

Abstract
Excess caloric intake combined with a sedentary lifestyle in the general population has greatly increased the prevalence of obesity and nonalcoholic fatty liver disease (NAFLD), which is defined as the accumulation of excess fat in the liver in the absence of alcohol abuse or other attributable causes such as infection with hepatitis C. Furthermore, NAFLD increases the risk for insulin resistance, type 2 diabetes (T2D), and cardiovascular disease, while currently having no approved therapy to counteract its pathology. Thus, increasing efforts to understand the mechanisms responsible for NAFLD have been pursued in preclinical studies, in the hopes of developing novel therapies that can prevent the progression of insulin resistance and/or T2D. The pathology of NAFLD is multifactorial, with proposed mechanisms including inflammation, oxidative stress, and mitochondrial dysfunction to name a few. The latter remains a subject of ongoing debate, but may be attributed to impaired hepatic fatty acid oxidation, thereby increasing the accumulation of triacylglycerol within hepatocytes. More recent studies have also demonstrated that the mitochondrial dysfunction in NAFLD may also encompass impairments in glucose oxidation, despite oxidative energy production having minimal contribution to overall glucose/pyruvate metabolism in the liver. Accordingly, strategies to reverse this defect in glucose oxidation can ameliorate hepatic steatosis and improve glucose homeostasis. We will review herein the evidence supporting impaired hepatic glucose oxidation as a mechanism of NAFLD, while discussing the validity of pyruvate dehydrogenase (PDH), the rate-limiting enzyme of glucose oxidation, as a potential target for NAFLD. In addition, we will discuss potential mechanisms of action by which increased hepatic PDH activity and subsequent glucose oxidation can reverse the pathology of obesity-induced NAFLD, as well as opportunities to target this pathway with clinical agents.
Keywords: Pyruvate dehydrogenase, glucose oxidation, obesity, type 2 diabetes, hepatic steatosis, nonalcoholic fatty liver disease, ranolazine
Nonalcoholic fatty liver disease (NAFLD)/hepatic steatosis is a condition in which excess fat (triacylglycerol [TAG]) accumulates (>5% within hepatocytes) in the liver of an individual in the absence of alcohol abuse or other attributable causes such as hepatitis C infection.1,2 Unfortunately, NAFLD is increasing exponentially and has become one of the leading causes of morbidity worldwide, where it is estimated to affect nearly one-third of the adult population in Western nations.3−5 The excess lipid/TAG storage in NAFLD does not necessarily worsen liver function, but in the presence of inflammation, it may progress to a more advanced form referred to as nonalcoholic steatohepatitis (NASH), which can also increase the risk for hepatocellular carcinoma.1 While NAFLD can often be relatively benign, the progression of NAFLD is tightly correlated with systemic insulin resistance and can also increase the risk for type 2 diabetes (T2D) and cardiovascular disease.1,6 One of the primary drivers of NAFLD is underlying obesity, and thus there has been extensive interrogation into understanding the mechanisms contributing to the pathology and natural progression of NAFLD, as this may lead to novel therapies for preventing insulin resistance and/or T2D. Whether NAFLD is necessarily causal toward insulin resistance and/or T2D is a complex issue and a topic of ongoing study, as clear dissociations between hepatic steatosis and insulin resistance have also been reported, whereas insulin resistance may also cause NAFLD.7
Regardless, there are a number of mediators that are proposed to contribute to obesity-related NAFLD, many of which have been identified through the completion of sophisticated preclinical studies using in vitro and in vivo models. This includes inflammation and oxidative stress, which may be influenced by alterations in the gut microbiome (extensively reviewed1,8,9). In addition, hyperinsulinemia may also contribute to the progression of NAFLD, since hepatic insulin signaling promotes de novo lipogenesis (DNL).10−12 Furthermore, selectivity with regard to insulin resistance has been documented in the liver, where insulin’s ability to suppress gluconeogenesis is lost, but not its ability to stimulate DNL.12 Hence, increases in insulin secretion often observed during the early stages of insulin resistance may contribute to hepatic steatosis by upregulating DNL.
Another proposed mediator of NAFLD involves mitochondrial dysfunction, with the majority of work in this area focused on impaired fatty acid oxidation rates. A decline in mitochondrial fatty acid oxidation in obesity results in subsequent increases in hepatic TAG accumulation as hepatic fatty acid supply and uptake outweigh rates of oxidation, but also drive increases in lipotoxic intermediates such as diacylglycerol (DAG) and ceramide. Numerous preclinical studies have demonstrated that increasing hepatic fatty acid oxidation rates can combat NAFLD, while also reducing the buildup of ceramide and DAG, thereby causing a potent insulin sensitizing effect and improvement in glucose homeostasis.2,13 In contrast, it has also been demonstrated that hepatic fatty acid oxidation rates are increased in both obese pigs and humans,14 and that hepatic mitochondrial dysfunction does not occur until NAFLD transitions into NASH.15 Despite the ongoing debate regarding mitochondrial dysfunction in NAFLD, more recent studies have observed that glucose oxidation rates are impaired in experimental models of obesity/T2D, and that increasing hepatic glucose oxidation may be a novel approach to attenuate obesity-related NAFLD.16 This may appear counterintuitive, as increasing the oxidation of glucose should lead to a corresponding reduction in fatty acid oxidation based on the glucose-fatty acid cycle originally described by Philip Randle and colleagues in the 1960s,17 thereby promoting hepatic lipid/TAG accumulation. Accordingly, the purpose of this review is to describe the role of glucose oxidation in the pathology of NAFLD while scrutinizing it as a potential therapeutic target based on these concerns. Furthermore, we will discuss the translational relevance of current pharmacotherapies that target glucose oxidation.
Pyruvate Dehydrogenase and Hepatic Glucose Oxidation
The liver plays a major role in whole-body glucose homeostasis, with glucose transporter 2 serving as the primary transporter for hepatic glucose uptake.18 Following transport into the liver, glucose is phosphorylated by glucokinase to glucose-6-phosphate and has a variety of metabolic fates. This includes (1) undergoing glycogenesis for storage as glycogen; (2) shuttling into the pentose phosphate pathway to generate reduced nicotinamide adenine dinucleotide phosphate for supporting DNL and the production of reduced glutathione; and (3) undergoing glycolysis to support small amounts of energy (ATP) production with the eventual generation of pyruvate (Figure 1). In order to maximize ATP production during glucose metabolism, glycolytically derived pyruvate is transported into the mitochondria via the mitochondrial pyruvate carrier (MPC), following which pyruvate is decarboxylated into acetyl CoA via the pyruvate dehydrogenase (PDH) complex (PDC) (Figure 2). It should be noted though that pyruvate oxidation only accounts for a minor fraction of glucose/pyruvate metabolism in the liver, even in the post-prandial state, as the vast majority of mitochondrial pyruvate is carboxylated into oxaloacetate via pyruvate carboxylase (PC), which contributes to supporting DNL.19
Figure 1.
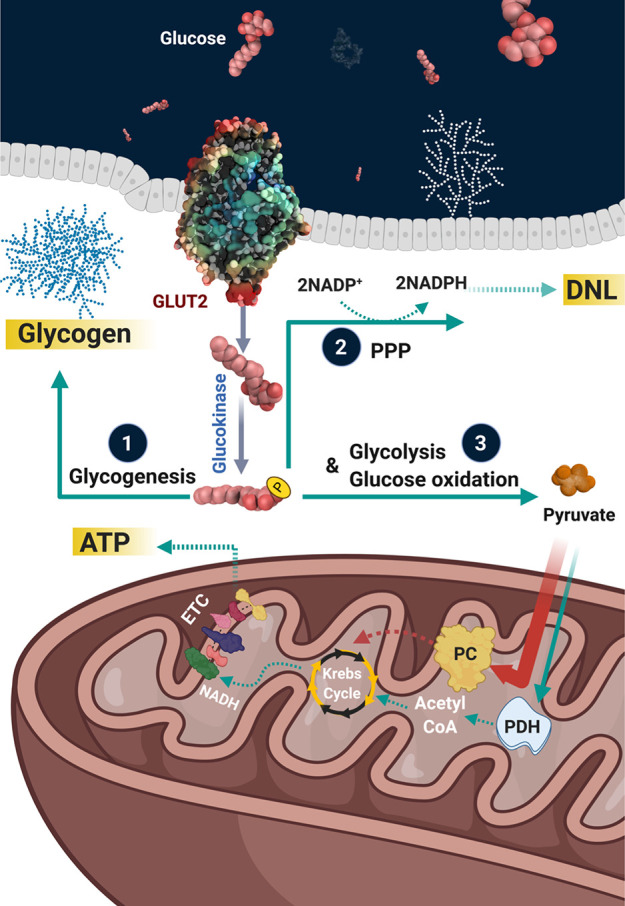
Hepatic glucose metabolism. Upon glucose transporter 2 (GLUT2) mediated entry into the hepatocyte, glucokinase phosphorylates glucose into glucose-6-phosphate, which subsequently undergoes 3 major metabolic fates. This includes (1) storage as glycogen via glycogenesis, (2) the pentose phosphate pathway (PPP) to generate important cofactors (NADPH) to support DNL and the production of reduced glutathione, and (3) to generate small amounts of energy (ATP) via aerobic glycolysis, which results in the formation of pyruvate as an end-product. This pyruvate may be transported into the mitochondria via the mitochondrial pyruvate carrier, following which the majority is carboxylated into oxaloacetate via PC, whereas a much smaller fraction is decarboxylated into acetyl CoA via PDH.
Figure 2.

Enzymatic regulation of the PDH complex. The PDH complex is multienzyme complex comprising three major enzymes, including PDH (enzyme 1 [E1]), dihydrolipoyl acetyltransferase (DLAT or E2), and dihydrolipoyl dehydrogenase (DLD or E3). PDH is responsible for the decarboxylation of pyruvate, and is tightly controlled by phosphorylation, whereby 4 isoforms of PDHK phosphorylate/deactivate PDH, and 2 isoforms of PDHP dephosphorylate/activate PDH. PDHK is positively regulated by increased acetyl CoA/CoA and NADH/NAD+ ratios, whereas increases in pyruvate inhibit PDHK but stimulate PDHP. In addition, increases in mitochondrial calcium and magnesium concentrations can also stimulate PDHP.
The PDC is a fundamental enzyme involved in cellular energy metabolism and the rate-limiting enzyme of glucose oxidation. The PDC is composed of three enzymes with distinct activities that act in a sequential fashion; PDH (or enzyme 1 [E1]), dihydrolipoyl acetyltransferase (DLAT or E2), and dihydrolipoyl dehydrogenase (DLD or E3) (for extensive review of the PDC please refer to refs (20,21)), which ultimately result in the formation of acetyl-CoA, CO2, and reduced nicotinamide adenine dinucleotide (NADH) (Figure 2). PDC generated acetyl-CoA feeds into the Krebs Cycle, subsequently resulting in formation of the reducing equivalents NADH and reduced flavin adenine dinucleotide, which act as electron donors to fuel oxidative phosphorylation in the mitochondrial electron transport chain for ATP production.
The oxidation of pyruvate through the PDH component of the PDC is tightly regulated by a variety of post-translational modifications, with phosphorylation/dephosphorylation of specific serine residues of PDH being the most extensively characterized to date.20,21 Two tightly bound regulatory enzymes, pyruvate dehydrogenase kinase (PDHK), of which there are four isoforms, and pyruvate dehydrogenase phosphatase (PDHP), of which there are two isoforms, catalyze the deactivating phosphorylation and activating dephosphorylation of PDH, respectively (Figure 2). Moreover, PDC-derived metabolic byproducts and byproduct ratios, including acetyl CoA/free CoA and the NADH/NAD+, can regulate PDH activity secondary to regulating the activity of PDHK and PDHP. Recent studies have also identified that PDH can be regulated by both acetylation22,23 and glutathionylation,24,25 both of which inhibit PDH activity and subsequent glucose oxidation.
In the liver, PDH activity is stimulated post-prandially, whereby PDH-derived acetyl CoA is used not only to support ATP production but also to support DNL.26 In contrast, during fasting or prolonged starvation, PDH activity is decreased as a consequence of elevated fatty acid oxidation rates and corresponding increases in the acetyl CoA/free CoA and the NADH/NAD+ ratios. In addition, increases in the acetyl CoA/free CoA ratio also stimulate the activity of PC, which converts pyruvate into oxaloacetate. This is an essential metabolic adaptation from a physiological homeostasis standpoint during fasting/starvation, as this allows pyruvate to be used as a substrate for gluconeogenesis, thereby allowing the body to maintain normoglycemia.
PDH Activity in Obesity
Pivotal studies by Jeoung and Harris demonstrated that targeting PDH activity might have therapeutic value in obesity, as they observed that mice harboring a whole-body deletion of PDHK4 were protected against insulin resistance when fed a high-fat diet (HFD; 59.5% kcal from lard) for 18 weeks.27 Reasons for this protection have primarily been attributed to actions in skeletal muscle, as experimental obesity increases muscle PDHK4 expression, thereby impairing muscle PDH activity and subsequent glucose oxidation rates. Moreover, a multitude of studies have now demonstrated that increasing muscle PDH activity and glucose oxidation imparts a potent glucose-lowering response in obesity. For example, provision of mangiferin (a primary constituent of mango tree extract) in the diet increases soleus muscle PDH activity and glucose oxidation rates, likely due to a significant reduction in PDHK4 expression, ultimately improving glucose tolerance and insulin sensitivity in obese mice.28 In addition, it has also been demonstrated that increases in muscle PDH activity are key mechanisms by which either increasing muscle carnitine acetyltransferase activity or decreasing muscle ketone body oxidation rates result in an overall improvement in glucose homeostasis in experimental obesity.29,30 Illustrating the clinical relevance of PDH as a target, supplementation for 6 months with l-carnitine (2 g/day) increased vastus lateralis muscle PDH activity (secondary to reductions in the acetyl CoA/free CoA ratio31), and lowered both circulating glucose and insulin levels in overweight/obese, insulin-resistant human subjects.30
Hepatic PDH Activity in NAFLD
While the previous section alluded to increases in muscle glucose oxidation representing the mechanism by which stimulating PDH activity imparts beneficial actions in obesity, the majority of these studies have not considered whether increases in PDH activity/glucose oxidation in other organs could be responsible. Reasons for hepatic PDH activity not being considered likely involve the minimal contribution that pyruvate oxidation has in terms of overall pyruvate metabolism in the liver.19 Furthermore, liver-specific PDH deficient mice exhibit robust reductions in hepatic glucose production and an improvement in whole-body insulin sensitivity,26 which would raise concern with regard to stimulating hepatic PDH activity in the setting of NAFLD. However, these studies were not performed in the context of HFD supplementation and obesity.
The prototypical pan-PDHK inhibitor is dichloroacetate,32 but this agent is limited by a short half-life, and thus a series of novel PDHK inhibitors were recently developed by the Chuang laboratory, with the strongest candidate being 2-[(2,4-dihydroxyphenyl)sulfonyl]isoindoline-4,6-diol (PS10).33 While acute treatment of mice with PS10 appears to stimulate PDH activity in multiple organs, prolonged PS10 treatment appears to harbor selectivity toward increasing hepatic PDH activity.33 Moreover, treatment of male C57BL/6J mice fed a HFD (60% kcal from lard) for 14 weeks with PS10 (70 mg/kg once daily) during the final 4 weeks improved glucose tolerance, which was associated with a robust reduction in hepatic steatosis as indicated by decreased Oil Red O staining.33,34 As NAFLD is a major risk factor for insulin resistance/T2D, this suggests that systemic activation of PDH to increase glucose oxidation rates may also improve glucose homeostasis via reductions in hepatic steatosis. Indeed, despite mangiferin treatment increasing muscle PDH activity/glucose oxidation in obese mice, it also caused marked reductions in adiposity,28 suggesting that reductions in hepatic steatosis may have been present. Observations from Go et al. engendered further interest along this perspective, as mice with a whole-body PDHK2 deficiency demonstrated protection against insulin resistance when subjected to experimental obesity via chronic HFD (60% kcal from lard) supplementation, which they specifically attributed to a reduction in hepatic steatosis.16 Furthermore, they observed that experimental obesity increased hepatic PDHK2 mRNA/protein expression, thereby decreasing hepatic PDH activity, whereas obese PDHK2 deficient mice exhibited normal hepatic PDH activity, which was associated with decreased liver weights and hepatic steatosis compared to their obese wild-type littermates. Because PDHK2 deficient mice also demonstrated reductions in adiposity in response to experimental obesity, the reductions in hepatic steatosis could once again be secondary to weight loss. However, liver-specific knockdown of PDHK2 via tail vein injection of an adenovirus also lowered hepatic TAG content and improved glucose tolerance, alluding to effects in the liver being a major driver of the phenotype observed in PDHK2 deficient mice.
Additional support for increasing hepatic PDH activity and subsequent glucose oxidation rates to mitigate obesity-related NAFLD have been observed with ranolazine, a second-line therapy used for the treatment of angina. While ranolazine’s mechanism of action for improving angina stems from its ability to inhibit the late inward sodium current by blocking the voltage-gated sodium channel subunit 1.5,35 ranolazine has also been demonstrated to increase glucose oxidation rates in both isolated working heart and muscle preparations.36,37 Accordingly, male C57BL/6J mice fed a HFD (60% kcal from lard) for 10 weeks were subsequently treated with ranolazine (50 mg/kg daily) for 30 days while remaining on the HFD, which led to marked reductions in the liver weight/body weight ratio and hepatic TAG content, as well as an overall improvement in glycemia.38 The ranolazine mediated improvement in hepatic steatosis was associated with decreased hepatic PDH phosphorylation (indicative of increased PDH activity), which may involve a direct effect, as ranolazine treatment of HepG2 cells also decreased PDHK4 mRNA expression and subsequent PDH phosphorylation. Importantly, reductions in hepatic steatosis are necessary for the glucose-lowering actions of ranolazine, as a single treatment of obese male C57BL/6J mice with ranolazine failed to lower hepatic TAG content and glucose levels during a pyruvate tolerance test. Conversely, a 1-week treatment of obese male C57BL/6J mice with ranolazine was sufficient to lower hepatic TAG content, which was now associated with improved glycemia during a pyruvate tolerance test. As obese individuals are often at risk for angina/ischemic heart disease, it may prove worthwhile for future studies to assess the prevalence of NAFLD in subjects treated with ranolazine in this patient population.
Potential Mechanisms by Which Increasing Hepatic PDH Activity Attenuates NAFLD
Although a number of studies support the premise that increases in hepatic PDH activity and glucose oxidation protect against obesity-related NAFLD, the specific mechanisms involved remain to be elucidated. It has been proposed that the mechanism by which PDHK2 deficiency in the liver protects against hepatic steatosis is an increased rate of hepatic ketogenesis.16 Ketogenesis is the physiological process by which fatty acids are metabolized and converted into the ketone bodies acetoacetate and β-hydroxybutyrate, which primarily takes place in the liver and can dispose of approximately two-thirds of hepatic fatty acid uptake.39,40 Increases in hepatic PDH activity divert pyruvate away from supporting anaplerosis through PC mediated entry into the Krebs Cycle, thereby lowering Krebs Cycle intermediates and promoting ketogenesis, all of which was observed in obese PDHK2 deficient mice.16 Furthermore, hepatic mRNA expression of 3-hydroxymethyl-3-methylglutaryl CoA synthase 2, the fate-committing enzyme of ketogenesis, was also increased in obese PDHK2 deficient mice, while hepatocytes isolated from these mice demonstrated decreased formation of 14CO2 from [14C1]-octanoate and an increased rate of β-hydroxybutyrate formation. These findings are compatible with studies in liver-specific PC deficient mice, whereby these animals also demonstrated robust increases in hepatic ketogenesis, though only macrovesicular and not microvesicular steatosis were improved in their livers when fed a HFD.41 In contrast, pharmacological PDHK inhibition with PS10 did not influence hepatic Krebs Cycle flux or increase circulating ketone bodies in obese C57BL/6J mice,34 but instead may decrease hepatic steatosis by decreasing nuclear levels of carbohydrate-responsive element binding protein, thereby decreasing hepatic DNL. Such observations, however, are incompatible with findings in liver-specific PDH deficient mice, demonstrating that PDH-derived acetyl CoA plays an important role in supporting hepatic DNL.26,42 It also needs to be considered whether increases in hepatic PDH activity protect against NAFLD by simply diverting pyruvate away from PC. Because glycolytically derived pyruvate enters the mitochondria via the actions of the MPC, reduced MPC activity would limit pyruvate flux through both PDH and PC in the liver. Of interest, mice with a liver-specific MPC2 deficiency exhibit reductions in hepatic mRNA expression of collagens and overall fibrosis scoring, as well as decreased liver injury when placed on a HFD (40% kcal primarily from trans-fats) that also contains high fructose (20%) and cholesterol (2%).43 Such observations support the notion that diverting pyruvate away from PC may be more important than increasing its flux through PDH in the liver, though hepatic TAG content was not decreased in these animals.
As previously mentioned, increases in glucose oxidation often result in a corresponding reduction of fatty acid oxidation via the glucose-fatty acid cycle described by Randle et al.17 Thus, on the surface, one might predict that stimulating hepatic PDH activity and glucose oxidation would inhibit fatty acid oxidation through Randle’s glucose-fatty acid cycle, thereby shuttling fatty acids toward lipogenic pathways and increasing hepatic TAG accumulation. Observations in obese PDHK2 deficient mice or obese mice treated with either PS10 or ranolazine clearly indicate that this is not the case, even though ranolazine has also been shown to decrease fatty acid oxidation rates in both cardiac and skeletal muscle.36,37 Of interest, it has been suggested that the glucose-fatty acid cycle is most relevant to cardiac and skeletal muscle, while being less applicable in the liver,44 which may explain why stimulating hepatic PDH activity does not promote hepatic steatosis and can surprisingly mitigate obesity-related NAFLD.16,34,38 It is also possible that since flux through PDH accounts for only a minor fraction of overall pyruvate metabolism in the liver, any increase in hepatic glucose oxidation offset by a decrease in fatty acid oxidation is minimal. On the contrary, if the glucose-fatty acid cycle is relevant in the liver and increasing hepatic PDH activity does result in a corresponding and significant reduction in hepatic fatty acid oxidation, it is possible this may induce an energy deficit that stimulates the energy sensor, 5′AMP activated protein kinase (AMPK). Indeed, decreasing fatty acid oxidation can stimulate AMPK activity,45 and increased AMPK phosphorylation (indicative of increased AMPK activity46) was observed in obese male C57BL/6J mice treated with PS10.34 Moreover, numerous studies have demonstrated that augmenting AMPK activity can prevent and/or reverse hepatic steatosis and the progression of NAFLD.47
Taken together, a number of proposed mechanisms including increases in hepatic ketogenesis or AMPK activity, as well as decreases in hepatic DNL or flux through PC may explain how stimulating PDH activity and glucose oxidation confers benefit against obesity-related NAFLD (Figure 3). Nonetheless, it remains to be conclusively determined whether these mechanisms are truly required for the salutary actions against NAFLD in response to hepatic PDH activation, which will need to be addressed in future preclinical studies.
Figure 3.
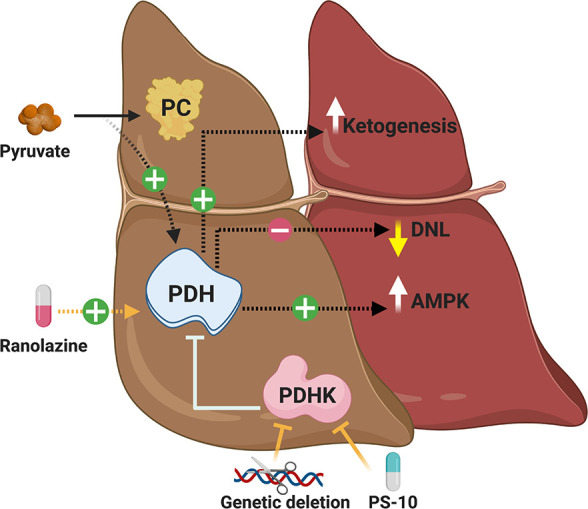
Proposed mechanisms explaining how increased hepatic PDH activity ameliorates NAFLD. Increases in hepatic PDH activity and subsequent glucose oxidation rates may reduce hepatic steatosis and the progression of NAFLD by possibly increasing hepatic ketogenesis, decreasing hepatic DNL, stimulating AMPK activity, or diverting pyruvate away from PC.
Summary
NAFLD is a major cause of morbidity worldwide that increases the risk for nonalcoholic steatohepatitis and hepatocellular carcinoma, while also increasing the risk for insulin resistance, T2D, and cardiovascular disease.1,6 Hence, it is critical that we identify the mechanisms of action responsible for NAFLD, especially in the context of obesity, as that may lead to the development of new treatments that can prevent and/or reverse the progression of insulin resistance/T2D. Although a controversial topic of ongoing scrutiny, a plethora of evidence supports that mitochondrial dysfunction may be a key mechanism responsible for obesity-induced NAFLD, with impaired hepatic fatty acid oxidation posited to be the primary culprit of this mitochondrial dysfunction. On the contrary, more recent studies have suggested that impaired hepatic glucose oxidation may also contribute to mitochondrial dysfunction in the pathology of obesity-induced NAFLD. As such, genetic and pharmacological strategies that increase hepatic PDH activity and subsequent glucose oxidation result in an amelioration of hepatic steatosis, while also improving glucose homeostasis. Moreover, ranolazine is an anti-anginal therapy used clinically, which has secondary actions to increase hepatic PDH activity, providing a unique translational opportunity to confirm whether increasing hepatic glucose oxidation is a feasible approach to treat NAFLD.
Acknowledgments
All the schematics in this review were created with BioRender.com. This review was supported by an Operating Grant from the Canadian Liver Foundation to JRU, and JRU is a Tier 2 Canada Research Chair (Pharmacotherapy of Energy Metabolism in Obesity).
The authors declare no competing financial interest.
References
- Haas J. T.; Francque S.; Staels B. (2016) Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu. Rev. Physiol. 78, 181–205. 10.1146/annurev-physiol-021115-105331. [DOI] [PubMed] [Google Scholar]
- Perry R. J.; Samuel V. T.; Petersen K. F.; Shulman G. I. (2014) The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 510 (7503), 84–91. 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Zhong G. C.; Tan H. Y.; Hao F. B.; Hu J. J. (2019) Nonalcoholic fatty liver disease and mortality from all causes, cardiovascular disease, and cancer: a meta-analysis. Sci. Rep. 9 (1), 11124. 10.1038/s41598-019-47687-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik J. M.; Golabi P.; Younossi Y.; Mishra A.; Younossi Z. M. (2020) Changes in the Global Burden of Chronic Liver Diseases From 2012 to 2017: The Growing Impact of NAFLD. Hepatology 72 (5), 1605–1616. 10.1002/hep.31173. [DOI] [PubMed] [Google Scholar]
- Younossi Z.; Anstee Q. M.; Marietti M.; Hardy T.; Henry L.; Eslam M.; George J.; Bugianesi E. (2018) Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 15 (1), 11–20. 10.1038/nrgastro.2017.109. [DOI] [PubMed] [Google Scholar]
- Mantovani A.; Byrne C. D.; Bonora E.; Targher G. (2018) Nonalcoholic Fatty Liver Disease and Risk of Incident Type 2 Diabetes: A Meta-analysis. Diabetes Care 41 (2), 372–382. 10.2337/dc17-1902. [DOI] [PubMed] [Google Scholar]
- Monetti M.; Levin M. C.; Watt M. J.; Sajan M. P.; Marmor S.; Hubbard B. K.; Stevens R. D.; Bain J. R.; Newgard C. B.; Farese R. V. Sr.; Hevener A. L.; Farese R. V. Jr. (2007) Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 6 (1), 69–78. 10.1016/j.cmet.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Bessone F.; Razori M. V.; Roma M. G. (2019) Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 76 (1), 99–128. 10.1007/s00018-018-2947-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal A. J. (2019) Past, present and future perspectives in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 16 (6), 377–386. 10.1038/s41575-019-0144-8. [DOI] [PubMed] [Google Scholar]
- Czech M. P.; Tencerova M.; Pedersen D. J.; Aouadi M. (2013) Insulin signalling mechanisms for triacylglycerol storage. Diabetologia 56 (5), 949–64. 10.1007/s00125-013-2869-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodson L.; Karpe F. (2019) Hyperinsulinaemia: does it tip the balance toward intrahepatic fat accumulation?. Endocr. Connect. 8 (10), R157–R168. 10.1530/EC-19-0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen M. C.; Shulman G. I. (2018) Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 98 (4), 2133–2223. 10.1152/physrev.00063.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel V. T.; Shulman G. I. (2012) Mechanisms for insulin resistance: common threads and missing links. Cell 148 (5), 852–71. 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iozzo P.; Bucci M.; Roivainen A.; Nagren K.; Jarvisalo M. J.; Kiss J.; Guiducci L.; Fielding B.; Naum A. G.; Borra R.; Virtanen K.; Savunen T.; Salvadori P. A.; Ferrannini E.; Knuuti J.; Nuutila P. (2010) Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology 139 (3), 846–56. 10.1053/j.gastro.2010.05.039. [DOI] [PubMed] [Google Scholar]
- Koliaki C.; Szendroedi J.; Kaul K.; Jelenik T.; Nowotny P.; Jankowiak F.; Herder C.; Carstensen M.; Krausch M.; Knoefel W. T.; Schlensak M.; Roden M. (2015) Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 21 (5), 739–46. 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Go Y.; Jeong J. Y.; Jeoung N. H.; Jeon J. H.; Park B. Y.; Kang H. J.; Ha C. M.; Choi Y. K.; Lee S. J.; Ham H. J.; Kim B. G.; Park K. G.; Park S. Y.; Lee C. H.; Choi C. S.; Park T. S.; Lee W. N.; Harris R. A.; Lee I. K. (2016) Inhibition of Pyruvate Dehydrogenase Kinase 2 Protects Against Hepatic Steatosis Through Modulation of Tricarboxylic Acid Cycle Anaplerosis and Ketogenesis. Diabetes 65 (10), 2876–87. 10.2337/db16-0223. [DOI] [PubMed] [Google Scholar]
- Randle P. J.; Garland P. B.; Hales C. N.; Newsholme E. A. (1963) The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 281 (7285), 785–9. 10.1016/S0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- Thorens B.; Mueckler M. (2010) Glucose transporters in the 21st Century. Am. J. Physiol Endocrinol Metab 298 (2), E141–5. 10.1152/ajpendo.00712.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt M. E.; Harrison C.; Sherry A. D.; Malloy C. R.; Burgess S. C. (2011) Flux through hepatic pyruvate carboxylase and phosphoenolpyruvate carboxykinase detected by hyperpolarized 13C magnetic resonance. Proc. Natl. Acad. Sci. U. S. A. 108 (47), 19084–9. 10.1073/pnas.1111247108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M. S.; Korotchkina L. G. (2006) Regulation of the pyruvate dehydrogenase complex. Biochem. Soc. Trans. 34, 217–22. 10.1042/BST20060217. [DOI] [PubMed] [Google Scholar]
- Patel M. S.; Nemeria N. S.; Furey W.; Jordan F. (2014) The pyruvate dehydrogenase complexes: structure-based function and regulation. J. Biol. Chem. 289 (24), 16615–23. 10.1074/jbc.R114.563148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing E.; O’Neill B. T.; Rardin M. J.; Kleinridders A.; Ilkeyeva O. R.; Ussar S.; Bain J. R.; Lee K. Y.; Verdin E. M.; Newgard C. B.; Gibson B. W.; Kahn C. R. (2013) Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 62 (10), 3404–17. 10.2337/db12-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Ji R.; Liao X.; Castillero E.; Kennel P. J.; Brunjes D. L.; Franz M.; Mobius-Winkler S.; Drosatos K.; George I.; Chen E. I.; Colombo P. C.; Schulze P. C. (2018) MicroRNA-195 Regulates Metabolism in Failing Myocardium Via Alterations in Sirtuin 3 Expression and Mitochondrial Protein Acetylation. Circulation 137 (19), 2052–2067. 10.1161/CIRCULATIONAHA.117.030486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almutairi M.; Gopal K.; Greenwell A. A.; Young A.; Gill R.; Aburasayn H.; Al Batran R.; Chahade J. J.; Gandhi M.; Eaton F.; Mailloux R. J.; Ussher J. R. (2021) The GLP-1 Receptor Agonist Liraglutide Increases Myocardial Glucose Oxidation Rates via Indirect Mechanisms and Mitigates Experimental Diabetic Cardiomyopathy. Can. J. Cardiol. 37, 140–150. 10.1016/j.cjca.2020.02.098. [DOI] [PubMed] [Google Scholar]
- O’Brien M.; Chalker J.; Slade L.; Gardiner D.; Mailloux R. J. (2017) Protein S-glutathionylation alters superoxide/hydrogen peroxide emission from pyruvate dehydrogenase complex. Free Radical Biol. Med. 106, 302–314. 10.1016/j.freeradbiomed.2017.02.046. [DOI] [PubMed] [Google Scholar]
- Choi C. S.; Ghoshal P.; Srinivasan M.; Kim S.; Cline G.; Patel M. S. (2010) Liver-specific pyruvate dehydrogenase complex deficiency upregulates lipogenesis in adipose tissue and improves peripheral insulin sensitivity. Lipids 45 (11), 987–95. 10.1007/s11745-010-3470-8. [DOI] [PubMed] [Google Scholar]
- Jeoung N. H.; Harris R. A. (2008) Pyruvate dehydrogenase kinase-4 deficiency lowers blood glucose and improves glucose tolerance in diet-induced obese mice. Am. J. Physiol Endocrinol Metab 295 (1), E46–54. 10.1152/ajpendo.00536.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apontes P.; Liu Z.; Su K.; Benard O.; Youn D. Y.; Li X.; Li W.; Mirza R. H.; Bastie C. C.; Jelicks L. A.; Pessin J. E.; Muzumdar R. H.; Sauve A. A.; Chi Y. (2014) Mangiferin stimulates carbohydrate oxidation and protects against metabolic disorders induced by high-fat diets. Diabetes 63 (11), 3626–36. 10.2337/db14-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Batran R.; Gopal K.; Capozzi M. E.; Chahade J. J.; Saleme B.; Tabatabaei-Dakhili S. A.; Greenwell A. A.; Niu J.; Almutairi M.; Byrne N. J.; Masson G.; Kim R.; Eaton F.; Mulvihill E. E.; Garneau L.; Masters A. R.; Desta Z.; Velazquez-Martinez C. A.; Aguer C.; Crawford P. A.; Sutendra G.; Campbell J. E.; Dyck J. R. B.; Ussher J. R. (2020) Pimozide Alleviates Hyperglycemia in Diet-Induced Obesity by Inhibiting Skeletal Muscle Ketone Oxidation. Cell Metab. 31 (5), 909–919. 10.1016/j.cmet.2020.03.017. [DOI] [PubMed] [Google Scholar]
- Muoio D. M.; Noland R. C.; Kovalik J. P.; Seiler S. E.; Davies M. N.; DeBalsi K. L.; Ilkayeva O. R.; Stevens R. D.; Kheterpal I.; Zhang J.; Covington J. D.; Bajpeyi S.; Ravussin E.; Kraus W.; Koves T. R.; Mynatt R. L. (2012) Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 15 (5), 764–77. 10.1016/j.cmet.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick T. L.; Quinney H. A.; Lopaschuk G. D. (1992) Carnitine stimulation of glucose oxidation in the fatty acid perfused isolated working rat heart. J. Biol. Chem. 267 (6), 3758–63. 10.1016/S0021-9258(19)50590-7. [DOI] [PubMed] [Google Scholar]
- Stacpoole P. W.; Henderson G. N.; Yan Z.; James M. O. (1998) Clinical pharmacology and toxicology of dichloroacetate. Environ. Health Perspect 106 (4), 989–94. 10.2307/3434142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tso S. C.; Qi X.; Gui W. J.; Wu C. Y.; Chuang J. L.; Wernstedt-Asterholm I.; Morlock L. K.; Owens K. R.; Scherer P. E.; Williams N. S.; Tambar U. K.; Wynn R. M.; Chuang D. T. (2014) Structure-guided development of specific pyruvate dehydrogenase kinase inhibitors targeting the ATP-binding pocket. J. Biol. Chem. 289 (7), 4432–43. 10.1074/jbc.M113.533885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. Y.; Tso S. C.; Chuang J. L.; Gui W. J.; Lou M.; Sharma G.; Khemtong C.; Qi X.; Wynn R. M.; Chuang D. T. (2018) Targeting hepatic pyruvate dehydrogenase kinases restores insulin signaling and mitigates ChREBP-mediated lipogenesis in diet-induced obese mice. Mol. Metab. 12, 12–24. 10.1016/j.molmet.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossalla S.; Maier L. S. (2012) Role of ranolazine in angina, heart failure, arrhythmias, and diabetes. Pharmacol. Ther. 133 (3), 311–23. 10.1016/j.pharmthera.2011.11.003. [DOI] [PubMed] [Google Scholar]
- McCormack J. G.; Baracos V. E.; Barr R.; Lopaschuk G. D. (1996) Effects of ranolazine on oxidative substrate preference in epitrochlearis muscle. J. Appl. Physiol. 81 (2), 905–10. 10.1152/jappl.1996.81.2.905. [DOI] [PubMed] [Google Scholar]
- McCormack J. G.; Barr R. L.; Wolff A. A.; Lopaschuk G. D. (1996) Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation 93 (1), 135–42. 10.1161/01.CIR.93.1.135. [DOI] [PubMed] [Google Scholar]
- Al Batran R.; Gopal K.; Aburasayn H.; Eshreif A.; Almutairi M.; Greenwell A. A.; Campbell S. A.; Saleme B.; Court E. A.; Eaton F.; Light P. E.; Sutendra G.; Ussher J. R. (2019) The antianginal ranolazine mitigates obesity-induced nonalcoholic fatty liver disease and increases hepatic pyruvate dehydrogenase activity. JCI Insight 4 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchalska P.; Crawford P. A. (2017) Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 25 (2), 262–284. 10.1016/j.cmet.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson J. R.; Scholz R.; Browning E. T. (1969) Control mechanisms of gluconeogenesis and ketogenesis. II. Interactions between fatty acid oxidation and the citric acid cycle in perfused rat liver. J. Biol. Chem. 244 (17), 4617–27. 10.1016/S0021-9258(18)93669-0. [DOI] [PubMed] [Google Scholar]
- Cappel D. A.; Deja S.; Duarte J. A. G.; Kucejova B.; Inigo M.; Fletcher J. A.; Fu X.; Berglund E. D.; Liu T.; Elmquist J. K.; Hammer S.; Mishra P.; Browning J. D.; Burgess S. C. (2019) Pyruvate-Carboxylase-Mediated Anaplerosis Promotes Antioxidant Capacity by Sustaining TCA Cycle and Redox Metabolism in Liver. Cell Metab. 29 (6), 1291–1305. 10.1016/j.cmet.2019.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood S.; Birkaya B.; Rideout T. C.; Patel M. S. (2016) Lack of mitochondria-generated acetyl-CoA by pyruvate dehydrogenase complex downregulates gene expression in the hepatic de novo lipogenic pathway. Am. J. Physiol Endocrinol Metab 311 (1), E117–27. 10.1152/ajpendo.00064.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCommis K. S.; Hodges W. T.; Brunt E. M.; Nalbantoglu I.; McDonald W. G.; Holley C.; Fujiwara H.; Schaffer J. E.; Colca J. R.; Finck B. N. (2017) Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology 65 (5), 1543–1556. 10.1002/hep.29025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage D. B.; Petersen K. F.; Shulman G. I. (2007) Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 87 (2), 507–20. 10.1152/physrev.00024.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmers S.; Nabben M.; Bosma M.; van Bree B.; Lenaers E.; van Beurden D.; Schaart G.; Westerterp-Plantenga M. S.; Langhans W.; Hesselink M. K.; Schrauwen-Hinderling V. B.; Schrauwen P. (2012) Augmenting muscle diacylglycerol and triacylglycerol content by blocking fatty acid oxidation does not impede insulin sensitivity. Proc. Natl. Acad. Sci. U. S. A. 109 (29), 11711–6. 10.1073/pnas.1206868109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg G. R.; Kemp B. E. (2009) AMPK in Health and Disease. Physiol. Rev. 89 (3), 1025–78. 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- Day E. A.; Ford R. J.; Steinberg G. R. (2017) AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 28 (8), 545–560. 10.1016/j.tem.2017.05.004. [DOI] [PubMed] [Google Scholar]