

Abstract
Transmissible spongiform encephalopathies (TSEs) or prion diseases of animals notably include scrapie in small ruminants, chronic wasting disease (CWD) in cervids and classical bovine spongiform encephalopathy (C‐BSE). As the transmission barrier phenomenon naturally limits the propagation of prions from one species to another, and the lack of epidemiological evidence for an association with human prion diseases, the zoonotic potential of these diseases was for a long time considered negligible. However, in 1996, C‐BSE was recognized as the cause of a new human prion disease, variant Creutzfeldt‐Jakob disease (vCJD), which triggered an unprecedented public health crisis in Europe. Large‐scale epidemio‐surveillance programs for scrapie and C‐BSE that were implemented in the EU after the BSE crisis revealed that the distribution and prevalence of prion diseases in the ruminant population had previously been underestimated. They also led to the recognition of new forms of TSEs (named atypical) in cattle and small ruminants and to the recent identification of CWD in Europe. At this stage, the characterization of the strain diversity and zoonotic abilities associated with animal prion diseases remains largely incomplete. However, transmission experiments in nonhuman primates and transgenic mice expressing human PrP clearly indicate that classical scrapie, and certain forms of atypical BSE (L‐BSE) or CWD may have the potential to infect humans. The remaining uncertainties about the origins and relationships between animal prion diseases emphasize the importance of the measures implemented to limit human exposure to these potentially zoonotic agents, and of continued surveillance for both animal and human prion diseases.
Keywords: BSE, CWD, prion, scrapie, TSE, zoonotic
Introduction
Prion diseases, or transmissible spongiform encephalopathies (TSE), are fatal neurodegenerative diseases in which a key feature of the pathogenesis is the accumulation of a misfolded form (PrPSc) of a normal host glycoprotein (PrPC). The term prion (derived from proteinaceous infectious particle) arises from the hypothesis that infectious or contagious forms of these diseases are caused solely by the transmission of PrPSc 108.
Animal prion diseases include scrapie in sheep, bovine spongiform encephalopathy (C‐BSE) in cattle, chronic wasting disease (CWD) in cervids and transmissible mink encephalopathy (TME) in farmed mink, which all have an infectious etiology. The origin of new outbreaks or forms of animal prion disease is often obscure and therefore unpredictable, as evidenced by the recent emergence of CWD in Scandinavia and recognition of a novel prion disease of camels in Algeria 11, 17. In humans, as well as acquired prion diseases such as iatrogenic and variant Creutzfeldt‐Jakob disease (iCJD, vCJD), there are genetic/inherited prion diseases, but the most common form of disease is sporadic CJD (sCJD) for which the etiology is not fully understood 28, 57.
The first evidence for the infectious nature of prion diseases came from experiments in which scrapie was transmitted to healthy sheep by inoculation of brain extracts from a diseased animal 45, 46. Following this seminal result, experimental inoculation of scrapie and other prion diseases (kuru, Creutzfeldt‐Jakob disease) was attempted in a variety of species, including many commonly used laboratory rodents, as well as nonhuman primates 58. It was quickly recognized that attempts to transmit infection from one species to another are limited by a transmission barrier, also named “species barrier.” Transmission barriers often result in a lack of propagation of prion in the new host species. In other cases, inoculation of a prion into a new host species produces a low or inconsistent disease incidence, and prolonged incubation periods or subclinical infection. After one or more sub‐passages in the same host, the clinical incidence rate increases and incubation periods become shorter, and are ultimately very predictable for a defined dose and route of infection 21.
The molecular mechanisms that determine the permeability of transmission barrier to a prion in a particular host are still not fully understood. However, there is now a wealth of evidence that important factors influencing cross‐species transmission include the nature of the prion strain, compatibility between the primary amino acid sequence of donor and host PrP, and the dose/exposure route in the recipient host. Despite this progress, it still remains impossible to predict a priori the capacity of a prion to propagate in a new host species.
Concerns and speculation about the possibility of cross‐species transmission of animal prion diseases to humans have existed since the infectious nature of these diseases was demonstrated.
It has been known for several centuries that scrapie is endemic in sheep used for human food production 48. However, despite the likely exposure of certain individuals to infected sheep, no link could be established between scrapie and TSE occurrence in humans 132. This lack of evidence of zoonotic transmission led to the general opinion that a high transmission barrier protects humans from animal prion diseases. However, the emergence of vCJD in 1996 provided incontrovertible evidence that inter‐species transmission barriers are not sufficient to protect the human population from prion agents circulating in domesticated animals and wildlife 139. The resulting public health crisis provided the impetus for the development of novel experimental techniques and models, which have also been employed to estimate the risk to humans from other animal prion diseases. Using the example of BSE and vCJD, this paper will review and discuss the available evidence for associations between human and animal prion diseases.
Classical BSE and Variant CJD
History and Epidemiology
Classical BSE (C‐BSE) was first recognized in 1984–1985 as novel prion disease affecting cattle in the UK 136. The origin of C‐BSE has still not been clearly established, but the number of cases was amplified by the recycling of infected carcasses into cattle feed in the form of meat and bone meal (MBM) 138.
BSE was disseminated to at least 28 countries, mostly in Europe but also in the USA, Canada and Japan, through the export of infected live animals and/or contaminated MBM and livestock feed. However, the numbers of cattle affected were much lower than in the UK, with a total of 6193 recorded cases in EU countries between 1989 and 2016.
In the UK, the C‐BSE epidemic peaked in 1992, with more than 37 000 confirmed cases in that year (Figure 1). Legislation prohibiting the inclusion of MBM and other animal proteins in livestock feed in the UK (1988, 1996) and EU (2001) was instrumental in controlling the transmission of BSE and exposure of other farmed animals. The incidence of C‐BSE has now declined to very low levels, with no cases or a single‐digit number of cases reported each year since 2011 (Table 1, Figure 1). It has been estimated that globally over 1 million BSE‐infected cattle entered the human food chain, resulting in potential dietary exposure to C‐BSE for millions of consumers 6. Even if the number of C‐BSE cases that occurred into the UK exceeded by several orders of magnitude than those observed in other affected countries, international trade of food commodities could have resulted in exposure of people even in regions that did not experience autochthonous C‐BSE cases.
Figure 1.
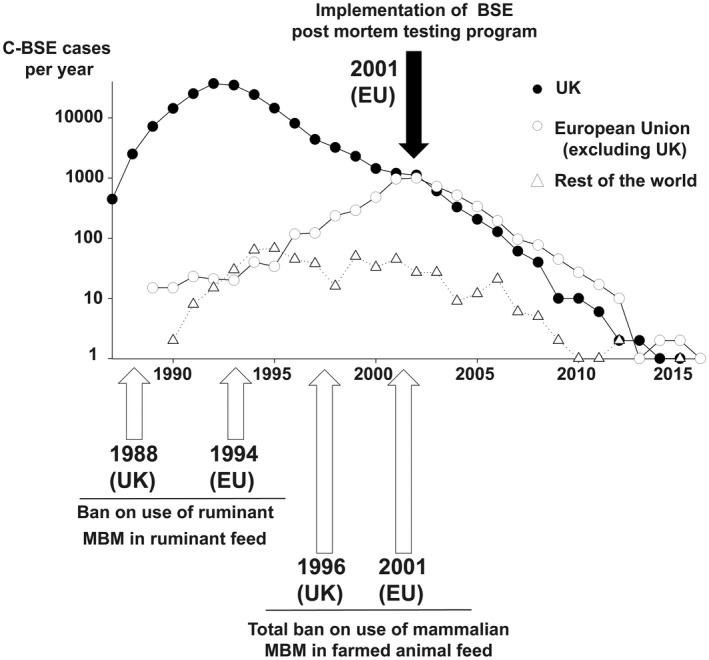
Number of confirmed C‐BSE cases (Logarithmic scale) reported to the OIE (Office International des Epizooties) by the UK, the European Union (excluding the UK) and the rest of the world, by year. Implementation dates of key control measures (bans on the use of meat and bone meal in farm animal feedstuffs) and active surveillance system (postmortem testing of slaughtered cattle and fallen stock) in the UK and the EU (also applicable to UK) are indicated on the graph.
Table 1.
Number of cattle tested postmortem and number of confirmed classical and atypical BSE cases in the European Union per year.
| 2001 | 2002 | 2003 | 2004 | 2005 | 2006 | 2007 | 2008 | |
|---|---|---|---|---|---|---|---|---|
| Total number of tested cattle (in millions) | 8.516 | 10.423 | 10.041 | 11.049 | 10.113 | 10.047 | 9.692 | 10.051 |
| C‐BSE cases | 2174 | 2129 | 1334 | 848 | 542 | 323 | 157 | 117 |
| H‐BSE cases | 2 | 3 | 4 | 2 | 3 | 3 | 5 | 5 |
| L‐BSE cases | 0 | 5 | 4 | 4 | 4 | 4 | 8 | 6 |
| 2009 | 2010 | 2011 | 2012 | 2013 | 2014 | 2015 | 2016 | |
| Total number of tested cattle (in millions) | 7.467 | 7.504 | 6.361 | 4.795 | 3.135 | 2.287 | 1.423 | 1.352 |
| C‐BSE cases | 55 | 37 | 23 | 12 | 3 | 3 | 3 | 1 |
| H‐BSE cases | 6 | 4 | 4 | 1 | 4 | 2 | 2 | 4 |
| L‐BSE cases | 5 | 4 | 3 | 6 | 1 | 6 | 1 | 0 |
The occurrence of a large epidemic of a novel prion disease in cattle rapidly raised concerns about the risk C‐BSE might represent for humans. The first protective measures for the food chain were implemented in November 1989, with a ban on the use of certain specified bovine offal in human food (Figure 2). Concomitantly, the UK Department of Health set up the National CJD Surveillance Unit in 1990, whose mission was to monitor the incidence and study the epidemiology of human prion diseases.
Figure 2.
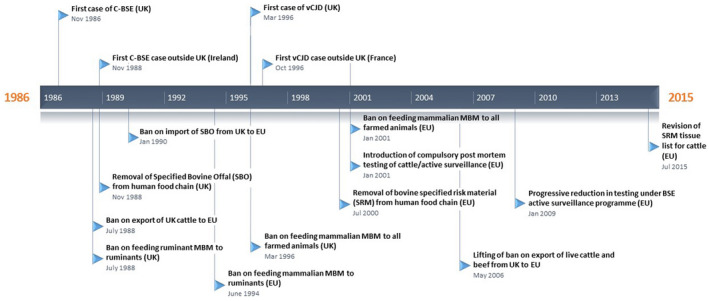
Timeline of major events and control measures for protection of animal and human health during the C‐BSE epidemics in the UK and European Union.
The detection of a novel prion disease (feline spongiform encephalopathy) in cats in the UK, and the experimental transmission of the C‐BSE agent to nonhuman primates (marmoset) further reinforced concerns regarding the ability of C‐BSE to cross interspecies transmission barriers 1, 12. In 1995, two cases of CJD were reported in teenagers in the UK, an unusually early onset of disease 14, 27. These patients also displayed atypical clinical symptoms and distinctly different neuropathological changes compared to known human prion diseases, and the emergence of a new form of prion disease, named variant CJD (vCJD), naturally pointed to C‐BSE as the probable causative agent 139.
Definitive evidence for the link between BSE and vCJD was provided by transmission studies in inbred mouse lines, in which mice injected with vCJD exhibited phenotypes (incubation periods and lesion profile) indistinguishable from those obtained following transmission of BSE or feline spongiform encephalopathy (FSE), indicating that all three diseases were caused by the same prion agent 32. Similarly, C‐BSE isolates from individual affected cattle in different farms and locations displayed identical phenotypes following strain typing in mice, which confirmed that C‐BSE was caused by a single prion strain 33, 38.
To date, out of the 231 vCJD cases identified worldwide, 178 cases have occurred in UK residents, and a number of other cases have occurred in individuals with a history of residence in the UK during the high risk period 1980–1996 (http://www.cjd.ed.ac.uk/surveillance/data-and-reports). All definite clinical cases of vCJD that have undergone PRNP genetic analysis are homozygous for methionine at codon 129 (129 MM), apart from the latest UK case, who was heterozygous (129 MV) 95.
After a peak in 2001–2002, the number of vCJD cases recorded in the UK has declined. While the limited number of cases is consistent with inefficient transmission of the C‐BSE agent to humans, many uncertainties remain concerning the number of individuals incubating vCJD in the exposed population 49. To address this issue, several studies have been performed to estimate the prevalence of vCJD infection in the UK population. As abnormal PrP deposits (PrPd) can be detected in the lymphoid organs of vCJD patients during preclinical and clinical phases of the disease 64, 106, prevalence studies were based on anonymized surveys of more than 45 000 appendix and tonsil samples removed and archived following routine surgery. Targeted patients belonged to age cohorts most likely to have experienced dietary exposure to C‐BSE, and the tissues were tested for the presence of PrPd by immunohistochemistry (IHC). In two separate surveys, a total of 19 appendix samples were positive for abnormal PrP accumulation 59, 67. Strikingly, positive samples were found to have codon 129 methionine‐methionine, methionine‐valine and valine‐valine PRNP genotypes, suggesting that the BSE agent may infect individuals of all codon 129 genotypes 59. These findings led to an estimated global prevalence of abnormal PrP of up to 1 in 2000 of the UK population (95% confidence interval 1/3500–1/1250) 59. Whether this represents the true prevalence of vCJD infection is still a matter of debate, particularly as the prevalence estimates are not consistent with the small observed numbers of clinical vCJD cases. One interpretation is that there are subclinically infected individuals in the population who may never develop vCJD themselves, but who may be a source of iatrogenic transmission of infection, for example, by blood or organ donation. In light of these unresolved concerns, it is likely that vCJD will remain a public health issue for the foreseeable future.
Pathogenesis of C‐BSE
Experimental oral challenges of cattle with C‐BSE have been used to study disease pathogenesis in the natural host. In cattle receiving a range of oral doses, transmission was observed in all dose groups, including one of 15 animals given the lowest dose of 1 mg of BSE‐infected cattle brain, demonstrating the sensitivity of cattle to C‐BSE 76. In experiments where groups of cattle were culled at different intervals following oral infection with BSE, infectivity was first detected in the distal ileal Peyer’s patch (PP), and later in the central nervous system and specific sensory ganglia a few months before the onset of clinical disease 51, 68, 135. The distribution of infectivity and PrPSc in the CNS in preclinical animals suggests that neuroinvasion from the gastrointestinal tract occurs via the autonomic innervation 68, 123. Studies to date have failed to find evidence of infectivity in lymphoid tissues other than the ileal PP and tonsils of preclinical or clinically affected cattle, or in blood and milk 51, 135, 137. At late stages of the disease, there is growing evidence for centrifugal spread of infection along neuronal pathways to tissues such as the tongue and nasal mucosa 13.
Information from these experiments on the distribution of the C‐BSE agent in the tissues of infected cattle underpinned the development of a list of tissues defined as Specified Risk Material (SRM). Legislation to enforce the systematic retrieval of SRM from all slaughtered cattle was introduced in the UK (1996) and EU (2001), preventing the entry of significant amounts of C‐BSE infectivity into the human food chain (Figure 2). This measure was decisive in limiting the occurrence of human dietary exposure to the C‐BSE agent 114.
Experimental Modeling of the Human Transmission Barrier to BSE
Careful surveillance and epidemiological studies were instrumental in the identification of zoonotic transmission of classical BSE. However, the success of this approach depended on the occurrence of a large BSE epidemic in the UK cattle population followed by the emergence of a new human prion disease phenotype (variant CJD) in the same country 139. Given the low total numbers of vCJD cases, if transmission of BSE had resulted in a disease similar or indistinguishable from sporadic CJD, it is unlikely that surveillance for human prion diseases in the UK alone would have been able to identify its zoonotic properties. Moreover, epidemiology necessarily relies upon the retrospective analysis of events, and therefore obviously offers no possibility of preventing human transmission before it occurs.
In that context, a number of in vivo and in vitro experimental models have been developed, using C‐BSE/vCJD as a benchmark, with the aim of assessing the relative ability of animal prion agents to cross the human transmission barrier 21, 37, 112, 113. Among these models, transmission in nonhuman primates and transgenic mice expressing human PrP (Table 2) is currently considered as the most informative and reliable approach 114.
Table 2.
Transmissibility of animal prion diseases in animal models of the human species barrier.
| Prion diseases | Positive transmission reported in animal models of the human species barrier | |||
|---|---|---|---|---|
| New World Primates* | Old world Primates* | Human PrP expressing transgenic mice‡ | ||
| Cattle | C‐BSE | Yes (IC and oral route) (12) | Yes (IC, intravenous and oral route) | Yes (10, 36, 54,66, 69, 104) |
| (65, 84, 87) | No (25) | |||
| Atypical H‐BSE | – | still ongoing (IC route) (41) | No (20, 142) | |
| Atypical L‐BSE | – | Yes (IC route) (40, 103) | Yes (20, 69, 75) | |
| No (142) | ||||
| Small Ruminants | Classical Scrapie§ | Yes (IC route) (12) | Yes (IC Route) (41) | Yes (36) |
| Atypical Scrapie | – | Ongoing experiment (IC route) (41) | No (107, 141, 142) | |
| Cervids | CWD§ (North American isolates) | Yes (IC and oral route) (109) | No (IC and oral route) (109) | No (74, 125, 142) |
| Yes (IC and oral route)¶ | ||||
Squirrel monkeys or Marmoset.
Cynomolgus Macaques –.
Mice were all inoculated by the intracerebral route (IC) –.
These prion diseases are associated with multiple prion strains.
Czub et al personal communication: https://www.cdc.gov/prions/cwd/transmission.html.
BSE transmits efficiently to cynomolgous macaques by intracerebral and intravenous routes (including blood transfusion), producing similar clinical and neuropathological features to those observed in human patients affected by vCJD 65, 85, 87. The relative efficacy of C‐BSE transmission observed in macaques exposed by the oral route to low infectious doses of C‐BSE also fits well with the limited number of vCJD cases that occurred in dietary‐exposed human populations 84. BSE has also been successfully transmitted to other nonhuman primates including marmosets, squirrel monkeys and lemurs (Table 2) 12, 26. The degree to which different nonhuman primate species are evolutionarily related to humans may influence the interpretation of zoonotic risk following experimental transmission of animal prions. As Old World monkeys (including macaques) are more closely related to humans than New World monkeys (eg, squirrel monkeys) and lemurs, cynomolgus macaques are regarded as a better model to assess the permeability of the human transmission barrier to C‐BSE and other animal prion diseases.
The major drawback of using nonhuman primates in prion research are ethical issues and the long incubation periods following infection, which make such experiments very expensive and severely limit their use in characterizing the zoonotic potential of animal prions.
Following the demonstration that the transmission barrier preventing infection of mice with hamster‐adapted scrapie could be removed by the expression of hamster PrPC in transgenic mice 117, several transgenic mouse lines expressing human PrPC (TgHu) have been developed. These include lines that over‐express PrP 30, 118, 126, 134 and gene‐targeted lines that express PrP at physiological levels under the control of mouse PRNP regulatory elements 24. These mouse models express different variants of the human PRNP gene including the methionine/valine (M/V) di‐morphism at codon 129, which is a major determinant of susceptibility to human prion disease 92, 143. Most TgHu mouse lines have been shown to propagate human prion diseases, for example, sCJD, without an apparent species barrier 10, 24, 66, 104.
Transmission of C‐BSE to transgenic mice over‐expressing human 129 MM, 129 VV or 129 MV PrP resulted in low attack rates and long incubation times in 129 MM and apparent absence of transmission of the agent in 129 MV and VV PrP expressing mice (Table 2) 10, 66, 104. Iterative passages of C‐BSE into 129 MM mice led to an increase in the attack rate and a slight reduction of the incubation period, but did not result in disease transmission to 129 MV‐ and 129 VV‐expressing mice 36, 54, 69. In contrast, vCJD isolates transmitted to all three humanized lines, producing a gradation in transmission efficiency from MM (most efficient) to VV (less efficient) 54. In gene‐targeted 129 MM, 129 MV and 129 VV mouse lines, inoculation with BSE failed to transmit the infection, while vCJD transmitted to all three lines with a similar gradation in efficiency to that seen in over‐expressing transgenic models 24.
Taken together, the results of experimental transmission of C‐BSE to TgHu mice converge to indicate a high transmission barrier to C‐BSE in humans, and a higher susceptibility of 129 MM individuals to infection, which is consistent with the vCJD epidemiological features observed in exposed human populations. The relative efficiency of transmission of C‐BSE, vCJD and sCJD to TgHu mice strongly supports the view that these models provide good predictive value of the capacity of prion agents to cross the human transmission barrier. However, these models also have their limitations, including the short lifespan (2–3 years) of mice in comparison to the long incubation periods (several decades) reported in patients accidentally exposed to human prions (like sCJD‐contaminated growth hormones) 28.
Scrapie in Small Ruminants
Classical scrapie is the archetype of prion disease. It was reported for the first time in sheep in the UK in 1732 and few years later (1759) in Germany. Over the following centuries, scrapie spread to many countries in the world through the export of living animals, and is still endemic in most of these regions 47, 48.
In 1998, a new form of sheep prion disease (named Nor98) was identified in Norway. The epidemiological features and biochemical properties of PrPSc associated with Nor98 cases clearly differed from scrapie cases that had been previously reported (termed “classical” scrapie), and the disease was therefore considered to be an “atypical” form of scrapie 16.
Classical scrapie can be caused by several prion agent strains. Historically, identification of scrapie strains has relied on characteristic biological phenotypes (incubation period and lesion profile) observed following transmission to a panel of inbred mouse lines. However, the propagation of natural prion isolates into inbred mice lines requires passage through a transmission barrier, which can result in the non‐propagation of certain isolates and/or a radical evolution (mutation) of the prion agent they contain 31, 55. Therefore, transmission to conventional mouse models is unlikely to provide a comprehensive and reliable picture of the diversity of prion agents in small ruminants. Transgenic mice expressing ovine or caprine Prnp genes display a lower transmission barrier to scrapie agents 61, 117. Serial transmission of about 80 scrapie isolates from Europe in such mouse lines has so far permitted the identification of at least four phenotypically distinct classes of classical scrapie agents 18, 21, 127, 128, 129. In contrast to classical scrapie, no strain variability has been observed among isolates of atypical scrapie 9, 60, 86.
For the most part, identification of scrapie cases has relied on clinical suspicion (passive surveillance). Starting in 2001, active surveillance for prion diseases of small ruminants was implemented in EU countries, and more recently in a number of other countries, for example, the USA and Canada. Active surveillance relies on systematic testing of a proportion of the slaughtered or found‐dead animals for the detection PrPSc in the posterior brainstem. These surveillance programs resulted in the identification of atypical scrapie cases in most EU member states, the USA and Canada, and indeed, atypical scrapie has been incidentally detected in many countries across the world, including those considered free of classical scrapie, that is, Australia and New Zealand 16. Data from the EU active surveillance program clearly demonstrated that earlier evaluations based on passive surveillance had significantly underestimated the prevalence of prion diseases in small ruminants and their geographical distribution 53.
A review of the epidemiology of classical and atypical scrapie in EU member states, using active surveillance data collected between 2002 and 2012, was published by the European Food Standards Agency (EFSA) in 2014 63. Over this period, a total of 4.7 million sheep and 1.4 million goats were tested, and classical and/or atypical scrapie cases were identified in 25 EU countries. The annual crude prevalence of classical scrapie in the EU was equivalent to about 9 cases per 10 000 tested animals, but prevalence estimates differed considerably among different individual member states. For example, in Cyprus, the annual crude prevalence of classical scrapie was between 10 and 800 times higher than that observed in other affected countries. For affected flocks, the within‐flock prevalence was on average 20 times higher than the apparent prevalence in the general population identified by active surveillance. Over the same period, atypical scrapie was detected with an overall prevalence of about 5.8 cases per 10 000 tested animals. In contrast with classical scrapie, atypical scrapie displayed a similar prevalence over time and in different countries 53.
In both sheep and goats, susceptibility to classical scrapie is strongly influenced by polymorphisms of the gene (Prnp) encoding for PrP protein. In sheep, the A136R154R171 (denoting amino acids encoded at Prnp codons 136, 154 and 171) haplotype is associated with a very high resistance to infection in homozygous individuals. Heterozygous ARR individuals also have a reduced susceptibility to infection 80, 91. In goats, the K222, S146 and D146 polymorphisms of the Prnp gene are also associated with a strong resistance to infection by classical scrapie 43, 105. The EU, the USA and several other countries have implemented breeding policies in sheep, with the aim of control/eradication of classical scrapie by increasing the frequency of the ARR allele in affected flocks, and in the general sheep population. Similarly, several countries are now considering the development of Prnp genotype selection programs for the control and eradication of scrapie in goats.
The association between genetic Prnp variations and susceptibility to atypical scrapie is totally different from that observed in classical scrapie. Susceptibility to atypical scrapie is linked to polymorphisms at Prnp codons 141 and 154, and strikingly, ARR allele carriers (both homozygous and heterozygous) that are resistant to classical scrapie can develop the disease 9, 97, 98. This difference means that genetic selection for the ARR allele, which has been the basis of successful control programs for classical scrapie, will not be effective in controlling atypical scrapie at the flock/herd or population level.
According to the EFSA expert group, the eradication and control measures that were implemented at the EU level from 2001 (including selective breeding for scrapie resistance) were very effective in controlling classical scrapie outbreaks at the flock/herd level. However, over the studied period, a statistically significant reduction in classical scrapie prevalence/incidence was demonstrated in only six EU countries. These results demonstrate the difficulties in monitoring the epidemiology of animal prion diseases and the effect of control measures at a population level.
Classical scrapie is a prion disease that, under natural conditions, is acquired from the environment and/or other infected animals by the oral route 48. The within‐host dissemination of the classical scrapie agent in naturally exposed small ruminants has been carefully characterized by the study of animals born and raised in endemically infected flocks.
These studies have established that infection usually occurs during the first weeks of life. The scrapie agent enters the host animal via gut‐associated lymphoid tissues (GALT), for example, Peyer’s patches, before rapid spread to draining mesenteric lymph nodes and later to all secondary lymphoid organs 3, 133. The amount of infectivity and PrPSc in lymphoid tissues increases with age before reaching a plateau level. Prnp genotype appears to influence the extent of scrapie replication in lymphoid tissues, for example, sheep heterozygous for the ARR allele have much less detectable PrPSC in the lymphoreticular system 70. The scrapie agent disseminates to the CNS (brain and spinal cord) about halfway through the incubation period, apparently via axonal transport through the enteric (autonomic) nervous system 3, 133. In later stages, the agent appears to redistribute (centrifugally) from the central to the peripheral nervous system and skeletal muscle 5. In blood, the infectious agent can be detected as early as three months of age and persists throughout the incubation period 83. In clinically normal ewes infected with scrapie, the placenta accumulates large amounts of infectivity, and plays a major role in the dissemination of the prion into the environment and to other individuals 81, 111, 131. Similarly, colostrum and milk were shown to contain infectivity, and their capacity to transmit disease to suckling lambs was demonstrated 78, 82.
The pathogenesis of atypical scrapie has not been characterized to the same extent. Initial investigations failed to identify PrPSc accumulation in peripheral (non‐neuronal) tissues collected from field cases or experimental atypical scrapie cases 4, 16. The apparent restriction of the agent to the CNS was interpreted to support the hypothesis that atypical scrapie could be a spontaneous disorder of PrP folding and metabolism, occurring in aged animals without external cause 16. In addition, there was no statistical difference in the prevalence of atypical scrapie between the general population and flocks where a positive case had been identified, providing further support for the idea that atypical scrapie may not be a contagious disease 52. However, low levels of infectivity have been detected in skeletal muscle, peripheral nerves and lymphoid tissues of animals infected naturally or experimentally with atypical scrapie 4. In the same study, brain samples containing very high levels of infectivity were negative for PrPSc using the most sensitive current diagnostic tests. Moreover, atypical scrapie can be experimentally transmitted via the oral route in small ruminants, resulting in a similar clinicopathological phenotype to that observed in natural cases 122. These findings mean that the origin and etiology of atypical scrapie (spontaneous disorder vs. acquired disease) remain an open question.
Zoonotic Potential of Classical and Atypical Scrapie
Several epidemiological studies, generally based on case‐control approaches, have failed to identify exposure to small ruminants or small ruminant products as a risk factor for developing CJD 29, 132. Countries considered to be scrapie‐free, such as Australia and New Zealand, display a similar sCJD prevalence to countries affected with scrapie. In addition, there is no apparent difference in the range of clinical and pathological manifestations of human TSE cases between scrapie‐affected countries and those designated scrapie free. Together, these different lines of evidence have led to the general conclusion that small ruminant prion diseases were of negligible risk to humans.
Recently, cases of atypical scrapie were confirmed in both Australia and New Zealand, casting serious doubt on the validity of one of the most popular arguments used to reject the zoonotic potential of small ruminant prion diseases 42, 73. More generally, active surveillance programs for TSE in small ruminants revealed how inaccurate knowledge was on the prevalence and geographical distribution of TSEs in small ruminants 53, and highlighted the limited value of past epidemiological studies aiming at assessing the zoonotic potential of animal TSEs.
Data collected through active surveillance programs offer an opportunity to reassess the zoonotic potential of small ruminant TSEs through informed and modern epidemiological investigations. However, the incubation period for prion disease in humans after exposure to prions via peripheral routes, such as in cases of iatrogenic CJD and kuru, can exceed several decades 28, 39. In this context, it will be a challenge to combine epidemiological data collected contemporaneously in animal and human populations to determine the existence of a causative link between prion disease occurrences in these different hosts.
Seminal early experiments failed to demonstrate the transmissibility of classical scrapie in nonhuman primates 58. However, subsequently one sheep classical scrapie isolate was transmitted to two intracerebrally challenged marmosets (Table 2). The incubation periods observed with this scrapie isolate were slightly shorter than with a cattle BSE isolate, suggesting that that transmission barrier for both isolates might not be different 12. More recently, successful transmission of a classical scrapie isolate to a cynomolgus macaque has been described. The incubation period in this animal was prolonged (>10 years following intracerebral challenge) and the neuropathology observed was unique in comparison to other animal prion diseases (C‐BSE, L‐BSE) transmitted in this model 41. To date, there are no available results concerning the experimental transmission of atypical scrapie to nonhuman primates (Table 2).
The transmission of both classical and atypical scrapie isolates to TgHu mice has been tested in several studies (Table 2). In gene‐targeted transgenic mouse lines expressing physiological levels of human PrP and challenged intracerebrally with a number of natural sheep and goat scrapie isolates, there was no evidence of infection or clinical disease following primary passage 107, 141, 142. However, serial passages in the same transgenic mouse lines, which could allow the identification of subclinical infection, were not performed in these studies. The same mouse lines were also shown to be resistant to cattle BSE 25, and therefore this model does not allow the assessment of whether the zoonotic potential of classical or atypical scrapie is lower or higher than that of BSE.
In a more recent study, a panel of classical scrapie isolates were tested in transgenic mouse lines that over‐express human PrP 36. The mouse lines represented 129 MM (tg340), 129 VV (tg361) and 129 MV (F1 cross of tg340 and tg361) PRNP genotypes. Following intracerebral inoculation of the three mouse lines with a panel of six natural sheep scrapie isolates (collected in EU countries between 1994 and 2002), clinical disease was not identified in any inoculated mice, but PrPSc accumulation was observed in the brain of two out of six 129 MV mice infected with a single scrapie isolate. Serial passage of the isolates in the same mouse lines led after second passage to positive transmission, resulting in clinical signs in mice inoculated with three of the six isolates. Interestingly, the sheep scrapie prions that propagated in humanized transgenic mouse models displayed a transmission efficiency (attack rate on first and second passage) that was comparable to that of cattle BSE. After third passage, the propagated prions displayed a phenotype (incubation periods and PrPSc distribution in the brain) that was identical to those causing sporadic CJD (sCJD) in humans. This last finding raised important questions about the possible link between TSE in small ruminants and occurrence of human TSE cases. These transmission experiments unambiguously showed that sheep scrapie prions propagate in mice that express variants of human PrP. While the efficiency of transmission at primary passage was low, subsequent passages resulted in a highly virulent prion disease in human PrP expressing mice.
Atypical BSE
The implementation by the EU of an active surveillance system in ruminant livestock in 2001, involving testing of healthy slaughtered cattle and fallen stock (Figure 2), led to the discovery of “atypical” cases of BSE in cattle, which were often not associated with overt neurological abnormalities/disease. Two atypical phenotypes were observed, categorized on the basis of low and high apparent molecular masses of unglycosylated protease‐resistant PrP on western blots, and were termed as bovine amyloidotic spongiform encephalopathy (BASE) or L‐BSE and H‐BSE, respectively 22, 35.
Transmission of atypical BSE isolates in various mouse models clearly demonstrated that H‐ and L‐type BSE cases are caused by specific prion strains that differ from the classical BSE/vCJD agent 18, 19, 34, 75. The transmission of H‐BSE field isolates to transgenic mice expressing bovine PrP resulted in the propagation of an agent with phenotypic characteristics of classical BSE in some animals 130. This suggested that a low level of C‐BSE agent could be present in a proportion of the H‐BSE isolates and might represent a source for C‐BSE re‐emergence in cattle.
The L‐BSE and H‐BSE cases reported so far were mainly detected in asymptomatic cattle 8 years of age or older, in contrast to C‐BSE, where the majority of cases were 4–6 years old. Since 2001, a total of 60 cases of L‐BSE and 44 cases of H‐BSE were reported in the whole EU (Table 1). Epidemiological studies in the French cattle population indicated that the apparent prevalence of atypical BSE cases is very low (1.9 cases H‐BSE and 1.7 cases L‐BSE per million tested cattle over 8 years old) 23. Outside the EU, rare atypical BSE cases have been reported in Japan, the USA, Canada, Switzerland and Brazil 50, 88, 115.
The origin of H‐BSE and L‐BSE cases is unknown. It has been argued that the low prevalence and the advanced age of positive animals provide evidence for a spontaneous origin of atypical BSE, but an infectious etiology cannot be definitively ruled out.
Limited tissues are available from L‐BSE and H‐BSE cases identified through surveillance; therefore, experimental transmission studies in cattle are the main source of information concerning the pathogenesis of atypical BSE agents. Following IC challenge, abnormal prion protein accumulation was identified in the central nervous system (brain, spinal cord and retina), the peripheral nervous system (autonomic and motor) and at lower levels in skeletal muscle (muscle spindles) of affected animals. No consistent prion accumulation was detected in the lymphoid organs 13, 77, 100, 124. Following oral challenge of cattle with a range of doses of L‐BSE‐infected cattle brain homogenate, only one animal that received the highest dose (50 g) developed neurological clinical signs. This animal showed a similar tissue distribution of PrPSc to that observed in IC‐challenged animals (central and peripheral nervous systems), although there were subtle differences in the distribution of PrPSc within the brain regions 101.
Other experiments in which cattle were orally challenged with L‐BSE and H‐BSE are still ongoing, and the final analysis of the pathogenesis of atypical BSE in comparison to C‐BSE awaits their results. However, the examination of a limited panel of tissues collected from natural L‐BSE cases in Italy confirmed the apparent restriction of the prion to the central and peripheral nervous systems 124.
Due to the low apparent prevalence of atypical BSE and the design of cattle TSE surveillance programs, it is unlikely that atypical BSE cases are efficiently detected. Therefore, it should be assumed that small numbers of atypical BSE incubating cattle are entering the human food chain. The many uncertainties related to the distribution of atypical BSE cases in the cattle population mean that epidemiological approaches are unlikely to be informative in assessing the zoonotic abilities of these prions.
Intracerebral injection of L‐BSE isolates from European and Japanese cattle into cynomolgus macaques resulted in disease transmission with shorter survival times than in C‐BSE‐infected macaques (23–25 months for L‐BSE vs. 38–40 months C‐BSE) (Table 2) 40, 103. In contrast, no positive transmission was reported in macaques that were inoculated with H‐type BSE 41. Inoculation of L‐BSE, H‐BSE and C‐BSE isolates into transgenic mice over‐expressing human PrP demonstrated that L‐BSE transmitted more efficiently than C‐BSE on primary transmission, with 100% attack rates and no shortening of incubation periods on subsequent sub‐passage (Table 2). In contrast, H‐BSE isolates failed to transmit in the same mouse model 20. Another study using a different transgenic mouse model expressing human 129 M PrP also showed efficient primary transmission of L‐BSE 75. Biological strain typing of L‐BSE and a panel of sCJD subtypes in human PrP transgenic mice failed to find any evidence that L‐BSE causes a recognized form of sCJD 69.
Collectively, these results support the view that H‐BSE agent has a low zoonotic potential. In contrast, L‐BSE displays an equal or greater virulence than C‐BSE in primate and TgHu mouse models, and may therefore pose a higher risk of zoonotic infection.
Chronic Wasting Disease
Chronic wasting disease (CWD) was first recognized as a disease of captive mule deer in Colorado (USA) during the 1960s, and confirmed to be a prion disease in 1980 140. Since then, the CWD‐affected area of North America has considerably expanded, with the disease now having been identified in 20 US states and two Canadian provinces (Saskatchewan and Alberta). CWD has become endemic in wild and captive cervid populations in most of these regions. It affects the majority of North American endemic cervid species, with the notable exception of free‐ranging caribou in Canada. The prevalence of CWD in free‐ranging cervids varies across North America, but can be as high as 30% in some areas, and the spread of CWD in North America is likely to be irreversible 140. Outside North America, CWD infection has been confirmed in captive cervids in South Korea as a result of importation of subclinically infected animals from Canada 72. Despite the implementation of vigorous control and eradication measures, CWD is still present in South Korea.
In April 2016, CWD was found in a free‐ranging reindeer (Rangifer tarandus) population in the Nordfjella region of Norway, and then later (June 2016) in two European moose (Alces alces), in a different area of the country 17. This led to intensive active surveillance across Norway, and the implementation of a culling program in the affected wild reindeer population. At the time of writing, 17 additional cases of CWD have been identified in reindeer from Nordfjella. One additional moose and one red deer from Norway also tested positive, and a further CWD‐infected moose was identified in Finland. At this stage, it is still unclear whether the discovery of CWD in Europe has any relationship with the epidemic observed in North America. Epidemiological studies based on large‐scale testing of free‐ranging cervids were recently implemented by several northern European countries, and should be helpful in documenting the geographical distribution and prevalence of CWD in European cervid populations.
There is clear evidence demonstrating that several prion strains are responsible for CWD in North American cervids. Inoculation of a panel of CWD isolates from various species and geographic locations in North America into transgenic mice over‐expressing cervid PrP indicated the presence of at least two CWD prion strains (referred to as CWD1 and CWD2) that circulate either independently or as a strain mixture 7. Bioassays in heterologous PrP transgenic mouse models or in conventional rodent models are consistent with these results 44, 125. However, it is unlikely that the strain typing work carried out so far has provided a definitive picture of the diversity of CWD strains that are circulating in North American cervid populations. Transmission experiments for strain typing of Norwegian CWD isolates are still ongoing, and thus definitive results are not yet available. Preliminary data presented by several teams in the 2018 International Prion Congress suggest that the phenotypic characteristics of the Norwegian isolates are different from those observed so far in North American isolates.
In North America, CWD pathogenesis has been investigated using both naturally exposed and experimentally challenged animals. Under natural exposure, the infection apparently occurs by the oral route following contact with an infected individual or contaminated environment 96. The pathogenesis and PrPSc distribution in CWD are very similar to that reported in classical scrapie in small ruminants. Initial entry of the agent occurs through tonsils and GALT with rapid involvement of the lymphoreticular system (LRS) and the enteric nervous system, followed by neuroinvasion of the CNS via autonomic nervous system pathways 56, 110, 119, 121. Involvement of the LRS seems to vary between deer and wapiti, with less abnormal PrP deposition in the lymphoid tissues of wapiti compared with deer 110. PrPSc has been detected in a large number tissues of affected deer, including those commonly consumed as venison (heart, skeletal muscles, tongue, liver, kidneys) or used as “natural medicine” (antler velvet) 6, 8, 94, 120.
In both captive and wild cervids, CWD has been demonstrated to be highly contagious 93. During the preclinical phase of infection, the CWD agent has been demonstrated in placenta, saliva, faeces and urine, which are all likely to contribute both to inter‐individual transmission and contamination of the environment 62, 89.
So far, 16 amino acid polymorphisms have been described in the PrP sequence of different cervid species, some of which are associated with lower rates of CWD infection and slower progression of the disease in natural hosts 116. For instance, in wapiti (Cervus canadensis nelsoni), the L132 allele (vs. M132) is associated with partial protection against CWD infection 99, while in mule deer (Odocoileus hemionus), the S/F dimorphism at codon 225 influences susceptibility to the disease, the 225F allele being partially protective against infection under natural exposure conditions 71. As in small ruminants affected with classical scrapie, the selection of PrP CWD‐resistant alleles could be an effective means to control the disease and prevent cervid depopulation in endemic areas, at least in captive herds. However, none of the polymorphisms identified so far seems to provide a sufficient level of disease protection to make genetic selection a feasible proposition for control of CWD.
No new or unusual form of human TSE has been identified so far in countries where CWD epidemics have developed 2. Comparison of the CJD prevalence rates between North America and other countries (Europe) does not suggest that CWD is responsible for a detectable increase in human prion disease prevalence. A retrospective study carried out using State death registry data collected between 1979 and 2001 in Colorado failed to identify any statistical difference in CJD prevalence between CWD endemic vs. CWD non‐endemic counties 90. In the USA, retrospective investigations identified several sCJD cases who had a history of potential or demonstrated exposure to CWD prions through venison consumption or hunting cervids in affected areas 15, but longitudinal studies related to known dietary exposure to CWD failed to demonstrate an increased risk of developing a prion disease in these individuals 102. Nevertheless, considering the limited duration of the observation period in these studies (in comparison with the potentially very long incubation periods for TSE in man), the significance of these observations remains uncertain. Collectively, the epidemiological data suggest a lack of causative link between CWD epidemics in North America and the occurrence of human TSEs. However, because of the considerable limitations of the data supporting this statement, it would probably be unwise at this stage to conclude an absence of zoonotic risk associated with CWD agents.
Although CWD was relatively recently identified, a number of studies specifically aiming at clarifying the capacity of CWD agent(s) to cross the human species barrier have already been carried out through experimental inoculation of TgHu mice and primates. A total of seven CWD isolates have been inoculated into different conventional (wild‐type) and TgHu mouse models, including models that expressed the 129 M and/or the 129 V variants of the PRNP gene (Table 2). The lack of clinical disease or PrPSc accumulation in the brains of the inoculated mice indicates the existence of a substantial transmission barrier 74, 79, 125, 142. However, it should be noted that these studies did not include secondary passage in TgHu mice, which was necessary for revealing the transmissibility of scrapie isolates and certain C‐BSE isolates in similar models 36.
Two separate studies have attempted to transmit CWD isolates to cynomolgus macaques (Table 2). In the first study, cynomolgus macaques and squirrel monkeys were challenged by intracerebral and oral routes with CWD‐infected brain homogenate. Although squirrel monkeys proved to be susceptible to CWD, there has been no evidence of transmission in macaques after observation periods ranging from 1.5 to over 13 years post‐inoculation 109. In the second study, 18 cynomolgus macaques were challenged with CWD by different routes, including oral inoculation with muscle tissue from CWD‐infected deer. The results of this study are still unpublished, but presentations at recent international scientific conferences and meetings (https://www.cdc.gov/prions/cwd/transmission.html) described the occurrence of neurodegenerative disease 4.5–6.3 years post‐inoculation in 5 out of 18 of the cynomolgus macaques that had been exposed by either the intracerebral (n = 2) or the oral route (n = 3). Faint but consistent PrPSc accumulation and amyloid seeding activity were observed in the central nervous system of clinically affected animals, which strongly supports the view that the propagation of a prion disease was the cause of the neurodegenerative disorder. The reasons for the discrepancy between the results from these two macaque transmission experiments are not clear, but may relate to differences in the oral dosing regimen and/or differences in CWD strains present in the inocula used. A more definitive analysis awaits completion and publication of the second study, but these preliminary data have prompted renewed concern about the potential for transmission of CWD to humans.
Conclusions
Thirty years ago, the BSE crisis brought the previously obscure “prion diseases” into the world spotlight. Since then, our comprehension of the properties and biology of prions has progressed remarkably. However, despite the massive efforts that were deployed, there are still major unanswered questions about animal prion diseases and the risk they might represent for the human population.
The recent discovery of atypical forms of BSE and scrapie, the identification of CWD cases in Europe and the recognition of a novel prion disease of camels within the past year illustrate beyond any words the limits of our knowledge in this field.
There is no epidemiological evidence that classical or atypical scrapie, atypical forms of BSE or chronic wasting disease (CWD) is associated with human prion disease but the limitations of the epidemiological data should be taken into account when interpreting these results. Transmission experiments in nonhuman primates and human PrP transgenic mice clearly illustrate that classical scrapie, L‐type atypical BSE (L‐BSE) and CWD may have zoonotic potential. However, it is still difficult to predict from these results the likelihood that an animal prion disease will transmit to humans under conditions of field exposure.
The profound uncertainties we are still facing concerning the zoonotic abilities of prion diseases of livestock emphasize the importance of maintaining the effective but expensive measures (SRM retrieval, surveillance program, feed ban on animal proteins) that were implemented during the BSE crisis (Figure 2) to prevent human exposure to these pathogens.
References
- 1. Aldhous P (1990) BSE: spongiform encephalopathy found in cat. Nature 345:194. [DOI] [PubMed] [Google Scholar]
- 2. Anderson CA, Bosque P, Filley CM, Arciniegas DB, Kleinschmidt‐Demasters BK et al (2007) Colorado surveillance program for chronic wasting disease transmission to humans: lessons from 2 highly suspicious but negative cases. Arch Neurol 64:439–441. [DOI] [PubMed] [Google Scholar]
- 3. Andreoletti O, Berthon P, Marc D, Sarradin P, Grosclaude J, van Keulen L et al (2000) Early accumulation of PrP(Sc) in gut‐associated lymphoid and nervous tissues of susceptible sheep from a Romanov flock with natural scrapie. J Gen Virol 81(Pt 12):3115–3126. [DOI] [PubMed] [Google Scholar]
- 4. Andreoletti O, Orge L, Benestad SL, Beringue V, Litaise C, Simon S et al (2011) Atypical/Nor98 scrapie infectivity in sheep peripheral tissues. PLoS Pathog 7:e1001285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Andreoletti O, Simon S, Lacroux C, Morel N, Tabouret G, Chabert A et al (2004) PrPSc accumulation in myocytes from sheep incubating natural scrapie. Nat Med 10:591–593. [DOI] [PubMed] [Google Scholar]
- 6. Angers RC, Browning SR, Seward TS, Sigurdson CJ, Miller MW, Hoover EA, Telling GC (2006) Prions in skeletal muscles of deer with chronic wasting disease. Science 311:1117. [DOI] [PubMed] [Google Scholar]
- 7. Angers RC, Kang HE, Napier D, Browning S, Seward T, Mathiason C et al (2010) Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science 328:1154–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Angers RC, Seward TS, Napier D, Green M, Hoover E, Spraker T et al (2009) Chronic wasting disease prions in elk antler velvet. Emerg Infect Dis 15:696–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arsac JN, Andreoletti O, Bilheude JM, Lacroux C, Benestad SL, Baron T (2007) Similar biochemical signatures and prion protein genotypes in atypical scrapie and Nor98 cases, France and Norway. Emerg Infect Dis 13:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Asante EA, Linehan JM, Desbruslais M, Joiner S, Gowland I, Wood AL et al (2002) BSE prions propagate as either variant CJD‐like or sporadic CJD‐like prion strains in transgenic mice expressing human prion protein. Embo J 21:6358–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Babelhadj B, Di Bari MA, Pirisinu L, Chiappini B, Gaouar SBS, Riccardi G et al (2018) Prion disease in dromedary camel, Algeria. Emerg Infect Dis 24:1029–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baker HF, Ridley RM, Wells GA (1993) Experimental transmission of BSE and scrapie to the common marmoset. Vet Rec 132:403–406. [DOI] [PubMed] [Google Scholar]
- 13. Balkema‐Buschmann A, Fast C, Kaatz M, Eiden M, Ziegler U, McIntyre L et al (2011) Pathogenesis of classical and atypical BSE in cattle. Prev Vet Med 102:112–117. [DOI] [PubMed] [Google Scholar]
- 14. Bateman D, Hilton D, Love S, Zeidler M, Beck J, Collinge J (1995) Sporadic Creutzfeldt‐Jakob disease in a 18‐year‐old in the UK. Lancet 346:1155–1156. [DOI] [PubMed] [Google Scholar]
- 15. Belay ED, Gambetti P, Schonberger LB, Parchi P, Lyon DR, Capellari S et al (2001) Creutzfeldt‐Jakob disease in unusually young patients who consumed venison. Arch Neurol 58:1673–1678. [DOI] [PubMed] [Google Scholar]
- 16. Benestad SL, Arsac JN, Goldmann W, Noremark M (2008) Atypical/Nor98 scrapie: properties of the agent, genetics, and epidemiology. Vet Res 39:19. [DOI] [PubMed] [Google Scholar]
- 17. Benestad SL, Mitchell G, Simmons M, Ytrehus B, Vikoren T (2016) First case of chronic wasting disease in Europe in a Norwegian free‐ranging reindeer. Vet Res 47:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beringue V, Andreoletti O, Le Dur A, Essalmani R, Vilotte JL, Lacroux C et al (2007) A bovine prion acquires an epidemic bovine spongiform encephalopathy strain‐like phenotype on interspecies transmission. J Neurosci 27:6965–6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beringue V, Bencsik A, Le Dur A, Reine F, Lai TL, Chenais N et al (2006) Isolation from cattle of a prion strain distinct from that causing bovine spongiform encephalopathy. PLoS Pathog 2:e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beringue V, Herzog L, Reine F, Le Dur A, Casalone C, Vilotte JL, Laude H (2008) Transmission of atypical bovine prions to mice transgenic for human prion protein. Emerg Infect Dis 14:1898–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Beringue V, Vilotte JL, Laude H (2008) Prion agent diversity and species barrier. Vet Res 39:47. [DOI] [PubMed] [Google Scholar]
- 22. Biacabe AG, Laplanche JL, Ryder S, Baron T (2004) Distinct molecular phenotypes in bovine prion diseases. EMBO Rep 5:110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Biacabe AG, Morignat E, Vulin J, Calavas D, Baron TG (2008) Atypical bovine spongiform encephalopathies, France, 2001‐2007. Emerg Infect Dis 14:298–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bishop MT, Hart P, Aitchison L, Baybutt HN, Plinston C, Thomson V et al (2006) Predicting susceptibility and incubation time of human‐to‐human transmission of vCJD. Lancet Neurol 5:393–398. [DOI] [PubMed] [Google Scholar]
- 25. Bishop MT, Will RG, Manson JC (2010) Defining sporadic Creutzfeldt‐Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A 107:12005–12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bons N, Mestre‐Frances N, Belli P, Cathala F, Gajdusek DC, Brown P (1999) Natural and experimental oral infection of nonhuman primates by bovine spongiform encephalopathy agents. Proc Natl Acad Sci U S A 96:4046–4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Britton TC, Al‐Sarraj S, Shaw C, Campbell T, Collinge J (1995) Sporadic Creutzfeldt‐Jakob disease in a 16‐year‐old in the UK. Lancet 346:1155. [DOI] [PubMed] [Google Scholar]
- 28. Brown P, Brandel JP, Sato T, Nakamura Y, MacKenzie J, Will RG et al (2012) Iatrogenic Creutzfeldt‐Jakob disease, final assessment. Emerg Infect Dis 18:901–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brown P, Cathala F, Raubertas RF, Gajdusek DC, Castaigne P (1987) The epidemiology of Creutzfeldt‐Jakob disease: conclusion of a 15‐year investigation in France and review of the world literature. Neurology 37:895–904. [DOI] [PubMed] [Google Scholar]
- 30. Browning SR, Mason GL, Seward T, Green M, Eliason GA, Mathiason C et al (2004) Transmission of prions from mule deer and elk with chronic wasting disease to transgenic mice expressing cervid PrP. J Virol 78:13345–13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bruce ME, Boyle A, Cousens S, McConnell I, Foster J, Goldmann W, Fraser H (2002) Strain characterization of natural sheep scrapie and comparison with BSE. J Gen Virol 83(Pt 3):695–704. [DOI] [PubMed] [Google Scholar]
- 32. Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A et al (1997) Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 389:498–501. [DOI] [PubMed] [Google Scholar]
- 33. Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A et al (1997) Transmissions to mice indicate that 'new variant' CJD is caused by the BSE agent [see comments]. Nature 389:498–501. [DOI] [PubMed] [Google Scholar]
- 34. Buschmann A, Gretzschel A, Biacabe AG, Schiebel K, Corona C, Hoffmann C et al (2006) Atypical BSE in Germany–proof of transmissibility and biochemical characterization. Vet Microbiol 117:103–116. [DOI] [PubMed] [Google Scholar]
- 35. Casalone C, Zanusso G, Acutis P, Ferrari S, Capucci L, Tagliavini F et al (2004) Identification of a second bovine amyloidotic spongiform encephalopathy: molecular similarities with sporadic Creutzfeldt‐Jakob disease. Proc Natl Acad Sci U S A 101:3065–3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cassard H, Torres JM, Lacroux C, Douet JY, Benestad SL, Lantier F et al (2014) Evidence for zoonotic potential of ovine scrapie prions. Nat Commun 5:5821. [DOI] [PubMed] [Google Scholar]
- 37. Castilla J, Gonzalez‐Romero D, Saa P, Morales R, De Castro J, Soto C (2008) Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell 134:757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Castilla J, Gutierrez Adan A, Brun A, Pintado B, Ramirez MA, Parra B et al (2003) Early detection of PrPres in BSE‐infected bovine PrP transgenic mice. Arch Virol 148:677–691. [DOI] [PubMed] [Google Scholar]
- 39. Collinge J, Whitfield J, McKintosh E, Beck J, Mead S, Thomas DJ, Alpers MP (2006) Kuru in the 21st century–an acquired human prion disease with very long incubation periods. Lancet 367:2068–2074. [DOI] [PubMed] [Google Scholar]
- 40. Comoy EE, Casalone C, Lescoutra‐Etchegaray N, Zanusso G, Freire S, Marce D et al (2008) Atypical BSE (BASE) transmitted from asymptomatic aging cattle to a primate. PLoS ONE 3:e3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Comoy EE, Mikol J, Luccantoni‐Freire S, Correia E, Lescoutra‐Etchegaray N, Durand V et al (2015) Transmission of scrapie prions to primate after an extended silent incubation period. Sci Rep 5:11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cook RW, Bingham J, Besier AS, Bayley CL, Hawes M, Shearer PL et al (2016) Atypical scrapie in Australia. Aust Vet J 94:452–455. [DOI] [PubMed] [Google Scholar]
- 43. Corbiere F, Perrin‐Chauvineau C, Lacroux C, Costes P, Thomas M, Bremaud I et al (2013) PrP‐associated resistance to scrapie in five highly infected goat herds. J Gen Virol 94(Pt 1):241–245. [DOI] [PubMed] [Google Scholar]
- 44. Crowell J, Hughson A, Caughey B, Bessen RA (2015) Host Determinants of Prion Strain Diversity Independent of Prion Protein Genotype. J Virol 89:10427–10441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cuillé J, Chelle PL (1936) La maladie dite tremblante du mouton est‐elle inoculable? C R Acad Sci Paris 203:1552–1554. [Google Scholar]
- 46. Cuillé J, Chelle PL (1938) La tremblante du mouton est bien inoculable. C R Acad Sci Paris 206:1687–1688. [Google Scholar]
- 47. Detwiler LA (1992) Scrapie. Rev Sci Tech 11:491–537. [DOI] [PubMed] [Google Scholar]
- 48. Detwiler LA, Baylis M (2003) The epidemiology of scrapie. Rev Sci Tech 22(1):121–143. [DOI] [PubMed] [Google Scholar]
- 49. Diack AB, Head MW, McCutcheon S, Boyle A, Knight R, Ironside JW et al (2014) Variant CJD. 18 years of research and surveillance. Prion 8:286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dudas S, Yang J, Graham C, Czub M, McAllister TA, Coulthart MB, Czub S (2010) Molecular, biochemical and genetic characteristics of BSE in Canada. PLoS ONE 5:e10638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Espinosa JC, Morales M, Castilla J, Rogers M, Torres JM (2007) Progression of prion infectivity in asymptomatic cattle after oral bovine spongiform encephalopathy challenge. J Gen Virol. 88(Pt 4):1379–1383. [DOI] [PubMed] [Google Scholar]
- 52. Fediaevsky A, Maurella C, Noremark M, Ingravalle F, Thorgeirsdottir S, Orge L et al (2010) The prevalence of atypical scrapie in sheep from positive flocks is not higher than in the general sheep population in 11 European countries. BMC Vet Res 6:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fediaevsky A, Tongue SC, Noremark M, Calavas D, Ru G, Hopp P (2008) A descriptive study of the prevalence of atypical and classical scrapie in sheep in 20 European countries. BMC Vet Res 4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fernandez‐Borges N, Espinosa JC, Marin‐Moreno A, Aguilar‐Calvo P, Asante EA, Kitamoto T et al (2017) Protective effect of Val129‐PrP against Bovine Spongiform Encephalopathy but not Variant Creutzfeldt‐Jakob Disease. Emerg Infect Dis 23:1522–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Foster JD, Dickinson AG (1988) The unusual properties of CH1641, a sheep‐passaged isolate of scrapie. Vet Rec 123:5–8. [DOI] [PubMed] [Google Scholar]
- 56. Fox KA, Jewell JE, Williams ES, Miller MW (2006) Patterns of PrPCWD accumulation during the course of chronic wasting disease infection in orally inoculated mule deer (Odocoileus hemionus). J Gen Virol 87:3451–3461. [DOI] [PubMed] [Google Scholar]
- 57. Gambetti P, Kong Q, Zou W, Parchi P, Chen SG (2003) Sporadic and familial CJD: classification and characterisation. Br Med Bull 66:213–239. [DOI] [PubMed] [Google Scholar]
- 58. Gibbs CJ Jr, Gajdusek DC (1971) Transmission and characterization of the agents of spongiform virus encephalopathies: kuru, Creutzfeldt‐Jakob disease, scrapie and mink encephalopathy. Res Publ Assoc Res Nerv Ment Dis 49:383–410. [PubMed] [Google Scholar]
- 59. Gill ON, Spencer Y, Richard‐Loendt A, Kelly C, Dabaghian R, Boyes L et al (2013) Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ 347:f5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Griffiths PC, Spiropoulos J, Lockey R, Tout AC, Jayasena D, Plater JM et al (2010) Characterisation of atypical scrapie cases from Great Britain in transgenic ovine PrP mice. J Gen Virol 91:2132–2138. [DOI] [PubMed] [Google Scholar]
- 61. Groschup MH, Buschmann A (2008) Rodent models for prion diseases. Vet Res 39:32. [DOI] [PubMed] [Google Scholar]
- 62. Haley NJ, Hoover EA (2015) Chronic wasting disease of cervids: current knowledge and future perspectives. Ann Rev Ani Biosci 3:305–325. [DOI] [PubMed] [Google Scholar]
- 63. Hazards EPoB (2014) Scientific Opinion on the scrapie situation in the EU after 10 years of monitoring and control in sheep and goats. EFSA J 12:3781‐n/a. [Google Scholar]
- 64. Head MW, Ritchie D, Smith N, McLoughlin V, Nailon W, Samad S et al (2004) Peripheral tissue involvement in sporadic, iatrogenic, and variant Creutzfeldt‐Jakob disease: an immunohistochemical, quantitative, and biochemical study. Am J Pathol 164:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Herzog C, Riviere J, Lescoutra‐Etchegaray N, Charbonnier A, Leblanc V, Sales N et al (2005) PrPTSE distribution in a primate model of variant, sporadic, and iatrogenic Creutzfeldt‐Jakob disease. J Virol 79:14339–14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J (1997) The same prion strain causes vCJD and BSE. Nature 389:448–450, 526. [DOI] [PubMed] [Google Scholar]
- 67. Hilton DA, Ghani AC, Conyers L, Edwards P, McCardle L, Ritchie D et al (2004) Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J Pathol 203:733–739. [DOI] [PubMed] [Google Scholar]
- 68. Hoffmann C, Ziegler U, Buschmann A, Weber A, Kupfer L, Oelschlegel A et al (2007) Prions spread via the autonomic nervous system from the gut to the central nervous system in cattle incubating bovine spongiform encephalopathy. J Gen Virol 88(Pt 3):1048–1055. [DOI] [PubMed] [Google Scholar]
- 69. Jaumain E, Quadrio I, Herzog L, Reine F, Rezaei H, Andreoletti O et al (2016) Absence of evidence for a causal link between bovine spongiform encephalopathy strain variant L‐BSE and known forms of sporadic Creutzfeldt‐Jakob disease in human PrP transgenic mice. J Virol 90:10867–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jeffrey M, Gonzalez L (2007) Classical sheep transmissible spongiform encephalopathies: pathogenesis, pathological phenotypes and clinical disease. Neuropathol Appl Neurobiol 33:373–394. [DOI] [PubMed] [Google Scholar]
- 71. Jewell JE, Conner MM, Wolfe LL, Miller MW, Williams ES (2005) Low frequency of PrP genotype 225SF among free‐ranging mule deer (Odocoileus hemionus) with chronic wasting disease. J Gen Virol 86(Pt 8):2127–2134. [DOI] [PubMed] [Google Scholar]
- 72. Kim TY, Shon HJ, Joo YS, Mun UK, Kang KS, Lee YS (2005) Additional cases of Chronic Wasting Disease in imported deer in Korea. J Vet Med Sci 67:753–759. [DOI] [PubMed] [Google Scholar]
- 73. Kittelberger R, Chaplin MJ, Simmons MM, Ramirez‐Villaescusa A, McIntyre L, MacDiarmid SC et al (2010) Atypical scrapie/Nor98 in a sheep from New Zealand. J Vet Diagn Invest 22:863–875. [DOI] [PubMed] [Google Scholar]
- 74. Kong Q, Huang S, Zou W, Vanegas D, Wang M, Wu D et al (2005) Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci 25:7944–7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kong Q, Zheng M, Casalone C, Qing L, Huang S, Chakraborty B et al (2008) Evaluation of the human transmission risk of an atypical bovine spongiform encephalopathy prion strain. J Virol 82:3697–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Konold T, Arnold ME, Austin AR, Cawthraw S, Hawkins SA, Stack MJ et al (2012) Bovine spongiform encephalopathy: the effect of oral exposure dose on attack rate and incubation period in cattle ‐ an update. BMC Res Notes 5:674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Konold T, Bone GE, Clifford D, Chaplin MJ, Cawthraw S, Stack MJ, Simmons MM (2012) Experimental H‐type and L‐type bovine spongiform encephalopathy in cattle: observation of two clinical syndromes and diagnostic challenges. BMC Vet Res. 8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Konold T, Moore SJ, Bellworthy SJ, Simmons HA (2008) Evidence of scrapie transmission via milk. BMC Vet Res 4:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kurt TD, Jiang L, Fernandez‐Borges N, Bett C, Liu J, Yang T et al (2015) Human prion protein sequence elements impede cross‐species chronic wasting disease transmission. J Clin Invest 125:2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lacroux C, Cassard H, Simmons H, Yves Douet J, Corbiere F, Lugan S et al (2017) Classical scrapie transmission in ARR/ARR genotype sheep. J Gen Virol 98:2200–2204. [DOI] [PubMed] [Google Scholar]
- 81. Lacroux C, Corbiere F, Tabouret G, Lugan S, Costes P, Mathey J et al (2007) Dynamics and genetics of PrPSc placental accumulation in sheep. J Gen Virol 88(Pt 3):1056–1061. [DOI] [PubMed] [Google Scholar]
- 82. Lacroux C, Simon S, Benestad SL, Maillet S, Mathey J, Lugan S et al (2008) Prions in milk from ewes incubating natural scrapie. PLoS Pathog 4(12):e1000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lacroux C, Vilette D, Fernandez‐Borges N, Litaise C, Lugan S, Morel N et al (2012) Prionemia and leukocyte‐platelet‐associated infectivity in sheep transmissible spongiform encephalopathy models. J Virol 86:2056–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lasmezas CI, Comoy E, Hawkins S, Herzog C, Mouthon F, Konold T et al (2005) Risk of oral infection with bovine spongiform encephalopathy agent in primates. Lancet 365:781–783. [DOI] [PubMed] [Google Scholar]
- 85. Lasmezas CI, Deslys JP, Demaimay R, Adjou KT, Lamoury F, Dormont D et al (1996) BSE transmission to macaques. Nature 381:743–744. [DOI] [PubMed] [Google Scholar]
- 86. Le Dur A, Beringue V, Andreoletti O, Reine F, Lai TL, Baron T et al (2005) A newly identified type of scrapie agent can naturally infect sheep with resistant PrP genotypes. Proc Natl Acad Sci U S A 102:16031–16036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lescoutra‐Etchegaray N, Jaffre N, Sumian C, Durand V, Correia E, Mikol J et al (2015) Evaluation of the protection of primates transfused with variant Creutzfeldt‐Jakob disease‐infected blood products filtered with prion removal devices: a 5‐year update. Transfusion 55:1231–1241. [DOI] [PubMed] [Google Scholar]
- 88. Masujin K, Shu Y, Yamakawa Y, Hagiwara K, Sata T, Matsuura Y et al (2008) Biological and biochemical characterization of L‐type‐like bovine spongiform encephalopathy (BSE) detected in Japanese black beef cattle. Prion 2:123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mathiason CK, Powers JG, Dahmes SJ, Osborn DA, Miller KV, Warren RJ et al (2006) Infectious prions in the saliva and blood of deer with chronic wasting disease. Science 314:133–136. [DOI] [PubMed] [Google Scholar]
- 90. Mawhinney S, Pape WJ, Forster JE, Anderson CA, Bosque P, Miller MW (2006) Human prion disease and relative risk associated with chronic wasting disease. Emerg Infect Dis 12:1527–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. McGovern G, Martin S, Jeffrey M, Bellworthy SJ, Spiropoulos J, Green R et al (2015) Influence of breed and genotype on the onset and distribution of infectivity and disease‐associated prion protein in sheep following oral infection with the bovine spongiform encephalopathy agent. J Comp Pathol 152:28–40. [DOI] [PubMed] [Google Scholar]
- 92. Mead S, Poulter M, Uphill J, Beck J, Whitfield J, Webb TE et al (2009) Genetic risk factors for variant Creutzfeldt‐Jakob disease: a genome‐wide association study. Lancet Neurol 8:57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Miller MW, Williams ES, McCarty CW, Spraker TR, Kreeger TJ, Larsen CT, Thorne ET (2000) Epizootiology of chronic wasting disease in free‐ranging cervids in Colorado and Wyoming. J Wildl Dis 36:676–690. [DOI] [PubMed] [Google Scholar]
- 94. Mitchell GB, Sigurdson CJ, O'Rourke KI, Algire J, Harrington NP, Walther I et al (2012) Experimental oral transmission of chronic wasting disease to reindeer (Rangifer tarandus tarandus). PLoS ONE 7:e39055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mok T, Jaunmuktane Z, Joiner S, Campbell T, Morgan C, Wakerley B et al (2017) Variant Creutzfeldt‐Jakob Disease in a Patient with Heterozygosity at PRNP Codon 129. N Engl J Med 376:292–294. [DOI] [PubMed] [Google Scholar]
- 96. Moore SJ, Kunkle R, Greenlee MH, Nicholson E, Richt J, Hamir A et al (2016) Horizontal Transmission of Chronic Wasting Disease in Reindeer. Emerg Infect Dis 22:2142–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Moreno CR, Moazami‐Goudarzi K, Laurent P, Cazeau G, Andreoletti O, Chadi S et al (2007) Which PrP haplotypes in a French sheep population are the most susceptible to atypical scrapie? Arch Virol 152:1229–1232. [DOI] [PubMed] [Google Scholar]
- 98. Moum T, Olsaker I, Hopp P, Moldal T, Valheim M, Moum T, Benestad SL (2005) Polymorphisms at codons 141 and 154 in the ovine prion protein gene are associated with scrapie Nor98 cases. J Gen Virol 86(Pt 1):231–235. [DOI] [PubMed] [Google Scholar]
- 99. O'Rourke KI, Besser TE, Miller MW, Cline TF, Spraker TR, Jenny AL et al (1999) PrP genotypes of captive and free‐ranging Rocky Mountain elk (Cervus elaphus nelsoni) with chronic wasting disease. J Gen Virol 80(Pt 10):2765–2679. [DOI] [PubMed] [Google Scholar]
- 100. Okada H, Iwamaru Y, Imamura M, Masujin K, Matsuura Y, Shimizu Y et al (2011) Experimental H‐type bovine spongiform encephalopathy characterized by plaques and glial‐ and stellate‐type prion protein deposits. Vet Res 42:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Okada H, Masujin K, Miyazawa K, Iwamaru Y, Imamura M, Matsuura Y et al (2017) Experimental infection of cattle with a novel prion derived from atypical H‐type bovine spongiform encephalopathy. Vet Pathol 54:892–900. [DOI] [PubMed] [Google Scholar]
- 102. Olszowy KM, Lavelle J, Rachfal K, Hempstead S, Drouin K, Darcy JM 2nd et al (2014) Six‐year follow‐up of a point‐source exposure to CWD contaminated venison in an Upstate New York community: risk behaviours and health outcomes 2005‐2011. Public Health 128:860–868. [DOI] [PubMed] [Google Scholar]
- 103. Ono F, Tase N, Kurosawa A, Hiyaoka A, Ohyama A, Tezuka Y et al (2011) Atypical L‐type bovine spongiform encephalopathy (L‐BSE) transmission to cynomolgus macaques, a non‐human primate. Jpn J Infect Dis 64:81–84. [PubMed] [Google Scholar]
- 104. Padilla D, Beringue V, Espinosa JC, Andreoletti O, Jaumain E, Reine F et al (2011) Sheep and goat BSE propagate more efficiently than cattle BSE in human PrP transgenic mice. PLoS Pathog 7:e1001319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Papasavva‐Stylianou P, Kleanthous M, Toumazos P, Mavrikiou P, Loucaides P (2007) Novel polymorphisms at codons 146 and 151 in the prion protein gene of Cyprus goats, and their association with natural scrapie. Vet J 173:459–462. [DOI] [PubMed] [Google Scholar]
- 106. Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW (2004) Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 364:527–529. [DOI] [PubMed] [Google Scholar]
- 107. Plinston C, Hart P, Chong A, Hunter N, Foster J, Piccardo P et al (2011) Increased susceptibility of human‐PrP transgenic mice to bovine spongiform encephalopathy infection following passage in sheep. J Virol 85:1174–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216:136–144. [DOI] [PubMed] [Google Scholar]
- 109. Race B, Meade‐White KD, Phillips K, Striebel J, Race R, Chesebro B (2014) Chronic wasting disease agents in nonhuman primates. Emerg Infect Dis 20:833–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Race BL, Meade‐White KD, Ward A, Jewell J, Miller MW, Williams ES et al (2007) Levels of abnormal prion protein in deer and elk with chronic wasting disease. Emerg Infect Dis 13:824–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Race R, Jenny A, Sutton D (1998) Scrapie infectivity and proteinase K‐resistant prion protein in sheep placenta, brain, spleen, and lymph node: implications for transmission and antemortem diagnosis. J Infect Dis 178:949–953. [DOI] [PubMed] [Google Scholar]
- 112. Raymond GJ, Bossers A, Raymond LD, O'Rourke KI, McHolland LE, Bryant PK et al (2000) Evidence of a molecular barrier limiting susceptibility of humans, cattle and sheep to chronic wasting disease. Embo J 19:4425–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Raymond GJ, Hope J, Kocisko DA, Priola SA, Raymond LD, Bossers A et al (1997) Molecular assessment of the potential transmissibilities of BSE and scrapie to humans. Nature 388:285–288. [DOI] [PubMed] [Google Scholar]
- 114. Requena JR, Kristensson K, Korth C, Zurzolo C, Simmons M, Aguilar‐Calvo P et al (2016) The Priority position paper: Protecting Europe's food chain from prions. Prion 10:165–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Richt JA, Kunkle RA, Alt D, Nicholson EM, Hamir AN, Czub S et al (2007) Identification and characterization of two bovine spongiform encephalopathy cases diagnosed in the United States. J Vet Diagn Invest 19:142–154. [DOI] [PubMed] [Google Scholar]
- 116. Robinson SJ, Samuel MD, O'Rourke KI, Johnson CJ (2012) The role of genetics in chronic wasting disease of North American cervids. Prion 6:153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Scott M, Foster D, Mirenda C, Serban D, Coufal F, Walchli M et al (1989) Transgenic mice expressing hamster prion protein produce species‐ specific scrapie infectivity and amyloid plaques. Cell 59:847–857. [DOI] [PubMed] [Google Scholar]
- 118. Scott MR, Safar J, Telling G, Nguyen O, Groth D, Torchia M et al (1997) Identification of a prion protein epitope modulating transmission of bovine spongiform encephalopathy prions to transgenic mice. Proc Natl Acad Sci U S A 94:14279–14284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sigurdson CJ, Barillas‐Mury C, Miller MW, Oesch B, Van Keulen LJ, Langeveld JP, Hoover EA (2002) PrP(CWD) lymphoid cell targets in early and advanced chronic wasting disease of mule deer. J Gen Virol 83(Pt 10):2617–2628. [DOI] [PubMed] [Google Scholar]
- 120. Sigurdson CJ, Spraker TR, Miller MW, Oesch B, Hoover EA (2001) PrP(CWD) in the myenteric plexus, vagosympathetic trunk and endocrine glands of deer with chronic wasting disease. J Gen Virol 82(Pt 10):2327–2334. [DOI] [PubMed] [Google Scholar]
- 121. Sigurdson CJ, Williams ES, Miller MW, Spraker TR, O'Rourke KI, Hoover EA (1999) Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus). J Gen Virol 80(Pt 10):2757–2764. [DOI] [PubMed] [Google Scholar]
- 122. Simmons MM, Konold T, Thurston L, Bellworthy SJ, Chaplin MJ, Moore SJ (2010) The natural atypical scrapie phenotype is preserved on experimental transmission and sub‐passage in PRNP homologous sheep. BMC Vet Res 6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Stack MJ, Moore SJ, Vidal‐Diez A, Arnold ME, Jones EM, Spencer YI et al (2011) Experimental bovine spongiform encephalopathy: detection of PrP(Sc) in the small intestine relative to exposure dose and age. J Comp Pathol 145:289–301. [DOI] [PubMed] [Google Scholar]
- 124. Suardi S, Vimercati C, Casalone C, Gelmetti D, Corona C, Iulini B et al (2012) Infectivity in skeletal muscle of cattle with atypical bovine spongiform encephalopathy. PLoS ONE 7:e31449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Tamguney G, Giles K, Bouzamondo‐Bernstein E, Bosque PJ, Miller MW, Safar J et al (2006) Transmission of elk and deer prions to transgenic mice. J Virol 80:9104–9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE et al (1995) Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83:79–90. [DOI] [PubMed] [Google Scholar]
- 127. Thackray AM, Hopkins L, Klein MA, Bujdoso R (2007) Mouse‐adapted ovine scrapie prion strains are characterized by different conformers of PrPSc. J Virol 81:12119–12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Thackray AM, Lockey R, Beck KE, Spiropoulos J, Bujdoso R (2012) Evidence for co‐infection of ovine prion strains in classical scrapie isolates. J Comp Pathol 147:316–329. [DOI] [PubMed] [Google Scholar]
- 129. Tixador P, Herzog L, Reine F, Jaumain E, Chapuis J, Le Dur A et al (2010) The physical relationship between infectivity and prion protein aggregates is strain‐dependent. PLoS Pathog 6:e1000859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Torres JM, Andreoletti O, Lacroux C, Prieto I, Lorenzo P, Larska M et al (2011) Classical bovine spongiform encephalopathy by transmission of H‐type prion in homologous prion protein context. Emerg Infect Dis 17:1636–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Tuo W, O'Rourke KI, Zhuang D, Cheevers WP, Spraker TR, Knowles DP (2002) Pregnancy status and fetal prion genetics determine PrPSc accumulation in placentomes of scrapie‐infected sheep. Proc Natl Acad Sci U S A 99:6310–6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. van Duijn CM, Delasnerie‐Laupretre N, Masullo C, Zerr I, de Silva R, Wientjens DP et al (1998) Case‐control study of risk factors of Creutzfeldt‐Jakob disease in Europe during 1993‐95. European Union (EU) Collaborative Study Group of Creutzfeldt‐Jakob disease (CJD). Lancet 351:1081–1085. [DOI] [PubMed] [Google Scholar]
- 133. van Keulen LJ, Schreuder BE, Vromans ME, Langeveld JP, Smits MA (2000) Pathogenesis of natural scrapie in sheep. Arch Virol Suppl 16:57–71. [DOI] [PubMed] [Google Scholar]
- 134. Vilotte JL, Soulier S, Essalmani R, Stinnakre MG, Vaiman D, Lepourry L et al (2001) Markedly increased susceptibility to natural sheep scrapie of transgenic mice expressing ovine prp. J Virol 75:5977–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Wells GA, Hawkins SA, Green RB, Austin AR, Dexter I, Spencer YI et al (1998) Preliminary observations on the pathogenesis of experimental bovine spongiform encephalopathy (BSE): an update. Vet Rec 142:103–106. [DOI] [PubMed] [Google Scholar]
- 136. Wells GA, Scott AC, Johnson CT, Gunning RF, Hancock RD, Jeffrey M et al (1987) A novel progressive spongiform encephalopathy in cattle. Vet Rec 121:419–420. [DOI] [PubMed] [Google Scholar]
- 137. Wells GA, Spiropoulos J, Hawkins SA, Ryder SJ (2005) Pathogenesis of experimental bovine spongiform encephalopathy: preclinical infectivity in tonsil and observations on the distribution of lingual tonsil in slaughtered cattle. Vet Rec 156:401–407. [DOI] [PubMed] [Google Scholar]
- 138. Wilesmith JW, Wells GA, Cranwell MP, Ryan JB (1988) Bovine spongiform encephalopathy: epidemiological studies. Vet Rec 123:638–644. [PubMed] [Google Scholar]
- 139. Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A et al (1996) A new variant of Creutzfeldt‐Jakob disease in the UK. Lancet 347:921–925. [DOI] [PubMed] [Google Scholar]
- 140. Williams ES (2005) Chronic wasting disease. Vet Pathol 42:530–549. [DOI] [PubMed] [Google Scholar]
- 141. Wilson R, King D, Hunter N, Goldmann W, Barron RM (2013) Characterization of an unusual transmissible spongiform encephalopathy in goat by transmission in knock‐in transgenic mice. J Gen Virol 94(Pt 8):1922–1932. [DOI] [PubMed] [Google Scholar]
- 142. Wilson R, Plinston C, Hunter N, Casalone C, Corona C, Tagliavini F et al (2012) Chronic wasting disease and atypical forms of bovine spongiform encephalopathy and scrapie are not transmissible to mice expressing wild‐type levels of human prion protein. J Gen Virol 93(Pt 7):1624–1629. [DOI] [PubMed] [Google Scholar]
- 143. Windl O, Dempster M, Estibeiro JP, Lathe R, de Silva R, Esmonde T et al (1996) Genetic basis of Creutzfeldt‐Jakob disease in the United Kingdom: a systematic analysis of predisposing mutations and allelic variation in the PRNP gene. Hum Genet 98:259–264. [DOI] [PubMed] [Google Scholar]