

Abstract
Huntington's disease (HD) belongs to the group of inherited polyglutamine (PolyQ) diseases caused by an expanded CAG repeat in the coding region of the Huntingtin (HTT) gene that results in an elongated polyQ stretch. Abnormal function and aggregation of the mutant protein has been typically delineated as the main molecular cause underlying disease development. However, the most recent advances have revealed novel pathogenic pathways directly dependent on an RNA toxic gain‐of‐function. Expanded CAG repeats within exon 1 of the HTT mRNA induce toxicity through mechanisms involving, at least in part, gene expression perturbations. This has important implications not only for basic and translational research in HD, but also for other types of diseases carrying the expanded CAG in other genes, which likely share pathogenic aspects. Here I will review the evidence and mechanisms underlying RNA toxicity in CAG repeat expansions, with particular focus on HD. These comprise abnormal subcellular localization of the transcripts containing the expanded CAG repeats; sequestration of several types of proteins by the expanded CAG repeat which results in defects of alternative splicing events and gene expression; and aberrant biogenesis and detrimental activity of small CAG repeated RNAs (sCAG) that produce altered gene silencing. Although these altered pathways have been detected in HD models, their contribution to disease development and progress requires further study.
Keywords: CAG repeats, polyglutamine disorders, RNA toxicity, RNA binding proteins
INTRODUCTION
Trinucleotide repeats (TNR), short tandem repeats and microsatellites are frequent in the genome 14, 24, 59, with CAG‐TNR being among the most common type 30. CAG repeats (and other types of CNG repeats, where N is any nucleotide) are overrepresented in the exons, and the majority of them are present in the open reading frame (ORF) 18, 24. The number of CAG repeats decreases exponentially with length and the longest tracts are below 20 repeats, suggesting their detrimental effect.
In normal individuals, TNRs are found with polymorphic variation in their number and with no apparent clinical or phenotypic effects. However, abnormal TNR expansions in certain transcribed genes that commonly present long and highly polymorphic repeats underlie toxicity in more than 30 neurodegenerative and neuromuscular disorders 25, 32, 36.
In TNR expansion diseases (TREDs), expanded TNRs occur in translated and untranslated regions (UTR) of the causative gene, with pathogenesis typically involving mutant protein and/or RNA toxic gain of function. In addition, the toxic effects of different types of expanded TNR may co‐exist, due to the recently described bidirectional transcription in most TRED loci 4. For instance, in myotonic dystrophy type 1, Huntington's disease‐like 2 and Spinocerebellar ataxia 8 caused by a CTG expansion in the non‐coding regions of DMPK, JPH3, and ATXN8 genes, respectively, antisense transcription in these loci results in the production of an expanded CAG transcript 38, 40, 67. Similarly, in the majority of CAG‐TREDs, including HD, antisense transcripts have been described for most of the causative genes, resulting in CUG expanded tracks. Although antisense transcripts are expressed at lower levels, their contribution to the disease outcome should be specifically addressed in each TRED.
Another layer of complexity in TRED pathogenicity has recently been uncovered. The repeat associated non‐ATG (RAN) translation 73 results in several types of homopolymeric proteins from different expanded TNRs, including those embedded in non‐coding transcripts. Recently, the accumulation of four types of expanded homopolymeric proteins (polyAla, polySer, polyLeu, and polyCys) were described in HD human brains 2. However, the toxic activity and relevance of this process in disease evolution needs to be examined in each TRED.
A total of nine CAG TREDs have been described so far, with most of them presenting the expansion in a translated region of the causative gene. The CAG expansion leads to an elongated glutamine fragment in the encoded protein, resulting in the group of TREDs globally termed polyglutamine (polyQ) diseases 42. Although rare, these diseases constitute the most common form of inherited neurodegenerative disorders, with Huntington's disease (HD) being the most prevalent.
In polyQ diseases, pathogenesis has been traditionally linked to abnormal function of the mutant protein which, through misfolding and aggregation, promotes aberrant interactions with other proteins 8, 33, 57. The systematic search for hits binding to the mutant HTT protein has revealed preferential interaction with proteins involved in cell stress, protein trafficking, RNA modification, and cell death 47. Detrimental effects of the mutant HTT protein include impairment of neurotransmitter release and mitochondrial function, altered transcriptional activity, and proteome disruption 33.
Several lines of evidence indicate that expanded CAG repeats show toxicity at the RNA level, including the cell degeneration and dysfunction associated with in vivo models expressing untranslated transcripts with expanded CAG repeats 23, 35, 65. In addition, SCA12 is caused by a CAG expansion in 5′UTR, suggesting that RNA species with expanded CAG contribute to disease in the absence of polyQ 22. Long CAG‐TNRs form stable hairpin structures in transcripts 25, with the stem portion presenting protein‐binding properties 18, 32. These secondary RNA structures and the mislocalization of the mutant transcript, produce abnormal interactions with proteins that result in alterations of gene expression and alternative splicing. An important question that has not been thoroughly addressed in PolyQ diseases is the relative contribution of protein and RNA dependent mechanisms to pathogenesis. The correct assessment of these questions has potentially important translational implications.
Here, I will review the evidence accounting for expanded CAG RNA toxicity, the mechanisms at play and the RNA structural requirements for the deleterious effects of expanded CAG. Next, I will summarize the known RNA toxic mechanisms (Figure 1), including the transcriptional alterations linked to the sequestration of RNA‐binding proteins by expanded CAG repeats and the biogenesis and activity of small repeated CAG RNA molecules.
Figure 1.
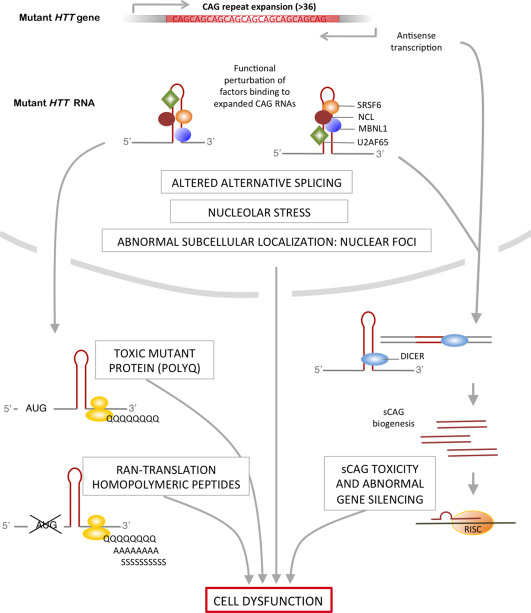
Expanded CAG pathogenic mechanisms.
EVIDENCE FOR RNA TOXICITY TRIGGERED BY CAG TRINUCLEOTIDE REPEATS
The concept of “RNA toxicity” denotes the direct ability of the mutant transcript to induce pathogenesis. This effect is straightforward in the cases where the expanded TNR is located in a non‐coding region of the gene and does not affect the protein structure. However, RNA toxicity is more difficult to assess in PolyQ diseases, where diverse protein and RNA toxic effects coexist.
The toxicity triggered by expanded CAG repeats at the RNA level has been demonstrated by expressing several CAG repeat lengths in different model systems. Neurodegeneration was first observed in Drosophila melanogaster expressing an elongated track of translated and untranslated CAG repeats 35. Drosophila expressing CAG repeats interrupted with CAA showed a clearly less pronounced effect. While both CAG and CAA encode for glutamine, the secondary RNA structures linked to each TNR differ. Whereas expanded CAG repeats produce a hairpin‐like structure, CAA tracks or CAG stretches interrupted with CAA do not fold into a hairpin 55. Therefore, differences in toxicity are not due to the expanded polyQ track and may in fact depend on the formation of an expanded CAG repeat hairpin‐like structure that is toxic at the RNA level. A similar result was found in human neuronal cells expressing translated and untranslated versions of the HTT exon‐1 harboring expanded CAG repeats 3, 56. While these constructs decreased cell viability, the effect of expanded CAA was clearly milder. In these studies, no RAN translation was detected in the SH‐SY5Y neuronal cells expressing untranslated HTT exon 1 with expanded CAG repeats, suggesting a pure RNA toxic mechanism. In addition, the toxic effect of untranslated, long CAG repeats has recently been shown in in vivo worm 65 and mouse 23 models, with extended CAG repeats being expressed in the 3′UTR of a marker protein. Caenorhabditis elegans expressing untranslated CAG repeats presented a shortened life span and motility difficulties in direct correlation with the extent of the CAG repeat 65. In mice, transgenic expression of extended CAG repeats in the muscle was associated with profound histological and functional defects of muscle cell as well as behavioral abnormalities 23. In these models, however, possible pathogenesis associated with RAN‐translation should be specifically addressed.
INSIGHTS INTO THE STRUCTURE AND SUBCELLULAR LOCALIZATION OF RNAS CONTAINING EXPANDED CAG REPEATS
Biochemical and biophysical approaches have revealed the ability of TNR to form complex structures 18, 55. CNG repeats, where N is any nucleotide (A, C, G, or U), form semi‐stable hairpins 30, with this structure having the potential to interact with different RNA‐binding proteins that generally modulate gene expression. Depending on the subcellular localization of the RNA, different functions for the TNR have been proposed, including splicing modulation in the nuclear compartment and regulation of translation in the cytoplasm 15, 46, 48. Although the normal function of non‐pathogenic TNR has not been resolved, expanded TNRs have effects in both the subcellular localization of the transcript carrying the repeat and the binding dynamics of specific protein having affinity for the TNR.
It has been shown that most of the variants in the normal gene contain base interruptions within the CAG repeat that are not present in the expanded gene 18. Differing from the non‐interrupted expanded CAG repeat that forms a long hairpin, interruptions in the normal gene destabilize the smaller hairpin structure 53, 54. These data and functional in vivo approaches (see below) suggest a relevant role for the RNA structure in inducing a toxic effect. In addition, RNA structures are context‐dependent and may vary in each host transcript carrying the TNR 18. In the HTT gene, another nearby CCG polymorphic TNR provides extra stability to the CAG hairpin structure 13. However, this stability is not strong enough, as the RNAi machinery is able to efficiently target the double‐stranded stem within the hairpin 13.
A characteristic feature of TREDs is the presence of nuclear RNA inclusions or nuclear foci that typically sequester several proteins having affinity for the TNR hairpin 69. The type of TNR sequence, its secondary structure and the host cell protein environment have all been proposed as modulators of nuclear foci morphology 69. Presumably, RNA repeat inclusions are trapped in the nucleus as a consequence of RNA overloading with proteins. However, the precise mechanisms leading to the formation of nuclear foci have been only partially tackled. Recent studies have shown that the dosage of the RNA‐binding proteins MBNL1 and U2AF65 contribute to the abnormal subcellular localization of the mutant HTT transcripts 29, 56, 62.
Nuclear RNA foci have been extensively studied in myotonic dystrophy type 1 (MD‐1) caused by a CUG expansion in the 3′UTR of the DMPK gene 12, 58. The number of foci per cell and the fraction of cells carrying foci depend on the CUG repeat length and the expression levels of the mutant transcript 69. In PolyQ diseases, analogous inclusions have recently been described in HTT and ATX3 mutant transcripts and in diverse models ectopically expressing long CAG repeats 13, 21, 23, 35, 56. Similarly to expanded CUG repeat, the number of foci per nucleus correlates with CAG repeat expansion 23.
Although nuclear foci formation is linked to pathology development in TREDs and contributes to the functional disruption of TNR‐binding proteins, it is still a matter of debate whether nuclear accumulation of the mutant transcript is pathogenic or is an epiphenomenon running in parallel with clinical and pathological features.
FUNCTIONAL PERTURBATION OF PROTEINS BINDING TO RNAS WITH CAG REPEATS
More than 50% of the multifunctional RNA‐binding proteins are expressed in the brain, where they modulate a plethora of essential biological processes including alternative splicing, transport, localization, stability, and translation of RNAs 7. It is not surprising that functional perturbation of RNA‐protein complexes underlies neurological and neurodegenerative conditions, including TREDs. This section will summarize current knowledge about the proteins whose binding to expanded CAG repeats results in alterations of their normal function.
Sequestration of the muscleblind‐like 1 (MBNL1) disrupts alternative splicing
MBNL1 regulates the alternative splicing of target mRNAs, thus modulating the expression of specific protein isoforms 21, 64. MBNL1 binding to the target mRNAs acts both as an activator and an inhibitor of splicing. This diverse activity is exemplified in the target gene insulin receptor (IR) and cardiac Tropin‐T (TNNT2): while MBNL1 induces IR pre‐mRNA exon inclusion, it inhibits exon inclusion in the TNNT2 mRNA.
In addition to modulating the splicing of target genes, MBNL1 has comparable affinity for TNR, especially CUG and CAG repeats 71. Binding affinity to TNR increases with repeat length, and the decreased stability of double‐stranded structures in TNR below a certain threshold (<20 repeats) results in a loss of MBNL1 binding 71.
Abnormal localization or sequestration of MBNL1 to nuclear foci was first observed in MD1 muscle cells 16, 39. Sequestration linked to MBNL1 loss of function was demonstrated by defects in the alternative splicing of developmentally regulated MBNL1 targets, including insulin receptor, the chloride channel, sarcoplasmic/endoplasmic reticulum Ca2+ ATPase cardiac troponin T and Tau 9, 26, 28, 45, 52. Perturbed expression of these splicing isoforms has been linked to several clinical symptoms in DM1.
MBNL1 sequestration by expanded CAG repeats was first demonstrated in a monkey cell line expressing a long CAG repeat stretch 21. Nuclear RNA foci co‐localizing MBNL1 were found in fibroblasts of patients with HD and SCA3 41. Furthermore, alternative splicing defects of MBNL1 target genes were detected in HD and SCA3 fibrobalsts and other human cell lines expressing expanded CAG repeats 41.
Studies in non‐human model organisms argue for a role for MBNL1 sequestration and alternative splicing defects in polyQ diseases. In the muscle of R6/2 HD transgenic mice, an altered splicing pattern of the MBNL1 target Clcn1 potassium channel results in skeletal muscle hyperexcitability 66. In addition, in Drosophila, Mbnl increases the neurodegeneration induced by expanded CAG in a SCA3 model 35. These model organisms show long CAG repeat expansions. However, alternative splicing defects were detected in human cells expressing 70 CAG repeats but not 45 CAG repeats 41, which calls into question the contribution of altered MBNL1 function in this low range of pathogenic CAG repeats.
Binding of SRSF6 by CAG repeats alters HTT alternative splicing
The serine/arginine‐rich (SR) protein SRSF6 (SRp55) belongs to a family of highly conserved RNA‐binding splicing‐factor proteins. Perturbed alternative splicing of the HTT gene has been described in HD depending on alterations of SRSF6 activity 19, 50. SRSF6 binding to the CAG repeats underlies the CAG repeat length‐dependent aberrant splicing of the HTT exon‐1, resulting in a short polyadenylated mRNA that is translated into an exon 1 HTT protein. This may be relevant to understanding of the pathogenesis in HD, since HTT exon 1 alone is highly toxic in diverse model systems. However, in these models it remains to be determined what the specific contribution of the aberrantly spliced HTT‐exon 1 RNA and/or protein toxic mechanisms is.
Similarly to MBNL1, SRSF6 binding to the CAG repeats could lead to SRSF6 loss of function and subsequent splicing defects in specific targets. Although this mechanism has not been specifically addressed, pathogenic tau isoforms produced by the abnormal expression and function of SRSF6 have recently been described in HD 17. An imbalance in the relative amount of tau isoforms containing either three or four microtubule‐binding repeats (3R and 4R, respectively) in favor of the 4R isoform is observed in the striatum and cortex of HD patients. Altered expression of the 3R/4R isoforms could underlie pathophysiological aspects of HD. It may be produced by increased abundance and or phosphorylation of SRSF6. However, further studies are needed to link SRSF6 sequestration by CAG repeats and alternative splicing defects in tau, and other targets.
Sequestration of nucleolin by expanded CAG repeats induces nucleolar stress
Nucleolin (NCL) is a multifunctional protein that is mainly localized in the nucleolus. It is involved in various steps of ribosome biogenesis, including rRNA transcription, rRNA maturation, and ribosome assembly 61. Perturbations of these processes lead to nucleolar stress, a signaling pathway producing apoptosis that has been linked to neurodegeneration 27, 44, 49, 63.
NCL normally binds to an upstream control element of the rRNA promoter, thus protecting this region from CpG hypermethylation. Recently, it has been shown that expanded CAG repeats compete with rRNA promoter for NCL binding, leading to decreased association with rRNA promoter and subsequent hypermethylation and reduction of rRNA expression 60, 63. Expanded CAG repeat‐mediated rRNA decrease in transgenic animal models and PolyQ patients induces nucleolar stress and apoptosis via p53 stabilization and activation.
Levels of P53 modulate the detrimental effects of expanded HTT, since P53 silencing or inhibition is neuroprotective in models of HD 1, 20. In addition, P53 is consistently upregulated in several cell and animal models of HD, as well as in human lymphoblasts and brain 6. Increased levels of p53 imply its increased activity, since genes downstream of P53 signaling are upregulated in both cell and mouse models of HD. Overall, these data highlight how pathways activated by p53 play a role in HD pathogenesis, with NCL sequestration by expanded CAG representing an upstream factor that contributes to increased p53 levels.
U2AF65 binds directly to CAG repeats and perturbs nuclear export
U2 small nuclear ribonucleoprotein auxiliary factor 65 (U2AF65) protein is involved in both splicing 43 and nuclear export of RNAs 5, 72. It facilitates the formation of a messenger ribonucleoprotein export complex that contains both the NXF1 receptor and the RNA substrate 5, 72.
The NXF1/U2AF65 RNA export pathway is linked to expanded CAG RNA‐mediated toxicity 62. In a Drosophila model of Machado‐Joseph disease (MJD), an MDJ transcript carrying 78 CAG repeats (MJD78CAG) induced nuclear localization of the expanded transcript and eye degeneration, with both phenomena being enhanced by U2AF65 knockdown. The authors proved that expanded MJDCAG78 RNA directly interacts with U2AF65 protein, which depends on the expanded and continuous nature of CAG repeats in the RNA. U2AF65 serves as an adaptor to couple expanded CAG RNA to NXF1 for its export 62.
U2AF65 dosage modulates the nuclear export of MJDCAG78 RNA and other types of transcripts carrying expanded CAG repeats, including mutant ATX3 and HTT 56, 62. The physiological drop of U2AF65 expression in symptomatic adult R6/2 mouse was coupled with the nuclear accumulation of the mutant HTT‐exon1 transcript, suggesting that U2AF65 dosage is also relevant in CAG expanded‐HTT transcript nuclear export in vivo 62. The relevance of U2AF65 dosage in mutant HTT export was also demonstrated in human neuroblastoma cell lines expressing pathogenic CAG repeat expansions 56.
While these studies explicitly demonstrate that U2AF65 dosage correlates with nuclear accumulation of TREDS transcripts, a parallel readout with the toxicity driven by expanded CAG RNAs has not been totally demonstrated. Downregulation of U2AF65 results in increased toxicity 62, but the expected protective effect of its upregulation coupled with expanded CAG transcript export has not been evaluated. The effect of U2AF65 dosage in the toxicity driven by expanded CAG repeats may not be straightforward, since U2AF65 modulation of alternative splicing of specific transcripts may provide an additional source of toxicity.
Dicer binds to expanded CAG repeats, inducing the formation of small RNAs containing CAG repeats with toxic activity
Dicer is a type III endonuclease that cleaves an miRNA precursor hairpin‐like structure (pre‐miRNA) or long double‐stranded RNAs to produce miRNAs and small interfering RNAs (siRNA), respectively. The resulting products are double‐stranded short RNAs (20‐25 nucleotides) that incorporate into the RNA‐induced silencing complex (RISC) and induce gene silencing 68.
Uninterrupted CNG repeats (above 17 CNG repeats) forming hairpin‐like structures are an appropriate substrate of recombinant Dicer 31 that cleaves them to form short double‐stranded CNG repeated RNAs of 21 nucleotides. Furthermore, these short products are increased in fibroblasts of patients with TREDs 31, and their abundance is dependent on the activity of endogenous Dicer. A possible additional source of small repeated TNR RNAs may be provided by Dicer activity on long double‐stranded RNAs that result from the interaction of sense and anti‐sense transcripts with expanded CNG repeats. Bi‐directional transcription producing anti‐sense transcripts is a widespread phenomenon 10 that has been observed in many TRED loci, including HD 11. However, the contribution of bi‐directional transcription to increased production of short TNR‐RNAs may be limited, since anti‐sense transcripts are normally expressed at low rates.
Several studies have shown that short RNAs composed of TNR are neurotoxic. Dicer‐2 dependent neurodegeneration is detected in flies co‐expressing long CAG and CUG tracks 34, 70. The long double‐stranded transcripts are cleaved to 21 nt CAG/CUG siRNAs that are toxic in Drosophila. In addition, CAG/CUG siRNAs impair the viability of diverse human cell lines 3. However, studies that force the overexpression of CAG/CUG siRNAs do not address their real pathogenic relevance in CAG or CUG TREDs.
Sequencing of the short 21 nt RNAs produced in the Drosophila models co‐expressing long CAG and CUG tracks demonstrated that the CAG strand of the hybrid short RNA was preferentially stabilized and loaded into RISC 34. Increased levels of short repeated CAG (sCAG) are also detected in fibroblasts of patients with HD and SCA1 31 and in the frontal cortex and striatum of patients with HD 3. sCAG biogenesis is length dependent, with increased amounts being observed in cells expressing HTT‐exon‐1 with larger CAG repeats 3.
Transfection of small RNAs (sRNA) isolated from cells expressing mutant HTT‐exon‐1 and from the brain of patients with HD is neurotoxic 3. This toxicity is abolished if sRNAs are co‐transfected with an oligonlucleotide complementary to the CAG repeat, suggesting that the detrimental effect is caused by sCAG. Furthermore, deleterious effects of sCAG depend on Ago2, indicating participation of gene‐silencing pathways. Interestingly, toxicity driven by sCAG differs depending on the cell type, with special involvement of differentiated neuronal cells 3. A cell‐type specific transcriptomic profile may provide diverse scenarios for deleterious sCAG gene silencing that could explain differential tissue vulnerability in HD and other CAG‐TREDs.
However, the rules governing sCAG‐mediated gene silencing and how this is linked to cell dysfunction remain poorly understood. In cell models expressing expanded HTT‐exon 1, downregulation of transcripts containing complementary CUG repeats was mild 3, and general evaluation of brain HD transcriptomic profiles has not revealed enrichment in the downregulation of targets containing complementary CUG or CAG target sites (unpublished observations). This is in line with studies in Drosophila showing a neurodegeneration phenotype depending on CAG/CUG siRNAs, as no changes in the expression of targets containing repeated CAG or CUG tracks were detected 34. This differs from an analogous study in Drosophila 70, showing strong downregulation of two endogenous targets containing CAG repeats but no involvement of genes containing CUG repeats.
A possible explanation for the detrimental effect of sCAG is competition of the expanded CAG repeat or double‐stranded CAG/CUG repeat for the biogenesis machinery, which could result in an impairment of both endogeneous Dicer activity and downstream pathways. Supporting the decreased function of Dicer‐2, most deregulated miRNAs in Drosophila showing CAG/CUG siRNA‐dependent neurodegeneration were downregulated 34. However, in another analogous study in flies, the RNA interference pathways were largely intact 70.
Similar amounts of up‐ and downregulated miRNAs are detected in HD brains 37, suggesting little involvement of Dicer activity and indicating that diverse mechanisms are involved in miRNA deregulation. An additional layer of complexity is observed in HD, since impaired gene silencing is detected, resulting from abnormal interaction of mutant HTT protein with Ago2 51. Overall these data indicate that perturbed gene silencing in HD may occur through diverse mechanisms, involving at least altered miRNA expression and abnormal activity of mutant HTT protein.
CONCLUDING REMARKS
The evidence summarized in this review unequivocally shows that pathogenic mechanisms in HD and other polyQ diseases are more complex than anticipated. Altered function of the mutant protein carrying the expanded glutamine track coexists with detrimental effects of expanded CAG RNA. Major future challenges include increasing our understanding of pathogenesis driven by each of these processes, of how they crosstalk, and, more importantly, how they explain specific pathologic and clinical outcomes.
A number of model organisms expressing untranslated expanded CAG repeats have provided a proof of concept for RNA toxicity. However, these models do not reflect the real situation in polyQ diseases, in which both mutant transcripts and proteins co‐exist. In addition, the pathogenic bases in polyQ model organisms that express the translated version of the expanded gene should be re‐examined. While these models have traditionally been used to explain mutant protein‐based mechanisms, the possible contribution of expanded RNA to pathogenesis calls for further examination.
In HD, RNA pathogenic mechanisms include perturbation of alternative splicing, altered gene silencing, aberrant subcellular localization of transcripts, and nucleolar stress. Future proteomic studies to fully characterize the proteins directly or indirectly binding to expanded CAG will likely provide additional pathogenic insights. While the RNA alterations discovered so far have been detected in models expressing mutant HTT protein, their real relevance in disease evolution is far from being understood.
Unraveling of the deleterious mechanisms of RNA vs. protein is not a trivial task. It requires novel strategies to specifically target each effect. These approaches may shed light on the extent to which both the inhibition of mutant protein synthesis and the blockage or degradation of mutant transcript will be required to overcome or block disease evolution. Therefore, understanding of the relevance of RNA and/or protein mechanisms may provide both mechanistic insights into polyQ disease development and progression, and novel scenarios for therapeutic approaches.
AKCNOWLEDGMENTS
This work was supported by the Spanish Government, the Spanish Ministry of Economy and Competitiveness and the grant number: SAF2014‐60551‐R, The European fund for regional development (FEDER) and The Spanish Ministry of Economy and Competitiveness ‘Centro de Excelencia Severo Ochoa 2013‐2017’
The author has declared that no conflict of interest exists.
REFERENCES
- 1. Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y et al (2005) p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron 47:29–41. [DOI] [PubMed] [Google Scholar]
- 2. Banez‐Coronel M, Ayhan F, Tarabochia AD, Zu T, Perez BA, Tusi SK et al (2015) RAN Translation in Huntington disease. Neuron 88:667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Banez‐Coronel M, Porta S, Kagerbauer B, Mateu‐Huertas E, Pantano L, Ferrer I et al (2012) A pathogenic mechanism in Huntington's disease involves small CAG‐repeated RNAs with neurotoxic activity. PLoS Genet 8:e1002481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Batra R, Charizanis K, Swanson MS (2010) Partners in crime: bidirectional transcription in unstable microsatellite disease. Hum Mol Genet 19:R77–R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blanchette M, Labourier E, Green RE, Brenner SE, Rio DC (2004) Genome‐wide analysis reveals an unexpected function for the Drosophila splicing factor U2AF50 in the nuclear export of intronless mRNAs. Mol Cell 14:775–786. [DOI] [PubMed] [Google Scholar]
- 6. Bowles KR, Jones L (2014) Kinase signalling in Huntington's disease. J Huntingtons Dis 3:89–123. [DOI] [PubMed] [Google Scholar]
- 7. Bryant CD, Yazdani N (2016) RNA‐binding proteins, neural development and the addictions. Genes Brain Behav 15:169–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cattaneo E, Zuccato C, Tartari M (2005) Normal huntingtin function: an alternative approach to Huntington's disease. Nat Rev Neurosci 6:919–930. [DOI] [PubMed] [Google Scholar]
- 9. Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA (2002) Loss of the muscle‐specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell 10:45–53. [DOI] [PubMed] [Google Scholar]
- 10. Chen J, Sun M, Kent WJ, Huang X, Xie H, Wang W et al (2004) Over 20% of human transcripts might form sense‐antisense pairs. Nucleic Acids Res 32:4812–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chung DW, Rudnicki DD, Yu L, Margolis RL (2011) A natural antisense transcript at the Huntington's disease repeat locus regulates HTT expression. Hum Mol Genet 20:3467–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE (1997) Expansion of a CUG trinucleotide repeat in the 3' untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci U S A 94:7388–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Mezer M, Wojciechowska M, Napierala M, Sobczak K, Krzyzosiak WJ (2011) Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res 39:3852–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ellegren H (2004) Microsatellites: simple sequences with complex evolution. Nat Rev Genet 5:435–445. [DOI] [PubMed] [Google Scholar]
- 15. Fabre E, Dujon B, Richard GF (2002) Transcription and nuclear transport of CAG/CTG trinucleotide repeats in yeast. Nucleic Acids Res 30:3540–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fardaei M, Larkin K, Brook JD, Hamshere MG (2001) In vivo co‐localisation of MBNL protein with DMPK expanded‐repeat transcripts. Nucleic Acids Res 29:2766–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fernandez‐Nogales M, Cabrera JR, Santos‐Galindo M, Hoozemans JJ, Ferrer I, Rozemuller AJ et al (2014) Huntington's disease is a four‐repeat tauopathy with tau nuclear rods. Nat Med 20:881–885. [DOI] [PubMed] [Google Scholar]
- 18. Galka‐Marciniak P, Urbanek MO, Krzyzosiak WJ (2012) Triplet repeats in transcripts: structural insights into RNA toxicity. Biol Chem 393:1299–1315. [DOI] [PubMed] [Google Scholar]
- 19. Gipson TA, Neueder A, Wexler NS, Bates GP, Housman D (2013) Aberrantly spliced HTT, a new player in Huntington's disease pathogenesis. RNA Biol 10:1647–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly‐Rosen D, Qi X (2013) Inhibition of mitochondrial fragmentation diminishes Huntington's disease‐associated neurodegeneration. J Clin Invest 123:5371–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, Cooper TA (2004) Muscleblind proteins regulate alternative splicing. Embo J 23:3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Holmes SE, O'Hearn EE, McInnis MG, Gorelick‐Feldman DA, Kleiderlein JJ, Callahan C et al (1999) Expansion of a novel CAG trinucleotide repeat in the 5' region of PPP2R2B is associated with SCA12. Nat Genet 23:391–392. [DOI] [PubMed] [Google Scholar]
- 23. Hsu RJ, Hsiao KM, Lin MJ, Li CY, Wang LC, Chen LK et al (2011) Long tract of untranslated CAG repeats is deleterious in transgenic mice. PLoS One 6:e16417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jasinska A, Krzyzosiak WJ (2004) Repetitive sequences that shape the human transcriptome. FEBS Lett 567:136–141. [DOI] [PubMed] [Google Scholar]
- 25. Jasinska A, Michlewski G, de Mezer M, Sobczak K, Kozlowski P, Napierala M et al (2003) Structures of trinucleotide repeats in human transcripts and their functional implications. Nucleic Acids Res 31:5463–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA (2004) Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet 13:3079–3088. [DOI] [PubMed] [Google Scholar]
- 27. Kalita K, Makonchuk D, Gomes C, Zheng JJ, Hetman M (2008) Inhibition of nucleolar transcription as a trigger for neuronal apoptosis. J Neurochem 105:2286–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kimura T, Nakamori M, Lueck JD, Pouliquin P, Aoike F, Fujimura H et al (2005) Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+‐ATPase in myotonic dystrophy type 1. Hum Mol Genet 14:2189–2200. [DOI] [PubMed] [Google Scholar]
- 29. Kino Y, Washizu C, Kurosawa M, Oma Y, Hattori N, Ishiura S et al (2015) Nuclear localization of MBNL1: splicing‐mediated autoregulation and repression of repeat‐derived aberrant proteins. Hum Mol Genet 24:740–756. [DOI] [PubMed] [Google Scholar]
- 30. Kozlowski P, de Mezer M, Krzyzosiak WJ (2010) Trinucleotide repeats in human genome and exome. Nucleic Acids Res 38:4027–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krol J, Fiszer A, Mykowska A, Sobczak K, de Mezer M, Krzyzosiak WJ (2007) Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol Cell 25:575–586. [DOI] [PubMed] [Google Scholar]
- 32. Krzyzosiak WJ, Sobczak K, Wojciechowska M, Fiszer A, Mykowska A, Kozlowski P (2012) Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic Acids Res 40:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Labbadia J, Morimoto RI (2013) Huntington's disease: underlying molecular mechanisms and emerging concepts. Trends Biochem Sci 38:378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lawlor KT, O'Keefe LV, Samaraweera SE, van Eyk CL, McLeod CJ, Maloney CA et al (2011) Double‐stranded RNA is pathogenic in Drosophila models of expanded repeat neurodegenerative diseases. Hum Mol Genet 20:3757–3768. [DOI] [PubMed] [Google Scholar]
- 35. Li LB, Yu Z, Teng X, Bonini NM (2008) RNA toxicity is a component of ataxin‐3 degeneration in Drosophila. Nature 453:1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lopez Castel A, Cleary JD, Pearson CE (2010) Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol 11:165–170. [DOI] [PubMed] [Google Scholar]
- 37. Marti E, Pantano L, Banez‐Coronel M, Llorens F, Minones‐Moyano E, Porta S et al (2010) A myriad of miRNA variants in control and Huntington's disease brain regions detected by massively parallel sequencing. Nucleic Acids Res 38:7219–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Michel L, Huguet‐Lachon A, Gourdon G (2015) Sense and antisense DMPK RNA foci accumulate in DM1 tissues during development. PLoS One 10:e0137620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miller JW, Urbinati CR, Teng‐Umnuay P, Stenberg MG, Byrne BJ, Thornton CA et al (2000) Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. Embo J 19:4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS et al (2006) Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet 38:758–769. [DOI] [PubMed] [Google Scholar]
- 41. Mykowska A, Sobczak K, Wojciechowska M, Kozlowski P, Krzyzosiak WJ (2011) CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res 39:8938–8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Orr HT, Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30:575–621. [DOI] [PubMed] [Google Scholar]
- 43. Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR (2004) Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci U S A 101:15974–15979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Parlato R, Kreiner G, Erdmann G, Rieker C, Stotz S, Savenkova E et al (2008) Activation of an endogenous suicide response after perturbation of rRNA synthesis leads to neurodegeneration in mice. J Neurosci 28:12759–12764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Philips AV, Timchenko LT, Cooper TA (1998) Disruption of splicing regulated by a CUG‐binding protein in myotonic dystrophy. Science 280:737–741. [DOI] [PubMed] [Google Scholar]
- 46. Raca G, Siyanova EY, McMurray CT, Mirkin SM (2000) Expansion of the (CTG)(n) repeat in the 5'‐UTR of a reporter gene impedes translation. Nucleic Acids Res 28:3943–3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ratovitski T, Chighladze E, Arbez N, Boronina T, Herbrich S, Cole RN et al (2012) Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle 11:2006–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Richards RI, Holman K, Yu S, Sutherland GR (1993) Fragile X syndrome unstable element, p(CCG)n, and other simple tandem repeat sequences are binding sites for specific nuclear proteins. Hum Mol Genet 2:1429–1435. [DOI] [PubMed] [Google Scholar]
- 49. Rieker C, Engblom D, Kreiner G, Domanskyi A, Schober A, Stotz S et al (2011) Nucleolar disruption in dopaminergic neurons leads to oxidative damage and parkinsonism through repression of mammalian target of rapamycin signaling. J Neurosci 31:453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK et al (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci U S A 110:2366–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Savas JN, Makusky A, Ottosen S, Baillat D, Then F, Krainc D et al (2008) Huntington's disease protein contributes to RNA‐mediated gene silencing through association with Argonaute and P bodies. Proc Natl Acad Sci U S A 105:10820–10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Savkur RS, Philips AV, Cooper TA (2001) Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet 29:40–47. [DOI] [PubMed] [Google Scholar]
- 53. Sobczak K, Krzyzosiak WJ (2004) Imperfect CAG repeats form diverse structures in SCA1 transcripts. J Biol Chem 279:41563–41572. [DOI] [PubMed] [Google Scholar]
- 54. Sobczak K, Krzyzosiak WJ (2005) CAG repeats containing CAA interruptions form branched hairpin structures in spinocerebellar ataxia type 2 transcripts. J Biol Chem 280:3898–3910. [DOI] [PubMed] [Google Scholar]
- 55. Sobczak K, Michlewski G, de Mezer M, Kierzek E, Krol J, Olejniczak M et al (2010) Structural diversity of triplet repeat RNAs. J Biol Chem 285:12755–12764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sun X, Li PP, Zhu S, Cohen R, Marque LO, Ross CA et al (2015) Nuclear retention of full‐length HTT RNA is mediated by splicing factors MBNL1 and U2AF65. Sci Rep 5:12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Takahashi T, Katada S, Onodera O (2010) Polyglutamine diseases: where does toxicity come from? what is toxicity? where are we going? J Mol Cell Biol 2:180–191. [DOI] [PubMed] [Google Scholar]
- 58. Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH (1995) Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol 128:995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Toth G, Gaspari Z, Jurka J (2000) Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res 10:967–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tsoi H, Chan HY (2013) Expression of expanded CAG transcripts triggers nucleolar stress in Huntington's disease. Cerebellum 12:310–312. [DOI] [PubMed] [Google Scholar]
- 61. Tsoi H, Chan HY (2014) Roles of the nucleolus in the CAG RNA‐mediated toxicity. Biochim Biophys Acta 1842:779–784. [DOI] [PubMed] [Google Scholar]
- 62. Tsoi H, Lau CK, Lau KF, Chan HY (2011) Perturbation of U2AF65/NXF1‐mediated RNA nuclear export enhances RNA toxicity in polyQ diseases. Hum Mol Genet 20:3787–3797. [DOI] [PubMed] [Google Scholar]
- 63. Tsoi H, Lau TC, Tsang SY, Lau KF, Chan HY (2012) CAG expansion induces nucleolar stress in polyglutamine diseases. Proc Natl Acad Sci U S A 109:13428–13433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ et al (2012) Transcriptome‐wide regulation of pre‐mRNA splicing and mRNA localization by muscleblind proteins. Cell 150:710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang LC, Chen KY, Pan H, Wu CC, Chen PH, Liao YT et al (2011) Muscleblind participates in RNA toxicity of expanded CAG and CUG repeats in Caenorhabditis elegans. Cell Mol Life Sci 68:1255–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Waters CW, Varuzhanyan G, Talmadge RJ, Voss AA (2013) Huntington disease skeletal muscle is hyperexcitable owing to chloride and potassium channel dysfunction. Proc Natl Acad Sci U S A 110:9160–9165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wilburn B, Rudnicki DD, Zhao J, Weitz TM, Cheng Y, Gu X et al (2011) An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease‐like 2 mice. Neuron 70:427–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wilson RC, Doudna JA (2013) Molecular mechanisms of RNA interference. Annu Rev Biophys 42:217–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wojciechowska M, Krzyzosiak WJ (2011) Cellular toxicity of expanded RNA repeats: focus on RNA foci. Hum Mol Genet 20:3811–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yu Z, Teng X, Bonini NM (2011) Triplet repeat‐derived siRNAs enhance RNA‐mediated toxicity in a Drosophila model for myotonic dystrophy. PLoS Genet 7:e1001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yuan Y, Compton SA, Sobczak K, Stenberg MG, Thornton CA, Griffith JD et al (2007) Muscleblind‐like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res 35:5474–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zolotukhin AS, Tan W, Bear J, Smulevitch S, Felber BK (2002) U2AF participates in the binding of TAP (NXF1) to mRNA. J Biol Chem 277:3935–3942. [DOI] [PubMed] [Google Scholar]
- 73. Zu T, Gibbens B, Doty NS, Gomes‐Pereira M, Huguet A, Stone MD et al (2011) Non‐ATG‐initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A 108:260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]