

Abstract
The term long‐term epilepsy associated tumor (LEAT) encompasses lesions identified in patients investigated for long histories (often 2 years or more) of drug‐resistant epilepsy. They are generally slowly growing, low grade, cortically based tumors, more often arising in younger age groups and in many cases exhibit neuronal in addition to glial differentiation. Gangliogliomas and dysembryoplastic neuroepithelial tumors predominate in this group. LEATs are further united by cyto‐architectural changes that may be present in the adjacent cortex which have some similarities to developmental focal cortical dysplasias (FCD); these are now grouped as FCD type IIIb in the updated International League Against Epilepsy (ILAE) classification. In the majority of cases, surgical treatments are beneficial from both perspectives of managing the seizures and the tumor. However, in a minority, seizures may recur, tumors may show regrowth or recurrence, and rarely undergo anaplastic progression. Predicting and identifying tumors likely to behave less favorably are key objectives of the neuropathologist. With immunohistochemistry and modern molecular pathology, it is becoming increasingly possible to refine diagnostic groups. Despite this, some LEATs remain difficult to classify, particularly tumors with “non‐specific” or diffuse growth patterns. Modification of LEAT classification is inevitable with the goal of unifying terminological criteria applied between centers for accurate clinico‐pathological‐molecular correlative data to emerge. Finally, establishing the epileptogenic components of LEAT, either within the lesion or perilesional cortex, will elucidate the cellular mechanisms of epileptogenesis, which in turn will guide optimal surgical management of these lesions.
Keywords: brain, dysplasia, epilepsy, neoplasm, neuropathology, seizure
INTRODUCTION
Any tumor type of any grade involving the brain can cause seizures. The concept of a long‐term epilepsy associated tumor (LEAT) was introduced by Luyken et al in the recognition of tumors more commonly encountered in surgical series of patients who have been investigated and treated for drug‐resistant seizure episodes for 2 years or longer (172). Characteristics and properties of LEATs that distinguish them from more conventional primary brain tumors include a young age of onset of symptoms (with epilepsy usually as the primary and often only neurological symptom), slow growth, typically a neocortical localization and often, a temporal lobe predominance. In addition, many LEATs exhibit neuronal differentiation or incorporate native cortical neurones. Their slow growth rate, compatible with long survival, is likely a critical factor in the development of secondary cellular and network reorganization in the adjacent cortex or even remote sites, such as the hippocampus. All these elements may be fundamental to the patient's propensity for, and mediation of, focal seizures.
The prognosis for LEATs following surgical resection is, in the majority of cases, favorable from both perspectives of improving seizures and treating the tumor. However, in a proportion of cases, seizures continue despite surgical resection. Similarly, a minority of apparently low‐grade World Health Organization (WHO) I LEATs can behave more unpredictably and aggressively with local tumor regrowth or recurrence and rarely manifesting as anaplastic transformation to a higher‐grade lesion. Histological or molecular biomarkers that can identify such tumors likely to behave less favorably is one the key objectives of the neuropathologist's review, in order to inform appropriate patient management.
In recent years, within the framework of LEATs and the revised WHO classification of central nervous system (CNS) tumors (167), there has been recognition of new, often rare, tumor types. In addition, occasional tumors have hybrid or mixed features that are more difficult to categorize, for example, tumors with mixed features of ganglioglioma and dysembryoplastic neuroepithelial tumor (DNT), suggestive of a histological spectrum. Other neoplasms, which meet the clinical and radiological criteria for a LEAT, lack specific pathological or diagnostic features of currently recognized entities in the WHO classification, for example, diffusely growing cortical tumors with cytological similarities to DNT. One aim of this review is to summarize the current tumor terminology and the pathological features of LEATs with attention to the newer entities and to highlight current limitations in the classification systems. In addition, we review the putative stem cell origins of LEATs and associated adjacent cortical and reorganizational changes that characterize some of these lesions. Identification of pro‐epileptogenic cellular mechanisms (either within the tumor or perilesional cortex) has been a fundamental research area in recent years and remains critical to our understanding of the processes of epileptogenesis in LEAT and to guiding appropriate surgical management.
LEATS: GLIONEURONAL TUMORS (GNT)
Gangliogliomas and gangliocytomas
Gangliogliomas are one of the more common tumors in epilepsy surgical series (Table 1) and the best studied of the glioneuronal tumor group; gangliocytomas, by comparison, are much less frequently reported (Table 1). By definition, both contain nodular or compact aggregates of dysplastic neurones (Figure 1A). Ganglion cell tumors with more diffuse growth patterns have occasionally been reported (230) (Figure 1C) where the differential diagnosis may include cortical dysplasia. Although arising in any location, gangliogliomas overall strongly favor the temporal lobe with a slight male predominance (302) (Figure 2). The dysplastic neurone has overt ganglioid morphology often with an enlarged, distorted and multipolar cell body, vesicular nucleus with prominent nucleolus, demonstrable Nissl substance and sometimes, bi‐ or multinucleation. Immunohistochemistry typically demonstrates synaptophysin (often peri‐membranous), neuronal nuclear antigen (NeuN) (weak to negative), neurofilament, chromogranin and microtubule‐associated protein 2 (MAP2) positivity to varying degrees (Table 2). CD34‐positive multipolar cells and processes typically aggregate and surround the abnormal neurones in up to 80% of cases (Figure 1B) and frequent calbindin labelling of dysmorphic ganglion cells has been noted (272). The proliferative glial component of ganglioglioma varies in proportion, type [more often astrocytic but oligodendroglial in up to 23% (221)] and distribution (30). Degenerative changes, such as calcification, are often observed in ganglioglioma (Figure 3) and lymphocytic infiltrates are particularly notable.
Table 1.
The relative incidence of tumor types in reported epilepsy surgical series. Abbreviation: DNT = dysembryoplastic neuroepithelial tumor.
| Glial tumors | Glioneuronal tumors | Other tumors | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number in series [%] | Number in series [%] | Number in series [%] | ||||||||||
| Series (n = number of cases) Era of collection | Low‐grade glioma ≡ diffuse astrocytoma Grade II | Angiocentric glioma | Pilocytic astrocytoma | Pleomorhic xanthoastrocytoma | Oligodendroglioma Grade II | DNT (all types) | Ganglioglioma | Gangliocytoma | DNT/Gangliogliomas | Glioneuronal tumor uncertain type | All GNT % of series | Other tumors |
| NHNN, London*; Adults (n = 155) 1994–2010 | 16 [10.3] | 4 [2.5] | 8 (7 GII; 1 GIII) [5.1] | 88 [56] | 12 [7.7] | 5 [3.2] | 10 [6.4] | 10†[6.4] | 79.7 | 1 ependymoma | ||
| 1 meningioma | ||||||||||||
| [1.2] | ||||||||||||
| Kings, London; (n = 92) 1975–1999 (116) | 7 [7.6] | 74 [80] | 6 [6.5] | 86.5 | 1 oligoastrocytoma | |||||||
| 4 hamartoma | ||||||||||||
| [5.4] | ||||||||||||
| Grenoble; all ages (n = 94) 1990–2000 (206) | 4 [4.2] | 61 [64.8] | 29 [30.8] | 95 | ||||||||
| Cleveland; adults (219) (n = 141) 1989–2009 (219) | 24 [17%] | 1 [0.7] | 2 [1.4] | 2 [1.4] | 22 [15.6} | 10 [7.1] | 38 [27] | 3 [2.1] | 14 [9.9] | 46.1 | Low‐grade mixed glioma (13), hamartoma (5), Meningioangiomatosis (2), gliofibroma (1), anaplastic ganglioglioma (1), anaplastic astrocytoma (1) [16] | |
| Cleveland; pediatric (218) (n = 129) 1989–2009 (218) | 15 [11.6%] | 3 [2.3] | 1 [0.8] | 2 [1.6] | 5 [3.9] | 17 [13.2] | 48 [37] | 3 [2.3] | 18 [14] | 66.4 | Low‐grade mixed glioma (8), hamartoma (2), meningioangiomatosis (4), other low‐grade glioma (1) [11.6] | |
| Beijing; all ages (n = 51) 2004–2008 (215) | 1 [2] | 4 [7.8] | 10 [19.6] | 19 [37.7] | 13 [25] | 82.3 | Meningioangiomatosis (1) [2] | |||||
| Illinois; (n = 39) 1981–2005 (240) | 4 [10.5] | 3 [7.9] | 3 [7.9] | 10 [26.3] | 14 [36] | 62.3 | Mixed oligoastrocytoma (3) | |||||
| Astroblastoma (1) [10.5] | ||||||||||||
| Erlangen, German epilepsy database‡ (n = 1354) | 63 (4.7) | 3 (0.2) | 76 (5.6) | 35 (2.6) | 29 (2.1) | 246 (18.2) | 669 (49.4) | 5 (0.4) | 52 (3.8) | 71.8 | Meningiomas (21) [1.6] | |
| Cysts (22) [1.6] | ||||||||||||
| Anaplastic gliomas (97) [7.2] | ||||||||||||
| Isomorphic astrocytomas (19) [1.4] | ||||||||||||
| Ependymomas (4) [0.3] SEGA (13) [1.0] | ||||||||||||
| Summary data (ranges between centers) (%) | 5–17 | 0–2 | 1–6 | 1–8 | 2–16 | 7–80 | 6–49 | 0–3 | 2–6 | 6–25 | 46–95 | |
These are unreported data taken from the epilepsy database kept at the National Hospital for Neurology and Neurosurgery London (NHNN) and includes patients who have been investigated through the epilepsy surgical program during adulthood for drug‐resistant focal epilepsy and where a surgical procedure was carried out.
In these cases, the glioneuronal tumor was difficult to unequivocally classify, mainly because of the small size of specimen.
Unreported data from the German Epilepsy Brain Bank, Erlangen. The DNT and ganglioglioma group form the main tumor categories in most series, albeit with marked variation between centers as highlighted in bottom row. In the first three series, the category of diffuse DNT is included in the DNT numbers. If all glioneuronal tumor types (all DNT and ganglioglioma) are considered together (column all GNT as % of series) the data of relative incidence appear more consistent.
Figure 1.
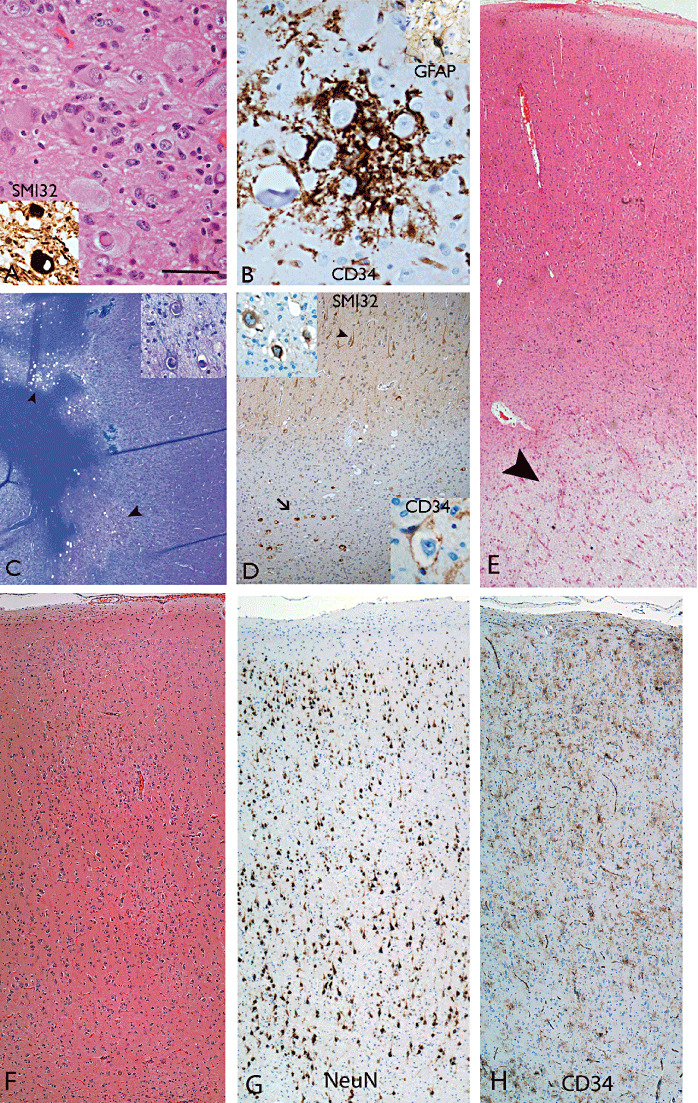
Ganglioglioma. A. Aggregates of atypical ganglion cells of varying size typically with neurofilament positivity demonstrated (inset) are diagnostic criteria. B. Clusters of CD34 positivity cells around dysplastic neurones are often identified with scattered intermingled astrocytic cells (inset). C and D. LEAT with features of diffuse ganglion cell tumor in the temporal lobe with cortex on right side comprising diffuse and subcortical nodular aggregates (arrowhead in C) of neuronal islands with atypical neurones (inset in C) within islands but no glial component (Luxol fast blue/cresyl violet preparation). In the same case (D), neurofilament highlights normal orientation of cortical neurones (arrowhead) as well as the single dysmorphic white matter neurones (arrow in main picture and top inset); CD34 was focally expressed around abnormal neurones (bottom inset). E. A mixed glioneuronal tumor with rarefaction and pallor of the white matter beneath a diffusely infiltrated cortex (arrowhead) but without cavitation or cystic change. (F–H) Cortex adjacent to a ganglioglioma. F. H&E shows a disrupted cortical architecture which may mimic a cortical dysplasia. G. NeuN reveals the vestiges of an overrun cortex with residual lamination, including layers II and IV, recognizable. H. CD34 staining reveals scattered multipolar cells, including in layer I. Scale bar A, B = 40 µm; C = 100 µm; D–H = 115 µm. H&E = hematoxylin and eosin; LEAT = long‐term epilepsy associated tumor; NeuN = neuronal nuclear antigen.
Figure 2.
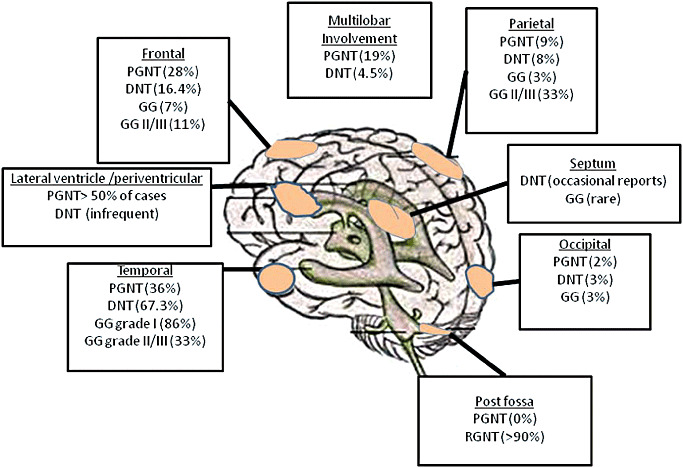
Diagram of the relative distribution of glioneuronal long‐term epilepsy associated tumors (LEATs). DNT = dysembryoplastic neuroepithelial tumor; GG = Ganglioglioma; PGNT = papillary glioneuronal tumor; RGNT = Rosette‐forming glioneuronal tumor.
Table 2.
Some of the more common and useful immunohistochemical patterns reported in subtypes of long‐term epilepsy associated tumors (LEATs). Abbreviations: EMA = epithelial membrane antigen; GFAP = glial fibrillary acidic protein; MAP2 = microtubule‐associated protein 2; NeuN = neuronal nuclear antigen.
| GFAP | Nestin | S100 | NeuN | Synaptophysin | Neurofilament | Chromogranin A | Beta III tubulin | NCAM | Calbindin | Olig2 | PDGFRα | MAP2 | CD34 | CD133 | EMA | cytokeratins | p53 | bcl2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Papillary glioneuronal tumor | + | +(97) | + | + | + | + | +(148) | + | +(278) | +(270) | +(97) | + | − | +(105) | − | − | −125, 188 | −(125) | |
| Rosette forming glioneuronal tumor of the IVth ventricle | + | + | −/+4, 248, 257 | + | −4, 257 | −(4) | +170, 299 | +/−(4) | −(279) | −(4) | |||||||||
| Glioneuronal tumor with neuropil islands | + | + | + | +(271) | −(142) | −(142) | −(142) | +20, 220 | |||||||||||
| Ganglioglioma | + | + | +/− | + | + | + | +(272) | + | + | − | − | ||||||||
| Dysembryoplastic neuroepithelial tumor | + | + | +/− | +/− | +/− | +/− | +/− | +(272) | + | + | − | − | |||||||
| Pleomorphic xanthoastrocytoma | + | + | +/− | + | + | ||||||||||||||
| Angiocentric glioma | + | + | − | − | − | + | − | ||||||||||||
| Neurocytic oligodendroglioma | + | + | + | + | + | + | +/− |
Selective cellular labeling within the tumor may be observed (see text for details of staining patterns). References are included for the more unusual or less commonly reported observations. (+) = positive labeling has been demonstrated in tumor elements; (−) = tumor usually negative; (+/−) = labeling noted in some, but not all tumors/reports.
Figure 3.
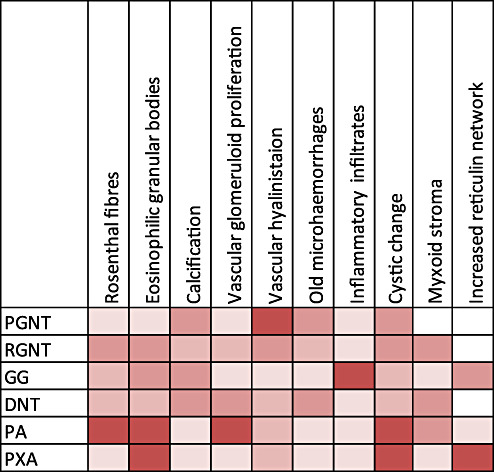
The relative occurrence of degenerative pathological features in long‐term epilepsy associated tumors (LEATs). These common features attest to the slow tumor growth and progressive accumulation of more “ancient” features. The chart is shown as a five‐tier system from: 1 (palest white shade) = never reported; 2 (next lightest shade) = rare reports; 3 (mid shade) = infrequent feature; 4 (darker shade) = often (observed in around half of cases approximately); 5 (darkest shade) = observed in majority of cases. DNT = dysembryoplastic neuroepithelial tumor; GG = Ganglioglioma; PA = pilocytic astrocytoma; PGNT = papillary glioneuronal tumor; PXA = pleomorphic xanthoastrocytoma; RGNT = Rosette‐forming glioneuronal tumor.
Gangliogliomas are largely WHO grade I neoplasms with a favorable prognosis following surgery and over 90% of patients have recurrence‐free, long survivals. It is therefore important to consider this diagnosis in the examination of a low‐grade tumor in a patient with a long history of epilepsy and not to overlook a minor ganglion cell component. Equally important, however, is that overdiagnosis of ganglioglioma can arise because of misinterpretation of a conventional glioma overrunning and distorting (often enlarging) cortical neurones. It has been recently shown that there is poor inter‐observer reliability, even between experienced pathologists, in differentiating infiltrative gliomas from classic gangliogliomas on hematoxylin and eosin (H&E) sections, with agreement rates of only 55% (118). NeuN immunohistochemistry is advocated as superior to H&E in accentuating relatively evenly, intermittently distributed neurones, often with vestiges of laminar ordering, in a cortex overrun by tumor.
A small proportion of gangliogliomas display a more aggressive biology with recurrence and progression. Histological diagnosis of atypical and anaplastic ganglioglioma is based on the identification of significant mitotic activity and elevated Ki67 index (typically >10%) or other features, including microvascular proliferation and necrosis (168). The incidence of de novo anaplastic change (WHO grade III) was 5% in one large series, involving the glial but not the neuronal component (30). Gangliogliomas that are anaplastic at the time of presentation less frequently involve the temporal lobe than their grade I counterparts (Figure 2), and are more common in young males 179, 256, but seizures are still the most frequent presenting sign (139). The rate of anaplastic transformation of previous grade I ganglioglioma was 1 in 177 tumors (0.6%) in one series (179) and 1 in 38 (2.6%) in a pediatric series (77). Predicting those gangliogliomas that are more likely to progress is imperative. Clinico‐pathological risk factors include age over 40, a nontemporal lobe tumor, incomplete resection and histological atypia. IDH1 R132H mutations were observed in 8% of a cohort of 98 gangliogliomas and associated with advanced age, increased atypia or anaplasia and worse outcome (118). It remains possible that IDH1 mutant‐positive cases actually represent conventional gliomas misdiagnosed as gangliogliomas. Nevertheless, this study highlights that IDH1 screening, as well as being a histological modifier, provides a useful adjunct and important prognostic information for management guidance (118).
Overall, regarding treatment, there is evidence of improved seizure control following early surgical intervention of gangliogliomas (302). Gross total resection is recommended where feasible and a recent review of 402 patients supported a role for radiotherapy following partial resection, even for low‐grade ganglioglioma (225). By contrast, other studies have shown that radiotherapy does not influence overall survival in anaplastic ganglioglioma (256) and good outcome following gross tumor resection alone (139).
Regarding diagnostic molecular‐genetic signatures, BRAF mutations have been identified in up to 50% of gangliogliomas 71, 251 linking this tumor with pleomorphic xanthoastrocytoma (PXA) and pilocytic astrocytoma (Table 3). Recent reports of oligodendrogliomas that harbor foci of ganglion cell differentiation have been described 213, 301. In order to distinguish these from oligodendroglial gangliogliomas, analysis of tumoral CD34 expression and 1p/19q status may be helpful (see section on oligodendrogliomas). Gangliogliomas have been reported in patients with neurofibromatosis type 1 (238). Despite similarities to cortical tubers, molecular analysis of TSC1&2 genes have not identified mutations in gangliogliomas (205). Common genetic aberrations included loss of 9p and gain of 7q (306) in one study. Gene‐expression studies in gangliogliomas have the potential to highlight differences between other glioneuronal tumors and to identify intercellular signaling pathways relevant to tumor formation and seizures as well as distinct signatures for tumor types (83). Abnormalities of the reelin‐signaling pathway in gangliogliomas, involving Cdk5, DCX and dab1, has been shown (25), although no mutations in dab1 or p35 (137) were identified. Gene expression profiles in gangliogliomas utilizing microarray showed altered expression of inflammatory proteins, cell adhesion and proliferation molecules compared with control tissues (12). Furthermore, a recent study demonstrated enhanced Pi3K‐mTOR signaling pathway activation in gangliogliomas, critical to cell growth, which was not seen in DNT (37).
Table 3.
Selected molecular genetic abnormalities with diagnostic utility in the investigation of LEAT (%). Abbreviations: DNT = dysembryoplastic neuroepithelial tumor; MGMT = O6‐methylguanine‐DNA methyltransferase; IDH1; IDH2 = isocitrate dehydrogenase.
| Molecular abnormalities | DNT | Ganglioglioma | Papillary GNT | Rosette‐forming GNT | Pilocytic astrocytoma | Astrocytoma Grade II | Glioneuronal tumor with neuropil islands | Oligodendroglioma Grade II | Pleomorphic xanthoastrocytoma | Angiocentric glioma |
|---|---|---|---|---|---|---|---|---|---|---|
| 1p/19q co‐deletion | 0%–13% 36, 93, 222, 272 | 0% (223) | 0% 1p (n = 10) (270) | 0% (n = 1) (299) | 0% (223) | 0%–10% 19, 40, 262 | 12% (n=8) (20); 0% (n=1) (2) | 39%–80% 27, 40, 46, 62, 129, 144. | 0% 140, 223 | 0% (223) |
| 0% 19q (n = 4) (188) | 100% (n=1) (91) | |||||||||
| P53 mutation | 0% 71, 93 | 0% 71, 294 | 0% 57, 195 14% | ∼50% 49, 87, 135, 144, 197 | 5%–13% 130, 144, 197. | 2%–25% *, 101, 140, 208, 309. | _ | |||
| (207) | ||||||||||
| MGMT hypermethylation | 75% (n = 4) cases (188) | 0% 104, 234 | 11%–45% 40, 80, 147, 190, 193, 289. | 60%–93% 40, 70, 81, 185, 193, 288. | 18% (182) | _ | ||||
| BRAF V600E mutation | 0% 71, 251 | 50% (n=18) (71) | 0% (n = 2) (188) | Absent BRAF KIAA 1549 fusion (98) | 9% (n = 97) (251) 1 | 0% (n = 57) (251) | 2% (n = 64) (251) | 66% (n=64) (251) | _ | |
| 18% (n=77) (251) | 70% (n = 70); BRAF fusion (152). | 28% (n = 18) BRAF fusion (89) | ||||||||
| 0% (n=18) BRAF fusion (71) | ||||||||||
| IDH1 mutations | 0%–4% 36, 272 | 5% (n = 18) (71) | 0% †, *, (188) | 0% (n = 2) 263, 299, | 0% 40, 152. | 60%–90% 17, 51, 110, 144, 152 | 100% (n=12) (121) | 50%–100% 17, 51, 52, 110, 144. | 0% 17, 84. | _ |
| 8.2% (n = 98) (118) | ||||||||||
| EGFR gene amplification | O% (272) | 0% (4/4) (188) | 0% (n = 1) (2) | |||||||
| PTEN loss | 4% (n = 74) (272) | 50%*, †, (188) |
Including tumors with anaplastic features.
Detected by immunohistochemistry.
n = number of cases in referenced study.
Dysembryoplastic neuroepithelial tumours (DNT)
This tumor virtually always presents with seizures, particularly drug‐resistant complex partial seizures and is frequently, albeit variably, represented in LEAT tumor series (Table 1). The term DNT was coined in the recognition of their early presentation and slow progression in a quasi‐hamartomatous fashion with a composition of mixed neuroepithelial cell types including astrocytes and oligodendrocyte‐like cells (OLCs). The tumors have an intracortical, nodular growth pattern and a predilection for temporal lobe (Figure 2). A pathognomonic feature present in simple and complex DNT is the glioneuronal element in which a specific cortical growth pattern of microcolumns of OLC often aligned along vessels with an intervening myxoid matrix and floating neurones are observed (Figure 4A,B). It is arguable whether the identification of the glioneuronal element, although a relatively specific feature, is an absolute criterion for the diagnosis of DNT. It was noted in 59% of cases in the original series and 73% of all reported DNT, as recently reviewed (272). Differences in diagnostic criteria between centers may go some way to explain the variation in the relative incidence of DNT between epilepsy surgical centers (Table 1). Furthermore, there is long‐standing controversy in the acceptance of a diffuse form of DNT which lacks the nodular architecture of conventional DNT but is otherwise clinically and immunophenotypically similar. Tumor with diffuse growth pattern, where included in classifications, represent between 28% and 35% of all DNT 36, 64, 116, 272. One of the main reservations for its recognition is the potential overinclusion of conventional gliomas, including oligodendrogliomas and astrocytomas. However, lack of IDH1 mutation was recently shown in a study of nine cases and in only 2 of 29 diffuse DNT in another study which suggests that the majority are molecularly distinct from conventional low grade gliomas 36, 272.
Figure 4.
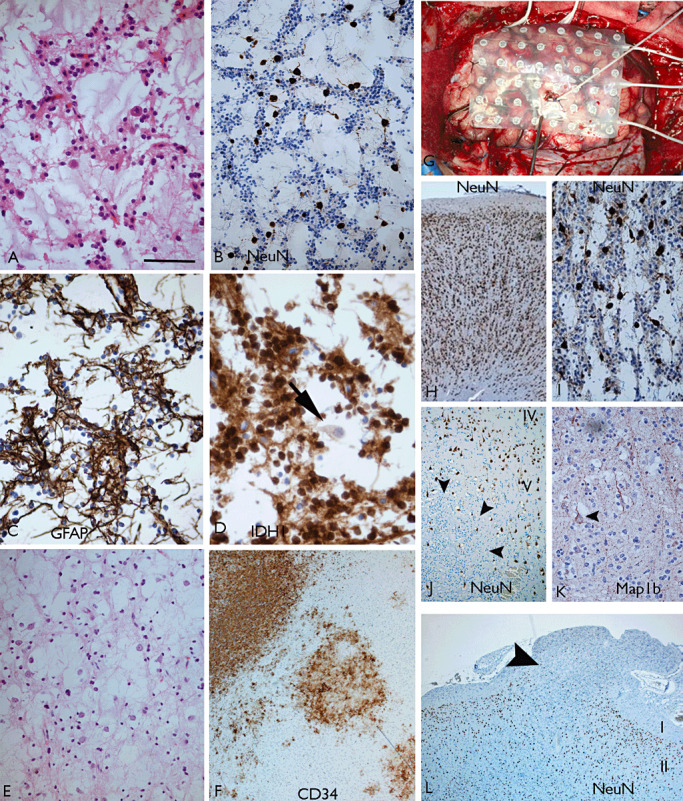
Dysembryoplastic neuroepithelial tumor (DNT). Simple‐type DNT with a typical glioneuronal element as visualized with (A) H&E, (B) NeuN and (C) GFAP; in this case, IDH1 mutation was present with positivity for mutant protein in oligodendroglial‐like cells (OLCs), while floating neurones (arrow in D) were immunonegative. This tumor occurred in a patient with a 5‐year history of seizures and a temporal lobe lesion on neuroimaging compatible with a DNT. E. Another simple DNT which had 1p 19q LOH, which is a rare finding. F. Complex multinodular DNT with 1p19q LOH and CD34 highlighting the nodular architecture. G–I illustrate a simple DNT in which intracranial recordings were carried out using subdural grids. The cortical sample illustrated in (H) was from an area with rapid firing discharges and showed normal architecture and no neoplasm whereas a more electrically quiet area (illustrated in I) revealed the DNT. J. A DNT intracortical nodule of NeuN‐negative OLCs (arrowheads) in which entrapped layer V neurones are identified with NeuN. In addition, the entrapped “floating” neurones within DNT‐specific elements display layer‐appropriate cortical laminar markers and normal morphology and orientation, as shown here with Map1b (arrowhead) (K). L. Where a complex DNT overruns the superficial cortex and spills into the overlying leptomeninges, NeuN reveals the underlying residual cortical neurones with occasional NeuN‐positive cells within the meningeal tumor extension (arrowhead, Layers I and II shown). Scale bar = A–E 70 µm; F 80 µm; H,L 120 µm; I–K = 75 µm. GFAP = glial fibrillary acidic protein; H&E = hematoxylin and eosin; NeuN = neuronal nuclear antigen.
The lineage markers commonly reported in OLC of DNT are arguably more supportive of neurocytic than oligodendroglial differentiation 116, 206, 272, 296 (Table 2). However, it is not infrequent to encounter an individual case where immunohistochemical support of neuronal differentiation using conventional markers is not secured, the diagnosis nevertheless based on the distinctive histological features. The original hypothesis of DNT arising from residual multipotential precursor cell types in the subpial or marginal zone niche remains attractive, but as yet unproven, with inconclusive evidence for expression of developmentally regulated proteins or progenitor cell markers such as Pax6, Tbr1, doublecortin and delta‐glial fibrillary acidic protein (δ‐GFAP) (272). Frequent tumor localization in regions known to harbor multipotential cells in adulthood, including the dentate gyrus, amygdala, superficial temporal cortex and occasionally in periventricular regions 61, 287, would seem more than a tantalizing coincidence but progenitor cells with DNT‐initiating properties remain to be identified.
Degenerative features, such as tumoral calcification, are relatively common (Figure 3) and an underrecognized feature is dysmyelination or rarefaction of the white matter in the vicinity of a DNT 36, 272. The mature neuronal components of DNT, including the “floating” neurones, lack the dysmorphism exhibited in gangliogliomas, tend to express markers appropriate to corresponding cortical layer (107) and retain normal orientation suggesting they are “entrapped” (Figure 4J,K). This is supported by a rare case of simple DNT that was immunopositive for mutant IDH1, where the floating neurons were perceived as negative compared with positive OLC (Figure 4D), implying that they are non‐neoplastic elements. By contrast, in glioneuronal tumors with neuropil islands (see below), both the neuronal and glial elements show positive immunolabeling for mutant IDH1, in support of divergent differentiation (121). The “neuronal entrapment” hypothesis of DNT, however, becomes more difficult to validate when mature neurones are incorporated in extracortical tumor extensions into the leptomeninges (Figure 4L) and ventricles (16).
The majority of DNT behave in a benign fashion as slowly growing WHO grade I tumors. However, tumors with more accelerated growth rates have long been recognized (191). In addition, recurrence or regrowth of tumors has been reported following surgical removal, more often when the initial tumors are partially resected but also in a few cases following gross total resection 85, 177, 183, 232. Of interest, recurrence of DNT has been reported more often with extratemporal tumors. There are just six reported cases documenting progressive changes accompanied by accumulative atypical or anaplastic features to a higher‐grade tumor 74, 103, 241, 252, 272. These include oligoastrocytoma grade II, anaplastic astrocytoma and glioblastoma with one report of an anaplastic glioneuronal tumor (272). Overall, the chance of malignant transformation of a DNT is likely to be less than 1%. Transformation was associated with postoperative adjuvant treatment in two cases following the initial resection 232, 241 and in another case with late initial surgical resection and focally elevated Ki67 index (272). Extended follow‐up periods in the postoperative periods are therefore probably justified, particularly following partial tumor resection, extratemporal DNT and cases with elevated Ki67.
Reported seizure‐free outcomes following surgery vary from 62% to 100% 116, 268. There are reports of late seizure recurrences, even in the absence of tumor regrowth (50), and in a recent study with longer follow‐up periods of 10 years, 42% of patients had remained seizure free (68). Partial resections have been associated with continued seizures 55, 194 often necessitating further surgery. In terms of histological predictors of a seizure‐free outcome, there is no evidence that tumor type (simple, complex, vs. diffuse) influences outcome. Comparison between pediatric and adult series does not disclose obvious outcome differences, although single reports suggest that older age and longer duration of epilepsy are associated with poorer outcomes (194). There are also inconsistent reports regarding the influence of associated cortical dysplasia in predicting seizure freedom 159, 242 and extratemporal DNT has been associated with greater risk of continued seizures (55).
Papillary glioneuronal tumor (PGNT)
PGNT is a rare glioneuronal tumor first described by Kim and Suh in 1997 (143). There have since been 58 cases of PGNT reported in the literature, appearing mainly as single case reports 1, 15, 22, 39, 42, 43, 44, 53, 56, 69, 78, 82, 97, 105, 124, 125, 128, 131, 148, 150, 176, 188, 192, 217, 226, 265, 270, 274, 278, 282, 298. The PGNT shows consistent histological features and was recognized in the WHO 2007 classification as an entity distinct from ganglioglioma (168). The PGNT arises in a supratentorial location. The most common sites (based on information from published reports and the unreported case illustrated in Figure 5) are temporal (36%), frontal (28%), parietal (9%) and occipital lobe (2%) with 19% of cases showing multilobar involvement on magnetic resonance imaging (MRI) at presentation (Figure 2). In over half the cases, a periventricular location was noted with an intraventricular origin in three cases (trigone and third ventricle) 97, 270.Typical MRI features show a cystic enhancing lesion with solid areas and often a mural nodule.
Figure 5.
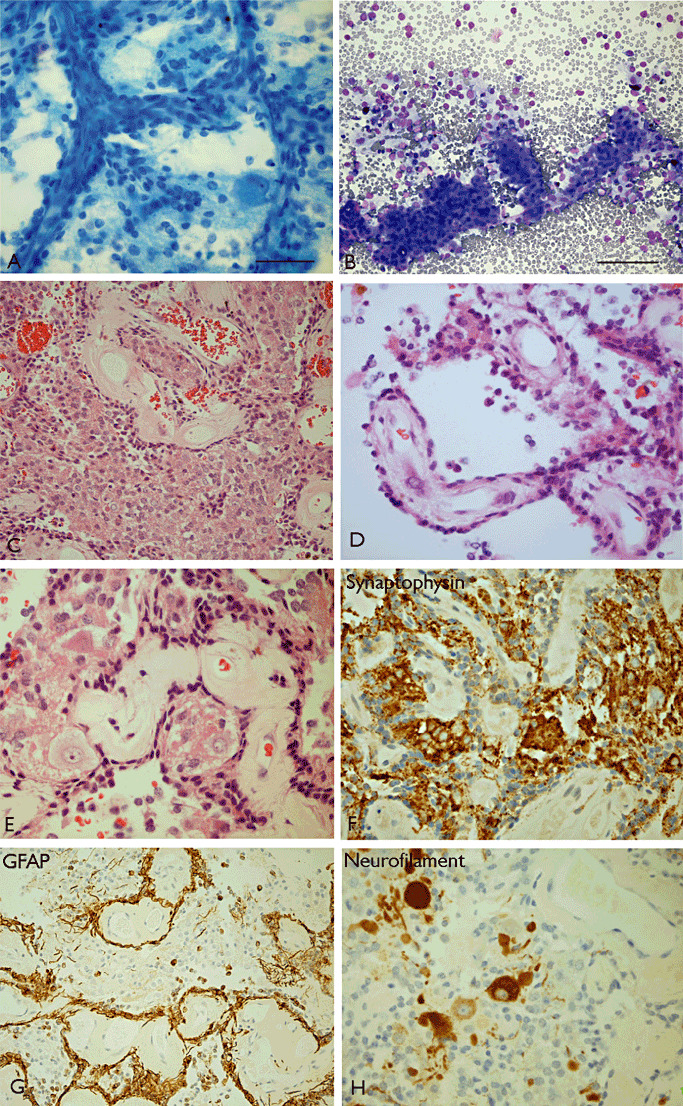
Papillary glioneuronal tumor (PGNT). A cystic temporal lobe tumor in a 32‐year‐old male which has shown no recurrence 11 years later. A. Shows papillary projections visible on smear with adherent cells and intervening ganglion cells (toludine blue) and similar composition was noted in cytospin preparations of aspirated cyst fluid (Giemsa) (B). C. Histology showed solid areas composed of mixed glial‐papillary structures and intervening neurocytes. In area D, pseudopapillary architecture was visible. E. Ganglion cells admixed with smaller neurocytes were clearly identified in interpapillary regions and (F) extensive synaptophysin labeling was visible. (G) GFAP expression was restricted to the perivascular layer of cells immediately adherent to the papillae and (H) neurofilament stain highlighted the ganglion cells only. Scale bar = 35 µm. GFAP = glial fibrillary acidic protein.
PGNTs tend to be tumors of young adults with mean age at presentation of 25.9 years (range 4–75 years). A history of seizures was recorded in 34.5% of the reported PGNT, increasing to 53% of patients where the tumor was localized to the temporal lobe. However, in the majority of the reports where information is provided, the seizure history was short (less than 1 month), with epilepsy of 2 years or more reported in only three cases (and therefore strictly qualifying as a LEAT) 56, 105. Several reports in fact highlight a relatively sudden onset of neurological symptoms in the presentation of PGNT 148, 274. There is no evidence to support that seizures are more often seen with pediatric PGNT (105).
Histologically the two components, represented in varying proportion, include the papillary glial elements composed of a central hyalinized vascular core with an outer layer of plump astroglia and an intervening solid neuronal component composed of sheets of neurocytes, ganglioid (features intermediate between neurocytes and ganglion cells) and ganglion cells set within a neuropil matrix (148)(Figure 5). The papillae, or more correctly pseudopapillae, are formed by artifactual tissue dehiscence between the neuronal and glial components and lack the orientation of glial cellular processes observed in ependymal pseudo‐rosettes (Figure 5G). Homer Wright rosettes are generally absent in PGNT with larger “neuropil‐rosette” structures only occasionally reported 188, 282. The tumor is often, but not always (148), sharply demarcated from adjacent parenchyma 125, 217. In contrast to other glioneuronal tumors, cortical dysplasia has not been described in association with PGNT.
The astrocytic component lining the papillae are GFAP and nestin positive, with some reports of co‐expression of synaptophysin 105, 143, 278. However, neuronal proteins are largely demonstrated in the interpapillary regions with diffuse synaptophysin staining, and neurofilament expression restricted to the ganglioid and full‐blown ganglion cells (Table 2; Figure 5H). Cytokeratin and epithelial membrane antigen (EMA) are negative. A “non‐specific” cell type has been described in PGNT, distinct from astrocytes and neurocytes with a mini‐gemistocyte or rhabdoid‐like appearance. Olig2 and PDGFRα labeling has also been demonstrated in a subset of cells suggesting oligodendroglial differentiation 97, 125, 270. Recent reports of co‐expression of synaptophysin and Olig2 in PGNT (82) have been used to propose an origin of these tumors from a common multipotential, oligodendroglial progenitor cell type residing in the subventricular zone. Inhibition of PDGFRα signaling has been suggested as a potential therapeutic target. Labeling of PGNT with CD133 (105) and Galectin‐3 has also been reported (188).
In general, PGNT appears to be a pure entity without hybrid tumor forms, apart from two cases which support pilocytic‐like areas in the vicinity 148, 274. Occasional eosinophilic granular bodies and Rosenthal fibers are observed in PGNT. Capillary knots of endothelial vascular proliferations have occasionally been noted (22) and in one case, an angioma‐like proliferation of small capillaries (226). Degenerative calcification and old blood pigment may be present (Figure 3). In one patient, the tumor presented with a brain hemorrhage (44) and in another, was associated with development of superficial siderosis (150) attesting to a predilection for bleeds which may be relevant to development of seizures (see section on mechanisms of epileptogenesis).
Atypical histological features are usually not evident in PGNT, with a lack of mitotic activity and Ki67 labeling index of 1%–2% in the majority of cases. In over 80% of reported cases with postsurgical follow‐up data (mean follow‐up period 3.5 years; range 0.2–19 years), there were no tumor recurrences, whether gross total or partial resections, and it is regarded as a WHO grade I neoplasm. However, recent reports have highlighted cases with atypical histological features 1, 124 and recurrent behavior requiring adjuvant treatment (192). Although it has been stated that increased mitotic activity is not directly predictive of adverse prognosis (282), of note, in five reported progressive, recurrent or fatal PGNT, elevated Ki67 indices of between 5% and 16% were observed 128, 131, 188.
The limited molecular genetic studies of PGNT (Table 3) report O6‐methylguanine‐DNA methyltransferase (MGMT) hypermethylation and phosphatase and tensin homolog (PTEN) loss but no. 1p/19q loss, isocitrate dehydrogenase (IDH1) or BRAF mutation, or epidermal growth factor (EGFR) amplification 188, 270. Gains and structural aberrations of chromosome 7 have been shown (82). A recently reported recurrent and fatal PGNT in a child was associated with PMS2 germline mutation (131).
Rosette‐forming glioneuronal tumors (RGNTs)
This rare GNT type arising near the fourth ventricle/posterior fossa is not typically associated with clinical seizures, but included here for its intriguing histological similarity to supratentorial DNT 257, 263. In fact, the first report of RGNT referred to this as “DNT of the cerebellum”(157) with its composition of a round neurocytes (resembling OLCs of DNT), formation of delicate rosettes (Figure 6A–C), alveolar growth patterns (Figure 6D), pilocytic astrocytoma‐like components and degenerative features (Figure 3). Histological differences between DNT and RGNT are cited as a lack of true synaptophysin‐positive neurocytic rosettes in DNT and the infrequency of the specific glioneuronal element with “floating neurones” in RGNT 149, 257. If floating neurones in DNT represent trapped cortical neurones rather than neoplastic elements, this could explain this difference. RGNTs have been reported in the pineal region, optic chiasm, septum pellucidum and spine 4, 92, 248, 299. Reports of DNT‐like neoplasms arising in the septum pellucidum, some presenting with seizures (16), as well as tumors with coexisting features of DNT and RGNT (169), suggest that morphologically similar tumors arising in divergent locations may be united by origin from a common progenitor cell type residing in the residual niches of the periventricular germinal matrix or marginal zone.
Figure 6.
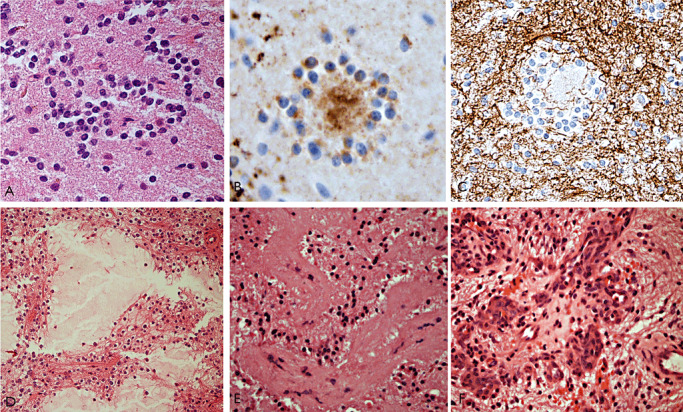
Rosette‐forming glioneuronal tumor (RGNT) of the fourth ventricle. A–C. This was the third recurrence of an RGNT in 23 years. No anaplastic features were noted but there was vascular thrombosis and hemorrhage in this specimen (not shown): Typical neurocytic rosettes are seen with (A) H&E, (B) synaptophysin and (C) GFAP. (D–F) First resection of a cerebellar vermis RGNT: D. Shows an alveolar arrangement of neurocytes around mucin‐filled cystic spaces. E. Hyalinization of vessels is often a striking feature and (F) proliferating knots of capillaries. Scale bar = 45 µm. GFAP = glial fibrillary acidic protein; H&E = hematoxylin and eosin.
Glioneuronal tumor with neuropil islands (GTNI)
GTNI, first described in 1999 (271), has histological features of diffuse fibrillary or gemistocytic astrocytic gliomas, or occasionally oligodendrogliomas, punctuated by islands or nodules of neuronal differentiation. These are characterized by synaptophysin‐positive neuropil rosettes with a circumferential rim of NeuN‐positive small neurocytes; occasional ganglion cells may be interspersed and the overall morphology is distinct from rosettes of other glial and glioneuronal neoplasms. A significant proportion of patients with GTNI present with seizures (271) either of short or long duration of several years 2, 142, 220, raising possible functional properties of the neuronal component. In these tumors, the neuronal differentiation represents the minor component and is more often 20, 271, but not always (142), nonproliferative. Some cases with proliferating neuronal islands are more reminiscent of PNET‐like differentiation pathways (276). In general, though, the progressive, anaplastic behavior reported in GTNI appears largely dictated by the astroglial element. GTNI are mainly supratentorial, with three cases showing spinal involvement which may represent disseminated tumor (91). Molecular profiles of GTNI are similar to diffuse astrocytoma with frequent IDH1 mutation (Table 3).
GTNI draws comparison to the well‐reported observation of neuronal differentiation in conventional oligodendrogliomas, including cases with clear neurocytic differentiation and Homer Wright rosettes as reported by Perry et al (212). All four patients in Perry's study presented with seizures and three of four had loss of heterozygosity (LOH) for 1p/19q. Similarly, both of the unpublished neurocytic oligodendrogliomas shown in (Figure 7) also had 1p/19q LOH, although neither presented with seizures. In conclusion, there is limited data as to whether neurocytic oligodendrogliomas more often present with seizures than conventional oligodendrogliomas and whether they should be reclassified as glioneuronal tumors.
Figure 7.
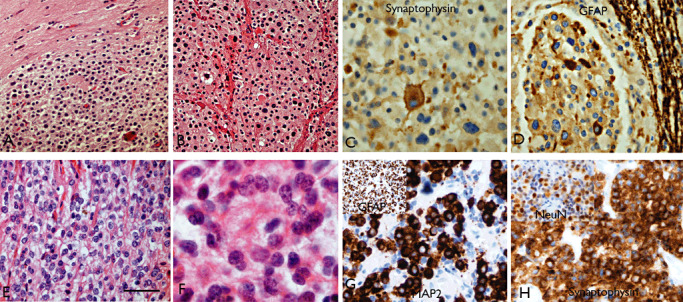
Neurocytic differentiation in oligodendrogliomas. Case 1 (A–D): Tumor composed of nodules of round cells (A,B) in areas resembling both extraventricular neurocytoma and typical oligodendroglioma, including both focal synaptophysin (C) and GFAP (D) positivity. Case 2 (E–H): oligodendroglioma‐like areas (E) and regions with neurocytic morphology and rosettes (F). There was focal expression of MAP2 (G) and GFAP (inset), in addition to neuronal markers, synaptophysin (H) and NeuN (inset). Both cases showed combined 1p/19q LOH and in patient 2, there was an additional IDH1 mutation, but in neither case was there a seizure history. Scale bar A,B,E 45 µm; C,D,F,G,H 30 µm. GFAP = glial fibrillary acidic protein; MAP2 = microtubule‐associated protein 2; NeuN = neuronal nuclear antigen.
LEATS: GLIAL TUMORS
Pilocytic astrocytoma (PA)
PAs are circumscribed, slowly growing tumors that correspond to WHO grade I (167) and they occur throughout the neuraxis. The most common symptoms are associated with increased intracranial pressure; however, supratentorial PAs have been shown to be frequently associated with chronic epilepsy (90). Accordingly, PAs are included within the common histological entities encountered in series of tumor‐associated epilepsy cases 172, 218, 293 (Table 1). The radiological (21) and histological features of PAs are characteristic, the latter most often including biphasic growth patterns, bipolar astrocytes and Rosenthal fibers. Other features that are variably present in PAs are degenerative changes, including hyalinized blood vessels, calcification, infarct‐like necrosis and chronic inflammation with lymphocytic infiltrates (Figure 8). In addition, there is evidence of microglial activation (120), together with an up‐regulation of immune system‐related genes (145).
Figure 8.
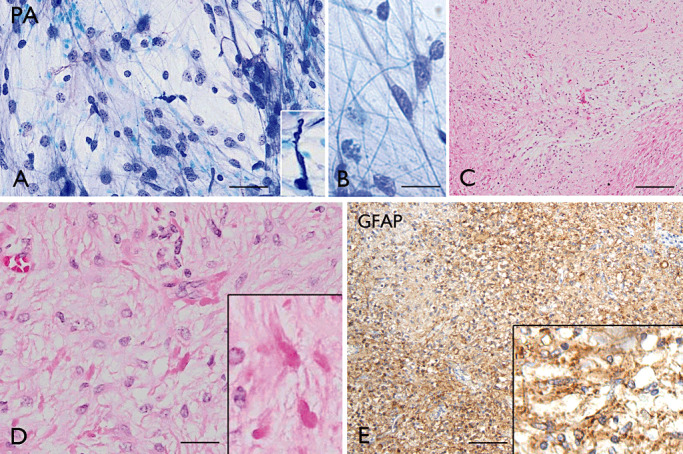
Pilocytic astrocytoma (PA). A–B. (Toluidine blue): intraoperative smear preparations of PA showing cells with fine pilocytic (hair‐like) processes and regular round‐oval nuclei; insert in A: a Rosenthal fiber. C–E: Typical biphasic histological pattern consisting of compact areas and loose, hypocellular areas (C–D, H&E); Rosenthal fibers are shown in D and GFAP immunoreactivity in E. Scale bars: A, D 40 µm; B, 20 µm; C, 160 µm; E, 80 µm. GFAP = glial fibrillary acidic protein.
Overlapping histologic features between PAs and the glial component of ganglioglioma (GG) can make the differential diagnosis difficult. PAs may occasionally contain neurons that appear to be part of the neoplasm 30, 47. In these cases, the discrimination between preexisting neuronal cells and dysplastic neurons can be critical and may require additional immunocytochemical analysis; for example, the absence of CD34 immunoreactivity combined with MAP2‐postive neoplastic glial cells supports the diagnosis of PA (30).
Classical PAs have generally a favourable prognosis after gross total resection alone (172); long‐term survival and good seizure outcome of supratentorial PAs have also been reported within epilepsy‐associated tumor cases 172, 218, 293; for reviews, see 29, 228 (Table 1). Histologic progression represents a rare finding in PAs; however, PAs with anaplastic features have been recently reported and these tumors have been associated with decreased patient survival times when compared with typical PA 201, 239. Worse prognosis is also associated with pilomyxoid astrocytoma (WHO grade II), a variant of PA mainly localized to the hypothalamic/chiasmatic region [although may also occur in other regions, such as in the temporal lobe; (273)] of infants and characterized by a monomorphic cytology, prominent myxoid matrix, lack of a compact component and an angiocentric growth pattern, generally without Rosenthal fibers or eosinophilic granular bodies.
Understanding the histogenesis and molecular pathogenesis of PAs, as well as the genetic changes associated with tumor recurrence or malignant transformation, represents a major challenge in the field of pediatric neuro‐oncology. Several studies have demonstrated clear differences in gene expression profiles between PAs and diffusely infiltrating gliomas, suggesting similarities with fetal astrocytes, as well as with oligodendrocyte progenitor cells 41, 59, 106, 120. A more recent study suggests that PAs may arise from a single population of glial progenitors during fetal or early postnatal life (209).
Most of the genetic changes commonly encountered in diffuse astrocytomas are rarely detected in PAs 57, 195, 207. Previous cytogenetic studies have shown a normal karyotype in the vast majority of both adult and paediatric cases 199, 244. Jones et al (133) showed a nonrandom pattern of genetic alteration in PAs, demonstrating chromosomal gains which increased with age and more frequently affected chromosomes 5 and 7 (followed by 6, 11, 15, and 20).
However, over the last few years, rapid progress has also been made in elucidating the molecular pathogenesis and recent studies have highlighted the importance of MAPK pathway activation in PAs [for reviews, see 73, 75, 108]. Rearrangements or mutations of the BRAF oncogene (a member of the RAF family of serine/threonine proteins involved in the RAS‐RAF‐MEK‐ERK‐MAP kinase signaling pathway) are believed to represent the mechanism leading to MAPK pathway activation in PAs 58, 134, 214, 251. Several studies have shown that BRAF fusion is more frequent in cerebellar than in extra‐cerebellar tumors 89, 117, 127, 134, but the study of Schindler et al (251) indicates that, contrary to BRAF fusion, BRAF V600E mutation was strongly associated with extracerebellar location (251). Interestingly, BRAF has been shown to be mutated in a subgroup of pediatric low‐grade glial tumors, including not only PAs, but also desmoplastic infantile ganglioglioma, ganglioglioma and pleomorphic xanthoastrocytoma 71, 251 (Table 3). BRAF rearrangements do not seem to be associated with PAs with different behavior and they remain to be determined if clinical outcome is influenced by BRAF status 112, 117. Although BRAF rearrangement or mutation appears to predominate in sporadic PAs (308), a BRAF V600E mutation has been reported in an NF1 patient, suggesting that more than one RAS/ERK pathway component can be affected in PAs (76). A recent study suggests that activation of the PI3K/AKT, in addition to MAPK/ERK signaling pathways, may influence the biological behavior of PAs, mediating the increased proliferative activity observed in anaplastic tumors (236).Mutations in the isocitrate dehydrogenase (IDH1 and IDH2) genes were not detected in PA. Thus, combined molecular analysis of BRAF and IDH1 may represent a sensitive and highly specific approach to separate pilocytic astrocytoma from diffuse astrocytoma 40, 152.
Pleomorphic xanthoastrocytoma (PXA)
PXA is an astrocytic tumor that predominantly occurs in children and young adults and usually has a relatively favorable behavior, corresponding histologically to WHO grade II (167). PXAs account for less than 1% of all astrocytic tumors. PXA has a typical superficial meningo‐cerebral location (Figure 9A), often with involvement of the temporal lobe (167). Patients with PAs often present with seizures and are frequently represented in epilepsy surgery series within the spectrum of LEATs (172, 218, 293; for reviews, see 29, 228 (Table 1).
Figure 9.
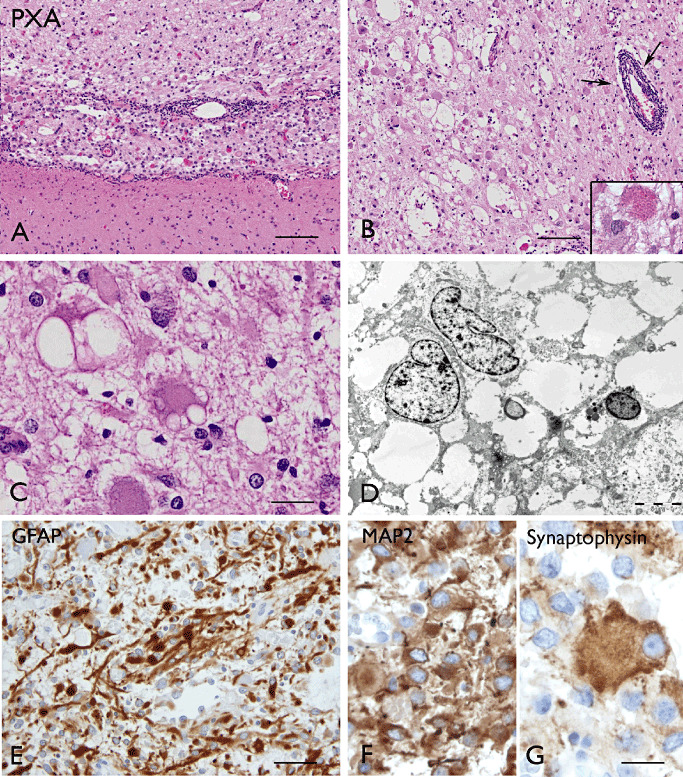
Pleomorphic xanthoastrocytoma (PXA). A–C (H&E): A: superficially located PXA well demarcated from the underlying cerebral cortex. Panels B–C: PXA with cellular pleomorphism, xanthomatous changes and eosinophilic granular bodies (insert in B); arrows in B show perivascular lymphocytic infiltrates. The xanthomatous changes consist of large cells with accumulation of lipid droplets (C). Panel D.(electron microscopy) shows vacuolated cytoplasm in PXA cells. GFAP (E), MAP2 (F)and synaptophysin (G) immunostaining in PXA cells. Scale bars: A–B, 160 µm; C, F 40 µm; D, 5 µm; E, 80 µm; G, 20 µm. GFAP = glial fibrillary acidic protein; MAP2 = microtubule‐associated protein 2.
Most of the lesions are cystic and nodular, often consisting of a mural nodule within the cyst. They are usually superficially located and attached to the meninges and the cyst wall is often well demarcated from the surrounding cerebral cortex (167). Histopathologically, PXAs are characterized by cellular pleomorphism and xanthomatous changes (167) (Figure 9C). Typical tumor cells in PXAs are large astroglial cells with abundant eosinophilic cytoplasm and large, often bizarre and sometimes multilobated nuclei; nuclear inclusions can be also observed. The xanthomatous changes consist in the presence of large cells with accumulation of lipid droplets and these cells are also GFAP positive; eosinophilic granular bodies are frequent (Figure 9). The prominent reticulin network is also characteristic and can be demonstrated with silver impregnation staining, as well as by immunocytochemistry with collagen IV and by electron microscopy (EM) as pericellular basal lamina; lymphocytic infiltrates may be present in PXA (Figure 9B). Mitoses are rarely seen and the MIB‐1 labeling index is generally lower than 1% (167). High mitotic activity (five or more mitoses per 10HPF) and necrosis are uncommon at presentation and the term of “PXA with anaplastic features” has been used 24, 99, 171.
The tumor cells in PXA display the ultrastructural features of astroglial cells, along with immunoreactivity for GFAP and S‐100. However, a neuronal pattern of differentiation has been reported in a subset of PXAs, including with expression of neuronal markers such as synaptophysin and MAP‐2 102, 122, 158, as well as neuronal features ultrastructurally (114). CD34 expression has also been observed (30). Thus, the differential diagnosis with GG may be particularly difficult on stereotactic biopsy. The lack of a clear neuronal dysplastic component, together with the MAP2‐positivity of neoplastic astrocytes, may help to differentiate these two entities (Table 2).
A possible origin from subpial astrocytes was suggested in the past (141), based on the superficial location of the tumor and the ultrastructural features of tumor cells with prominent pericellular basal lamina. However, the features suggesting neuronal differentiation, as well as the reported association with cortical dysplasia, support a possible developmental origin from multipotential neuroectodermal precursor cells or from a preexisting region of cortical dysplasia 122, 158, 218. PXA has been reported in NF1 patients and can be included in the broad spectrum of astroglial tumors that can occur in NF1 109, 156, 202. A case of temporal lobe PXA has been recently reported in a patient with DiGeorge syndrome/velocardiofacial syndrome (DGS/VCFS), a rare congenital genetic disorder resulting from a constitutional microdeletion at chromosome 22q11.2 (187).
Previous cytogenetic studies have shown complex karyotypes in PXA, with translocations involving the long arm of chromosome 1, gains on chromosomes 3, 4, 5, 7, 19, 20 and X, and losses on chromosomes 9, 17, 18, 20 and 22 164, 247, 305. Kaulich et al (140) showed that CDK4, MDM2 and EGFR genes were not amplified in any of the 62 tumors analyzed, indicating differences between the chromosomal and genetic aberrations of PXAs and those typically associated with diffuse astrocytomas. Weber et al (290) reported homozygous 9p21.3 deletions, involving the CDKN2A/p14(ARF)/CDKN2B loci. TP53 mutations were also reported in some PXAs 101, 208, 309. The methylation of the MGMT gene promoter does not appear to be frequent in PXA (182). Mutations in the IDH1 gene were not detected in PXAs 17, 84. In contrast, BRAF V600E mutations are common 71, 89, 251 (Table 3).
Surgery remains the mainstay of treatment and PXAs have generally a favorable prognosis, as indicated by overall survival rates of 81% at 5 years and 70% at 10 years 100, 203, 269. Long‐term survival and good seizure outcome of PXAs have also been reported within epilepsy‐associated tumor cases (172). However, recurrence and anaplastic transformation may occur and close follow‐up is recommended because of their relatively unpredictable biological behavior (181).
Diffuse astrocytoma
Diffuse astrocytomas represent slowly growing, diffusely infiltrating tumors that correspond histologically to WHO grade II. They occur throughout the CNS, but most commonly arise in the cerebral hemispheres of young adults (frontal and temporal cerebral lobes). Diffuse astrocytomas account for 10%–15% of all astrocytic tumors (167). Seizures represent a common presenting symptom and these tumors are represented in epilepsy surgery series within the spectrum of LEATs 172, 218, 293; for reviews, see 29, 228 (Table 1). There is evidence that an initial presentation with seizures can influence long‐term survival (23). Histopathologically, diffuse astrocytomas consist of well‐differentiated tumor astrocytes (fibrillary, protoplasmic or gemistocytic). The cellularity is moderately increased compared with control cortex, but mitotic activity, necrosis, and microvascular proliferation are lacking (167).
Previous clinico‐neuropathological studies have shown differences between astrocytomas with a very long history of seizures and those with a very short history of seizure, proposing a subtype presenting with better prognosis in patients with chronic epilepsy, the so‐called “isomorphic astrocytoma”34, 253. The latter were characterized by low cellularity, lack of mitotic activity and highly differentiated astroglial elements infiltrating the brain parenchyma; the MIB‐1 labeling was less than 1% and there was no evidence of p53, glial MAP2 or CD34 expression (34). Moreover, this subgroup of astrocytomas showed significantly better survival and lower recurrence rate, similar to those of PAs (253).
Diffuse astrocytoma has been reported in patients with Li–Fraumeni syndrome, characterized by a p53 germline point mutation (267). Accordingly, TP53 mutations are frequently encountered in diffuse astrocytomas 49, 87, 135, 144 and shorter survival has been reported for patients with low‐grade gliomas harboring such mutations (144).Comparative genomic hybridization (CGH) analysis has additionally revealed frequent gains of 7q, 5p, 8q, 9 and 19p, and losses of 1p, 19q and Xp in diffuse astrocytomas 14, 115, 254. A recent study using high‐resolution CGH points to genetically distinct subsets of diffuse low‐grade gliomas and suggests that tumors with complex chromosomal aberrations are often more aggressive (62). Methylation of MGMT gene promoter has been reported in diffuse astrocytoma and has been considered a prognostic and predictive marker 147, 190, 193, 289. IDH1 mutations are common in diffuse low‐grade glioma, with high frequency in diffuse astrocytomas 17, 51, 110, 144. The predictive value of IDH1 mutation in WHO grade II gliomas is, however, still a matter of debate; such mutations have been associated with a more favorable outcome in grade II gliomas 72, 245; however, another study indicates that the presence of IDH1/2 mutations are not prognostic on either univariate or multivariate analyses (144). Visualization of the most frequent IDH1 mutation with an antibody specifically detecting mutant IDH1‐R132H protein has been shown to be useful in differentiating reactive from neoplastic cells in grade II gliomas (49). In a recent study, IDH1 mutations were detected in 76% of WHO grade II astrocytomas (but no BRAF fusions), whereas in PA, BRAF fusion was observed in 70% of cases (but no IDH1 mutations), suggesting the combined molecular analysis of BRAF and IDH1 to differentiate PA from diffuse astrocytoma (152). BRAF V600E mutations were found in PA, PXA and GG, whereas glioblastomas and other diffuse gliomas, including WHO grade II astrocytomas, were characterized by a low frequency or absence of mutations 250, 251 (Table 3).
Oligodendroglioma
Oligodendrogliomas represent diffusely infiltrating tumors that correspond to WHO grade II and most commonly arise in the cerebral hemispheres of adults in the frontal, temporal, parietal and occipital lobes (with a ratio of about 3:2:2:1); they account for 5%–6% of all gliomas (167). Seizures represent a common presenting symptom and these tumors are represented in epilepsy surgery series within the spectrum of LEATs 172, 218, 293; for reviews, see 29, 228 (Table 1).
Oligodendrogliomas have an infiltrative growth patterns that often produce an expansion of the involved gyrus with some cases infiltrating the leptomeninges. Calcification and intratumoral hemorrhages are not uncommon. Histopathologically, oligodendrogliomas are moderately cellular, diffuse infiltrating tumors composed of glial cells with uniform nuclei, small amounts of cytoplasm and minimal cell processes. In paraffin sections, these cells display characteristic perinuclear halos (“honeycomb” morphology). Also characteristic is the capillary vascular pattern with thin‐walled and branching capillaries (“chicken wire” pattern). Microcalcifications are common and microcystic changes can also be observed. Subpial accumulations of tumor cells, as well as perineuronal satellitosis, are commonly observed. Prominent mitotic activity, microvascular proliferation and/or necrosis are not features of WHO grade II oligodendrogliomas; the MIB‐1 labeling is generally less than 5% (167).
EM examinations may detect cells types with different ultrastructural features, including typical oligodendroglial cells with large round nuclei and scant cytoplasm, cells with abundant cytoplasm rich in ribosomes and glycogen granules, and the minigemistocyte containing abundant intermediate filaments 154, 165, 167, 246.
Immunocytochemistry has a limited role in the identification of oligodendroglial tumor cells. GFAP can be expressed in both reactive astrocytes and in some oligodendroglial tumor cells (ie, minigemistocytes and gliofibrillary oligodendrocytes). The oligodendroglial lineage‐associated markers, Olig‐1 and Olig‐2, are in fact expressed in different types of gliomas (196), including astrocytomas; as such, they represent good glioma markers in general, but are not specific to oligodendrogliomas as was originally hoped. The MAP2 is expressed in both astrocytic and oligodendroglial neoplasms, although a distinct pattern of immunoreactivity has been reported in oligodendroglioma (33). In addition, the immunocytochemical profile of oligodendroglial tumor cells is further complicated by the expression of neuronal markers 285, 291, 296 (Table 2). Whether these tumors arise from the neoplastic transformation of mature oligodendrocytes or, as suggested by experimental data, from progenitor cells, is still a matter of discussion 41, 260.
Familial clustering of oligodendrogliomas is rare and only a few cases have been reported 26, 88, 155. Point mutations of TP53 are not common in familial oligodendrogliomas (267). TP53 mutations have been detected in sporadic oligodendrogliomas, but with a lower frequency compared with other low‐grade gliomas (diffuse astrocytomas and oligoastrocytomas) 130, 144, 197. LOH studies have demonstrated that most oligodendrogliomas lose both the 1p and 19q chromosomal arms (up to 80% of tumors), which is considered the hallmark alteration in these tumors. Loss of 1p/19q has been shown to represent not only a diagnostic marker, but also a well‐recognized prognostic marker associated with prolonged survival and chemosensitivity in patients with oligodendroglial tumors 27, 40, 46, 62, 129, 144. In addition, 10q LOH has been suggested to decrease survival in patients with oligodendroglial tumors, irrespective of 1p/19q LOH status (229). Whether some of these cases represent the small cell variant of glioblastoma rather than true oligodendrogliomas remains controversial. Hypermethylation of multiple genes, including the MGMT gene promoter, has also been reported in oligodendrogliomas 70, 81, 185, 288. In terms of seizures predicting outcome in oligodendroglioma, it was associated with better survival in one series (184)
IDH1 mutations are common in oligodendrogliomas, with high frequency in both oligodendrogliomas and oligoastrocytomas 17, 51, 52, 110, 144. Thus, IDH1 mutation may represent a useful marker for the diagnosis of diffuse WHO grade II glioma when the differential diagnosis includes other considerations, such as pilocytic astrocytomas, ependymomas, glioneuronal tumors and non‐neoplastic brain tissue. Moreover, immunocytochemistry with the antibody that specifically detects R132H‐mutated IDH1 protein has been shown to be useful in the diagnosis of oligodendrogliomas with neurocytic differentiation and in the difficult discrimination of these tumors from extraventricular neurocytomas (52). A recent study, addressing the frequency of various combinations of genetic alterations in low‐grade diffuse glioma, showed that survival in patients with 1p/19q ± IDH1/2 mutation was significantly longer than that of patients with TP53 mutation ± IDH1/2 mutation (144).
BRAF V600E mutations were found in PA, PXA and GG, whereas glioblastomas and other diffuse gliomas, including WHO grade II oligodendroglioma, were characterized by a low frequency or absence of mutations 250, 251 (Table 3).
Angiocentric gliomas (AG)
AG represents a rare slowly growing cerebral tumor that has been recognized by the WHO as a grade I tumor (167). AGs often occur in children and young adults and are more frequently identified in the setting of chronic epilepsy 13, 89, 94, 119, 174, 178, 180, 186, 218, 223, 224, 258, 275, 286 (Table 1). AGs have a cerebrocortical location, often with involvement of the fronto‐parietal and temporal lobe (167).
On MRI, AGs appear as well‐circumscribed solid, hyperintense, nonenhancing cortical lesions often with involvement of the subcortical white matter; intrinsically high signal on T1‐weighted images and a stalk‐like extension to the ventricle are characteristics of AGs; necrosis and calcification are rare findings 162, 174, 178, 224, 275, 286.Macroscopically, AGs may produce an expansion of the involved region with blurring of anatomical structures. Histopathologically, AGs consist of monomorphic, bipolar spindled cells with elongated nuclei and with a characteristic angiocentric arrangements around cortical blood vessels, forming perivascular pseudorosettes with an ependymoma‐like appearance (167) (Figure 10). However, variations in the histopathological features (cell shape and arrangement) have been reported (13). Mitoses, necrosis and microvascular proliferation are usually not observed.
Figure 10.
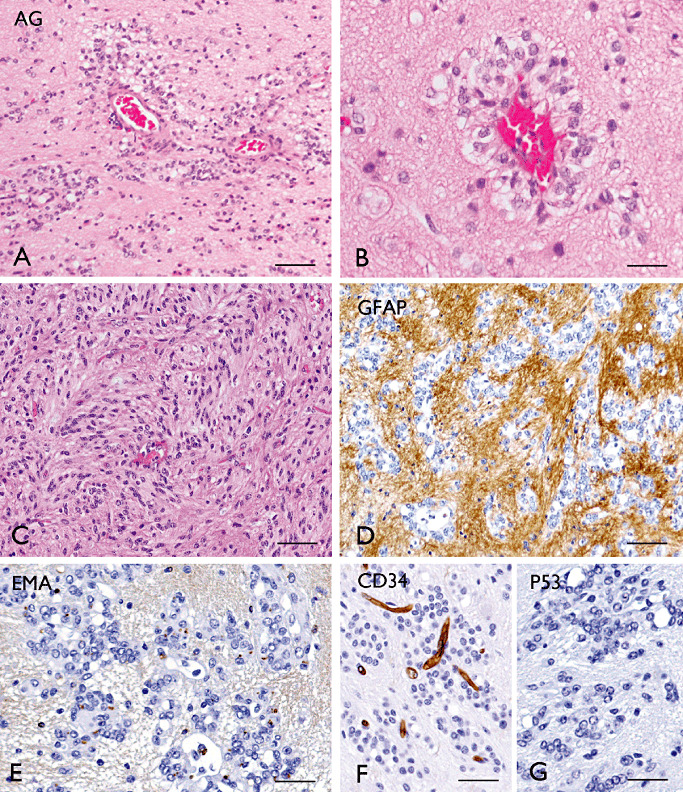
Angiocentric gliomas (AG). A–B (H&E): AG with characteristic perivascular pseudorosette growth pattern. C. Compact areas with elongated tumor cells. GFAP (D), EMA (E; “dot‐like” staining), CD34 (F, negative in tumor cells), p53 (G) immunostaining in AG; (images A, B, D–G, kindly provided by Dr. A.J. Becker Department of Neuropathology; University of Bonn Medical Centre, Germany). Scale bars: A: 160 µm; B: 40 µm. C–G: 80 µm. EMA = epithelial membrane antigen; GFAP = glial fibrillary acidic protein.
The MIB‐1 labeling ranges from 1% or less to 5% (generally 1% or less) (167). A case with anaplastic features on recurrence, including an increased labeling index of 10%, has been previously reported (286). More recently, Li et al (163) reported a series of three AGs with elevated proliferative indices (∼10%) and increased mitotic activity at presentation. Nevertheless, these cases followed a benign clinical behavior with protracted recurrence‐free survival after 6 years of follow‐up (163).
Immunohistochemistry for GFAP, S‐100 and vimentin are positive, while neuronal markers (such as synaptophysin, chromogranin and NeuN) and p53 are negative (Table 2). Expression of EMA with a “dot‐like” cytoplasmic pattern is frequently detected in the tumor cells forming rosettes (Figure 10E). Immunohistochemistry and EM provide evidence of mixed astrocytic and ependymal differentiation 224, 286.
The histogenesis of AGs is still unclear and a matter of debate. The morphological, immunohistochemical and EM features, described previously, have supported the hypothesis that AGs could arise from an ependymal precursor cell (161). However, the typical cortical location of these tumors argues against an origin from native ependymocytes or tanycytes. Another hypothesis suggests that AGs may arise from bipolar radial glial during the early stages of corticogenesis 162, 186. Accordingly, the reported association with cortical dysplasia supports the developmental origin of these tumors, possibly from multipotential neuroectodermal precursor cells (218). CGH has revealed a focal chromosomal loss involving 6q24‐q25 as the only alteration in 1 of 8 cases; in addition, high‐resolution screen by array‐CGH has identified, in 1 of 3 cases, a copy number gain at 11p11.2 containing the protein‐tyrosine phosphatase receptor‐type gene (224). In the few AG cases evaluated, loss of 1p/19q has not been shown (223). Surgery represents the treatment of choice and AGs have a favorable prognosis with long‐term survival and good seizure outcome 174, 178, 186, 218, 224.
IMMUNOHISTOCHEMISTRY IN THE ASSESSMENT OF LEAT
A broad panel of neuronal and glial markers is recommended in the evaluation of low‐grade tumors in epilepsy, particularly for the disclosure of any neuronal differentiation (Table 2). One marker that has been recently widely employed in the evaluation of low‐grade and malformative lesions in epilepsy is the vascular and stem cell marker, CD34. Although not uniquely expressed in LEATs, having been reported in the giant cell variant of GBM (95) and in the sarcomatous portion of gliosarcomas 237, 292, it appears to be expressed in a high proportion (70%–80%) of epilepsy‐associated tumors (31). CD34 positivity is noted in up to 80% of gangliogliomas (although typically sparing the dysplastic neurones) (30), some DNT [typically lacking in the myxoid tumor components but more often in diffuse DNT 36, 189, 272] and 84% of PXA (233). Moreover, the pattern of CD34 labeling, highlighting nodular architecture, satellite cortical peri‐tumoral micronodules and small mutipolar cells, is very typical and helpful in their evaluation, particularly in a small biopsy or marginal sample. CD34‐positive cells more often co‐express neuronal markers, such as NeuN (31) and nestin (272), than astrocytic markers. Abnormal calbindin expression has been noted in a high proportion of glioneuronal tumors in epilepsy including gangliogliomas (272), although its expression in non‐LEAT CNS tumors is relatively unexplored. The phosphorylated ribosomal S6 protein (PS6) may represent an additional marker, which can be used to detect the neuronal component of ganglioglioma, indicating activation of the Pi3K‐mTOR pathway in this tumor (37). Immunocytochemical detection of adhesion molecule on glia (AMOG) shows a pattern of immunoreactivity similar to that observed for CD34 and AMOG co‐localizes with CD34 (37). Thus, both CD34 and AMOG may help to identify a population of glioneuronal precursor cells in ganglioglioma.
MOLECULAR DIAGNOSTICS IN LEAT
Although the current gold standard method for the diagnosis of LEATs still relies on histological evaluation, the recent knowledge in the underlying molecular pathogenesis of these tumors provides new interesting tools, not only for understanding their histogenesis, but also to improve their diagnosis. Thus, assessment of specific molecular markers could represent a useful approach to separate the different entities (Table 3).
Mutations in IDH1 and IDH2 genes are common in grade II diffuse gliomas (∼70% to 80%) regardless of cell type 17, 51, 52, 110, 144. IDH1 and IDH2 mutations are generally not detected in PA and gangliogliomas 40, 71, 152, although a mutation has been reported in one of 18 gangliogliomas (with prominent astrocytic component) analyzed (71). Thus, IDH1 mutations may represent a useful marker for the differential diagnosis between WHO grade II astrocytoma or oligodendroglioma and grade I gliomas, such as PA and ganglioglioma. In contrast to IDH1 and IDH2 mutations, BRAF V600E mutations were found in PA and GG, whereas glioblastomas and other gliomas, including diffuse astrocytomas WHO grade II, were characterized by a low frequency or absence of mutations 250, 251. Thus, combined molecular analysis of BRAF and IDH1 may represent a sensitive and highly specific approach to separate a PA or ganglioglioma from diffuse astrocytoma 40, 152. Molecular analysis of BRAF and IDH1 can also be useful in the differential diagnosis between diffuse astrocytoma and PXA, as in the latter tumor, mutations in the IDH1 gene were not detected 17, 84, but analysis of BRAF V600E mutation has revealed high mutation frequencies (71, 89, 251.
Very little is known about the molecular pathogenesis of DNTs. The few genetic studies performed on limited numbers have argued against the contribution of mutational events in genes with pathogenic relevance in other glial tumors (e.g. TP53, EGFR, PTEN, BRAF) 71, 132, 235, 251. A more recent study in a large series of DNTs shows PTEN and IDH1 mutations in only 3 of 73 cases (272) (Figure 4). As outlined previously (see paragraph on oligodendroglioma), LOH 1p/19q represents the hallmark alteration in oligodendrogliomas (up to 80% of tumors). There is some evidence that oligodendrogliomas with 1p/19q LOH present more often with seizures (3). Previous LOH studies in DNT have failed to detect 1p/19q loss, suggesting that the 1p/19q LOH favors a diagnosis of oligodendroglioma, particularly in cases in which the quality or size of the biopsy raises does not allow a definitive diagnosis based on routine histology 93, 222, 232. One should keep in mind, however, that pediatric oligodendrogliomas typically lack 1p/19q codeletions such that genetic testing is of limited assistance in this age group 153, 227. Combined 1p/19q loss has also been reported in two DNTs with evolution to grade II oligodendroglioma (103). However, more recent studies indicate the occurrence of 1p/19q loss or isolated 9q loss in a subset of DNTs, displaying a favorable biologic behavior, similar to typical grade I tumor 71, 272.
A practical limitation regarding the molecular analysis of LEATs, particularly the rarer types, is that a single center can collect relatively few cases of a particular entity compared with more common gliomas. Furthermore, tumor groups may be selected based on specific histological features which may introduce bias. Different methodologies used (with different sensitivities), as polymerase chain reaction (PCR)‐based methods and fluorescence in situ hybridization (FISH) may also influence results. Finally, a key issue in the analysis of low‐cellularity, cortically based tumors is the frequent admixture of contaminating non‐neoplastic cells, potentially leading to false‐negative findings. In summary, molecular and histological diagnoses in LEATs should be carried out as parallel assessments with neither one necessarily trumping the other. Future studies carried out in central referral centers amalgamating large collections of LEATs are likely to advance our knowledge of this area.
MECHANISMS OF EPILEPTOGENESIS
The association between epilepsy and brain tumors has been observed for over a century. In 1882, the neurologist Hughlings Jackson made the important observation that epilepsy often represents the initial and only clinical manifestation of glial tumors. In addition, Jackson was the first to recognize the relationship between tumor epileptogenicity and involvement of cortical gray matter in patients with brain tumors 126, 307. Jackson's ideas on the epileptogenicity of brain tumors have subsequently been reinforced by clinical studies emphasizing that pharmacologically intractable epilepsy critically affects the daily quality of life of patients with brain tumors, even if the tumor is otherwise under control [for review, see 67, 228, 280]. The incidence of brain tumors in patients with epilepsy is about 4% and the frequency of epilepsy in patients with brain tumors is 30% or more depending on the type of the tumor (280).
In principle, any tumor (extra‐axial, intra‐axial, benign or malignant, common or uncommon) can cause seizures. However, patients with supratentorial low‐grade glial and glioneuronal tumors are more likely to develop epilepsy 67, 280. In particular, the entities discussed previously (LEATs) are associated with long histories of pharmacologically intractable seizures, which represent the first, and in some cases, the only clinical manifestation of the tumor 30, 173, 280.
Understanding the mechanisms that underlie epileptogenesis in LEATs is essential to identify new treatment targets and to develop effective therapy. A number of hypotheses have been put forward during the past decades that could explain increased excitability in patients with brain tumors. It is likely that multiple mechanisms are involved, including both tumor‐related factors (tumor size, tumor location), as well as peri‐tumoral changes 67, 228, 280.
Tumor‐related factors
An MRI‐based morphometric study indicates that the influence of tumor size on the propensity to cause seizures varies with the grade of the tumor (160). High‐grade tumors that present with seizures are usually smaller in size than high‐grade tumors without seizures, whereas low‐grade tumors that present with epilepsy are often larger than those without associated epilepsy. The propensity to develop seizures is higher in patients with temporal, frontal or insular low‐grade tumors 160, 280. Thus, the usual localization of LEATs in the temporal and frontal lobe, often with cortical involvement, may partially explain the raised likelihood of epileptic activity in this group of tumors.
The strong association of specific histological tumor types (such as the glioneuronal tumors, ganglioglioma and DNTs) with chronic seizures suggests that the cellular composition and neurochemical profile of these tumors may be relevant for epileptogenesis. Several studies have supported the intrinsic epileptogenicity of glioneuronal tumors, indicating the presence of a hyperexcitable neuronal component in these tumors [for review, see 29, 30]. Analysis of electrocorticographic patterns in glioneuronal tumors (86) reveals that a relatively high neuronal density in the lesion is associated with highly epileptiform discharge patterns, such as continuous spiking or recruiting discharges, sustaining the hypothesis of a neuronal tumor component functionally integrated into excitatory circuitries. Immunocytochemical studies have demonstrated high expression of specific glutamate receptor (GluR) subtypes, including both ionotropic and metabotropic glutamate receptors (mGluR) in the neuronal component of ganglioglioma and DNT 7, 295, 296. These findings suggest a central role for glutamatergic transmission in the mechanisms underlying the intrinsically high epileptogenicity of glioneuronal tumors. More recent studies analyzing the gene expression profile of ganglioglioma support the existence of developmental alterations in the balance between excitation and inhibition within the lesion 12, 83, 243. Single cell analysis of the dysplastic neurons in gangliogliomas demonstrates a prominent expression of the mGluR5 (243). In contrast, several GABAa receptor (GABAAR) subunits (including α1, α5, β1, β3 and δ subunit) are down‐regulation, suggesting impairment of GABAergic inhibition 12, 243.
Increase of the sodium‐potassium chloride co‐transporter (NKCC1) expression and a reduced expression of the potassium‐chloride co‐transporter KCC2 has also been reported in gangliogliomas 10, 12. The expression patterns of NKCC1 and KCC2 resemble the expression patterns observed in immature brain and support the hypothesis of a failure of developmental maturation in the pathogenesis of these tumors. Moreover, the reported deregulation of these transporters could actively contribute to the epileptogenicity of ganglioglioma via modulation of GABA receptors (300).
Glutamate appears to play a central role in the biology of glial tumors and their epileptogenicity (65). Glial tumor cells respond to glutamate through activation of ionotropic and metabotropic glutamate receptors which have been shown to be expressed in both astroglial and oligodendroglial tumors 7, 65, 295. Glutamate receptor activation has been suggested to trigger electrical activity in human glioma cells (295). Underediting of glutamate receptor GluR2, associated with increased alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) receptor Ca2+ permeability, has been observed in gliomas and proposed as epileptogenic mechanisms in these tumor types (175). However, in high‐grade glial tumors such as glioblastoma, a down‐regulation and mislocalization of AMPA receptor has been reported, suggesting a possible mechanism to explain the lower excitability in a glutamate‐rich environment, of high‐grade tumors compared with low‐grade tumors (281).
Attention has also been focused on the deregulation of glutamate uptake and release 65, 255. Decreased expression of glial glutamate transporters has been observed in both glial and glioneuronal tumors and may contribute to tumor epileptogenesis, increasing the extracellular glutamate concentration 243, 304. Interestingly, activation of mGluR5 which is expressed in human glioma cells in vivo, as well as in vitro, has been shown to down‐regulate the expression of glial glutamate transporters in vitro 8, 96. In addition, it has been recently demonstrated that glutamate, produced and released from glioma cells via the Xc ‐ system (a Na+‐independent cysteine‐glutamate transporter), may contribute to neuronal hyperexcitability and epileptic activity 45, 65, 303.
Disturbed communication between cells, resulting from an altered expression of gap‐junction channels (connexins), may also contribute to epileptogenesis (264). Low‐grade gliomas demonstrate overexpression of connexin 43, while oligodendroglioma and glioneuronal tumors show overexpression of connexin 32 (6). An additional potential mechanism underlying tumor‐associated epilepsy is represented by an altered potassium homeostasis resulting from both reduced glial inwardly rectifying K+ channel currents and mislocalization of Kir4.1 channels 28, 38, 198. Also, the gene expression profile of ganglioglioma with low expression of several potassium channel genes suggests a disturbed ion homeostasis and transport that could lead to increased excitability in these tumor types (12).
Neuron‐glia interactions involving pro‐inflammatory molecules may also play a critical role in the generation of seizures in glial and glioneuronal tumors [for review, see 5, 67]. Both gene expression and immunocytochemical studies provide evidence for a prominent activation of the inflammatory response in glioneuronal tumors (9). In particular, consistent findings in GG include up‐regulation of inflammatory interleukins (such IL‐1β) and activation of both the complement cascade and the Toll‐like receptor pathway 12, 231. Interestingly, several observations strongly support the proconvulsant and ictogenic properties of inflammatory molecules and suggest a pathogenic role for inflammation in epilepsy [reviewed in 283, 284]. The prominent increased expression of the immune response with activation of the complement cascade and the production of pro‐inflammatory cytokines observed in brain tumors may induce alterations of the blood–brain barrier (BBB), which could play an additional role in the tumor epileptogenicity (259).
Alterations in Pi3K‐mTOR pathway components in GG have been recently reported 243, 249. Interestingly, recent studies have demonstrated the critical role of this pathway in epileptogenesis and the anti‐epileptogenic potential of mTOR inhibitors 297, 311.
Peri‐tumoral changes
The peri‐tumoral region may also be relevant for the generation and propagation of seizure activity (280). Both functional and immunocytochemical studies support the epileptogenicity of the peri‐tumoral zone, demonstrating network alterations and revealing cytoarchitectural and neurochemical changes in the cortex resected from patients with tumor‐associated intractable epilepsy 228, 280.
Abrupt tissue damage with isolation of cortical area (deafferentation) and the deposition of hemosiderin have been potentially implicated as causes of seizure activity in rapidly progressive high‐grade tumors (259). Experimental studies have shown that glioma invasion may alter the discharge properties of neighboring neurons, converting them to bursting cells, hence providing “pacemaker” cells driving the neuronal networks surrounding the tumor (146). In addition, a more recent study through system xc− activates glutamate receptors on peri‐tumoral neurons, leading to neuronal hyperexcitability and epileptic activity. Interestingly, treatment with sulfasalazine (SAS, a drug that blocks system xc−) has been shown to reduce epileptic activity in glioma‐bearing mice (45). Whether this mechanism could also play a role in the epileptogenicity of slowly growing, diffusely infiltrating, grade II glial tumors (such as diffuse astrocytoma or oligodendroglioma), requires further studies.
Hypoxia and acidosis, ionic changes, enhanced intercellular communication through increased expression of gap junction channels in the peri‐tumoral region have also been suggested as potential mechanisms affecting epileptogenesis in gliomas 6, 228, 280. Perilesional changes, involving both excitatory and inhibitory pathways, have been reported in glioneuronal tumors with, in particular, a complex alteration of the GABAergic system in patients with ganglioglioma (11). In addition, it has been recently shown that an altered neuronal expression of NKCC1 and KCC2 in peri‐tumoral tissue of glial tumors may lead to a positive shift of EGABA (depolarized reversal potential), supporting the key role of these Cl(‐) transporters in tumor‐related epilepsy (60).
Enzymatic changes have been demonstrated in peri‐tumoral tissue (259). These changes may impair neurotransmitter synthesis and storage, leading to increased neuronal excitability. In particular, the dysregulation of adenosine kinase (ADK; key metabolic enzyme for the regulation of extracellular adenosine levels) observed in peri‐tumoral infiltrated tissue of glioma patients with epilepsy supports the role of this enzyme in tumor‐associated epilepsy (66).
Finally, the potential contribution of peri‐tumoral cortical disorganization (coexistence with cortical dysplasia) has to be considered in the evaluation of the epileptogenicity of LEAT and in particular of glioneuronal tumors (35).
ASSOCIATED PATHOLOGIES INCLUDING CORTICAL DYSPLASIA
The main types of pathology coexisting with LEATs include hippocampal sclerosis and malformation, in particular focal cortical dysplasia (FCD) (223). The presence of hippocampal sclerosis in association with a LEAT is considered a dual pathology whereas FCD is not. In one large series of 30 DNTs, where mesial temporal structures were resected, the more common observation was an absence of hippocampal sclerosis; when sclerosis was present, atypical patterns of end folium sclerosis predominated over classical hippocampal sclerosis (272).
Cytoarchitectural abnormalities in the cortex adjacent to low grade glial and glioneuronal tumors 180, 242 in epilepsy have long been recognized, albeit variably reported. Previously classified as Palmini type I or II FCD (204), in the new International League Against Epilepsy (ILAE) classification they are grouped together as FCD Type IIIb (35). One interpretation of this association was that a preexisting region of cortical malformation forms the “dysembryoplastic” bed from which tumors arise. In support of this temporal relationship, however, the observation of tumors developing from a preexisting cortical dysplasia is infrequent (200) and furthermore cortical dysplasia is not always present with LEATs. The new ILAE classification of cortical dysplasias now recognizes that acquired, “re‐organisational” cortical changes can occur in the maturing postnatal brain, simulating a developmental dysplasia. It seems more likely that peri‐tumoral FCD‐like changes develop in synchrony with or secondary to the slowly growing tumor.
Common cytoarchitectural changes reported in the adjacent cortex, which may extend into the adjacent gyrus, are layer I hypercellularity, cortical (and white matter) satellite clusters of small immature cells (both can be highlighted with CD34 and may be indistinct on H&E) and a paucicellular cortex with less distinct lamination (Figure 1E). In addition, white matter rarefaction may be present, reminiscent of the developmental hypomyelination observed in FCD IIb (50) (Figure 1E). The interpretation of some of these histological features, as tumor precursor lesions or secondary tumor structures, is still speculative and further harmonization of the criteria for FCD IIIb is required. This is exemplified by the marked variation in the observation of dysplasia adjacent to tumors. For example with DNT, FCD was reported in 47% of tumors in the original series (63), but in subsequent series its recognition varies between 0% (48) and 83% of cases (268), which likely relates to observer diagnostic criteria (54). Incorporating NeuN as a recommended practice (35) in addition to H&E in the evaluation of cyto‐architecture is likely to improve accuracy in distinction of FCD from cortical disruption at tumor infiltration margins (Figure 1G). Also of fundamental importance, as raised previously, the contribution of perilesional (dysplastic) cortex to epileptogenesis is pertinent to surgical planning, with evidence both for and against this strategy (18) (see Figure 1F–H) in individual cases. This question will be best addressed through studies of large tumor series, rather than anecdotal cases, with accurate pathological and electrophysiological correlation.
FUTURE DIRECTIONS IN LEAT TERMINOLOGY
Future modifications to the current classification system, based on further advances in molecular and histological delineation of such tumors, are inevitable. This need is highlighted by the marked variation in the relative incidence in the reporting of gangliogliomas and DNT between centers (Table 1). The explanation is likely to be differences in interobserver diagnostic criteria, as discussed previously, rather than true geographical variation. Modifications are also necessary in order to incorporate currently difficult to classify tumors including mixed and nonspecific tumor types.
LEATs with mixed tumor features
“Mixed” tumors in this context refer not to evidence of both glial and neuronal differentiation at the cellular level but to “hybrid” tumors which bear typical growth patterns of different tumor entities. Transition or mixed forms of ganglioglioma and DNT have long been recognized, representing up to 2%–6% of tumors in epilepsy surgical series 36, 113, 216, 219, 261, 272. There are also reports of composite tumors with features of both PXA and ganglioglioma 79, 151, 166, 210, 266, 277, PXA with DNT (123) and PXA with an oligodendroglioma 111, 211. Cases where a PA developed within a DNT 136, 310, as well as PA in combination with a low‐grade oligodendroglioma (138), have been noted. Such combinations suggest intriguing links between these low‐grade tumors and that they might represent a phenotypic spectrum. Although the accurate grading and classification of mixed tumors are challenging, there is no clear evidence to suggest enhanced atypical behavior or adverse outcomes.
LEATs with diffuse growth pattern
It is increasingly clear that a proportion of tumors in patients with long‐standing epilepsy has diffuse (predominantly cortical) growth patterns but fall short of the classical diagnostic criteria and features of ganglioglioma, DNT or other recognized tumor type. “Non‐specific” or “diffuse” DNTs have been proposed for these tumors; however, this terminology has not been universally accepted by all centers, and many pathologists cautiously reserve the term DNT for only those cases with a “specific” glioneuronal component. Furthermore, as detailed previously, diffuse astrocytic “isomorphic gliomas” with grade I behavior have been described in epilepsy patients. This leaves us with a gap in the current WHO classification for such diffusely growing, biologically grade I LEATs.
Proposals for future LEAT terminology
Any revisions to the current classification system for LEATs will follow consensus agreement across disciplines (following the ILAE FCD taskforce format (35)), based primarily on histological criteria, but incorporating immunohistochemistry markers and novel molecular genetics data that may aid in tumor categorization. New classifications should be unambiguous and evidence based, enabling improved agreements in LEAT nomenclature between centers which, in turn, will allow reliable prognostic information to emerge. A model for future LEAT terminology is presented in Figure 11. A further future consideration is regional reorganization, including establishing centers or mechanisms for central pathology review, particularly for less conventional LEATs.
Figure 11.
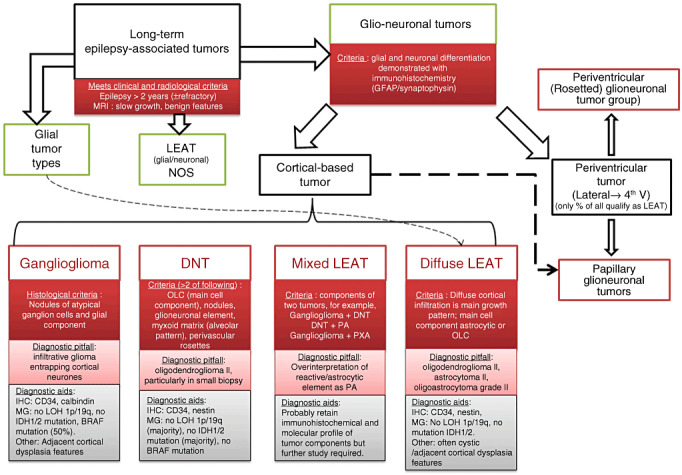
Model for a proposed future modification of the current classification of LEATs. This is based on histological criteria with addition of “diffuse LEAT” and “mixed LEAT” entities, as well as a LEAT “N.O.S.” category for tumors that remain difficult to classify. Regarding the diagnosis of DNT, these are no longer split into simple and complex subtypes, as there is no evidence to confirm clinical relevance (either in terms of tumor progression or seizure outcome) of categorizing DNT into these two groups. Furthermore, the diagnostic criteria for DNT is based on a constellation of two or more histological features and a glioneuronal element is therefore not an essential criterion. In respect to the presence of cortical dysplasia, this is regarded as a “diagnostic aid” but not a histological criterion. Similarly, molecular genetics and immunohistochemistry (other than GFAP and synaptophysin for assessment of glioneuronal differentiation) are diagnostic aids. The relative contribution of tumor components in the “Mixed LEAT” requires further clarity; however, the additional finding of PA‐like regions in DNT should no longer be regarded as a component of “complex” DNT but diagnosed as a mixed tumor type. “DNT like” tumors arising in the periventricular region are distinct from the cortical based DNTs. DNT = dysembryoplastic neuroepithelial tumor; GFAP = glial fibrillary acidic protein; IHC = immunohistochemistry; LEAT = long‐term epilepsy associated tumor; LOH = loss of heterozygosity; MG = molecular genetics; OLC = oligodendroglial‐like cells; PA = pilocytic astrocytoma; PXA = pleomorphic xanthoastrocytoma.
CONCLUSIONS
In the last several years, there have been major advances in the recognition of a wide spectrum of tumors associated with epilepsy. We have highlighted some of the current controversies in their classification. Analogous to establishing consensus criteria in the diagnosis of focal cortical dysplasias in epilepsy (32), it is imperative that similar uniformity in diagnosis is a target for LEATs and will be the subject of future ILAE taskforce groups. Identification of molecular genetic signatures that are unique or contrary to a tumor type or that unite a group of tumors, will further advance our diagnostic accuracy, terminology and biological understanding. Furthermore, establishing markers which identify cases likely to behave in a more aggressive manner, as well as mechanisms that generative seizures, is a primary research goal in our understanding of these neoplasms. A refinement of the histopathological classification of LEATs would have a great value for diagnosis and create the basis for new therapeutic strategies in refractory epilepsy.
ACKNOWLEDGMENTS
We acknowledge the support of Jane de Tisi, Shu An and Lillian Martinian of UCL, Institute of Neurology London, in the preparation of this paper. EA is supported by the National Epilepsy Funds (NEF 09‐05) and KIKA.Stichting Kinderen Kankervrij.
REFERENCES
- 1. Adam C, Polivka M, Carpentier A, George B, Gray F (2007) Papillary glioneuronal tumor: not always a benign tumor? Clin Neuropathol 26:119–124. [DOI] [PubMed] [Google Scholar]
- 2. Agarwal S, Suri V, Rishi A, Shukla B, Garg A, Sharma MC et al (2009) Glioneuronal tumor with neuropil‐like islands: a new entity. Neuropathology 29:96–100. [DOI] [PubMed] [Google Scholar]
- 3. Aldape K, Burger PC, Perry A (2007) Clinicopathologic aspects of 1p/19q loss and the diagnosis of oligodendroglioma. Arch Pathol Lab Med 131:242–251. [DOI] [PubMed] [Google Scholar]
- 4. Anan M, Inoue R, Ishii K, Abe T, Fujiki M, Kobayashi H et al (2009) A rosette‐forming glioneuronal tumor of the spinal cord: the first case of a rosette‐forming glioneuronal tumor originating from the spinal cord. Hum Pathol 40:898–901. [DOI] [PubMed] [Google Scholar]
- 5. Aronica E, Crino PB (2011) Inflammation in epilepsy: clinical observations. Epilepsia 52(Suppl. 3):26–32. [DOI] [PubMed] [Google Scholar]
- 6. Aronica E, Gorter JA, Jansen GH, Leenstra S, Yankaya B, Troost D (2001) Expression of connexin 43 and connexin 32 gap‐junction proteins in epilepsy‐associated brain tumors and in the perilesional epileptic cortex. Acta Neuropathol (Berl) 101:449–459. [DOI] [PubMed] [Google Scholar]
- 7. Aronica E, Yankaya B, Jansen GH, Leenstra S, van Veelen CW, Gorter JA, Troost D (2001) Ionotropic and metabotropic glutamate receptor protein expression in glioneuronal tumours from patients with intractable epilepsy. Neuropathol Appl Neurobiol 27:223–237. [DOI] [PubMed] [Google Scholar]
- 8. Aronica E, Gorter JA, Ijlst‐Keizers H, Rozemuller AJ, Yankaya B, Leenstra S, Troost D (2003) Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur J Neurosci 17:2106–2118. [DOI] [PubMed] [Google Scholar]
- 9. Aronica E, Gorter JA, Redeker S, Ramkema M, Spliet WG, van Rijen PC et al (2005) Distribution, characterization and clinical significance of microglia in glioneuronal tumours from patients with chronic intractable epilepsy. Neuropathol Appl Neurobiol 31:280–291. [DOI] [PubMed] [Google Scholar]
- 10. Aronica E, Boer K, Redeker S, van Rijen PC, Troost D, Gorter JA (2007) Differential expression patterns of Chloride transporters, NKCC1 and KCC2, in epilepsy‐associated malformations of cortical development. Neuroscience 145:185–196. [DOI] [PubMed] [Google Scholar]
- 11. Aronica E, Redeker S, Boer K, Spliet WG, van Rijen PC, Gorter JA, Troost D (2007) Inhibitory networks in epilepsy‐associated gangliogliomas and in the perilesional epileptic cortex. Epilepsy Res 74:33–44. [DOI] [PubMed] [Google Scholar]
- 12. Aronica E, Boer K, Becker A, Redeker S, Spliet WG, van Rijen PC et al (2008) Gene expression profile analysis of epilepsy‐associated gangliogliomas. Neuroscience 151:272–292. [DOI] [PubMed] [Google Scholar]
- 13. Arsene D, Ardeleanu C, Ogrezeanu I, Danaila L (2008) Angiocentric glioma: presentation of two cases with dissimilar histology. Clin Neuropathol 27:391–395. [DOI] [PubMed] [Google Scholar]
- 14. Arslantas A, Artan S, Oner U, Muslumanoglu MH, Ozdemir M, Durmaz R et al (2007) Genomic alterations in low‐grade, anaplastic astrocytomas and glioblastomas. Pathol Oncol Res 13:39–46. [DOI] [PubMed] [Google Scholar]
- 15. Atri S, Sharma MC, Sarkar C, Garg A, Suri A (2007) Papillary glioneuronal tumour: a report of a rare case and review of literature. Childs Nerv Syst 23:349–353. [DOI] [PubMed] [Google Scholar]
- 16. Baisden BL, Brat DJ, Melhem ER, Rosenblum MK, King AP, Burger PC (2001) Dysembryoplastic neuroepithelial tumor‐like neoplasm of the septum pellucidum: a lesion often misdiagnosed as glioma: report of 10 cases. Am J Surg Pathol 25:494–499. [DOI] [PubMed] [Google Scholar]
- 17. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol (Berl) 116:597–602. [DOI] [PubMed] [Google Scholar]
- 18. Barba C, Coras R, Giordano F, Buccoliero AM, Genitori L, Blümcke I, Guerrini R (2011) Intrinsic epileptogenicity of gangliogliomas may be independent from co‐occurring focal cortical dysplasia. Epilepsy Res 97:208–213. [DOI] [PubMed] [Google Scholar]
- 19. Barbashina V, Salazar P, Holland EC, Rosenblum MK, Ladanyi M (2005) Allelic losses at 1p36 and 19q13 in gliomas: correlation with histologic classification, definition of a 150‐kb minimal deleted region on 1p36, and evaluation of CAMTA1 as a candidate tumor suppressor gene. Clin Cancer Res 11:1119–1128. [PubMed] [Google Scholar]
- 20. Barbashina V, Salazar P, Ladanyi M, Rosenblum MK, Edgar MA (2007) Glioneuronal tumor with neuropil‐like islands (GTNI): a report of 8 cases with chromosome 1p/19q deletion analysis. Am J Surg Pathol 31:1196–1202. [DOI] [PubMed] [Google Scholar]
- 21. Barkovich AJ (2000) Pediatric Neuroimaging. Lippincott Williams & Wilkins: Philadelphia. [Google Scholar]
- 22. Barnes NP, Pollock JR, Harding B, Hayward RD (2002) Papillary glioneuronal tumour in a 4‐year‐old. Pediatr Neurosurg 36:266–270. [DOI] [PubMed] [Google Scholar]
- 23. Bauman G, Fisher B, Watling C, Cairncross JG, Macdonald D (2009) Adult supratentorial low‐grade glioma: long‐term experience at a single institution. Int J Radiat Oncol Biol Phys 75:1401–1407. [DOI] [PubMed] [Google Scholar]
- 24. Bayindir C, Balak N, Karasu A, Kasaroglu D (1997) Anaplastic pleomorphic xanthoastrocytoma. Childs Nerv Syst 13:50–56. [DOI] [PubMed] [Google Scholar]
- 25. Becker AJ, Blümcke I, Urbach H, Hans V, Majores M (2006) Molecular neuropathology of epilepsy‐associated glioneuronal malformations. J Neuropathol Exp Neurol 65:99–108. [DOI] [PubMed] [Google Scholar]
- 26. Bhatt AR, Mohanty S, Sharma V, Shukla PK (1999) Familial gliomas: a case report. Neurol India 47:136–138. [PubMed] [Google Scholar]
- 27. Bigner SH, Rasheed BK, Wiltshire R, McLendon RE (1999) Morphologic and molecular genetic aspects of oligodendroglial neoplasms. Neuro Oncol 1:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Binder DK, Steinhauser C (2006) Functional changes in astroglial cells in epilepsy. Glia 54:358–368. [DOI] [PubMed] [Google Scholar]
- 29. Blümcke I (2009) Neuropathology of focal epilepsies: a critical review. Epilepsy Behav 15 34–39. [DOI] [PubMed] [Google Scholar]
- 30. Blümcke I, Wiestler OD (2002) Gangliogliomas: an intriguing tumor entity associated with focal epilepsies. J Neuropathol Exp Neurol 61:575–584. [DOI] [PubMed] [Google Scholar]
- 31. Blümcke I, Giencke K, Wardelmann E, Beyenburg S, Kral T, Sarioglu N et al (1999) The CD34 epitope is expressed in neoplastic and malformative lesions associated with chronic, focal epilepsies. Acta Neuropathol (Berl) 97:481–490. [DOI] [PubMed] [Google Scholar]
- 32. Blümcke I, Lobach M, Wolf HK, Wiestler OD (1999) Evidence for developmental precursor lesions in epilepsy‐associated glioneuronal tumors. Microsc Res Tech 46:53–58. [DOI] [PubMed] [Google Scholar]
- 33. Blümcke I, Becker AJ, Normann S, Hans V, Riederer BM, Krajewski S et al (2001) Distinct expression pattern of microtubule‐associated protein‐2 in human oligodendrogliomas and glial precursor cells. J Neuropathol Exp Neurol 60:984–993. [DOI] [PubMed] [Google Scholar]
- 34. Blümcke I, Luyken C, Urbach H, Schramm J, Wiestler OD (2004) An isomorphic subtype of long‐term epilepsy‐associated astrocytomas associated with benign prognosis. Acta Neuropathol (Berl) 107:381–388. [DOI] [PubMed] [Google Scholar]
- 35. Blümcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A et al (2011) The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 52:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bodi I, Selway R, Bannister P, Doey L, Mullatti N, Elwes R, Honavar M (2011) Diffuse form of dysembryoplastic neuroepithelial tumour: the histological and immunohistochemical features of a distinct entity showing transition to dysembryoplastic neuroepithelial tumour and ganglioglioma. Neuropathol Appl Neurobiol doi: 10.1111/j.1365‐2990.2011.01225.x. [DOI] [PubMed] [Google Scholar]
- 37. Boer K, Troost D, Timmermans W, van Rijen PC, Spliet WG, Aronica E (2010) Pi3K‐mTOR signaling and AMOG expression in epilepsy‐associated glioneuronal tumors. Brain Pathol 20:234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bordey A, Sontheimer H (1998) Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res 32:286–303. [DOI] [PubMed] [Google Scholar]
- 39. Borges G, Bonilha L, Menezes AS, Queiroz Lde S, Carelli EF, Zanardi V, Menezes JR (2004) Long term follow‐up in a patient with papillary glioneuronal tumor. Arq Neuropsiquiatr 62:869–872. [DOI] [PubMed] [Google Scholar]
- 40. Bourne TD, Schiff D (2010) Update on molecular findings, management and outcome in low‐grade gliomas. Nat Rev Neurol 6:695–701. [DOI] [PubMed] [Google Scholar]
- 41. Bouvier C, Bartoli C, Aguirre‐Cruz L, Virard I, Colin C, Fernandez C et al (2003) Shared oligodendrocyte lineage gene expression in gliomas and oligodendrocyte progenitor cells. J Neurosurg 99:344–350. [DOI] [PubMed] [Google Scholar]
- 42. Bouvier‐Labit C, Daniel L, Dufour H, Grisoli F, Figarella‐Branger D (2000) Papillary glioneuronal tumour: clinicopathological and biochemical study of one case with 7‐year follow up. Acta Neuropathol (Berl) 99:321–326. [DOI] [PubMed] [Google Scholar]
- 43. Broholm H, Madsen FF, Wagner AA, Laursen H (2002) Papillary glioneuronal tumor—a new tumor entity. Clin Neuropathol 21:1–4. [PubMed] [Google Scholar]
- 44. Buccoliero AM, Giordano F, Mussa F, Taddei A, Genitori L, Taddei GL (2006) Papillary glioneuronal tumor radiologically mimicking a cavernous hemangioma with hemorrhagic onset. Neuropathology 26:206–211. [DOI] [PubMed] [Google Scholar]
- 45. Buckingham SC, Campbell SL, Haas BR, Montana V, Robel S, Ogunrinu T, Sontheimer H (2011) Glutamate release by primary brain tumors induces epileptic activity. Nat Med 17:1269–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Buckley PG, Alcock L, Heffernan J, Woods J, Brett F, Stallings RL, Farrell MA (2011) Loss of chromosome 1p/19q in oligodendroglial tumors: refinement of chromosomal critical regions and evaluation of internexin immunostaining as a surrogate marker. J Neuropathol Exp Neurol 70:177–182. [DOI] [PubMed] [Google Scholar]
- 47. Burger P, Scheithauer B (2007) Atlas of Tumor Pathology. Patholog AARo: (ed.), American Registry of Pathology: Washington, DC. [Google Scholar]
- 48. Burneo JG, Tellez‐Zenteno J, Steven DA, Niaz N, Hader W, Pillay N, Wiebe S (2008) Adult‐onset epilepsy associated with dysembryoplastic neuroepithelial tumors. Seizure 17:498–504. [DOI] [PubMed] [Google Scholar]
- 49. Camelo‐Piragua S, Jansen M, Ganguly A, Kim JC, Cosper AK, Dias‐Santagata D et al (2011) A sensitive and specific diagnostic panel to distinguish diffuse astrocytoma from astrocytosis: chromosome 7 gain with mutant isocitrate dehydrogenase 1 and p53. J Neuropathol Exp Neurol 70:110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Campos AR, Clusmann H, von Lehe M, Niehusmann P, Becker AJ, Schramm J, Urbach H (2009) Simple and complex dysembryoplastic neuroepithelial tumors (DNT) variants: clinical profile, MRI, and histopathology. Neuroradiology 51:433–443. [DOI] [PubMed] [Google Scholar]
- 51. Capper D, Weissert S, Balss J, Habel A, Meyer J, Jager D et al (2010) Characterization of R132H mutation‐specific IDH1 antibody binding in brain tumors. Brain Pathol 20:245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Capper D, Reuss D, Schittenhelm J, Hartmann C, Bremer J, Sahm F et al (2011) Mutation‐specific IDH1 antibody differentiates oligodendrogliomas and oligoastrocytomas from other brain tumors with oligodendroglioma‐like morphology. Acta Neuropathol (Berl) 121:241–252. [DOI] [PubMed] [Google Scholar]
- 53. Celli P, Caroli E, Giangaspero F, Ferrante L (2006) Papillary glioneuronal tumor. Case report and literature review. J Neurooncol 80:185–189. [DOI] [PubMed] [Google Scholar]
- 54. Chamberlain WA, Cohen ML, Gyure KA, Kleinschmidt‐DeMasters BK, Perry A, Powell SZ et al (2009) Interobserver and intraobserver reproducibility in focal cortical dysplasia (malformations of cortical development). Epilepsia 50:2593–2598. [DOI] [PubMed] [Google Scholar]
- 55. Chang EF, Christie C, Sullivan JE, Garcia PA, Tihan T, Gupta N et al (2010) Seizure control outcomes after resection of dysembryoplastic neuroepithelial tumor in 50 patients. J Neurosurg Pediatr 5:123–130. [DOI] [PubMed] [Google Scholar]
- 56. Chen L, Piao YS, Xu QZ, Yang XP, Yang H, Lu DH (2006) Papillary glioneuronal tumor: a clinicopathological and immunohistochemical study of two cases. Neuropathology 26:243–248. [DOI] [PubMed] [Google Scholar]
- 57. Cheng Y, Pang JC, Ng HK, Ding M, Zhang SF, Zheng J et al (2000) Pilocytic astrocytomas do not show most of the genetic changes commonly seen in diffuse astrocytomas. Histopathology 37:437–444. [DOI] [PubMed] [Google Scholar]
- 58. Cin H, Meyer C, Herr R, Janzarik WG, Lambert S, Jones DT et al (2011) Oncogenic FAM131B‐BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol (Berl) 121:763–774. [DOI] [PubMed] [Google Scholar]
- 59. Colin C, Voutsinos‐Porche B, Nanni I, Fina F, Metellus P, Intagliata D et al (2009) High expression of cathepsin B and plasminogen activator inhibitor type‐1 are strong predictors of survival in glioblastomas. Acta Neuropathol (Berl) 118:745–754. [DOI] [PubMed] [Google Scholar]
- 60. Conti L, Palma E, Roseti C, Lauro C, Cipriani R, de Groot M et al (2011) Anomalous levels of Cl(‐) transporters cause a decrease of GABAergic inhibition in human peritumoral epileptic cortex. Epilepsia 52:1635–1644. [DOI] [PubMed] [Google Scholar]
- 61. Costa MR, Kessaris N, Richardson WD, Gotz M, Hedin‐Pereira C (2007) The marginal zone/layer I as a novel niche for neurogenesis and gliogenesis in developing cerebral cortex. J Neurosci 27:11376–11388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dahlback HS, Gorunova L, Brandal P, Scheie D, Helseth E, Meling TR, Heim S (2011) Genomic aberrations in diffuse low‐grade gliomas. Genes Chromosomes Cancer 50:409–420. [DOI] [PubMed] [Google Scholar]
- 63. Daumas‐Duport C, Scheithauer BW, Chodkiewicz JP, Laws ER Jr, Vedrenne C (1988) Dysembryoplastic neuroepithelial tumor: a surgically curable tumor of young patients with intractable partial seizures. Report of thirty‐nine cases. Neurosurgery 23:545–556. [DOI] [PubMed] [Google Scholar]
- 64. Daumas‐Duport C, Varlet P, Bacha S, Beuvon F, Cervera‐Pierot P, Chodkiewicz JP (1999) Dysembryoplastic neuroepithelial tumors: nonspecific histological forms—a study of 40 cases. J Neurooncol 41:267–280. [DOI] [PubMed] [Google Scholar]
- 65. de Groot J, Sontheimer H (2011) Glutamate and the biology of gliomas. Glia 59:1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. de Groot M, Iyer A, Zurolo E, Anink J, Heimans JJ, Boison D et al (2012) Overexpression of ADK in human astrocytic tumors and peritumoral tissue is related to tumor‐associated epilepsy. Epilepsia 53:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. de Groot M, Reijneveld JC, Aronica E, Heimans JJ (2011) Epilepsy in brain tumour patients: focal epilepsy requires focused treatment. Brain doi:10.1093/brain/awr310 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 68. de Tisi J, Bell GS, Peacock JL, McEvoy AW, Harkness WF, Sander JW, Duncan JS (2011) The long‐term outcome of adult epilepsy surgery, patterns of seizure remission, and relapse: a cohort study. Lancet 378:1388–1395. [DOI] [PubMed] [Google Scholar]
- 69. Dim DC, Lingamfelter DC, Taboada EM, Fiorella RM (2006) Papillary glioneuronal tumor: a case report and review of the literature. Hum Pathol 37:914–918. [DOI] [PubMed] [Google Scholar]
- 70. Dong SM, Pang JC, Poon WS, Hu J, To KF, Chang AR, Ng HK (2001) Concurrent hypermethylation of multiple genes is associated with grade of oligodendroglial tumors. J Neuropathol Exp Neurol 60:808–816. [DOI] [PubMed] [Google Scholar]
- 71. Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ et al (2010) Activating mutations in BRAF characterize a spectrum of pediatric low‐grade gliomas. Neuro Oncol 12:621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dubbink HJ, Taal W, van Marion R, Kros JM, van Heuvel I, Bromberg JE et al (2009) IDH1 mutations in low‐grade astrocytomas predict survival but not response to temozolomide. Neurology 73:1792–1795. [DOI] [PubMed] [Google Scholar]
- 73. Dubuc AM, Northcott PA, Mack S, Witt H, Pfister S, Taylor MD (2010) The genetics of pediatric brain tumors. Curr Neurol Neurosci Rep 10:215–223. [DOI] [PubMed] [Google Scholar]
- 74. Duggal N, Taylor R, Zou GY, Hammond RR (2008) Dysembryoplastic neuroepithelial tumours: clinical, proliferative and apoptotic features. J Clin Pathol 61:127–131. [DOI] [PubMed] [Google Scholar]
- 75. Dunham C (2010) Pediatric brain tumors: a histologic and genetic update on commonly encountered entities. Semin Diagn Pathol 27:147–159. [DOI] [PubMed] [Google Scholar]
- 76. Eisenhardt AE, Olbrich H, Roring M, Janzarik W, Anh TN, Cin H et al (2010) Functional characterization of a BRAF insertion mutant associated with pilocytic astrocytoma. Int J Cancer 129:2297–2303. [DOI] [PubMed] [Google Scholar]
- 77. El Khashab M, Gargan L, Margraf L, Koral K, Nejat F, Swift D et al (2009) Predictors of tumor progression among children with gangliogliomas. Clinical article. J Neurosurg Pediatr 3:461–466. [DOI] [PubMed] [Google Scholar]
- 78. Epelbaum S, Kujas M, Van Effenterre R, Poirier J (2006) Two cases of papillary glioneuronal tumours. Br J Neurosurg 20:90–93. [DOI] [PubMed] [Google Scholar]
- 79. Evans AJ, Fayaz I, Cusimano MD, Laperriere N, Bilbao JM (2000) Combined pleomorphic xanthoastrocytoma‐ganglioglioma of the cerebellum. Arch Pathol Lab Med 124:1707–1709. [DOI] [PubMed] [Google Scholar]
- 80. Everhard S, Kaloshi G, Criniere E, Benouaich‐Amiel A, Lejeune J, Marie Y et al (2006) MGMT methylation: a marker of response to temozolomide in low‐grade gliomas. Ann Neurol 60:740–743. [DOI] [PubMed] [Google Scholar]
- 81. Ezaki T, Sasaki H, Hirose Y, Miwa T, Yoshida K, Kawase T (2011) Molecular characteristics of pediatric non‐ependymal, nonpilocytic gliomas associated with resistance to temozolomide. Mol Med Report 4:1101–1105. [DOI] [PubMed] [Google Scholar]
- 82. Faria C, Miguens J, Antunes JL, Barroso C, Pimentel J, Martins Mdo C et al (2008) Genetic alterations in a papillary glioneuronal tumor. J Neurosurg Pediatr 1:99–102. [DOI] [PubMed] [Google Scholar]
- 83. Fassunke J, Majores M, Tresch A, Niehusmann P, Grote A, Schoch S, Becker AJ (2008) Array analysis of epilepsy‐associated gangliogliomas reveals expression patterns related to aberrant development of neuronal precursors. Brain 131:3034–3050. [DOI] [PubMed] [Google Scholar]
- 84. Felsberg J, Wolter M, Seul H, Friedensdorf B, Goppert M, Sabel MC, Reifenberger G (2010) Rapid and sensitive assessment of the IDH1 and IDH2 mutation status in cerebral gliomas based on DNA pyrosequencing. Acta Neuropathol (Berl) 119:501–507. [DOI] [PubMed] [Google Scholar]
- 85. Fernandez C, Girard N, Paz Paredes A, Bouvier‐Labit C, Lena G, Figarella‐Branger D (2003) The usefulness of MR imaging in the diagnosis of dysembryoplastic neuroepithelial tumor in children: a study of 14 cases. AJNR Am J Neuroradiol 24:829–834. [PMC free article] [PubMed] [Google Scholar]
- 86. Ferrier CH, Aronica E, Leijten FS, Spliet WG, van Huffelen AC, van Rijen PC, Binnie CD (2006) Electrocorticographic discharge patterns in glioneuronal tumors and focal cortical dysplasia. Epilepsia 47:1477–1486. [DOI] [PubMed] [Google Scholar]
- 87. Figarella‐Branger D, Maues de Paula A, Colin C, Bouvier C (2011) Histomolecular classification of adult diffuse gliomas: the diagnostic value of immunohistochemical markers. Rev Neurol (Paris) 167:683–690. [DOI] [PubMed] [Google Scholar]
- 88. Flannery T, Cawley D, Zulfiger A, Alderazi Y, Heffernan J, Brett F et al (2008) Familial occurrence of oligodendroglial tumours. Br J Neurosurg 22:436–438. [DOI] [PubMed] [Google Scholar]
- 89. Forshew T, Tatevossian RG, Lawson AR, Ma J, Neale G, Ogunkolade BW et al (2009) Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol 218:172–181. [DOI] [PubMed] [Google Scholar]
- 90. Forsyth PA, Shaw EG, Scheithauer BW, O'Fallon JR, Layton DD Jr, Katzmann JA (1993) Supratentorial pilocytic astrocytomas. A clinicopathologic, prognostic, and flow cytometric study of 51 patients. Cancer 72:1335–1342. [DOI] [PubMed] [Google Scholar]
- 91. Fraum TJ, Barak S, Pack S, Lonser RR, Fine HA, Quezado M, Iwamoto FM (2011) Spinal cord glioneuronal tumor with neuropil‐like islands with 1p/19q deletion in an adult with low‐grade cerebral oligodendroglioma. J Neurooncol 107:421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Frydenberg E, Laherty R, Rodriguez M, Ow‐Yang M, Steel T (2010) A rosette‐forming glioneuronal tumour of the pineal gland. J Clin Neurosci 17:1326–1328. [DOI] [PubMed] [Google Scholar]
- 93. Fujisawa H, Marukawa K, Hasegawa M, Tohma Y, Hayashi Y, Uchiyama N et al (2002) Genetic differences between neurocytoma and dysembryoplastic neuroepithelial tumor and oligodendroglial tumors. J Neurosurg 97:1350–1355. [DOI] [PubMed] [Google Scholar]
- 94. Fulton SP, Clarke DF, Wheless JW, Ellison DW, Ogg R, Boop FA (2009) Angiocentric glioma‐induced seizures in a 2‐year‐old child. J Child Neurol 24:852–856. [DOI] [PubMed] [Google Scholar]
- 95. Galloway M (2010) CD34 expression in glioblastoma and giant cell glioblastoma. Clin Neuropathol 29:89–93. [DOI] [PubMed] [Google Scholar]
- 96. Gegelashvili G, Dehnes Y, Danbolt NC, Schousboe A (2000) The high‐affinity glutamate transporters GLT1, GLAST, and EAAT4 are regulated via different signalling mechanisms. Neurochem Int 37:163–170. [DOI] [PubMed] [Google Scholar]
- 97. Gelpi E, Preusser M, Czech T, Slavc I, Prayer D, Budka H (2007) Papillary glioneuronal tumor. Neuropathology 27:468–473. [DOI] [PubMed] [Google Scholar]
- 98. Gessi M, Waha A, Setty P, Pietsch T (2011) Analysis of KIAA1549‐BRAF fusion status in a case of rosette‐forming glioneuronal tumor of the fourth ventricle (RGNT). Neuropathology 31:654–657. [DOI] [PubMed] [Google Scholar]
- 99. Giannini C, Scheithauer BW (1997) Classification and grading of low‐grade astrocytic tumors in children. Brain Pathol 7:785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Giannini C, Scheithauer BW, Burger PC, Brat DJ, Wollan PC, Lach B, O'Neill BP (1999) Pleomorphic xanthoastrocytoma: what do we really know about it? Cancer 85:2033–2045. [PubMed] [Google Scholar]
- 101. Giannini C, Hebrink D, Scheithauer BW, Dei Tos AP, James CD (2001) Analysis of p53 mutation and expression in pleomorphic xanthoastrocytoma. Neurogenetics 3:159–162. [DOI] [PubMed] [Google Scholar]
- 102. Giannini C, Scheithauer BW, Lopes MB, Hirose T, Kros JM, VandenBerg SR (2002) Immunophenotype of pleomorphic xanthoastrocytoma. Am J Surg Pathol 26:479–485. [DOI] [PubMed] [Google Scholar]
- 103. Gonzales M, Dale S, Susman M, Nolan P, Ng WH, Maixner W, Laidlaw J (2007) Dysembryoplastic neuroepithelial tumor (DNT)‐like oligodendrogliomas or DNTs evolving into oligodendrogliomas: two illustrative cases. Neuropathology 27:324–330. [DOI] [PubMed] [Google Scholar]
- 104. Gonzalez‐Gomez P, Bello MJ, Lomas J, Arjona D, Alonso ME, Aminoso C et al (2003) Epigenetic changes in pilocytic astrocytomas and medulloblastomas. Int J Mol Med 11:655–660. [PubMed] [Google Scholar]
- 105. Govindan A, Mahadevan A, Bhat DI, Arivazhagan A, Chakraborti S, Suja MS et al (2009) Papillary glioneuronal tumor‐evidence of stem cell origin with biphenotypic differentiation. J Neurooncol 95:71–80. [DOI] [PubMed] [Google Scholar]
- 106. Gutmann DH, Hedrick NM, Li J, Nagarajan R, Perry A, Watson MA (2002) Comparative gene expression profile analysis of neurofibromatosis 1‐associated and sporadic pilocytic astrocytomas. Cancer Res 62:2085–2091. [PubMed] [Google Scholar]
- 107. Hadjivassiliou G, Martinian L, Squier W, Blümcke I, Aronica E, Sisodiya SM, Thom M (2010) The application of cortical layer markers in the evaluation of cortical dysplasias in epilepsy. Acta Neuropathol (Berl) 120:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hargrave D (2009) Paediatric high and low grade glioma: the impact of tumour biology on current and future therapy. Br J Neurosurg 23:351–363. [DOI] [PubMed] [Google Scholar]
- 109. Hariharan S, Donahue JE, Garre C, Origone P, Grewal RP (2006) Clinicopathologic and genetic analysis of siblings with NF1 and adult‐onset gliomas. J Neurol Sci 247:105–108. [DOI] [PubMed] [Google Scholar]
- 110. Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A et al (2009) Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1010 diffuse gliomas. Acta Neuropathol (Berl) 118:469–474. [DOI] [PubMed] [Google Scholar]
- 111. Hattab EM, Martin SE, Shapiro SA, Cheng L (2011) Pleomorphic xanthoastrocytoma and oligodendroglioma: collision of 2 morphologically and genetically distinct anaplastic components. J Neurosurg 114:1648–1653. [DOI] [PubMed] [Google Scholar]
- 112. Hawkins C, Walker E, Mohamed N, Zhang C, Jacob K, Shirinian M et al (2011) BRAF‐KIAA1549 fusion predicts better clinical outcome in pediatric low‐grade astrocytoma. Clin Cancer Res 17:4790–4798. [DOI] [PubMed] [Google Scholar]
- 113. Hirose T, Scheithauer BW (1998) Mixed dysembryoplastic neuroepithelial tumor and ganglioglioma. Acta Neuropathol (Berl) 95:649–654. [DOI] [PubMed] [Google Scholar]
- 114. Hirose T, Giannini C, Scheithauer BW (2001) Ultrastructural features of pleomorphic xanthoastrocytoma: a comparative study with glioblastoma multiforme. Ultrastruct Pathol 25:469–478. [DOI] [PubMed] [Google Scholar]
- 115. Hirose Y, Aldape KD, Chang S, Lamborn K, Berger MS, Feuerstein BG (2003) Grade II astrocytomas are subgrouped by chromosome aberrations. Cancer Genet Cytogenet 142:1–7. [DOI] [PubMed] [Google Scholar]
- 116. Honavar M, Janota I, Polkey CE (1999) Histological heterogeneity of dysembryoplastic neuroepithelial tumour: identification and differential diagnosis in a series of 74 cases. Histopathology 34:342–356. [DOI] [PubMed] [Google Scholar]
- 117. Horbinski C, Hamilton RL, Nikiforov Y, Pollack IF (2010) Association of molecular alterations, including BRAF, with biology and outcome in pilocytic astrocytomas. Acta Neuropathol (Berl) 119:641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Horbinski C, Kofler J, Yeaney G, Camelo‐Piragua S, Venneti S, Louis DN et al (2011) Isocitrate dehydrogenase 1 analysis differentiates gangliogliomas from infiltrative gliomas. Brain Pathol 21:564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Hu XW, Zhang YH, Wang JJ, Jiang XF, Liu JM, Yang PF (2010) Angiocentric glioma with rich blood supply. J Clin Neurosci 17:917–918. [DOI] [PubMed] [Google Scholar]
- 120. Huang H, Hara A, Homma T, Yonekawa Y, Ohgaki H (2005) Altered expression of immune defense genes in pilocytic astrocytomas. J Neuropathol Exp Neurol 64:891–901. [DOI] [PubMed] [Google Scholar]
- 121. Huse JT, Nafa K, Shukla N, Kastenhuber ER, Lavi E, Hedvat CV et al (2011) High frequency of IDH‐1 mutation links glioneuronal tumors with neuropil‐like islands to diffuse astrocytomas. Acta Neuropathol (Berl) 122:367–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Im SH, Chung CK, Kim SK, Cho BK, Kim MK, Chi JG (2004) Pleomorphic xanthoastrocytoma: a developmental glioneuronal tumor with prominent glioproliferative changes. J Neurooncol 66:17–27. [DOI] [PubMed] [Google Scholar]
- 123. Ishizawa K, Terao S, Kobayashi K, Yoshida K, Hirose T (2007) A neuroepithelial tumor showing combined histological features of dysembryoplastic neuroepithelial tumor and pleomorphic xanthoastrocytoma—a case report and review of the literature. Clin Neuropathol 26:169–175. [DOI] [PubMed] [Google Scholar]
- 124. Ishizawa T, Komori T, Shibahara J, Ishizawa K, Adachi J, Nishikawa R et al (2006) Papillary glioneuronal tumor with minigemistocytic components and increased proliferative activity. Hum Pathol 37:627–630. [DOI] [PubMed] [Google Scholar]
- 125. Izycka‐Swieszewska E, Majewska H, Szurowska E, Mazurkiewicz‐Beldzinska M, Drozynska E (2008) Papillary glioneuronal tumour of the precentral gyrus. Folia Neuropathol 46:158–163. [PubMed] [Google Scholar]
- 126. Jackson JH (1882) Localized convulsions from tumor of the brain. Brain 5:364–374. [Google Scholar]
- 127. Jacob K, Albrecht S, Sollier C, Faury D, Sader E, Montpetit A et al (2009) Duplication of 7q34 is specific to juvenile pilocytic astrocytomas and a hallmark of cerebellar and optic pathway tumours. Br J Cancer 101:722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Javahery RJ, Davidson L, Fangusaro J, Finlay JL, Gonzalez‐Gomez I, McComb JG (2009) Aggressive variant of a papillary glioneuronal tumor. Report of 2 cases. J Neurosurg Pediatr 3:46–52. [DOI] [PubMed] [Google Scholar]
- 129. Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M et al (2006) A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 66:9852–9861. [DOI] [PubMed] [Google Scholar]
- 130. Jeuken JW, von Deimling A, Wesseling P (2004) Molecular pathogenesis of oligodendroglial tumors. J Neurooncol 70:161–181. [DOI] [PubMed] [Google Scholar]
- 131. Johannesma PC, van der Klift HM, van Grieken NC, Troost D, Te Riele H, Jacobs MA et al (2011) Childhood brain tumours due to germline bi‐allelic mismatch repair gene mutations. Clin Genet 80:243–255. [DOI] [PubMed] [Google Scholar]
- 132. Johnson MD, Vnencak‐Jones CL, Toms SA, Moots PM, Weil R (2003) Allelic losses in oligodendroglial and oligodendroglioma‐like neoplasms: analysis using microsatellite repeats and polymerase chain reaction. Arch Pathol Lab Med 127:1573–1579. [DOI] [PubMed] [Google Scholar]
- 133. Jones DT, Ichimura K, Liu L, Pearson DM, Plant K, Collins VP (2006) Genomic analysis of pilocytic astrocytomas at 0.97 Mb resolution shows an increasing tendency toward chromosomal copy number change with age. J Neuropathol Exp Neurol 65:1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, Collins VP (2008) Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68:8673–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Jones DT, Mulholland SA, Pearson DM, Malley DS, Openshaw SW, Lambert SR et al (2011) Adult grade II diffuse astrocytomas are genetically distinct from and more aggressive than their paediatric counterparts. Acta Neuropathol (Berl) 121:753–761. [DOI] [PubMed] [Google Scholar]
- 136. Josan V, Smith P, Kornberg A, Rickert C, Maixner W (2007) Development of a pilocytic astrocytoma in a dysembryoplastic neuroepithelial tumor. Case report. J Neurosurg 106(Suppl.):509–512. [DOI] [PubMed] [Google Scholar]
- 137. Kam R, Chen J, Blümcke I, Normann S, Fassunke J, Elger CE et al (2004) The reelin pathway components disabled‐1 and p35 in gangliogliomas—a mutation and expression analysis. Neuropathol Appl Neurobiol 30:225–232. [DOI] [PubMed] [Google Scholar]
- 138. Kan P, Gottfried O, Blumenthal DT, Townsend JJ, Drozd‐Borysiuk E, Brothman AR, Jensen RL (2004) Oligodendroglioma and juvenile pilocytic astrocytoma presenting as synchronous primary brain tumors. Case report with histological and molecular differentiation of the tumors and review of the literature. J Neurosurg 100:700–705. [DOI] [PubMed] [Google Scholar]
- 139. Karremann M, Pietsch T, Janssen G, Kramm CM, Wolff JE (2009) Anaplastic ganglioglioma in children. J Neurooncol 92:157–163. [DOI] [PubMed] [Google Scholar]
- 140. Kaulich K, Blaschke B, Numann A, von Deimling A, Wiestler OD, Weber RG, Reifenberger G (2002) Genetic alterations commonly found in diffusely infiltrating cerebral gliomas are rare or absent in pleomorphic xanthoastrocytomas. J Neuropathol Exp Neurol 61:1092–1099. [DOI] [PubMed] [Google Scholar]
- 141. Kepes JJ, Rubinstein LJ, Eng LF (1979) Pleomorphic xanthoastrocytoma: a distinctive meningocerebral glioma of young subjects with relatively favorable prognosis. A study of 12 cases. Cancer 44:1839–1852. [DOI] [PubMed] [Google Scholar]
- 142. Keyvani K, Rickert CH, von Wild K, Paulus W (2001) Rosetted glioneuronal tumor: a case with proliferating neuronal nodules. Acta Neuropathol (Berl) 101:525–528. [DOI] [PubMed] [Google Scholar]
- 143. Kim DH, Suh YL (1997) Pseudopapillary neurocytoma of temporal lobe with glial differentiation. Acta Neuropathol (Berl) 94:187–191. [DOI] [PubMed] [Google Scholar]
- 144. Kim YH, Nobusawa S, Mittelbronn M, Paulus W, Brokinkel B, Keyvani K et al (2010) Molecular classification of low‐grade diffuse gliomas. Am J Pathol 177:2708–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Klein R, Roggendorf W (2001) Increased microglia proliferation separates pilocytic astrocytomas from diffuse astrocytomas: a double labeling study. Acta Neuropathol (Berl) 101:245–248. [DOI] [PubMed] [Google Scholar]
- 146. Kohling R, Senner V, Paulus W, Speckmann EJ (2006) Epileptiform activity preferentially arises outside tumor invasion zone in glioma xenotransplants. Neurobiol Dis 22:64–75. [DOI] [PubMed] [Google Scholar]
- 147. Komine C, Watanabe T, Katayama Y, Yoshino A, Yokoyama T, Fukushima T (2003) Promoter hypermethylation of the DNA repair gene O6‐methylguanine‐DNA methyltransferase is an independent predictor of shortened progression free survival in patients with low‐grade diffuse astrocytomas. Brain Pathol 13:176–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Komori T, Scheithauer BW, Anthony DC, Rosenblum MK, McLendon RE, Scott RM et al (1998) Papillary glioneuronal tumor: a new variant of mixed neuronal‐glial neoplasm. Am J Surg Pathol 22:1171–1183. [DOI] [PubMed] [Google Scholar]
- 149. Komori T, Scheithauer BW, Hirose T (2002) A rosette‐forming glioneuronal tumor of the fourth ventricle: infratentorial form of dysembryoplastic neuroepithelial tumor? Am J Surg Pathol 26:582–591. [DOI] [PubMed] [Google Scholar]
- 150. Konya D, Peker S, Ozgen S, Kurtkaya O, Necmettin Pamir M (2006) Superficial siderosis due to papillary glioneuronal tumor. J Clin Neurosci 13:950–952. [DOI] [PubMed] [Google Scholar]
- 151. Kordek R, Biernat W, Sapieja W, Alwasiak J, Liberski PP (1995) Pleomorphic xanthoastrocytoma with a gangliomatous component: an immunohistochemical and ultrastructural study. Acta Neuropathol (Berl) 89:194–197. [DOI] [PubMed] [Google Scholar]
- 152. Korshunov A, Meyer J, Capper D, Christians A, Remke M, Witt H et al (2009) Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol (Berl) 118:401–405. [DOI] [PubMed] [Google Scholar]
- 153. Kreiger PA, Okada Y, Simon S, Rorke LB, Louis DN, Golden JA (2005) Losses of chromosomes 1p and 19q are rare in pediatric oligodendrogliomas. Acta Neuropathol (Berl) 109:387–392. [DOI] [PubMed] [Google Scholar]
- 154. Kros JM, de Jong AA, van der Kwast TH (1992) Ultrastructural characterization of transitional cells in oligodendrogliomas. J Neuropathol Exp Neurol 51:186–193. [DOI] [PubMed] [Google Scholar]
- 155. Kros JM, Lie ST, Stefanko SZ (1994) Familial occurrence of polymorphous oligodendroglioma. Neurosurgery 34:732–736. [DOI] [PubMed] [Google Scholar]
- 156. Kubo O, Sasahara A, Tajika Y, Kawamura H, Kawabatake H, Takakura K (1996) Pleomorphic xanthoastrocytoma with neurofibromatosis type 1: case report. Noshuyo Byori 13:79–83. [PubMed] [Google Scholar]
- 157. Kuchelmeister K, Demirel T, Schlorer E, Bergmann M, Gullotta F (1995) Dysembryoplastic neuroepithelial tumour of the cerebellum. Acta Neuropathol (Berl) 89:385–390. [DOI] [PubMed] [Google Scholar]
- 158. Lach B, Duggal N, DaSilva VF, Benoit BG (1996) Association of pleomorphic xanthoastrocytoma with cortical dysplasia and neuronal tumors. A report of three cases. Cancer 78:2551–2563. [PubMed] [Google Scholar]
- 159. Lee J, Lee BL, Joo EY, Seo DW, Hong SB, Hong SC et al (2009) Dysembryoplastic neuroepithelial tumors in pediatric patients. Brain Dev 31:671–681. [DOI] [PubMed] [Google Scholar]
- 160. Lee JW, Wen PY, Hurwitz S, Black P, Kesari S, Drappatz J et al (2010) Morphological characteristics of brain tumors causing seizures. Arch Neurol 67:336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Lehman NL (2008) Central nervous system tumors with ependymal features: a broadened spectrum of primarily ependymal differentiation? J Neuropathol Exp Neurol 67:177–188. [DOI] [PubMed] [Google Scholar]
- 162. Lellouch‐Tubiana A, Boddaert N, Bourgeois M, Fohlen M, Jouvet A, Delalande O et al (2005) Angiocentric neuroepithelial tumor (ANET): a new epilepsy‐related clinicopathological entity with distinctive MRI. Brain Pathol 15:281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Li JY, Langford LA, Adesina A, Bodhireddy SR, Wang M, Fuller GN (2011) The high mitotic count detected by phospho‐histone H3 immunostain does not alter the benign behavior of angiocentric glioma. Brain Tumor Pathol 29:68–72. [DOI] [PubMed] [Google Scholar]
- 164. Li YS, Ramsay DA, Fan YS, Armstrong RF, Del Maestro RF (1995) Cytogenetic evidence that a tumor suppressor gene in the long arm of chromosome 1 contributes to glioma growth. Cancer Genet Cytogenet 84:46–50. [DOI] [PubMed] [Google Scholar]
- 165. Liberski PP (1996) The ultrastructure of oligodendroglioma: personal experience and the review of the literature. Folia Neuropathol 34:206–211. [PubMed] [Google Scholar]
- 166. Lindboe CF, Cappelen J, Kepes JJ (1992) Pleomorphic xanthoastrocytoma as a component of a cerebellar ganglioglioma: case report. Neurosurgery 31:353–355. [DOI] [PubMed] [Google Scholar]
- 167. Louis DN, Ohgaki H, Wiestler OD, Cavanee WK (2007) WHO Classification of Tumours of the Central Nervous System. IARC: Lyon. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Louis DN, World Health Organization , International Agency for Research on Cancer , Deutsches Krebsforschungszentrum Heidelberg (2007) WHO Classification of Tumours of the Central Nervous System, 4th edn. IARC (International Agency for Research on Cancer): Lyon. [Google Scholar]
- 169. Lu JQ, Scheithauer BW, Sharma P, Scott JN, Parney IF, Hader W et al (2009) Multifocal complex glioneuronal tumor in an elderly man: an autopsy study: case report. Neurosurgery 64:E1193–E1195; discussion E5. [DOI] [PubMed] [Google Scholar]
- 170. Luan S, Zhuang D, Sun L, Huang FP (2010) Rosette‐forming glioneuronal tumor (RGNT) of the fourth ventricle: case report and review of literature. Clin Neurol Neurosurg 112:362–364. [DOI] [PubMed] [Google Scholar]
- 171. Lubansu A, Rorive S, David P, Sariban E, Seligmann R, Brotchi J, Pirotte B (2004) Cerebral anaplastic pleomorphic xanthoastrocytoma with meningeal dissemination at first presentation. Childs Nerv Syst 20:119–122. [DOI] [PubMed] [Google Scholar]
- 172. Luyken C, Blümcke I, Fimmers R, Urbach H, Elger CE, Wiestler OD, Schramm J (2003) The spectrum of long‐term epilepsy‐associated tumors: long‐term seizure and tumor outcome and neurosurgical aspects. Epilepsia 44:822–830. [DOI] [PubMed] [Google Scholar]
- 173. Luyken C, Blümcke I, Fimmers R, Urbach H, Wiestler OD, Schramm J (2004) Supratentorial gangliogliomas: histopathologic grading and tumor recurrence in 184 patients with a median follow‐up of 8 years. Cancer 101:146–155. [DOI] [PubMed] [Google Scholar]
- 174. Ma X, Ge J, Wang L, Xia C, Liu H, Li Y et al (2010) A 25‐year‐old woman with a mass in the hippocampus. Brain Pathol 20:503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Maas S, Patt S, Schrey M, Rich A (2001) Underediting of glutamate receptor GluR‐B mRNA in malignant gliomas. Proc Natl Acad Sci U S A 98:14687–14692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Mahajan H, Varikatt W, Dexter M, Boadle R, Ng T (2010) Papillary glioneuronal tumor of the frontal lobe. J Clin Neurosci 17:534–536. [DOI] [PubMed] [Google Scholar]
- 177. Maher CO, White JB, Scheithauer BW, Raffel C (2008) Recurrence of dysembryoplastic neuroepithelial tumor following resection. Pediatr Neurosurg 44:333–336. [DOI] [PubMed] [Google Scholar]
- 178. Majores M, Niehusmann P, von Lehe M, Blümcke I, Urbach H (2007) Angiocentric neuroepithelial tumor mimicking Ammon's horn sclerosis—case report. Clin Neuropathol 26:311–316. [DOI] [PubMed] [Google Scholar]
- 179. Majores M, von Lehe M, Fassunke J, Schramm J, Becker AJ, Simon M (2008) Tumor recurrence and malignant progression of gangliogliomas. Cancer 113:3355–3363. [DOI] [PubMed] [Google Scholar]
- 180. Marburger T, Prayson R (2011) Angiocentric glioma: a clinicopathologic review of 5 tumors with identification of associated cortical dysplasia. Arch Pathol Lab Med 135:1037–1041. [DOI] [PubMed] [Google Scholar]
- 181. Marton E, Feletti A, Orvieto E, Longatti P (2007) Malignant progression in pleomorphic xanthoastrocytoma: personal experience and review of the literature. J Neurol Sci 252:144–153. [DOI] [PubMed] [Google Scholar]
- 182. Marucci G, Morandi L (2011) Assessment of MGMT promoter methylation status in pleomorphic xanthoastrocytoma. J Neurooncol 105:397–400. [DOI] [PubMed] [Google Scholar]
- 183. Minkin K, Klein O, Mancini J, Lena G (2008) Surgical strategies and seizure control in pediatric patients with dysembryoplastic neuroepithelial tumors: a single‐institution experience. J Neurosurg Pediatr 1:206–210. [DOI] [PubMed] [Google Scholar]
- 184. Mirsattari SM, Chong JJ, Hammond RR, Megyesi JF, Macdonald DR, Lee DH, Cairncross JG (2011) Do epileptic seizures predict outcome in patients with oligodendroglioma? Epilepsy Res 94:39–44. [DOI] [PubMed] [Google Scholar]
- 185. Mollemann M, Wolter M, Felsberg J, Collins VP, Reifenberger G (2005) Frequent promoter hypermethylation and low expression of the MGMT gene in oligodendroglial tumors. Int J Cancer 113:379–385. [DOI] [PubMed] [Google Scholar]
- 186. Mott RT, Ellis TL, Geisinger KR (2010) Angiocentric glioma: a case report and review of the literature. Diagn Cytopathol 38:452–456. [DOI] [PubMed] [Google Scholar]
- 187. Murray JC, Donahue DJ, Malik SI, Dzurik YB, Braly EZ, Dougherty MJ et al (2011) Temporal lobe pleomorphic xanthoastrocytoma and acquired BRAF mutation in an adolescent with the constitutional 22q11.2 deletion syndrome. J Neurooncol 102:509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Myung JK, Byeon SJ, Kim B, Suh J, Kim SK, Park CK et al (2011) Papillary glioneuronal tumors: a review of clinicopathologic and molecular genetic studies. Am J Surg Pathol 35:1794–1805. [DOI] [PubMed] [Google Scholar]
- 189. Nagaishi M, Arai M, Osawa T, Yokoo H, Hirato J, Yoshimoto Y, Nakazato Y (2011) An immunohistochemical finding in glioneuronal lesions associated with epilepsy: the appearance of nestin‐positive, CD34‐positive and tau‐accumulating cells. Neuropathology 31:468–475. [DOI] [PubMed] [Google Scholar]
- 190. Nakasu S, Fukami T, Jito J, Matsuda M (2007) Prognostic significance of loss of O6‐methylguanine‐DNA methyltransferase expression in supratentorial diffuse low‐grade astrocytoma. Surg Neurol 68:603–608. [DOI] [PubMed] [Google Scholar]
- 191. Nakatsuka M, Mizuno S, Kimura T, Hara K (2000) A case of an unclassified tumor closely resembling dysembryoplastic neuroepithelial tumor with rapid growth. Brain Tumor Pathol 17:41–45. [DOI] [PubMed] [Google Scholar]
- 192. Newton HB, Dalton J, Ray‐Chaudhury A, Gahbauer R, McGregor J (2008) Aggressive papillary glioneuronal tumor: case report and literature review. Clin Neuropathol 27:317–324. [DOI] [PubMed] [Google Scholar]
- 193. Nikiforova MN, Hamilton RL (2011) Molecular diagnostics of gliomas. Arch Pathol Lab Med 135:558–568. [DOI] [PubMed] [Google Scholar]
- 194. Nolan MA, Sakuta R, Chuang N, Otsubo H, Rutka JT, Snead OC et al (2004) Dysembryoplastic neuroepithelial tumors in childhood: long‐term outcome and prognostic features. Neurology 62:2270–2276. [DOI] [PubMed] [Google Scholar]
- 195. Ohgaki H, Eibl RH, Schwab M, Reichel MB, Mariani L, Gehring M et al (1993) Mutations of the p53 tumor suppressor gene in neoplasms of the human nervous system. Mol Carcinog 8:74–80. [DOI] [PubMed] [Google Scholar]
- 196. Ohnishi A, Sawa H, Tsuda M, Sawamura Y, Itoh T, Iwasaki Y, Nagashima K (2003) Expression of the oligodendroglial lineage‐associated markers Olig1 and Olig2 in different types of human gliomas. J Neuropathol Exp Neurol 62:1052–1059. [DOI] [PubMed] [Google Scholar]
- 197. Okamoto Y, Di Patre PL, Burkhard C, Horstmann S, Jourde B, Fahey M et al (2004) Population‐based study on incidence, survival rates, and genetic alterations of low‐grade diffuse astrocytomas and oligodendrogliomas. Acta Neuropathol (Berl) 108:49–56. [DOI] [PubMed] [Google Scholar]
- 198. Olsen ML, Sontheimer H (2004) Mislocalization of Kir channels in malignant glia. Glia 46:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Orr LC, Fleitz J, McGavran L, Wyatt‐Ashmead J, Handler M, Foreman NK (2002) Cytogenetics in pediatric low‐grade astrocytomas. Med Pediatr Oncol 38:173–177. [DOI] [PubMed] [Google Scholar]
- 200. Ortiz‐Gonzalez XR, Venneti S, Biegel JA, Rorke‐Adams LB, Porter BE (2011) Ganglioglioma arising from dysplastic cortex. Epilepsia 52:e106–e108. [DOI] [PubMed] [Google Scholar]
- 201. Otero‐Rodriguez A, Sarabia‐Herrero R, Garcia‐Tejeiro M, Zamora‐Martinez T (2010) Spontaneous malignant transformation of a supratentorial pilocytic astrocytoma. Neurocirugia (Astur) 21:245–252. [DOI] [PubMed] [Google Scholar]
- 202. Ozek MM, Sav A, Pamir MN, Ozer AF, Ozek E, Erzen C (1993) Pleomorphic xanthoastrocytoma associated with von Recklinghausen neurofibromatosis. Childs Nerv Syst 9:39–42. [DOI] [PubMed] [Google Scholar]
- 203. Pahapill PA, Ramsay DA, Del Maestro RF (1996) Pleomorphic xanthoastrocytoma: case report and analysis of the literature concerning the efficacy of resection and the significance of necrosis. Neurosurgery 38:822–828; discussion 8–9. [PubMed] [Google Scholar]
- 204. Palmini A, Najm I, Avanzini G, Babb T, Guerrini R, Foldvary‐Schaefer N et al (2004) Terminology and classification of the cortical dysplasias. Neurology 62(Suppl. 3):S2–S8. [DOI] [PubMed] [Google Scholar]
- 205. Parry L, Maynard JH, Patel A, Hodges AK, von Deimling A, Sampson JR, Cheadle JP (2000) Molecular analysis of the TSC1 and TSC2 tumour suppressor genes in sporadic glial and glioneuronal tumours. Hum Genet 107:350–356. [DOI] [PubMed] [Google Scholar]
- 206. Pasquier B, Peoc HM, Fabre‐Bocquentin B, Bensaadi L, Pasquier D, Hoffmann D et al (2002) Surgical pathology of drug‐resistant partial epilepsy. A 10‐year‐experience with a series of 327 consecutive resections. Epileptic Disord 4:99–119. [PubMed] [Google Scholar]
- 207. Patt S, Gries H, Giraldo M, Cervos‐Navarro J, Martin H, Janisch W, Brockmoller J (1996) p53 gene mutations in human astrocytic brain tumors including pilocytic astrocytomas. Hum Pathol 27:586–589. [DOI] [PubMed] [Google Scholar]
- 208. Paulus W, Lisle DK, Tonn JC, Wolf HK, Roggendorf W, Reeves SA, Louis DN (1996) Molecular genetic alterations in pleomorphic xanthoastrocytoma. Acta Neuropathol (Berl) 91:293–297. [DOI] [PubMed] [Google Scholar]
- 209. Payton JE, Schmidt J, Yu J, Lusis EA, Watson MA, Gutmann DH (2011) Genome‐wide polymorphism analysis demonstrates a monoclonal origin of pilocytic astrocytoma. Neuropathol Appl Neurobiol 37:321–325. [DOI] [PubMed] [Google Scholar]
- 210. Perry A, Giannini C, Scheithauer BW, Rojiani AM, Yachnis AT, Seo IS et al (1997) Composite pleomorphic xanthoastrocytoma and ganglioglioma: report of four cases and review of the literature. Am J Surg Pathol 21:763–771. [DOI] [PubMed] [Google Scholar]
- 211. Perry A, Scheithauer BW, Szczesniak DM, Atkinson JL, Wald JT, Hammak JE (2001) Combined oligodendroglioma/pleomorphic xanthoastrocytoma: a probable collision tumor: case report. Neurosurgery 48:1358–1361. [DOI] [PubMed] [Google Scholar]
- 212. Perry A, Scheithauer BW, Macaulay RJ, Raffel C, Roth KA, Kros JM (2002) Oligodendrogliomas with neurocytic differentiation. A report of 4 cases with diagnostic and histogenetic implications. J Neuropathol Exp Neurol 61:947–955. [DOI] [PubMed] [Google Scholar]
- 213. Perry A, Burton SS, Fuller GN, Robinson CA, Palmer CA, Resch L et al (2010) Oligodendroglial neoplasms with ganglioglioma‐like maturation: a diagnostic pitfall. Acta Neuropathol (Berl) 120:237–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N et al (2008) BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low‐grade astrocytomas. J Clin Invest 118:1739–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Piao YS, Lu DH, Chen L, Liu J, Wang W, Liu L et al (2010) Neuropathological findings in intractable epilepsy: 435 Chinese cases. Brain Pathol 20:902–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Prayson RA (1999) Composite ganglioglioma and dysembryoplastic neuroepithelial tumor. Arch Pathol Lab Med 123:247–250. [DOI] [PubMed] [Google Scholar]
- 217. Prayson RA (2000) Papillary glioneuronal tumor. Arch Pathol Lab Med 124:1820–1823. [DOI] [PubMed] [Google Scholar]
- 218. Prayson RA (2010) Tumours arising in the setting of paediatric chronic epilepsy. Pathology 42:426–431. [DOI] [PubMed] [Google Scholar]
- 219. Prayson RA (2011) Brain tumors in adults with medically intractable epilepsy. Am J Clin Pathol 136:557–563. [DOI] [PubMed] [Google Scholar]
- 220. Prayson RA, Abramovich CM (2000) Glioneuronal tumor with neuropil‐like islands. Hum Pathol 31:1435–1438. [PubMed] [Google Scholar]
- 221. Prayson RA, Khajavi K, Comair YG (1995) Cortical architectural abnormalities and MIB1 immunoreactivity in gangliogliomas: a study of 60 patients with intracranial tumors. J Neuropathol Exp Neurol 54:513–520. [DOI] [PubMed] [Google Scholar]
- 222. Prayson RA, Castilla EA, Hartke M, Pettay J, Tubbs RR, Barnett GH (2002) Chromosome 1p allelic loss by fluorescence in situ hybridization is not observed in dysembryoplastic neuroepithelial tumors. Am J Clin Pathol 118:512–517. [DOI] [PubMed] [Google Scholar]
- 223. Prayson RA, Fong J, Najm I (2010) Coexistent pathology in chronic epilepsy patients with neoplasms. Mod Pathol 23:1097–1103. [DOI] [PubMed] [Google Scholar]
- 224. Preusser M, Hoischen A, Novak K, Czech T, Prayer D, Hainfellner JA et al (2007) Angiocentric glioma: report of clinico‐pathologic and genetic findings in 8 cases. Am J Surg Pathol 31:1709–1718. [DOI] [PubMed] [Google Scholar]
- 225. Rades D, Zwick L, Leppert J, Bonsanto MM, Tronnier V, Dunst J, Schild SE (2010) The role of postoperative radiotherapy for the treatment of gangliogliomas. Cancer 116:432–442. [DOI] [PubMed] [Google Scholar]
- 226. Radotra BD, Kumar Y, Bhatia A, Mohindra S (2007) Papillary glioneuronal tumor: a new entity awaiting inclusion in WHO classification. Diagn Pathol 2:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Raghavan R, Balani J, Perry A, Margraf L, Vono MB, Cai DX et al (2003) Pediatric oligodendrogliomas: a study of molecular alterations on 1p and 19q using fluorescence in situ hybridization. J Neuropathol Exp Neurol 62:530–537. [DOI] [PubMed] [Google Scholar]
- 228. Rajneesh KF, Binder DK (2009) Tumor‐associated epilepsy. Neurosurg Focus 27:E4. [DOI] [PubMed] [Google Scholar]
- 229. Ramirez C, Bowman C, Maurage CA, Dubois F, Blond S, Porchet N, Escande F (2010) Loss of 1p, 19q, and 10q heterozygosity prospectively predicts prognosis of oligodendroglial tumors—towards individualized tumor treatment? Neuro Oncol 12:490–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Ratilal B, McEvoy A, Sisodiya S, Thom M, Toma A (2007) Diffuse cerebral gangliocytoma in an adult with late‐onset refractory epilepsy. Neuropathol Appl Neurobiol 33:706–709. [DOI] [PubMed] [Google Scholar]
- 231. Ravizza T, Boer K, Redeker S, Spliet WG, van Rijen PC, Troost D et al (2006) The IL‐1beta system in epilepsy‐associated malformations of cortical development. Neurobiol Dis 24:128–143. [DOI] [PubMed] [Google Scholar]
- 232. Ray WZ, Blackburn SL, Casavilca‐Zambrano S, Barrionuevo C, Orrego JE, Heinicke H et al (2009) Clinicopathologic features of recurrent dysembryoplastic neuroepithelial tumor and rare malignant transformation: a report of 5 cases and review of the literature. J Neurooncol 94:283–292. [DOI] [PubMed] [Google Scholar]
- 233. Reifenberger G, Kaulich K, Wiestler OD, Blümcke I (2003) Expression of the CD34 antigen in pleomorphic xanthoastrocytomas. Acta Neuropathol (Berl) 105:358–364. [DOI] [PubMed] [Google Scholar]
- 234. Riemenschneider MJ, Reifenberger G (2010) Molecular neuropathology of low‐grade gliomas and its clinical impact. Adv Tech Stand Neurosurg 35:35–64. [DOI] [PubMed] [Google Scholar]
- 235. Roberts P, Chumas PD, Picton S, Bridges L, Livingstone JH, Sheridan E (2001) A review of the cytogenetics of 58 pediatric brain tumors. Cancer Genet Cytogenet 131:1–12. [DOI] [PubMed] [Google Scholar]
- 236. Rodriguez EF, Scheithauer BW, Giannini C, Rynearson A, Cen L, Hoesley B et al (2011) PI3K/AKT pathway alterations are associated with clinically aggressive and histologically anaplastic subsets of pilocytic astrocytoma. Acta Neuropathol (Berl) 121:407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Rodriguez FJ, Scheithauer BW, Jenkins R, Burger PC, Rudzinskiy P, Vlodavsky E et al (2007) Gliosarcoma arising in oligodendroglial tumors (“oligosarcoma”): a clinicopathologic study. Am J Surg Pathol 31:351–362. [DOI] [PubMed] [Google Scholar]
- 238. Rodriguez FJ, Perry A, Gutmann DH, O'Neill BP, Leonard J, Bryant S, Giannini C (2008) Gliomas in neurofibromatosis type 1: a clinicopathologic study of 100 patients. J Neuropathol Exp Neurol 67:240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Rodriguez FJ, Scheithauer BW, Burger PC, Jenkins S, Giannini C (2010) Anaplasia in pilocytic astrocytoma predicts aggressive behavior. Am J Surg Pathol 34:147–160. [DOI] [PubMed] [Google Scholar]
- 240. Ruban D, Byrne RW, Kanner A, Smith M, Cochran EJ, Roh D, Whisler WW (2009) Chronic epilepsy associated with temporal tumors: long‐term surgical outcome. Neurosurg Focus 27:E6. [DOI] [PubMed] [Google Scholar]
- 241. Rushing EJ, Thompson LD, Mena H (2003) Malignant transformation of a dysembryoplastic neuroepithelial tumor after radiation and chemotherapy. Ann Diagn Pathol 7:240–244. [DOI] [PubMed] [Google Scholar]
- 242. Sakuta R, Otsubo H, Nolan MA, Weiss SK, Hawkins C, Rutka JT et al (2005) Recurrent intractable seizures in children with cortical dysplasia adjacent to dysembryoplastic neuroepithelial tumor. J Child Neurol 20:377–384. [DOI] [PubMed] [Google Scholar]
- 243. Samadani U, Judkins AR, Akpalu A, Aronica E, Crino PB (2007) Differential cellular gene expression in ganglioglioma. Epilepsia 48:646–653. [DOI] [PubMed] [Google Scholar]
- 244. Sanoudou D, Tingby O, Ferguson‐Smith MA, Collins VP, Coleman N (2000) Analysis of pilocytic astrocytoma by comparative genomic hybridization. Br J Cancer 82:1218–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F et al (2009) Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 27:4150–4154. [DOI] [PubMed] [Google Scholar]
- 246. Sarkar C, Roy S, Tandon PN (1988) Oligodendroglial tumors. An immunohistochemical and electron microscopic study. Cancer 61:1862–1866. [DOI] [PubMed] [Google Scholar]
- 247. Sawyer JR, Roloson GJ, Chadduck WM, Boop FA (1991) Cytogenetic findings in a pleomorphic xanthoastrocytoma. Cancer Genet Cytogenet 55:225–230. [DOI] [PubMed] [Google Scholar]
- 248. Scheithauer BW, Silva AI, Ketterling RP, Pula JH, Lininger JF, Krinock MJ (2009) Rosette‐forming glioneuronal tumor: report of a chiasmal‐optic nerve example in neurofibromatosis type 1: special pathology report. Neurosurgery 64:E771–E772; discussion E2. [DOI] [PubMed] [Google Scholar]
- 249. Schick V, Majores M, Koch A, Elger CE, Schramm J, Urbach H, Becker AJ (2007) Alterations of phosphatidylinositol 3‐kinase pathway components in epilepsy‐associated glioneuronal lesions. Epilepsia 48(Suppl. 5):65–73. [DOI] [PubMed] [Google Scholar]
- 250. Schiffman JD, Hodgson JG, VandenBerg SR, Flaherty P, Polley MY, Yu M et al (2010) Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res 70:512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold‐Mende C et al (2011) Analysis of BRAF V600E mutation in 1320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra‐cerebellar pilocytic astrocytoma. Acta Neuropathol (Berl) 121:397–405. [DOI] [PubMed] [Google Scholar]
- 252. Schittenhelm J, Mittelbronn M, Wolff M, Truebenbach J, Will BE, Meyermann R, Beschorner R (2007) Multifocal dysembryoplastic neuroepithelial tumor with signs of atypia after regrowth. Neuropathology 27:383–389. [DOI] [PubMed] [Google Scholar]
- 253. Schramm J, Luyken C, Urbach H, Fimmers R, Blümcke I (2004) Evidence for a clinically distinct new subtype of grade II astrocytomas in patients with long‐term epilepsy. Neurosurgery 55:340–347; discussion 7‐8. [DOI] [PubMed] [Google Scholar]
- 254. Schrock E, Blume C, Meffert MC, du Manoir S, Bersch W, Kiessling M et al (1996) Recurrent gain of chromosome arm 7q in low‐grade astrocytic tumors studied by comparative genomic hybridization. Genes Chromosomes Cancer 15:199–205. [DOI] [PubMed] [Google Scholar]
- 255. Seifert G, Carmignoto G, Steinhauser C (2010) Astrocyte dysfunction in epilepsy. Brain Res Rev. 63:212–221. [DOI] [PubMed] [Google Scholar]
- 256. Selvanathan SK, Hammouche S, Salminen HJ, Jenkinson MD (2011) Outcome and prognostic features in anaplastic ganglioglioma: analysis of cases from the SEER database. J Neurooncol 105:539–545. [DOI] [PubMed] [Google Scholar]
- 257. Shah MN, Leonard JR, Perry A (2010) Rosette‐forming glioneuronal tumors of the posterior fossa. J Neurosurg Pediatr 5:98–103. [DOI] [PubMed] [Google Scholar]
- 258. Shakur SF, McGirt MJ, Johnson MW, Burger PC, Ahn E, Carson BS, Jallo GI (2009) Angiocentric glioma: a case series. J Neurosurg Pediatr 3:197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Shamji MF, Fric‐Shamji EC, Benoit BG (2009) Brain tumors and epilepsy: pathophysiology of peritumoral changes. Neurosurg Rev 32:275–284; discussion 84–6. [DOI] [PubMed] [Google Scholar]
- 260. Shih AH, Holland EC (2004) Developmental neurobiology and the origin of brain tumors. J Neurooncol 70:125–136. [DOI] [PubMed] [Google Scholar]
- 261. Shimbo Y, Takahashi H, Hayano M, Kumagai T, Kameyama S (1997) Temporal lobe lesion demonstrating features of dysembryoplastic neuroepithelial tumor and ganglioglioma: a transitional form? Clin Neuropathol 16:65–68. [PubMed] [Google Scholar]
- 262. Smith JS, Perry A, Borell TJ, Lee HK, O'Fallon J, Hosek SM et al (2000) Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol 18:636–645. [DOI] [PubMed] [Google Scholar]
- 263. Solis OE, Mehta RI, Lai A, Farchoukh LO, Green RM, Cheng JC et al (2011) Rosette‐forming glioneuronal tumor: a pineal region case with IDH1 and IDH2 mutation analyses and literature review of 43 cases. J Neurooncol 102:477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Steinhauser C, Seifert G (2002) Glial membrane channels and receptors in epilepsy: impact for generation and spread of seizure activity. Eur J Pharmacol 447:227–237. [DOI] [PubMed] [Google Scholar]
- 265. Stosic‐Opincal T, Peric V, Gavrilovic S, Gavrilov M, Markovic Z, Sener RN (2005) Papillary glioneuronal tumor. AJR Am J Roentgenol 185:265–267. [DOI] [PubMed] [Google Scholar]
- 266. Sugita Y, Irie K, Ohshima K, Hitotsumatsu T, Sato O, Arimura K (2009) Pleomorphic xanthoastrocytoma as a component of a temporal lobe cystic ganglioglioma: a case report. Brain Tumor Pathol 26:31–36. [DOI] [PubMed] [Google Scholar]
- 267. Tachibana I, Smith JS, Sato K, Hosek SM, Kimmel DW, Jenkins RB (2000) Investigation of germline PTEN, p53, p16(INK4A)/p14(ARF), and CDK4 alterations in familial glioma. Am J Med Genet 92:136–141. [DOI] [PubMed] [Google Scholar]
- 268. Takahashi A, Hong SC, Seo DW, Hong SB, Lee M, Suh YL (2005) Frequent association of cortical dysplasia in dysembryoplastic neuroepithelial tumor treated by epilepsy surgery. Surg Neurol 64:419–427. [DOI] [PubMed] [Google Scholar]
- 269. Tan TC, Ho LC, Yu CP, Cheung FC (2004) Pleomorphic xanthoastrocytoma: report of two cases and review of the prognostic factors. J Clin Neurosci 11:203–207. [DOI] [PubMed] [Google Scholar]
- 270. Tanaka Y, Yokoo H, Komori T, Makita Y, Ishizawa T, Hirose T et al (2005) A distinct pattern of Olig2‐positive cellular distribution in papillary glioneuronal tumors: a manifestation of the oligodendroglial phenotype? Acta Neuropathol (Berl) 110:39–47. [DOI] [PubMed] [Google Scholar]
- 271. Teo JG, Gultekin SH, Bilsky M, Gutin P, Rosenblum MK (1999) A distinctive glioneuronal tumor of the adult cerebrum with neuropil‐like (including “rosetted”) islands: report of 4 cases. Am J Surg Pathol 23:502–510. [DOI] [PubMed] [Google Scholar]
- 272. Thom M, Toma A, An S, Martinian L, Hadjivassiliou G, Ratilal B et al (2011) One hundred and one dysembryoplastic neuroepithelial tumors: an adult epilepsy series with immunohistochemical, molecular genetic, and clinical correlations and a review of the literature. J Neuropathol Exp Neurol 70:859–878. [DOI] [PubMed] [Google Scholar]
- 273. Tihan T, Fisher PG, Kepner JL, Godfraind C, McComb RD, Goldthwaite PT, Burger PC (1999) Pediatric astrocytomas with monomorphous pilomyxoid features and a less favorable outcome. J Neuropathol Exp Neurol 58:1061–1068. [DOI] [PubMed] [Google Scholar]
- 274. Tsukayama C, Arakawa Y (2002) A papillary glioneuronal tumor arising in an elderly woman: a case report. Brain Tumor Pathol 19:35–39. [DOI] [PubMed] [Google Scholar]
- 275. Urbach H, Binder D, von Lehe M, Podlogar M, Bien CG, Becker A et al (2007) Correlation of MRI and histopathology in epileptogenic parietal and occipital lobe lesions. Seizure 16:608–614. [DOI] [PubMed] [Google Scholar]
- 276. Vajtai I, Reinert MM (2007) Malignant glioneuronal tumor of the adult cerebrum with neuropil‐like islands involving “proliferating nodules”: confirmatory report of an unusual variant. Acta Neuropathol (Berl) 113:711–713. [DOI] [PubMed] [Google Scholar]
- 277. Vajtai I, Varga Z, Aguzzi A (1997) Pleomorphic xanthoastrocytoma with gangliogliomatous component. Pathol Res Pract 193:617–621. [DOI] [PubMed] [Google Scholar]
- 278. Vajtai I, Kappeler A, Lukes A, Arnold M, Luthy AR, Leibundgut K (2006) Papillary glioneuronal tumor. Pathol Res Pract 202:107–112. [DOI] [PubMed] [Google Scholar]
- 279. Vajtai I, Arnold M, Kappeler A, Jeless O, Lukes A, Mariani L, Paulus W (2007) Rosette‐forming glioneuronal tumor of the fourth ventricle: report of two cases with a differential diagnostic overview. Pathol Res Pract 203:613–619. [DOI] [PubMed] [Google Scholar]
- 280. van Breemen MS, Wilms EB, Vecht CJ (2007) Epilepsy in patients with brain tumours: epidemiology, mechanisms, and management. Lancet Neurol 6:421–430. [DOI] [PubMed] [Google Scholar]
- 281. van Vuurden DG, Yazdani M, Bosma I, Broekhuizen AJ, Postma TJ, Heimans JJ et al (2009) Attenuated AMPA receptor expression allows glioblastoma cell survival in glutamate‐rich environment. PLoS ONE 4:e5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Vaquero J, Coca S (2007) Atypical papillary glioneuronal tumor. J Neurooncol 83:319–323. [DOI] [PubMed] [Google Scholar]
- 283. Vezzani A, Granata T (2005) Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia 46:1724–1743. [DOI] [PubMed] [Google Scholar]
- 284. Vezzani A, Maroso M, Balosso S, Sanchez MA, Bartfai T (2011) IL‐1 receptor/Toll‐like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav Immun 25:1281–1289. [DOI] [PubMed] [Google Scholar]
- 285. Vyberg M, Ulhoi BP, Teglbjaerg PS (2007) Neuronal features of oligodendrogliomas—an ultrastructural and immunohistochemical study. Histopathology 50:887–896. [DOI] [PubMed] [Google Scholar]
- 286. Wang M, Tihan T, Rojiani AM, Bodhireddy SR, Prayson RA, Iacuone JJ et al (2005) Monomorphous angiocentric glioma: a distinctive epileptogenic neoplasm with features of infiltrating astrocytoma and ependymoma. J Neuropathol Exp Neurol 64:875–881. [DOI] [PubMed] [Google Scholar]
- 287. Wang YZ, Plane JM, Jiang P, Zhou CJ, Deng W (2011) Concise review: quiescent and active states of endogenous adult neural stem cells: identification and characterization. Stem Cells 29:907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Watanabe T, Nakamura M, Kros JM, Burkhard C, Yonekawa Y, Kleihues P, Ohgaki H (2002) Phenotype versus genotype correlation in oligodendrogliomas and low‐grade diffuse astrocytomas. Acta Neuropathol (Berl) 103:267–275. [DOI] [PubMed] [Google Scholar]
- 289. Watanabe T, Katayama Y, Yoshino A, Yachi K, Ohta T, Ogino A et al (2007) Aberrant hypermethylation of p14ARF and O6‐methylguanine‐DNA methyltransferase genes in astrocytoma progression. Brain Pathol 17:5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Weber RG, Hoischen A, Ehrler M, Zipper P, Kaulich K, Blaschke B et al (2007) Frequent loss of chromosome 9, homozygous CDKN2A/p14(ARF)/CDKN2B deletion and low TSC1 mRNA expression in pleomorphic xanthoastrocytomas. Oncogene 26:1088–1097. [DOI] [PubMed] [Google Scholar]
- 291. Wharton SB, Chan KK, Hamilton FA, Anderson JR (1998) Expression of neuronal markers in oligodendrogliomas: an immunohistochemical study. Neuropathol Appl Neurobiol 24:302–308. [DOI] [PubMed] [Google Scholar]
- 292. Wharton SB, Whittle IR, Collie DA, Bell HS, Ironside JW (2001) Gliosarcoma with areas of primitive neuroepithelial differentiation and extracranial metastasis. Clin Neuropathol 20:212–218. [PubMed] [Google Scholar]
- 293. Wolf HK, Campos MG, Zentner J, Hufnagel A, Schramm J, Elger CE, Wiestler OD (1993) Surgical pathology of temporal lobe epilepsy. Experience with 216 cases. J Neuropathol Exp Neurol 52:499–506. [DOI] [PubMed] [Google Scholar]
- 294. Wolf HK, Muller MB, Spanle M, Zentner J, Schramm J, Wiestler OD (1994) Ganglioglioma: a detailed histopathological and immunohistochemical analysis of 61 cases. Acta Neuropathol (Berl) 88:166–173. [DOI] [PubMed] [Google Scholar]
- 295. Wolf HK, Birkholz T, Wellmer J, Blümcke I, Pietsch T, Wiestler OD (1995) Neurochemical profile of glioneuronal lesions from patients with pharmacoresistant focal epilepsies. J Neuropathol Exp Neurol 54:689–697. [DOI] [PubMed] [Google Scholar]
- 296. Wolf HK, Buslei R, Blümcke I, Wiestler OD, Pietsch T (1997) Neural antigens in oligodendrogliomas and dysembryoplastic neuroepithelial tumors. Acta Neuropathol (Berl) 94:436–443. [DOI] [PubMed] [Google Scholar]
- 297. Wong M (2010) Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: from tuberous sclerosis to common acquired epilepsies. Epilepsia 51:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Xiao H, Ma L, Lou X, Gui Q (2011) Papillary glioneuronal tumor: radiological evidence of a newly established tumor entity. J Neuroimaging 21:297–302. [DOI] [PubMed] [Google Scholar]
- 299. Xiong J, Liu Y, Chu SG, Chen H, Chen HX, Mao Y, Wang Y (2011) Rosette‐forming glioneuronal tumor of the septum pellucidum with extension to the supratentorial ventricles: rare case with genetic analysis. Neuropathology doi: 10.1111/j.1440‐1789.2011.01261.x. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 300. Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A (2004) Cl‐ uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol 557:829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Yamashita S, Yokogami K, Niibo T, Takeishi G, Ikeda T, Miyata S et al (2011) Oligodendroglial ganglioglioma. Brain Tumor Pathol 28:311–316. [DOI] [PubMed] [Google Scholar]
- 302. Yang I, Chang EF, Han SJ, Barry JJ, Fang S, Tihan T et al (2011) Early surgical intervention in adult patients with ganglioglioma is associated with improved clinical seizure outcomes. J Clin Neurosci 18:29–33. [DOI] [PubMed] [Google Scholar]
- 303. Ye ZC, Sontheimer H (1999) Glioma cells release excitotoxic concentrations of glutamate. Cancer Res 59:4383–4391. [PubMed] [Google Scholar]
- 304. Ye ZC, Rothstein JD, Sontheimer H (1999) Compromised glutamate transport in human glioma cells: reduction‐ mislocalization of sodium‐dependent glutamate transporters and enhanced activity of cystine‐glutamate exchange. J Neurosci 19:10767–10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Yin XL, Hui AB, Liong EC, Ding M, Chang AR, Ng HK (2002) Genetic imbalances in pleomorphic xanthoastrocytoma detected by comparative genomic hybridization and literature review. Cancer Genet Cytogenet 132:14–19. [DOI] [PubMed] [Google Scholar]
- 306. Yin XL, Hui AB, Pang JC, Poon WS, Ng HK (2002) Genome‐wide survey for chromosomal imbalances in ganglioglioma using comparative genomic hybridization. Cancer Genet Cytogenet 134:71–76. [DOI] [PubMed] [Google Scholar]
- 307. York GK 3rd, Steinberg DA (2011) Hughlings Jackson's neurological ideas. Brain 134:3106–3113. [DOI] [PubMed] [Google Scholar]
- 308. Yu J, Deshmukh H, Gutmann RJ, Emnett RJ, Rodriguez FJ, Watson MA et al (2009) Alterations of BRAF and HIPK2 loci predominate in sporadic pilocytic astrocytoma. Neurology 73:1526–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Zakrzewska M, Szybka M, Biernat W, Papierz T, Rieske P, Liberski PP, Zakrzewski K (2009) Prevalence of mutated TP53 on cDNA (but not on DNA template) in pleomorphic xanthoastrocytoma with positive TP53 immunohistochemistry. Cancer Genet Cytogenet 193:93–97. [DOI] [PubMed] [Google Scholar]
- 310. Zakrzewski K, Biernat W, Liberski PP, Polis L, Nowoslawska E (2009) Pilocytic astrocytoma as a predominant component of a recurrent complex type DNT. Folia Neuropathol 47:284–288. [PubMed] [Google Scholar]
- 311. Zeng LH, Xu L, Gutmann DH, Wong M (2008) Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol 63:444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]