

Abstract
To clarify whether long‐term oral ingestion of aluminum (Al) can increase tau aggregation in mammals, we examined the effects of oral Al administration on tau accumulation, apoptosis in the central nervous system (CNS) and motor function using tau transgenic (Tg) mice that show very slowly progressive tau accumulation. Al‐treated tau Tg mice had almost twice as many tau‐positive inclusions in the spinal cord as tau Tg mice without Al treatment at 12 months of age, a difference that reached statistical significance, and the development of pretangle‐like tau aggregates in the brain was also significantly advanced from 9 months. Al exposure did not induce any tau pathology in wild‐type (WT) mice. Apoptosis was observed in the hippocampus in Al‐treated tau Tg mice, but was virtually absent in the other experimental groups. Motor function as assessed by the tail suspension test was most severely impaired in Al‐treated tau Tg mice. Given our results, chronic oral ingestion of Al may more strongly promote tau aggregation, apoptosis and neurological dysfunction if individuals already had a pathological process causing tau aggregation. These findings may also implicate chronic Al neurotoxicity in humans, who frequently have had mild tau pathology from a young age.
Keywords: aluminum, Alzheimer's disease, apoptosis, astrocyte, tau
Introduction
Aluminum (Al) is the third most abundant element in the environment and is widely used in daily life as pharmacological and cosmetic compounds. However, Al is not an essential chemical element for humans, and no biological reaction that requires Al is known. Many epidemiological studies 34, 41, experiments using cell culture 3, 32 and biochemical studies 13, 21 have demonstrated that Al exposure may be a potential risk factor for Alzheimer's disease (AD). Since the pioneering report by Klatzo et al 29, who showed that injection of Al into the brains of rabbits induced neurofibrillary tangle (NFT)‐like inclusions, many researchers have examined the effects of Al on the development of AD pathology, including tau aggregation and Aβ deposition 37, 48. However, because several subsequent studies failed to demonstrate a significant effect of Al exposure 1, 16, 18, 35, 40, the possible linkage of Al and AD has been a matter of controversy.
In previous animal studies focusing on the association between Al and tau accumulation, as well as the historic study by Klatzo et al 29, Al was often injected directly into the central nervous system because the procedure bypasses the blood–brain barrier 44. On the other hand, for evaluation of the more natural effect of Al as an environmental risk factor, oral administration of a low dose of Al to animals for a long period may be better. However, because oral administration of Al has a smaller effect on tau accumulation than injection, there are only a few studies that used that route. Further, the effect of orally administered Al on tau aggregation was not consistently observed even in studies using rabbits 27, 50, which are considered to be particularly sensitive to Al toxicity 44. The relative paucity of evidence regarding oral administration may make it difficult to estimate the actual risk of Al exposure on humans under natural conditions.
In previous animal studies, the risk of Al toxicity was discussed mainly on the basis of results demonstrating the presence or absence of de novo development of tau aggregation caused by Al exposure. On the other hand, whether Al exposure can advance tau pathology that has been started by other causes may be also important for humans because surprisingly, it was recently demonstrated that about 25% of young people under 20 years and up to 90% of individuals under 30 years already have mild abnormal tau accumulation that is characteristic of very early stage AD 6, 7. However, there are only a few previous studies that attempted to examine this possible effect of Al employing tau transgenic (Tg) mice 1, 35, and an acceleration of tau pathology was not observed in these studies.
The aim of this study was to examine whether chronic oral administration of Al can increase the severity of tau pathology and hasten its development in vivo. To address this issue, we orally administered Al to tau Tg mice and wild‐type (WT) littermate mice for 12 months, and compared them to tau Tg and WT mice without Al treatment. In these four experimental groups, we first examined the influence of Al exposure on (i) the number of tau‐positive axonal inclusions in the spinal cord, (ii) the number of flame‐shaped NFT‐like tau‐positive inclusions in the brain and (iii) motor function. In these examinations, we obtained several lines of evidence that Al can accelerate the formation of tau pathology. Therefore, we also evaluated (iv) the activation of astrocytes in the hippocampus and (v) apoptotic changes in the hippocampus to explore the neurodegenerative process caused by Al exposure.
Materials and Methods
Tau transgenic mice
T44 tau Tg mice were employed in this study to evaluate the change in the severity of tau pathology after long‐term Al exposure because T44 tau Tg mice show very slowly progressive tau pathology and exceptionally good survival for tau Tg mice: in our previous studies, about 85% of T44 Tg mice lived over 24 months and about 25% lived up to 32 months 22, 23. T44 was created using a transgene including cDNA of the shortest human tau isoform driven by the mouse PrP promoter and 3′ untranslated sequences on a B6D2/F1 background 22. WT littermate mice were used as control mice. Heterozygous T44 Tg mice overexpress human tau proteins at levels approximately five times higher than endogenous mouse tau. We do not find gender differences among the phenotypes in these Tg mice, nor does the fact that the transgene is inherited from father or mother affect features of the phenotype in these mice 22, 23, 36. In T44, filamentous tau aggregates are induced in axons in the spinal cord and neurons in the cerebrum and increase in number with age. They are composed of insoluble hyperphosphorylated tau, and show argyrophilia with Bodian and Gallyas‐Braak silver stains 22, 23, features that are consistent with those of NFTs observed in human brains.
Aluminum maltolate preparation, treatment and measurement
Aluminum nitrate (Al(NO3)3·9H2O) was dissolved in distilled water to the final concentration of 10 mM. Maltol (3‐hydroxy‐2‐methyl‐4‐pyrone, Sigma‐Aldrich, Tokyo, Japan) was dissolved (by warming slightly) in distilled water to make a 10 mM solution whose pH was finally adjusted to 7.4 with NaOH. Then, an aluminum‐maltol solution was prepared by mixing equal volumes of the 10 mM stock solutions of aluminum and maltol to give a final concentration of 2 mM for each substance 32. The pH was then adjusted to 7.4 with NaOH, and the resulting Al‐maltolate was used for each experiment. Mice were randomly divided into four experimental groups: (i) T44 tau Tg mice administered Al solution (Tg‐Al), (ii) T44 tau Tg mice without Al treatment (Tg‐CTL), (iii) WT mice administered Al solution (WT‐Al), and (iv) WT mice without Al treatment (WT‐CTL). Mice in the Tg‐Al and WT‐Al groups received drinking water containing 2 mM Al‐maltolate, and mice in the Tg‐CTL and WT‐CTL groups received untreated drinking water throughout their lifetimes. It has been reported that Al is ingested by suckling mammals through maternal milk 51. Therefore, the drinking water provided to the mothers of the Al‐treated groups (ie, Tg‐Al and WT‐Al groups) was changed to Al‐maltolate water immediately after the pups were born. Then, after weaning, they were given the same Al‐maltolate water. Food and water were provided ad libitum. The concentration of Al in food was 157 μg/g, while that of Al in distilled water was below measurement sensitivity (measured by Japan Food Research Laboratories, Tokyo, Japan, and SRL Inc., Tokyo, Japan, respectively). Mice were maintained on a 12‐h light/dark cycle in a temperature‐controlled (20 ± 1°C) room. All animals were treated in accordance with the Guidelines for Animal Experimentation of Okayama University.
For measurement of the Al concentrations in the central nervous system, brain tissues (0.1 g) from 12‐month‐old mice in Tg‐Al, Tg‐CTL, WT‐Al and WT‐CTL groups (n = 3, respectively) were digested with nitric acid (60%) of high purity grade (6.0 mL). The heating program consisted of the following steps: 0–600 W for 10 minutes and holding at 600 W for 20 minutes. After digestion, the solution was filtered through a polytetrafluoroethylene membrane filter (ADVANTEC DISMIC‐25HP, pore size of 0.20 μm, ADVANTEC, Ltd, Tokyo, Japan). The filtrate was diluted to 100 mL in a measuring flask with ultrapure water. The concentrations of Al were measured by inductively coupled plasma mass spectrometry (SPQ9700, Seiko Instruments Inc., Chiba, Japan).
Immunohistochemistry and conventional histopathological staining
At the ages of 3, 6, 9 and 12 months mice from each of the four experimental groups were anesthetized and perfused transcardially with 20 mL of phosphate‐buffered saline (PBS), followed by 20 mL of 70% ethanol in isotonic saline. Six‐μm‐thick paraffin sections of Tg and WT mouse brain and spinal cord were immunostained by standard streptavidin‐biotin‐peroxidase methods described previously 22, using a panel of antibodies (Table 1). Gallyas‐Braak silver, thioflavin‐S and Congo red staining were also done on paraffin sections of the brain in representative mice having tau pathology.
Table 1.
Antibodies used in this study. Abbreviations: R = rabbit polyclonal; M = mouse monoclonal.
| Antibody | Species | Dilution | Antigen | Reference |
|---|---|---|---|---|
| 17026 | R | 1:1500 | Tau | Ishihara et al 22 |
| Tau5 | M | 1:2000 | Tau | Chemicon |
| T14 | M | 1:500 | Human tau | Zymed |
| T1 | M | 1:500 | Nonphosphorylated tau | Roche Boehringer Mannheim |
| AT8 | M | 1:1000 | Tau phosphorylated at Ser 202 | Innogenetics |
| AT100 | M | 1:500 | Tau phosphorylated at Ser 212 and Thr 214 | Innogenetics |
| AT180 | M | 1:500 | Tau phosphorylated at Thr 231 | Innogenetics |
| AT270 | M | 1:500 | Tau phosphorylated at Thr 181 | Innogenetics |
| PHF‐1 | M | 1:250 | Tau phosphorylated at Ser 396 and Ser 404 | Greenberg and Davies 19 |
| PT231 | R | 1:500 | Tau phosphorylated at Thr 231 | Biosource |
| PS396 | R | 1:500 | Tau phosphorylated at Ser 396 | Biosource |
| PS404 | R | 1:500 | Tau phosphorylated at Ser 404 | Biosource |
| PS422 | R | 1:500 | Tau phosphorylated at Ser 422 | Biosource |
| TG3 | M | 1:100 | Tau phosphorylated at Thr 231 | Dickson et al 11 |
| Alz‐50 | M | 1:250 | Tau epitope at aa 5–15 | Bowser et al 5 |
| MC‐1 | M | 1:100 | Conformation‐dependent tau epitope within aa 312–322 | Jicha et al 25 |
| NFL | R | 1:100 | Neurofilament L | Chemicon |
| NFH | R | 1:100 | Neurofilament H | Novus Biologicals |
| GFAP | M | 1:200 | GFAP | Dakocytomation |
| GFAP | R | 1:100 | GFAP | Dakocytomation |
| Ubiquitin | M | 1:500 | Ubiquitin | Chemicon |
Analysis of tau‐positive spheroids in spinal cord
Axonal tau pathology immunolabeled with 17 026 anti‐tau antibody in sections of the spinal cord was quantitatively evaluated at 3, 6, 9 and 12 months in WT‐CTL (n = 5, 5, 5 and 6), WT‐Al (n = 12, 10, 13 and 10), Tg‐CTL (n = 7, 7, 10 and 6), and Tg‐Al groups (n = 10, 13, 12 and 10). The number of tau‐positive spheroids in 24 sections per mouse of the lumbar cord was counted, and the mean value was calculated. The quantitative examinations were performed by two investigators who were blind to the treatment.
Analysis of cytoplasmic tau accumulation in hippocampus and cortex
Sections of the cerebrum in Tg‐Al mice were immunostained with anti‐tau antibodies, MC‐1, PHF1, and AT270. MC‐1 recognizes the conformational change of tau protein into pretangles and NFTs 25, 49. Tau‐positive neuronal cytoplasmic inclusions in the hippocampus and cortex at 3, 6, 9 and 12 months in Tg‐CTL (n = 9, 10, 11 and 9 respectively), and Tg‐Al (n = 10, 13, 13 and 11, respectively) mice were counted. Likewise, tau‐positive inclusions in 12‐month‐old WT‐CTL mice (n = 6) and 12‐month‐old WT‐Al mice (n = 6) were also counted. The total number of inclusions per section in the hippocampus and cortex was used as the representative value of each mouse.
Analysis of GFAP‐positive astrocytes in hippocampus
Activated astrocytes immunolabeled with anti‐glial fibrillary acidic protein (GFAP) antibody in the hippocampal dentate gyrus were quantitatively evaluated at 3, 6, 9 and 12 months in WT‐CTL (n = 5, 6, 6 and 7), WT‐Al (n = 12, 9, 10 and 9), Tg‐CTL (n = 7, 7, 10 and 6) and Tg‐Al (n = 10, 12, 12 and 11) groups. The number of GFAP‐positive astrocytes was counted in three ×400 microscopic fields selected in the dorsal, lateral and ventral portions of the hippocampus on coronal sections through the decussatio supramammillaris. The mean value calculated was employed as the representative value of each mouse.
TUNEL staining and quantitative analysis of TUNEL‐positive cells in hippocampus
To assess DNA fragmentation in neurons in the hippocampus of WT‐CTL, WT‐Al, Tg‐CTL and Tg‐Al mice at 3, 6, 9 and 12 months (n = 5 in each experimental group by age, respectively), terminal deoxynucleotidyl transferase‐mediated dUTP‐digoxigenin end‐labeling (TUNEL) was performed (Roche in situ cell death detection kit POD, Roche, Basel, Switzerland). Paraffin sections (6 μm thick) were deparaffinized and digested with proteinase K (20 μg/mL, Qiagen, Tokyo, Japan) in PBS at 37°C for 15 minutes. Then, the sections were incubated with TdT buffer at 37°C for 60 minutes. The reaction was stopped using the Stop/Wash buffer. TUNEL‐positive cells were manually counted in three microscopic fields in the hippocampus at ×400 magnification using a fluorescence microscope. The proportion of positive cells was calculated per microscopic field, and the mean value calculated was employed as the representative value of each mouse.
Analysis of co‐localization of TUNEL, tau and GFAP immunoreactivity in hippocampus
Whether apoptosis is induced in tau‐positive cells was first examined by using mirror sections. Two 6‐μm‐thick serial paraffin sections of 12‐month‐old Tg‐Al mice were obtained with the cut surfaces facing each other. Each section was stained by the TUNEL method or MC‐1 immunohistochemistry 25, respectively. The converter‐POD solution was applied to the TUNEL‐stained section, and finally, it was stained with diaminobenzidine (DAB)/hydrogen peroxide. The other section that had been reacted with MC‐1 was immunostained by standard streptavidin‐biotin‐peroxidase methods, then the distribution of TUNEL‐positive cells was compared that of MC‐1‐positive neurons. Then, we also attempted to examine the colocalization of tau‐positive inclusions and TUNEL‐positive cells by double‐labeled immunofluorescence using the TUNEL method and MC‐1 or other anti‐tau antibodies. However, because the shape of tau‐positive inclusions was remarkably changed (eg, to a granular shape) after the TUNEL method was superimposed, only immunoreactivities on mirror sections were examined.
To examine the presence or absence of the co‐localization of GFAP and TUNEL immunoreactivities, double‐labeling immunofluorescence was performed on 12‐month‐old Tg‐Al mice with a combination of TUNEL staining and anti‐GFAP antibody (GFAP, rabbit, polyclonal, 1:100, DakoCytomation, Glostrup, Denmark). For these double stains, sections were first stained with TUNEL, and subsequently, incubated overnight at 4°C with anti‐GFAP primary antibody. Sections were incubated in fluorescence‐labeled secondary antibodies (Alexa Fluor 594‐labeled anti‐rabbit IgG (H + L)) for 1 h, and were viewed using a Zeiss MPM400 microscope (Carl Zeiss Japan, Tokyo, Japan) and Olympus DP‐72 CCD imaging system (Olympus, Tokyo, Japan).
Tail suspension test
Details of the tail suspension test were noted in our previous report 24. Briefly, mice at 3, 6, 9 or 12 months of age in the four experimental groups (n = 11 in each group) were videotaped while being suspended by their tails for 15 s, and assessed for clasping behavior. The test period was divided into 2‐s segments. An animal received one point for each abnormal movement displayed during each time segment. An abnormal movement was defined as dystonic movements of the hind limbs or a combination of hind limbs, forelimbs and trunk, during which the limbs were pulled into the body in a manner usually not observed in WT mice.
Statistical analysis
Two‐tailed unpaired Student's t‐test was used to compare the data between two groups. Comparison among three or more groups was made using one‐way analysis of variance (ANOVA), followed by Tukey's post hoc test to determine significant differences among the means of the data groups. The relationship between Al ingestion and death during the experimental period was examined by Fisher's exact test. Statistical analysis was performed using SPSS 10.0J (SPSS Inc., Chicago, IL, USA). A P‐value <0.05 was accepted as significant.
Results
The presence of Al in the drinking water did not affect the volume consumed and did not lead to overt systemic toxicity. The mean body weight at 12 months was not significantly different between Tg‐Al mice and Tg‐CTL mice (n = 13 and 12, 25.48 ± 3.59 g vs. 28.54 ± 5.53 g) (Student's t‐test, P = 0.055) and between WT‐Al and WT‐CTL (n = 11 and 13, 30.04 ± 5.91 g vs. 33.50 ± 8.23 g; Student's t‐test, P = 0.15). The survival rate at 12 months (the percentage of surviving mice at 9 months) was not significantly different between Tg‐Al and Tg‐CTL (n = 13 and 17; 85% vs. 71%) and between WT‐Al and WT‐CTL (n = 10 and 15; 100% vs. 86%), respectively (Fisher's exact test). However, the mean concentration of Al in brain tissue was significantly different between Tg‐Al and Tg‐CTL (16.27 ± 2.86 mg/kg and 8.57 ± 1.97 mg/kg; Student's t‐test, P = 0.019), and between WT‐Al and WT‐CTL (12.39 ± 0.34 mg/kg and 5.96 ± 3.25 mg/kg; Student's t‐test, P = 0.030), respectively. There was no significant difference between Al‐treated and ‐untreated groups in blood cell counts (ie, red blood cells, white blood cells, platelets, hemoglobin and hematocrit) or plasma levels of Na+, Cl− and K+ (data not shown).
Tau‐positive spheroids in spinal cord
The tau‐positive spheroids in the spinal cord are shown in Figure 1A–F. In mice in the Tg‐CTL group, a small number of tau‐positive spheroids appeared in the spinal cord at 3 months (Figure 1C). The number of labeled spheroids had almost doubled at 6 months, but then decreased slightly up to 12 months (Figures 1D and 2). The number of spheroids in Tg‐Al mice was significantly larger than that in the Tg‐CTL group at 12 months (Student's t‐test, P = 0.0073, Figures 1A,B and 2). In our old tau Tg mice, vacuolar lesions in the spinal cord were commonly observed. Although not quantitatively evaluated, these lesions tended to be more frequent in the Tg‐Al group than in the Tg‐CTL group (Figure 1B,D). To determine whether Al treatment alters tau phosphorylation, anti‐tau antibodies that recognize different phosphorylation sites (Table 1) were tried, but no obvious difference in immunoreactivity between anti‐tau antibodies was observed at any observation time (data not shown). No tau‐positive inclusion was noted in any WT‐CTL or WT‐Al mouse (Figure 1E,F).
Figure 1.
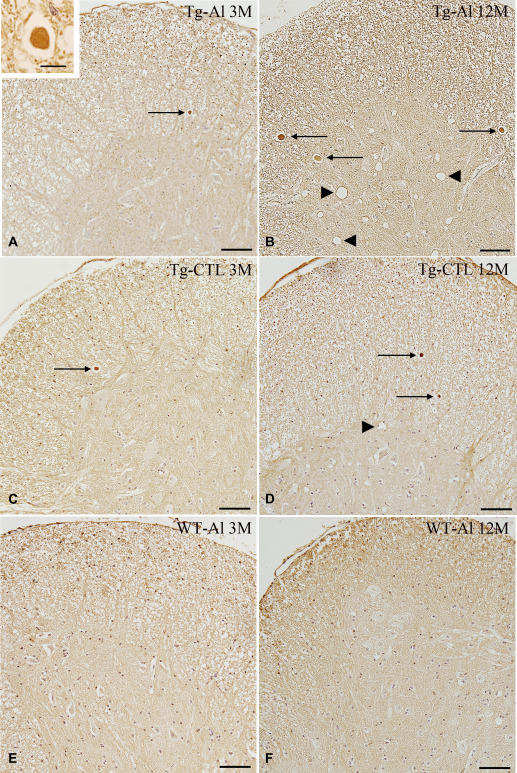
Tau‐positive spheroids and vacuolar lesions in spinal cords of representative tau Tg mice with and without Al treatment and WT mice with Al treatment. A. An Al‐treated tau Tg mouse (ie, Tg‐Al) at 3 months. Small tau‐positive spheroids (arrow) are apparent, but vacuolar lesion was absent. B. An Al‐treated tau Tg mouse (Tg‐Al) at 12 months. Tau‐positive spheroids were increased in number and size. Many large vacuolar lesions also occurred (arrowheads). C. A tau Tg mouse without Al treatment (Tg‐CTL) at 3 months. A few tau‐positive spheroids are seen (arrow). D. A tau Tg mouse without Al treatment (Tg‐CTL) at 12 months. Tau‐positive inclusions gradually increased in number with age (arrows) along with vacuolar lesions (arrowhead). However, it was less severe than tau Tg mice treated with Al. E,F. WT mice treated with Al. Neither a tau‐positive spheroid nor vacuolar lesion was observed in the spinal cords at 3 (E) or 12 months (F). Tau (17026) immunohistochemistry. Scale bars = 100 μm (A–F) and 10 μm (A, inset).
Figure 2.
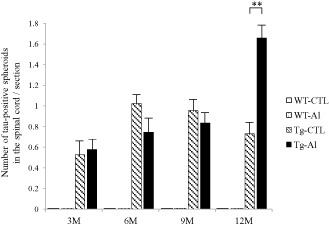
Numbers of tau‐positive spheroids in spinal cords of tau Tg mice with and without Al treatment. The mean counts of spheroids in each experimental group are shown by age. Tau‐positive spheroids in Al‐treated tau Tg mice (ie, Tg‐Al) at 3, 6 and 9 months are comparable in number to those in tau Tg mice without Al treatment (Tg‐CTL), respectively. However, the mean number of labeled spheroids in Tg‐Al mice was significantly larger than that in Tg‐CTL mice (Student's t‐test, P = 0.0073). A slight decrease of the number of inclusions in 6‐ to 12‐month‐old Tg‐CTL mice was also observed in our previous study that used T44 mice 22. The data of WT mice are not shown because no mouse had tau‐positive inclusions. A significant difference between experimental groups is indicated by asterisks (**P < 0.01). The error bars represent SEM.
Cytoplasmic tau aggregation in hippocampus and entorhinal cortex
In the Tg‐Al group, tau‐positive cytoplasmic inclusions were not found at 3 or 6 months. However, at 9 months, six of 13 mice (46%) had several tau‐positive inclusions labeled with MC‐1 (Figure 3A,E), and the proportion of mice having tau‐positive inclusions reached 63% (seven of 11 mice) at 12 months (Figure 3B,C,E). In contrast, in the Tg‐CTL group, tau‐positive inclusions were found in only one of nine mice (11%) at 12 months (Figure 3D). Thioflavin‐S, Congo red and Gallyas‐Braak silver staining did not demonstrate any lesions in mice in the Tg‐CTL or Tg‐Al groups. These tau‐positive inclusions were not stained by anti‐neurofilament antibodies. No mouse in a WT‐CTL or WT‐Al group had tau aggregates in neurons in the hippocampus and cortex. Because MC‐1 immunoreactivity was stronger than those of PHF1 and AT270, tau‐positive inclusions were counted on sections stained with MC‐1. The mean number of MC‐1‐positive inclusions in Tg‐Al mice was significantly larger than that in Tg‐CTL mice at 9 and 12 months, respectively (Student's t‐test, P = 0.012 and 0.048, Figure 3E).
Figure 3.
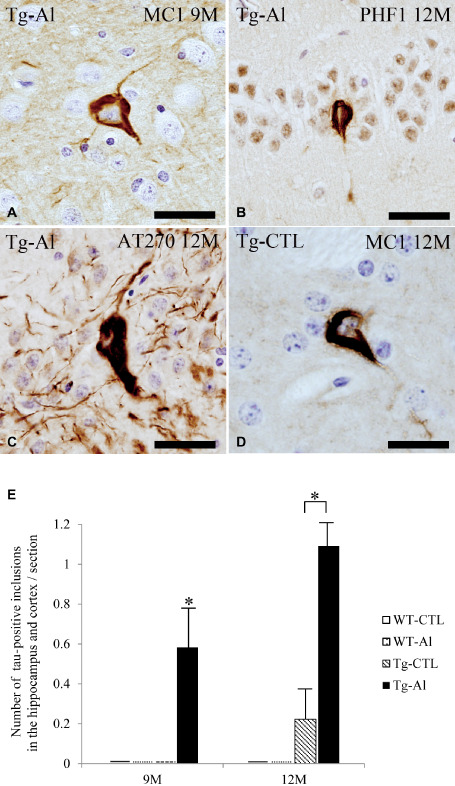
Cytoplasmic tau accumulation in cortex and hippocampus in representative tau Tg mice with and without Al treatment. A–C. NFT‐like tau‐positive inclusions in the hippocampus in Al‐treated 9‐ (A) and 12‐month‐old tau Tg mice (B,C). D. NFT‐like cytoplasmic tau accumulation in the hippocampus in the brain in a 12‐month‐old tau Tg mouse without Al treatment. (A,D) MC‐1 immunohistochemistry, (B) PHF1 immunohistochemistry, and (C) AT270 immunohistochemistry. All scale bars = 30 μm. E. The numbers of tau‐positive cytoplasmic inclusions in the cortex and hippocampus in tau Tg mice with (Tg‐Al) and without (Tg‐CTL) Al treatment at 9 and 12 months. The mean numbers of inclusions in the cortex and hippocampus in Tg‐Al mice were significantly larger than those in Tg‐CTL mice at 9 and 12 months, respectively (Student's t‐test, P = 0.012 and 0.048). The data of WT at all experimental time points and Tg mice at 3 and 6 months are not shown because no mouse had tau‐positive inclusions at that age. A significant difference between experimental groups is indicated by asterisks (*P < 0.05). The error bars represent SEM.
GFAP‐positive astrocytes in hippocampus
In the hippocampus of WT‐CTL mice, only a small number of astrocytes immunopositive for GFAP were scattered at 3 months, and they tended to increase in number with age (Figures 4A,B and 5). The number of GFAP‐positive astrocytes in WT‐Al and Tg‐CTL groups tended to be slightly larger than that in the WT‐CTL group, respectively (Figures 4C–F and 5).
Figure 4.
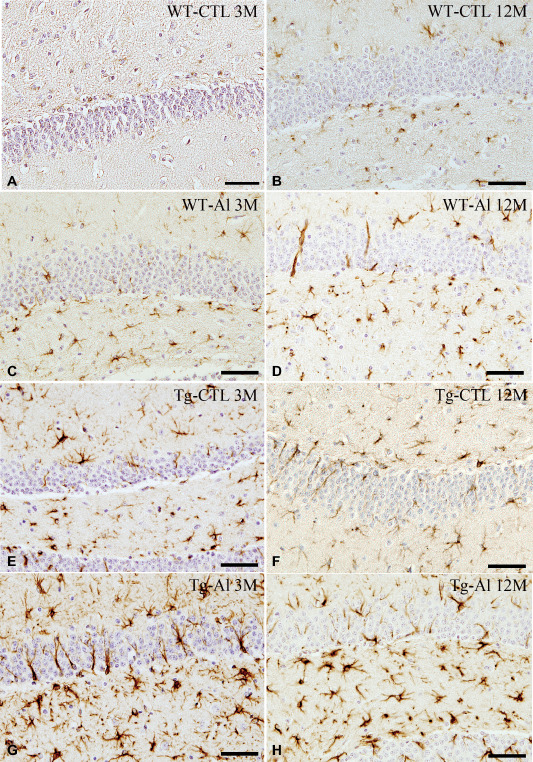
GFAP‐positive astrocytes in hippocampus in representative tau Tg and WT mice with and without Al treatment. A,B. WT mice without Al treatment (WT‐CTL). At 3 months, only a few, scattered astrocytes were weakly immunopositive for GFAP (A). They were slightly increased in number at 12 months (B). C,D. Al‐treated WT mice (WT‐Al). A moderate number of GFAP‐positive astrocytes were noted at 3 months (C). The number of labeled astrocytes slightly increased with age (D), and it was also larger than that in a WT‐CTL mouse (B). E,F. Tau Tg mice without Al treatment (Tg‐Al). A moderate number of GFAP‐positive astrocytes were seen at 3 months (E), and they increased slightly in number up to 12 months (F). G,H. Al‐treated tau Tg mice (Tg‐Al). Abundant intensely labeled astrocytes were already noted at 3 months (G). The number of labeled astrocytes at 3 months was comparable to that at 12 months (H). GFAP immunohistochemistry. All scale bar = 50 μm.
Figure 5.
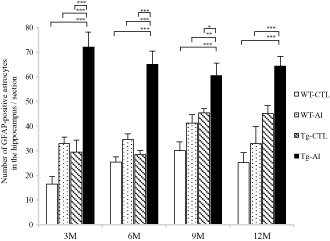
Numbers of GFAP‐positive astrocytes in brains of tau Tg and WT mice with and without Al treatment. In Al‐treated tau Tg mice (Tg‐Al), abundant GFAP‐positive astrocytes were already seen at 3 months, and similar numbers of labeled astrocytes were also observed at 6, 9 and 12 months. The numbers of labeled astrocytes in tau Tg mice without Al treatment (Tg‐CTL) and WT mice with and without Al treatment (WT‐Al and WT‐CTL) tended to increase with age, although they were smaller than those in Tg‐Al mice at any observation time. The mean numbers of labeled astrocytes in Tg‐Al mice at 3, 6 and 9 months were significantly larger than those in the other three groups, respectively (P < 0.05). The mean number of GFAP‐positive astrocytes in the Tg‐Al group at 12 months was also significantly higher than those in WT‐CTL and WT‐Al groups at 12 months, respectively (P < 0.001). Statistical analysis was performed by one‐way ANOVA and Tukey post hoc test. A significant difference between experimental groups is indicated by asterisks (*P < 0.05, **P < 0.01, ***P < 0.001). The error bars represent SEM.
In contrast, in a Tg‐Al group, abundant intensely labeled astrocytes were already noted at 3 months (Figure 4G), and many activated astrocytes were also found throughout the experimental period (Figures 4H and 5). The mean numbers of GFAP‐positive astrocytes in Tg‐Al mice at 3, 6 and 9 months were significantly higher than those in the other three groups (ie, WT‐CTL, WT‐Al and Tg‐CTL), respectively (one‐way ANOVA and Tukey post hoc test, P < 0.05). The mean number of GFAP‐positive astrocytes in the Tg‐Al group at 12 months was also significantly higher than those in WT‐CTL and WT‐Al groups at 12 months (one‐way ANOVA and Tukey post hoc test, P < 0.001; Figures 4G and 5).
TUNEL assay in hippocampus
TUNEL‐positive cells were only rare or virtually absent in WT‐CTL, WT‐Al and Tg‐CTL mice throughout the observation period. However, TUNEL‐positive cells were often observed only in Tg mice treated with Al for 12 months (Figure 6A,F). At 12 months, the mean number of TUNEL‐positive cells in Tg‐Al mice was significantly larger than that in the other three groups (Figure 6F, one‐way ANOVA and Tukey post hoc test, P < 0.001).
Figure 6.
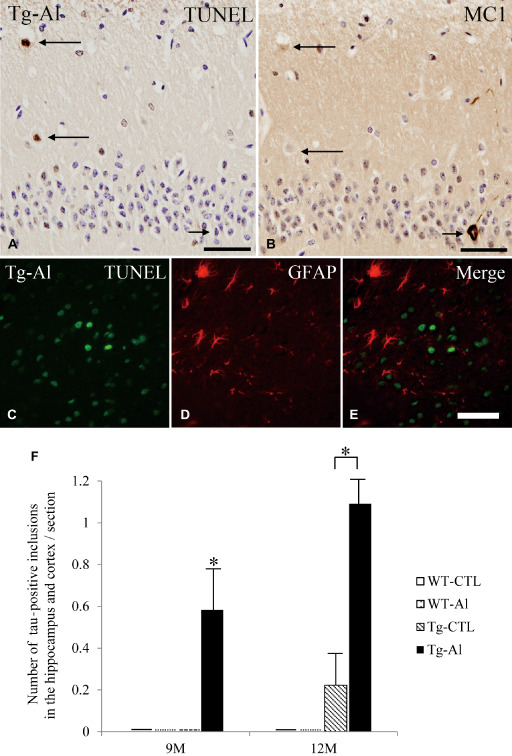
TUNEL‐positive cells in hippocampus, and its relationship to tau‐positive inclusions and GFAP‐positive astrocytes. A. TUNEL‐positive cells (long arrows) in the hippocampus in a 12‐month‐old Tg mouse treated with Al. B. This picture was taken from the identical region of a mirror section of (A) and reversed. TUNEL‐positive cells shown in (A) do not show tau (MC‐1) immunoreactivity (long arrows). Conversely, a neuron containing tau aggregation (short arrow in B) is TUNEL‐negative (short arrow in A). C–E. Double‐labeled immunofluorescence in the hippocampus in 12‐month‐old Tg‐Al mice did not show co‐localization of TUNEL and GFAP immunoreactivities. (A,B) scale bars = 50 μm, (C–E) scale bars = 30 μm. F. Mean numbers of TUNEL‐positive cells in the hippocampus of four groups (ie, WT‐CTL, WT‐Al, Tg‐CTL, Tg‐Al) at 3 months and 12 months. The data at 6 months and 9 months are not shown because no mouse had a TUNEL‐positive cell. The mean number of TUNEL‐positive cells in the hippocampus in Tg‐Al mice was significantly larger than in the other three groups. Statistical analysis were performed by one‐way ANOVA and Tukey post hoc test (***P < 0.001). The error bars represent SEM.
On mirror sections, the distribution of TUNEL‐positive cells was not consistent with that of MC‐1‐positive neurons in 12‐month‐old Tg‐Al mice (Figure 6A,B). Double‐labeled immunofluorescence in the hippocampus in 12‐month‐old Tg‐Al mice did not show co‐localization of TUNEL and GFAP immunoreactivities (Figure 6C–E).
Motor symptoms
The most severe impairment of motor function was noted in Tg‐Al mice, followed by Tg‐CTL, WT‐Al and WT‐CTL mice. The mean clasping score in Tg‐Al mice was significantly higher than those in WT‐CTL and WT‐Al groups at 3 and 6 months (Figure 7). At 9 and 12 months, the mean clasping score in Tg‐Al mice was significantly higher than that in other three groups (one‐way ANOVA and Tukey's post hoc test, P = 0.0077). In addition, motor function was significantly more severely impaired in WT mice after long‐term Al treatment (12 months) than in WT mice without it.
Figure 7.
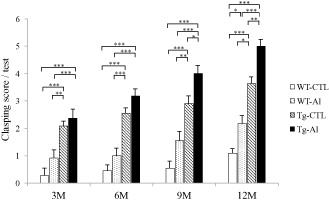
Motor function in tau Tg and WT mice with and without Al treatment. Motor function was evaluated by the tail suspension test and indicated by the clasping score (see text). Motor function in Al‐treated tau Tg mice (Tg‐Al) was more severely impaired than those in other groups at any observation time. In all experimental groups, the impairment gradually worsened with age. The mean clasping score in the Tg‐Al group was significantly higher than those in WT‐CTL and WT‐Al groups at 3 and 6 months. At 9 and 12 months, the score in a Tg‐Al group was significantly higher than that in all of the other groups. Statistical analysis was performed by one‐way ANOVA and Tukey post hoc test (*P < 0.05, **P < 0.01, ***P < 0.001). The error bars represent SEM.
Discussion
In this study, we demonstrated that tau aggregation as well as apoptosis in the central nervous system in tau Tg mice was significantly increased by chronic oral administration of Al. Motor function in tau Tg mice was also significantly impaired by chronic exposure to Al. To our knowledge, this is the first report that demonstrated the effect of chronic oral ingestion of Al on tau aggregation, apoptosis and neurological dysfunction in a mouse model of tauopathies.
There is only one previous study that examined the effect of long‐term oral intake of Al on tau pathology in tau Tg mice 1. In that study, Al was administered to APP/tau double Tg mice (Tg2576/tau) for 9 months. However, although the concentration of Al administered was eight times as high as the concentration we used, tau pathology in the Tg mice was not increased by Al exposure. Although these findings appear to be inconsistent with our results, there are several fundamental differences between the experimental settings of the two studies. First is the duration of the administration of Al: the duration in the previous study was only 9 months, which is two‐thirds of that in our study. Second is the time that Al administration was started: Al was administered from 5 months old in the previous study, while in our study, Al exposure started immediately after birth by ingesting milk from mothers that took Al in water. Although two studies cannot be simply compared, considering that Al is transferred from lactating rats to suckling rats through the mother's milk 51, it is possible that tau accumulation in mice might be more readily accelerated when Al exposure begins from the younger age and/or continues for the longer duration. Third, a ceiling effect regarding the acceleration of tau accumulation caused by Al exposure might be also associated with the negative results in the previous study because tau pathology in the double Tg mice shown in the paper appears to be far more severe than that in the T44 mice that we used 1.
The effect of Al exposure on tau accumulation in human mutant (V337M) tau Tg mice was also evaluated in another previous study, although Al was injected into the peritoneal cavity 35. That study did not demonstrate a significant increase of the level of tau in the brains of the Tg mice after a 2‐week administration of Al from the age of 3 months, and the authors concluded that Al may not be a risk factor for AD. However, although Al exposure for 2 weeks may be sufficient for the evaluation of acute or subacute toxicity, it is probably insufficient to demonstrate its chronic effect. In addition, considering that the tau Tg mice used in that study usually develop tau pathology from 11 months old 47, it may be difficult to deny the effect of Al based on the lack of the change of the tau level at 3 months old.
Morphologically, NFT‐like tau aggregates observed in the brain in our Al‐treated tau Tg mice at 9 months were labeled with MC‐1, which recognizes the early conformational change of tau protein and several phosphorylation‐dependent anti‐tau antibodies, but they were not stained by Gallyas‐Braak silver stain. These are histological features of the early pathological process before the aggregates are converted into argyrophilic NFTs, being also consistent with those of pretangles observed in human brains. A previous study demonstrated that Al treatment induced neurofilament‐positive but tau‐negative cytoplasmic aggregates in cultured neurons 31. In contrast, NFT‐like tau‐positive inclusions in the cerebrum in our tau Tg mice treated with Al were not labeled with anti‐neurofilament antibodies. Although this inconsistency might be associated with many differences in experimental settings, our findings suggest that tau aggregation induced by Al ingestion precedes neurofilament accumulation. In our tau Tg mice without Al treatment, NFT‐like tau aggregates had not developed at 9 months age (Figure 3E). These findings suggest that chronic oral exposure to Al may accelerate tau aggregation with respect to not only the quantity but also the time of its development in our tau Tg mice. On the other hand, Al exposure did not induce a de novo abnormal accumulation of tau in our WT mice. These findings led us to consider that, at least in mice, Al may promote the occurrence of tau pathology more readily when a certain pathological process causing tau aggregation already exists than when it does not. If Al actually had this potential effect, it would seem to be more important in humans because tau accumulation as a very early stage of AD begins at a far younger age than considered previously: a recent pathological study demonstrated that about 25% of teens, 90% of people in their twenties, 95% of those in their thirties and 100% of those in their forties already have pretangles in the locus coeruleus and other brain stem nuclei, and that the tau pathology was gradually progressing to the cerebrum 7. Thus, the possibility that tau pathology is present almost universally in the central nervous system in young and middle‐aged people caused by Al probably leads to an actual risk of the onset of AD in later life.
Previous studies demonstrated that motor function was impaired after administration of Al to WT rats 12 and WT mice 17. It was reported that the motor dysfunction caused by Al was developed through tau accumulation 46 and was associated with the impairment of neurofilament transport in the axons 4. In our study, because motor dysfunction, as well as tau‐positive inclusions were increased in severity during the period of Al treatment, it is plausible that Al neurotoxicity may induce motor dysfunction through abnormal tau accumulation. On the other hand, because the increase of motor dysfunction caused by Al exposure was seen not only in tau Tg mice but also in WT mice in our study, it is evident that Al exposure can promote motor impairment regardless of the formation of tau‐positive inclusions. Whether the neurological disturbance is a simple additive result of tau accumulation‐dependent and ‐independent processes or a synergistic consequence of them remains unclear.
It was reported that both Al exposure 20, 38, 42, 43, 45 and tau accumulation 14, 26 may induce apoptosis. Our study suggests that apoptosis may be induced more efficiently when both Al exposure and the pathological process of tau accumulation occur than when only one is present. On the other hand, TUNEL‐positive cells in our Al‐treated tau Tg mice consistently lacked tau aggregates in the cytoplasm, suggesting that apoptosis in our mice is not always induced directly by aggregated tau protein. These findings led us to consider that some pathological molecule prior to fibrillar tau aggregates, such as soluble tau, might play an important role in the induction of apoptosis caused by Al exposure. This view appears to be supported by recent findings that soluble tau species rather than fibrillar tau contribute to impairment of neuronal function and neurodegeneration 15, 30.
It is difficult to interpret the pathophysiological significance of early activation of astrocytes observed only in Al‐treated tau Tg mice. The activation evidently preceded the occurrence of tau‐positive inclusions, suggesting that the astrocytic change may not be a secondary reactive phenomenon caused by tau aggregation. Regarding the relationship between astrocyte activation, Al exposure and tau aggregation, it was reported that activation of astrocytes after Al exposure was observed in the cortex in WT rats 12 and basal ganglia in WT mice 10. It is also known that Al is preferentially taken up by glial cells 8, 52, and that Al exposure can trigger the production of inflammatory cytokines including IL‐6 by activated astrocytes 2, 9, which induces activation of the cdk5/p35 system 39. Whether astrocyte activation by Al exposure plays an important role in the acceleration of an early process in the formation of tau pathology, eg, before NFT‐like tau aggregates occur, needs to be examined by further studies.
There are several limitations to this study. First, we did not assess the effect of maltol on tau aggregation in this study. However, no previous study demonstrated that maltol induced aggregation of tau protein 28, although a previous study demonstrated that maltol induced a small number of neurofilament‐positive cytoplasmic inclusions in cultured neurons. However, the lesions were not labeled with an anti‐tau antibody, suggesting that the inclusions differed from NFTs 32. Second, although we assessed apoptosis by a TUNEL assay, TUNEL‐positive cells are found not only in apoptotic processes but several other pathological conditions involving DNA breakage and its repair 33.
In conclusion, the present study demonstrated a potential linkage between chronic oral ingestion of Al, tau accumulation, apoptosis and neurological dysfunction. Although our findings should be confirmed by further experiments, whether these relationships are also involved in the pathogenic process involved in the onset of AD in humans may be the next topic that should be considered.
Acknowledgments
We thank Prof. J.Q. Trojanowski and Prof. V.M.Y. Lee for their generous gift of tau transgenic mice T44, and Dr. P. Davies for PHF‐1, MC1, Alz50, TG3 antibody. We also thank M. Onbe and R. Wada for technical assistance. This work was supported by grants from the Japanese Ministry of Education, Culture, Sports, Science and Technology (no. 18590942, no. 21591517, no. 23591708), Zikei Institute of Psychiatry and Kobayashi Magobe Memorial Medical Foundation.
References
- 1. Akiyama H, Hosokawa M, Kametani F, Kondo H, Chiba M, Fukushima M, Tabira T (2012) Long‐term oral intake of aluminium or zinc does not accelerate Alzheimer pathology in AβPP and AβPP/tau transgenic mice. Neuropathology 32:390–397. [DOI] [PubMed] [Google Scholar]
- 2. Arai KI, Lee F, Miyajima A, Miyatake S, Arai N, Yokota T (1990) Cytokines: coordinators of immune and inflammatory responses. Annu Rev Biochem 59:783–836. [DOI] [PubMed] [Google Scholar]
- 3. Aremu DA, Meshitsuka S (2005) Accumulation of aluminum by primary cultured astrocytes from aluminum amino acid complex and its apoptotic effect. Brain Res 1031:284–296. [DOI] [PubMed] [Google Scholar]
- 4. Bizzi A, Crane RC, Autilio‐Gambetti L, Gambetti P (1984) Aluminum effect on slow axonal transport: a novel impairment of neurofilament transport. J Neurosci 4:722–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bowser R, Giambrone A, Davies P (1995) FAC1, a novel gene identified with the monoclonal antibody Alz50, is developmentally regulated in human brain. Dev Neurosci 17:20–37. [DOI] [PubMed] [Google Scholar]
- 6. Braak H, Tredici KD (2011) The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol 121:171–181. [DOI] [PubMed] [Google Scholar]
- 7. Braak H, Thal DR, Ghebremedhin E, Tredici KD (2011) Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70:960–969. [DOI] [PubMed] [Google Scholar]
- 8. Campbell A, Prasad KN, Bondy SC (1999) Aluminum‐induced oxidative events in cell lines: glioma are more responsive than neuroblastoma. Free Radic Biol Med 26:1166–1171. [DOI] [PubMed] [Google Scholar]
- 9. Campbell A, Yang EY, Tsai‐Turton M, Bondy SC (2002) Pro‐inflammatory effects of aluminum in human glioblastoma cells. Brain Res 933:60–65. [DOI] [PubMed] [Google Scholar]
- 10. Campbell A, Becaria A, Lahiri DK, Sharman K, Bondy SC (2004) Chronic exposure to aluminum in drinking water increases inflammatory parameters selectively in the brain. J Neurosci Res 75:565–572. [DOI] [PubMed] [Google Scholar]
- 11. Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P (1995) Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging 16:285–304. [DOI] [PubMed] [Google Scholar]
- 12. Erazi H, Sansar W, Ahboucha S, Gamrani H (2010) Aluminum affects glial system and behavior of rats. C R Biol 333:23–27. [DOI] [PubMed] [Google Scholar]
- 13. Exley C (2006) Aluminium and iron, but neither copper nor zinc, are key to the precipitation of beta‐sheets of Abeta42 in senile plaque cores in Alzheimer's disease. J Alzheimers Dis 10:173–177. [DOI] [PubMed] [Google Scholar]
- 14. Fath T, Eidenmüller J, Brandt R (2002) Tau‐mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer's disease. J Neurosci 22:9733–9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fox LM, William CM, Adamowicz DH, Pitstick R, Carlson GA, Spires‐Jones TL, Hyman BT (2011) Soluble tau species, not neurofibrillary aggregates, disrupt neural system integration in a tau transgenic model. J Neuropathol Exp Neurol 70:588–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gillette‐Guyonnet S, Andrieu S, Nourhashemi F, de La Guéronnière V, Grandjean H, Vellas B (2005) Cognitive impairment and composition of drinking water in women: findings of the EPIDOS Study. Am J Clin Nutr 81:897–902. [DOI] [PubMed] [Google Scholar]
- 17. Golub MS, Germann SL (2001) Long‐term consequences of developmental exposure to aluminum in a suboptimal diet for growth and behavior of Swiss Webster mice. Neurotoxicol Teratol 23:365–372. [DOI] [PubMed] [Google Scholar]
- 18. Gómez M, Esparza JL, Cabré M, García T, Domingo JL (2008) Aluminum exposure through the diet: metal levels in AbetaPP transgenic mice, a model for Alzheimer's disease. Toxicology 249:214–219. [DOI] [PubMed] [Google Scholar]
- 19. Greenberg SG, Davies P (1990) A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci U S A 87:5827–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Griffioen KJ, Ghribi O, Fox N, Savory J, DeWitt DA (2004) Aluminum maltolate‐induced toxicity in NT2 cells occurs through apoptosis and includes cytochrome c release. Neurotoxicology 25:859–867. [DOI] [PubMed] [Google Scholar]
- 21. House E, Collingwood J, Khan A, Korchazkina O, Berthon G, Exley C (2004) Aluminium, iron, zinc and copper influence the in vitro formation of amyloid fibrils of Abeta42 in a manner which may have consequences for metal chelation therapy in Alzheimer's disease. J Alzheimers Dis 6:291–301. [DOI] [PubMed] [Google Scholar]
- 22. Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM (1999) Age‐dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron 24:751–762. [DOI] [PubMed] [Google Scholar]
- 23. Ishihara T, Zhang B, Higuchi M, Yoshiyama Y, Trojanowski JQ, Lee VM (2001) Age‐dependent induction of congophilic neurofibrillary tau inclusions in tau transgenic mice. Am J Pathol 158:555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ishihara T, Higuchi M, Zhang B, Yoshiyama Y, Hong M, Trojanowski JQ, Lee VM (2001) Attenuated neurodegenerative disease phenotype in tau transgenic mouse lacking neurofilaments. J Neurosci 21:6026–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jicha GA, Bowser R, Kazam IG, Davies P (1997) Alz‐50 and MC‐1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res 48:128–132. [DOI] [PubMed] [Google Scholar]
- 26. Khurana V, Lu Y, Steinhilb ML, Oldham S, Shulman JM, Feany MB (2006) TOR‐mediated cell‐cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr Biol 16:230–241. [DOI] [PubMed] [Google Scholar]
- 27. Kihira T, Yoshida S, Uebayashi Y, Wakayana I, Yase Y (1994) Experimental model of motor neuron disease: oral aluminum neurotoxicity. Biomed Res Tokyo 15:27–36. [Google Scholar]
- 28. Kihira T, Yoshida S, Yase Y, Ono S, Kondo T (2002) Chronic low‐Ca/Mg high‐Al diet induces neuronal loss. Neuropathology 22:171–179. [DOI] [PubMed] [Google Scholar]
- 29. Klatzo I, Wisniewski H, Streicher E (1965) Experimental production of neurofibrillary degeneration. I. Light microscopic observations. J Neuropathol Exp Neurol 24:187–199. [DOI] [PubMed] [Google Scholar]
- 30. Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI et al (2011) Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer's disease brain. Am J Pathol 179:2071–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Langui D, Anderton BH, Brion JP, Ulrich J (1988) Effects of aluminium chloride on cultured cells from rat brain hemispheres. Brain Res 438:67–76. [DOI] [PubMed] [Google Scholar]
- 32. Langui D, Probst A, Anderton B, Brion JP, Ulrich J (1990) Aluminium‐induced tangles in cultured rat neurones. Enhanced effect of aluminium by addition of maltol. Acta Neuropathol 80:649–655. [DOI] [PubMed] [Google Scholar]
- 33. Loo DT (2011) In situ detection of apoptosis by the TUNEL assay: an overview of techniques. Methods Mol Biol 682:3–13. [DOI] [PubMed] [Google Scholar]
- 34. McLachlan DR, Bergeron C, Smith JE, Boomer D, Rifat SL (1996) Risk for neuropathologically confirmed Alzheimer's disease and residual aluminum in municipal drinking water employing weighted residential histories. Neurology 46:401–405. [DOI] [PubMed] [Google Scholar]
- 35. Mizoroki T, Meshitsuka S, Maeda S, Murayama M, Sahara N, Takashima A (2007) Aluminum induces tau aggregation in vitro but not in vivo. J Alzheimers Dis 11:419–427. [DOI] [PubMed] [Google Scholar]
- 36. Nakashima H, Ishihara T, Suguimoto P, Yokota O, Oshima E, Kugo A et al (2005) Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol 110:547–556. [DOI] [PubMed] [Google Scholar]
- 37. Praticò D, Uryu K, Sung S, Tang S, Trojanowski JQ, Lee VM (2002) Aluminum modulates brain amyloidosis through oxidative stress in APP transgenic mice. FASEB J 16:1138–1140. [DOI] [PubMed] [Google Scholar]
- 38. Prolo P, Chiappelli F, Grasso E, Rosso MG, Neagos N, Dovio A et al (2007) Aluminium blunts the proliferative response and increases apoptosis of cultured human cells: putative relationship to Alzheimer's disease. Bioinformation 2:24–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Quintanilla RA, Orellana DI, González‐Billault C, Maccioni RB (2004) Interleukin‐6 induces Alzheimer‐type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp Cell Res 295:245–257. [DOI] [PubMed] [Google Scholar]
- 40. Reusche E, Koch V, Lindner B, Harrison AP, Friedrich HJ (2001) Alzheimer morphology is not increased in dialysis‐associated encephalopathy and long‐term hemodialysis. Acta Neuropathol 101:211–216. [DOI] [PubMed] [Google Scholar]
- 41. Rondeau V, Jacqmin‐Gadda H, Commenges D, Helmer C, Dartigues JF (2009) Aluminum and silica in drinking water and the risk of Alzheimer's disease or cognitive decline: findings from 15‐year follow‐up of the PAQUID cohort. Am J Epidemiol 169:489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Savory J, Rao JK, Huang Y, Letada PR, Herman MM (1999) Age‐related hippocampal changes in Bcl‐2:Bax ratio, oxidative stress, redox‐active iron and apoptosis associated with aluminum‐induced neurodegeneration: increased susceptibility with aging. Neurotoxicology 20:805–817. [PubMed] [Google Scholar]
- 43. Savory J, Ghribi O, Forbes MS, Herman MM (2001) Aluminium and neuronal cell injury: inter‐relationships between neurofilamentous arrays and apoptosis. J Inorg Biochem 87:15–19. [DOI] [PubMed] [Google Scholar]
- 44. Savory J, Ghribi O, Forbes MS, Herman MM (2001) The rabbit model system for studies of aluminum‐induced neurofibrillary degeneration: relevance to human neurodegenerative disorders. In: Aluminum and Alzheimer's Disease. The Science That Describes the Link, Exley C (ed.), pp. 203–219. Elsevier: Amsterdam. [Google Scholar]
- 45. Savory J, Herman MM, Ghribi O (2003) Intracellular mechanisms underlying aluminum‐induced apoptosis in rabbit brain. J Inorg Biochem 97:151–154. [DOI] [PubMed] [Google Scholar]
- 46. Shaw CA, Petrik MS (2009) Aluminum hydroxide injections lead to motor deficits and motor neuron degeneration. J Inorg Biochem 103:1555–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tanemura K, Murayama M, Akagi T, Hashikawa T, Tominaga T, Ichikawa M et al (2002) Neurodegeneration with tau accumulation in a transgenic mouse expressing V337M human tau. J Neurosci 22:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Walton JR (2007) An aluminum‐based rat model for Alzheimer's disease exhibits oxidative damage, inhibition of PP2A activity, hyperphosphorylated tau, and granulovacuolar degeneration. J Inorg Biochem 101:1275–1284. [DOI] [PubMed] [Google Scholar]
- 49. Weaver CL, Espinoza M, Kress Y, Davies P (2000) Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging 21:719–727. [DOI] [PubMed] [Google Scholar]
- 50. Wills MR, Hewitt CD, Savory J, Herman MM (1993) Long‐term oral aluminum administration in rabbits. II. Brain and other organs. Ann Clin Lab Sci 1:17–23. [PubMed] [Google Scholar]
- 51. Yumoto S, Nagai H, Kobayashi K, Tamate A, Kakimi S, Matsuzaki H (2003) 26Al incorporation into the brain of suckling rats through maternal milk. J Inorg Biochem 97:155–160. [DOI] [PubMed] [Google Scholar]
- 52. Zielke HR, Jackson MJ, Tildon JT, Max SR (1993) A glutamatergic mechanism for aluminum toxicity in astrocytes. Mol Chem Neuropathol 19:219–233. [DOI] [PubMed] [Google Scholar]