

Abstract
We aimed to test the hypothesis that in spontaneously hypertensive stroke‐prone rats (SHRSP), non‐amyloid cerebral small vessel disease/hypertensive arteriopathy (HA) results in vessel wall injury that may promote cerebral amyloid angiopathy (CAA). Our study comprised 21 male SHRSP (age 17–44 weeks) and 10 age‐ and sex‐matched Wistar control rats, that underwent two‐photon (2PM) imaging of the arterioles in the parietal cortex using Methoxy‐X04, Dextran and cerebral blood flow (CBF) measurements. Our data suggest that HA in SHRSP progresses in a temporal and age‐dependent manner, starting from small vessel wall damage (stage 1A), proceeding to CBF reduction (stage 1B), non‐occlusive (stage 2), and finally, occlusive thrombi (stage 3). Wistar animals also demonstrated small vessel wall damage, but were free of any of the later HA stages. Nearly half of all SHRSP additionally displayed vascular Methoxy‐X04 positivity indicative of cortical CAA. Vascular β‐amyloid deposits were found in small vessels characterized by thrombotic occlusions (stage 2 or 3). Post‐mortem analysis of the rat brains confirmed the findings derived from intravital 2PM microscopy. Our data thus overall suggest that advanced HA may play a role in CAA development with the two small vessel disease entities might be related to the same pathological spectrum of the aging brain.
Keywords: cerebral amyloid angiopathy, cerebral small vessel disease, hypertensive arteriopathy, intravital imaging, spontaneously hypertensive stroke‐prone rat
Introduction
Sporadic cerebral small vessel disease (CSVD) is frequently found in the aging brain and represents one of the major causes of stroke and (vascular) dementia 13, 29, 54, 88. The term CSVD thereby stands for a mixture of clinical, cognitive, neuroimaging, and neuropathological findings arising from the integrity loss of the cerebral small vasculature 29, 60, 88. There are two major forms of CSVD comprising non‐amyloid CSVD (also called hypertensive arteriopathy, HA) and cerebral amyloid angiopathy (CAA) 13, 29, 54. HA mainly develops as a result of an early endothelial failure, basement membrane changes, and associated blood brain barrier (BBB) breakdown and later stages progress to overall small vessel wall damage 6, 13, 28, 80, 85, 88. In CAA, however, tunica media destruction caused by β‐amyloid (Aβ) accumulation precedes the later disease stages characterized by endothelial and adventitial damage 6, 7, 19.
There is evidence from human post‐mortem and imaging studies that HA and CAA occur together in the aging brain 1, 20, 39, 63, 69. This raises the question whether this observation is simply explained by the common co‐occurrence of the two CSVD entities, eg, due to shared (risk) factors such as aging and apolipoprotein (APO) Eε4 genotype 29, 82, 88 or if there is a more causal relationship.
The consideration of a causal relationship is based on experimental data demonstrating the accumulation of β‐amyloid in the small vessel walls due to a failure of intramural periarterial protein drainage and endothelial transport mechanisms 31, 91, 97. Both require small vessel wall integrity, in particular that of endothelial and basement membranes. HA‐related small vessel wall damage may therefore be susceptible to the deposition of β‐amyloid. Investigating this hypothesis, however, demands for the definition of suitable animal models revealing (pure) HA without developing Aβ deposits as a result of, eg, transgenic modifications.
The spontaneously hypertensive stroke‐prone rat (SHRSP) develops malignant hypertension and displays the broad spectrum of HA‐related pathologies comprising, eg, BBB breakdown 9, 10, 33, 44, 66. That rodent model additionally shows parenchymal and (peri)vascular amyloid depositions, albeit no transgenic modifications of any genes involved in the Aβ turnover have been conducted 11, 33, 61, 68. One may thus argue, that in SHRSP HA‐related small vessel wall pathology itself could play a leading (causal) role when considering the etiology of vascular amyloid accumulations.
In the present study conducted in a set of male SHRSP and age‐ and sex‐matched Wistar control rats, we intended to investigate the hypothesis that HA represents a risk factor for the development of CAA. We firstly aimed to define temporal stages of HA progression based on the findings from intravital 2‐photon microscopy (2PM) small vessel imaging, comprising cerebral blood flow (CBF) measurements. We then examined whether HA progression relates to CAA development.
Materials and Methods
Animals
All experiments were performed in compliance with the German regulations on the welfare of animals used for experiments or for other scientific purposes (Tierschutz‐Versuchstierverordnung, August 2013). Animal procedures were conducted after obtaining the approval of the local Animal Care Committee of Saxony‐Anhalt (reference number of license for animal testing 42502‐2‐1148 DZNE) and in compliance with the ARRIVE guidelines for animal experiments. N = 26 male SHRSP (Charles River Laboratories International Inc., Wilmington, MA, USA) aged from 17 to 44 weeks and n = 15 male Wistar rats (Charles River Laboratories, Research Models and Services, Germany GmbH, Sulzfeld, GER) aged from 17 to 44 weeks were used in the study. All animals were housed under standard conditions including free access to food and water and a natural light–dark cycle. To record the health status of the animals, their neurological function (such as decreased spontaneous activity, coordination failure, falling to one side and hunched posture) was assessed daily and body weight was monitored weekly.
Two‐photon imaging
Before surgery, Pentobarbital (1 mL per 100 g body weight) was injected intraperitoneally. During surgery the body temperature was controlled and kept constant at 37°C with a heating pad. A 4 × 6 mm2 sized craniotomy was performed over the parietal cortex, and the dura was then removed carefully. The cranial window was located between −2 mm and −8 mm relative to Bregma and between 1 mm and 5 mm lateral from the sagittal suture. After surgery, the cranial window was filled with sterile irrigation and sealed by a 7‐mm cover glass. Out of n = 26 SHRSP n = 5 (19%) and out of n = 15 Wistar rats n = 2 (13%) animals died during surgery (eg, from subdural or intracranial bleeding). Thus, n = 21 SHRSP and n = 13 Wistar rats remained for intravital 2PM conducted using a LSM 7 MP microscope (Carl Zeiss, Oberkochen, GER) with the animals’ heads fixed in a head holder to ensure a stable position during the imaging sessions. N = 3 Wistar rats aged 44 weeks, however, did not fit into the head holder due to too large head sizes, leaving n = 10 Wistar rats aged from 17 to 35 weeks for final analysis using 2PM.
First, in each out of n = 8 SHRSP (aged 18 weeks (w)—[n = 2], 28–30 w—[n = 3], 32–33 w—[n = 3]) and n = 5 Wistar rats (aged 18 w—[n = 2], 32 w—[n = 3]) red blood cell (RBC) flow velocity was determined in 9 parietal arterioles on average, and arteriolar luminal diameter ranged from 10 to 60 μm. RBC flow velocity was assessed using the line scan technique recently described by Kleinfeld and colleagues 40.
Dextran (Dextran, Tetramethylrhodamine, 70 kDa, 10 mg/mL, Life Technologies, Darmstadt, GER) was injected intraorbitally to mark the blood plasma. Since this dye does not bind to the RBCs, they appear as dark moving shadows within the Dextran‐enhanced blood plasma.
Using a Radon transform algorithm (Radon function in the MATLAB Image Processing Toolbox) 24 evaluated in MATLAB R2013a (Mathworks, Natick, MA) RBC flow velocity [in mm/s] was then determined from space time images (line scans) generated from repeated scanning along the central vessel axis. Starting from the upper part of the image for each vessel those line scans were created along the y‐time‐axis, while the respective vessel was scanned for 10.000 msec. Line scans thus indicate the non‐fluorescent RBCs’ time‐dependent movement from the upper left to the lower right corner of the image with the x‐axis displaying the spatial position of the red blood cells (Figure 1). The movement of RBCs observed as the change in the RBCs’ spatial position ∆x during a certain time period ∆t results in dark streaks whose slopes (∆t/∆x) are inversely related to the RBC flow velocity (∆x/∆t) 21, 40, 65. Arterioles without pathological alterations display line scans characterized by a large number of nearly non‐sloping streaks indicative of plenty of RBCs moving unhampered from the upper left to the lower right corner of the image (Figure 1A). In this case, ∆x exceeds ∆t resulting in high values for RBC flow velocity. Conversely, line scans of arterioles affected by pathological alterations such as non‐occlusive erythrocyte thrombi or luminal Dextran accumulations exhibit a smaller number of streaks characterized by steep slopes. The latter indicate slowly to nearly not moving RBCs resulting in a reduced number of red blood cells passing the scanned vessel segment during the same course of time (Figure 1B–D). While time is passing by, there is limited change of the RBCs’ spatial position resulting in low RBC flow velocity values.
Figure 1.
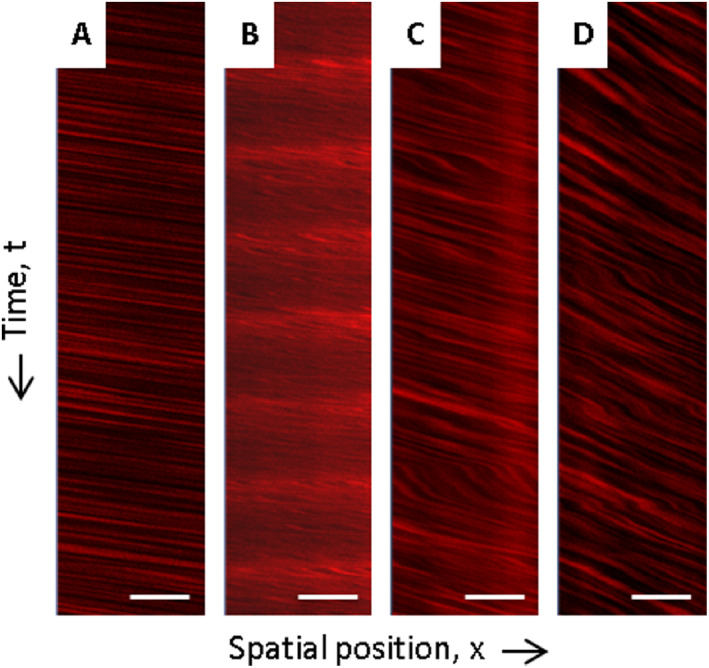
Intravital RBC flow velocity measures using 2‐photon microscopy. A shows an exemplary line scan of an arteriole without any HA phenomena displaying unhampered RBC flow velocity. For comparison, B—D demonstrate line scans of arterioles characterized by RBC flow velocity disturbances. B thereby shows a line scan of an exemplary arteriole with luminal Dextran accumulations and C and D display line scans of vessels filled with non‐occlusive erythrocyte thrombi. HA, hypertensive arteriopathy; RBC, red blood cell. Scale bars = 20 μm; x‐axis—spatial position of RBCs; y‐axis—time t (10.000 ms from top to bottom of the line scan).
The remaining n = 13 SHRSP (aged 17 w—[n = 1], 28 w—[n = 2], 32–33 w—[n = 7], 39 w—[n = 1], 44 w—[n = 2]) underwent Aβ imaging of the parietal cortex with an imaging depth up to 300 μm. For this purpose, the fluorescent Congo red derivate Methoxy‐X04 23, 42 (2 mg/mL, TOCRIS bioscience, Bristol, UK) together with Dextran (70 kDa, 10 mg/mL) were injected intraorbitally allowing for the detection of vascular amyloid as well as the integrity and structure of the microvasculature. Methoxy‐X04 was injected 80 minutes prior to imaging, and Dextran was applied immediately before 2PM took place. After crossing the BBB, Methoxy‐X04 binds up to 72 h after injection to fibrillary Aβ including CAA 4, 23, 41, 64. It is, however, cleared within seconds from brain tissue that does not contain fibrillary amyloid 42. For Wistar rats, however, no Methoxy imaging was performed as our previous studies showed that age‐matched Wistar rats do not exhibit any Aβ accumulation 11, 33, 68.
Tissue preparation and histology
After imaging, all n = 21 SHRSP and n = 10 Wistar rats were transcardially perfused using 120 mL phosphate‐buffered saline (PBS) followed by fixation through 120 mL paraformaldehyde (PFA, 4%) within 8 minutes. Brains were removed, fixed in 4% PFA for 48 h, cryoprotected in 30% sucrose for 6 days and frozen in methylbutane at −80°C. Using a cryostat the brains were then sliced and coronal slices were taken from 11 planes comprising all brain regions extending from the frontal to the occipital pole.
The surgical area was located between the fifth and the ninth sectional plane and covered an area of around 4 × 6 mm2 of the parietal cortex. For all histological analysis (and immunohistochemical analysis, see below) the researchers (SJ, CG) were blind with respect to group and age of the animals. For each animal, three slices (30 μm slice thickness) per plane within the surgical area were stained with hematoxylin and eosin (HE, used for the detection of HA features that is non‐occlusive erythrocyte thrombi and occlusive erythrocyte thrombi/totally obstructed vessel segments). For the n = 13 SHRSP that were used for Methoxy imaging one slice per plane within the surgical area was stained with Congo red (CR)/Prussian blue and Thioflavin T/Prussian blue (used for the detection of (peri)vascular Aβ accumulations). The brains of Wistar rats were not stained with CR or Thioflavin T since they were not used for intravital Methoxy imaging (see above).
Immunohistochemistry
In SHRSP, besides conventional histological β‐amyloid detection, (peri)vascular Aβ was assessed immunohistochemically in n = 5 brain slices within the surgical area per rat; this was undertaken for all n = 13 animals that underwent in vivo Methoxy‐X04 imaging. Moreover, n = 5 brain slices of n = 12 SHRSP were additionally stained with Aβ and IgG to investigate whether vascular β‐amyloid accumulations relate to BBB breakdown. Wall‐adherent and perivascular IgG depositions thereby indicate small vessel wall/BBB damage 51. In short, tissue was pretreated with citrate buffer (70°C, 30 minutes), slices were repeatedly washed in PBS and blocked with 10% donkey serum. Subsequently, slices were stained with STL‐FITC (solanum tuberosum lectin‐fluorescein isothiocyanate, endothelial marker, 1:500; Axxora, Enzo Life Sciences GmbH, Lörrach, GER) and anti‐rodent Aβ (1:500; Covance, Dedham, MA, USA; specific for rodent Aβ) overnight at 4°C. Moreover, in all n = 21 SHRSP in vivo applied Dextran (Dextran, Tetramethylrhodamine, 70 kDa; Life Technologies, Darmstadt, GER) was visualized post‐mortem by its Tetramethylrhodamine labeling without any further antibody. The co‐localization between IgG and wall‐adherent and perivascular Dextran indicate small vessel wall damage and BBB breakdown 53. For IgG detection, slices were then incubated with Cy3‐donkey anti rat IgG (1:200, Jackson Immuno Research, West Grove, PA, USA) and Cy5‐donkey anti rabbit IgG (1:500, detection of Aβ; Jackson Immuno Research) for 2 h at room temperature. Finally, DAPI (4′.6‐diamidino‐2‐phenylindol, nuclear staining, 1:10.000; MoBiTec GmbH, Göttingen, GER) staining was performed for 20 minutes at room temperature. After dehydration with increasing concentrations of alcohol, slices were mounted on slides with Histomount (Fisher Scientific GmbH, Schwerte, GER).
Data analysis and quantification
2PM data analysis
For analysis, 2PM data were recorded using the ZEN 2010 software (Carl Zeiss Microscopy GmbH, Jena, Germany) and saved as lsm‐files for later data evaluation.
HA phenomena, HA cascade, and HA staging
Lsm‐files of all n = 21 SHRSP and n = 10 Wistar rats were then used to quantify the presence of distinct HA phenomena, that are arteriolar (luminal diameter 10–60 μm) erythrocyte accumulations in the vessel walls, accumulations of Dextran in the vessel walls and vessel lumina, non‐occlusive and occlusive luminal erythrocyte thrombi as well as totally obstructed vessel segments. Wall‐adherent erythrocyte accumulations became obvious through non‐moving dark spots in the vessel walls. Dextran accumulations were defined through extraordinary brightly fluorescent vessel lumina or walls; slowly and/or non‐moving RBCs in the vessel lumina were indicative of non‐occlusive or occlusive luminal erythrocyte thrombi, while long‐distant Dextran gaps displayed totally obstructed vessel segments (see Figure 2). Quantification of each phenomenon took place in a binary manner (existent or not existent) in all available vessels from on average of 8 fields of view (FOVs) per animal. In the SHRSP, an additional evaluation of the different HA phenomena was carried out in young (17–28 weeks, n = 7), middle‐aged (30–32 weeks, n = 7), and old (33–44 weeks, n = 7) animals.
Figure 2.

Phenomena of cortical small vessel pathology in SHRSP demonstrated using intravital 2‐photon microscopy. B shows small vessel wall damage indicated by Dextran accumulations in the small vessel walls (arrows). C demonstrates erythrocyte accumulations within an arteriolar wall (arrows) and D shows Dextran accumulations within the small vessel lumen (arrow). In E (arrow) the luminal formation of non‐occlusive erythrocyte thrombi is demonstrated together with intraluminal Dextran accumulations (arrowhead). Non‐occlusive erythrocyte thrombi then transform to occlusive erythrocyte thrombi (F, arrow) together with luminal Dextran accumulations (arrowhead) or later to totally obstructed vessel segments (F, asterisk). A refers to HA stage 0 (no HA features), B and C refer to HA stage 1A (small vessel wall damage), D indicates HA stage 1B (RBC flow reduction), E refers to stage 2 (non‐occlusive thrombus formation) and F refers to HA stage 3 (occlusive thrombus formation). Data refer to the investigation of 21 male SHRSP aged 17–44 weeks. HA, hypertensive arteriopathy; RBC, red blood cell. A–F: scale bars = 50 μm; age of the animals: D and E—18 weeks (w), C—32w, A—33w, B and F—44w.
The different phenomena characteristic of HA were subsequently taken into account to define an intravital HA cascade. Based on the cascade we outlined the existence of 5 HA stages. Assignment of each rat to a certain HA stage was based on the most advanced HA pathology derived from the HA cascade detectable in the individual animal.
Measurement of RBC flow velocity
On average 9 line scans per animal were taken into account with each line scan mapping one arteriole. Each arteriole was assigned to exhibit one of the following features: (i) no HA phenomena, (ii) luminal Dextran accumulations, or (iii) non‐occlusive erythrocyte thrombi. RBC flow velocity was assessed in all available arterioles in FOVs randomly placed into the surgical area. For statistical analysis, per animal for each arteriolar HA entity RBC flow velocity was averaged across all line scans of vessels displaying that entity.
In vivo assessment of (peri)vascular β‐amyloid accumulations
In the n = 13 SHRSP that underwent Methoxy imaging, 3 FOVs per animal were investigated semi‐quantitatively for the occurrence of (peri)vascular amyloid deposits defined as accumulations of Methoxy‐X04 surround or within the arteriolar or small artery walls (existent or not existent). Furthermore, in all Aβ/CAA‐positive SHRSP the number of CAA‐positive and totally obstructed vessel segments was counted (within 3 FOVs).
Histological and immunohistochemical analysis
Within each slice covering the parietal cortex we first assessed the occurrence of HA phenomena (for all SHRSP and Wistar rats) that is BBB breakdown, non‐occlusive erythrocyte thrombi, occlusive erythrocyte thrombi/totally obstructed vessel segments and the occurrence of (peri)vascular amyloid deposits (for SHRSP only) in a binary manner (existent, not existent). In order to examine the % of small vessels per FOV affected by HA phenomena (see above) and CAA in SHRSP, we counted the number of all arterioles (diameter 10 to 60 μm) as well as the number of (i) HA‐positive arterioles and (ii) CAA‐positive arterioles; within the arterioles displaying CAA we also assessed the concurrent occurrence of small vessel wall damage, non‐occlusive erythrocyte thrombi and occlusive erythrocyte thrombi/totally obstructed vessel segments. Analysis took account of 27 FOVs within 9 brain slices per rat with 3 FOVs per slice (for HE staining); 20 FOVs, 5 brain slices, 3 FOVs per slice (for Dextran/IgG immunohistochemistry); and of 15 FOVS, 5 brain slices, 3 FOVs per slice (for β‐amyloid staining). The mean of FOVs was taken for statistical analysis.
Statistics
For binary variables, group comparisons (Wistar rats vs. SHRSP or various SHRSP age groups) were conducted using a χ2‐test or a binary logistic regression analysis. To compare RBC flow between (a) arterioles with luminal Dextran accumulations and arterioles without any HA pathology, and between (b) arterioles with non‐occlusive erythrocyte thrombi and arterioles without any HA pathology a paired‐samples t‐test was performed. P‐values ≤ 0.05 were considered to be significant.
Results
In vivo HA phenomena
Intravital fluorescent Dextran imaging showed both, arterioles without any HA feature (Figure 2A) and with different HA phenomena. N = 4 (40%) Wistar rats did not display any HA, while only n = 1 (5%) SHRSP was devoid of any HA, resulting in significant group differences (χ2(1) = 6.2, P = 0.03). In contrast, the prevalence of various HA phenomena was significantly higher in SHRSP compared to Wistar rats: (1) accumulation of Dextran in the small vessel walls (SHRSP n = 19 [90%] vs. Wistar n = 3 [30%], χ2(1) = 6.1, P = 0.02) (Figure 2B), (2) accumulation of erythrocytes in the small vessel walls (n = 16 [76%] vs. n = 4 [40%], χ2(1) = 9.0, P = 0.006), or (3) a combination of (1) and (2) (n = 11 [52%] vs. n = 1 [10%], χ2(1) = 5.1, P = 0.05) (Figure 2C), (4) accumulation of Dextran in the small vessel lumina (n = 16 [76%] vs. n = 1 [10%], χ2(1) = 12.0, P = 0.001) (Figure 2D), (5) non‐occlusive luminal erythrocyte thrombi (n = 15 [71%] vs. n = 0 [0%], χ2(1) = 13.8, P < 0.001), (6) occlusive luminal erythrocyte thrombi (n = 9 [43%] vs. n = 0 [0%], χ2(1) = 6.0, P = 0.03), or (5a) a combination of (4) and (5) (n = 14 [67%] vs. n = 0 [0%], χ2(1) = 12.2, P < 0.001) (Figure 2E) (supplemental movie 1) or (6a) a combination of (4) and (6) (n = 6 [29%] vs. n = 0 [0%], not significant) (Figure 2F, arrow) and (7) totally obstructed vessel segments, indicated by long distant Dextran gaps (n = 9 [43%] vs. n = 0 [0%], χ2(1) = 6.0, P = 0.03) (Figure 2F, asterisk, supplemental movie 2). Detailed prevalence of all phenomena in each individual animal is given in Figure 3. On a group‐level, frequencies of all HA phenomena for SHRSP and Wistar rats are demonstrated in Figure 4A.
Figure 3.
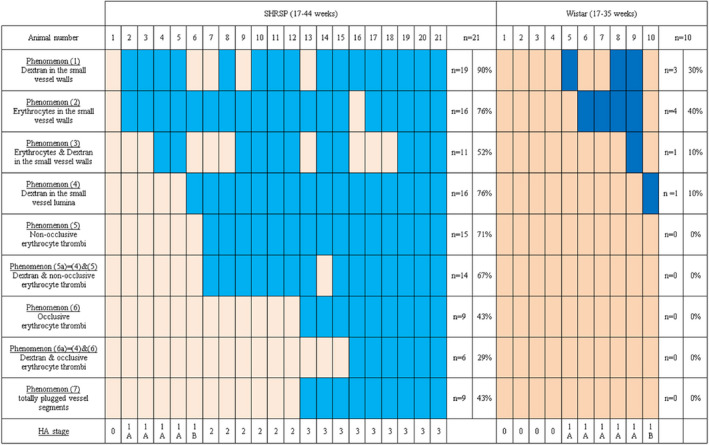
Prevalence of HA phenomena and HA stages in each individual SHRSP and Wistar rat detected by in vivo imaging. All n = 21 SHRSP and n = 10 Wistar rats that underwent in vivo imaging were evaluated for the occurrence of HA phenomena (1)–(7) and the resulting HA stages. The light and dark blue boxes indicate the existence of single HA phenomena, while the light and dark beige boxes display that a certain HA phenomenon was not detectable in one individual rat. Absolute and relative numbers of rats that were positive for the distinct HA phenomena are given in the outer right columns. HA, hypertensive arteriopathy.
Figure 4.
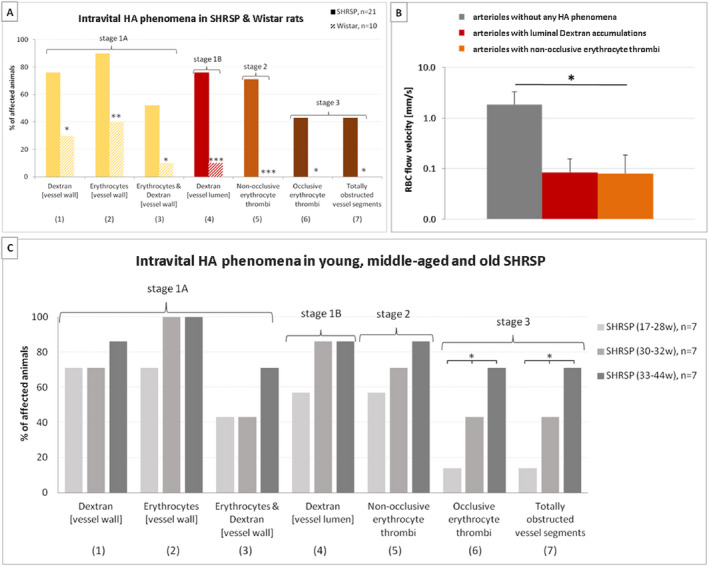
Frequencies of intravital HA phenomena and RBC flow velocity. A. Frequencies of various small vessel pathology phenomena detected by in vivo imaging (2PM) of the parietal cortex in 21 male SHRSP aged 17–44 weeks (filled bars) and 10 Wistar rats aged 17–35 weeks (striped bars). The yellow bars indicate small vessel wall damage (HA stage 1A), the red ones refer to RBC flow reduction (HA stage 1B), the orange one shows non‐occlusive thrombus formation (HA stage 2), and the brown bars are indicative of occlusive thrombus formation (HA stage 3). Asterisks show significant group differences between SHRSP and Wistar rats (*P < 0.05, ** P < 0.01, *** P < 0.001). B. RBC flow velocity measures revealed lower RBC flow in arterioles with luminal Dextran accumulations (red bar) and significantly reduced RBC flow in arterioles with non‐occlusive erythrocyte thrombi (orange bar) compared to arterioles without any HA phenomena (gray bar). Error bars indicate the standard deviation, *P < 0.05. RBC, red blood cell; RBC flow data refer to the investigation of 8 male SHRSP aged 18–33 weeks; y‐axis—logarithmic scale. C. A separate consideration of young (17–28 weeks, light gray bars), middle‐aged (30–32 weeks, middle gray bars) and old (33–44 weeks, dark gray bars) SHRSP revealed that phenomena (1)–(5) (x‐axis) occurred rather frequently in both age groups while phenomena (6) and (7) showed a significantly higher prevalence in the old compared to the middle‐aged and young animals. * P < 0.05. HA, hypertensive arteriopathy.
When considering the SHRSP group alone, phenomena (1) to (5) were equally distributed across all SHRSP age groups (P > 0.05, respectively); eg, phenomenon (1) was detectable in 71% of the young rats (aged 17–28 weeks), in 71% of the middle‐aged SHRSP (30–32 weeks) and in 86% of the old animals (33–44 weeks). The prevalence of occlusive erythrocyte thrombi (6) and totally obstructed vessel segments (7) was, however, significantly different between the different age groups (B(1) = 1.3, P = 0.05, respectively): 14% (17–28 weeks) vs. 43% (30–32 weeks) vs. 71% (33–44 weeks), respectively (Figure 4C).
In vivo HA cascade
We consider the existence of a temporal HA cascade in SHRSP initiated by the accumulation of erythrocytes and Dextran within the small vessel walls (phenomena (1) to (3)), indicative of small vessel wall damage. Wall damage is accompanied by the reduction of RBC flow which becomes obvious through luminal Dextran depositions (phenomenon (4)). The latter is supported by the finding that arterioles characterized by luminal Dextran accumulations compared to arterioles without any HA feature exhibit lower RBC flow velocity (mean [SD] 0.08 [0.10] mm/s vs. 1.83 [1.36] mm/s, t(2) = 2.4, P = 0.1 [trend‐level], Figure 4B). RBC flow velocity reduction initiates the formation of incomplete small vessel thrombi (phenomenon (5)), which is supported by the fact that arterioles comprising non‐occlusive erythrocyte thrombi reveal significantly decreased RBC flow velocity when compared to arterioles without any HA feature (mean [SD] 0.08 [0.07] mm/s vs. 1.83 [1.36] mm/s, t(4) = 3.2, P = 0.03, Figure 4B). Incomplete thrombi progress to complete small vessel occlusions (phenomena (6) to (7)). In Wistar rats, small vessel wall damage as indicated by phenomena (1) to (3) was the predominant finding. Only one of the control animals displayed luminal Dextran accumulations (phenomenon (4)), representing the most advanced HA phenomenon detectable in the age‐matched Wistar rats, while none of the controls exhibited any thrombus formation (phenomena (5) to (7)). This prevented us from comparing RBC flow velocity between arterioles without HA features against arterioles with luminal Dextran accumulations or non‐occlusive erythrocyte thrombi in the control group. Overall, our results suggest that the proposed HA cascade is unique to the SHRSP.
In vivo HA stages
Considering the demonstrated HA cascade we secondly defined the existence of 5 distinct HA stages: stage 0—no HA features, stage 1A—small vessel wall damage, stage 1B—RBC flow reduction, stage 2—non‐occlusive/incomplete thrombus formation, stage 3—occlusive/complete thrombus formation. Out of all n = 21 SHRSP compared to n = 10 Wistar rats, n = 1 (5%) vs. n = 4 (40%) were assigned to stage 0 (χ2(1) = 6.2, P = 0.03); n = 4 (19%) vs. n = 5 (50%) to stage 1A (not significant); n = 1 (5%) vs. n = 1 (10%) to stage 1B (not significant); n = 6 (29%) vs. n = 0 (0%) to stage 2 (not significant), and n = 9 (43%) vs. n = 0 (0%) to stage 3 (χ2(1) = 5.1, P = 0.03). These results indicate that SHRSP and Wistar rats do not differ much with regard to initial HA characterized by small vessel wall damage, while the late HA stages characterized by thrombus formations are unique to the SRHSP. The HA stage of each individual animal is demonstrated in Figure 3.
When considering the SHRSP alone, early HA stages (stage 0, 1A, 1B) were equally distributed across all SHRSP age groups (P > 0.05, respectively), while the prevalence of advanced HA (stage 3) was significantly different between the different age groups (B(1) = 1.3, P = 0.05): 14% (17‐28 weeks) vs. 43% (30–32 weeks) vs. 86% (33–44 weeks).
In vivo vascular Aβ pathology
Six out of 13 SHRSP (46%) that underwent in vivo Methoxy imaging displayed intravital β‐amyloid positivity of the cerebral small vessel walls indicative of CAA. Half of those Methoxy‐positive rats (50%) exhibited periarteriolar amyloid deposits (Figure 5A) indicating Aβ deposition spreading from the small vessel walls into the surrounding tissue and referred to as dyshoric CAA (2), while all Methoxy‐positive animals displayed brightly fluorescent arteriolar/small artery wall adherent plane‐ or circular‐shaped amyloid accumulations (Figure 5B–D). Since those green vessel segments could also be interpreted as intraluminal Aβ staining, we checked whether Aβ accumulates within the tunica media of small vessel walls as found in CAA. In this instance, Figure 5E and F demonstrates the histological/immunohistochemical data of three exemplary SHRSP (animal number 19, 21, and 14, age 32, 33, and 39 weeks, Methoxy‐positive; see Figure 3), proving that amyloid is not accumulating in the vessel lumen but in the vessel wall, especially in the tunica media. Considering all CAA‐positive vessels, 66% displayed complete small vessel occlusions. Out of the Methoxy‐positive rats, none was in HA stage 0, 1A or 1B; one animal (17%) was assigned to HA stage 2, while the remaining 5 (83%) were in HA stage 3. Supplementary Table 1 demonstrates the assignment to the various HA stages of each individual SHRSP that had an intravital Aβ status available.
Figure 5.
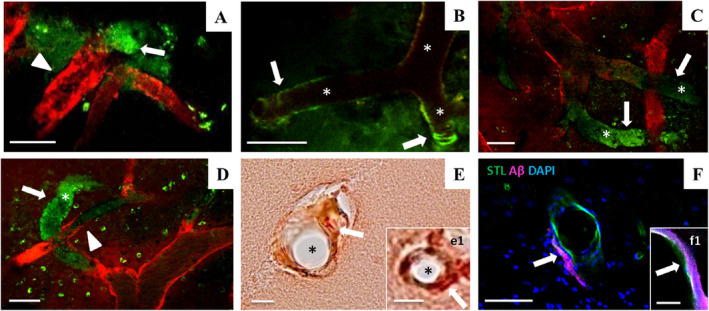
Intravital two‐photon microscopy of amyloid pathology in the parietal cortex of SHRSP. Vascular Methoxy‐positivity (green) indicative of CAA comprises periarteriolar or dysphoric CAA (A, arrow) and wall‐adherent amyloid deposits (arrows in B–D). CAA was detectable next to non‐occlusive erythrocyte thrombi (A and D, arrowheads) or surround totally obstructed vessel segments characterized by long distant Dextran gaps (asterisks in B–D) leading to the assignment of all Methoxy‐positive SHRSP to either HA stage 2 or stage 3. E and e1 (arrows) show CAA in the small vessel wall but no Aβ accumulation in the lumen of the vessel (asterisks in E and e1). STL (green in F and f1) marks the endothelium and therefore the inner layer of the vessel wall; the vascular amyloid deposits (magenta) are found in the second layer of the vessel wall called tunica media (F and f1). Data refer to the investigation of 13 male SHRSP aged 17–44 weeks. Small vessels and vessel walls are visualized by Dextran (red). CAA, cerebral amyloid angiopathy; HA, hypertensive arteriopathy. STL ‐ solanum tuberosum lectin (endothelial marker), DAPI ‐ 4′.6‐diamidino‐2‐phenylindole (nuclear staining); A–D—2PM, E and e1 ‐ CR/Prussian blue staining, F and f1—Immunohistochemistry; scale bars: A–D, F: = 50 μm, E = 20 μm, e1, f1 = 10 μm; age of the animals: A—28 weeks (w), B, E, F—32w, C–E—33w, e1 and f1 = 39w
Post‐mortem analysis of HA phenomena and vascular β‐amyloid positivity
Intravital HA phenomena (see Figure 2) were verified by histology and immunohistochemistry, as demonstrated in Figure 6: intravital phenomenon (1) consisting of the accumulation of Dextran in the small vessel walls was also detectable using immunohistochemistry (Figure 2B vs. Figure 6A); the accumulation of erythrocytes in the small vessel walls (phenomenon (2), Figure 2C vs. Figure 6B), non‐occlusive erythrocyte thrombi (phenomenon (5), Figure 2E vs. Figure 6C), and occlusive erythrocyte thrombi/totally obstructed vessel segments (phenomenon (7), Figure 2F vs. Figure 6D) were proven post‐mortem as well.
Figure 6.
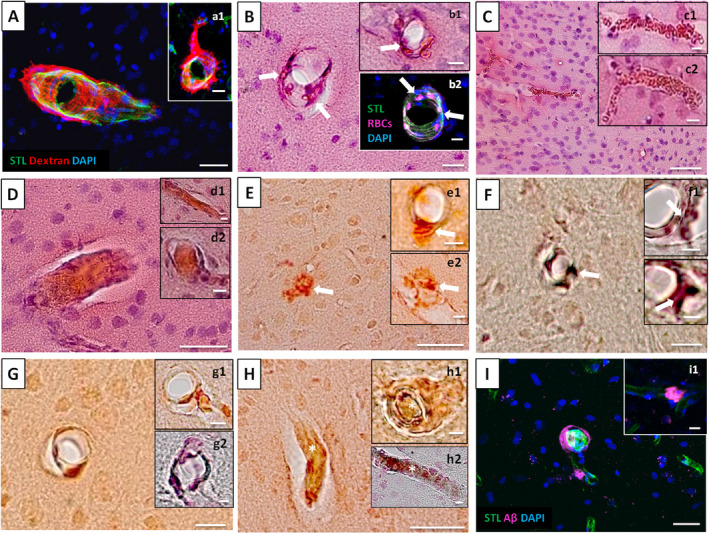
Spectrum of post‐mortem HA and amyloid pathology in SHRSP. A and a1 display Dextran accumulations in and surround the small vessel walls indicative of BBB breakdown and B, b1 and b2 show the accumulation of erythrocytes in the small vessel walls (arrows). C, c1 and c2 demonstrate non‐occlusive erythrocyte thrombi and D, d1 and d2 show totally obstructed vessel segments. E, e1, e2, F, f1, f2 demonstrate dyshoric CAA with amyloid spreading from the vessel wall into the perivascular tissue (arrows in E, e1, e2, F, f1, f2), while G, g1, g2, H, h1, h2, I, and i1 display wall adherent Aβ accumulations both indicating CAA. Totally obstructed vessel segments within CAA‐positive small vessels are shown in H, h1, and h2 (asterisks). CAA, cerebral amyloid angiopathy; HA, hypertensive arteriopathy. HA data refer to the investigation of 21 and amyloid data to the investigation of 13 male SHRP aged 17–44 weeks. STL—solanum tuberosum lectin (endothelial marker), DAPI—4′.6‐diamidino‐2‐phenylindole (nuclear staining); A, a1, b2, I, i1—Immunohistochemistry, B, b1, C, c1, c2, D, d1, d2—HE staining, E, e1, e2, G, g1, H, h1—CR/Prussian blue staining, F, f1, f2, g2, h2—Thioflavin T/Prussian blue staining; scale bars: C‐E, H = 50 μm, A, a1, B, F, G, I = 20 μm, b1, b2, c1, c2, d1, d2, e1, e2, f1, g1, g2, h1, h2, i1 = 10 μm, f2 = 5 μm; age of the animals: C, c1, e1, G—18 weeks (w), H—28w, a1, c2, E, e2, h2, i1—32w, f2, g2, I—33w, B, b2—39w, A, b1, D, d1, d2, F, f1, g1, h1—44w
Quantitative tissue analysis of the surgical area of SHRSP vs. Wistar rats revealed that n = 19 (90%) vs. n = 8 (80%) (χ2(1) = 0.6, P = 0.6) of the animals displayed small vessel wall damage as indicated by Dextran accumulations. Non‐occlusive erythrocyte thrombi were found in n = 16 (76%) vs. n = 3 (30%) (χ2(1) = 6.1, P = 0.02); mean [SD] number of arterioles per FOV displaying non‐occlusive erythrocyte thrombi was, moreover, significantly higher in SHRSP compared to Wistar rats (0.7 [0.7] vs. 0.04 [0.1]; Mann‐Whitney U test, Z = −3.2, P = 0.001). Occlusive erythrocyte thrombi/totally obstructed vessel segments were seen in SHRSP only, but not in the controls (n = 8 (38%) vs. n = 0 (0%); χ2(1) = 5.1, P = 0.03). In SHRSP, vascular amyloid positivity was found in n = 7 (54%) animals with n = 5 displaying dyshoric CAA and n = 6 revealing wall‐adherent β‐amyloid deposits (Figure 6 E‐I).
When considering the % of small vessels per FOV affected by HA phenomena or CAA in SHRSP, the distribution was as follows: 22% of the arterioles per FOV showed one or more of the HA phenomena (27% of the arterioles/FOV displayed small vessel wall damage, 18% non‐occlusive erythrocyte thrombi, 2% occlusive erythrocyte thrombi/totally obstructed vessel segments) and 9% of the arterioles per FOV were affected by CAA. Out of the arterioles displaying CAA, 100% also revealed small vessel wall damage, 24% were filled with non‐occlusive thrombi and 56% showed occlusive erythrocyte thrombi/totally obstructed vessel segments. Since all CAA‐positive arterioles also revealed any HA phenomena, both pathologies, HA and CAA, occurred together in 9% of all arterioles. Out of those 22% of arterioles affected by HA, there was thus a remainder of 13% displaying HA without CAA.
To summarize, the post‐mortem and intravital features of HA were correlated with each other. The higher frequencies of HA compared to CAA‐positive arterioles and the fact that the majority of CAA vessels also displayed any HA phenomenon overall support our hypothesis that HA creates a state that may facilitate CAA development.
HA‐related cascade of CAA development in SHRSP
Figure 7 provides a graphical overview of the results depicted in the present study. HA is thereby initiated by small vessel wall damage (Figure 7B) and cerebral blood flow reductions (Figure 7C) followed by the formation of non‐occlusive erythrocyte thrombi (Figure 7D) to finally result in complete small vessel occlusions (Figure 7E). Within that cascade, CAA development is related to advanced HA stages and mainly takes place in the plugged small vessels (Figure 7D and E).
Figure 7.
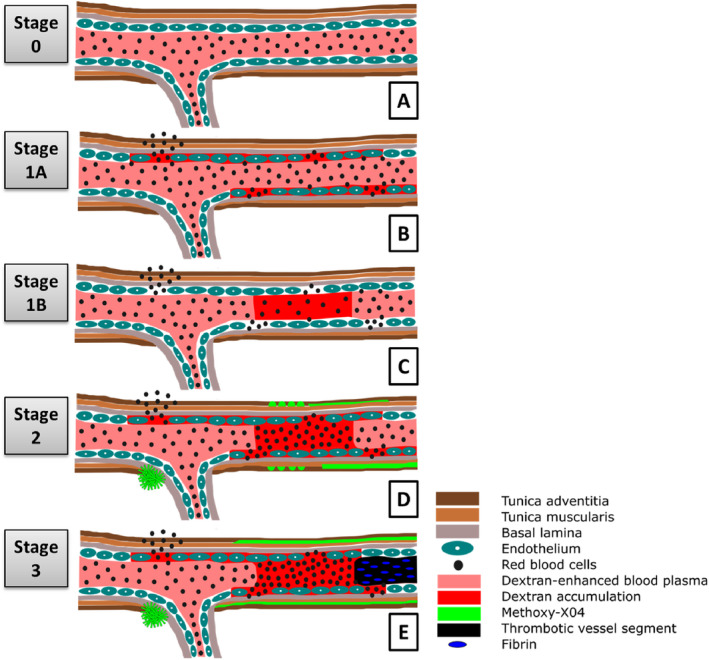
Non‐amyloid HA cascade related CAA development. A demonstrates non‐pathological vessels without any HA phenomena (A, stage 0). Non‐amyloid HA‐related changes are initiated by small vessel wall damage and blood flow reductions indicated by the accumulation of Dextran and erythrocytes in the small vessel wall (B, stage 1A) or Dextran in the small vessel lumen (C, stage 1B). The walls of small vessels with subsequent formation of non‐occlusive (D) or occlusive thrombi (E) accompanied by a significant reduction of RBC flow velocity are prone to accumulate (peri)vascular amyloid deposits. CAA, cerebral amyloid angiopathy; HA, hypertensive arteriopathy; RBC, red blood cell.
Discussion
We here present an in vivo cascade of hypertensive cerebral small vessel disease/arteriopathy in SHRSP starting from small vessel wall damage and ending with occlusive small vessel thrombosis. Small vessel wall damage initiating the HA cascade is also found in Wistar control rats, but the later disease stages characterized by thrombus formation are unique to the SHRSP. In SHRSP, in around 50% of the rodents ongoing small vessel damage was related to wall‐adherent Aβ accumulations (cerebral amyloid angiopathy). The findings from intravital observations in SHRSP and Wistar rats were in line with subsequent histological and immunohistochemical analysis of the post‐mortem rat brains, supporting our hypothesis. While exploratory in nature, our experimental data provides support for the working hypothesis that there is a continuous spectrum of hypertensive small vessel disease and CAA, pointing toward the idea, that CAA initiation and progression could take place (faster) in the presence of (severe) HA.
Historically, HA has mainly been related to advanced vascular lesions such as arteriosclerosis, hyalinosis and small vessel thromboses 26. During the last years, however, our understanding of HA has substantially changed, as early endothelial dysfunction and BBB breakdown have become an important starting point of hypertensive small vessel wall damage accelerating the development of overall small vessel pathology comprising the loss of smooth muscle cells or vessel wall thickening 36, 54, 66, 67, 71, 85, 87, 88. The disruption of the BBB appears to favor the depositions of vascular fibrin and clot formation 22, 71. Those findings are in line with our temporal HA cascade overall supporting the existence of a circle of events comprising a pro‐inflammatory thrombotic state related to blood flow reductions, hypercoagulability and finally, small vessel occlusions 25, 45, 77, 78, 79. That circle obviously depends on age, since progressing disease pathology in terms of small vessel occlusions was found more frequently in the older SHRSP. Furthermore, age‐ and sex‐matched control animals also exhibited the initial HA stages, emphasizing that (HA‐related) small vessel wall damage likewisely occurs in the aging brain, even in the absence of vascular risk factors. The proposed HA cascade likely promotes the degeneration of the neurovascular unit which in turn results in cognitive decline in terms of vascular dementia and/or ischemic and hemorrhagic stroke 74, 95, 98.
Recently, Ihara and Yamamoto described a hypothetical HA cascade derived from several studies in spontaneously hypertensive (stroke‐prone) rats (SHR and SHRSP) that is likewisely initiated by endothelial dysfunction leading to BBB breakdown, but finally resulting in white matter damage, cerebral microbleeds and enlarged perivascular spaces 36. The latter phenomena have, however, not been addressed in our study as white matter pathologies and perivascular space enlargement are not detectable by intravital imaging of the rat cortex. White matter damage, microbleeds and enlarged perivascular spaces may indeed indicate breakdown of the BBB and/or failure of intramural periarterial drainage 5, 35, 84, 86, 90. The findings of Ihara and Yamamoto are in line with our previous post‐mortem studies in SHRSP reporting on BBB dysfunction, subsequent small vessel wall rupture and consecutive small perivascular bleeds 67, 68. Future studies should address the temporal interconnection between white matter, subcortical and cortical as well as between thrombotic and hemorrhagic pathologies in HA in more detail by using intravital imaging together with post‐mortem tissue analysis.
Our experimental data emphasizes the common involvement of cortical areas in HA, counteracting the widely accepted view that human HA is mainly restricted to subcortical regions 13, 54. Current perspectives are, however, changing and magnetic resonance imaging (MRI) studies report on the existence of cortical pathologies such as gray matter atrophy, cerebral microinfarcts or CBF reduction in HA patients 46, 58, 70, 81.
Our data demonstrates that HA, ie, small vessel thrombosis seems to create a state that facilitates periarteriolar and wall‐adherent amyloid accumulations in SHRSP. They replicate the results derived from previous animal studies conducted in amyloid β precursor protein (AβPP) transgenic mice and human autopsy studies in Alzheimer's disease or stroke reporting the frequent occurrence of CAA surround small vessel occlusions characterized, eg, by vascular fibrin(ogen) accumulations 16, 61. Fibrin(ogen) accumulating in the vessel lumen has thereby been hypothesized to “entrap” Aβ impeding its vascular clearance across the compromised endothelium 18. An intraluminal accumulation of Aβ in occluded vessels was also reported in mice 38 and might also be possible in SHRSP. Nevertheless, our histological and immunohistochemical analyses clearly showed that in SHRSP the Aβ is accumulating within the tunica media, which is indicative of CAA 32. Since Wistar control rats showed no thrombus formation, which in turn seems to favor the deposition of vascular amyloid, our current findings may also explain the absence of any small vessel wall Aβ pathology/CAA in Wistar animals 11, 33, 68.
HA is one possible mechanism for CAA development in the SHRSP, but other mechanisms may exist as well: SHRSP is a non‐transgenic, but genetically hypertensive rat that was selectively bred from the spontaneously hypertensive rat (SHR), which in turn was developed by selective cross‐breeding of outbred Wistar Kyoto rats 6, 45, 52. The SHRSP shows malignant hypertension by 12 weeks of age, which has been related to polygenic alterations of several candidate genes interacting with one another and involved in, eg, blood pressure regulation, salt‐induced blood pressure variability, activation of the renin‐angiotensin system, adrenergic mechanisms, prostaglandin metabolism and renal pathology 43, 50, 52. Beside HA‐related changes, ie, BBB breakdown, endothelial disturbances and hemorrhagic/ischemic stroke‐like lesions, this hypertensive rat model additionally develops parenchymal and vascular Aβ deposits 11, 33, 61, 68, 96, which, to the best of our knowledge, should not rely on the genetic alterations resulting in hypertension. There is, indeed, thus far no genome sequencing study available, that would definitely have ruled out any (spontaneous) mutations or specific risk alleles of AβPP, β‐site APP cleaving enzyme, presenilin‐1 or ‐2, or APOE, potentially explaining an altered Aβ turnover in SHRSP. We, however, conducted some preliminary analysis in SHRSP that did not reveal altered neuronal RNA expression of any proteins involved in the Aβ metabolism (unpublished data). Recent studies performed by our group, nevertheless, depicted some upregulation of neuronal AβPP protein expression in SHRSP, which might rely on HA‐related chronic hypoperfusion and subsequent neuronal injury rather than on any AβPP gene mutation 47, 68. Indeed, in case of neuronal AβPP overexpression, CAA could evolve as well as a consequence of an increased clearance of Aβ via the perivascular drainage channels 14, 34, potentially leading to a kind of “exhaustion” of vascular amyloid transport mechanisms. Further studies are needed to in‐depth address the question, to what proportion CAA in SHRSP is related to HA (only), and to what extent small vessel pathology and neuronal AβPP overexpression act together to promote vascular amyloid deposits.
Moreover, age and arterial hypertension themselves (and together) could further create a state where Aβ transport or degradation mechanisms fail. In line with this, arterial hypertension is indeed quiet commonly found in human CAA affecting around 30% to nearly 70% of patients fulfilling the Boston criteria 8, 17, 18, 37. Experimental data thereby relate hypertension (and age) to an upregulation of the receptor for advanced glycation end products (RAGE), providing greater Aβ influx, and to downregulation of the low‐density lipoprotein receptor related protein 1 (LRP1), resulting in lower Aβ efflux, and thus in vascular Aβ overload 15, 30, 62, 76. Arterial hypertension further promotes plasma protein leakage into the vessel wall, that in turn could accelerate BBB dysfunction, which has not only been discussed as a direct risk factor for CAA development but should further facilitate CAA‐related hemorrhage 28, 57. Indeed, distinction of “hypertension‐related” and “HA‐related” CAA formation is somewhat artificial, as both are inseparably interwoven. Overall, as for HA, it remains an unsolved question whether there is just some co‐existence between CAA and hypertension, or whether one could assume there to be a more causal relationship 17, 93. As there is no direct treatment of CAA, it would be a huge benefit if an indirect treatment via HA and blood pressure management would be possible. However, while in transgenic Alzheimer mice antihypertensive drugs have been already shown to be capable to reduce amyloid levels 55, 83, studies focusing on the understanding how HA restoration (through, eg, antihypertensive treatment) could mediate the lowering of vascular Aβ depositions are still missing.
Human autopsy studies reveal that 20% to 40% of the non‐demented elderly aged 75 years and older display CAA 16, 19, 59, 75, 92. Frequencies are even higher when considering HA with prevalences of 50% to up to around 70% from an age of 65 years on 3, 38. The majority of neuropathology studies has, however, been focusing either on the description of the extent of hypertensive CSVD pathologies, or CAA severity assessment, or the evaluation of general HA and CAA frequencies in the aged human brain 1, 20, 39, 63, 69. Only few human autopsy studies give indeed some more precise overlap prevalence of the two CSVD entities, which would better argue for the common occurrence and probable transition of HA and CAA. Ritter et al. described a weak relationship between deep hemorrhages (indicative of HA) and CAA with 18% of the patients showing that constellation 63. Multiple vascular disease was, moreover, found in 7% to 26% of population samples of the non‐demented elderly 1, 48. One recent autopsy study identified much higher overlap prevalences with 75% of the patients with pathologically defined CAA also displayed HA‐related arteriolosclerosis 20. Similarly, there are nearly no consistent estimates of the overlap between HA‐ and CAA‐related pathologies on neuroimaging, but two studies reported on mixed patterns of HA and CAA in 19% to 23% of patients suffering from intracerebral hemorrhages 56, 73. Based on the literature one may overall assume, that several aged human subjects display CAA without HA, and vice versa, having HA without CAA. Additionally, there should be, however, a certain proportion in the aged population (especially that proportion that also experiences a hemorrhagic stroke) that has both, CAA together with HA, but thus far little is known about its precise prevalence. In that instance, our data supports the frequent common occurrence of HA and CAA (that constellation was found in 50% of our SHRSP animals), and points toward the idea, that CAA initiation and progression could take place (faster) in the presence of (severe) HA.
One may further argue, that once CAA has developed vascular Aβ (which is vasoactive) could promote further disturbances of endothelial properties resulting in a failure of vascular autoregulation and BBB leakage 27. In the first instance, increased vascular Aβ thereby further impairs solute clearance and endothelial protein transport, faciliating the accumulation of potentially toxic products and its accompanying local inflammation in the small vessel walls. The latter should forward ongoing endothelial failure and finally small vessel wall reorganization, as it is typically found in HA, closing the circle. We, however, did not focus on the hypothesis that CAA also promotes HA progression, which thus needs confirmation by future studies.
The strength of our study is the use of an animal model displaying spontaneous HA development without additional induction of further vessel pathologies such as large vessel occlusions, allowing for the investigation of the spontaneous course of hypertensive CSVD. We here, moreover, demonstrate the applicability of intravital β‐amyloid microscopy in a HA rat model as previous studies using 2PM Methoxy‐X04 imaging have solely been conducted in transgenic Alzheimer's disease mouse models 12, 23, 32, 42, 49, 94. Our study thus provides a unique in vivo characterization of mixed vascular and amyloid pathologies in a non‐transgenic rat model opening the door for further studies combining in vivo Aβ together with small vessel imaging.
Our study is, however, limited by its rather descriptive approach and future studies are needed to understand the underlying mechanisms relating HA and vascular β‐amyloid deposits. Furthermore, this study is limited by the use of one HA rodent model only, demanding for the replication of our findings in further animal models of hypertensive arteriopathy. We, moreover, did not treat the rats with antihypertensive drugs to document that the effect of HA progression is indeed related to hypertension‐associated small vessel changes. Additionally, in few Wistar rats scattered non‐occlusive erythrocyte thrombi were found by histological analysis that could not be detected by 2PM. This indicates that post‐mortem analysis cannot totally replicate intravital findings and intravital imaging might miss some HA features. In this study, HA definition was based upon functional data received from 2PM imaging of living animals, comprising very initial small vessel (wall) alterations. This is different from human autopsy or in vivo imaging (MRI) studies, which identify HA through more advanced small vessel wall (eg, arteriolosclerosis) 72 or downstream pathologies (eg, white matter hyperintensities or lacunes) 89. Additionally, at human autopsy, fresh agonal/post‐mortem blood clots cannot be distinguished from very fresh thrombi. Those aspects somewhat limit the comparability (eg, of the HA/CAA overlap prevalences) of our experimental and existent human studies.
In conclusion, our data arisen in a non‐transgenic HA rodent model (SHRSP) using intravital imaging suggests small vessel thromboses to play a role for CAA development relating HA and CAA to the same disease spectrum. SHRSP could fill the gap of thus far missing animal models suitable to understand the interplay between the two small vessel disease entities of the aging brain. As there is currently no CAA treatment with affected patients sustaining severe hemorrhagic stroke and dementia, HA involvement could provide new therapeutic opportunities such as the rigorous control of vascular risk factors.
Author Contribution Statement
SJ: study concept and design, data acquisition, data analysis, data interpretation, drafting the manuscript.
CG, DS: data acquisition, data analysis, critical revision of the manuscript.
MS: proof‐read and critical revision of the manuscript.
HJH: critical revision of the manuscript.
ROC: proof‐read and critical revision of the manuscript.
SS: study concept and design, data analysis, data interpretation, drafting the manuscript, critical revision of the manuscript.
Disclosure/Conflict of interest
The authors declare no conflict of interest.
Supporting information
Supplemental Movie 1: HA stage 2—Non‐occlusive thrombus formation. Dextran (red) was used to mark the blood plasma and erythrocytes are detected as dark spots in front of the bright background. In the movie the formation of a non‐occlusive thrombus is presented. In the lower part of the vessel normal blood flow is detectable but after the vessel is branching, the upper left part of the vessel shows a significant reduction of the red blood cell flow velocity. Here, single blood cells are accumulating in the vessel (yellow arrow) and Dextran and erythrocytes are slowing down their flow velocity (white arrow). Even though three is still blood flow detectable, a non‐occlusive thrombus is formed.
Supplemental Movie 2: HA stage 3—Occlusive thrombus formation. Dextran (red) was used to mark the blood plasma and erythrocytes are detected as dark spots in front of the bright background. In the movie the formation of an occlusive thrombus is presented. In the upper part of the vessel, normal blood flow is detectable and the original vessel then leaves the level of the video. At this branching point of the vessel, a free‐floating thrombus is formed (arrow). Here, dextran and erythrocytes are accumulating in the lumen of the vessel and cannot flow through it because the following vessel segment is totally plugged (asterisk). This is indicated by long‐distant Dextran gaps and referred to as totally plugged vessel segment.
Supplemental Table 1: HA phenomena and HA stages of SHRSP that underwent Methoxy imaging. All 13 SHRSP that underwent Methoxy imaging were classified as Methoxy‐positive or –negative and were evaluated for the occurrence of the following HA phenomena: (1) accumulation of Dextran in the small vessel walls, (2) accumulation of erythrocytes in the small vessel walls, (3) a combination of (1) and (2, 4) accumulation of Dextran in the small vessel lumina, (5) non‐occlusive erythrocyte thrombi, (5a) a combination of (4) and (5, 6) occlusive erythrocyte thrombi, (6a) a combination of (4) and (6) and (7) totally plugged vessel segments. Additionally all rats were assigned to one of the following HA stages: stage 0—no HA features, stage 1A—small vessel wall damage, stage 1B—RBC flow reduction, stage 2—non‐occlusive thrombus formation, stage 3—occlusive thrombus formation. Animal numbers refer to Table 1. Methoxy positive SHRSP are marked in gray. HA, hypertensive arteriopathy; RBC, red blood cell.
Acknowledgments
We thank Andre Weber, Leibniz Institute for Neurobiology in Magdeburg, for writing the MATLAB script used for line scan analysis and Dr. Stoyan Stoyanov, German Center for Neurodegenerative Diseases in Magdeburg, for introducing us to the data evaluation using the above mentioned MATLAB script.
References
- 1. Arvanitakis Z, Capuano AW, Leurgans SE, Buchman AS, Bennett DA, Schneider JA (2017) The relationship of cerebral vessel pathology to brain microinfarcts. Brain Pathol (Zurich, Switzerland) 27:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Attems J, Jellinger K, Thal DR, van Nostrand W (2011) Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 37:75–93. [DOI] [PubMed] [Google Scholar]
- 3. Attems J, Jellinger KA (2014) The overlap between vascular disease and Alzheimer's disease—lessons from pathology. BMC Med 12: 10.1186/s12916-014-0206-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bacskai BJ, Klunk WE, Mathis CA, Hyman BT (2002) Imaging amyloid‐beta deposits in vivo . J Cerebral Blood Flow and Metabolism Official J Int Soc Cerebral Blood Flow Metabolism ;22:1035–1041. [DOI] [PubMed] [Google Scholar]
- 5. Bailey EL, McCulloch J, Sudlow C, Wardlaw JM (2009) Potential animal models of lacunar stroke: a systematic review. Stroke 40:e451–e458. [DOI] [PubMed] [Google Scholar]
- 6. Banerjee G, Wilson D, Jäger HR, Werring DJ (2016) Novel imaging techniques in cerebral small vessel diseases and vascular cognitive impairment. Biochim Biophys Acta 1862:926–938. [DOI] [PubMed] [Google Scholar]
- 7. Biffi A, Greenberg SM (2011) Cerebral amyloid angiopathy: a systematic review. J Clin Neurol (Seoul, Korea) 7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boulouis G, Charidimou A, Pasi M, Roongpiboonsopit D, Xiong L, Auriel E et al (2017) Hemorrhage recurrence risk factors in cerebral amyloid angiopathy: comparative analysis of the overall small vessel disease severity score versus individual neuroimaging markers. J Neurolog Sci 380:64–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Braun H, Bueche CZ, Garz C, Oldag A, Heinze H‐J, Goertler M et al (2012) Stases are associated with blood‐brain barrier damage and a restricted activation of coagulation in SHRSP. J Neurol Sci 322:71–76. [DOI] [PubMed] [Google Scholar]
- 10. Braun H, Schreiber S (2013) Microbleeds in cerebral small vessel disease. Lancet Neurol 12:735–736. [DOI] [PubMed] [Google Scholar]
- 11. Bueche CZ, Hawkes C, Garz C, Vielhaber S, Attems J, Knight RT et al (2014) Hypertension drives parenchymal β‐amyloid accumulation in the brain parenchyma. Ann Clin Trans Neurol 1:124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burgold S, Bittner T, Dorostkar MM, Kieser D, Fuhrmann M, Mitteregger G et al (2011) In vivo multiphoton imaging reveals gradual growth of newborn amyloid plaques over weeks. Acta Neuropathol 121:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cai Z, Wang C, He W, Tu H, Tang Z, Xiao M et al (2015) Cerebral small vessel disease and Alzheimer's disease. Clin Interventions Aging 10:1695–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold K‐H et al (1999) Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA 96:14088–14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carnevale D, Mascio G, D'Andrea I, Fardella V, Bell RD, Branchi I et al (2012) Hypertension induces brain β‐amyloid accumulation, cognitive impairment and memory deterioration through activation of rage in brain vasculature. Hypertension (Dallas, Tex. 1979) 60:188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Charidimou A, Boulouis G, Gurol ME, Ayata C, Bacskai BJ, Frosch MP et al (2017) Emerging concepts in sporadic cerebral amyloid angiopathy. Brain J Neurol 140:1829–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Charidimou A, Boulouis G, Pasi M, Auriel E, van Etten ES, Haley K et al (2017) MRI‐visible perivascular spaces in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology 88:1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Charidimou A, Boulouis G, Xiong L, Jessel MJ, Roongpiboonsopit D, Ayres A et al (2017) Cortical superficial siderosis and first‐ever cerebral hemorrhage in cerebral amyloid angiopathy. Neurology 88:1607–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Charidimou A, Gang Q, Werring DJ (2012) Sporadic cerebral amyloid angiopathy revisited: Recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiat 83:124–137. [DOI] [PubMed] [Google Scholar]
- 20. Charidimou A, Martinez‐Ramirez S, Reijmer YD, Oliveira‐Filho J, Lauer A, Roongpiboonsopit D et al (2016) Total magnetic resonance imaging burden of small vessel disease in cerebral amyloid angiopathy: an imaging‐pathologic study of concept validation. JAMA Neurol 73:994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chhatbar PY, Kara P (2013) Improved blood velocity measurements with a hybrid image filtering and iterative Radon transform algorithm. Front Neurosci 7:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cortes‐Canteli M, Mattei L, Richards AT, Norris EH, Strickland S (2015) Fibrin deposited in the Alzheimer's disease brain promotes neuronal degeneration. Neurobiol Aging 36:608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dong J, Revilla‐Sanchez R, Moss S, Haydon PG (2010) Multiphoton in vivo imaging of amyloid in animal models of Alzheimer's disease. Neuropharmacology 59:268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Drew PJ, Blinder P, Cauwenberghs G, Shih AY, Kleinfeld D (2010) Rapid determination of particle velocity from space‐time images using the Radon transform. J Comput Neurosci 29:5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Esmon CT (2009) Basic mechanisms and pathogenesis of venous thrombosis. Blood Rev 23:225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fisher CM (1968) The arterial lesions underlying lacunes. Acta Neuropathol 12:1–15. [DOI] [PubMed] [Google Scholar]
- 27. Grinberg LT, Korczyn AD, Heinsen H (2012) Cerebral amyloid angiopathy impact on endothelium. Exp Gerontol 47:838–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grinberg LT, Thal DR (2010) Vascular pathology in the aged human brain. Acta Neuropathol 119:277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haffner C, Malik R, Dichgans M (2016) Genetic factors in cerebral small vessel disease and their impact on stroke and dementia. J Cerebral Blood Flow Metabolism Official J Int Soc Cerebral Blood Flow Metabolism 36:158–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hawkes CA, Härtig W, Kacza J, Schliebs R, Weller RO, Nicoll JA et al (2011) Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol 121:431–443. [DOI] [PubMed] [Google Scholar]
- 31. Hawkes CA, Jayakody N, Johnston DA, Bechmann I, Carare RO (2014) Failure of perivascular drainage of β‐amyloid in cerebral amyloid angiopathy. Brain Pathol (Zurich, Switzerland) 24:396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hefendehl JK, Wegenast‐Braun BM, Liebig C, Eicke D, Milford D, Calhoun ME et al (2011) Long‐term in vivo imaging of β‐amyloid plaque appearance and growth in a mouse model of cerebral β‐amyloidosis. J Neurosci Official J Soc Neurosci 31:624–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Held F, Morris AWJ, Pirici D, Niklass S, Sharp MMG, Garz C et al (2017) Vascular basement membrane alterations and β‐amyloid accumulations in an animal model of cerebral small vessel disease. Clin Sci (London, England 1979) 131:1001–1013. [DOI] [PubMed] [Google Scholar]
- 34. Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD et al (2004) Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci 7:954–960. [DOI] [PubMed] [Google Scholar]
- 35. Huisa BN, Caprihan A, Thompson J, Prestopnik J, Qualls CR, Rosenberg GA (2015) Long‐term blood‐brain barrier permeability changes in binswanger disease. Stroke 46:2413–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ihara M, Yamamoto Y (2016) Emerging evidence for pathogenesis of sporadic cerebral small vessel disease. Stroke 47:554–560. [DOI] [PubMed] [Google Scholar]
- 37. Jang YK, Kim HJ, Lee JS, Kim YJ, Kim KW, Kim Y et al (2017) Distinctive clinical effects of haemorrhagic markers in cerebral amyloid angiopathy. Sci Rep 7: 10.1038/s41598-017-16298-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jellinger KA, Attems J (2012) Neuropathology and general autopsy findings in nondemented aged subjects. Clin Neuropathol 31:87–98. [DOI] [PubMed] [Google Scholar]
- 39. Kim YJ, Kim HJ, Park J‐H, Kim S, Woo S‐Y, Kwak K‐C et al (2016) Synergistic effects of longitudinal amyloid and vascular changes on lobar microbleeds. Neurology 87:1575–1582. [DOI] [PubMed] [Google Scholar]
- 40. Kleinfeld D, Mitra PP, Helmchen F, Denk W (1998) Fluctuations and stimulus‐induced changes in blood flow observed in individual capillaries in layers 2 through 4 of rat neocortex. Proc Natl Acad Sci USA 95:15741–15746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Klohs J, Rudin M, Shimshek DR, Beckmann N (2014) Imaging of cerebrovascular pathology in animal models of Alzheimer's disease. Front Aging Neurosci 6:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP et al (2002) Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy‐X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol 61:797–805. [DOI] [PubMed] [Google Scholar]
- 43. Koh‐Tan HHC, Dashti M, Wang T, Beattie W, Mcclure J, Young B et al (2017) Dissecting the genetic components of a quantitative trait locus for blood pressure and renal pathology on rat chromosome 3. J Hypertens 35:319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Krafft PR, Bailey EL, Lekic T, Rolland WB, Altay O, Tang J et al (2012) Etiology of stroke and choice of models. Int J Stroke Off J Int Stroke Soc 7:398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kumar DR, Hanlin E, Glurich I, Mazza JJ, Yale SH (2010) Virchow's contribution to the understanding of thrombosis and cellular biology. Clin Med Res 8:168–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lambert C, Sam Narean J, Benjamin P, Zeestraten E, Barrick TR, Markus HS (2015) Characterising the grey matter correlates of leukoaraiosis in cerebral small vessel disease. Neuroimage Clin 9:194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin B, Schmidt‐Kastner R, Busto R, Ginsberg MD (1999) Progressive parenchymal deposition of beta‐amyloid precursor protein in rat brain following global cerebral ischemia. Acta Neuropathol 97:359–368. [DOI] [PubMed] [Google Scholar]
- 48. Matthews FE, Brayne C, Lowe J, McKeith I, Wharton SB, Ince P (2009) Epidemiological pathology of dementia: attributable‐risks at death in the medical research council cognitive function and ageing study. PLoS Med 6:e1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McCarter JF, Liebscher S, Bachhuber T, Abou‐Ajram C, Hübener M, Hyman BT et al (2013) Clustering of plaques contributes to plaque growth in a mouse model of Alzheimer's disease. Acta Neuropathol 126:179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McGiff JC, Quilley CP (1981) The rat with spontaneous genetic hypertension is not a suitable model of human essential hypertension. Circ Res 48:455–464. [DOI] [PubMed] [Google Scholar]
- 51. Michalski D, Grosche J, Pelz J, Schneider D, Weise C, Bauer U et al (2010) A novel quantification of blood‐brain barrier damage and histochemical typing after embolic stroke in rats. Brain Res 1359:186–200. [DOI] [PubMed] [Google Scholar]
- 52. Nabika T, Ohara H, Kato N, Isomura M (2012) The stroke‐prone spontaneously hypertensive rat: Still a useful model for post‐GWAS genetic studies? Hypertens Res Off J Jpn Soc Hypertens 35:477–484. [DOI] [PubMed] [Google Scholar]
- 53. Niklass S, Stoyanov S, Garz C, Bueche CZ, Mencl S, Reymann K et al (2014) Intravital imaging in spontaneously hypertensive stroke‐prone rats‐a pilot study. Exp Transl Stroke Med 6:1. 10.1186/2040-7378-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pantoni L (2010) Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol 9:689–701. [DOI] [PubMed] [Google Scholar]
- 55. Paris D, Bachmeier C, Patel N, Quadros A, Volmar C‐H, Laporte V et al (2011) Selective antihypertensive dihydropyridines lower Aβ accumulation by targeting both the production and the clearance of Aβ across the blood‐brain barrier. Mol Med (Cambridge, Mass.) 17:149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pasi M, Charidimou A, Boulouis G, Auriel E, Ayres A, Schwab KM et al (2018) Mixed‐location cerebral hemorrhage/microbleeds: Underlying microangiopathy and recurrence risk. Neurology 90:e119–e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Passos GF, Kilday K, Gillen DL, Cribbs DH, Vasilevko V (2016) Experimental hypertension increases spontaneous intracerebral hemorrhages in a mouse model of cerebral amyloidosis. J Cereb Blood Flow Metab 36:399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Peres R, Guio F, de Chabriat H, Jouvent E (2016) Alterations of the cerebral cortex in sporadic small vessel disease: a systematic review of in vivo MRI data. J Cereb Blood Flow Metab 36:681–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ (2002) Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology 58:1629–1634. [DOI] [PubMed] [Google Scholar]
- 60. Pinter D, Enzinger C, Fazekas F (2015) Cerebral small vessel disease, cognitive reserve and cognitive dysfunction. J Neurol 262:2411–2419. [DOI] [PubMed] [Google Scholar]
- 61. Pirici D, Stanaszek L, Garz C, Niklass S, Heinze H‐J, Kalinski T et al (2017) Common impact of chronic kidney disease and brain microhemorrhages on cerebral Aβ pathology in SHRSP. Brain Pathol (Zurich, Switzerland) 27:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ramanathan A, Nelson AR, Sagare AP, Zlokovic BV (2015) Impaired vascular‐mediated clearance of brain amyloid beta in Alzheimer's disease: the role, regulation and restoration of LRP1. Front Aging Neurosci 7: 10.3389/fnagi.2015.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ritter MA, Droste DW, Hegedüs K, Szepesi R, Nabavi DG, Csiba L et al (2005) Role of cerebral amyloid angiopathy in intracerebral hemorrhage in hypertensive patients. Neurology 64:1233–1237. [DOI] [PubMed] [Google Scholar]
- 64. Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A et al (2013) Pericyte loss influences Alzheimer‐like neurodegeneration in mice. Nat Commun 4:2932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65. Schaffer CB, Friedman B, Nishimura N, Schroeder LF, Tsai PS, Ebner FF et al (2006) Two‐photon imaging of cortical surface microvessels reveals a robust redistribution in blood flow after vascular occlusion. PLoS Biol 4:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schreiber S, Bueche CZ, Garz C, Braun H (2013) Blood brain barrier breakdown as the starting point of cerebral small vessel disease?—new insights from a rat model. Exp Transl Stroke Med 5:4. 10.1186/2040-7378-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schreiber S, Bueche CZ, Garz C, Kropf S, Angenstein F, Goldschmidt J et al (2012) The pathologic cascade of cerebrovascular lesions in SHRSP: is erythrocyte accumulation an early phase? J Cerebral Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 32:278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schreiber S, Drukarch B, Garz C, Niklass S, Stanaszek L, Kropf S et al (2014) Interplay between age, cerebral small vessel disease, parenchymal amyloid‐β, and tau pathology: longitudinal studies in hypertensive stroke‐prone rats. J Alzheimer's Dis JAD 42:S205–S215. [DOI] [PubMed] [Google Scholar]
- 69. Shams S, Granberg T, Martola J, Charidimou A, Li X, Shams M et al (2017) Cerebral microbleeds topography and cerebrospinal fluid biomarkers in cognitive impairment. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 37:1006–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shi Y, Thrippleton MJ, Makin SD, Marshall I, Geerlings MI, de Craen AJ, et al (2016) Cerebral blood flow in small vessel disease: a systematic review and meta‐analysis. J Cereb Blood Flow Metab 36:1653–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shi Y, Wardlaw JM (2016) Update on cerebral small vessel disease: a dynamic whole‐brain disease. Stroke Vasc Neurol 1:83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Skrobot OA, Attems J, Esiri M, Hortobágyi T, Ironside JW, Kalaria RN et al (2016) Vascular cognitive impairment neuropathology guidelines (VCING): the contribution of cerebrovascular pathology to cognitive impairment. Brain J Neurol 139:2957–2969. [DOI] [PubMed] [Google Scholar]
- 73. Smith EE, Nandigam KRN, Chen Y‐W, Jeng J, Salat D, Halpin A et al (2010) MRI markers of small vessel disease in lobar and deep hemispheric intracerebral hemorrhage. Stroke 41:1933–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Stanimirovic DB, Friedman A (2012) Pathophysiology of the neurovascular unit: disease cause or consequence? J Cereb Blood Flow Metab 32:1207–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tanskanen M, Lindsberg PJ, Tienari PJ, Polvikoski T, Sulkava R, Verkkoniemi A et al (2005) Cerebral amyloid angiopathy in a 95+ cohort: Complement activation and apolipoprotein E (ApoE) genotype. Neuropathol Appl Neurobiol 31:589–599. [DOI] [PubMed] [Google Scholar]
- 76. Tarasoff‐Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E et al (2015) Clearance systems in the brain‐implications for Alzheimer disease. Nat Rev Neurol 11:457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Undas A (2014) Fibrin clot properties and their modulation in thrombotic disorders. Thromb Haemost 112:32–42. [DOI] [PubMed] [Google Scholar]
- 78. Undas A, Ariëns RAS (2011) Fibrin clot structure and function: a role in the pathophysiology of arterial and venous thromboembolic diseases. Arterioscler Thromb Vasc Biol 31:e88–e99. [DOI] [PubMed] [Google Scholar]
- 79. Undas A, Slowik A, Wolkow P, Szczudlik A, Tracz W (2010) Fibrin clot properties in acute ischemic stroke: relation to neurological deficit. Thromb Res 125:357–361. [DOI] [PubMed] [Google Scholar]
- 80. van Norden AG, Laat KF, de Gons RA, van Uden IW, van Dijk EJ, van Oudheusden LJ et al (2011) Causes and consequences of cerebral small vessel disease. The RUN DMC study: a prospective cohort study. Study rationale and protocol. BMC Neurol 2011:1. 10.4061/2011/647869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. van Veluw SJ, Jolink WMT, Hendrikse J, Geerlings MI, Luijten PR, Biessels GJ et al (2014) Cortical microinfarcts on 7T MRI in patients with spontaneous intracerebral hemorrhage. J Cereb Blood Flow Metab 34:1104–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Viswanathan A, Greenberg SM (2011) Cerebral amyloid angiopathy in the elderly. Ann Neurol 70:871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wang J, Ho L, Chen L, Zhao Z, Zhao W, Qian X et al (2007) Valsartan lowers brain beta‐amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J Clin Investig 117:3393–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang X, Chappell FM, Valdes Hernandez M, Lowe G, Rumley A, Shuler K et al (2016) Endothelial function, inflammation, thrombosis, and basal ganglia perivascular spaces in patients with stroke. J Stroke Cerebrovascular Dis Off J Nat Stroke Assoc 25:2925–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wardlaw JM (2010) Blood‐brain barrier and cerebral small vessel disease. J Neurol Sci 299:66–71. [DOI] [PubMed] [Google Scholar]
- 86. Wardlaw JM, Doubal F, Armitage P, Chappell F, Carpenter T, Muñoz Maniega S et al (2009) Lacunar stroke is associated with diffuse blood‐brain barrier dysfunction. Ann Neurol 65:194–202. [DOI] [PubMed] [Google Scholar]
- 87. Wardlaw JM, Makin SJ, Valdés V, Hernández MC, Armitage PA, Heye AK et al (2017) Blood‐brain barrier failure as a core mechanism in cerebral small vessel disease and dementia: evidence from a cohort study. Alzheimer's Dementia 13:634–643. [Google Scholar]
- 88. Wardlaw JM, Smith C, Dichgans M (2013) Mechanisms of sporadic cerebral small vessel disease: insights from neuroimaging. Lancet Neurol 12:483–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R et al (2013) Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12:822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Weller RO, Hawkes CA, Kalaria RN, Werring DJ, Carare RO (2015) White matter changes in dementia: role of impaired drainage of interstitial fluid. Brain Pathol (Zurich, Switzerland) 25:63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Weller RO, Subash M, Preston SD, Mazanti I, Carare RO (2007) Perivascular drainage of amyloid‐beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol (Zurich, Switzerland) 18:253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Xuereb JH, Brayne C, Dufouil C, Gertz H, Wischik C, Harrington C et al (2000) Neuropathological findings in the very old. Results from the first 101 brains of a population‐based longitudinal study of dementing disorders. Ann N Y Acad Sci 903:490–496. [DOI] [PubMed] [Google Scholar]
- 93. Yamada M (2015) Cerebral amyloid angiopathy: emerging concepts. J Stroke 17:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yan P, Bero AW, Cirrito JR, Xiao Q, Hu X, Wang Y et al (2009) Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J Neurosci Off J Soc Neurosci 29:10706–10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zhang L, Zhang ZG, Chopp M (2012) The neurovascular unit and combination treatment strategies for stroke. Trends Pharmacol Sci 33:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhang L, Zheng H, Luo J, Li L, Pan X, Jiang T et al (2018). Inhibition of endothelial nitric oxide synthase reverses the effect of exercise on improving cognitive function in hypertensive rats. Hypertens Res Off J Jpn Soc Hypertens 41:414. [DOI] [PubMed] [Google Scholar]
- 97. Zlokovic BV (2004) Clearing amyloid through the blood‐brain barrier. J Neurochem 89:807–811. [DOI] [PubMed] [Google Scholar]
- 98. Zlokovic BV (2010) Neurodegeneration and the neurovascular unit. Nat Med 16:1370–1371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Movie 1: HA stage 2—Non‐occlusive thrombus formation. Dextran (red) was used to mark the blood plasma and erythrocytes are detected as dark spots in front of the bright background. In the movie the formation of a non‐occlusive thrombus is presented. In the lower part of the vessel normal blood flow is detectable but after the vessel is branching, the upper left part of the vessel shows a significant reduction of the red blood cell flow velocity. Here, single blood cells are accumulating in the vessel (yellow arrow) and Dextran and erythrocytes are slowing down their flow velocity (white arrow). Even though three is still blood flow detectable, a non‐occlusive thrombus is formed.
Supplemental Movie 2: HA stage 3—Occlusive thrombus formation. Dextran (red) was used to mark the blood plasma and erythrocytes are detected as dark spots in front of the bright background. In the movie the formation of an occlusive thrombus is presented. In the upper part of the vessel, normal blood flow is detectable and the original vessel then leaves the level of the video. At this branching point of the vessel, a free‐floating thrombus is formed (arrow). Here, dextran and erythrocytes are accumulating in the lumen of the vessel and cannot flow through it because the following vessel segment is totally plugged (asterisk). This is indicated by long‐distant Dextran gaps and referred to as totally plugged vessel segment.
Supplemental Table 1: HA phenomena and HA stages of SHRSP that underwent Methoxy imaging. All 13 SHRSP that underwent Methoxy imaging were classified as Methoxy‐positive or –negative and were evaluated for the occurrence of the following HA phenomena: (1) accumulation of Dextran in the small vessel walls, (2) accumulation of erythrocytes in the small vessel walls, (3) a combination of (1) and (2, 4) accumulation of Dextran in the small vessel lumina, (5) non‐occlusive erythrocyte thrombi, (5a) a combination of (4) and (5, 6) occlusive erythrocyte thrombi, (6a) a combination of (4) and (6) and (7) totally plugged vessel segments. Additionally all rats were assigned to one of the following HA stages: stage 0—no HA features, stage 1A—small vessel wall damage, stage 1B—RBC flow reduction, stage 2—non‐occlusive thrombus formation, stage 3—occlusive thrombus formation. Animal numbers refer to Table 1. Methoxy positive SHRSP are marked in gray. HA, hypertensive arteriopathy; RBC, red blood cell.