

Abstract
Platelet‐derived growth factors (PDGFs) are important mitogens for various types of mesenchymal cells, and as such, they exert critical functions during organogenesis in mammalian embryonic and early postnatal development. Increased or ectopic PDGF activity may also cause or contribute to diseases such as cancer and tissue fibrosis. Until recently, no loss‐of‐function (LOF) mutations in PDGF or PDGF receptor genes were reported as causally linked to a human disease. This changed in 2013 when reports appeared on presumed LOF mutations in the genes encoding PDGF‐B and its receptor PDGF receptor‐beta (PDGF‐Rβ) in familial idiopathic basal ganglia calcification (IBGC), a brain disease characterized by anatomically localized calcifications in or near the blood microvessels. Here, we review PDGF‐B and PDGF‐Rβ biology with special reference to their functions in brain–blood vessel development, pericyte recruitment and the regulation of the blood–brain barrier. We also discuss various scenarios for IBGC pathogenesis suggested by observations in patients and genetically engineered animal models of the disease.
Keywords: Pericytes
Platelet‐Derived Growth Factors (PDGFs) and PDGF Receptors
PDGF was one of the first growth factors to become characterized structurally and functionally [reviewed in 24]. Its release from platelets in conjunction with the platelet release reaction, its presence in serum and its growth factor activity on fibroblasts and smooth muscle cells provided the rationale for an early assumption that PDGF functions as a wound hormone 24. Although this role has yet to be formally demonstrated, tumor biology studies, performed mainly in the 1980s, and subsequent analyses by transgenic and gene targeting techniques in mice from the 1990s and 2000s, established that PDGF has several important functions in health, such as in organogenesis during embryonic and postnatal development [reviewed in 3], and in disease, such as cancer 23 and tissue fibrosis 35.
As the original isolation and characterization of what in the late 1970s appeared to be a homogenous PDGF protein 25, subsequent cloning work, and the more recent analysis of genomes and transcriptomes, have completed the genetic and molecular PDGF landscape in mammals. Human platelet‐derived PDGF was first characterized as a dimer of two polypeptide chains, A and B, the complementary DNAs (cDNAs) of which were subsequently cloned and shown to be the products of two distinct genes, PDGFA and PDGFB in human (Pdgfa and Pdgfb in mouse). In the 2000s, the family of PDGF genes expanded further to encompass PDGFC and PDGFD. The four encoded polypeptides, PDGF‐A, PDGF‐B, PDGF‐C and PDGF‐D, make up biologically active dimers through disulfide bridging. In addition to the originally characterized PDGF‐AB heterodimer, four homo‐dimers (PDGF‐AA, PDGF‐BB, PDGF‐CC and PDGF‐DD) have been demonstrated and functionally compared. They bind to and exert their activity through two receptor tyrosine kinases (RTKs), PDGF receptor‐α (PDGF‐Rα) and PDGF‐Rβ. These too are subunits of biologically active receptor dimers (αα, αβ or ββ) and are encoded by two different genes, PDGFRA and PDGFRB, in the human genome (Pdgfra and Pdgfrb in mouse). The biochemical and signaling properties of PDGF‐Rα and PDGF‐Rβ have been reviewed elsewhere 24.
Tumorigenic Roles of PDGF‐B and PDGF‐Rβ
The present review focuses on PDGFB, PDGFRB and their newly discovered roles in idiopathic basal ganglia calcification (IBGC). Therefore, the other PDGFs and PDGF‐Rα will not be further introduced here. The interested reader is referred to other recent reviews. Early biochemical characterization of the PDGF receptor (later known as PDGF‐Rβ) established it as the second known receptor to be associated with tyrosine kinase activity 18 (after epidermal growth factor receptor), which at the time (early 1980s) was an enzymatic activity abundantly associated with retroviral carcinogenesis. Also, PDGF‐B came into the limelight at approximately the same time (1983) through the discovery that it was homologous to (and in fact encoded by) c‐sis, the cellular homologue of the simian sarcoma virus oncogene, v‐sis 17, 91. Subsequent work established that the oncogenic properties of v‐sis reflected the autocrine activation of PDGF receptors by a seemingly normal PDGF‐BB molecule 37. Hence, PDGF‐B was the first oncogene product with an elucidated biological function, namely a growth factor activity. The nature of sis/PDGF‐B oncogenic transformation is well illustrated by the human skin tumor dermatofibrosarcoma protuberans (DPs). The primary cause of DP is a chromosomal translocation, resulting in the placement of PDGF‐B‐encoding DNA sequences under the transcriptional control of Collagen‐1 (COL1A1) gene sequences 77. Thus, the COL1A1/PDGFB gene fusion results in PDGF‐BB synthesis in cells normally expressing type 1 collagen, that is, fibroblasts/fibrocytes. Such cells also express PDGF receptors, including PDGF‐Rb, and thus, the basis for an autocrine growth stimulatory loop is established 78. DP is a slow‐growing non‐metastatic skin fibrosarcoma, in which autocrine PDGF‐B synthesis is probably not only an initiating lesion but likely also maintains the growth of the established tumor. DP, similar to some other cancers where PDGFs and or PDGFRs have been implicated as transforming protein, is thus based on somatic gain‐of‐function (GOF) mutations. Recently, putative activating germline mutations in PDGFRB were reported as a genetic cause of infantile myofibromatosis, a syndrome characterized by the formation of multiple connective tissue tumors, in skin and in visceral organs 13, 52. In summary, at present, there is evidence for both somatic and inherited GOF mutations in PDGFB and PDGFRB in human cancer and cancer‐like syndromes.
Physiological Roles of PDGF‐B and PDGF‐Rβ
Many of the known functions of PDGF‐B and PDGF‐Rβ have been revealed by targeted knockout of Pdgfb and Pdgfrb in mice 3, 29. Whereas initial publications described renal, cardiovascular, placental and hematological defects in Pdgfb and Pdgfrb null mice 42, 83, later analyses established at least one common cellular denominator of these pathologies: the failure of expansion of vascular mural cells in conjunction with angiogenic growth of blood vessels during embryonic and early postnatal life 27, 45. In this process, PDGF‐B is synthesized and released from angiogenic endothelial cells to act on neighboring PDGF‐Rβ positive mural cells [pericytes and vascular smooth muscle cells (VSMCs)] to allow their proliferation and/or co‐migration along newly forming angiogenic sprouts. In the absence of either PDGF‐B or PDGF‐Rβ, this process fails, and the resulting vasculature is largely devoid of pericytes. Some mural cell formation nevertheless occurs in these mutants, particularly around the major trunk vessels (eg, the aorta and its proximal branches) 27, which probably explains why these mutants survive early embryogenic development and develop lethal cardiovascular problems first around birth.
In order to allow the analysis of postnatal functions of PDGF‐B and PDGF‐Rβ, other mutations in Pdgfb and Pdgfrb have been generated that change the function of the respective gene 10, 28, 46, 65, 86, or are inducible null mutations 10, 19. For Pdgfb, one such mutation that has been extensively used to analyze postnatal roles of PDGF‐B and pericytes is the Pdgfb retention motif knockout mouse (Pdgfbret/ret), in which a targeted mutation at the Pdgfb locus deletes a C‐terminal motif (the retention motif). This motif binds heparan sulfate proteoglycans and its loss leads to the formation of a PDGF‐BB molecule with retained receptor binding/activating ability. However, its diffusible nature likely leads to lower concentrations of PDGF‐BB near the producer cell 1, 46. Endothelial cells have proteoglycans on their cell surface, which likely retains PDGF‐BB near to its site of secretion. Pdgfbret/ret mice show reduced pericyte coverage of their blood vessels, and moreover, the remaining pericytes appear partially detached from the endothelial cells. The interpretation of these results is that the PDGF‐B retention motif is needed for the proper presentation of endothelium‐derived PDGF‐BB to the nearby pericytes.
Usage of Pdgfbret/ret mice, tissue‐specific Pdgfb knockouts, compound transgenics (Pdgfb null mice complemented with endothelial‐specific Pdgfb expression transgenes) or Pdgfrb mutant mice where individual amino acid residues or entire domains have been replaced, have pinpointed roles of PDGF‐B and PDGF‐Rβ in the postnatal development and/or function of the kidneys 46, heart 10, retina 19, 46 and brain 6. At present, all of these roles appear to involve pericytes in the respective organs. For example, the role for PDGF‐B‐PDGF‐Rβ in the kidney appears to be primarily to stimulate the recruitment of mesangial cells—the pericytes of the kidney glomeruli—into the glomerular tuft 44. However, it should be remembered, and will be discussed further below, that there may be other functions of PDGF‐B and PDGF‐Rβ that have gone undetected in studies of vascular development. For example, both PDGF‐B and PDGF‐Rβ have been reported to be expressed in neurons, and PDGF‐Rb has been shown to play a role in neural stem/progenitor cells in vitro 80, 92. Whereas the physiological significance of this expression pattern is presently unclear, several studies have implicated a role for PDGF‐B and PDGF‐Rβ in neuroprotection in vivo 34, 73, as well as in memory, cognitive function and socio‐emotional activity 33, 67, 92.
Functions of Pericytes
Clearly, one of the key physiological roles of the PDGF‐B/PDGF‐Rβ signaling axis is to regulate pericyte formation and/or recruitment during blood vessel morphogenesis. During the development, pericytes are required for the formation of quiescent and durable vessels. In early Pdgfb or Pdgfrb null embryos, the absence of pericytes leads to endothelial hyperplasia 26. As embryonic development proceeds, this hyperplasia persists, eventually causing the formation of increased diameter capillaries, which are also irregularly shaped and locally bulging into micro‐aneurysms. Extensive vascular leakage probably explains the edema and multiple microvascular hemorrhages observed at this age. Similar vascular abnormalities, albeit milder, develop in postnatal viable Pdgfb mutants. These mutants have not yet been excessively explored, but published work demonstrates abnormalities in cardiovascular physiology 63, renal filtration 10, liver sinusoidal permeability 68, retinal morphology 19, 46 and blood–brain barrier (BBB) function 5. Pdgfbret/ret mice have also been assessed for tumor growth and tumor vessel formation and function 2, 60.
A number of functions have been attributed to pericytes in the adult vasculature, including blood flow regulation, stem/progenitor cell function in tissue repair, the formation of fibrogenic cells in tissue fibrosis and scarring [reviewed in 5], in immune cell trafficking across the vessel wall 84, 85 and in the regulation of the BBB 6, 9, 15. The reader is referred to other review literature for a more comprehensive overview of the many proposed roles for pericytes 5, but it is worth pointing out that the identification and definition of pericytes remains a problem in the field. Several types of mesenchymal cells are present in the blood vessel wall, and it would not be meaningful to assign the term pericyte to all of them. The classical definition of a pericyte is a cell embedded in the endothelial basement membrane. However, this definition is not practical as it requires transmission electron microscopy analysis. In recent years, therefore, the pericyte concept has evolved to designate capillary wall cells expressing certain markers, including PDGF‐Rβ, desmin, NG2, CD13 and a few others 4, 5. However, none of these markers are specific for pericytes. PDGF‐Rβ and CD13, for example, are also expressed by fibroblasts; desmin is expressed by other types of muscle cells; and NG2 by certain glia and epithelial cell types. Therefore, pericytes are commonly defined (or described) by a combination of features, including anatomical location and molecular marker expression. A recent example of nomenclature ambiguity concerns the origin of scar‐forming cells in traumatic spinal cord injury in the mouse. Whereas one study designated the origin of the scar‐forming cell to a subpopulation of pericytes (“type A pericytes”) using a combination of GLAST‐Cre fate mapping, and PDGF‐Rβ and PDGF‐Rα expression 22, another study concluded based on expression of Col1a1, PDGF‐Rβ, CD13 and the absence of NG2, that the spinal cord scar‐forming cells have the hallmarks of a perivascular fibroblast 82.
Pericytes and the BBB
Using several models of Pdgfb and Pdfrb mouse mutants, it has been demonstrated that pericytes play a critical and specific role in the BBB 6, 9, 15. Although some uncertainties still exist concerning which modalities of the BBB that are regulated by pericytes, available evidence point to a possible role in endothelial junction formation 9, 15, a prominent role in the (negative) regulation of endothelial transcytosis 6, 15, a specific role in astrocyte end‐foot polarization 6, a (likely negative) role in immune cell transmigration across the BBB 15 and a limited (yet specific) role in the regulation of endothelial expression of certain transporters 6. Importantly, pericytes do not appear to regulate the major central nervous system (CNS)‐specific physical barrier and molecular properties of the BBB. The pericyte‐deficient brain vessels express the general molecular hallmarks of BBB endothelium, and their endothelial junctions appear normal by ultrastructural analysis 6. However, a wide range of tracers differing by molecular size and chemical composition were shown to pass from blood to brain by vesicular transport in pericyte‐deficient mice. The size of the vesicles and their non‐selectivity with regard to cargo suggest that they are engaged in pinocytosis. Intriguingly, the treatment of pericyte‐deficient mice with the tyrosine kinase inhibitor imatinib led to vesicle trapping inside the brain endothelial cells, suggesting a specific block of the exocytosis step of the transcytosis 6. How pericytes control endothelial transcytosis in the brain is unclear, but conceivably, it involves a paracrine or juxtacrine signal. Likewise, the molecular machinery involved in transendothelial transport of pinocytotic vesicles is not known but should involve a molecular target of imatinib.
CNS pericytes are sandwiched between endothelial cells and astrocyte end‐feet, and as such, they are strategically positioned to regulate the function of both cell types. Indeed, astrocyte end‐feet are abnormally polarized in pericyte‐deficient vessels 6. Normally, certain astrocyte proteins become uniquely localized at the end‐foot–vessel interface, including the water channel aquaporin 4 and the basement membrane component laminin alpha 2 (Lama2). In the absence of pericytes, aquaporin 4 was mis‐localized to appear throughout the astrocytes, and Lama2 was largely absent from the vessels devoid of pericyte contact 6. The significance of these astrocyte abnormalities for the BBB dysfunction in pericyte‐deficient mice is unclear.
IBGCs
Ectopic intracranial brain calcification is the most common incidental finding in patients undergoing computed tomography (CT) scans 50. These calcifications may not be associated with clinical findings; however, in some cases, they are critical for the diagnosis of disease. Familial idiopathic basal ganglia calcification (FIBGC/IBGC) is a rare autosomal neurodegenerative disease with dominant inheritance, which manifests with calcifications in different brain regions. Typically, radiological neuroimaging demonstrates bilateral basal ganglia calcifications. Other affected regions include the cerebellar gyri, thalamus, cortical white and gray matter and the brain stem 51. IBGC is clinically heterogeneous and clinical features include Parkinsonism, psychosis, seizures, migraine, cognitive decline and impairment, and cerebellar involvement (ataxia) 51. Although brain calcifications are present, some affected individuals are asymptomatic. However, radiological penetrance of the disease is 100% at the age of 50 81.
Importantly, disturbances in systemic mineral metabolism are not seen in IBGC, and calcium, phosphorus and parathyroid hormone levels are normal. Historically, this rare disease was first described by Delacour in 1850 16. Even though IBGC is a rare disease (the prevalence is not known), there are 35 different names in literature that have been used to describe this condition 51. The most common is “Fahr's disease”; however, this name is now considered a misnomer. Fahr was not the first to describe the disease and the patient he described likely did not suffer from IBGC 51. Recently, “familiar primary brain calcification” was suggested as a new name for this neurodegenerative disease 81. However, in order to avoid confusion, we continue to use “FIBGC” or “IBGC” in the present review, as “familiar primary brain calcification” is not yet established, and most of the current literature and databases on human genetic diseases (eg, omim.org) continue to apply the terms “FIBGC” or “IBGC.”
Pathophysiology of IBGC
The pathological findings obtained from rare autopsy cases of IBGC reveal evidence of microvascular insufficiency. Calcified nodules can be observed not only along capillaries but also in the neuropil 40, 54, 62. In addition, calcium precipitates have been reported on neurons and astrocytes 54. The precipitates contain hydroxyapatite, which is the main mineral component of the bone 79. In the case of normal bone formation, hydroxyapatite crystals accumulate on a permissive matrix rich in collagen fibrils. Interestingly, neuronal excitotoxicity has been shown to induce intracellular calcium precipitation in neurons 49. Although gliosis and neuronal loss in severely affected areas have been reported 40, neurons generally remain preserved in IBGC 54. In addition, extravasation of plasma proteins and signs of neuroinflammation are observed in IBGC 54. The use of non‐invasive imaging techniques such as magnetic resonance imaging (MRI) has confirmed disturbances in microvascular function, showing the presence of vasogenic edema 21. Also, dilatation of the lateral ventricles has been reported 40. Regional blood flow changes have been reported in patients with severe calcifications in basal ganglia 66, 76, 87. Reduction in glucose metabolism in the basal ganglia by fludeoxyglucose‐positron emission tomography imaging has been demonstrated 69, 76. In addition, pre‐ and post‐synaptic nigrostriatal dopaminergic dysfunction has been indicated in IBGC patient with psychiatric symptoms, which may contribute to some of the clinical manifestations 69. There is a considerable heterogeneity of the pathological signs and also of the genetic origin of the IBGC (discussed below). Interestingly, several cases of sporadic and familiar IBGC have been described to have neurofibrillary tangles, Lewy bodies or α‐synucleinopathy 31, 61, 74. However, it should be remembered that much of the reports on IBGC pathology and neurology appeared before the any genetic causes of IBGC were known, and may hence represent diverse disease etiologies. Recently, Baker and co‐workers reported two new mutations in one of the IBGC genes, SLC20A2 (see below), in a collection of autopsy samples where excessive basal ganglia calcification was seen in a setting of another neurodegenerative disease (asymmetric Parkinsonism and autosomal‐dominant dystonia) 8.
Genetics of IBGC
During the last years, a tremendous progress has been made in identifying the underlying genetic causes of FIBGC. The first FIBGC gene was reported in 2012. Seven different mutations in the sodium‐dependent P(i) transporter SLCA20A2 (also called PiT2) were reported in different FIBGC families 90. The involvement of SLC20A2 implicates that changes in intracellular phosphate transport may cause FIBGC 90. Mutations in the SLCA20A2 gene are a major cause of FIBGC, representing approximately 50% of investigated families 12, 30, 90, 93, 95. Although not extensively supported by experiments, available data nevertheless suggest that SLCA20A2 mutations in FIBGC are loss‐of‐function (LOF) mutations, and the proposed disease mechanism is the loss of a functional copy of the SLC20A2 gene resulting in insufficient production of SLC20A2/Pit2 protein (haploinsufficiency). The recent identification in an IBGC case of a large genomic deletion in SLC20A2 supports this conclusion 8.
In 2013, two additional FIBGC genes were reported. A mutation (p.Leu658Pro) in PDGFRB was identified in one family with FIBGC and was predicted to be damaging for PDGF‐Rβ function 58. Two additional mutations in PDGFRB were found in sporadic IBGC cases (p.Arg987Trp and p.Glu1071Val) 57, 58, but the functional consequences of these mutations are less clear (Figure 1A). The third identified FIBGC gene was PDGFB 39 (Figure 1B). PDGFB mutations were found in six different families, all predicted to result in LOF effects 39. A recently described heterozygous intragenic deletion in PDGFB involving exons 2‐5 (which encode critical parts of PDGFB protein) in an IBGC case supports haploinsufficiency as a pathogenic mechanism 59. A de novo nonsense mutation in PDGFB exon 4 (p.Gln147*) has further been described in one IBGC patient 56. Until now, PDGFB or PDGFRB mutations (Figure 1) have been identified in approximately 20% of IBGC cases, suggesting that additional IBGC genes exist beyond SLC20A2, PDGFRB and PDGFB.
Figure 1.
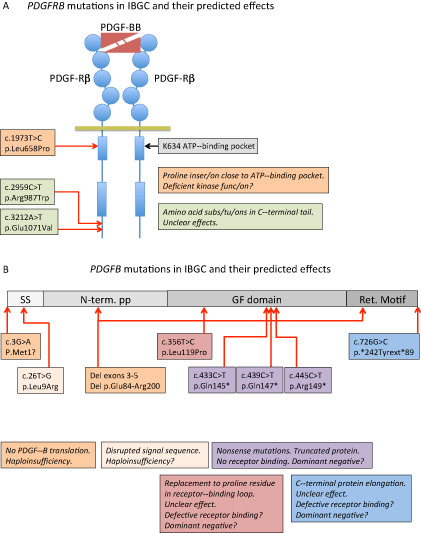
Schematic representation of the mutations reported to date in PDGFB and PDGFRB and their predicted effects on protein function. A. A schematic representation of PDGF‐Rβ with PDGF‐BB bound to the extracellular ligand‐binding domain. Circles in PDGF‐Rβ indicate immunoglobulin domains and boxes represent kinase domains. The three idiopathic basal ganglia calcification (IBGC) mutations reported to date are indicated by type and approximate position. Note that one of the mutations is predicted to be damaging to the receptor functions, as it introduces a proline residue close to the ATP‐binding pocket of the receptor kinase. Proline residues are known to have strong effects on the three‐dimensional (3D) structure in this region. B. The PDGF‐B precursor protein is indicated in different shades of gray. SS, signal sequence for secretion; N‐term. PP, amino‐terminal pro‐peptide; GF domain, growth factor domain; Ret. Motif, heparan sulfate proteoglycan‐binding extracellular matrix retention motif. The nature and approximate position of the mutations are indicated and their effects described in boxes with the same color. Haploinsufficiency indicates that mere loss of one functional copy of the PDGFB gene is disease causing. For the mutants with putative residual cysteine residues engaged in ligand dimerization, a dominant‐negative effect might be conceived, which in theory predicts a 75% reduction of the amount produced functional PDGF‐BB.
As mentioned earlier, the IBGC is a genetically and clinically heterogeneous disease with the variable clinical penetrance and it is unclear as to what extent different genetic, pathological and neurological features are causally correlated. In one study, calcifications were found to be more severe in symptomatic vs. asymptomatic individuals, but the quantification of the calcifications by analyzing CT images by visual examination was found to be insufficient to predict the clinical severity 57.
Mouse Models of IBGC and the Pathogenic Mechanism of IBGC
Currently, it is unclear how haploinsufficiency of SLCA20A2, PDGFB or PDGFRB leads to ectopic brain calcification. It is also not known why mutations in genes belonging to structurally and functionally different protein families—growth factor and its receptor (PDGFB/PDGFRB), on the one hand, and the inorganic phosphate transporter (SLC20A2), on the other hand—lead to the same pathology. As discussed previously, the PDGF‐B/PDGF‐Rβ signaling pathway is important for pericyte recruitment and the integrity of the BBB; however, very little is known about the function and expression pattern of SLC20A2 in the brain.
Analysis of mouse models that mimic certain aspects of IBGC will advance our understanding of pathogenesis of this neurodegenerative disease. Currently, three mouse lines with genetically modified expression of IBGC genes have reported to develop intracranial brain calcifications 36, 39. Two of these are Pdgfb hypomorphs (pdgfb ret/ret and pdgfb−/−, Tie2CreR26hPDGFB). These mice develop brain calcifications with similar anatomical location, appearance and composition as those in human IBGC 39. In further similarity to human IBGC, brain calcifications progress with time in the mentioned mice. It was also demonstrated that endothelial expression of PDGF‐B is protective of brain calcifications. In addition, the severity of calcification correlates with the degree of pericyte deficiency and BBB dysfunction 39. Thus, the analysis of mouse Pdgfb hypomorphs indicates that IBGC may be caused by pericyte deficiency, leading to BBB disruption and the subsequent formation of calcified lesions. As noted earlier, evidence of a defective BBB has also been reported in IBGC autopsy cases. Other connections between BBB deficiency and brain calcification have been noted. The human autosomal recessive neurodevelopmental disorder band‐like calcification with simplified gyration and polymicrogyria, caused by mutations in the OCLN gene, which encoding the endothelial tight junction protein occludin, is one such example 64. Blood vessel‐associated calcifications are also seen in brains of occludin knockout mice 70. Thus, loss of the occludin gene in humans and mice leads to the formation of brain calcifications that are potentially mediated by changes in BBB permeability. Another human genetic disease—cerebral cavernous malformation (CCM)—caused by mutations in the genes encoding endothelial junctional proteins CCM1, CCM2 or CCM3 affects the venous capillary bed and is also associated with BBB breakdown and calcifications 20.
Thus, whereas it is possible that the BBB dysfunction and increased vascular permeability in the brain contributes to the formation of brain calcifications in Pdgfb hypomorphs, it is currently not known by which mechanism brain calcifications form in Slc20a2 knockout, the third mouse model of IBGC 36. There are three families of sodium phosphate co‐transporters in mammals. The type III family comprises PiT2 and PiT1, which are thought to play a less important role in systemic phosphate homeostasis but seem to be involved in maintaining cellular phosphate homeostasis 41. The expression pattern of SLC20A2/PiT2 has been reported to be ubiquitous, but very few studies have investigated PiT2 expression at the cell type level. A recent immunohistochemical study reported PiT2 expression in mouse brain in neurons, astrocytes and endothelial cells 32. However, the specificity of the used polyclonal antibody staining was not proven. PiT2 has been shown to be expressed at high level also in VSMC 89. As SLC20A2 expression is ubiquitous, it is puzzling why SLC20A2 loss‐of‐mutations cause vessel‐associated calcifications restricted to certain brain regions. Adding to the complexity, PiT1 has been shown to be necessary for pathological vascular calcification induced by high P(i)‐induced, a process in which VSMC acquired an osteochondrogenic phenotype in vitro 43. It is paradoxical why PiT1 GOF causes a similar phenotype as Pit2 LOF.
Thus, there are many unanswered questions about the pathogenesis of IBGC. As mentioned previously, it is presently unclear if the severity of calcified lesions correlates with the clinical manifestations of the disease 57. Are calcified lesions causal for progressive neurodegeneration, or do they represent an innocuous consequence of another, primary, pathogenic process? IBGC is also accompanied by a strong neuroinflammatory reaction. It is noteworthy that inflammation is associated with calcification also in the peripheral nervous system 11, 14, 48, 71. Neuroinflammation might therefore cause changes in the extracellular environment, favoring the generation of bone‐forming cells and the subsequent formation of a calcified matrix (Figure 2). While it is unknown if bone‐forming cells are at all generated in association with IBGC lesions, both pericytes and endothelial appear to have the capacity to differentiate into bone‐forming cells 7, 75. It is therefore not farfetched to speculate that mutations in IBGC genes may trigger the generation of bone‐forming cells locally in the brain vasculature. Pericytes can form extracellular calcifications containing hydroxyapatite under in vitro conditions, a process that is dependent on culture conditions 72. Whether pericytes can become bone‐forming in vivo is not known. On the contrary, recent studies on fibrodysplasia ossificans progressiva, a fatal disease characterized by extensive extraskeletal bone formation, have demonstrated the endothelium as a source of bone‐forming cells 53, 94. In addition to endothelial cells and pericytes, one may consider the possibility that also other cell types at neurovascular unit might be able to transdifferentiate into bone‐forming cells, such as microglia or perivascular fibroblasts. Microglia are the resident macrophages of the CNS, and macrophage‐derived matrix vesicles have been shown to contribute to accelerated microcalcification in chronic renal disease 55. Interestingly, GLAST‐Cre expressing cells were also shown to give rise to bone‐forming cell in a mouse model of heterotopic calcification 38. Although the identity of the GLAST‐Cre positive cells in the periphery remains unclear, GLAST‐Cre positive cells in the CNS have been reported to be pericytes, or possibly blood vessel‐associated fibroblasts 22, 82.
Figure 2.
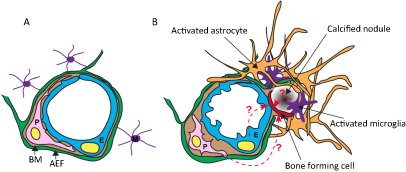
Cartoon illustrating the changes at the neurovascular unit (NVU) in idiopathic basal ganglia calcification (IBGC). The illustration takes into account observations made in genetically engineered mouse models of IBGC. A. Cross section of a normal NVU. Endothelial cells (E) are surrounded by a basement membrane (BM), pericytes (P) and astrocyte end‐feet (AEF). Microglia (M) make contacts to the NVU through cytoplasmic processes. B. Altered features of the NVU in IBGC. Calcified nodules are associated with blood vessels and are surrounded by reactive astrocytes and microglia. A hypothetical bone‐forming cell (red) initiates and propagates the formation and growth of the calcified nodule. The origin of the bone‐forming cell is unclear, but available literature provides precedence for several possible origins, as indicated (dashed line with question mark). Mouse models of IBGC provide a correlation between pericyte deficiency and BBB disruption, on the one hand, and brain calcification, on the other hand. The altered properties of endothelial cells and pericytes are indicated by their changed shape.
It is also unclear why presumable haploinsufficency of SLC20A2, PDGFB or PDGFRB leads to development of calcifications and progressive neurodegeneration in humans, whereas mice possessing only a single copy of functional allele of Slc20a2, Pdgfb or Pdgrfb (ie, heterozygous knockouts) do not develop brain calcifications 36, 39 and (Lebouvier, Keller and Betsholtz, unpub. obs.). Thus, more work is needed to characterize the IBGC mutations. Are these mutations LOF mutations or have mutated proteins acquired a new dominant‐negative trait? For example, a truncated form of parkin (Q311X), the most frequently mutated gene causing early onset autosomal recessive Parkinsonism, has acquired a toxic neomorphic function, leading to death of dopaminergic neurons in mice 47. However, as already discussed earlier, two recent studies have reported heterozygotes genetic deletions in SLC20A2 and PDGFB gene, confirming the haploinsufficiency as causal of IBGC 8, 59. Furthermore, the IBGC case with PDGFB deletion presented with severe leukoencephalopathy in addition to brain calcification, indicating that a pure LOF allele may produce the worst outcome 59, hence arguing against an acquisition of a toxic neomorphic functions as a disease mechanism.
Conclusion
Further analysis of mouse models of IBGC will most likely bring new insights into the pathogenic mechanism of formation of ectopic brain calcifications and lead to strategies on how to interfere with the formation of ectopic brain calcifications. Such studies should also be directed in understanding the cause of neurodegeneration in this disease. In addition to providing insights into IBGC pathogenesis, the mouse models might also shed light on other causes of brain calcification. Certain brain regions commonly become calcified during aging, including the pineal gland, choroid plexus and basal ganglia. Calcifications in the pineal gland are very common and are often used as anatomical landmarks in radiological analysis. The calcified structures in the pineal gland are also called “corpora arenacea” or “brain sand” 88. Although brain calcification during aging is considered nonpathological, brain calcifications are nevertheless associated with various brain pathologies in addition to IBGC 50, 51. These include infections (eg, viral encephalitis), metabolic changes (eg, chronic kidney disease, thyroid hormone imbalance), vasculopathies (eg, atherosclerosis, cavernomas), tumors (eg, oligodendroglioma, astrocytoma), neurodegeneration (eg, Parkinsonism, Huntington and Alzheimer's disease) and familial diseases (eg, neurofibromatosis, Gorlin's syndrome, MELAS syndrome). Brain calcifications can also form as a result of toxic injury (eg, carbon monoxide poisoning, radio‐ and chemotherapy). The pathogenic mechanisms involved in brain calcifications in these diverse conditions are not understood, except in the cases of systemic imbalance of calcium and phosphate caused by different diseases (eg, kidney failure, hypo‐ or hyperparathyroidism). It is possible, and even likely, that in spite of wide‐ranging etiologies, brain calcification engages common pathogenic mechanisms, which may also constitute ideal targets for therapy.
Acknowledgments
We thank Dr Elisabeth Rushing for valuable comments. We are also indebted to Uppsala University and Karolinska Institutet for financial support. Cited own work was supported by an advanced grant from the European Research Council (BBBARRIER), the Knut and Alice Wallenberg Foundation, the Torsten and Ragnar Söderberg Foundation, the Leducq Foundation and the Swedish Cancer Society.
References
- 1. Abramsson A, Kurup S, Busse M, Yamada S, Lindblom P, Schallmeiner E et al (2007) Defective N‐sulfation of heparan sulfate proteoglycans limits PDGF‐BB binding and pericyte recruitment in vascular development. Genes Dev 21:316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abramsson A, Lindblom P, Betsholtz C (2003) Endothelial and nonendothelial sources of PDGF‐B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest 112:1142–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andrae J, Gallini R, Betsholtz C (2008) Role of platelet‐derived growth factors in physiology and medicine. Genes Dev 22:1276–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Armulik A, Abramsson A, Betsholtz C (2005) Endothelial/pericyte interactions. Circ Res 97:512–523. [DOI] [PubMed] [Google Scholar]
- 5. Armulik A, Genové G, Betsholtz C (2011) Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21:193–215. [DOI] [PubMed] [Google Scholar]
- 6. Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C et al (2010) Pericytes regulate the blood‐brain barrier. Nature 468:557–561. [DOI] [PubMed] [Google Scholar]
- 7. Armulik A, Genove G, Betsholtz C (2011) Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21:193–215. [DOI] [PubMed] [Google Scholar]
- 8. Baker M, Strongosky A, Sanchez‐Contreras M, Yang S, Ferguson W, Calne D et al (2014) SLC20A2 and THAP1 deletion in familial basal ganglia calcification with dystonia. Neurogenetics 15:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R et al (2010) Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68:409–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bjarnegård M, Enge M, Norlin J, Gustafsdottir S, Fredriksson S, Abramsson A et al (2004) Endothelium‐specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development 131:1847–1857. [DOI] [PubMed] [Google Scholar]
- 11. Chakrabarty P, Ceballos‐Diaz C, Lin W‐L, Beccard A, Jansen‐West K, McFarland N et al (2011) Interferon‐γ induces progressive nigrostriatal degeneration and basal ganglia calcification. Nat Neurosci 14:694–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen W‐J, Yao X‐P, Zhang Q‐J, Ni W, He J, Li H‐F et al (2013) Novel SLC20A2 mutations identified in southern Chinese patients with idiopathic basal ganglia calcification. Gene 529:159–162. [DOI] [PubMed] [Google Scholar]
- 13. Cheung YH, Gayden T, Campeau PM, LeDuc CA, Russo D, Nguyen VH et al (2013) A recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am J Hum Genet 92:996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crow Y, Hayward B, Parmar R, Robins P, Leitch A, Ali M et al (2006) Mutations in the gene encoding the 3'‐5’ DNA exonuclease TREX1 cause Aicardi‐Goutières syndrome at the AGS1 locus. Nat Genet 38:917–920. [DOI] [PubMed] [Google Scholar]
- 15. Daneman R, Zhou L, Kebede AA, Barres BA (2010) Pericytes are required for blood‐brain barrier integrity during embryogenesis. Nature 468:562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Delacour A (1850) Ossification des capillaires du cerveau. Ann Med Psychol 2:458–461. [Google Scholar]
- 17. Doolittle RF, Hunkapiller MW, Hood LE, Devare SG, Robbins KC, Aaronson SA et al (1983) Simian sarcoma virus onc gene, v‐sis, is derived from the gene (or genes) encoding a platelet‐derived growth factor. Science 221:275–277. [DOI] [PubMed] [Google Scholar]
- 18. Ek B, Westermark B, Wasteson A, Heldin CH (1982) Stimulation of tyrosine‐specific phosphorylation by platelet‐derived growth factor. Nature 295:419–420. [DOI] [PubMed] [Google Scholar]
- 19. Enge M, Bjarnegård M, Gerhardt H, Gustafsson E, Kalén M, Asker N et al (2002) Endothelium‐specific platelet‐derived growth factor‐B ablation mimics diabetic retinopathy. EMBO J 21:4307–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fischer A, Zalvide J, Faurobert E, Albiges‐Rizo C, Tournier‐Lasserve E (2013) Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends Mol Med 19:302–308. [DOI] [PubMed] [Google Scholar]
- 21. Gomez C, Luque A, Horenstein S (1989) Microvasculopathy may precede idiopathic cerebral calcifications—case report. Angiology 40:67–72. [DOI] [PubMed] [Google Scholar]
- 22. Goritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisen J (2011) A pericyte origin of spinal cord scar tissue. Science 333:238–242. [DOI] [PubMed] [Google Scholar]
- 23. Heldin CH (2013) Targeting the PDGF signaling pathway in tumor treatment. Cell Commun Signal 11:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heldin CH, Westermark B (1999) Mechanism of action and in vivo role of platelet‐derived growth factor. Physiol Rev 79:1283–1316. [DOI] [PubMed] [Google Scholar]
- 25. Heldin CH, Westermark B, Wasteson A (1979) Platelet‐derived growth factor: purification and partial characterization. Proc Natl Acad Sci USA 76:3722–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hellström M, Gerhardt H, Kalén M, Li X, Eriksson U, Wolburg H et al (2001) Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol 153:543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hellström M, Kalén M, Lindahl P, Abramsson A, Betsholtz C (1999) Role of PDGF‐B and PDGFR‐beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 126:3047–3055. [DOI] [PubMed] [Google Scholar]
- 28. Heuchel R, Berg A, Tallquist M, Ahlén K, Reed RK, Rubin K et al (1999) Platelet‐derived growth factor beta receptor regulates interstitial fluid homeostasis through phosphatidylinositol‐3’ kinase signaling. Proc Natl Acad Sci USA 96:11410–11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hoch RV, Soriano P (2003) Roles of PDGF in animal development. Development 130:4769–4784. [DOI] [PubMed] [Google Scholar]
- 30. Hsu S, Sears R, Lemos R, Quintáns B, Huang A, Spiteri E et al (2013) Mutations in SLC20A2 are a major cause of familial idiopathic basal ganglia calcification. Neurogenetics 14:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ikeda M, Tanabe H, Mori T, Komori K, Nakagawa Y, Tanimukai S et al (1994) [A case of atypical presenile dementia]. Nō to Shinkei 46:175–181. [PubMed] [Google Scholar]
- 32. Inden M, Iriyama M, Takagi M, Kaneko M, Hozumi I (2013) Localization of type‐III sodium‐dependent phosphate transporter 2 in the mouse brain. Brain Res 1531:75–83. [DOI] [PubMed] [Google Scholar]
- 33. Ishii Y, Matsumoto Y, Watanabe R, Elmi M, Fujimori T, Nissen J et al (2008) Characterization of neuroprogenitor cells expressing the PDGF beta‐receptor within the subventricular zone of postnatal mice. Mol Cell Neurosci 37:507–518. [DOI] [PubMed] [Google Scholar]
- 34. Ishii Y, Oya T, Zheng L, Gao Z, Kawaguchi M, Sabit H et al (2006) Mouse brains deficient in neuronal PDGF receptor‐beta develop normally but are vulnerable to injury. J Neurochem 98:588–600. [DOI] [PubMed] [Google Scholar]
- 35. Iwayama T, Olson LE (2013) Involvement of PDGF in fibrosis and scleroderma: recent insights from animal models and potential therapeutic opportunities. Curr Rheumatol Rep 15:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jensen N, Schrøder H, Hejbøl E, Füchtbauer E‐M, de Oliveira J, Pedersen L (2013) Loss of function of Slc20a2 associated with familial idiopathic basal ganglia calcification in humans causes brain calcifications in mice. J Mol Neurosci 51:994–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johnsson A, Betsholtz C, Heldin CH, Westermark B (1985) Antibodies against platelet‐derived growth factor inhibit acute transformation by simian sarcoma virus. Nature 317:438–440. [DOI] [PubMed] [Google Scholar]
- 38. Kan LX, Peng CY, McGuire TL, Kessler JA (2013) Glast‐expressing progenitor cells contribute to heterotopic ossification. Bone 53:194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Keller A, Westenberger A, Sobrido M, García‐Murias M, Domingo A, Sears R et al (2013) Mutations in the gene encoding PDGF‐B cause brain calcifications in humans and mice. Nat Genet 45:1077–1082. [DOI] [PubMed] [Google Scholar]
- 40. Kozik M, Kulczycki J (1978) Laser‐spectrographic analysis of the cation content in Fahr's syndrome. Archiv für Psychiatrie und Nervenkrankheiten 225:135–142. [DOI] [PubMed] [Google Scholar]
- 41. Lederer E, Miyamoto K (2012) Clinical consequences of mutations in sodium phosphate cotransporters. Clin J Am Soc Nephrol 7:1179–1187. [DOI] [PubMed] [Google Scholar]
- 42. Levéen P, Pekny M, Gebre‐Medhin S, Swolin B, Larsson E, Betsholtz C (1994) Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev 8:1875–1887. [DOI] [PubMed] [Google Scholar]
- 43. Li X, Yang H‐Y, Giachelli C (2006) Role of the sodium‐dependent phosphate cotransporter, Pit‐1, in vascular smooth muscle cell calcification. Circ Res 98:905–912. [DOI] [PubMed] [Google Scholar]
- 44. Lindahl P, Hellström M, Kalén M, Karlsson L, Pekny M, Pekna M et al (1998) Paracrine PDGF‐B/PDGF‐Rbeta signaling controls mesangial cell development in kidney glomeruli. Development 125:3313–3322. [DOI] [PubMed] [Google Scholar]
- 45. Lindahl P, Johansson BR, Levéen P, Betsholtz C (1997) Pericyte loss and microaneurysm formation in PDGF‐B‐deficient mice. Science 277:242–245. [DOI] [PubMed] [Google Scholar]
- 46. Lindblom P, Gerhardt H, Liebner S, Abramsson A, Enge M, Hellström M et al (2003) Endothelial PDGF‐B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev 17:1835–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lu X‐H, Fleming S, Meurers B, Ackerson L, Mortazavi F, Lo V et al (2009) Bacterial artificial chromosome transgenic mice expressing a truncated mutant parkin exhibit age‐dependent hypokinetic motor deficits, dopaminergic neuron degeneration, and accumulation of proteinase K‐resistant alpha‐synuclein. J Neurosci 29:1962–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maetzler W, Berg D, Funke C, Sandmann F, Stünitz H, Maetzler C et al (2010) Progressive secondary neurodegeneration and microcalcification co‐occur in osteopontin‐deficient mice. Am J Pathol 177:829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maetzler W, Stünitz H, Bendfeldt K, Vollenweider F, Schwaller B, Nitsch C (2009) Microcalcification after excitotoxicity is enhanced in transgenic mice expressing parvalbumin in all neurones, may commence in neuronal mitochondria and undergoes structural modifications over time. Neuropathol Appl Neurobiol 35:165–177. [DOI] [PubMed] [Google Scholar]
- 50. Kıroğlu Y, Çallı C, Karabulut N, Öncel C (2010) Intracranial calcifications on CT. Diagn Interv Radiol 16:263–269. [DOI] [PubMed] [Google Scholar]
- 51. Manyam B (2005) What is and what is not “Fahr's disease”. Parkinsonism Relat Disord 11:73–80. [DOI] [PubMed] [Google Scholar]
- 52. Martignetti JA, Tian LF, Li D, Ramirez MCM, Camacho‐Vanegas O, Camacho SC et al (2013) Mutations in PDGFRB cause autosomal‐dominant infantile myofibromatosis. Am J Hum Genet 92:1001–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Medici D, Shore E, Lounev V, Kaplan F, Kalluri R, Olsen B (2010) Conversion of vascular endothelial cells into multipotent stem‐like cells. Nat Med 16:1400–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Miklossy J, Mackenzie I, Dorovini‐Zis K, Calne D, Wszolek Z, Klegeris A et al (2005) Severe vascular disturbance in a case of familial brain calcinosis. Acta Neuropathol 109:643–653. [DOI] [PubMed] [Google Scholar]
- 55. New SEP, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K et al (2013) Macrophage‐derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res 113:72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nicolas G, Jacquin A, Thauvin‐Robinet C, Rovelet‐Lecrux A, Rouaud O, Pottier C et al (2014) A de novo nonsense PDGFB mutation causing idiopathic basal ganglia calcification with laryngeal dystonia. Eur J Hum Genet [Epub ahead of print; doi: 10.1038/ejhg.2014.9.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nicolas G, Pottier C, Charbonnier C, Guyant‐Maréchal L, Le Ber I, Pariente J et al (2013) Phenotypic spectrum of probable and genetically‐confirmed idiopathic basal ganglia calcification. Brain 136 (Pt 11):3395–3407. [DOI] [PubMed] [Google Scholar]
- 58. Nicolas G, Pottier C, Maltête D, Coutant S, Rovelet‐Lecrux A, Legallic S et al (2013) Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology 80:181–187. [DOI] [PubMed] [Google Scholar]
- 59. Nicolas G, Rovelet‐Lecrux A, Pottier C, Martinaud O, Wallon D, Vernier L et al (2014) PDGFB partial deletion: a new, rare mechanism causing brain calcification with leukoencephalopathy. J Mol Neurosci 53:171–175. [DOI] [PubMed] [Google Scholar]
- 60. Nisancioglu MH, Betsholtz C, Genové G (2010) The absence of pericytes does not increase the sensitivity of tumor vasculature to vascular endothelial growth factor‐A blockade. Cancer Res 70:5109–5115. [DOI] [PubMed] [Google Scholar]
- 61. Nomoto N, Sugimoto H, Iguchi H, Kurihara T, Wakata N (2002) [A case of Fahr's disease presenting “diffuse neurofibrillary tangles with calcification”]. Rinshō Shinkeigaku 42:745–749. [PubMed] [Google Scholar]
- 62. Norman R, Urich H (1960) The influence of a vascular factor on the distribution of symmetrical cerebral calcifications. J Neurol Neurosurg Psychiatry 23:142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nystrom HC, Lindblom P, Wickman A, Andersson I, Norlin J, Faldt J et al (2006) Platelet‐derived growth factor B retention is essential for development of normal structure and function of conduit vessels and capillaries. Cardiovasc Res 71:557–565. [DOI] [PubMed] [Google Scholar]
- 64. O'Driscoll M, Daly S, Urquhart J, Black G, Pilz D, Brockmann K et al (2010) Recessive mutations in the gene encoding the tight junction protein occludin cause band‐like calcification with simplified gyration and polymicrogyria. Am J Hum Genet 87:354–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Olson LE, Soriano P (2011) PDGFRβ signaling regulates mural cell plasticity and inhibits fat development. Dev Cell 20:815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Paschali A, Lakiotis V, Messinis L, Markaki E, Constantoyannis C, Ellul J et al (2009) Dopamine transporter SPECT/CT and perfusion brain SPECT imaging in idiopathic basal ganglia calcinosis. Clin Nucl Med 34:421–423. [DOI] [PubMed] [Google Scholar]
- 67. Phuong THN, Nakamura T, Hori E, Urakawa S, Uwano T, Zhao JJ et al (2011) Cognitive and socio‐emotional deficits in platelet‐derived growth factor receptor‐beta gene knockout mice. PLoS ONE 6:e18004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Raines SM, Richards OC, Schneider LR, Schueler KL, Rabaglia ME, Oler AT et al (2011) Loss of PDGF‐B activity increases hepatic vascular permeability and enhances insulin sensitivity. Am J Physiol Endocrinol Metab 301:E517–E526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Saito T, Nakamura M, Shimizu T, Oda K, Ishiwata K, Ishii K et al (2010) Neuroradiologic evidence of pre‐synaptic and post‐synaptic nigrostriatal dopaminergic dysfunction in idiopathic basal ganglia calcification: a case report. J Neuroimaging 20:189–191. [DOI] [PubMed] [Google Scholar]
- 70. Saitou M, Furuse M, Sasaki H, Schulzke J, Fromm M, Takano H et al (2000) Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell 11:4131–4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Salisbury E, Rodenberg E, Sonnet C, Hipp J, Gannon F, Vadakkan T et al (2011) Sensory nerve induced inflammation contributes to heterotopic ossification. J Cell Biochem 112:2748–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schor A, Allen T, Canfield A, Sloan P, Schor S (1990) Pericytes derived from the retinal microvasculature undergo calcification in vitro . J Cell Sci 97 (Pt 3):449–461. [DOI] [PubMed] [Google Scholar]
- 73. Shen J, Ishii Y, Xu GH, Dang TC, Hamashima T, Matsushima T et al (2012) PDGFR‐beta as a positive regulator of tissue repair in a mouse model of focal cerebral ischemia. J Cereb Blood Flow Metab 32:353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shibayama H, Kobayashi H, Iwase S, Nakagawa M, Marui Y, Kayukawa Y et al (1986) Unusual cases of presenile dementia with Fahr's syndrome. Jpn J Psychiatry Neurol 40:85–100. [DOI] [PubMed] [Google Scholar]
- 75. Shore E, Xu M, Feldman G, Fenstermacher D, Cho T‐J, Choi I et al (2006) A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 38:525–527. [DOI] [PubMed] [Google Scholar]
- 76. Shoyama M, Shouyama M, Kitabata Y, Kaku T, Shinosaki K (2005) Evaluation of regional cerebral blood flow in Fahr disease with schizophrenia‐like psychosis: a case report. AJNR Am J Neuroradiol 26:2527–2529. [PMC free article] [PubMed] [Google Scholar]
- 77. Simon MP, Pedeutour F, Sirvent N, Grosgeorge J, Minoletti F, Coindre JM et al (1997) Deregulation of the platelet‐derived growth factor B‐chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant‐cell fibroblastoma. Nat Genet 15:95–98. [DOI] [PubMed] [Google Scholar]
- 78. Sjöblom T, Shimizu A, O'Brien KP, Pietras K, Dal Cin P, Buchdunger E et al (2001) Growth inhibition of dermatofibrosarcoma protuberans tumors by the platelet‐derived growth factor receptor antagonist STI571 through induction of apoptosis. Cancer Res 61:5778–5783. [PubMed] [Google Scholar]
- 79. Smeyers‐Verbeke J, Michotte Y, Pelsmaeckers J, Lowenthal A, Massart D, Dekegel D et al (1975) The chemical composition of idiopathic nonarteriosclerotic cerebral calcifications. Neurology 25:48–57. [DOI] [PubMed] [Google Scholar]
- 80. Smits A, Kato M, Westermark B, Nistér M, Heldin CH, Funa K (1991) Neurotrophic activity of platelet‐derived growth factor (PDGF): rat neuronal cells possess functional PDGF beta‐type receptors and respond to PDGF. Proc Natl Acad Sci USA 88:8159–8163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sobrido MJ, Coppola G, Oliveira J, Hopfer S, Geschwind DH (1993–2013) Primary familial brain calcification. In: GeneReviews(R). Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Smith RJH (eds). GeneReviews® [Internet]: Seattle (WA). [Google Scholar]
- 82. Soderblom C, Luo XT, Blumenthal E, Bray E, Lyapichev K, Ramos J et al (2013) Perivascular fibroblasts form the fibrotic scar after contusive spinal cord injury. J Neurosci 33:13882–13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Soriano P (1994) Abnormal kidney development and hematological disorders in PDGF beta‐receptor mutant mice. Genes Dev 8:1888–1896. [DOI] [PubMed] [Google Scholar]
- 84. Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, von Bruehl ML et al (2013) NG2(+) pericytes support interstitial migration and effector function of myeloid leukocytes during sterile inflammation. Eur J Clin Invest 43:3. [Google Scholar]
- 85. Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, von Bruhl ML et al (2013) Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and “instruct” them with pattern‐recognition and motility programs. Nat Immunol 14:41–51. [DOI] [PubMed] [Google Scholar]
- 86. Tallquist MD, French WJ, Soriano P (2003) Additive effects of PDGF receptor beta signaling pathways in vascular smooth muscle cell development. PLoS Biol 1:E52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Uygur G, Liu Y, Hellman R, Tikofsky R, Collier B (1995) Evaluation of regional cerebral blood flow in massive intracerebral calcifications. J Nucl Med 36:610–612. [PubMed] [Google Scholar]
- 88. Vígh B, Szél A, Debreceni K, Fejér Z, Manzano e Silva M, Vígh‐Teichmann I (1998) Comparative histology of pineal calcification. Histol Histopathol 13:851–870. [DOI] [PubMed] [Google Scholar]
- 89. Villa‐Bellosta R, Levi M, Sorribas V (2009) Vascular smooth muscle cell calcification and SLC20 inorganic phosphate transporters: effects of PDGF, TNF‐alpha, and Pi. Pflügers Arch 458:1151–1161. [DOI] [PubMed] [Google Scholar]
- 90. Wang C, Li Y, Shi L, Ren J, Patti M, Wang T et al (2012) Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat Genet 44:254–256. [DOI] [PubMed] [Google Scholar]
- 91. Waterfield MD, Scrace GT, Whittle N, Stroobant P, Johnsson A, Wasteson A et al (1983) Platelet‐derived growth factor is structurally related to the putative transforming protein p28sis of simian sarcoma virus. Nature 304:35–39. [DOI] [PubMed] [Google Scholar]
- 92. Xu G, Shen J, Ishii Y, Fukuchi M, Dang TC, Zheng Y et al (2013) Functional analysis of platelet‐derived growth factor receptor‐beta in neural stem/progenitor cells. Neuroscience 238:195–208. [DOI] [PubMed] [Google Scholar]
- 93. Yamada M, Tanaka M, Takagi M, Kobayashi S, Taguchi Y, Takashima S et al (2014) Evaluation of SLC20A2 mutations that cause idiopathic basal ganglia calcification in Japan. Neurology 82:705–712. [DOI] [PubMed] [Google Scholar]
- 94. Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly M, Boström K (2013) A role for the endothelium in vascular calcification. Circ Res 113:495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zhang Y, Guo X, Wu A (2013) Association between a novel mutation in SLC20A2 and familial idiopathic basal ganglia calcification. PLoS ONE 8:e57060. [DOI] [PMC free article] [PubMed] [Google Scholar]