

Abstract
Null mutations in progranulin gene (GRN) reduce the progranulin production resulting in haploinsufficiency and are tightly associated with tau‐negative frontotemporal lobar degeneration with TAR DNA‐binding protein 43‐positive inclusions (FTLD‐TDP). Missense mutations of GRN were also identified, but their effects are not completely clear, in particular unanswered is the question of what neuropathology they elicit, also considering that their occurrence has been reported in patients with typical clinical features of Alzheimer disease. They describe two fraternal twins carrying the missense GRN Cys139Arg mutation affected by late‐onset dementia and we report the neuropathological study of one of them. Both patients were examined by neuroimaging, neuropsychological assessment and genetic analysis of GRN and other genes associated with dementia. The brain of one was obtained at autopsy and examined neuropathologically. One sister presented clinical and MRI features leading to the diagnosis of Alzheimer disease. The other underwent autopsy and the brain showed neuropathological hallmarks of Alzheimer disease with abundant Aβ‐amyloid deposition and Braak stage V of neurofibrillary pathology, in the absence of the hallmark lesions of FTLD‐TDP. Their findings may contribute to better clarify the role of progranulin in neurodegenerative diseases indicating that some GRN mutations, in particular missense ones, may act as strong risk factor for Alzheimer disease rather than induce FTLD‐TDP.
Keywords: Progranulin, point mutation, Alzheimer disease, Frontotemporal lobar degeneration, genetics, neuropathology
Introduction
Frontotemporal lobar degeneration (FTLD) describes a group of neurodegenerative disorders characterized by predominant involvement of frontal and temporal lobes and by a broad range of clinical manifestations that can be subdivided in two main presentations: one with prominent behavioral changes and the other with language disturbances 9.
FTLD presents in sporadic or familial forms associated with genetic defects of three main genes: MAPT, GRN and C9ORF72 15. GRN is responsible of up to 22% of genetic cases, highly heterogeneous in terms of age of onset, disease duration and clinical presentations even among individuals carrying the same mutation 13.
About 150 GRN mutations have been reported. Most are non‐sense, frameshift or splicing null mutations giving rise to haploinsufficiency. They are associated with the neuropathological hallmarks consisting in neuronal cytoplasmic and lentiform intranuclear inclusions immunoreactive for ubiquitin and TAR DNA‐binding protein 43 (TDP‐43) and are indicated as FTLD‐TDP 15.
Although less common, missense GRN mutations were also found, whose pathogenicity has been demonstrated only in a limited number of cases. Beyond the mutations at the initial Methionine or in the leader peptide, that also induce premature mRNA degradation 2, 14, a few other mutations have been showed to interfere with normal structure and functions of progranulin (PGRN) by in vitro, in silico and in cell models. In particular, Cys139Arg has been demonstrated to derange PGRN folding by destroying one of the Cysteine‐bridges, to impair cleavage by elastase and to reduce neurites growth‐stimulating activity 5, 17. Also considering that a significant percentage of patients harboring missense GRN mutations exhibit clinical and neuroimaging features of AD, it has been suggested that they might rather act as more generic susceptibility factors for neurodegeneration because of their milder effect on PGRN structure and function compared with null mutations 5, but the issue is unresolved for the lack of neuropathological studies of these patients.
We here report two fraternal twins carrying GRN Cys139Arg mutation. In one of them, clinical and neuroimaging features were evocative of AD. In the other, fully expressed Alzheimer neuropathological changes in the absence of significant hallmarks of FTLD‐TDP were present.
Material and Methods
The family was studied following an outpatient evaluation of the proband for late‐onset dementia soon followed by her twin presenting similar symptoms. None of the eight other siblings presented dementia. The twins examined by us were the two youngest siblings. The eight other siblings were all dead and for six of them the age of death was below 65 years (Figure 1). Their parents had unrevealing clinical history.
Figure 1.
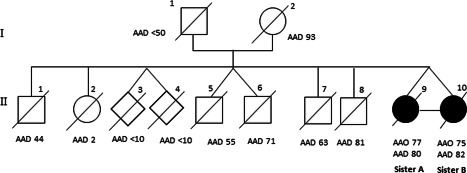
Pedigree of the family. Affected individuals are represented in black; diamonds represent unknown gender; diagonal slash denotes deceased individuals; AAO, age at onset; AAD, age at death.
After informed consent, the two twins underwent analysis of genomic DNA from peripheral lymphocytes. GRN (exons 0–12 and exon/intron boundaries), MAPT (exons 1–5, 7, 9–13 and intron 10), APP (exons 16, 17), PSEN1 (exons 3–12) and PSEN2 (exons 3–12) genes were amplified and subjected to sequencing; the region containing the hexanucleotide GGGGCC in the first intron of C9ORF72 gene was analyzed. APOE genotyping was performed.
Plasma progranulin dosage was only performed in sister B by an ELISA kit (Human Progranulin ELISA kit, Adipogen Inc., Seoul, Korea).
The brain of sister B was obtained at autopsy and fixed in formalin.
Coronal slices of the cerebral hemispheres were taken at several levels. Sections were stained by hematoxylin & eosin (H&E) and cresyl violet for Nissl substance, and immunostained with antibodies anti Aβ (monoclonal, 4G8, Signet, 1:2000), antiphosphorylated tau protein (AT8, monoclonal 1:300, Innogenetics, Belgium), anti‐TDP‐43 (monoclonal, 1:2000, Abnova; polyclonal, 1:100, ProteinTech Group), antiphosphorylated TDP‐43 (monoclonal, clone 1D3, 1:500, Millipore; monoclonal, pS409/410, 1:2000, Cosmo Bio), anti‐ubiquitin (1:500, DakoCytomation), anti p62 (monoclonal, 1:1000, Transduction Lab).
Results
Sister A (II‐9) presented at age 77 with memory decline and impairment of instrumental activities followed by spatial and temporal disorientation. At a global neuropsychological evaluation using the Milan Overall Dementia Assessment (MODA) 4 she showed temporal disorientation, deficit of memory, verbal intelligence, visuospatial abilities and verbal production (65.2/100 corrected by age and education, normal > 89/100). Cerebral MRI revealed cortical atrophy of temporal and frontal lobes (Figure 2A–C). Genetic analyses revealed a TGC to CGC mutation at codon 139 of GRN, leading to the Cys139Arg missense mutation. All the other genetic analyses were negative. APOE genotype was ε3/ε3. She died 5 years after disease onset and autopsy was not performed.
Figure 2.
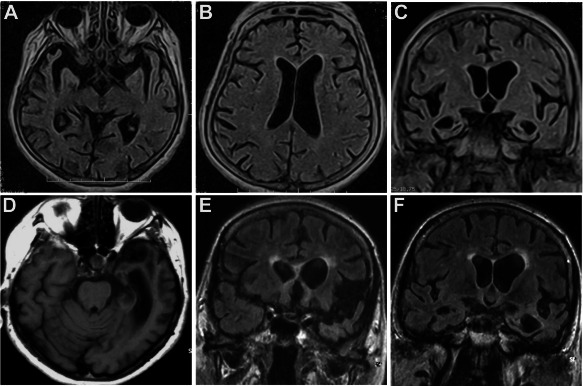
MRI findings. Sister A. FLAIR‐weighted axial and coronal sections show bilateral enlargement of sylvian fissures and temporal horns with mild asymmetrical enlargement of left lateral ventricle (A–C). Sister B. T1‐weighted axial image and FLAIR‐weighted coronal images show left temporal atrophy with asymmetrical (more severe on the left side) enlargement of temporal horn and temporal cortical sulci, sylvian fissure and frontal horn (D–F).
Sister B (II‐10) presented at age 75 with memory deficits and impairment in instrumental activities and language, followed by dyscalculia, spatial and temporal disorientation, prosopoagnosia. At the MODA, deficits of memory, verbal intelligence, visuo‐spatial abilities and verbal production emerged (81.8/100 corrected by age and education, normal > 89/100). Cerebral MRI revealed asymmetric cortical atrophy of frontal and temporal lobes, predominantly on the left side (Figure 2D–F). Genetic analyses demonstrated the GRN Cys139Arg mutation, without mutations in the other genes analyzed. APOE genotype was ε3/ε3. Plasma progranulin dosage (96 ng/mL) was intermediate between the values found in wild type control subjects and in null GRN mutation carriers. She died from pulmonary thromboembolism 5 years after disease onset and underwent autopsy.
Gross examination of the brain showed moderate brain atrophy of frontal, temporal and parietal lobes and lateral ventricular enlargement, more evident on the left side.
Microscopic examination revealed severe neuronal loss and gliosis throughout the cerebral cortex that presented the pathologic hallmarks of AD: Aβ‐amyloid deposits were abundant in all cortical lobes, in the hippocampus and in the striatum, while they were absent in the cerebellum and brainstem. Neurofibrillary degeneration in the form of NFT in the perikarya, neuropil threads dispersed in the cortex, and degenerating neurites surrounding Aβ‐amyloid deposits involved the mesial temporal structures and all neocortical areas examined with sparing of the primary motor and sensory fields and of calcarine cortex (stage V by Braak) (Figure 3A–C) 3.
Figure 3.
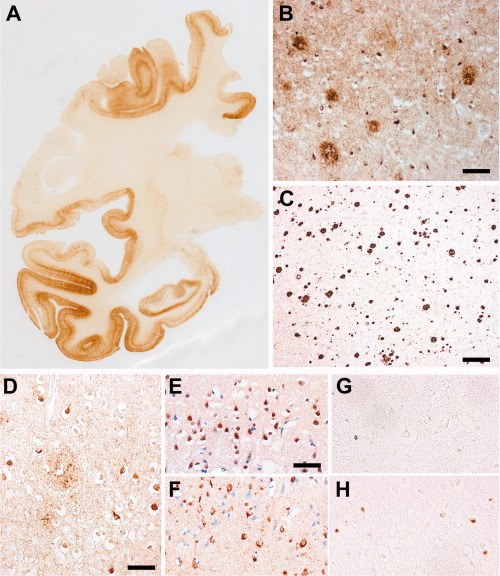
Neuropathological findings: sister B. Phospho‐tau positive neurofibrillary changes (AT8 immunohistochemistry, brown reaction product) were abundant in both cerebral hemispheres, in all the cortical fields with partial sparing of the primary motor and sensory areas (Braak stage V) (A, right hemisphere), taking the form of NFT, abnormal neurites of senile plaques and neuropil threads (B). Aβ deposits (4G8 immunohistochemistry) were numerous and widespread in the cerebral cortex (frontal cortex, C). Anti‐ubiquitin antibody immunolabeled many tangles and abnormal neurites of senile plaques, and occasionally thin thread‐like profiles (D). In our present case, the polyclonal anti‐TDP‐43 immunolabeled the nuclei of several neuronal cells, in particular in the upper cortical layers, representing the physiological nuclear staining of neurons. Abnormal neuronal or cytoplasmic inclusions were not detected (E), while in a control FTLD‐TDP case the polyclonal anti‐TDP‐43 immunolabeled the nuclei of different cells (physiological), but, in addition, abundant neuronal cytoplasmic structures (in neurons where the physiological nuclear staining is lacking) and thin threads mostly in the upper cortical layers (F). Anti‐phospho‐TDP‐43 antibody did not show pathologic immunoreactive inclusions in our present case (G), that were present in the positive control case of FTLD‐TDP (H). Bar in B = 100 μm; bar in C = 220 μm; bar in D = 40 μm; bar in E = 30 μm: E,F,G,H have the same magnification.
Anti‐ubiquitin and anti‐p62 antibodies immunolabeled NFT and dystrophic neurites of senile plaques but did not reveal neuronal compact oval or crescentic cytoplasmic inclusions nor neuronal intranuclear inclusions or scattered short dystrophic neurites. Accordingly, immunostaining for TDP‐43 was physiologically limited to nuclei and very occasionally to tangle structures, while immunostaining for phosphorylated TDP‐43 was absent (Figure 3D–H).
Discussion
While the null GRN mutations are closely associated with the typical clinico‐pathologic picture of FTLD‐TDP, the patients with missense GRN mutations presented with heterogeneous clinical phenotypes and very few data are available about their neuropathological features. In a large population‐based study, eight patients carrying GRN missense mutations, including one with Cys139Arg, were clinically diagnosed as late‐onset, probable AD; however, for none of the patients could the diagnosis be verified pathologically 5. In more recent studies, several patients with GRN Cys139Arg showed different clinical pictures: behavioral variant of FTD, semantic dementia, corticobasal syndrome, progressive non fluent‐aphasia 1, 10, again without neuropathological data available.
We report two fraternal twins carrying the GRN Cys139Arg mutation. In one (sister A), the clinical and neuroimaging features warranted the diagnosis of probable AD. In the other (sister B), the neuropathological study showed fully expressed AD pathology without FTLD‐TDP inclusions, representing an unprecedented association between GRN mutation, presence of fully expressed AD neuropathology and absence of FTLD‐TDP hallmark lesions. It is noteworthy that our patients APOE genotype was ε3/ε3 therefore ApoE4 risk factor for sporadic AD was absent in both of them.
The only cases of missense GRN mutations with neuropathological analysis were Ala324Thr 7 and Ser120Tyr 12, which displayed CBD and ALS‐FTD, suggesting the absence of a tight link between missense GRN mutations and FTLD‐TDP. However, these mutations might be benign polimorphisms, because their presence also in controls 8, 11. On the contrary, Cys139Arg is unlikely to be a polymorphism because of its ability to impair elastase cleavage 17, to reduce PGRN plasma levels 7, 10 and its absence in control populations of more than 1000 subjects 1, 10.
Thus, in this complex scenario, our cases are in compliance with the hypothesis that missense GRN mutations may induce or act as strong risk factor for neurodegenerative diseases other than FTLD‐TDP, in particular for AD. At present there is no clear explanation regarding the way on which modifications in progranulin may promote AD. However, the reduction of its physiological role in neuron protection and growth may be a factor favoring AD.
Although GRN was not found as a risk factor in GWAS studies on AD patients 6, changes of this ubiquitous, inflammatory protein and growth factor could give rise to different types of neurodegenerative processes, depending on the type of genetic defects. It is noteworthy in this regard that homozygous GRN mutation has been reported in neuroceroido‐lipofuscinosis 16.
Further studies correlating GRN genetic variations with the neuropathological phenotype will be crucial to clarify the role of PGRN and other modulator proteins in turning on FTLD‐TDP or AD or other neurodegenerative processes.
Acknowledgments
The study was funded by the Italian Ministry of Health.
References
- 1. Antonell A, Gil S, Sánchez‐Valle R, Balasa M, Bosch B, Prat MC et al (2012) Serum progranulin levels in patients with frontotemporal lobar degeneration and Alzheimer's disease: detection of GRN mutations in a Spanish cohort. J Alzheimers Dis 31:581–591. [DOI] [PubMed] [Google Scholar]
- 2. Baker M, Mackenzie IR, Pickering‐Brown SM, Gass J, Rademakers R, Lindholm C et al (2006) Mutations in progranulin cause tau‐negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919. [DOI] [PubMed] [Google Scholar]
- 3. Braak H, Braak E (1991) Neuropathological staging of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 4. Brazzelli M, Capitani E, Della Sala S, Spinnler H, Zuffi M (1994) A neuropsychological instrument adding to the description of patients with suspected cortical dementia: the Milan overall dementia assessment. J Neurol Neurosurg Psychiatry 57:1510–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brouwers N, Sleegers K, Engelborghs S, Maurer‐Stroh S, Gijselinck I, van der Zee J et al (2008) Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology 71:656–664. [DOI] [PubMed] [Google Scholar]
- 6. Van Cauwenberghe C, Van Broeckhoven C, Sleegers K (2016) The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med 18:421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Finch N, Baker M, Crook R, Swanson K, Kuntz K, Surtees R et al (2009) Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain 132:583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guerreiro RJ, Washecka N, Hardy J, Singleton A (2010) A thorough assessment of benign genetic variability in GRN and MAPT. Hum Mutat 31:E1126–E1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ, Work Group on Frontotemporal Dementia and Pick's Disease (2001) Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick's Disease. Arch Neurol 58:1803–1809. [DOI] [PubMed] [Google Scholar]
- 10. Piaceri I, Pradella S, Cupidi C, Nannucci S, Polito C, Bagnoli S et al (2014) Association of the variant Cys139Arg at GRN gene to the clinical spectrum of frontotemporal lobar degeneration. J Alzheimers Dis 40:679–685. [DOI] [PubMed] [Google Scholar]
- 11. Sassi C, Guerreiro R, Gibbs R, Ding J, Lupton MK, Troakes C et al (2014) Investigating the role of rare coding variability in Mendelian dementia genes (APP, PSEN1, PSEN2, GRN, MAPT, and PRNP) in late‐onset Alzheimer's disease. Neurobiol Aging 35:2881.e1–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schymick JC, Yang Y, Andersen PM, Vonsattel JP, Greenway M, Momeni P et al (2007) Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis‐frontotemporal dementia phenotypes. J Neurol Neurosurg Psychiatry 78:754–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seelaar H, Rohrer JD, Pijnenburg YA, Fox NC, van Swieten JC (2011) Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J Neurol Neurosurg Psychiatry 82:476–486. [DOI] [PubMed] [Google Scholar]
- 14. Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B, Haass C (2008) Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin‐immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem 283:1744–1753. [DOI] [PubMed] [Google Scholar]
- 15. Sieben A, Van Langenhove T, Engelborghs S, Martin JJ, Boon P, Cras P et al (2012) The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol 124:353–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M et al (2012) Strikingly different clinicopathological phenotypes determined by progranulin‐mutation dosage. Am J Hum Genet 90:1102–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang J, Van Damme P, Cruchaga C, Gitcho MA, Vidal JM, Seijo‐Martínez M et al (2010) Pathogenic cysteine mutations affect progranulin function and production of mature granulins. J Neurochem 112:1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]