

Abstract
Cerebral amyloid angiopathy (CAA) is of increasing clinical and research interest as the ability to detect it and its consequences by neuroimaging in living subjects has advanced. There is also increasing interest in understanding its possible role in the development of intracerebral hemorrhage, Alzheimer's disease (AD) and vascular dementia. In this article, the literature on this subject is reviewed and novel findings relating CAA to subcortical white matter damage in 224 subjects in the Oxford project to Investigate Memory and Ageing (OPTIMA) are reported. The relationship between CAA and subcortical tissue damage in the OPTIMA subjects was found to be critically dependent on ApoE genotype, there being a positive relationship between measures of CAA and subcortical small vessel disease in ApoE ε4 carriers and a significant negative relationship in ApoE ε2 carriers. These findings draw attention, as have many other studies, to the importance of ApoE genotype as a major risk factor not only for dementia but also for damage to blood vessels in the aging brain.
Keywords: Alzheimer's disease, ApoE, CAA, cerebral white matter, dementia
Introduction
Although cerebral amyloid angiopathy (CAA) has been well recognized since 1938 103, there remains considerable confusion about its contributions both to dementia and to subcortical white matter disease. Partly this confusion stems from the fact that CAA is closely associated with Alzheimer's disease (AD), which itself is the major cause of dementia in the elderly. Further difficulty arises because there has, until recently, been no widely adopted and validated system of scoring subcortical small vessel disease (SVD), although there have been moves made recently to try to rectify this (26, 106; Love et al unpublished). The literature on CAA has been substantially enlarged recently by studies of neuroimaging in living subjects in which some distinction has been made between those with CAA‐related intracerebral hemorrhage, superficial siderosis or microbleeds, used as a marker for CAA, and those with CAA complicating AD 3, 20, 21. A further potential source of additional information is the use of an amyloid‐binding marker such as Pittsburgh Compound B (PIB) to detect beta amyloid in living subjects with which it is possible to obtain an impression of the extent of CAA 43, 55, 63, 66. fMRI is also showing potential to provide another imaging biomarker of value in CAA 84. In the first part of this article, we review the literature on these topics. There are good recent reviews of more general aspects of CAA that are not covered here 4, 21, 118, 123. In the second part, we contribute new data derived from a study of CAA and its relationship to SVD in the Oxford project to Investigate Memory and Ageing (OPTIMA) cohort.
CAA and its contribution to subcortical white matter disease
The vessels affected by CAA mainly consist of leptomeningeal medium and small‐sized arteries and cortical arterioles. Some of the latter supply blood to not only the cerebral cortex but also the subcortical white matter. A smaller proportion of cases, mainly, but not entirely, restricted to those with fully developed AD pathology, also show CAA affecting cortical capillaries 4, 64, 109. Vessels affected by CAA show a wide range of alterations from very small deposits of amyloid with little or no alteration in vessel structure to loss of all the smooth muscle, elastic tissue (in arteries) and collagen, and alterations in the pattern of glycosaminoglycan expression 4, 94, 117. These structural changes impair the capacity of affected vessels to supply tissues with the blood they need and lead to cortical infarction and intracerebral hemorrhage in some subjects 15, 28, 50, 54, 81, 82. Therefore, it is logical to expect that the blood supply to the subcortical white matter is similarly compromised leading to an increased risk of ischemia, as suggested by Gray et al 38. In the great majority of cases of CAA, the peptides that are deposited as amyloid are amyloid beta (amyloidβ) peptides, the products of enzymatic breakdown of amyloid precursor protein (APP) and the peptides found also in the cortical amyloid plaques characteristic of AD. In a few inherited cases of CAA, different peptides are deposited in vessels walls, for example, cystatin C, transthyretin, gelsolin and BRI2 87, 93, 94. These forms of CAA are not covered by this review.
Ideally, studies of the importance of CAA require quantitation of the regional and overall severity of CAA for comparison with another parameter such as the extent of white matter disease or of cognitive impairment. So far there have been only semi‐quantitative scores used to assess CAA severity 4, 64, 83, 120. Studies that have examined the relationship between CAA and white matter ischemia at the pathological level in humans are limited. Ellis et al 28 found a significant relationship between CAA severity and SVD (classified as arteriosclerosis and arteriolosclerosis) as well as atherosclerosis in 117 autopsy‐confirmed AD cases in the CERAD study. Tomimoto et al 113 examined 39 cases of AD, 13 of Binswanger's disease and five elderly controls. They found more profound white matter damage in Binswanger's disease than in AD and in AD found more white matter damage than in controls, but it was more related to fibrohyalinosis in subcortical small vessels than to CAA. A study undertaken by Haglund and Englund 49 included 63 cases with varying degrees of Alzheimer‐type pathology. These cases were divided into 37 cases that had only AD pathology, 15 cases that had AD pathology severe enough to be designated AD by internationally recognized criteria, together with vascular disease, and 11 cases that had vascular disease judged sufficient to account for dementia and mild AD changes that were insufficient to merit a diagnosis of AD. Only in the cases of “pure” AD did the severity of white matter lesions correlate with the severity of CAA. Roher et al 97 studied 24 cases of AD and 17 controls. They quantified amyloidβ in gray matter and white matter by immunoassay and the amount of amyloidβ in isolated blood vessels with a thioflavin S stain. Dilatation of perivascular spaces was quantified histologically. They showed that the severity of this perivascular dilatation correlated with the amount of CAA and with the amyloidβ load in the cerebral cortex as well as with the ApoE ε4 genotype. Thal et al 110 studied the brains of 52 cases of AD with dementia or AD‐type pathology without dementia and reported that CAA affecting leptomeningeal and cortical vessels and arteriosclerosis/lipohyalinosis affecting subcortical small arteries and arterioles were all increased in AD with dementia compared with those with lesser degrees of AD pathology without dementia. Chalmers et al 19 studied frontal lobe white matter damage in 125 cases of pathologically confirmed AD and related it to the severity of CAA, cortical amyloidβ load in the form of plaques and ApoE genotype. Measures of white matter damage that were reported were the extent of immunolabeling for glial fibrillary acidic protein (GFAP), axonal accumulation of APP, axon density and myelin staining intensity. They found no relationship between atherosclerosis, arteriolosclerosis, CAA or ApoE genotype and measures of white matter damage. Instead, frontal lobe white matter damage, reflected in intensity and extent of GFAP immunostaining, was correlated with cortical amyloidβ load. Tian et al 112 studied 137 autopsy cases of pathologically confirmed AD and found that all cases had some degree of CAA. Eighty‐seven (63%) had some subcortical myelin loss that was worst in the occipital lobe, which also showed the most severe and frequent CAA. One hundred and twenty‐six (92%) of the cases also showed some arteriosclerotic changes that were not more common in the occipital lobe but were more severe at this site. Eighty‐seven cases (63%) showed both CAA and myelin loss with a weak significant relation between the two, and 47 cases showed myelin loss and arteriosclerotic pathology. The severity of the arteriosclerotic changes in these cases was correlated with CAA severity. These studies suggest a complex interplay of CAA with subcortical pathology in subjects with and without AD.
The favored explanation for the development of CAA is that it results from impaired drainage from the cerebral cortex of amyloidβ as a consequence of impaired clearance mechanisms for amyloidβ via the blood, impaired metabolic breakdown of amyloidβ in brain and impaired drainage along perivascular drainage pathways because of increased rigidity of vessels as they age 17, 121, 122, 123. Grinberg and Thal 46 put forward the interesting suggestion that SVD may exacerbate CAA by promoting cerebral edema that needs to compete with amyloidβ for clearance via perivascular drainage pathways.
This pathological evidence has now been supplemented by studies on neuroimaging. Gurol et al 47 examined T2 white matter hyperintensities (WMH) in magnetic resonance imagings (MRIs) on 54 AD or mild cognitive impairment (MCI) cases and 42 cases with probable CAA diagnosed using the Boston criteria that rely on a history of lobar intracerebral hemorrhage and absence of any other clear cause of intracerebral hemorrhage 60. They found more white matter damage in cases of CAA than of AD or MCI and found that the WMH and frequency of lacunar infarcts were correlated with plasma concentrations of amyloidβ 1–40, the shorter of the two common forms of amyloidβ and the one that forms the major deposits in CAA. A smaller MRI study of 11 cases of CAA‐associated intracerebral hemorrhage and 13 controls by Salat et al 102 found more white matter changes in temporal lobe in CAA. Holland et al 53 assessed WMH in 32 cases of probable CAA, 41 cases of clinically diagnosed AD or MCI and 29 healthy controls. WMH volumes were greater in the CAA and AD/MCI groups than in the controls. Chen et al 23 carried out an 1.1‐year median follow‐up on 26 patients fulfilling Boston criteria for possible (n = 3) or probable (n = 23) CAA and found a significant increase of 18% as percentage of baseline WMH that was also associated with an increase in cognitive impairment. In this study there were new lobar microbleeds seen in 46% of cases during follow‐up that were correlated with baseline WMH volume but not with the increase in WMH during follow‐up. Zhu et al 129 carried out a large MRI study of 102 subjects with lobar intracerebral hemorrhage diagnosed as possible or probable CAA together with 99 subjects with hypertension‐related intracerebral hemorrhage and 159 healthy elderly controls. They used a frontal‐occipital gradient to describe the difference in severity of WMH between frontal and occipital lobes. A higher proportion of occipital‐dominant WMH was found in cases with lobar intracerebral hemorrhage than in controls and those with clear occipital lobe‐dominant WMH had more WMH lesions overall and a higher prevalence of ApoE ε4 genotype consistent with CAA contributing to their white matter lesions. In a further study changes detected on diffusion tensor imaging were found to be a sensitive indicator of white matter damage related to CAA and to cognitive impairment 119.
Silent white matter infarcts have been shown to be associated with advanced CAA. Lesions detectable with diffusion‐weighted imaging have been found and are linked to the burden of cerebral micro‐bleeds (CMBs) 57 and leukoaraiosis and are common after CAA‐related intracerebral hemorrhage 45. Microbleeds are detectable on MRI as signal voids related to hemosiderin deposition resulting from minor previous hemorrhages 34, 44. Although not specific for CAA‐related hemorrhages, lobar microbleeds are associated with ApoE ε4 genotype which in turn is a risk factor for CAA 80. They also have a predilection for occurring in the parietal and occipital lobes where CAA is prevalent and have a higher frequency in cases with lobar intracerebral hemorrhage, which is itself closely linked to CAA 42, 44, 98. Moreover, lobar microbleeds are associated with white matter disease in AD, implicating CAA as the cause of both 77. In a study of amyloid imaging with PIB, CMBs were found to correspond to areas containing a high concentration of amyloid 27 and they were a risk factor for recurrent lobar hemorrhage 42. Yates et al 127 followed up 174 subjects with MRI and PIB over 3 years and found CMBs in 18.6% of controls, 24.3% of those with MCI and 40% of those with AD. The incidence of CMBs was sixfold higher in those with PIB+ amyloid than in those who were PIB negative for amyloid. Incident CMBs were associated with increasing age, ApoE ε4 genotype, PIB positivity for amyloid and baseline CMBs. Multiple CMBs were associated with lacunar infarction and WMH severity. In a further recent study using PIB and MRI, WMH was found to correlate with intensity of amyloid deposits in cases with CAA but not AD 48.
Investigation of the frequency with which dilated perivascular spaces by MRI are found in subcortical gray or white matter is beginning to yield valuable information about subcortical SVD. It has long been recognized that enlarged perivascular spaces around subcortical vessels, visible on imaging, are associated with SVD 52, 114. Martinez‐Ramirez et al 70 found more severely dilated perivascular spaces in white matter in a group of 89 subjects attending a memory clinic and having MCI if they also had evidence of lobar microbleeds, a marker of CAA. Age, a high volume of WMH and hypertension also increased the risk of finding severely dilated perivascular spaces in white matter. Severely dilated perivascular spaces in basal ganglia, in contrast, were only correlated with a history of hypertension. A separate clinicopathological study compared the presence of dilated perivascular spaces in the centrum ovale on MRI in 14 cases with CAA on pathology and 10 cases without CAA on pathology. There were dilated perivascular spaces in the centrum ovale in 10/14 cases with CAA and 0/10 cases without CAA 22.
Cortical subarachnoid hemorrhage is much less common than CMBs, but in elderly subjects it is another likely consequence of CAA 9, 92. It can be detected as acute bleeding on T2‐weighted MRI or as superficial siderosis, a linear gyriform pattern of hypointense signal 32, 61, 116. It was detected in 60% of elderly patients with a clinical diagnosis of CAA while being absent from controls 61.
These pathological and recent neuroimaging contributions to analyzing the significance of CAA for SVD, while not without some controversies, leave little doubt overall that these two features of the aging brain are related to each other.
CAA and its contribution to dementia
In their study of 117 cases of autopsy‐confirmed AD, Ellis et al 28 did not find any relationship between cognition and CAA severity. However, there is other increasing evidence that CAA does make at least a modest contribution to cognitive impairment in the elderly 37, 41, 125. In a useful systematic review of studies relating CAA, identified pathologically, to dementia Keage et al 56 found that CAA was consistently more frequent and more severe in elderly subjects with dementia than in those who were undemented. The UK MRC‐Cognitive Function and Ageing study, a community‐based study of elderly people, reported that severe congophilic angiopathy was strongly related to the presence of dementia 76. In the Honolulu‐Asia study of aging in Japanese‐American men, it was concluded that cases of AD with CAA had lower cognitive scores than those without CAA 85. In a study of community‐dwelling elderly subjects in the Religious Orders Study, CAA was found to be very common (84.9%) and related to the pathology of AD. After taking account of AD pathology, moderate‐to‐severe CAA was independently related to lower perceptual speed and episodic memory 2. In an imaging study, CMBs were found to be an independent predictor of cognitive impairment in multiple domains and also of severity of dementia in 83 subjects fulfilling criteria proposed by Erkinjuntti et al 30 for subcortical vascular dementia 105.
Complications of CAA and the role of Abeta vaccine treatment of AD in augmenting CAA
CAA has long been recognized to be associated with secondary structural changes of which inflammation, fibrinoid necrosis, microaneurysm formation, thrombosis and detachment of the tunica media from the tunica adventitia, giving an impression of duplication of the vessel lumen “double barreling,” are the most common 4, 21, 118. It is cases of severe CAA that are most likely to be associated with complications. Microaneurysms are the likely origin of lobar intracerebral hemorrhage, a serious and not uncommonly fatal complication. Hypertension and anti‐coagulant therapy are risk factors for hemorrhage associated with CAA 3, 11, 65, 96, 116. Inflammation leads to a vasculitis that is important to recognize in life as it may respond to high‐dose steroid or immunosuppressive therapy. It is clinically expressed by a relatively rapidly progressing cognitive impairment, seizures, behavioral changes, neurological deficits and headaches 24, 29, 59, 95, 104. It can only be diagnosed with certainty by a cortical and leptomeningeal biopsy. It has similarities to the form of meningoencephalitis associated with treatment of AD with a vaccine against amyloidβ and may be related also to the spontaneous development of anti‐amyloidβ antibodies 86. Such vaccine treatment was associated with an increase in CAA and evidence on MRI of white matter edema 13, 33, 79, now more fully characterized as amyloid‐related imaging abnormalities 8, 124.
The role of ApoE genotype
A role for ApoE genotype in the pathogenesis of CAA has been mentioned several times already in this review. To summarize, ApoE ε4, in both clinical and post‐mortem series, increases the risk of CAA and its severity in a dose‐dependent fashion 11, 18, 39, 62, 78, 83, 89, 90, 91, 115. The epsilon 2 variant also influences CAA 64, 78 and complications of CAA, possibly by promoting CAA‐related hemorrhage 12, 40, 71, 75 and the epsilon 4 and 2 variants interact so that subjects with both ApoE isoforms have earlier risk of CAA and a higher risk of recurrent hemorrhage 40, 80. It is unclear how ApoE has these effects, but the protein has important roles in lipoprotein metabolism and in amyloidβ clearance from the brain via the blood and the functional properties of the protein have been shown to be profoundly affected by the isoform 5, 6, 25, 67, 68, 115.
Contributions of animal studies
Animal studies of mouse models of AD and CAA have added considerably to our understanding of CAA and its influence on cognitive impairment and SVD. One important insight comes from a study of mice with the Swedish, Dutch and Iowa APP mutations. These mice developed more CAA when cerebral perfusion was chronically reduced by bilateral common carotid artery stenosis 81. In this context, it is noteworthy that human subjects with known risk factors for vascular disease develop reduced cortical cerebral blood flow as they age 7. The contribution of CAA to cognitive impairment has been shown in a mouse model of AD in which early CAA developed before cortical plaques were apparent. These mice had cognitive impairments while only affected by CAA 126. The importance of ApoE for the development of CAA has been studied in a series of experiments by Fryer et al 35, 36. Their study in 2003 showed that AD transgenic mice that lacked ApoE were protected from developing CAA. In a subsequent study Fryer et al 36 showed that AD mice transfected with the gene for human ApoE ε3 developed few cortical amyloidβ plaques and CAA. If, in contrast, the gene for human ApoE ε4 was transfected, more severe CAA developed and the ratio of amyloidβ 42:amyloidβ 40 in brain was reduced, suggesting that ApoE4 influenced the clearance of the two amyloidβ peptides in a differential manner. As amyloidβ 40 is the dominant species found in vessel walls in CAA, this altered ratio was likely to have had a role in promoting CAA in the mice carrying the gene for ApoE ε4. A study of AD transgenic mice treated with an anti‐amyloid vaccine has detailed changes in CAA that suggest that microhemorrhages develop as amyloid is cleared from vascular CAA deposits and is reduced when vessels then undergo a healing process 128. Finally, several animal experiments lend support to the model of CAA developing as a consequence of impaired clearance of cortical amyloid along vascular drainage pathways that has been put forward by Weller and colleagues 17, 121, 122, 123. These include a study showing that AD transgenic mice with APP overexpressed under the control of a neuronal promoter (thus providing only a neuronal source of amyloidβ) was sufficient to cause CAA 16, and a transgenic AD mouse model that showed that reduction of cerebral blood flow impaired drainage of interstitial fluid from the brain when CAA was present 1. A comparison of drainage of dextran from the brains of mice with and without CAA similarly showed impaired drainage of dextran much earlier in mice with AD pathology, including CAA, than in controls 51.
Conclusion
In conclusion, there is now evidence from a variety of sources that CAA can contribute to SVD and to cognitive impairment even in the presence of AD. Age and the ApoE ε4 genotype as well as AD have been shown to be strong risk factors for the development of CAA but much more remains to be uncovered before their roles are understood. Although the hypothesis that drainage of cortical amyloidβ in the aging brain leads to CAA is well supported (ie, CAA is a downstream consequence of AD pathology in the brain), there may still be an additional significance for CAA, perhaps as a consequence of altered cerebral blood flow with aging, in the promotion of sporadic AD pathology in the brain (ie, an upstream role for CAA as well). This potential upstream aspect of CAA merits further study and the new imaging methods for identifying it in living subjects will help the subject to progress.
CAA in Cases with and without AD in the OPTIMA Cohort
The relationships we have uncovered regarding semi‐quantitative scores for SVD, CAA and dementia in the OPTIMA cohort are described below.
Materials and methods
The OPTIMA project (Oxford Project to Investigate Memory and Ageing) (http://www.medsci.ox.ac.uk/optima) is a longitudinal study of elderly subjects with memory problems and of healthy controls in which a high proportion of enrolled subjects also agreed to donate their brain for research after their death. The project commenced in 1989 and received full NHS Ethics Committee approval (Frenchay REC Ref 09‐H0107/9 and Oxford REC Ref 07/H0606/85). Subjects agreed to undergo repeated cognitive evaluation [MMSE and CAMDEX 99 instruments], blood investigations and, in some cases, cerebrospinal fluid investigations as well as imaging studies. After death, brains were bisected and one‐half used for microscopy after fixation in 10% neutral formalin and the other half dissected and deep frozen at −80°C for genetic and biochemical studies.
Microscopical evaluation of the fixed side of the brain formed the basis of the pathological diagnosis that was made according to CERAD criteria 73, 74. Included in the present study were 154 cases fulfilling CERAD criteria for the pathological diagnosis of definite AD and 70 cases in which none of the criteria for possible, probable or definite AD were fulfilled and the Braak stage for tau pathology was no more than stage 2 14. There was no other pathology related to neurodegeneration in either group of cases except that 20 cases of definite AD and none of the other cases had a few Lewy bodies identified in catecholaminergic brain stem nuclei. The cases with definite AD were included in a study of 161 cases of AD in which the findings regarding subcortical vascular pathology (SVD) were reported 31, and all the other cases were included in an earlier study of SVD 106. The scoring system used to assess the extent and severity of SVD was the same in both studies and was reported in detail in Smallwood et al 106. Briefly, four paraffin sections (two frontal and occipital white matter and two basal ganglia and thalamus) were assessed from each case in adjacent H&E and Luxol‐fast blue/cresyl violet‐stained sections and given a semi‐quantitative score of 0–3 based on the extent of damage to subcortical white matter and deep gray matter. Therefore, each case had a maximum possible SVD score of 12. This scoring process was assisted by having look‐up images of stained sections showing pathology representative of each score.
In the present study, these SVD scores were compared with scores for severity and extent of CAA derived from an examination of five cortical paraffin sections (frontal, temporal, parietal and occipital lobes and hippocampus) stained with Congo Red. This assessment was similar to the recently published criteria of Love et al 64 and resulted in a total possible semi‐quantitative score for each case of 36. A score for CAA above 23 was arbitrarily rated as “severe” CAA for the purposes of analysis. All CAA scores were obtained blind to the clinical group of each case.
After the CAA scores had been assigned MMSE and CAMDEX scores for the last visit of each subject before death were retrieved from the OPTIMA database and the ApoE genotype status of each case, derived from analysis of DNA extracted from an in‐life blood sample, was ascertained. Blood pressure measures at the first visit of each subject were also retrieved. A systolic blood pressure >140 and a diastolic pressure >90 was regarded as evidence of a raised systolic or diastolic blood pressure, respectively.
Statistics
Statistical analysis was conducted using SPSS software, IBM, NY, USA (version 22.0). Demographic variables were tested by t‐tests for normally distributed continuous data (ie, age at death) and chi‐squared tests (χ2) for frequency data (eg, raised blood pressure). Measured variables (and potential covariates) were tested to determine if they met the general conditions for parametric analyses when grouped by the factors of interest. The main grouping factors were AD diagnosis, sex, blood pressure and ApoE status. SVD score and age at death passed the Shapiro–Wilke tests, indicating a normal distribution (as well as Levene's tests and Box's M‐tests for homogeneity of variance, unless otherwise stated). Therefore, these variables were analyzed with univariate analyses of variance (ANOVAs) for group differences and Pearson's correlation analysis for the relationship between variables. CAA score and cognitive scores (MMSE and CAMCOG) did not pass the Shapiro–Wilke test, mainly as a result of skew due to ceiling and floor effects, and were therefore analyzed with non‐parametric Mann–Whitney U‐tests for group differences (or Kruskal–Wallis tests for more than two groups; ie, for blood pressure or ApoE) and Spearman's correlations for the relationship between variables. In the results below, quantitative statistical details are generally confined to positive results and negative results that may be considered trends (P < 0.1) or demonstrating an important negative.
Results
Demographic variables, blood pressure and ApoE genotypes in relation to CAA
Demographic details of the cases included in the study are given in Table 1. Mean ages of AD and non‐AD groups were significantly different (t = 3.8, degrees of freedom 223, P < 0.01) (mean age of AD cases at death 78.3 ± 8.0 years; mean age of non‐AD cases 82.6 ± 7.7 years); therefore, the effect of age was investigated as a covariate in ANOVAs and further tests below. Eighty‐seven (56%) of the AD cases were female and 31 (44%) of the non‐AD cases were female (proportions were not significantly different between groups: χ2 = 0.1, P = 0.95). Ninety‐four (63%) of the AD cases were hypertensive at their first clinic visit. Fifty‐two (74%) of the non‐AD cases were hypertensive at their first clinic visit. The difference in prevalence of raised blood pressure between the AD and non‐AD groups was not quite significant (χ2 = 3.6, P = 0.06).
Table 1.
Demographic features and ApoE genotype of subjects included in this study
| AD cases (n = 154) | Non‐AD cases (n = 70) | Diff | |
|---|---|---|---|
| Age at death (years) | 78.3 ± 8.0 | 82.6 ± 7.7 | P < 0.01 |
| Gender F : M | 87:67 (56% females) | 31:39 (44% females) | NS |
| Cases with raised BP | 94 (63%) | 52 (74%) | NS |
| Cases with one or more | 116 (68%) | 11 (16%) | P < 0.0001 |
| ApoE4 allele | |||
| Cases with two ApoE4 alleles | 28 (19%) | 1 (1.4%) | P < 0.0001 |
| Cases with one or more | 115 (73%) | 67 (94%) | P < 0.0001 |
| ApoE3 alleles | |||
| Cases with two ApoE3 alleles | 36 (24%) | 45 (65%) | P < 0.0001 |
| Cases with one ApoE2 alleles | 4 (2.3%) | 12 (17%) | P < 0.0001 |
| Cases with two ApoE2 alleles | 0 | 0 |
Abbreviations: AD = Alzheimer's disease; BP = blood pressure; NS = not significant.
As expected, the ApoE genotype status of the two groups of cases differed greatly (χ2 = 64.2, P < 0.01) (Table 1). In the AD group and in the two groups combined, age was found not to be correlated with CAA score (Spearman's corr for both −0.03 to 0.05, P‐values < 0.5) but in the non‐AD group age was positively correlated with CAA score (Spearman's corr 0.3, P = 0.01). CAA scores were significantly higher in the AD group than in the non‐AD group (Mann–Whitney U‐test, P < 0.01) (Table 2). Thus, severe CAA occurred in 14 of the AD group (9%) but none of the non‐AD group while CAA was absent in 34 (48%) of non‐AD cases but in only 10 (6.4%) of AD cases. CAA scores were not influenced by gender (Mann–Whitney U‐test, P = 0.36) or by raised blood pressure (Kruskal–Wallis test, P = 0.09) in the combined groups or in either group of subjects (although noting a trend for CAA to be higher in males than females in subjects with AD (Mann–Whitney U‐test, P = 0.064).
Table 2.
Scores for CAA
| CAA absent | CAA present | CAA severe | |
|---|---|---|---|
| AD cases (n = 154) | 10 (6.5%) | 144 (93.5%) | 14 (9.1%) |
| AD cases with raised BP (n = 93)* | 7 (7.5%) | 86 (92.5%) | 10 (10.8%) |
| AD cases without raised BP (n = 55) | 3 (5.5%) | 52 (94.5%) | 3 (5.5%) |
| AD cases females (n = 87) | 6 (6.9%) | 81 (93.1%) | 5 (5.7%) |
| AD cases males (n = 67) | 4 (6.0%) | 63 (94.0%) | 9 (13.4%) |
| Non‐AD cases (n = 70) | 34 (49.0%) | 36 (51.0%) | 0 |
| Non‐AD cases with raised BP (n = 53) | 26 (49.1%) | 27 (50.9%) | 0 |
| Non‐AD cases without raised BP (n = 17) | 8 (47.0%) | 9 (53.0%) | 0 |
| Non‐AD cases females (n = 39) | 17 (43.6%) | 22 (56.4%) | 0 |
| Non‐AD cases males (n = 31) | 17 (54.8%) | 14 (45.2%) | 0 |
| All cases (n = 224) | 44 (19.6%) | 180 (80.4%) | 14 (6.3%) |
| All cases with raised BP (n = 146)* | 33 (22.6%) | 113 (77.4%) | 10 (10.8%) |
| All cases without raised BP (n = 72) | 11 (15.3%) | 61 (84.7%) | 3 (5.5%) |
| All cases females (n = 126) | 23 (18.3%) | 103 (81.7%) | 5 (5.7%) |
| All cases males (n = 98) | 21 (21.4%) | 77 (78.6%) | 9 (13.4%) |
| All cases CAA correlation with age? | N/A | r = −0.7 NS | r = −0.06 NS |
N/A not applicable due to zero values in cases with CAA absent. NS = non‐significant and no trend (P > 0.1).
*BP was not available for six cases.
Abbreviations: AD = Alzheimer's disease; BP = blood pressure; CAA = congophilic amyloid angiopathy; NS = not significant.
Relationship between scores for CAA and scores for SVD: influence of ApoE genotype status
Taking the AD and non‐AD groups together, there was no clear relationship between CAA scores and SVD scores. However, the relationship between scores for CAA and scores for SVD was strongly influenced by ApoE genotype as can be seen from Figure 1. In those subjects that had one or two ApoE ε4 alleles, there was a positive relationship between the two (Figure 1A,B) that was strongest for the ApoE ε4 homozygous group (Figure 1B). In those subjects that had ApoE ε3 alleles in the absence of an ApoE ε4 allele, the positive relationship between CAA scores and SVD scores was lost (Figure 1C) and for those with an ApoE ε2 allele there was a significantly negative correlation (Spearman's rho = −0.73, P < 0.01) (Figure 1D) (excluding the small number of heterozygous E2E4 cases). The contrast between the negative correlation in the ApoE ε2/3 genotype and the positive relationship in the ApoE ε4/4 genotype was particularly strong (different intercepts: F = 14.89, P < 0.01; different slopes: F = 3.85, P = 0.056).
Figure 1.
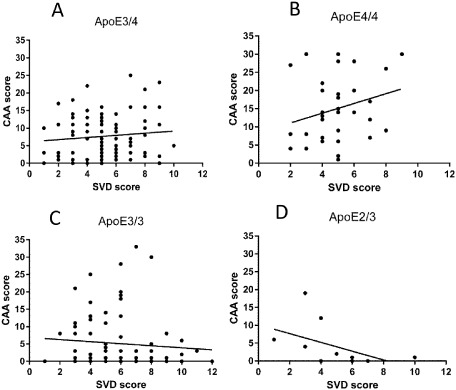
Relationship between congophilic amyloid angiopathy ( CAA ) score and small vessel disease ( SVD ) score in each of the main ApoE genotype groups [ A lzheimer's disease ( AD ) and non‐ AD combined]. A progressive shift in the direction of the relationship can be seen from the highest AD risk ε E 4 homozygous group through the intermediate risk groups toward the low AD risk ε E2/E 3 group. A. ε E4E 3 genotype (Spearman's correlation, rho = 0.06, P = 0.59). B. ε E4E 4 genotype (Spearman’ rho = 0.27, P = 0.12). C. ε E3E 3 genotype (Spearman's rho = −0.15, P = 0.20). D. ε E2E 3 genotype (Spearman's rho = −0.73, P < 0.01).
The severity and extent of CAA itself varied according to ApoE genotype overall (Figure 2) (Kruskal–Wallis test, P < 0.01), with 10 (8%) subjects with one or more ApoE ε4 alleles being classified as having severe CAA while severe CAA was seen in four (4%) subjects without an ApoE ε4 allele (P < 0.01). The effect of ApoE genotype was significant in each of the diagnostic groups separately (in the AD group: Kruskal–Wallis test, P < 0.01; in the non‐AD group: Kruskal–Wallis test, P < 0.05) (see Figure 2).
Figure 2.
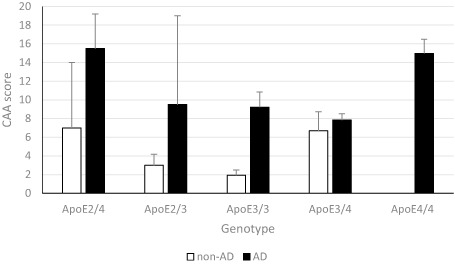
Congophilic amyloid angiopathy (CAA) scores in the Alzheimer's disease (AD) and non‐AD groups differ according to ApoE genotype. Graph shows means and standard error of the mean. CAA score is greater in ε E 4 cases and lower in ε E 3 homozygotes with a similar but relatively shifted pattern across genotypes in both AD and non‐AD groups.
For SVD scores, although there was only a trend for higher SVD scores in AD compared with non‐AD (ANOVA F = 3.2, P = 0.08), there were interactions with raised blood pressure and ApoE status. An AD × blood pressure interaction (F = 5.1, P < 0.01) was due to increased SVD associated with elevated systolic blood pressure in the non‐AD group. An ApoE × blood pressure interaction (F = 2.4, P < 0.05) was due to increased SVD score in ApoE ε2 carriers (both ApoE ε2 ε3 and ApoE ε2 ε4) with elevated blood pressure (both systolic and diastolic) and also increased SVD score in the ApoE ε3 ε3 group with elevated systolic blood pressure. Gender was not a significant factor for inclusion in the ANOVA (F = 0.56, P = 0.46) and therefore was not included. CAA also had no substantial effect if included. Age was a significant covariate and was included in the analysis (F = 17.5, P < 0.01). Age at death was significantly different in the different ApoE groups (Kruskal–Wallis test, P < 0.05) with a younger age at death in the ApoE ε4E ε4 homozygotes and an older age at death in the ApoE ε2 ε4 heterozygotes.
Overall, older subjects had higher SVD (Pearson's r = 0.29, P < 0.01). This was generally true for all subjects across diagnoses, genders and blood pressure groups as well as genotypes, although it was slightly less clear in the ApoE ε4 carriers, perhaps due to their earlier age at death.
Relationship between CAA scores and dementia
As expected, MMSE and CAMCOG scores were significantly lower in the AD group than the non‐AD group of cases (Mann–Whitney U‐test for MMSE P < 0.01, for CAMCOG P < 0.01). There was no significant difference of MMSE or CAMCOG scores dependent on gender (Mann–Whitney U for MMSE P = 0.73, for CAMCOG P = 0.78) or raised blood pressure (Kruskal–Wallis for MMSE P = 0.62, for CAMCOG P = 0.69).
There was a negative correlation between cognitive scores and CAA scores overall across all subjects combined (Spearman's corr for MMSE = −0.4, P < 0.01, for CAMCOG corr = −0.45, P < 0.01). However, this was largely due to the difference between groups defined by AD diagnosis. Separated by groups, in the AD group there was no correlation between CAA and cognitive scores and in the non‐AD group only CAMCOG score showed a non‐significant trend for a negative correlation with CAA (Spearman's corr = −0.22, P = 0.07).
Overall, ApoE genotype had a significant effect on cognitive scores (MMSE: Kruskal–Wallis test, P < 0.01; CAMCOG: Kruskal–Wallis test, P < 0.01) and this appeared to reflect the vulnerability of the genotypes to dementia as separation into AD and non‐AD groups resulted in no statistical effect of ApoE in either group.
Age was not correlated with cognitive scores overall, or within diagnostic groups, except for a statistical trend for a negative relationship between CAMCOG score and CAA in the non‐AD group (Spearman's corr = −0.23, P = 0.06).
Discussion
These findings regarding the relationship of CAA to SVD in the 224 OPTIMA cases reported here need to be considered in the light of the chief findings concerning SVD severity reported in Smallwood et al 106 and Esiri et al 31. In the Smallwood 106 study of the same non‐AD cases that are included in the present study, there was a positive relationship between the severity of SVD and cognitive impairment that was not seen in the AD cases reported in Esiri et al 31. This difference concerning the significance of SVD for cognition was due to the fact that the cases with AD had reached a much worse level of cognitive decline before death than those who had only vascular pathology so that in these cases any effect of SVD on cognition was overwhelmed by the effect of AD. The SVD scores themselves were significantly lower in the AD group than the non‐AD group and were slightly lower in females than males in the AD group. In the AD group, but not the non‐AD group, the SVD scores were positively related to increasing age. Raised blood pressure did not influence SVD scores in either group of cases. SVD scores were not influenced by the number of ApoEε4 alleles each subject possessed in either group of cases.
In the present study, there was no relationship between CAA scores and SVD scores when the total number of cases in this study was taken together. However, this overall correlation masked different correlations depending on the ApoE genotypes of the cases studied, regardless of whether they were suffering from AD or not. The strongest trend for a positive correlation was seen for cases that were ApoE ε4 homozygous. This positive correlation was lost in those cases that had ApoE ε3 alleles but no ApoE ε4 allele, while a significant negative correlation was present in those cases that had an ApoE ε2 allele combined with an ApoE ε3 allele. These variable findings, depending on ApoE genotype status, may explain why the literature (referred to in the review above) contains some inconsistent findings. The results would be expected to be different depending on the ApoE genotype mix in the population studied. Because ApoE ε4 is a risk factor for AD as well as for CAA, the findings might also be expected to be influenced by whether the population under study was one with AD or not.
To understand how these diverse effects on SVD severity might be produced in subjects with different ApoE genotypes, it is necessary to consider current understanding about the manner in which ApoE influences the fate of amyloidβ in the brain. The three human isoforms of ApoE differ only at two amino acid positions, but these differences influence their ability to bind lipids, their receptors and amyloidβ 62, 130. Thus, ApoE is known to bind to amyloidβ and to promote its fibrillogenesis, and this binding is tighter in the presence of ApoE4 than ApoE3 88, 115. Binding of amyloidβ to ApoE dramatically reduces the clearance of amyloidβ from the brain via the blood probably by influencing the manner in which amyloidβ interacts with receptors that facilitate clearance of amyloidβ from brain through the blood–brain barrier 10, 25, 69. The presence of ApoE disrupts binding of amyloidβ to its receptors [low‐density lipoprotein receptor (LDLR) and LDLR‐related protein 1] on vascular endothelial cells and this disruption is greater for ApoE4 than for ApoE3 or ApoE2. In addition, the acute phase protein, haptoglobin, has also been found to bind both to amyloidβ and to ApoE in an ApoE isoform‐specific manner 107. Thus, binding of amyloidβ to ApoE was facilitated by haptoglobin, particularly so for ApoE4. Further evidence of an influence of ApoE on clearance of amyloidβ across the blood–brain barrier has been reported by Bachmeier et al 6 who investigated shedding of lipoprotein receptors by ApoE from brain endothelial cells. There was an increase in lipoprotein receptor shedding in the presence of amyloidβ, but this was reduced if ApoE3 or ApoE2 was present, but not when ApoE4 was present. This effect of different ApoE isoforms mirrored the effect of intracranial administration of amyloidβ to mice transfected with different human ApoE genotypes in which shedding of lipoprotein receptors was greatest with ApoE ε4, followed by ApoE ε3 and then ApoE ε2. In an earlier study ApoE ε4 mice showed reduced clearance of amyloidβ compared with ApoE ε3 mice 5. These recent studies suggest that an important influence of ApoE ε4 on risk of AD and CAA may be mediated via the influence of ApoE on clearance of amyloidβ through the blood–brain barrier, a process that may be modulated by other proteins such as haptoglobin. Reducing the ability of ApoE to interact with amyloidβ by administration of an anti‐ApoE antibody or by overexpression of LDLR diminishes the build‐up of amyloidβ as cortical plaques in animal models of AD 58, 100, 101. Impaired clearance of amyloidβ via the blood and any impairment in the metabolic breakdown of amyloidβ by neprilysin, whose activity has been shown to wane with aging 72, is likely to render the vascular drainage route for the clearance of amyloidβ critically important. Hence, the enhanced risk of CAA in those with the ApoE ε4 genotype.
The increased severity of CAA in those with the ApoE ε4 genotype in the present study may allow for a stronger influence of CAA on SVD to be detected. However, the direction of causation in the correlation we have found between CAA and SVD severity is not clear. It should be noted that the method used to score SVD severity in this study and that of Smallwood et al 106 and Esiri et al 31 related to the extent of damage to the subcortical white and gray matter and not to the changes in the walls of the small subcortical vessels themselves. It could be that SVD is promoting CAA in ApoE ε4 carriers or CAA could be promoting SVD. The first possibility is raised by the suggestion of Grinberg and Thal 46 and Thal et al 111 that SVD may exert added pressure on the perivascular drainage system and lead to enhanced CAA. There is also evidence in a mouse model that elevated blood homocysteine, which leads to subcortical white matter damage, enhances CAA 108. The second possibility is supported by evidence that CAA impairs the blood supply to cortex and subcortical white matter, resulting in ischemia (reviewed above). In the case of those with the ApoE ε2 genotype, there may be other influences on SVD that tip the balance in favor of CAA protecting against SVD (or vice versa). It could be important for these influences to be identified as they may hold out opportunities for SVD to be reduced not only in those with ApoE ε2 genotype but also in others as well.
Other findings with respect to ApoE genotype and CAA between the AD and non‐AD groups are to some extent expected in the light of earlier studies 64, 78, 90, 91, although the size of the effects of ApoE genotype are, if anything, more marked in this cohort than some others that have been reported: much heavier representation of ApoE ε4 in the AD group than the non‐AD group; conversely, heavier representation of the ApoE ε3 and ApoE ε2 alleles in the non‐AD group; higher CAA scores in the AD group than the non‐AD group; no influence of gender on CAA scores in either group, lower age at death in the ApoE ε4 carriers but an increase in CAA scores with age in the non‐AD cases and an increase in SVD scores with age in both diagnostic groups. Elevated blood pressure had modest and complex effects in that in only the non‐AD group of cases elevated systolic blood pressure was correlated with increased SVD scores and in both diagnostic groups ApoE ε3 and ApoE ε2 carriers with elevated blood pressure had increased SVD scores.
There was no effect of CAA scores on dementia measures after the influence of an AD diagnosis had been taken into account. This result, which is at odds with some others in the literature (reviewed above), may in part have resulted from many of the subjects in the AD group being so demented as to be untestable in the period shortly before their deaths. Accordingly, these cases could not be entered into the statistical analysis relating cognition to CAA scores.
Conclusion
CAA has become a subject of increasing interest in part because it has recently become accessible for study by neuroimaging in living subjects, and also because of an increased awareness of its potential role in the pathogenesis of both AD and SVD. There is increasing evidence from both pathology and imaging that CAA contributes to SVD. Our novel finding of a significant relationship between the severity of CAA and that of SVD, when both these pathologies are semi‐quantitatively assessed, adds to this evidence. Because the relationships between CAA and SVD in the OPTIMA subjects reported here are critically dependent on ApoE genotype our findings draw attention, as have many other studies, to the importance of this major genetic risk factor not only for dementia but also for damage to the blood vessels in the aging brain.
Acknowledgments
We are most grateful to those who made the research reported here possible by donating their brains for research. Financial support for OPTIMA came from a number of sources including the UK Medical Research Council, the NIHR via the Oxford Biomedical Research Centre and the Charles Wolfson Charitable Trust. Accruals to the OPTIMA brain archive since 2009 have been incorporated into Brains for Dementia Research, funded by The UK Alzheimer's Society and Alzheimer's Research UK. GKW and MME have had support from the NIHR via the Oxford Biomedical Research Centre.
References
- 1. Arbel‐Ornath M, Hudry E, Eikermann‐Haerter K, Hou S, Gregory JL, Zhao L et al (2013) Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer's disease mouse models. Acta Neuropathol 126:353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA (2011) Cerebral amyloid angiopathy, pathology and cognitive domains in older persons. Ann Neurol 69:320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Attems J, Jellinger KA (2014) Pathologic aspects of the haemorrhagic consequences of small vessel disease on the brain. In: Cerebral Small Vessel Disease. Pantoni L, Gorelick PB (eds), pp. 29–41. Cambridge Univ Press: Cambridge. [Google Scholar]
- 4. Attems J, Jellinger K, Thal DR, Nostrand W (2011) Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 37:75–93. [DOI] [PubMed] [Google Scholar]
- 5. Bachmeier C, Paris D, Beaulieu‐Abdelahad D, Mouzon B, Mullan M, Crawford F (2013) A multifaceted role for apoE in the clearance of beta‐amyloid across the blood–brain barrier. Neurodegener Dis 11:13–21. [DOI] [PubMed] [Google Scholar]
- 6. Bachmeier C, Shackleton B, Ojo J, Paris D, Mullan M, Crawford F (2014) Apolipoprotein E isoform‐specific effects on lipoprotein receptor processing. Neuromolecular Med 868–696. doi: 10.1007/s12017-014-8318-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bangen K, Nation D, Clark L, Harmeli A, Wierenga C, Dev S et al (2014) Interactive effects of vascular risk burden and advanced age on cerebral blood flow. Front Aging Neurosci 6:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barakos J, Sperling R, Salloway S, Jack C, Gass A, Fiebach JB et al (2013) MR imaging features of amyloid‐related imaging abnormalities. AJNR Am J Neuroradiol 48:244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beitzke M, Gattringer T, Enzinger C, Wagner G, Niederkorn K, Fazekas F (2011) Clinical presentation, etiology and long term prognosis in patients with non‐traumatic convexal subarachnoid haemorrhage. Stroke 42:3055–3060. [DOI] [PubMed] [Google Scholar]
- 10. Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV (2007) Transport pathways for clearance of human Alzheimer's amyloid beta‐peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow and Metab 27:909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Biffi A, Sonni A, Anderson CD, Kissela B, Jagiella J, Schmidt H (2010) Variants at ApoE influence risk of deep and lobar intracerebral haemorrhage. Ann Neurol 68:934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Biffi A, Anderson CD, Jagiella JM, Schmidt J, Kissela B, Hansen B (2011) APOE genotype and extent of bleeding and outcome in lobar intracerebral haemorrhage: a genetic association study. Lancet Neurol 10:702–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boche D, Zotova E, Weller RO, Love S, Neal JW, Pickering RM et al (2008) Consequence of Abeta immunization on the vasculature of human Alzheimer's disease brain. Brain 131:3299–3310. [DOI] [PubMed] [Google Scholar]
- 14. Braak H, Braak E (1991) Neuropathological staging of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 15. Cadavid D, Mena H, Koeller K, Frommelt RA (2000) Cerebral beta amyloid angiopathy is a risk factor for cerebral ischemic infarction: a case control study in human brain biopsies. J Neuropathol Exp Neurol 59:768–773. [DOI] [PubMed] [Google Scholar]
- 16. Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold K‐H et al (1999) Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA 96:14088–14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carare RO, Hawkes CA, Jeffrey M, Kalaria RN, Weller RO (2013) Cerebral amyloid angiopathy, prion angiopathy, CADASIL and the spectrum of protein elimination protein results in prominent deposition of cerebrovascular amyloid. Neuropathol Appl Neurobiol 39:593–611. [DOI] [PubMed] [Google Scholar]
- 18. Chalmers K, Wilcock GK, Love S (2003) APOEe4 influences the pathological phenotype of Alzheimer's disease by favouring cerebrovascular over parenchymal accumulation of Ab protein. Neuropathol Appl Neurobiol 29:231–238. [DOI] [PubMed] [Google Scholar]
- 19. Chalmers K, Wilcock GK, Love S (2005) Contributors to white matter damage in the frontal lobe in Alzheimer's disease. Neuropathol Appl Neurobiol 31:623–631. [DOI] [PubMed] [Google Scholar]
- 20. Charimidou A, Werring DJ (2011) Cerebral microbleeds: detection, mechanisms and clinical challenges. Future Neurol 6:587–611. [Google Scholar]
- 21. Charimidou A, Gang Q, Werring DJ (2012) Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry 83:124–137. [DOI] [PubMed] [Google Scholar]
- 22. Charimidou A, Jaunmuktane Z, Baron J‐C, Burnell M, Varlet P, Peeters A et al (2014) White matter perivascular spaces: an MRI marker in pathology‐proven cerebral amyloid angiopathy? Neurology 82:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen YW, Gurol ME, Rosand J, Viswanathan A, Rakich SM, Groover TR et al (2006) Progression of white matter lesions and hemorrhages in cerebral amyloid angiopathy. Neurology 67:83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chung KK, Anderson NE, Hutchinson D, Synek B, Barber PA (2011) Cerebral amyloid angiopathy related inflammation: three case reports and a review. J Neurol Neurosurg Psychiatry 82:20–26. [DOI] [PubMed] [Google Scholar]
- 25. Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB et al (2008) apoE isoform‐specific disruption of amyloid β peptide clearance from mouse brain. J Clin Invest 118:4002–4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deramecourt V, Slade JY, Oakley AE, Perry RH, Ince PG, Maurage CA, Kalaria RN (2012) Staging and natural history of cerebrovascular pathology in dementia. Neurology 78:1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dierksen GA, Skehan ME, Khan MA, Jeng J, Nandigam RN, Becker JA et al (2010) Spatial relation between microbleeds and amyloid deposits in amyloid angiopathy. Ann Neurol 68:545–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, Heyman A (1996) Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, part XV. Neurology 46:1592–1596. [DOI] [PubMed] [Google Scholar]
- 29. Eng JA, Frosch MP, Choi K, Rebeck GW, Greenberg SM (2004) Clinical manifestations of cerebral amyloid angiopathy‐related inflammation. Ann Neurol 55:250–256. [DOI] [PubMed] [Google Scholar]
- 30. Erkinjuntti T, Inzitari D, Pantoni L, Wallin A, Scheltens P, Rockwood K et al (2000) Research criteria for subcortical vascular dementia in clinical trials. J Neural Transm Suppl 59:23–30. [DOI] [PubMed] [Google Scholar]
- 31. Esiri MM, Joachim C, Sloan C, Christie S, Agacinski G, Bridges LR et al (2014) Cerebral subcortical small vessel disease in subjects with pathologically confirmed Alzheimer's disease: a clinicopathologic study in the Oxford project to Investigate Memory and Ageing (OPTIMA). Alzheimer Dis Assoc Disord 28:35–40. [DOI] [PubMed] [Google Scholar]
- 32. Feldman HH, Maia LF, Mackenzie IR, Forster BB, Martzke J, Woolfenden A (2008) Superficial siderosis: a potential diagnostic marker of cerebral amyloid angiopathy in Alzheimer disease. Stroke 39:2894–2897. [DOI] [PubMed] [Google Scholar]
- 33. Ferrer I, Boada Rovira M, Sánchez Guerra ML, Rey MJ, Costa‐Jussá F (2004) Neuropathology and pathogenesis of encephalitis following amyloid‐beta immunization in Alzheimer's disease. Brain Pathol 14:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ferriero JA, Ansbacher LE, Vinters HV (1989) Stroke related to cerebral amyloid angiopathy: the significance of systemic vascular disease. J Neurol 236:267–272. [DOI] [PubMed] [Google Scholar]
- 35. Fryer JD, Taylor JW, DeMattos RB, Bales KR, Paul SM, Parsadanian M, Holtzman DM (2003) Apolipoprotein E markedly facilitates age‐dependent cerebral amyloid angiopathy and spontaneous haemorrhage in amyloid precursor protein transgenic mice. J Neurosci 23:7889–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, Holtzman DM (2005) Human apolipoprotein E4 alters the amyloid‐β 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci 25:2803–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gorelick PB, Scuteri A, Black SE, DeCarli C, Greenberg SM, Iadecola C et al (2011) Vascular contributions to cognitive impairment and dementia. A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 42:2672–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gray F, Dubas F, Roullet E, Escourolle R (1985) Leukoencephalopathy in diffuse hemorrhagic cerebral amyloid angiopathy. Ann Neurol 18:54–59. [DOI] [PubMed] [Google Scholar]
- 39. Greenberg SM, Rebeck GW, Vonsattel JP, Gomez‐Isla T, Hyman BT (1995) Apolipoprotein E ε4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol 50:961–965. [DOI] [PubMed] [Google Scholar]
- 40. Greenberg SM, Vonsattel JP, Segal AZ, Chiu RI, Clatworthy AE, Liao A et al (1998) Association of apolipoprotein E epsilon2 and vasculopathy in cerebral amyloid angiopathy. Neurology 50:961–965. [DOI] [PubMed] [Google Scholar]
- 41. Greenberg SM, Gurol E, Rosand J, Smith EE (2004a) Amyloid angiopathy‐related vascular cognitive impairment. Stroke 35:2616–2619. [DOI] [PubMed] [Google Scholar]
- 42. Greenberg SM, Eng JA, Ning M, Smith EE, Rosand J (2004b) Hemorrhage burden predicts recurrent intracerebral hemorrhage after lobar hemorrhage. Stroke 35:1415–1420. [DOI] [PubMed] [Google Scholar]
- 43. Greenberg SM, Grabowski T, Gurol E, Skehan ME, Nandigam K, Becker JA et al (2008) Detection of isolated cerebrovascular β‐amyloid with Pittsburgh compound B. Ann Neurol 64:587–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Greenberg SM, Vernooij MW, Cordonnier C, Viswanathan A, Al‐Shahi S, Warach S, Launer LJ (2009) Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol 8:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gregoire SM, Charimidou A, Gadapa N, Dolan E, Nagui A, Peeters A et al (2011) Acute ischaemic brain lesions in intracerebral haemorrhage: multicentre cross‐sectional magnetic resonance imaging study. Brain 134:2376–2386. [DOI] [PubMed] [Google Scholar]
- 46. Grinberg LT, Thal DR (2010) Vascular pathology in the aged human brain. Acta Neuropathol 119:277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gurol ME, Irizarry MC, Smith EE, Raju S, Diaz‐Arrastia R, Bottiglieri T et al (2006) Plasma β‐amyloid and white matter lesions in AD, MCI, and cerebral amyloid angiopathy. Neurology 66:23–29. [DOI] [PubMed] [Google Scholar]
- 48. Gurol ME, Viswanathan A, Gidicsin C, Hedden T, Martinez‐Ramirez S, Dumas A et al (2013) Cerebral amyloid angiopathy burden associated with leukoaraiosis: a positron emission tomography/magnetic resonance imaging study. Ann Neurol 73:529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Haglund M, Englund E (2002) Cerebral amyloid angiopathy, white matter lesions and Alzheimer encephalopathy—a histopathological assessment. Dement Geriatr Cogn Disord 14:161–166. [DOI] [PubMed] [Google Scholar]
- 50. Haglund M, Kalaria R, Slade JY, Englund E (2006) Differential deposition of amyloid b peptides in cerebral amyloid angiopathy associated with Alzheimer's disease and vascular dementia. Acta Neuropathol 111:430–435. [DOI] [PubMed] [Google Scholar]
- 51. Hawkes C, Sullivan PM, Hands S, Weller RO, Nicoll JAR, Carare RO (2012) Disruption of arterial perivascular drainage of amyloid b from the brains of mice expressing the human APOE e4 allele. PLoS ONE 7:e41636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heier LA, Bauer CJ, Schwartz L, Zimmerman RD, Morgello S, Deck MD (1989) Large Virchow‐Robin spaces: MR‐clinical correlation. AJNR Am J Neuroradiol 10:929–936. [PMC free article] [PubMed] [Google Scholar]
- 53. Holland CM, Smith EE, Csapo I, Gurol ME, Brylka DA, Killiany RJ et al (2008) Spatial distribution of white‐matter hyperintensities in Alzheimer disease, cerebral amyloid angiopathy and healthy aging. Stroke 39:1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jellinger KA (2002) Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm 109:813–836. [DOI] [PubMed] [Google Scholar]
- 55. Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK et al (2007) Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol 62:229–234. [DOI] [PubMed] [Google Scholar]
- 56. Keage HAD, Carare RO, Friedland RP, Ince PG, Love S, Nicoll JA et al (2009) Population studies of sporadic amyloid angiopathy and dementia: a systematic review. BMC Neurol 9:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kidwell CS, Greenberg SM (2009) Red meets white: do microbleeds link hemorrhagic and ischemic crebrovascular disease? Neurology 73:1614–1615. [DOI] [PubMed] [Google Scholar]
- 58. Kim J, Jiang H, Park S, Eltorai AEM, Stewart FR, Yoon H et al (2011) Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid β amyloidosis. J Neurosci 31:18007–18012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kinnecom C, Lev MH, Wendell L, Smith EE, Rosand J, Frosch MP, Greenberg SM (2007) Course of cerebral amyloid angiopathy‐related inflammation. Neurology 68:1411–1416. [DOI] [PubMed] [Google Scholar]
- 60. Knudsen KA, Rosand J, Karluk D, Greenberg SM (2001) Clinical diagnosis of cerebral amyloid angiopathy: validation of Boston criteria. Neurology 56:537–539. [DOI] [PubMed] [Google Scholar]
- 61. Linn J, Halpin A, Demaerel P, Ruhland J, Giese AD, Dichgans M et al (2010) Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 74:1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu C‐C, Kanakiyo T, Xu H, Bu G (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9:106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lockhart A, Lamb JR, Osredkar T, Sue LI, Joyce JN, Ye L et al (2007) PIB is a non‐specific imaging marker of amyloid‐beta (Ab) peptide‐related cerebral amyloidosis. Brain 130:2607–2615. [DOI] [PubMed] [Google Scholar]
- 64. Love S, Chalmers K, Ince P, Esiri M, Attems J, Jellinger K et al (2014) Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post mortem brain tissue. Am J Neurodegener Dis 3:19–32. [PMC free article] [PubMed] [Google Scholar]
- 65. Lovelock CE, Cordonnier C, Naka H, Salman RA, Sudlow CLM. (2010) Antithrombotic drug use, cerebral microbleeds, and intracerebral haemorrhage: a systematic review of published and unpublished studies. Stroke 41:1222–1228. [DOI] [PubMed] [Google Scholar]
- 66. Ly JV, Donnan GA, Villemagne VL, Zavala JA, Ma H, O'Keefe G et al (2010) 11 C‐PIB binding is increased in patients with cerebral amyloid angiopathy‐related haemorrhage. Neurology 74:487–493. [DOI] [PubMed] [Google Scholar]
- 67. Mahley RW, Huang Y (2009) Alzheimer disease: multiple causes, multiple effects of apolipoprotein E4, and multiple therapeutic approaches. Ann Neurol 65:623–625. [DOI] [PubMed] [Google Scholar]
- 68. Mahley RW, Huang Y (2012) Apolipoprotein E sets the stage: response to injury triggers neuropathology. Neuron 76:871–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Martel CL, Mackic JB, Matsubara E, Governale S, Miguel C, Miao W et al (1997) Isoform‐specific effects of apolipoproteins E2, E3, and E4 on cerebral capillary sequestration and blood‐brain barrier transport of circulating Alzheimer's amyloid beta. J Neurochem 69:1995–2004. [DOI] [PubMed] [Google Scholar]
- 70. Martinez‐Ramirez S, Pontes‐Neto OM, Dumas AP, Auriel E, Halpin A, Quimby M et al (2013) Topography of dilated perivascular spaces in subjects from a memory clinic cohort. Neurology 80:1551–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. McCarron MO, Nicoll JA, Stewart J, Ironside JW, Mann DMA, Love S et al (1999) The apolipoprotein E ε2 allele and the pathological features in cerebral amyloid angiopathy‐related cerebral hemorrhage. J Neuropathol Exp Neurol 58:711–718. [DOI] [PubMed] [Google Scholar]
- 72. Miners JS, Van Helmond Z, Chalmers K, Wilcock G, Love S, Kehoe PG (2006) Decreased expression and activity of neprilysin in Alzheimer's disease are associated with cerebral amyloid angiopathy. J Neuropathol Exp Neurol 65:1012–1021. [DOI] [PubMed] [Google Scholar]
- 73. Mirra SS (1997) The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer's disease: a commentary. Neurobiol Aging 18(Suppl. 4):S91–S94. [DOI] [PubMed] [Google Scholar]
- 74. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The consortium to establish a registry for Alzheimer's disease (CERAD) part II standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41:479–486. [DOI] [PubMed] [Google Scholar]
- 75. Montaner J (2011) Genetics of intracerebral haemorrhage: a tsunami effect of ApoE ε2 genotype on brain bleeding size? Lancet Neurol 10:673–675. [DOI] [PubMed] [Google Scholar]
- 76. MRC‐CFAS (2001) Pathological correlates of late‐onset dementia in a multicentre community‐based population in England and Wales. Lancet 357:169–175. [DOI] [PubMed] [Google Scholar]
- 77. Nakata‐Kudo Y, Mizuno T, Yamada K, Shiga K, Yoshikawa K, Mori S et al (2006) Microbleeds in Alzheimer disease are more related to cerebral amyloid angiopathy than cerebrovascular disease. Dement Geriatr Cogn Disord 22:8–14. [DOI] [PubMed] [Google Scholar]
- 78. Nelson PT, Pious NM, Jicha GA, Wilcock DM, Fardo DW, Estus S, Rebeck GW (2013) APOE‐ε2 and APOE‐ε4 correlate with increased amyloid accumulation in cerebral vasculature. J Neuropathol Exp Neurol 72:708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO (2003) Neuropathology of human Alzheimer disease after immunization with amyloid‐beta peptide: a case report. Nat Med 9:448–452. [DOI] [PubMed] [Google Scholar]
- 80. O'Donnell HC, Rosand J, Knudsen KA, Furie KL, Segal AZ, Chiu RI et al (2000) Apolipoprotein E genotype and the risk of recurrent lobar intracerebral haemorrhage. New Engl J Med 342:240–245. [DOI] [PubMed] [Google Scholar]
- 81. Okamoto Y, Yamamoto T, Kalaria RN, Senzaki H, Maki T, Hase Y et al (2012) Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol 123:381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Olichney JM, Hansen LA, Hofstetter CR, Grundman M, Katzman R, Thal LJ (1995) Cerebral infarction in Alzheimer's disease is associated with severe amyloid angiopathy and hypertension. Arch Neurol 52:702–708. [DOI] [PubMed] [Google Scholar]
- 83. Olichney JM, Hansen LA, Galasko D, Saitoh T, Hofstetter CR, Katzman R, Thal LJ (1996) The apolipoprotein E e4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer's disease and Lewy body variant. Neurology 47:190–196. [DOI] [PubMed] [Google Scholar]
- 84. Peca S, McCreary CR, Donaldson E, Kumarpillai G, Shobha N, Sanchez K et al (2013) Neurovascular decoupling is associated with severity of cerebral amyloid angiopathy. Neurology 81:1659–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ (2002) Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology 58:1629–1634. [DOI] [PubMed] [Google Scholar]
- 86. Piazza F, Greenberg SM, Savoiardo M, Gardinetti M, Chiapparini L, Raicher I et al (2013) Anti‐amyloid b autoantibodies in cerebral amyloid angiopathy‐related inflammation: Implications for amyloid‐modifying therapies. Ann Neurol 73:449–458. [DOI] [PubMed] [Google Scholar]
- 87. Plant GT, Ghiso J, Holton JL, Frangione B, Revesz T (2004) Familial and sporadic cerebral amyloid angiopathies associated with dementia and the BRI dementias. In: The Neuropathology of Dementia, 2nd edn. Esiri MM, Lee VM‐Y, Trojanowski JQ (eds), pp. 330–352. Cambridge University Press: Cambridge. [Google Scholar]
- 88. Potter H, Wisniewski T (2012) Apolipoprotein E: essential catalyst of the Alzheimer amyloid cascade. Int J Alzheimers Dis 2012:489428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Premkumar DR, Cohen DL, Hedera P, Friedland RP, Kalaria RN (1996) Apolipoprotein E‐epsilon 4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer's disease. Am J Pathol 148:2083–2095. [PMC free article] [PubMed] [Google Scholar]
- 90. Rannikmae K, Samarasekera N, Martînez‐Gonzâlez NA, Salman RA, Sudlow CLM (2013) Genetics of cerebral amyloid angiopathy: systematic review and meta‐analysis. J Neurol Neurosurg Psychiatry 84:901–908. [DOI] [PubMed] [Google Scholar]
- 91. Rannikmae K, Kalaria RN, Greenberg SM, Chui HC, Schmitt FA, Samarasekera N et al (2014) APOE associations with severe CAA‐associated vasculopathic changes: collaborative meta‐analysis. J Neurol Neurosurg Psychiatry 85:300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Raposo N, Viguier A, Cuvinciuc V (2011) Cortical subarachnoid haemorrhage in the elderly: a recurrent event probably related to cerebral amyloid angiopathy. Eur J Neurol 18:597–603. [DOI] [PubMed] [Google Scholar]
- 93. Revesz T, Holton JL, Lashley T, Plant G, Rostagno A, Ghiso J, Frangione B (2002) Sporadic and familial cerebral amyloid angiopathies. Brain Pathol 12:343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Revesz T, Holton JL, Lashley T, Plant G, Frangione B, Rostagno A, Ghiso J (2009) Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol 118:115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rigby H, Easton A, Bhan V (2011) Amyloid‐β related angiitis of the central nervous system: report of 3 cases. Can J Neurol Sci 38:626–630. [DOI] [PubMed] [Google Scholar]
- 96. Ritter MA, Droste DW, Hegedus K, Szepesi R, Nabavi DG, Csiba L, Ringelstein EB (2005) Role of cerebral amyloid angiopathy in intracerebral haemorrhage in hypertensive patients. Neurology 64:1233–1237. [DOI] [PubMed] [Google Scholar]
- 97. Roher AE, Kuo Y‐M, Esh C, Knebel C, Weiss N, Kalback W et al (2003) Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer's disease. Mol Med 9:112–1296. [PMC free article] [PubMed] [Google Scholar]
- 98. Roob G, Lechner A, Schmidt R, Flooh E, Hartung HP, Fazekas F (2000) Frequency and location of microbleeds in patients with primary intracerebral haemorrhage. Stroke 31:2665–2669. [DOI] [PubMed] [Google Scholar]
- 99. Roth M, Huppert F, Tim E, Mountjoy QC (1988) CAMDEX: The Cambridge Examination for Mental Disorders in the Elderly. Cambridge University Press: Cambridge. [Google Scholar]
- 100. Sadowski M, Pankiewicz J, Scholtzova H, Ripellino JA, Li YS, Schmidt SD et al (2004) A synthetic peptide blocking the apolipoprotein E/β‐amyloid binding mitigates β‐amyloid toxicity and fibril formation in vitro and reduces β‐amyloid plaques in transgenic mice. Am J Pathol 165:937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sadowski MJ, Pankiewicz J, Scholtzova H, Mehta PD, Prelli F, Quartermain D, Wisniewski T (2006) Blocking the apolipoprotein E/amyloid‐β interaction as a potential therapeutic approach for Alzheimer's disease. Proc Natl Acad Sci USA 103:18787–18792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Salat DH, Smith EE, Tuch DS, Benner T, Pappu V, Schwab KM et al (2006) White matter alterations in cerebral amyloid angiopathy measured by diffusion tensor imaging. Stroke 37:1759–1764. [DOI] [PubMed] [Google Scholar]
- 103. Scholz W (1938) Studienzurpathologiederhirngefabell: die drusige entartung der hirnarterien und capillaren. Gesamte Neurol Psychiatry 162:694–715. [Google Scholar]
- 104. Scolding NJ, Joseph F, Kirby PA, Mazanti I, Gray F, Mikol J et al (2005) Abeta‐related angiitis: primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain 128:500–515. [DOI] [PubMed] [Google Scholar]
- 105. Seo SW, Lee BH, Kim E‐J, Chin J, Cho YS, Yoon U, Na DL (2007) Clinical significance of microbleeds in subcortical vascular dementia. Stroke 38:1949–1951. [DOI] [PubMed] [Google Scholar]
- 106. Smallwood A, Oulhaj A, Joachim C, Christie C, Sloan C, Smith AD, Esiri M (2012) Cerebral subcortical small vessel disease and its relation to cognition in elderly subjects: a pathological study in the Oxford Project to Investigate Memory and Ageing (OPTIMA) cohort. Neuropathol Appl Neurobiol 38:337–343. [DOI] [PubMed] [Google Scholar]
- 107. Spagnuolo MS, Maresca B, La Marca V, Carrizzo A, Veronesi C, Cupidi C et al (2014) Haptoglobin interacts with apolipoprotein E and beta amyloid and influences their crosstalk. ACS Chem Neurosci 5:837–847. [DOI] [PubMed] [Google Scholar]
- 108. Suddath TL, Weekman EM, Brothers HM, Braun K, Wilcock DM (2014) β‐amyloid deposition is shifted to the vasculature and memory impairment is exacerbated when hyperhomocysteinemia is induced in APP/PS1 transgenic mice. Alzheimers Res Ther 6:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Thal DR, Ghebremedhin E, Rub U, Yamaguchi H, DelTredici K, Braak H (2002) Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 61:282–293. [DOI] [PubMed] [Google Scholar]
- 110. Thal DR, Ghebrenedhin E, Orantes M, Wiestler OD (2003) Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol 62:1287–1301. [DOI] [PubMed] [Google Scholar]
- 111. Thal DR, Grinberg LT, Attems J (2012) Vascular dementia: different forms of vessel disorders contribute to the development of dementia in the elderly brain. Exp Gerontol 47:816–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tian J, Shi J, Bailey K, Mann DMA (2004) Relationships between arteriosclerosis, cerebral amyloid angiopathy and myelin loss from cerebral cortical white matter in Alzheimer's disease. Neuropathol Appl Neurobiol 30:46–56. [DOI] [PubMed] [Google Scholar]
- 113. Tomimoto H, Akiguchi I, Akiyama H, Ikeda K, Wakita H, Lin JX, Budka H (1999) Vascular changes in white matter lesions of Alzheimer's disease. Acta Neuropathol 97:629–634. [DOI] [PubMed] [Google Scholar]
- 114. van Swieten JC, Geyskes GG, Derix MM, Peeck BM, Ramos LM, van Latum JC, van Gijn J (1991) Hypertension in the elderly is associated with white matter lesions and cognitive decline. Ann Neurol 30:825–830. [DOI] [PubMed] [Google Scholar]
- 115. Verghese PB, Castellano JM, Holtzman DM (2011) Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol 10:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Vernooij MW, van der Lugt A, Ikram MA, Wielopolski PA, Niessen WJ, Hofman A et al (2008) Prevalence and risk factors of cerebral microbleeds: the Rotterdam scan study. Neurology 70:1208–1214. [DOI] [PubMed] [Google Scholar]
- 117. Vinters HV, Ellis WG, Zarow C, Zaias BW, Jagust WJ, Mack WJ, Chui HC (2000) Neuropathologic substrates of ischemic vascular dementia. J Neuropathol Exp Neurol 59:931–945. [DOI] [PubMed] [Google Scholar]
- 118. Viswanathan A, Greenberg SM (2011) Cerebral amyloid angiopathy in the elderly. Ann Neurol 70:871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Viswanathan A, Patel P, Rahman R, Nandigam RNK, Kinnecom C, Bracoud L et al (2008) Tissue microstructural changes are independently associated with cognitive impairment in cerebral amyloid angiopathy. Stroke 39:1988–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. von Sattel PG, Myers RH, Hedley‐Whyte ET, Ropper AH, Bird ED, Richardson EP Jr (1991) Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 30:637–639. [DOI] [PubMed] [Google Scholar]
- 121. Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE (1998) Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. Am J Pathol 153:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Weller RO, Subash M, Preston SD, Mazanti I, Carare RO (2008) Perivascular drainage of amyloid‐beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol 18:253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Weller RO, Boche D, Nicholl JAR (2009) Microvasculature changes and cerebral amyloid angiopathy in Alzheimer's disease and their potential impact on therapy. Acta Neuropathol 118:87–102. [DOI] [PubMed] [Google Scholar]
- 124. Werring DJ, Sperling R (2013) Inflammatory cerebral amyloid angiopathy and amyloid‐modifying therapies: variations on the same ARIA? Ann Neurol 73:439–441. [DOI] [PubMed] [Google Scholar]
- 125. Xu D, Yang C, Wang L (2003) Cerebral amyloid angiopathy in aged Chinese: a clinico‐neuropathological study. Acta Neuropathol 106:89–91. [DOI] [PubMed] [Google Scholar]
- 126. Xu F, Grande AM, Robinson JK, Previti ML, Davis J, Van Nostrand WE (2007) Early‐onset subicular microvascular amyloid and neuroinflammation correlate with behavioural deficits in vasculotropic mutant amyloid beta‐protein precursor transgenic mice. Neuroscience 146:98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Yates PA, Desmond PM, Phal PM, Steward C, Szoeke C, Salvado O et al (2014) Incidence of cerebral microbleeds in preclinical Alzheimer's disease. Neurology 82:1266–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Zago W, Schroeter S, Guido T, Khan K, Seubert P, Yednock T et al (2013) Vascular alterations in PDAPP mice after anti‐Ab immunotherapy: implications for amyloid‐related imaging abnormalities. Alzheimers Dement 9:S105–S115. [DOI] [PubMed] [Google Scholar]
- 129. Zhu D, Zhu YY, Chabriat H, Godin O, Dufouil C, Rosand J et al (2012) Distribution of white matter hyperintensity in cerebral hemorrhage and healthy aging. J Neurol 259:530–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 12:723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]