

Abstract
Our previous studies showed that the transcription factor early growth response‐1 (EGR1) may play a role in keeping the brain cholinergic function intact in the preclinical stages of Alzheimer’s disease (AD). In order to elucidate the mechanisms involved, we first performed data mining on our previous microarray study on postmortem human prefrontal cortex (PFC) for the changes in the expression of EGR1 and acetylcholinesterase (AChE) and the relationship between them during the course of AD. The study contained 49 patients, ranging from non‐demented controls (Braak stage 0) to late AD patients (Braak stage VI). We found EGR1‐mRNA was high in early AD and decreased in late AD stages, while AChE‐mRNA was stable in preclinical AD and slightly decreased in late AD stages. A significant positive correlation was found between the mRNA levels of these two molecules. In addition, we studied the relationship between EGR1 and AChE mRNA levels in the frontal cortex of 3–12‐months old triple‐transgenic AD (3xTg‐AD) mice. EGR1‐ and AChE‐mRNA were lower in 3xTg‐AD mice compared with wild‐type (WT) mice. A significant positive correlation between these two molecules was present in the entire group and in each age group of either WT or 3xTg‐AD mice. Subsequently, AChE expression was determined following up‐ or down‐regulating EGR1 in cell lines and the EGR1 levels were found to regulate AChE at both the mRNA and protein levels. Dual‐luciferase assay and electrophoretic mobility shift assay in the EGR1‐overexpressing cells were performed to determine the functionally effective binding sites of the EGR1 on the AChE gene promoter. We conclude that the EGR1 can upregulate AChE expression by a direct effect on its gene promoter, which may contribute significantly to the changes in cholinergic function in the course of AD. The 3xTg‐AD mouse model only reflects later stage AD.
Keywords: Alzheimer’s disease, early growth response‐1, acetylcholinesterase
Abbreviations
- AChE
acetylcholinesterase
- ChAT
choline‐acetyl transferase
- CO2
carbon dioxide
- DMEM/F‐12
Dulbecco’s modified eagle medium: nutrient mixture F‐12
- EGR1
early growth response protein‐1
- EMSA
electrophoretic mobility shift assay
- FBS
fetal bovine serum
- MCD
mild cognitive decline
- MEM
minimum essential medium
- NCD
no cognitive decline
- NBM
Nucleus Basalis of Meynert
- PMD
post‐mortem delay
- PFC
prefrontal cortex
- SCD
severe cognitive decline
Introduction
The brain cholinergic system is crucial for cognitive functions, including learning and memory, which are severely impaired in Alzheimer’s disease (AD) 8, 9. In the human brain, the enzyme responsible for acetylcholine production, choline‐acetyl transferase (ChAT) is mainly produced in the cholinergic neurons of the Nucleus Basalis of Meynert (NBM), which have extensive projections to the neocortex including the prefrontal cortex (PFC). Studies of our group on postmortem human brain have shown that during the early, preclinical AD stages, in the PFC there is an activation of expression of 865 genes related to neuronal plasticity, synaptic activity and metabolism, followed by decreased activity in late AD stages. These alterations were described as “up‐down” pattern 2. The NBM was also found to become metabolically activated in early clinical AD stages 7. Our follow‐up study in the NBM suggested that the early growth response protein‐1 (EGR1) showed a significant positive correlation with ChAT expression 37 and might be involved in keeping the cholinergic function intact in early AD stages. EGR1 is a zinc finger transcription factor that belongs to the “up‐down” gene cluster 2 and it regulates a number of genes involved in synaptic activity and plasticity 14, 33. EGR1 can bind the ChAT gene promoter and increase its expression 29.
Acetylcholinesterase (AChE) is the enzyme degrading acetylcholine. It is decreased in the brain of late AD stages patients 10. An extensive network of cortical pyramidal neurons in the human brain with a high intensity of histochemically identified AChE activity show age‐dependent changes 20, 21, 22. Compared with the young adults, brains of cognitively normal elderly displayed a significant decrease in AChE‐staining intensity of these neurons. In addition, brains of elderly above age 80 with well‐preserved memory performance showed significantly lower staining intensity and density of these neurons when compared with age‐matched peers with declined memory. It was thus proposed that low levels of AChE activity could enhance the impact of acetylcholine on pyramidal neurons to counterbalance other factors that mediate the decline of memory capacity during average aging 13. AChE‐inhibitors are the first‐line medicine against AD 18. Interestingly, as a member of the EGR family, EGR1 has a high affinity for GC rich sequence, that is 5′‐GCG(G/T)GGGCG‐3′ 5, 28, 31, which is present multiple times in the promoter of the AChE gene, both in mouse 17, 25 and in human 11, 34.
Based upon the evidence mentioned above, we hypothesized that the regulation of AChE expression by EGR1 might play a significant role in the changes in cholinergic function during the course of AD. We aimed to test this hypothesis by (i) performing data mining using the data of our previous microarray study on postmortem PFC samples representing the course of AD 2, focusing on AChE‐mRNA changes in relation to EGR1‐mRNA; (ii) determining the expression of EGR1‐mRNA and AChE‐mRNA in the frontal cortex in three different age‐groups of the 3xTg‐AD mouse model harboring human PS1M146V, APPSwe and tau P301L transgenes 27 and their age/sex‐matched wild type (WT) controls; and (iii) testing AChE expression following up‐ or down‐regulating EGR1 in the SY5Y cells, performing a dual‐luciferase assay in 293T cells and performing electrophoretic mobility shift assay (EMSA) in SY5Y cells to determine whether EGR1 can directly regulate AChE expression by interacting with the AChE gene promoter.
Material and Methods
Part 1: Human postmortem AD brain study
49 snap‐frozen medial frontal gyrus samples, taken from Brodmann area 46 (Figure 1), were obtained from the Netherlands Brain Bank. For each Braak stage (n = 7, three or four female subjects), ranging from non‐demented controls (Braak stage = 0) to late AD stage patients (Braak stage = VI) 3, were matched as closely as possible for sex, age, postmortem delay (PMD), CSF pH and APOE genotype (see Supplementary Table S2). Three pooled clinical groups were distinguished in these patients on basis of cognitive functioning using the clinical Reisberg scale, that is the Global Deterioration Scale for the assessment of Degenerative Dementia 30. We distinguished Reisberg 1 (no cognitive decline group, NCD, n = 5), Reisberg 2–5 (mild cognitive decline group, MCD, including subjects with very mild to moderately severe cognitive decline, n = 5) and Reisberg 6–7 (severe cognitive decline group, SCD, including subjects with severe/very severe cognitive decline, n = 15). These three Reisberg groups were also matched for the confounding factors mentioned above (see Supplementary Table S2). Please note that 25 out 49 of the subjects’ medical histories lacked the information of the clinical Reisberg scale. All individuals were systematically neuropathologically investigated as described earlier 32. For each individual, neuropathological reports, including Braak staging for neurofibrillary changes and neuritic plaques 3 and clinical reports were available. No neuropathology other than that characteristics for AD or associated with normal aging was observed in any of the individuals in this study. No subjects were included from families with a history of familial AD (for more details for these subjects see Supplementary Table S1). For each sample, 20–30 sections of 50 µm were cut. Grey matter areas were dissected inside the cryostat using pre‐chilled scalpels, collected in pre‐chilled 2‐mL tubes that were immediately put on dry ice.
Figure 1.
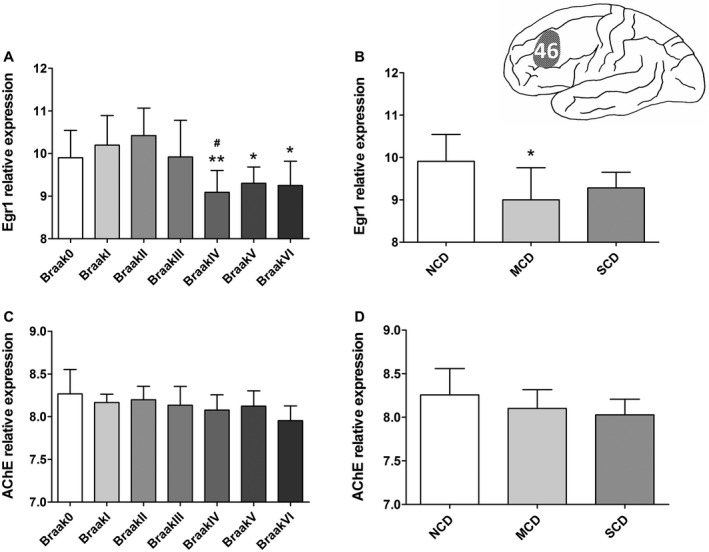
Expression of mRNA of early growth response 1 (EGR1) and acetylcholinesterase (AChE) in the prefrontal cortex (Brodmann area 46) during the course of Alzheimer’s disease (AD). A. EGR1‐mRNA levels showed significant changes (ANOVA, F(6,42) = 4.651, P = 0.001) among Braak stages. EGR1‐mRNA is especially decreased in Braak IV compared with Braak I and II, and further decreased in Braak V–VI (Bonferroni test, P ≤ 0.043); B. EGR1‐mRNA levels also showed a significant change among Reisberg groups (ANOVA, F(2,22) = 4.189, P = 0.0287), and in particular they significantly declined in the mild cognitive decline (MCD) group compared with the no cognitive decline (NCD) group (Bonferroni test, P = 0.033). C. AChE‐mRNA did not show a significant decrease along the Braak stages (ANOVA, F(6,42) = 1.877, P = 0.107). D. AChE‐mRNA also did not show significant changes among Reisberg groups (ANOVA, F(2,22) = 2.176, P = 0.137). The normalized data (log2‐transformation) are represented as mean (± standard deviation). (*) compared with (A) Braak II and (B) NCD, (#) compared with Braak I. *P < 0.05, **P < 0.01, # P < 0.05.
The weight of the tissue samples were measured and the samples were homogenized in an adapted volume ice‐cold Trizol (Life Technologies, Grand Island, NY, USA; 3 mL Trizol per 100 mg tissue). Total RNA was isolated using RNeasy Mini Kit (QIAGEN, Valencia, CA, USA) RNA isolation methods. RNA integrity number was determined by an Agilent 2100 bioanalyser (Agilent Technologies, Palo Alto, CA, USA). Isolated RNA was of high integrity (average RNA integrity number 8.3, range 6.5–9.6). For microarray analysis, Agilent 44K Whole Human Genome arrays (Agilent Technologies, Part Number G4112A, Palo Alto, CA, USA) were used. Sample labeling and microarray hybridization were performed according to manufacturer’s instructions. Microarrays were scanned using an Agilent DNA Microarray Scanner and quantified by using Agilent Feature Extraction software (version 8.5.1). For details, see reference 2.
Part 2: 3xTg‐AD mouse mode study
Two pairs of this 3xTg‐AD parent mice (Cat. number 34830) were obtained from the Jackson Laboratory (Bar Harbor, Maine, USA), maintained under a 12/12 h light/dark cycle at 25°C and provided with food and water ad libitum. All their neonatal mice were genotyped and their expression of PS1M146V, APPSwe and tau P301L was confirmed (see Supplementary Figure S1). Animal handling and experimental procedures were carried out in accordance with the Animal Experimental Committee of the Royal Netherlands Academy of Arts and Sciences.
In total, 55 (27 male, 28 female) 3xTg‐AD mice and 54 (28 male, 26 female) WT control mice were obtained. The mice were sacrificed by carbon dioxide (CO2) and cervical dislocation at three different ages that is 3–4‐months, 7–8‐months and 11–12‐months. Each age group contained six to ten male and six to ten female mice of equal sex number. The brains were rapidly removed and the frontal cortex was isolated following removal of the olfactory bulbs, cutting 1‐mm posterior to the bregma from both hemispheres and stored in −80°C until use.
Each frontal cortex from the left hemisphere was homogenized in 1 mL ice‐cold Trizol. RNA was isolated as described before 2. cDNA was synthesized from the RNA sample using QuantiTect reverse transcription kit (QIAGEN, Valencia, CA, USA), and a dilution of 1:10 cDNA was made for qPCR by 7300 Real Time PCR System (Thermo, Waltham, MA, USA). Two housekeeping genes, glyceraldehyde phosphate dehydrogenase (GAPDH) and hypoxanthine guanine phosphoribosyl transferase (HPRT), were used. EGR1‐mRNA and AChE‐mRNA were quantified. Sequences of primers are shown in Supplementary Table S3.
Part 3: In vitro studies in cell lines
293T cells were chosen for the luciferase assay since this cell line can efficiently be transfected with plasmids. In addition, the SY5Y cell, a frequently used neuroblastoma cell line which can be driven toward a variety of adult neuronal phenotypes including the cholinergic phenotype 15, 16 was chosen in the part of study that was related to the AChE gene and protein expression measurements.
Cell culture
293T cells were cultured in Minimum Essential Medium (MEM) (Thermo, 11095080, Waltham, MA, USA), while SY5Y cells were cultured in Dulbecco’s Modified Eagle Medium: Nutrient Mixture F‐12 (DMEM/F‐12) (Thermo, 10565018, Waltham, MA, USA). Both media were supplemented with 10% fetal bovine serum (FBS) (Thermo, 16000044, Waltham, MA, USA) together with 1% penicillin and streptomycin (Thermo, 15070063, Waltham, MA, USA). Cells were incubated at 37°C in a humidified chamber with 5% CO2.
Plasmid constructs
The EGR1 gene sequence was obtained from EGR1‐pRRL‐MCS+ (a gift from Dr. K Bossers, Netherlands Institute for Neuroscience) by restriction digestion with the enzymes Hind3 and EcoR1 (NEB, Hitchin, Hertfordshire, UK). The fragment was isolated by running a 1% agarose gel, followed by gel cleavage purification (Macherey‐Nagel, Düren, Germany), after which it was cloned into pcDNA3.1 vector (Thermo, Waltham, MA, USA). The plasmid contained short hairpin RNA (shRNA) with the target sequence of EGR1 (5′‐TCTCTCTGAACAACGAGAA‐3′) and the plasmid as the control contained shRNA with a scramble sequence of 5′‐TTCTCCGAACGTGTCACGT‐3′ were purchased from the GeneChem company (Shanghai, China).
A series of 5′ or 3′ deletion fragments of AChE gene promoter (−616/+707) (a gift from Dr. K Tsim, Hong Kong University of Science and Technology) were amplified by PCR with primers containing a Hind3 or Bgl2 restriction site (Figure 4A). These deletion‐fragments were isolated using a 1% agarose gel followed by gel cleavage purification, and were subsequently cloned into pGL3‐Basic Vector (Promega, Fitchburg, Wisconsin, USA) after cutting the amplified AChE fragments with the restriction enzyme Hind3 and Bgl2 (NEB, Hitchin, Hertfordshire, UK). The promoter constructs were −193/+707 pGL3, +108/+707 pGL3, −193/+107 pGL3, −98/+107 pGL3 and −60/+107 pGL3. The mutations in EGR1‐binding sites were generated using −193/+107 pGL3 as template, the resulting plasmids were named mutation 1, 2 and 3, see Figure 4A for details.
Figure 4.
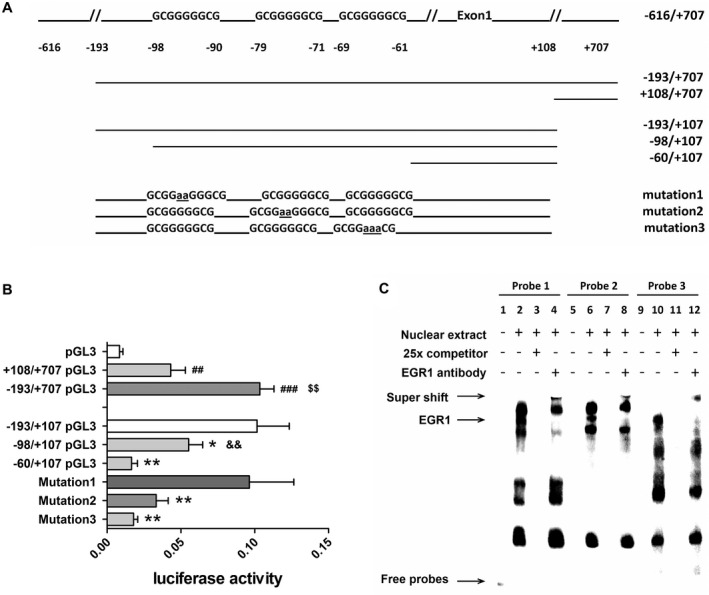
Early growth response 1 (EGR1) initiates acetylcholinesterase (AChE) transcription by binding to the potential EGR1‐binding sites in the gene promoter of AChE. A. Schematic illustration of the deletion constructs of AChE gene promoter. B. Luciferase activity of the deleted constructs in 293T cells. +108/+707 pGL3 and −193/+707 pGL3 were significantly increased compared with vector pGL3, −193/+707 pGL3 was significantly higher than +108/+707 pGL3; −60/+107 pGL3 and −98/+107 pGL3 were both significantly decreased compared with −193/+707 pGL3, while −98/+107 pGL3 was significantly higher than −60/+107 pGL3; mutation 2 and 3 were both significantly decreased compared with −193/+707 pGL3, while mutation 1 did not change. C. Electrophoretic mobility shift assay (EMSA) results of EGR1 overexpressing SY5Y cells. Incubation of nuclear extracts with the 3 oligonucleotides (probe 1–3) revealed bands (lanes 2, 6 and 10), which were abolished by an excess of appropriately related unlabeled oligonucleotide (lanes 3, 7 and 11); the specific bands that represented EGR1‐oligonucleotide complexes super‐shifted with the addition of EGR1 antibody (lane 4, 8 and 12). Student’s t‐test was applied. Luciferase assay was repeated twice in triplicates with a similar outcome and data are represented as mean (± standard deviation) of one of the experiments. Four to six pilot experiments had been performed before the final experiments. EMSA was performed three times with a similar outcome, a representative result was shown in C. (#) compared with pGL3, ($) compared with −108/+707 pGL3, (&) compared with −60/+107 pGL3 and (*) compared with −193/+107 pGL3. ## P < 0.01, ### P < 0.001, $$ P < 0.01, && P < 0.01, *P < 0.05, **P < 0.01.
Primers are shown in Supplementary Table S3 and all constructs were verified by corresponding enzyme digestions and sequencing.
Effect of up‐ or down‐regulation of EGR1 on the expression of AChE
SY5Y cells were seeded at 105 cells per well and cultured in 6‐well plate 24 h prior to transfection. EGR1‐pcDNA3.1 or EGR1‐shRNA, as well as their corresponding controls were transfected into SY5Y cells, by Viromer Red kit (Weinbergweg, Halle, Germany). The mRNA and protein levels of EGR1 and AChE were measured by qPCR and Western Blotting 72 h after transfection. Transfections were repeated three times, and within each time we had triplicates for the measurements of both mRNA and protein levels.
For qPCR measurement, the cell culture medium was removed from the 6‐well plate. About 1 mL ice‐cold Trizol was added per well and the cell lysate was collected into a 1.5 mL tube. The rest of the steps for EGR1‐ and AChE‐mRNA expressions quantification were the same as shown in Part 1 and 2. HPRT was used as housekeeping gene. Sequences of primers are shown in Supplementary Table S3.
For western blotting, cell culture medium was removed from the 6‐well plate. The total cell proteins were extracted by RIPA (Beyotime, Shanghai, China) and fractionated by SDS‐PAGE, after which they were transferred onto PVDF membranes according to the protocol offered by Abcam. Anti‐EGR1 (CST, 15F7, Boston, MA, USA; 1:1000) and AChE (Abcam, ab97299, Cambridge, UK; 1:3000) were used. Results were visualized with the Clarity Western ECL Substrate (Bio‐Rad, Hercules, California, USA) by ChemiDoc Touch Imaging System (Bio‐Rad, Hercules, California, USA).
Dual‐luciferase reporter assay
About 293T cells were cultured in 96‐well plate to a density of 106 per well before transfection. AChE promoter‐pGL3 constructs were co‐transfected, separately, with EGR1‐pcDNA3.1 by Lipofectamine 2000 (Invitrogen, Waltham, MA, USA) according to the manufacturer’s instruction. Luciferase activity was detected 48 h after transfection by using Dual‐Glo Luciferase Assay System (Promega, E2920, Fitchburg, Wisconsin, USA) on SpectraMax M5 Microplate Reader (Molecular Devices, Silicon Valley, CA, USA).
EMSA
Nuclear extracts were isolated from SY5Y cells which overexpressed EGR1 via EGR1‐pcDNA3.1 transfection (Viromer RED kit, Weinbergweg, Halle, Germany). EMSA was performed with the LightShift™ Chemiluminescent EMSA Kit (Thermo, Waltham, MA, USA) using biotin‐labeled double‐stranded oligonucleotides which target for the three binding sites in AChE gene promoter. Their respective nucleotide probe sequences of the forward strand are: position −110/−81 (5′‐GAGCGGGGAGGGgcgggggcgGAGACAGTG‐3′, probe 1), position −93/−64 (5′‐GGCGGAGACAGTGGgcgggggcgGGCGGGC‐3′, probe 2) and position −74/−45 (5′‐GGCGGgcgggggcgCTGTGAGGCCCGGAGG‐3′, probe 3).
About 80 fmol probes were incubated with 3‐μg nuclear extracts for 20 min at room temperature. For competition analysis, 25x unlabeled probe was added. For supershift analysis, 0.2 μL of anti‐EGR1 antibody (CST, 15F7, Boston, MA, USA) was added. The samples were then subjected to non‐denaturing 6% polyacrylamide gel electrophoresis. DNA‐protein complexes were electroblotted to positively charged nylon membranes and band shifts were visualized by ChemiDoc Touch Imaging System (Bio‐Rad, Hercules, California, USA).
Statistical analysis
For the human postmortem PFC study, the raw expression data were normally distributed following log2‐transformation. For the animal study, data were normally distributed following square root transformation. The data are presented as group means ± standard deviation. One‐way ANOVA was used for the comparison among more than two groups, followed by the post‐hoc Bonferroni test. Covariance analysis was performed with age, sex, PMD and APOE genotype as covariates. Student’s t‐test was applied for the comparison between 3xTg‐AD and WT mice, and between the experimental and the control group in cell line studies. Correlations between EGR1 and AChE were evaluated with the Pearson test. Correlation between Braak stages and Reisberg scales was evaluated with the Spearman test. Power analysis of the Braak stage‐ and Reisberg scale‐related observation was calculated. Statistical analyses were performed with SPSS version 17.0. P < 0.05 was considered to be significant.
Results
PFC EGR1‐ and AChE‐mRNA expression during the course of AD
PFC EGR1‐mRNA levels showed significant changes along the Braak stages (ANOVA, F(6,42) = 4.651, P = 0.001). EGR1 expression decreased significantly in Braak IV compared with Braak I and II, and further decreased in Braak V–VI (Bonferroni test, P ≤ 0.043, Figure 1A). In addition, a significant change was noticed around Braak III, that is compared with Braak 0‐III, Braak IV–VI showed a significant decrease of EGR1‐ (P < 0.0001) and AChE‐mRNA (P = 0.0145) expression. Moreover, PFC EGR1‐mRNA levels showed significant changes in the Reisberg groups (ANOVA, F(2,22) = 4.189, P = 0.0287), and particularly declined in the MCD group compared with the NCD group (Bonferroni test, P = 0.033, Figure 1B). AChE‐mRNA did not decrease significantly (ANOVA, F(6,42) = 1.877, P = 0.107) from Braak stage 0 to VI (Figure 1C), nor among the Reisberg groups (ANOVA, F(2,22) = 2.176, P = 0.137, Figure 1D). Covariance analysis showed that none of the confounding factors including age, sex, PMD and APOE genotype showed a significant effect on EGR1‐mRNA (P ≥ 0.245 and P ≥ 0.289, respectively) or AChE‐mRNA (P ≥ 0.212 and P ≥ 0.220, respectively) in the different Braak stages and Reisberg groups. Furthermore, a significant positive correlation was observed between the levels of EGR1‐mRNA and AChE‐mRNA in the entire group of subjects (r = 0.405, P < 0.01, n = 49), the Braak VI (r = 0.779, P < 0.05, n = 7) and in the SCD group (Reisburg 6–7, r = 0.519, P = 0.048, n = 15). Finally, Braak stages and Reisberg scales showed a significant positive correlation (ρ = 0.684, P < 0.001, n = 25). The statistical power of the Braak stage‐related observation was 0.975, while of Reisberg scale‐related observation was 0.675 (see Discussion section).
EGR1‐ and AChE‐mRNA expression in the frontal cortex of 3xTg‐AD mice
EGR1‐mRNA and AChE‐mRNA both significantly decreased in 3xTg‐AD mice compared with WT mice, both in the entire group (P = 0.031, and P < 0.001, Figure 2A, C, respectively) and in the 7–8‐months group (P = 0.025, and P = 0.0103, Figure 2B, D, respectively) and for AChE‐mRNA also in the 11–12‐months group (P = 0.041, Figure 2D). The WT mice showed stable EGR1‐ and AChE‐mRNA expression along the different months of age (ANOVA, F(2,51) = 0.0824, P = 0.921, Figure 2B; and F(2,50) = 0.125, P = 0.883, Figure 2D). The 3xTg‐AD mice, on the contrary, showed significantly changed EGR1‐mRNA levels among the various ages (ANOVA, F(2,52) = 4.355, P = 0.018, Figure 2B), especially when the 7–8‐months group was compared with the 3–4‐months group (Bonferroni test, P = 0.019, Figure 2B), while the AChE‐mRNA expression was stable for the different months of age (ANOVA, F(2,52) = 0.435, P = 0.65, Figure 2D). In addition, there was a significant positive correlation between the levels of EGR1‐mRNA and AChE‐mRNA in both WT and 3xTg‐AD mice, either in the entire group (r = 0.660, P < 0.001 and r = 0.668, P < 0.001), or in the individual age groups (r ≥ 0.583, P ≤ 0.05 and r ≥ 0.592, P ≤ 0.05).
Figure 2.
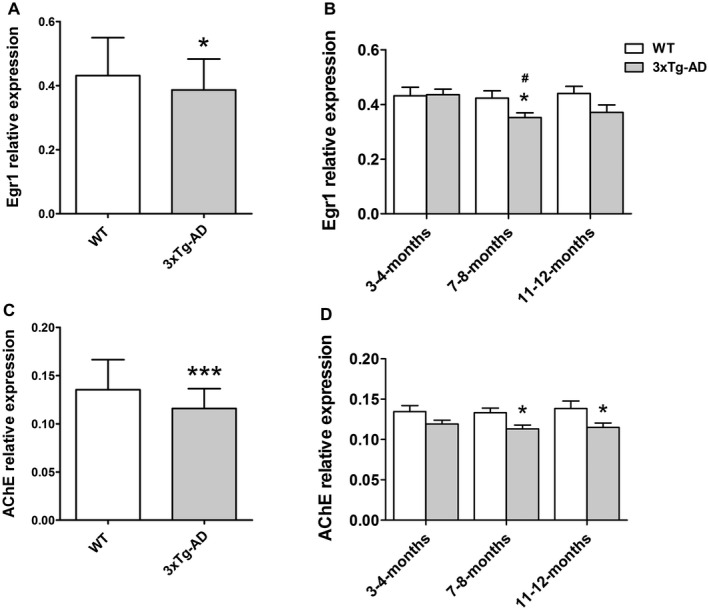
Expression of mRNA of early growth response 1 (EGR1) and acetylcholinesterase (AChE) in the frontal cortex of triple‐transgenic AD (3xTg‐AD) and wild type (WT) mice. A. EGR1‐mRNA and (C) AChE‐mRNA both significantly decreased in the entire group of 3xTg‐AD mice compared with WT mice (Student’s t‐test, P = 0.031 and P < 0.001). In addition, B. EGR1‐mRNA significantly decreased in the 7–8‐months group (Student’s t‐test, P = 0.025), while (D) AChE‐mRNA significantly decreased in the 7–8‐months group (Student’s t‐test, P = 0.0103) and the 11–12‐months group (Student’s t‐test, P = 0.041) compared with their respective month‐of‐age WT group. The WT mice showed stable (B) EGR1‐mRNA and (D) AChE‐mRNA expression among the different months of ages (ANOVA, F(2,51) = 0.0824, P = 0.921 and F(2,50) = 0.125, P = 0.883). The 3xTg‐AD mice, on the other hand, showed significant changes in (B) EGR1‐mRNA levels among ages (ANOVA, F(2,52) = 4.355, P = 0.018), especially in the 7–8‐months group compared with the 3–4‐months group (Bonferroni test, P = 0.019), while (D) AChE‐mRNA expression was stable among different months of age (ANOVA, F(2,52) = 0.435, P = 0.65). The normalized data (square root transformation) are represented as mean (± standard deviation). (*) compared with corresponding WT, (#) compared with the 3–4‐months group. *P < 0.05, ***P < 0.001, # P < 0.05.
EGR1 upregulates AChE gene transcription
Overexpression of EGR1 (P < 0.001) by transfecting EGR1‐pcDNA3.1 resulted in increased AChE expression both at the mRNA (P < 0.001) and protein levels (P < 0.01), while down‐regulating EGR1 (P < 0.01) by shRNA caused decreased AChE expression both at the mRNA (P < 0.05) and protein levels (P < 0.01) (Figure 3).
Figure 3.
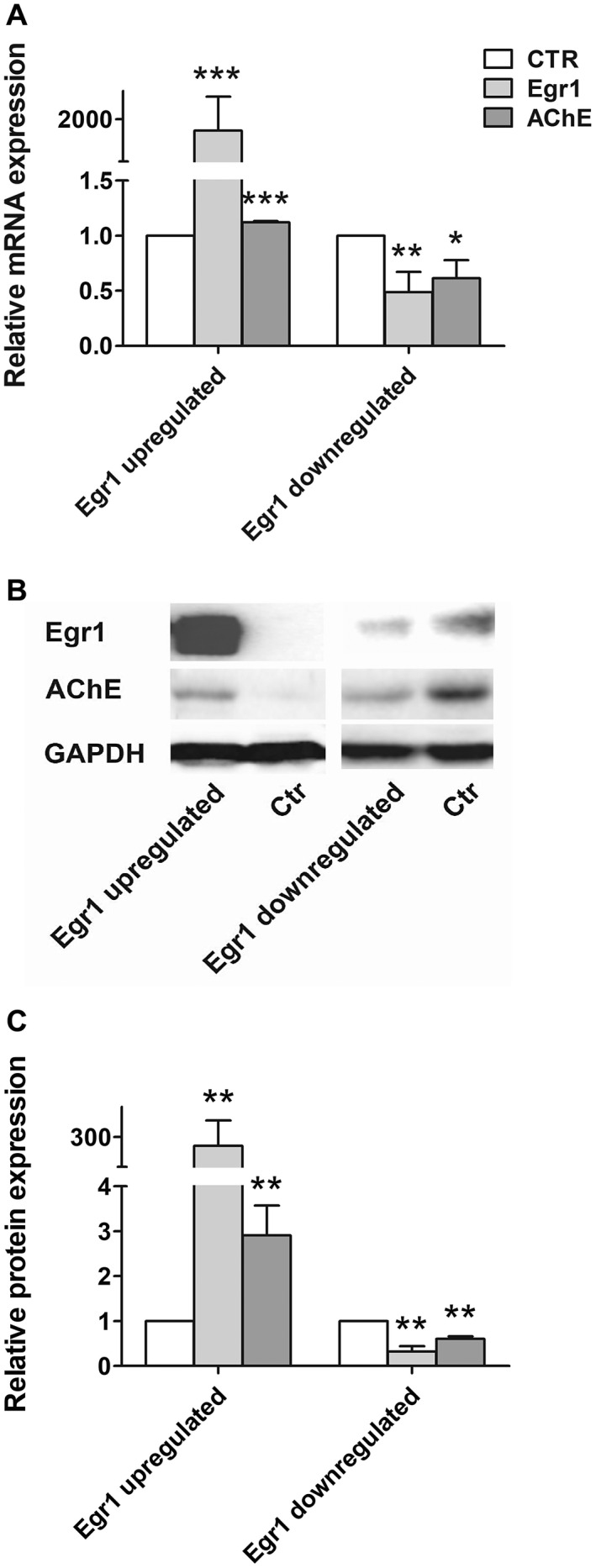
Early growth response 1 (EGR1) upregulates acetylcholinesterase (AChE) expression in the SY5Y cells. AChE expression was increased with the up‐regulation of EGR1 and decreased with down‐regulation of EGR1 in both (A) mRNA and (B–C) protein levels. Transfections of EGR1‐pcDNA3.1 or EGR1‐shRNA plasmids were repeated three times. Student’s t‐test was applied and data are represented as mean (± standard deviation) of the three times of transfection experiments, each of which was performed in triplicates. A representative result of the gel‐running results is presented in B. (*) compared with corresponding control. *P < 0.05, **P < 0.01, ***P < 0.001.
EGR1 upregulates AChE gene expression via binding AChE promoter
Following overexpression of EGR1 in 293T cells, luciferase activity was significantly increased in cell transfected with −193/+707 pGL3 compared with −108/+707 pGL3 (P < 0.01) or pGL3 (P < 0.001). In addition, there was a significant decrease in the luciferase activity in −60/+107 pGL3 compared with −98/+107 pGL3 (P < 0.01), which confirmed the prediction that the potential binding sites of EGR1 were located within −98/−60 region of AChE gene. Mutations in binding sites 2 and 3 both led to a dramatic decrease of the luciferase activity (P ≤ 0.01), while mutation in binding site 1 did not affect the fluorescence level (Figure 4B).
Protein‐oligonucleotides complexes were detected after the incubation of nuclear extracts from the EGR1‐overexpressing SY5Y cells with probe 1–3 separately (see Figure 4C, lanes 2, 6 and 10). The complexes were abolished by an excess of the related unlabelled oligonucleotide (see Figure 4C, lanes 3, 7 and 11). The specific bands that represented EGR1‐probe complexes super‐shifted after the addition of EGR1 antibody (see Figure 4C, lane 4, 8 and 12), which indicated that EGR1 physically binds to these three binding sites on the AChE promoter. Since mutation in binding site 1 did not change the luciferase activity, only site 2 and 3 were functionally involved in EGR1 regulating AChE gene expression.
Discussion
The present paper provides, for the first time, proof of a direct stimulating regulatory effect of EGR1 on the promotor of the AChE gene. A significantly positive correlation was observed between EGR1‐mRNA and AChE‐mRNA expression in both, the human PFC during the course of AD and the frontal cortex of the 3xTg‐AD mice of different ages, suggesting a stimulating regulating role of EGR1 on AChE expression. We confirmed such a role in cell culture and subsequently observed the underlying mechanism of EGR1 upregulating AChE expression via EGR1 binding to AChE gene promoter. These findings offer a novel mechanism for the characteristic up/stable‐down alteration pattern of brain cholinergic function in the course of AD.
Many studies showed that AChE enzymatic activity was depleted in AD 10. However, AChE‐protein levels are unexpectedly preserved in AD brain 4, which is in accordance with our finding that there is a relatively stable AChE‐mRNA expression in the PFC in AD. The finding that despite the reduction in PFC EGR1‐mRNA in the later Braak stages, AChE‐mRNA did not show significant changes at any Braak stage, even though EGR1 may stimulate AChE expression suggest that EGR1 defects may be a later AD stage event. Since AChE‐mRNA derives from AChE containing cortical neurons, which are in general ChAT negative, our current observations indicated that AChE expression is more related to the true cholinergic function at least in the PFC. It should be noted that although the statistical power of the neuropathological Braak stage‐related observation was 0.975, the statistic power of clinical Reisberg scale‐related observation was 0.675, which was mainly due to the fact that quite a number of subjects lacked Reisberg scale in their files. It should also be noted, though, that the Reisberg scales showed a significant positive correlation with Braak stages (ρ = 0.684, P < 0.001, n = 25), which was a confirmation of our earlier study 37. Finally, it may be worth to mention that PFC ChAT‐mRNA levels of these subjects were also available (data not shown) and we observed that PFC ChAT‐mRNA levels were stable among Braak stages (one‐way ANOVA, F(6,42) = 0.97, P = 0.46) and among the Reisberg groups (ANOVA, F(2,22) = 1.164, P = 0.3307). There was no significant correlation between the levels of EGR1‐mRNA and ChAT‐mRNA in the entire group of patients (r = 0.008, P = 0.959), although a significant negative correlation was found in the Braak IV between these 2 molecules (r = −0.85, P = 0.015). These data are in general in agreement with the findings of DeKosky et al 6 who observed that ChAT activity was unchanged in the inferior parietal‐, superior temporal‐ and anterior cingulate cortex in individuals with mild cognitive impairment (MCI) and mild AD, compared to no cognitive impairment (NCI), while ChAT activity in the superior frontal cortex (SFC) was significantly elevated above normal controls in MCI subjects. In addition, the mild AD group was not different from NCI or MCI patients. The difference between the upregulated ChAT activity in the SFC in MCI 6 and our observation that there was no significant change in PFC ChAT‐mRNA levels during the course of AD, indicates that the increased ChAT activity is not based upon enhanced ChAT production.
In our previous study, we found that EGR1 expression in the NBM remained stable during the early AD stages and decreased only during the late AD stages, while NBM ChAT started to decrease from middle Braak stage (~IV) or from Reisburg scale 2 onwards 37. We here propose that the up/stable‐down patterns of change in EGR1 in both the PFC and the NBM 37 in the course of AD may significantly contribute to the up/stable‐down patterns of cholinergic function, by regulating key factors of the cholinergic system, such as AChE. Although the regulating role of EGR1 on ChAT gene expression was observed in a cell line 29 and confirmed by our study on EGR1 and ChAT expression in the NBM during the course of AD 37, it remains to be elucidated how EGR1 is regulating the coupling of, or the balance between, ChAT and AChE during the course of this disease.
One of the inherent potential confounding factors in a postmortem study is medication use. In the framework of our present study, it is important to note that none of the subjects used anticholinesterase drugs during the last 3 months before death. In contrast, benzodiazepines were widely used in a balanced way over all Braak stages, so that they will not have confounded our data. In addition, opiates were widely used by subjects of every Braak stage except for Braak III. Concerning opiates such as morphine, Ziółkowska et al found in mouse forebrain that acute morphine administration might induce EGR1 expression 38, while morphine may also increase AChE activity 23. However, in our study the Braak III subjects did not show significant different EGR1‐ and AChE‐mRNA levels compared with any other single Braak stage subjects (P ≥ 0.376 and P = 1.00, respectively, Supplementary Figure S1, C–D). Therefore, we would not think that opiates will have interfered with our conclusions. It should also be noted that a number of subjects in Braak 0 (n = 4), Braak I (n = 2) and Braak II (n = 2) used corticosteroids, which were not used in Braak III‐VI. Weber et al found in Wistar rats that glucocorticoid did not change brain AChE activity 35. Other studies showed that glucocorticoid may increase 24, or not affect 12, EGR1 expression, depending on the brain area. The effect of glucocorticoid on EGR1 has not been studied in the frontal cortex. However, analysis on the values of EGR1‐mRNA and AChE‐mRNA of those subjects who had used glucocorticoids were generally within the same range as of those who had not used glucocorticoids within the same Braak stage (see Supplementary Figure S1, A–B). So we do not think that glucocorticoids have affected our main conclusions. In addition, antipsychotics were used over the different Braak stages, that is Braak II (n = 1), Braak III (n = 1), Braak IV (n = 3), Braak V (n = 4) and Braak VI (n = 2). There are data from animal studies showing that antipsychotics such as haloperidol may increase both EGR1 and AChE expression in the brain 19, 26, 36. However, on the basis of the overlapping values of subjects that used or had not used these medicines within the same Braak stage (Supplementary Figure S1, E–F), we do not think that antipsychotics may have interfered with our main conclusions. Moreover, the observation that EGR1‐mRNA and AChE‐mRNA are correlated not only in AD but also in 3xTg‐AD mice and decrease in later stages strengthens our conclusions.
The 3xTg‐AD mouse is considered to be a good AD model that expresses both β‐amyloid (Aβ) and hyperphosphorylated tau 27. In the present study, we found that there was a positive correlation between EGR1‐mRNA and AChE‐mRNA both in the WT and 3xTg‐AD mice, which is in line with the findings obtained in our cell‐line study (see below) and in Braak VI of AD patients in the PFC. However, the 3xTg‐AD mice showed significant lower levels of EGR1‐mRNA and AChE‐mRNA compared with WT mice in the entire group and in the 7–8‐month of age group for EGR1‐mRNA and in the 7–8‐month and 11–12‐month of age groups for AChE‐mRNA. This indicates that, concerning the changes in EGR1‐mRNA and AChE‐mRNA expression in frontal cortex, this 3xTg‐AD mouse model mainly reflects the late AD stages in human. In contrast, it did not mimic the brain activation in early AD as we observed in postmortem human brain material 2. Therefore, on the basis of our current data, this mouse model seems not suitable for further research concerning the activation of genes in the preclinical, early AD stages. It should also be noted that the earliest cognitive impairment in the 3xTg‐AD mouse model was found to begin at the age of 4 months without plaque‐ or tangle pathology 1 while we observed that EGR1‐mRNA is not different from the WT controls. In AD patients, however, the cognitive impairment usually begins during Braak III‐IV, when EGR1 starts to decrease in the PFC and when AD pathology in terms of Aβ, hyperphosphorylated tau and silver‐stained plaques are increased 3. Again this is an indication of the limitations of using the 3xTg‐AD mouse model for the study of this disease.
We proved, for the first time, the crucial and sufficiently positive regulating role of EGR1 on human AChE expression by showing that up‐ or down‐regulation of EGR1 may cause up‐ or down‐expression of AChE on the levels of mRNA and protein. This observation was followed by analyzing the functional EGR1 binding sites in the human AChE gene promoter by dual‐luciferase assay and EMSA assay. We showed three binding sites of EGR1 in the human AChE promoter region, from which two functioned. This is a confirmation of the study of Mutero et al who also used mutation of the mouse AChE promoter region and measurements of promoter activity, as well as gel mobility shift assays (Mutero, Camp and Taylor, JBC, 1995, 270(4):1866–1872) 25. In addition, we offered functional data of EGR1 regulating AChE expression. Although a previous study observed that human AChE gene expression can be activated by EGR1 via EGR1 binding to AChE promoter 34, it lacked the evidence on the direct effect on AChE expression and did not show decreased EGR1 leading to decreased AChE expression. Our findings that up‐ or down‐regulation of EGR1 respectively increased or decreased the AChE expression in both mRNA and protein levels in the SY5Y cell line has given novel evidence of a crucial and sufficiently positive regulating role of EGR1 on the expression of AChE. It also seems to offer a good explanation for the up/stable‐down pattern of cholinergic changes in the brain during the course of AD. Besides, via mutation analysis of the binding sites we observed two sites in −79/−71 and −69/−61 that are close to the transcription starting point and are functionally effective.
Concluding, during the early AD stages, up/stable EGR1 may play a protective role, maintaining cholinergic system function, which may contributes to stable cognition; while in late AD stages decreased EGR1 may lead to a decreased cholinergic system function, which contributes to dementia. Brain EGR1 thus could be a novel target for AD therapy. The 3xTg‐AD mouse may be a proper model to study EGR1 and AChE related pathways in late AD stages, but is not suitable for a study of the mechanisms that cause activation of expression in pre‐symptomatic AD stages.
Funding
This study was supported by the Nature Science Foundation of China (91849125, 31571048), Programme of Introducing Talents of Discipline to Universities of China (B13026) and the Non‐profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2017PT31038 and 2018PT31041). Miss Yu‐ting Hu was supported by the Fund for Cultivation of Innovative Talents, 985 Project of Zhejiang University (188310‐193840101/001). There are no financial or other relationships that might lead to a conflict of interest.
Supporting information
Figure S1 . Scatter plot of the data of early growth response 1 (EGR1)‐mRNA and acetylcholinesterase (AChE)‐mRNA levels along the Braak stages. Open circles represent the subjects who used corticosteroid (A–B), opiates (C–D) or antipsychotics (E–F) during the last 3 months before death. It was concluded that the data of the users and non‐users of these medicines overlap, and thus do not interfere with the main conclusions of the present study (for details see Discussion).
Figure S2 . 3xTg‐AD mice genotyping. Ear‐clipped tissue was obtained from each mouse. DNA was isolated according to the study of Oddo et al (2003). Subsequently qPCR was performed using ABsolute blue qPCR mix (AB4162B, Thermo, US). The products were checked by running a 2% agarose gel. PS1 = mutant human presenilin 1 gene (M146V); APP = mutant human amyloid beta precursor protein gene (K670N, M671L); Tau = mutant human microtubule associated protein tau gene (P301L).
Table S1 . Clinico‐pathological details of subjects. Braak stage: neuropathological progression in Alzheimer’s disease (AD) (Braak and Braak, 1991). Reisberg scale: clinical progression in AD (Reisberg et al, 1982). F = female; M = male; CSF = cerebrospinal fluid; PMD = postmortem delay (minutes); RIN = RNA integrity number.
Table S2 . Matching for confounding factors in Alzheimer’s disease (AD) patients. Braak stage: neuropathological progression in AD (Braak and Braak, 1991). Reisberg group: clinical progression in AD according to the Reisberg scale (Reisberg et al, 1982). Based on the Reisberg scale, three groups were distinguished in the present study, ie, no cognitive decline (NCD) group including Reisberg 1 subjects, mild cognitive decline (MCD) group including Reisberg 2–5 subjects, who had very mild to moderately severe cognitive decline, and severe cognitive decline (SCD) group including Reisberg 6–7 subjects, who had severe/very severe cognitive decline. F = female; M = male; CSF = cerebrospinal fluid; PMD = postmortem delay (minutes); P = ANOVA‐based P‐value between Braak stages or Reisberg groups. Data are represented as mean (± standard deviation).
Table S3 . Primer sequences for qPCR. GAPDH = glyceraldehyde phosphate dehydrogenase; HPRT = hypoxanthine guanine phosphoribosyl transferase; EGR1 = early growth response 1; AChE = acetylcholinesterase; F = forward primer; R = reverse primer.
Acknowledgments
The authors are grateful to Stichting Vrienden van het Herseninstituut for their support, to Dr. Koen Bossers (Netherlands Institute for Neuroscience) for providing data, to Dr. Zi‐Yi Wang for his technical help on EMSA, and to Ms. Wilma Verweij for secretarial help.
References
- 1. Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM (2005) Intraneuronal Abeta causes the onset of early Alzheimer’s disease‐related cognitive deficits in transgenic mice. Neuron 45:675–688. [DOI] [PubMed] [Google Scholar]
- 2. Bossers K, Wirz KT, Meerhoff GF, Essing AH, van Dongen JW, Houba P et al. (2010) Concerted changes in transcripts in the prefrontal cortex precede neuropathology in Alzheimer’s disease. Brain 133(Pt 12):3699–3723. [DOI] [PubMed] [Google Scholar]
- 3. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 4. Campanari ML, Garcia‐Ayllon MS, Blazquez‐Llorca L, Luk WK, Tsim K, Saez‐Valero J (2014) Acetylcholinesterase protein level is preserved in the Alzheimer’s brain. J Mol Neurosci: MN 53:446–453. [DOI] [PubMed] [Google Scholar]
- 5. Christy B, Nathans D (1989) DNA binding site of the growth factor‐inducible protein Zif268. Proc Natl Acad Sci U S A 86:8737–8741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, Bennett DA et al (2002) Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol 51:145–155. [DOI] [PubMed] [Google Scholar]
- 7. Dubelaar EJ, Mufson EJ, ter Meulen WG, Van Heerikhuize JJ, Verwer RW, Swaab DF (2006) Increased metabolic activity in nucleus basalis of Meynert neurons in elderly individuals with mild cognitive impairment as indicated by the size of the Golgi apparatus. J Neuropathol Exp Neurol 65:257–266. [DOI] [PubMed] [Google Scholar]
- 8. Ferreira‐Vieira TH, Guimaraes IM, Silva FR, Ribeiro FM (2016) Alzheimer’s disease: targeting the cholinergic system. Curr Neuropharmacol 14:101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Francis PT, Palmer AM, Snape M, Wilcock GK (1999) The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry 66:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garcia‐Ayllon MS, Small DH, Avila J, Saez‐Valero J (2011) Revisiting the role of acetylcholinesterase in Alzheimer’s disease: cross‐talk with P‐tau and beta‐amyloid. Front Mol Neurosci 4:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Getman DK, Mutero A, Inoue K, Taylor P (1995) Transcription factor repression and activation of the human acetylcholinesterase gene. J Biol Chem 270:23511–23519. [DOI] [PubMed] [Google Scholar]
- 12. Hansson AC, Fuxe K (2008) Time‐course of immediate early gene expression in hippocampal subregions of adrenalectomized rats after acute corticosterone challenge. Brain Res 1215:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Janeczek M, Gefen T, Samimi M, Kim G, Weintraub S, Bigio E et al (2018) Variations in acetylcholinesterase activity within human cortical pyramidal neurons across age and cognitive trajectories. Cereb Cortex 28:1329–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Knapska E, Kaczmarek L (2004) A gene for neuronal plasticity in the mammalian brain: Zif268/Egr‐1/NGFI‐A/Krox‐24/TIS8/ZENK? Prog Neurogibol 74:183–211. [DOI] [PubMed] [Google Scholar]
- 15. Korecka JA, van Kesteren RE, Blaas E, Spitzer SO, Kamstra JH, Smit AB et al (2013) Phenotypic characterization of retinoic acid differentiated SH‐SY5Y cells by transcriptional profiling. PLoS One 8:e63862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kovalevich J, Langford D (2013) Considerations for the use of SH‐SY5Y neuroblastoma cells in neurobiology. Methods Mol Biol 1078:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Y, Camp S, Rachinsky TL, Bongiorno C, Taylor P (1993) Promoter elements and transcriptional control of the mouse acetylcholinesterase gene. J Biol Chem 268:3563–3572. [PubMed] [Google Scholar]
- 18. Lleo A, Greenberg SM, Growdon JH (2006) Current pharmacotherapy for Alzheimer’s disease. Annu Rev Med 57:513–533. [DOI] [PubMed] [Google Scholar]
- 19. Mahadik SP, Mukherjee S (1995) Monosialoganglioside cotreatment prevents haloperidol treatment‐associated loss of cholinergic enzymes in rat brain. Biol Psychiatry 38:246–254. [DOI] [PubMed] [Google Scholar]
- 20. Mesulam MM, Geula C (1988) Acetylcholinesterase‐rich pyramidal neurons in the human neocortex and hippocampus: absence at birth, development during the life span, and dissolution in Alzheimer’s disease. Ann Neurol 24:765–773. [DOI] [PubMed] [Google Scholar]
- 21. Mesulam MM, Geula C (1988) Nucleus basalis (Ch4) and cortical cholinergic innervation in the human brain: observations based on the distribution of acetylcholinesterase and choline acetyltransferase. J Comp Neurol 275:216–240. [DOI] [PubMed] [Google Scholar]
- 22. Mesulam MM, Geula C (1991) Acetylcholinesterase‐rich neurons of the human cerebral cortex: cytoarchitectonic and ontogenetic patterns of distribution. J Comp Neurol 306:193–220. [DOI] [PubMed] [Google Scholar]
- 23. Mohanakumar KP, Sood PP (1983) Acetylcholinesterase changes in the central nervous system of mice during the development of morphine tolerance addiction and withdrawal. Brain Res Bull 10:589–596. [DOI] [PubMed] [Google Scholar]
- 24. Monsey MS, Boyle LM, Zhang ML, Nguyen CP, Kronman HG, Ota KT et al (2014) Chronic corticosterone exposure persistently elevates the expression of memory‐related genes in the lateral amygdala and enhances the consolidation of a Pavlovian fear memory. PLoS One 9:e91530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mutero A, Camp S, Taylor P (1995) Promoter elements of the mouse acetylcholinesterase gene. Transcriptional regulation during muscle differentiation. J Biol Chem 270:1866–1872. [PubMed] [Google Scholar]
- 26. Nguyen TV, Kosofsky BE, Birnbaum R, Cohen BM, Hyman SE (1992) Differential expression of c‐fos and zif268 in rat striatum after haloperidol, clozapine, and amphetamine. Proc Natl Acad Sci U S A 89:4270–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R et al (2003) Triple‐transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39:409–421. [DOI] [PubMed] [Google Scholar]
- 28. Pavletich NP, Pabo CO (1991) Zinc finger‐DNA recognition: crystal structure of a Zif268‐DNA complex at 2.1 A. Science 252:809–817. [DOI] [PubMed] [Google Scholar]
- 29. Quirin‐Stricker C, Mauvais C, Schmitt M (1997) Transcriptional activation of human choline acetyltransferase by AP2‐ and NGF‐induced factors. Mol Brain Res 49:165–174. [DOI] [PubMed] [Google Scholar]
- 30. Reisberg B, Ferris SH, de Leon MJ, Crook T (1982) The Global Deterioration Scale for assessment of primary degenerative dementia. Am J Psychiatry 139:1136–1139. [DOI] [PubMed] [Google Scholar]
- 31. Thiel G, Cibelli G (2002) Regulation of life and death by the zinc finger transcription factor Egr‐1. J Cell Physiol 193:287–292. [DOI] [PubMed] [Google Scholar]
- 32. van de Nes JA, Kamphorst W, Ravid R, Swaab DF (1998) Comparison of beta‐protein/A4 deposits and Alz‐50‐stained cytoskeletal changes in the hypothalamus and adjoining areas of Alzheimer’s disease patients: amorphic plaques and cytoskeletal changes occur independently. Acta Neuropathol 96:129–138. [DOI] [PubMed] [Google Scholar]
- 33. Veyrac A, Besnard A, Caboche J, Davis S, Laroche S (2014) The transcription factor Zif268/Egr1, brain plasticity, and memory. Prog Mol Biol Transl Sci 122:89–129. [DOI] [PubMed] [Google Scholar]
- 34. von der Kammer H, Mayhaus M, Albrecht C, Enderich J, Wegner M, Nitsch RM (1998) Muscarinic acetylcholine receptors activate expression of the EGR gene family of transcription factors. J Biol Chem 273:14538–14544. [DOI] [PubMed] [Google Scholar]
- 35. Weber U, Brank M, Grubic Z (1999) Glucocorticoids differentially control synthesis of acetylcholinesterase and butyrylcholinesterase in rat liver and brain. Chem‐Biol Interact 119–120:341–347. [DOI] [PubMed] [Google Scholar]
- 36. Wheeler AL, Creed MC, Voineskos AN, Nobrega JN (2014) Changes in brain functional connectivity after chronic haloperidol in rats: a network analysis. Int J Neuropsychopharmacol 17:1129–1138. [DOI] [PubMed] [Google Scholar]
- 37. Zhu QB, Unmehopa U, Bossers K, Hu YT, Verwer R, Balesar R et al (2016) MicroRNA‐132 and early growth response‐1 in nucleus basalis of Meynert during the course of Alzheimer’s disease. Brain 139(Pt 3):908–921. [DOI] [PubMed] [Google Scholar]
- 38. Ziolkowska B, Gieryk A, Solecki W, Przewlocki R (2015) Temporal and anatomic patterns of immediate‐early gene expression in the forebrain of C57BL/6 and DBA/2 mice after morphine administration. Neuroscience 284:107–124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 . Scatter plot of the data of early growth response 1 (EGR1)‐mRNA and acetylcholinesterase (AChE)‐mRNA levels along the Braak stages. Open circles represent the subjects who used corticosteroid (A–B), opiates (C–D) or antipsychotics (E–F) during the last 3 months before death. It was concluded that the data of the users and non‐users of these medicines overlap, and thus do not interfere with the main conclusions of the present study (for details see Discussion).
Figure S2 . 3xTg‐AD mice genotyping. Ear‐clipped tissue was obtained from each mouse. DNA was isolated according to the study of Oddo et al (2003). Subsequently qPCR was performed using ABsolute blue qPCR mix (AB4162B, Thermo, US). The products were checked by running a 2% agarose gel. PS1 = mutant human presenilin 1 gene (M146V); APP = mutant human amyloid beta precursor protein gene (K670N, M671L); Tau = mutant human microtubule associated protein tau gene (P301L).
Table S1 . Clinico‐pathological details of subjects. Braak stage: neuropathological progression in Alzheimer’s disease (AD) (Braak and Braak, 1991). Reisberg scale: clinical progression in AD (Reisberg et al, 1982). F = female; M = male; CSF = cerebrospinal fluid; PMD = postmortem delay (minutes); RIN = RNA integrity number.
Table S2 . Matching for confounding factors in Alzheimer’s disease (AD) patients. Braak stage: neuropathological progression in AD (Braak and Braak, 1991). Reisberg group: clinical progression in AD according to the Reisberg scale (Reisberg et al, 1982). Based on the Reisberg scale, three groups were distinguished in the present study, ie, no cognitive decline (NCD) group including Reisberg 1 subjects, mild cognitive decline (MCD) group including Reisberg 2–5 subjects, who had very mild to moderately severe cognitive decline, and severe cognitive decline (SCD) group including Reisberg 6–7 subjects, who had severe/very severe cognitive decline. F = female; M = male; CSF = cerebrospinal fluid; PMD = postmortem delay (minutes); P = ANOVA‐based P‐value between Braak stages or Reisberg groups. Data are represented as mean (± standard deviation).
Table S3 . Primer sequences for qPCR. GAPDH = glyceraldehyde phosphate dehydrogenase; HPRT = hypoxanthine guanine phosphoribosyl transferase; EGR1 = early growth response 1; AChE = acetylcholinesterase; F = forward primer; R = reverse primer.