apparent in the CA1‐CA3 pyramidal cell layer and in a few medium‐sized neurons scattered in the strata oriens and stratum radiatum but not in glial cells. Within the dentate gyrus, granule cell somata were outlined by a fine mesh of weakly stained puncta and occasional strongly labeled neurons. In comparison with the control hippocampus, a number of glial cells were found to express immunoreactive BACE1 (Figure 5C–F) and components of γ‐secretase complex in kainic acid‐treated rats from 2 day post‐treatment onwards (Figure 6C–F; Supporting Information Fig. S2C–F, S3C–F and S4C–F). Our double immunolabeling revealed that a subset of reactive astrocytes express immunoreactive BACE1 (Figure 5G,H), PS1 (Fig. 6G, H), nicastrin (Supporting Information Figure S2G,H), PEN2 (Supporting Information Figure S3G,H) and APH1 (Supporting Information Figure S4G,H) in the hippocampus of treated rats. Activated microglia, conversely, did not express either BACE1 (Figure 5I,J) or components of γ‐secretase, that is, PS1 (Figure 6I,J), nicastrin (Supporting Information Figure S2I,J), PEN2 (Supporting Information Figure S3I,J) and APH1 (Supporting Information Figure S4I,J) at any time points following kainic acid administration.
Figure 5.
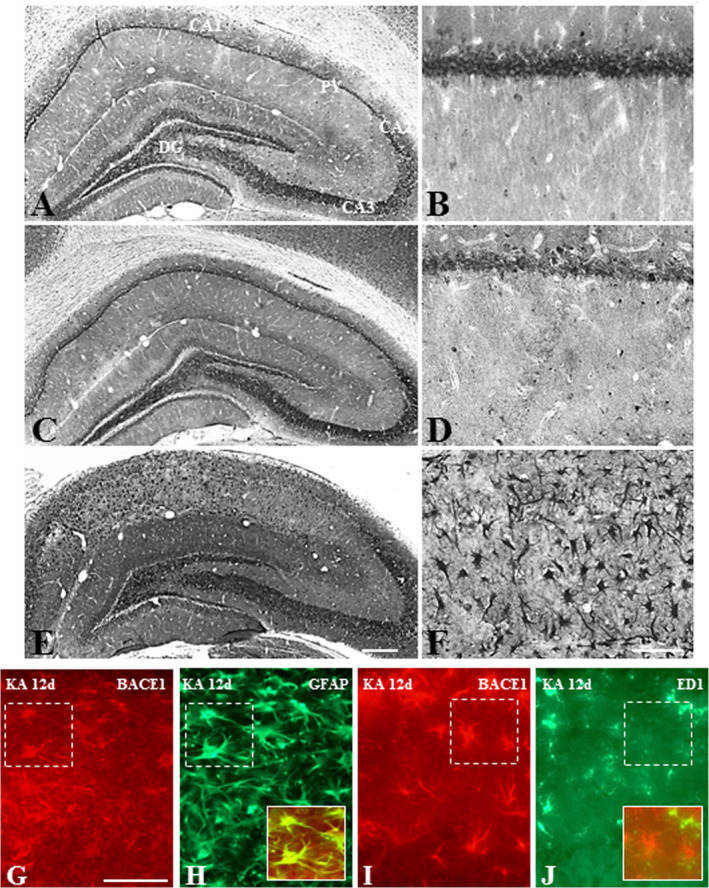
A–F. Photomicrographs depicting cellular distribution of BACE1 in the hippocampus of control (A, B) and following 12 h (C, D) and 12day (E, F) kainic acid‐treated rats at lower (A, C, E) and higher (B, D, F) magnifications. Note the expression of BACE1 in pyramidal and granule cell layers in control rats, whereas after 12 day kanic acid treatment BACE1 is evident mostly in glial cells of the hippocampus. G–J. Double immunofluorescence photomicrographs showing a subset of GFAP‐labeled astrocytes (G, H), but not ED1‐labeled microglia (I, J), exhibit immunoreactive BACE1 in 12 day kainic acid‐treated rat. Py, pyramidal cell layer; DG, dentate gyrus. Scale bar, 100 µM.
Figure 6.
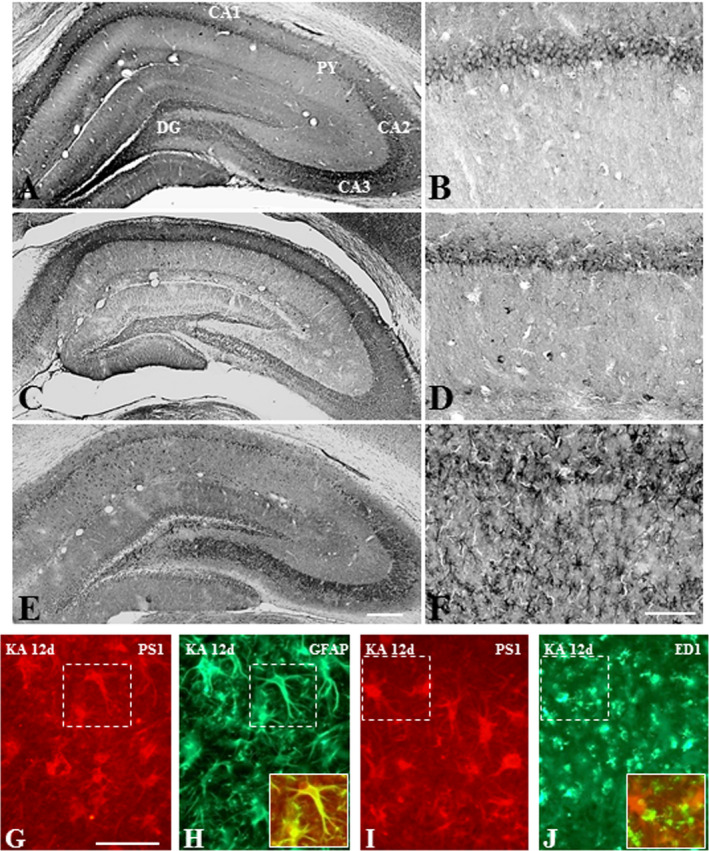
A–F. Photomicrographs depicting cellular distribution of PS1 in the hippocampus of saline‐treated control (A, B) and following 12 h (C, D) and 12 day (E, F) kainic acid‐treated rats at lower (A, C, E) and higher (B, D, F) magnifications. Note the expression of PS1 in pyramidal and granule cell layers in control rats, whereas after 12 day kanic acid treatment PS1 is evident mostly in glial cells of the hippocampus. G–J. Double immunofluorescence photomicrographs showing a subset of GFAP‐labeled astrocytes (G, H), but not ED1‐labeled microglia (I, J), exhibit immunoreactive PS1 in 12 day kainic acid‐treated rat. Py, pyramidal cell layer; DG, dentate gyrus. Scale bar, 100 µM.
APP processing and enhanced rat Aβ1–40/Aβ1–42 levels in kainic acid‐treated rats
To determine whether kainic acid treatment can influence APP processing, we first measured the activity of β‐ and γ‐secretases, as well as the steady state levels of APP‐cleaved products (sAPPα, sAPPβ, α‐CTF and β‐CTF) in the hippocampus of treated and control rats. Our results showed that BACE1 and γ‐secretase activities were significantly increased in time‐dependent manner following kainic acid administration (Figure 7A,B). Accompanying these data, levels of sAPPβ were increased from 2 day post‐treatment, while sAPPα levels were not altered at any time relative to the control rats (Figure 7C). The levels of α‐CTF and β‐CTF were also significantly increased after kainic acid treatment, but the changes observed for β‐CTF were found to be somewhat higher than α‐CTF (Figure 7D). In parallel, rat Aβ1–40 and Aβ1–42 levels, as detected by ELISA, were markedly increased from 2 day onwards in the hippocampus of kainic acid‐treated rats compared to control rats (Figure. 7E,F).
Figure 7.
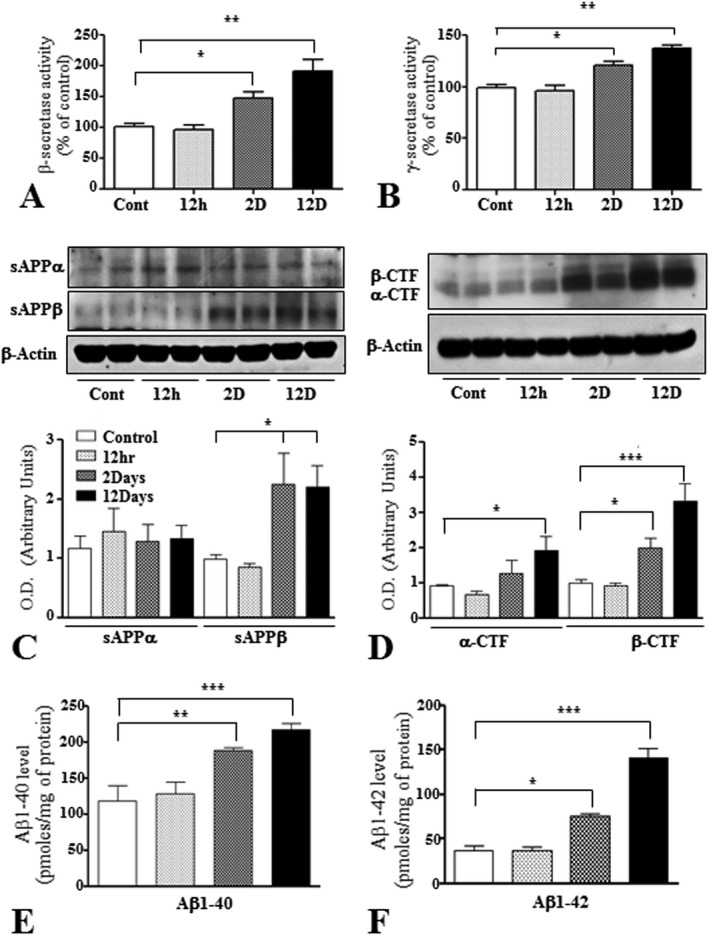
A and B. Histograms showing the activity of β‐ (A) and γ‐ (B) secretases in the hippocampus of control and kainic acid‐treated rats. Note the relative increase in the activity of both secretases following systemic administration of kainic acid compared to control rats. C and D. Representative immunoblots and histograms showing levels of sAPPα and sAPPβ (C), α‐CTF and β‐CTF (D) in the hippocampus of control and kainic acid‐treated rats. Note the relative increase in the levels of sAPPβ, α‐CTF and β‐CTF, but not sAPPα, following administration of kainic acid compared to control rats. E and F. Histograms showing the quantification of rat Aβ1–40 (E) and Aβ1–42 (F), as detected by ELISA, in the hippocampus of control and kainic acid‐treated rats. Note the significant increase in the levels of both Aβ peptides following kainic acid administration compared to control rats. All blots were re‐probed with anti‐β‐actin to monitor protein loading. Values represent means±SEM from 4–8 rats. *P < 0.05, **P < 0.01, ***P < 0.001.
Rat Aβ1–42 is toxic to primary rat hippocampal neurons
Consistent with earlier results 39, 75, chronic exposure to oligomeric human Aβ1–42 reduced viability of hippocampal neurons in a concentration‐dependent manner, as evident by a reduction in MTT values (Supporting Information Figure S6A). To determine whether rat Aβ1–42, which differs from the human Aβ1–42 by three amino acids 15, is toxic to cells, we first evaluated its dose‐dependent (1–20 µM) effect on hippocampal neurons. The viability of neurons, as indicated by MTT assay, was not affected by rat 1–10 µM Aβ1–42, but significantly decreased with 20 µM Aβ1–42 following 24 h treatment (Supporting Information Figure S6B). The toxic potency of rat Aβ1–42 was further substantiated by a time‐dependent decrease in MTT value, with ∼ 50% cell death observed at 72 h post‐treatment (Supporting Information Figure S6C). Our live‐dead assay also showed an increase in the number of dead cells following exposure to 20 µM rat Aβ1–42 for 24 h (Supporting Information Figure S6D–G).
Kainic acid treatment enhanced Aβ1–40/Aβ1–42 levels in cultured rat astrocytes
Our in vivo studies indicated that kainic acid‐induced neurotoxicity was accompanied by increased levels/processing of APP, leading to the enhanced production of Aβ‐related peptides. Since expression of APP and its processing enzymes were increased in a subset of reactive astrocytes, it is likely that Aβ peptides are partly generated from these astrocytes. To validate this notion, rat primary astrocytes were treated with 100 µM kainic acid for different durations (12 h–48 h) and then cellular/secretory Aβ levels were measured using ELISA. Our results showed that cellular levels of Aβ1–40 were significantly increased at 12 h and 24 h, whereas Aβ1–42 were increased only at 12 h after kainic acid treatment (Figure 8A). However, secretory levels of Aβ1–40 and Aβ1–42 were markedly increased with time following exposure to 100 µM kainic acid (Figure 8B). Pre‐treatment with the γ‐secretase inhibitor DAPT attenuated enhanced levels of Aβ peptides, suggesting that an increased production/secretion of the peptides is triggered by kainic acid treatment (Figure 8C). In parallel, we observed that the viability of cultured astrocytes was not compromised after 48 h exposure to either 100 µM kainic acid or 20 µM DAPT (Figure 8D).
Figure 8.
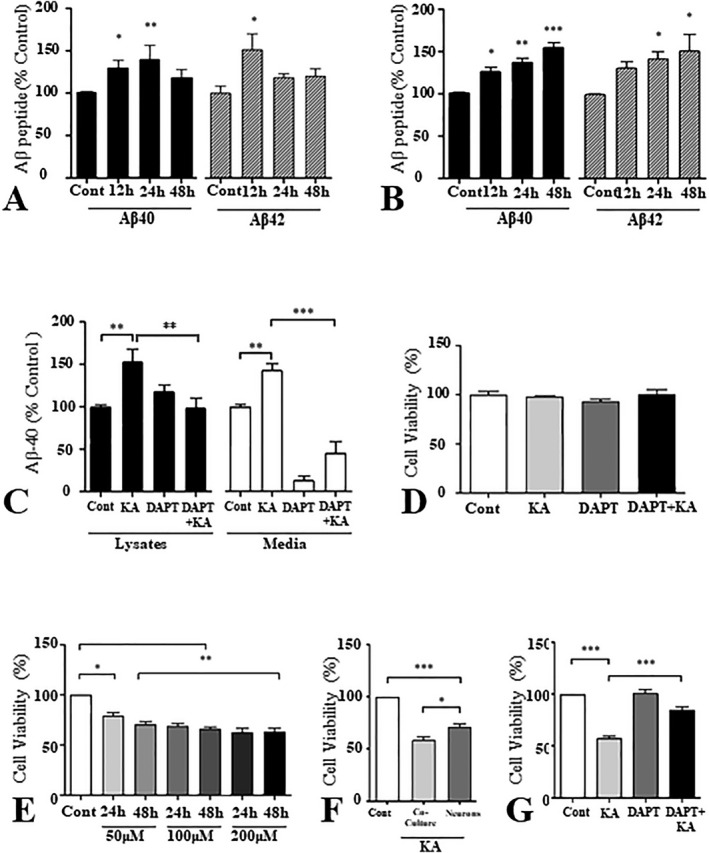
A and B. Histograms showing the alteration in Aβ1–40 and Aβ1–42 levels in cell lysates (A) and media (B) of rat primary astrocytes treated with 100 µM of kainic acid over 12 h, 24 h and 48 h relative to control (Cont). Both Aβ1–40 and Aβ1–42 show a significant increase in the cell lysates as well as in the media following kainic acid treatment. C. Histogram depicting the levels of Aβ1–40 in primary rat astrocyte cultures treated with either kainic acid, γ‐secretase inhibitor DAPT, or a combination of the two (DAPT + KA) relative to control. Note that DAPT drastically reverses the increase in Aβ1–40 levels triggered by kainic acid and this effect is especially prominent in the conditioned media. D. Histogram showing no change in the viability of cultured astrocytes following treatment with kainic acid, DAPT or DAPT + kainic acid, as measured by an MTT assay. E. Histogram showing the effect of kainic acid on the viability of rat primary hippocampal cultured neurons as assessed by an MTT assay. Kainic acid was found to be toxic to neurons in relation to control. F. Histogram showing viability of kainic acid‐treated neuron/astrocyte co‐cultures and pure neuronal cultures compared to control. G. Histogram showing the viability of neuron/astrocyte co‐cultures in relation to control following treatment with kainic acid, DAPT and DAPT + kainic acid. Kainic acid‐induced death of neurons was attenuated by DAPT pretreatment. All cultured data represent means±SEM from 3–4 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Kainic acid‐induced toxicity of mixed neuron/astrocyte cultures protected by γ‐secretase inhibitor
To determine the significance of Aβ peptides in kainic acid‐induced toxicity, pure rat hippocampal cultured neurons were first exposed to kainic acid (50–200 µM) for either 24 or 48 h, and then cell viability was assessed using MTT assay. As expected, viability of neurons was reduced significantly in most conditions, but it did not exhibit marked variation with either increasing concentrations or time following exposure to kainic acid (Figure 8E). Interestingly, treating mixed neuron/astrocyte co‐cultures with 100 µM kainic acid for 24 h was found to reduce cell viability significantly more than what was observed with pure neuronal cultures (Figure 8F). Since kainic acid did not affect the viability of cultured astrocytes (Figure 8D), we wanted to establish if Aβ peptides generated from cultured astrocytes in response to this treatment may be involved in triggering neurotoxicity. Thus, we pretreated mixed neuron/astrocyte co‐cultures with or without 20 µM DAPT, and then exposed them to 100 µM kainic acid for 24 h. Our results clearly showed that DAPT treatment can attenuate kainic acid‐induced toxicity (Figure 8G), indicating a role for Aβ peptides in reducing viability of neurons.
Discussion
This study uses a combination of experimental approaches to show that increased production and levels of Aβ peptides in reactive astrocytes have a role in neurotoxicity triggered by the systemic administration of kainic acid. This is supported by data which show that (i) kainic acid‐induced loss of hippocampal neurons is associated with higher expression of APP, BACE1 and γ‐secretase components in a subset of activated astrocytes, (ii) activity of β‐ and γ‐secretases are enhanced along with the levels of APP‐cleaved products including Aβ1–40 and Aβ1–42 in the hippocampus of kainic acid‐treated rats, (iii) rat Aβ1–42, like human Aβ1–42, is toxic to cultured hippocampal neurons, (iv) exposing rat cultured astrocytes to kainic acid can lead to enhanced production/secretion of Aβ1–40/Aβ1–42 without compromising their viability and (v) loss of rat cultured hippocampal neurons by kainic acid is enhanced in the presence of astrocytes, and can be attenuated by the γ‐secretase inhibitor DAPT. Collectively, these results suggest that activated astrocytes, by way of heightened APP expression and the increased production of Aβ peptides, play a critical role in reducing viability of neurons triggered by kainic acid. Since kainic acid treatment can lead to neuropathological changes resembling those observed in human TLE, it is possible that APP/Aβ peptides derived from astrocytes may also have a role in TLE pathogenesis.
Loss of neurons in kainic acid‐treated rats
Consistent with earlier studies, we observed that systemic administration of kainic acid can induce loss of neurons, along with glial hypertrophy and proliferation in the hippocampus 8, 45, 80, 84. These effects are largely mediated by kainate receptor‐induced glutamatergic currents that trigger an influx of Ca+2 and subsequent activation of downstream signaling cascades including ERK1/2, calpain and caspase as observed in the present study 4, 10, 34, 82, 93. Since neuronal vulnerability to excitotoxicity can be enhanced by Aβ peptides 53, and glutamate receptor activation can regulate amyloidogeneic processing of APP 13, 43, 64, it is of critical relevance to determine if Aβ peptides may have a role in reducing viability of neurons following kainic acid administration. This will not only refine the molecular pathways associated with seizure‐induced neuronal death mechanisms, but also may provide novel treatment targets for patients with TLE.
Influence of kainic acid administration on APP and its processing enzymes
Some earlier studies have shown that kainic acid can trigger APP expression 72, 91, but its significance in the development of pathological features and/or loss of neurons remains unclear. The present study clearly demonstrates that systemic administration of kainic acid increases not only the levels of APP, but also the expression/levels as well as activity of both BACE1 and the γ‐secretase complex. These alterations, which appeared first at 2 days and increased following 12 days post‐treatment, are accompanied by a time‐dependent increase in APP cleaved intermediates including sAPPβ and β‐CTF, and most importantly, Aβ1–40 and Aβ1–42 peptides. Collectively, these results suggest that increased levels and amyloidogenic processing of APP may have a critical role in the development of TLE pathology. Interestingly, certain neurodevelopmental disorders such as Fragile X syndrome (FXS) and autism spectrum disorders (ASD), unlike TLE, have been shown to be associated with increase processing of APP via non‐amyloidogenic pathway. While FXS is the most common form of inherited intellectual disability, ASD is characterized by persistent deficits in social communication/interactions and stereotypic repetitive behaviors 27, 88. Brain abnormalities common to FXS and ASD include deranged cortical pyramidal dendritic spines and increased disruption of white as well as grey matters contributing to macrocephaly 32, 33, 60. Pivotal work from two independent laboratories have demonstrated significantly elevated levels of sAPPα with a concomitant reduction in sAPPβ and Aβ levels in plasma and brain tissue samples of children with ASD 59, 60, 74. Similarly, it has been shown that levels of APP and sAPPα are increased in the plasma as well as brain samples of individuals with FXS 60, 88. Given the neurotrophic roles of sAPPα, it has been suggested that increased non‐amyloidogenic processing of APP may result in macrocephaly associated with FXS and ASD, whereas enhanced processing of APP via amyloidogenic pathway may be associated with neurodegeneration observed in AD and TLE 41, 88.
Under normal conditions, APP, BACE1 and γ‐secretase components are usually localized in neurons of the brain, but not in glial cells 9, 37, 71, 76. The present study showed that a subset of reactive astrocytes, which increased gradually with the progression of disease pathology 10, 25, 45, 90, express APP and its processing enzymes in the hippocampus of kainic acid‐treated rats. In fact, some earlier studies have reported that expression of APP, BACE1, PS1 and/or nicastrin can be induced in activated astrocytes of the brain following a variety of surgical/pathological lesions, such as cerebral ischemia, traumatic brain injury, excitotoxicity and cholesterol sequestration within the endosomal compartments 5, 7, 38, 54, 56, 72, 94. There is also evidence that activated astrocytes located in proximity to Aβ‐containing neuritic plaques in AD brains and in mutant APP transgenic mice express higher levels of APP and/or its processing enzymes 31, 55, 63, 73. Although the underlying mechanisms behind this occurrence remain unclear, it is possible that factors released from activated microglia or damaged neurons trigger astrocytic expression of APP and its processing enzymes 62.
Role of Aβ peptides in kainic acid‐treated rats
The administration of Aβ peptides has been implicated in the induction and propagation of seizures by possibly increasing extracellular glutamate levels caused by enhanced release and decreased uptake of glutamate 24, 36, 51, 66. The present study clearly shows that kainic acid can trigger an increased production and secretion of Aβ1–40/Aβ1–42 peptides, most likely in reactive astrocytes, which may partly be involved in the induction/propagation of seizures. In parallel, Aβ peptides may also have a role in the kainic acid‐driven loss of neurons. This is supported by three distinct lines of evidence—first, our results reveal that rat Aβ peptides, as observed earlier 15, can be toxic to neurons. Second, we demonstrated that kainic acid treatment can enhance cellular/secretory levels of both Aβ1–40 and Aβ1–42 from rat primary astrocytes without affecting their viability. Finally, using neuronal and neuronal/astrocyte mixed cultures we showed that the presence of astrocytes exacerbates the neuronal toxicity of kainic acid, which can be ameliorated by pretreatment with the γ‐secretase inhibitor DAPT. Thus, increased levels of Aβ derived from astrocytes may directly be involved in mediating toxicity induced by kainic acid. However, the underlying mechanism by which astrocytic Aβ decreased neuronal vulnerability remains unclear. It is possible that Aβ secreted from astrocytes following kainic acid treatment can act directly on neurons to decrease their vulnerability. Alternatively, Aβ peptide secreted from kainic acid‐treated astrocytes may act to increase the levels/processing of APP in neurons leading to enhanced intraneuronal Aβ level 57 which can then decrease neuronal vulnerability.
Significance of Aβ‐related peptides in TLE
Although Aβ‐containing plaques and increased levels of APP are apparent in the brain of some TLE patients 26, 46, 69, 70, the significance of these peptides in disease pathogenesis remain unclear. The predominant pathological feature associated with TLE is hippocampal sclerosis, which is characterized by atrophy, induration, gliosis, synaptic reorganization and loss of neurons in CA1, CA3 and the dentate hilar regions 12, 47, 77, 80. There is evidence that excitotoxic glutamatergic transmission resulting from neuronal hyperactivity has a role in triggering recurrent seizures and death of neurons in the sclerotic hippocampus. More recently, a number of studies have shown that activated astrocytes, by regulating the uptake and release of glutamate, can directly contribute to the generation of seizures and loss of neurons in the hippocampus 17, 23, 25. Our results, conversely, demonstrate that levels of Aβ‐related peptides, which increase gradually in kainic acid‐treated rats may originate, at least in part, from a subset of activated astrocytes exhibiting enhanced expression of APP and its processing enzymes. Since Aβ peptides can elevate extracellular glutamate levels by potentiating its release 36, 61, 66 and inhibiting its uptake by astrocytes 24, 30, our data indicate a role for Aβ in the development of seizures and subsequent loss of neurons in TLE brains. This is supported by the evidence that (i) Aβ peptide can directly induce neuronal hyperexcitability and trigger epilepsy 51, (ii) transgenic mice overexpressing Aβ peptide exhibit spontaneous as well as induced seizures more frequently than wild‐type mice 20, 58, 87, (iii) the prevalence of seizures is higher in AD cases than in control populations 2, 14, 42, 67 and the antiepileptic drugs can able to partially reverse AD‐related pathology 6, 65, 92, 95 and (iv) kainic acid‐induced neurotoxicity, as apparent from our results, can be attenuated by inhibition of Aβ synthesis. Additionally, it is reported that immunization of mutant APP transgenic mice with Aβ peptide protects them from seizures 52. These results, taken together, not only highlight the significance of astrocytic Aβ in TLE pathology, but also raise the possibility that lowering Aβ levels may be beneficial in attenuating seizures and neurodegeneration.
Supporting information
Figure S1. A–L. Double immunofluorescence photomicrographs showing the localization of APP (A‐D), BACE1 (E‐H) and PS1 (I‐L) with astrocyte marker GFAP and activated microglia marker ED1 in the hippocampal CA1 region of saline‐treated control rats. Note the localization of APP, BACE1 and PS1 in neurons, GFAP in astrocytes but the absence of ED1 labeling due to lack of activated microglia in control brains. Scale bar, 100 µM.
Figure S2. A–F. Photomicrographs depicting cellular distribution of nicastrin in the hippocampus of saline‐treated control (A, B) and following 12 h (C, D) and 12 day (E, F) kainic acid‐treated rats at lower (A, C, E) and higher (B, D, F) magnifications. Note the expression of nicastrin in pyramidal and granule cell layers in control rats, whereas after 12 day kainic acid treatment nicastrin is evident mostly in glial cells of the hippocampus. G–J. Double immunofluorescence photomicrographs showing a subset of GFAP‐labeled astrocytes (G, H), but not ED1‐labeled microglia (I, J), exhibit immunoreactive nicastrin in 12 day kainic acid‐treated rats. Py, pyramidal cell layer; DG, dentate gyrus. Scale bar, 100 µM.
Figure S3. A–F. Photomicrographs depicting cellular distribution of PEN2 in the hippocampus of saline‐treated control (A, B) and following 12 h (C, D) and 12 day (E, F) kainic acid‐treated rats at lower (A, C, E) and higher (B, D, F) magnifications. Note the expression of PEN2 in pyramidal and granule cell layers in control rats, whereas after 12day kainic acid treatment PEN2 is evident mostly in glial cells of the hippocampus. G–J. Double immunofluorescence photomicrographs showing a subset of GFAP‐labeled astrocytes (G, H), but not ED1‐labeled microglia (I, J), exhibit immunoreactive PEN2 in 12day kainic acid‐treated rats. Py, pyramidal cell layer; DG, dentate gyrus. Scale bar, 100 µM.
Figure S4. A–F. Photomicrographs depicting cellular distribution of APH1 in the hippocampus of saline‐treated control (A, B) and following 12hr (C, D) and 12day (E, F) kainic acid‐treated rats at lower (A, C, E) and higher (B, D, F) magnifications. Note the expression of APH1 in pyramidal and granule cell layers in control rats, whereas after 12 day kainic acid treatment APH1 is evident mostly in glial cells of the hippocampus. G–J. Double immunofluorescence photomicrographs showing a subset of GFAP‐labeled astrocytes (G, H), but not ED1‐labeled microglia (I, J), exhibit immunoreactive APH1 in 12 day kainic acid‐treated rats. Py, pyramidal cell layer; DG, dentate gyrus. Scale bar, 100 µM.
Figure S5. A–L. Double immunofluorescence photomicrographs showing the localization of nicastrin (A–D), PEN2 (E–H) and APH1 (I–L) with astrocyte marker GFAP and activated microglia marker ED1 in the hippocampal CA1 region of saline‐treated control rats. Note the localization of nicastrin, PEN2 and APH1 in neurons, GFAP in astrocytes but the absence of ED1 labeling due to lack of activated microglia in control brains. Scale bar, 100 µM.
Figure S6. A–C. Histograms showing the dose‐dependent effects of 24 h exposure to different concentrations of human Aβ1‐42 (A) and dose‐ as well as time‐dependent effects of rat Aβ1‐42 (B, C) on viability of hippocampal cultured neurons as revealed by MTT assay. Control cultures were treated with 10 µM human or rat Aβ42‐1 peptide. Note that human Aβ1‐42 was found to be more toxic than equimolar concentration of rat Aβ1‐42 on cultured neurons. D–G. Images from the live/dead assay depicting cell viability in control (D, E) and 20µM rat Aβ1‐42‐treated (F, G) hippocampal cultured neurons. Calcein AM staining (green) represents intracellular esterase activity in living neurons, while EthD‐1 (red) shows dead neurons with disintegrated plasma membrane. Note the toxic potency of rat Aβ1‐42 compared to control cultures treated with rat Aβ42‐1. MTT data presented as % of control (means ± SEM) were obtained from 3–5 separate experiments, each performed in triplicate. Cont, control. * P < 0.05 and ** P < 0.001 compared to control value. Scale bar, 50 µm.
Acknowledgments
This work is supported by a grant from the Canadian Institutes of Health Research to SK. MM is a recipient of a studentship award from the Alberta Innovates – Health Solutions (AIHS). YW is a recipient of Doctoral Awards from Alzheimer Society of Canada and a Graduate Studentship Award from AIHS. We would like to indicate that none of the authors included in this manuscript has had any actual or potential conflict of interest including financial, personal or other relationships with other people or organizations at any time that could inappropriately influence the work.
References
- 1. Alley GM, Bailey JA, Chen D, Ray B, Puli LK, Tanila H et al (2010) Memantine lowers amyloid‐beta peptide levels in neuronal cultures and in APP/PS1 transgenic mice. J Neurosci Res 88:143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amatniek JC, Hauser WA, DelCastillo‐Castaneda C, Jacobs DM, Marder K, Bell K et al (2006) Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia 47:867–872. [DOI] [PubMed] [Google Scholar]
- 3. Andrew RJ, Kellett KA, Thinakaran G, Hooper NM (2016) A greek tragedy: the growing complexity of Alzheimer amyloid precursor protein proteolysis. J Biol Chem 291:19235–19244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Araújo IM, Gil JM, Carreira BP, Mohapel P, Petersen A, Pinheiro PS et al (2008) Calpain activation is involved in early caspase‐independent neurodegeneration in the hippocampus following status epilepticus. J Neurochem 105:666–676. [DOI] [PubMed] [Google Scholar]
- 5. Avila‐Muñoz E, Arias C (2015) Cholesterol‐induced astrocyte activation is associated with increased amyloid precursor protein expression and processing. Glia 63:2010–2022. [DOI] [PubMed] [Google Scholar]
- 6. Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE et al (2012) Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 74:467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Banati RB, Gehrmann J, Wiessner C, Hossmann KA, Kreutzberg GW (1995) Glial expression of the β‐amyloid precursor protein (APP) in global ischemia. J Cereb Blood Flow Metab 15:647–654. [DOI] [PubMed] [Google Scholar]
- 8. Banerjee M, Sasse A, Wang Y, Maulik M, Kar S (2015) Increased levels and activity of cathepsins B and D in kainate‐induced toxicity. Neuroscience 284:360–373. [DOI] [PubMed] [Google Scholar]
- 9. Beeson JG, Shelton ER, Chan HW, Gage FH (1994) Differential distribution of amyloid protein precursor immunoreactivity in the rat brain studied by using five different antibodies. J Comp Neurol 342:78–96. [DOI] [PubMed] [Google Scholar]
- 10. Ben‐Ari Y, Cossart R (2000) Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci 23:580–587. [DOI] [PubMed] [Google Scholar]
- 11. Bi X, Chang V, Siman R, Tocco G, Baudry M (1996) Regional distribution and time‐course of calpain activation following kainate‐induced seizure activity in adult rat brain. Brain Res 726:98–108. [DOI] [PubMed] [Google Scholar]
- 12. Blümcke I, Coras R, Miyata H, Ozkara C (2012) Defining clinico‐neuropathological subtypes of mesial temporal lobe epilepsy with hippocampal sclerosis. Brain Pathol 22:402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bordji K, Becerril‐Ortega J, Nicole O, Buisson A (2010) Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid‐β production. J Neurosci 30:15927–15942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Born HA (2015) Seizures in Alzheimer's disease. Neuroscience 286:251–263. [DOI] [PubMed] [Google Scholar]
- 15. Boyd‐Kimball D, Sultana R, Mohmmad‐Abdul H, Butterfield DA (2004) Rodent Aβ (1–42) exhibits oxidative stress properties similar to those of human Aβ (1–42): implications for proposed mechanisms of toxicity. J Alzheimers Dis 6:515–525. [DOI] [PubMed] [Google Scholar]
- 16. Chishti MA, Yang DS, Janus C, Strome R, Horne P, Loukides J et al (2001) Early‐onset amyloid deposition in transgenic mice expressing a double mutant form of APP695. J Biol Chem 276:21562–21570. [DOI] [PubMed] [Google Scholar]
- 17. Coulter DA, Steinhäuser C (2015) Role of astrocytes in epilepsy. Cold Spring Harb Perspect Med 5:a022434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crépel V, Mulle C (2015) Physiopathology of kainate receptors in epilepsy. Curr Opin Pharmacol 20:83–88. [DOI] [PubMed] [Google Scholar]
- 19. de Lanerolle NC, Lee TS, Spencer DD (2010) Astrocytes and epilepsy. Neurotherapeutics 7:424–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Del Vecchio RA, Gold LH, Novick SJ, Wong G, Hyde LA (2004) Increased seizure threshold and severity in young transgenic CRND8 mice. Neurosci Lett 367:164–167. [DOI] [PubMed] [Google Scholar]
- 21. De Strooper B, Karran E (2016) The cellular phase of Alzheimer's disease. Cell 164:603–615. [DOI] [PubMed] [Google Scholar]
- 22. Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF et al (2007) Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci 27:10674–10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eid T, Williamson A, Lee TSW, Petroff OA, de Lanerolle NC (2008) Glutamate and astrocytes ‐ key players in human mesial temporal lobe epilepsy?. Epilepsia 49:42–52. [DOI] [PubMed] [Google Scholar]
- 24. Fernandez‐Tome P, Brera B, Arevalo M, de Ceballos ML (2004) β‐amyloid25–35 inhibits glutamate uptake in cultured neurons and astrocytes: modulation of uptake as a survival mechanism. Neurobiol Dis 15:580–589. [DOI] [PubMed] [Google Scholar]
- 25. Gibbons MB, Smeal RM, Takahashi DK, Vargas JR, Wilcox KS (2013) Contributions of astrocytes to epileptogenesis following status epilepticus: opportunities for preventive therapy?. Neurochem Int 63:660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gouras GK, Relkin NR, Sweeney D, Munoz DG, Mackenzie IR, Gandy S (1997) Increased apolipoprotein E epsilon 4 in epilepsy with senile plaques. Ann Neurol 41:402–404. [DOI] [PubMed] [Google Scholar]
- 27. Ha S, Sohn IJ, Kim N, Sim HJ, Cheon KA (2015) Characteristics of brains in autism spectrum disorder: structure, function and connectivity across the lifespan. Exp Neurobiol 24:273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Haass C, Kaether C, Thinakaran G, Sisodia S (2012) Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2:a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hadera MG, Eloqayli H, Jaradat S, Nehlig A, Sonnewald U (2015) Astrocyte‐neuronal interactions in epileptogenesis. J Neurosci Res 93:1157–1164. [DOI] [PubMed] [Google Scholar]
- 30. Harris ME, Wang Y, Pedigo NW, Hensley K, Butterfield DA, Carney JM (2002) Amyloid β peptide (25–35) inhibits Na+‐dependent glutamate uptake in rat hippocampal astrocyte cultures. J Neurochem 67:277–286. [DOI] [PubMed] [Google Scholar]
- 31. Hartlage‐Rubsamen M, Zeitschel U, Apelt J, Gartner U, Franke H, Stahl T et al (2003) Astrocytic expression of the Alzheimer's disease β‐secretase (BACE1) is stimulus‐dependent. Glia 41:169–179. [DOI] [PubMed] [Google Scholar]
- 32. Hutsler JJ, Zhang H (2010) Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res 1309:83–94. [DOI] [PubMed] [Google Scholar]
- 33. Irwin SA, Galvez R, Greenough WT (2000) Dendritic spine structural anomalies in fragile‐X mental retardation syndrome. Cereb Cortex 10:1038–1044. [DOI] [PubMed] [Google Scholar]
- 34. Jiang W, Van Cleemput J, Sheerin AH, Ji SP, Zhang Y, Saucier DM et al (2005) Involvement of extracellular regulated kinase and p38 kinase in hippocampal seizure tolerance. J Neurosci Res 81:581–588. [DOI] [PubMed] [Google Scholar]
- 35. Kar S, Seto D, Doré S, Chabot JG, Quirion R (1997) Systemic administration of kainic acid induces selective time dependent decrease in [125I]insulin‐like growth factor I, [125I]insulin‐like growth factor II and [125I]insulin receptor binding sites in adult rat hippocampal formation. Neuroscience 80:1041–1055. [DOI] [PubMed] [Google Scholar]
- 36. Kabogo D, Rauw G, Amritraj A, Baker G, Kar S (2010) ß‐amyloid‐related peptides potentiate K+‐evoked glutamate release from adult rat hippocampal slices. Neurobiol Aging 31:1164–1172. [DOI] [PubMed] [Google Scholar]
- 37. Kodam A, Vetrivel KS, Thinakaran G, Kar S (2008) Cellular distribution of gamma‐secretase subunit nicastrin in the developing and adult rat brains. Neurobiol Aging 29:724–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kodam A, Maulik M, Peake K, Amritraj A, Vetrivel K, Thinakaran G et al (2010) Altered levels and distribution of APP and its processing enzymes in Niemann‐Pick Type C1‐deficient mouse brains. Glia 58:1267–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kayed R, Lasagna‐Reeves CA (2013) Molecular mechanisms of amyloid oligomers toxicity. J Alzheimers Dis 33: S67–S78. [DOI] [PubMed] [Google Scholar]
- 40. Klyubin I, Wang Q, Reed MN, Irving EA, Upton N, Hofmeister J et al (2011) Protection against Aβ‐mediated rapid disruption of synaptic plasticity and memory by memantine. Neurobiol Aging 32:614–623. [DOI] [PubMed] [Google Scholar]
- 41. Lahiri DK, Sokol DK, Erickson C, Ray B, Ho CY, Maloney B (2013) Autism as early neurodevelopmental disorder: evidence for an sAPPα‐mediated anabolic pathway. Front Cell Neurosci 7:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Larner AJ (2010) Epileptic seizures in AD patients. Neuromol Med 12:71–77. [DOI] [PubMed] [Google Scholar]
- 43. Lesné S, Ali C, Gabriel C, Croci N, MacKenzie ET, Glabe CG et al (2005) NMDA receptor activation inhibits γ‐secretase and promotes neuronal amyloid β production. J Neurosci 25:9367–9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lévesque M, Avoli M (2013) The kainic acid model of temporal lobe epilepsy. Neurosci Biobehav Rev 37:2887–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lévesque M, Avoli M, Bernard C (2016) Animal models of temporal lobe epilepsy following systemic chemoconvulsant administration. J Neurosci Methods 260:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mackenzie IR, Miller LA (1994) Senile plaques in temporal lobe epilepsy. Acta Neuropathol 87:504–510. [DOI] [PubMed] [Google Scholar]
- 47. Majores M, Schoch S, Lie A, Becker AJ (2007) Molecular neuropathology of temporal lobe epilepsy: complementary approaches in animal models and human disease tissue. Epilepsia 48: 4–12. [DOI] [PubMed] [Google Scholar]
- 48. Masters CL, Selkoe DJ (2012) Biochemistry of amyloid β‐protein and amyloid deposits in Alzheimer disease. Cold Spring Harb Perspect Med 2:a006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maulik M, Peake K, Chung J, Wang Y, Vance JE, Kar S (2015) APP overexpression in absence of NPC1 excerbates metabolism of amyloidogenic proteins of Alzheimer's disease. Hum Mol Genetics 24:7132–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mehta A, Prabhakar M, Kumar P, Deshmukh R, Sharma PL (2013) Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur J Pharmacol 698:6–18. [DOI] [PubMed] [Google Scholar]
- 51. Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L et al (2009) Amyloid β‐induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci 29:3453–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mohajeri MH, Saini K, Schultz JG, Wollmer MA, Hock C, Nitsch RM (2002) Passive immunization against β‐amyloid peptide protects central nervous system (CNS) neurons from increased vulnerability associated with an Alzheimer's disease‐causing mutation. J Biol Chem 277:33012–33017. [DOI] [PubMed] [Google Scholar]
- 53. Morimoto K, Oda T (2003) Kainate exacerbates β‐amyloid toxicity in rat hippocampus. Neurosci Lett 340:242–244. [DOI] [PubMed] [Google Scholar]
- 54. Nadler Y, Alexandrovich A, Grigoriadis N, Hartmann T, Rao KS, Shohami E, Stein R (2008) Increased expression of the gamma‐secretase components presenilin‐1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia 56:552–567. [DOI] [PubMed] [Google Scholar]
- 55. Nagele RG, D'Andrea MR, Lee H, Venkataraman V, Wang HY (2003) Astrocytes accumulate Aβ42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res 971:197–209. [DOI] [PubMed] [Google Scholar]
- 56. Nihashi T, Inao S, Kajita Y, Kawai T, Sugimoto T, Niwa M et al (2001) Expression and distribution of beta amyloid precursor protein and beta amyloid peptide in reactive astrocytes after transient middle cerebral artery occlusion. Acta Neurochir (Wien) 143:287–295. [DOI] [PubMed] [Google Scholar]
- 57. Ourdev D, Foroutanpay BV, Wang Y, Kar S (2015) The effect of oligomers Aβ1–42 on APP processing and Aβ1–40 generation in cultured U373 astrocytes. Neurodegener Dis 15:361–368. [DOI] [PubMed] [Google Scholar]
- 58. Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien‐Ly N et al (2007) Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron 55:697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ray B, Long JM, Sokol DK, Lahiri DK (2011) Increased secreted amyloid precursor protein‐a (sAPPα) in severe autism: proposal of a specific, anabolic pathway and putative biomarker. PLoS One 6:e20405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ray B, Sokol DK, Maloney B, Lahiri DK (2016) Finding novel distinctions between the sAPPα‐mediated anabolic biochemical pathways in Autism Spectrum Disorder and Fragile X Syndrome plasma and brain tissue. Sci Rep 6:26052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Revett TJ, Baker GB, Jhamandas J, Kar S (2013) The glutamate system, amyloid β peptides and tau protein: functional interrelationships and relevance to Alzheimer's disease pathology. J Psychiat Neurosci 38:6–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rossner S, Lange‐Dohna C, Zeitschel U, Perez‐Polo JR (2005) Alzheimer's disease β‐secretase BACE1 is not a neuron‐specific enzyme. J Neurochem 92:226–234. [DOI] [PubMed] [Google Scholar]
- 63. Rossner S, Apelt J, Schliebs R, Perez‐Polo JR, Bigl V (2001) Neuronal and glial β‐secretase (BACE) protein expression in transgenic Tg2576 mice with amyloid plaque pathology. J Neurosci Res 64:437–446. [DOI] [PubMed] [Google Scholar]
- 64. Rush T, Buisson A (2014) Reciprocal disruption of neuronal signaling and Aβ production mediated by extrasynaptic NMDA receptors: a downward spiral. Cell Tissue Res 356:279–286. [DOI] [PubMed] [Google Scholar]
- 65. Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR et al (2012) Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc Natl Acad Sci USA 109:E2895–E2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sanz‐Blasco S, Piña‐Crespo JC, Zhang X, McKercher SR, Lipton SA (2016) Levetiracetam inhibits oligomeric Aβ‐induced glutamate release from human astrocytes. Neuroreport 27:705–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Scharfman HE (2012) “Untangling” Alzheimer's disease and epilepsy. Epilepsy Curr 12:178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schmued LC, Stowers CC, Scallet AC, Xu L (2005) Fluoro‐Jade C results in ultrahigh resolution and contrast labeling of degenerating neurons. Brain Res 1035:24–31. [DOI] [PubMed] [Google Scholar]
- 69. Sheng JG, Boop FA, Mrak RE, Griffin WS (1994) Increased neuronal beta‐amyloid precursor protein expression in human temporal lobe epilepsy: association with interleukin‐1 alpha immunoreactivity. J Neurochem 63:1872–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sima X, Xu J, Li J, Zhong W, You C (2014) Expression of β‐amyloid precursor protein in refractory epilepsy. Mol Med Rep 9:1242–1248. [DOI] [PubMed] [Google Scholar]
- 71. Siman R, Salidas S (2004) Gamma‐secretase subunit composition and distribution in the presenilin wild‐type and mutant mouse brain. Neuroscience 129:615–628. [DOI] [PubMed] [Google Scholar]
- 72. Siman R, Card JP, Nelson RB, Davis LG (1989) Expression of β‐amyloid precursor protein in reactive astrocytes following neuronal damage. Neuron 3:275–285. [DOI] [PubMed] [Google Scholar]
- 73. Simpson JE, Ince PG, Lace G, Forster G, Shaw PJ, Matthews F et al (2010) Astrocyte phenotype in relation to Alzheimer‐type pathology in the aging brain. Neurobiol Aging 31:578–590. [DOI] [PubMed] [Google Scholar]
- 74. Sokol DK, Chen D, Farlow MR, Dunn DW, Maloney B, Zimmer JA, Lahiri DK (2006) High levels of Alzheimer beta‐amyloid precursor protein (APP) in children with severely autistic behavior and aggression. J Child Neurol 21:444–449. [DOI] [PubMed] [Google Scholar]
- 75. Song MS, Rauw G, Baker GB, Kar S (2008) Memantine protects rat cortical cultured neurons against β‐amyloid‐induced toxicity by attenuating tau phosphorylation. Eur J Neurosci 28:1989–2002. [DOI] [PubMed] [Google Scholar]
- 76. Sun A, Koelsch G, Tang J, Bing G (2002) Localization of beta‐secretase memapsin 2 in the brain of Alzheimer's patients and normal aged controls. Exp Neurol 175:10–22. [DOI] [PubMed] [Google Scholar]
- 77. Thom M (2014) Hippocampal sclerosis in epilepsy: a neuropathology review. Neuropathol Appl Neurobiol 40:520–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tremblay R, Chakravarthy B, Hewitt K, Tauskela J, Morley P, Atkinson T, Durkin JP (2000) Transient NMDA receptor inactivation provides long‐term protection to cultured cortical neurons from a variety of death signals. J Neurosci 20:7183–7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Vetrivel KS, Kodam A, Gong P, Chen Y, Parent AT, Kar S, Thinakaran G (2008) Localization and regional distribution of p23/TMP21 in the brain. Neurobiol Dis 32:37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Vincent P, Mulle C (2009) Kainate receptors in epilepsy and excitotoxicity. Neuroscience 158:309–323. [DOI] [PubMed] [Google Scholar]
- 81. Wang Y, Han R, Liang ZQ, Wu JC, Zhang XD, Gu ZL, Qin ZH (2008) An autophagic mechanism is involved in apoptotic death of art striatal neurons induced by the non‐N‐methyl‐D‐aspartate receptor agonist kainic acid. Autophagy 4:214–226. [DOI] [PubMed] [Google Scholar]
- 82. Wang Y, Qin ZH (2010) Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 15:1382–1402. [DOI] [PubMed] [Google Scholar]
- 83. Wang Y, Buggia‐Prevot V, Zavorka ME, Bleackley RC, MacDonald RG, Thinakaran G, Kar S (2015) Overexpression of the insulin‐like growth factor‐II receptor increases β‐amyloid production and affects cell viability. Mol Cell Biol 35:2368–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang Q, Yu S, Simonyi A, Sun GY, Sun AY (2005) Kainic acid‐mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol 31:3–15. [DOI] [PubMed] [Google Scholar]
- 85. Waxman EA, Lynch DR (2005) N‐methyl‐D‐aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist 11:37–49. [DOI] [PubMed] [Google Scholar]
- 86. Wei Z, Song MS, MacTavish D, Jhamandas JH, Kar S (2008) Role of calpain and caspase in β‐amyloid‐induced cell death in rat primary septal cultured neurons. Neuropharmacology 54:721–733. [DOI] [PubMed] [Google Scholar]
- 87. Westmark CJ, Westmark PR, Beard AM, Hildebrandt SM, Malter JS (2008) Seizure susceptibility and mortality in mice that overexpress amyloid precursor protein. Int J Clin Exp Pathol 1:157–168. [PMC free article] [PubMed] [Google Scholar]
- 88. Westmark CJ, Sokol DK, Maloney B, Lahiri DK (2016) Novel roles of amyloid‐beta precursor protein metabolites in fragile X syndrome and autism. Mol Psychiatry 21:1333–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. White HS (2002) Animal model of epileptogenesis. Neurology 59:S7–S14. [DOI] [PubMed] [Google Scholar]
- 90. Wilcox KS, Gee JM, Gibbons MB, Tvrdik P, White JA (2015) Altered structure and function of astrocytes following status epilepticus. Epilepsy Behav 49:17–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Willoughby DA, Johnson SA, Pasinetti GM, Tocco G, Najm I, Baudry M, Finch CE (1992) Amyloid precursor protein mRNA encoding the Kunitz protease inhibitor domain is increased by kainic acid‐induced seizures in rat hippocampus. Exp Neurol 118:332–339. [DOI] [PubMed] [Google Scholar]
- 92. Xiao R (2016) Levetiracetam might act as an efficacious drug to attenuate cognitive deficits of Alzheimer's disease. Curr Top Med Chem 16:565–573. [DOI] [PubMed] [Google Scholar]
- 93. Yamagata Y, Nairn AC (2015) Contrasting features of ERK1/2 activity and synapsin I phosphorylation at the ERK1/2‐dependent site in the rat brain in status epilepticus induced by kainic acid in vivo. Brain Res 1625:314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yang H, Wang Y, Kar S (2017) U18666A‐induced cholesterol sequestration and APP metabolism in rat primary astrocytes. Glia 65:1728–1743. [DOI] [PubMed] [Google Scholar]
- 95. Zhang MY, Zheng CY, Zou MM, Zhu JW, Zhang Y, Wang J et al (2014) Lamotrigine attenuates deficits in synaptic plasticity and accumulation of amyloid plaques in APP/PS1 transgenic mice. Neurobiol Aging 35:2713–2725. [DOI] [PubMed] [Google Scholar]
- 96. Zheng WH, Bastianetto S, Mennicken F, Ma W, Kar S (2002) Amyloid β peptide induces tau phosphorylation and neuronal degeneration in rat primary septal cultured neurons. Neuroscience 115:201–211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. A–L. Double immunofluorescence photomicrographs showing the localization of APP (A‐D), BACE1 (E‐H) and PS1 (I‐L) with astrocyte marker GFAP and activated microglia marker ED1 in the hippocampal CA1 region of saline‐treated control rats. Note the localization of APP, BACE1 and PS1 in neurons, GFAP in astrocytes but the absence of ED1 labeling due to lack of activated microglia in control brains. Scale bar, 100 µM.
Figure S2. A–F. Photomicrographs depicting cellular distribution of nicastrin in the hippocampus of saline‐treated control (A, B) and following 12 h (C, D) and 12 day (E, F) kainic acid‐treated rats at lower (A, C, E) and higher (B, D, F) magnifications. Note the expression of nicastrin in pyramidal and granule cell layers in control rats, whereas after 12 day kainic acid treatment nicastrin is evident mostly in glial cells of the hippocampus. G–J. Double immunofluorescence photomicrographs showing a subset of GFAP‐labeled astrocytes (G, H), but not ED1‐labeled microglia (I, J), exhibit immunoreactive nicastrin in 12 day kainic acid‐treated rats. Py, pyramidal cell layer; DG, dentate gyrus. Scale bar, 100 µM.
Figure S3. A–F. Photomicrographs depicting cellular distribution of PEN2 in the hippocampus of saline‐treated control (A, B) and following 12 h (C, D) and 12 day (E, F) kainic acid‐treated rats at lower (A, C, E) and higher (B, D, F) magnifications. Note the expression of PEN2 in pyramidal and granule cell layers in control rats, whereas after 12day kainic acid treatment PEN2 is evident mostly in glial cells of the hippocampus. G–J. Double immunofluorescence photomicrographs showing a subset of GFAP‐labeled astrocytes (G, H), but not ED1‐labeled microglia (I, J), exhibit immunoreactive PEN2 in 12day kainic acid‐treated rats. Py, pyramidal cell layer; DG, dentate gyrus. Scale bar, 100 µM.
Figure S4. A–F. Photomicrographs depicting cellular distribution of APH1 in the hippocampus of saline‐treated control (A, B) and following 12hr (C, D) and 12day (E, F) kainic acid‐treated rats at lower (A, C, E) and higher (B, D, F) magnifications. Note the expression of APH1 in pyramidal and granule cell layers in control rats, whereas after 12 day kainic acid treatment APH1 is evident mostly in glial cells of the hippocampus. G–J. Double immunofluorescence photomicrographs showing a subset of GFAP‐labeled astrocytes (G, H), but not ED1‐labeled microglia (I, J), exhibit immunoreactive APH1 in 12 day kainic acid‐treated rats. Py, pyramidal cell layer; DG, dentate gyrus. Scale bar, 100 µM.
Figure S5. A–L. Double immunofluorescence photomicrographs showing the localization of nicastrin (A–D), PEN2 (E–H) and APH1 (I–L) with astrocyte marker GFAP and activated microglia marker ED1 in the hippocampal CA1 region of saline‐treated control rats. Note the localization of nicastrin, PEN2 and APH1 in neurons, GFAP in astrocytes but the absence of ED1 labeling due to lack of activated microglia in control brains. Scale bar, 100 µM.
Figure S6. A–C. Histograms showing the dose‐dependent effects of 24 h exposure to different concentrations of human Aβ1‐42 (A) and dose‐ as well as time‐dependent effects of rat Aβ1‐42 (B, C) on viability of hippocampal cultured neurons as revealed by MTT assay. Control cultures were treated with 10 µM human or rat Aβ42‐1 peptide. Note that human Aβ1‐42 was found to be more toxic than equimolar concentration of rat Aβ1‐42 on cultured neurons. D–G. Images from the live/dead assay depicting cell viability in control (D, E) and 20µM rat Aβ1‐42‐treated (F, G) hippocampal cultured neurons. Calcein AM staining (green) represents intracellular esterase activity in living neurons, while EthD‐1 (red) shows dead neurons with disintegrated plasma membrane. Note the toxic potency of rat Aβ1‐42 compared to control cultures treated with rat Aβ42‐1. MTT data presented as % of control (means ± SEM) were obtained from 3–5 separate experiments, each performed in triplicate. Cont, control. * P < 0.05 and ** P < 0.001 compared to control value. Scale bar, 50 µm.