

Abstract

A series of bone-targeting EP4 receptor agonist conjugate prodrugs were prepared wherein a potent EP4 receptor agonist was bound to a biologically inactive, bisphosphonate-based bone-targeting moiety. Singly and doubly radiolabeled conjugates were synthesized and were shown to be stable in blood, to be rapidly eliminated from the bloodstream, and to be effectively taken up into bone in vivo after intravenous dosing. From these preliminary studies a preferred conjugate 4 (also known as C3 and Mes-1007) was selected for follow up biodistribution and elimination studies. Doubly radiolabeled conjugate 4 was found to partition largely to the liver and bones, and both labels were eliminated from liver at the same rate indicating the conjugate was eliminated intact. Quantification of the labels in bones indicated that free EP4 agonist (EP4a)(2a) was released from bone-bound 4 with a half-time of about 7 days. When dosed orally, radiolabeled 4 was not absorbed and passed through the gastrointestinal tract essentially unchanged, and only traces of radiolabeled 4 were found in the liver, blood, or bones. 4 was found to bind rapidly and completely to powdered bone mineral or to various forms of calcium phosphate, forming a stable matrix suitable for implant and that could made into powders or solid forms and be sterilized without decomposition or release of 4. Basic hydrolysis released free EP4 agonist 2a quantitatively from the material.
Keywords: EP4 receptor, EP4 agonist, osteoporosis, bisphosphonate, bone anabolic, bone-targeting, prostaglandin E2
Introduction
Osteoporosis is a disease characterized by progressive loss of bone mass through an imbalance in the bone remodeling process such that bone resorption outstrips compensatory bone formation leading to deterioration of bone tissue, bone fragility, morbidity, and fractures. Statistically, osteoporosis-related fractures are more common than heart attack, stroke, and breast cancer combined, affecting half of women and one-fifth of men in their lifetime. A number of drugs, such as bisphosphonates, have been developed to suppress excessive resorption, but there remains an important medical need for effective agents to induce healthy bone formation that might reverse the effects of osteoporosis. Many other bone conditions such as fracture repair could also benefit from a safe and effective bone anabolic therapy. One available therapy has used analogs of parathyroid hormone (PTH) such as teriparatide (Forteo, but teriparatide’s use has been limited by its relative expense, a daily injection protocol, and its association with an increase in bone tumors in animal safety studies although no direct association with osteosarcoma has yet been established in humans.1
Prostaglandin E2 (1) has been shown to be stimulate bone growth in vivo in animals and in humans and to exert this effect through agonism at the EP4 receptor subtype.2 This effect has also been recapitulated, thus far in animals, with a number of highly potent and selective EP4 receptor agonists (e.g., compound 2a (Figure 1)),3,4 but unfortunately, even highly selective EP4 agonists such as 2a (like PGE2) are also associated with gastrointestinal (GI) and hypotension side effects that have thus far precluded them from development for treatment of osteoporosis.
Figure 1.

Prostaglandin E2 (1), EP4 receptor agonist 2 and the conjugate drug C1 (3).
To address these issues, we developed several EP4 receptor agonist prodrugs, wherein the agonist is conjugated via a differentially hydrolyzable linker group to a bone-targeting moiety derived from an ω-amino-bisphosphonate (such as alendronate).5 The synthesis, pharmacokinetics, and efficacy (in a rat model of osteoporosis) of one such conjugate, known as C1 (3) (Figure 1), have been reported.6−8 These prodrugs were designed to be inactive until hydrolyzed and, to be useful, need to be reasonably stable in the blood such that after administration the intact conjugate is either rapidly taken up into bones (mediated by the avidity of the bisphosphonate component for apatite mineral in bone) or eliminated from the bloodstream, thus avoiding systemic side effects. Once bound to bone, the conjugate prodrugs were designed to slowly liberate both the active EP4 agonist and bone-bound alendronate, thus exerting both local anabolic and anti-resorption effects. Compound 3 was shown to meet these criteria in rats where about 5% of the administered dose was taken up into bone within a few hours, and the majority was rapidly eliminated from the bloodstream via the liver and feces. Through the use of doubly radioactive-labeled conjugate we were able to show that 3 bound to bone largely intact and liberated the EP4 agonist with a half-time of about 5 days, while subsequent removal of the intermediate linker element liberated the alendronic acid component with a half-time of 28 days (Figure 2). Thus, 3 was compatible with infrequent, once a week, intravenous (IV) dosing.
Figure 2.
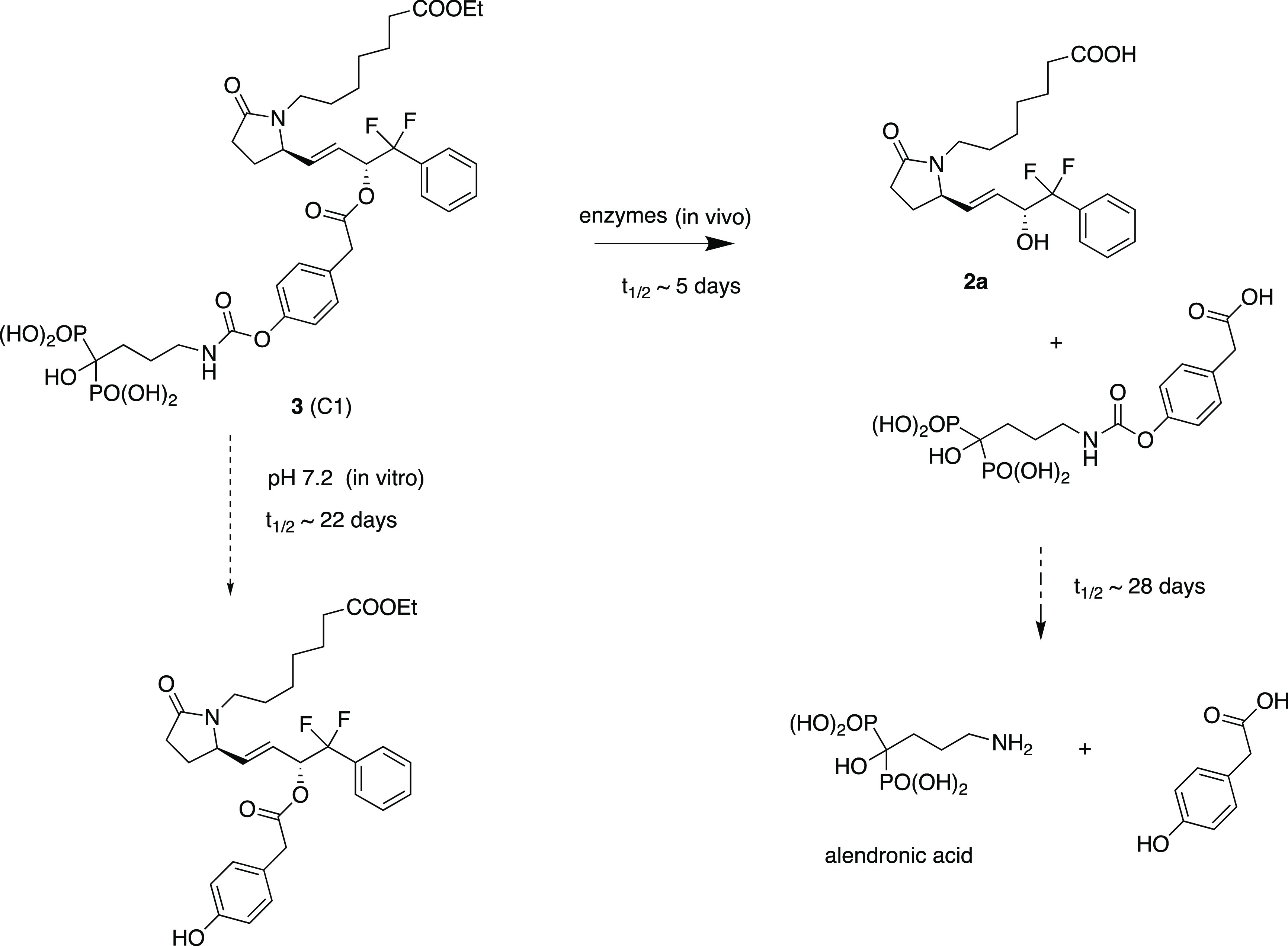
Conjugate C1 (3) and its conversion to liberate components.
While 3 proved to be efficacious in the ovariectomized rat model (OVX) of osteoporosis,7,8 several issues were encountered that suggested the conjugate was not optimal and could be improved. One issue relates to the stability of 3. Compound 3 links the EP4 agonist via an ester bond to 4-hydroxyphenylacetic acid and the relative stability of the linkage in blood and rate of cleavage once bound to bone (presumably via local esterase activities) is ideal for once-a-week dosing. The alendronic acid component is linked via a carbamate that was expected to be a chemically stable linkage and to be slowly and enzymatically hydrolyzed in vitro. While 3 was found to be stable in the solid form, it was found to spontaneously hydrolyze at the carbamate linkage (and not at the ester linkage) to yield the corresponding phenol (Figure 2) at a rate of about 2–3% per day when stored at pH 7.2 in buffered solution.6 The mechanism behind this surprising observation is not known, but it may involve some sort of self-catalysis involving the hydroxybisphosphonate moiety. This instability suggested that a more stable replacement for the carbamate should be sought. In addition, while the degree of uptake of 3 into bones (∼5% of administered dose) was sufficient to provide efficacy with a dose of 5 mg/kg per week, it was felt that a greater level of uptake would be desirable and might serve to allow the dose, or the dose frequency, to be reduced. Finally, we speculated that the “dual action” of conjugates such as 3 might in fact be less than optimal in certain situations. Normal bone turnover and remodeling involves both resorption and formation, and it is the balance of these processes that is important. If our drug was uniquely anabolic, then it could still reverse a bone formation deficit while leaving the resorption component unchanged and potentially lead to more normal healthy bone. In addition, the anti-resorptive effects of bisphosphonates and other drugs9 have been associated with rare but significant side effects such as necrosis of the jaw which is related to anti-resorptive potency; thus, an inactive bisphosphonate should not have such side effects.
For these reasons we set out to redesign our conjugate drugs with an aim to (1) improve bone uptake, (2) improve chemical stability, and (3) to use a bisphosphonate as the bone-targeting component that was inactive as an anti-resorptive agent at the doses used. We proposed that replacement of the carbamate in 3 by an amide would provide a greater stability both in solution and in vivo, and such a prodrug would retain the bone-targeting properties of the hydroxybisphosphonate moiety. However, after release of the EP4 agonist, the conjugate would leave the intact targeting moiety bound to bone, a molecule designed to bind effectively to bones but to be essentially inactive as an inhibitor of osteoclast action (Figure 3). It is known that with ω-aminoalkylbisphosphonates such as alendronate, the basic amino function is very important for anti-resorptive potency10 as is the alkyl chain length. Analogs with shorter chains (such as in pamidronate) or longer chains (such as neridronate) are considerably less active than alendronate11 and bisphosphonates lacking a basic nitrogen are also less potent.12 The N-acetyl analog of neridronate was reported to be essentially inactive in a model of hypercalcemia induced in TPTX rats by 1,25-dihydroxyvitamin D3.10 Also, alendronic acid benzoyl amides have been shown to be very stable chemically.13 However, some N-benzoyl analogs of alendronate and pamidronate have shown inhibitory activity on osteoclasts, at least in vitro, while notably, some of these same compounds were shown to stimulate osteoblast maturation in vitro.14 It is not clear if one can easily predict the potency (or lack of activity) of such analogs.
Figure 3.
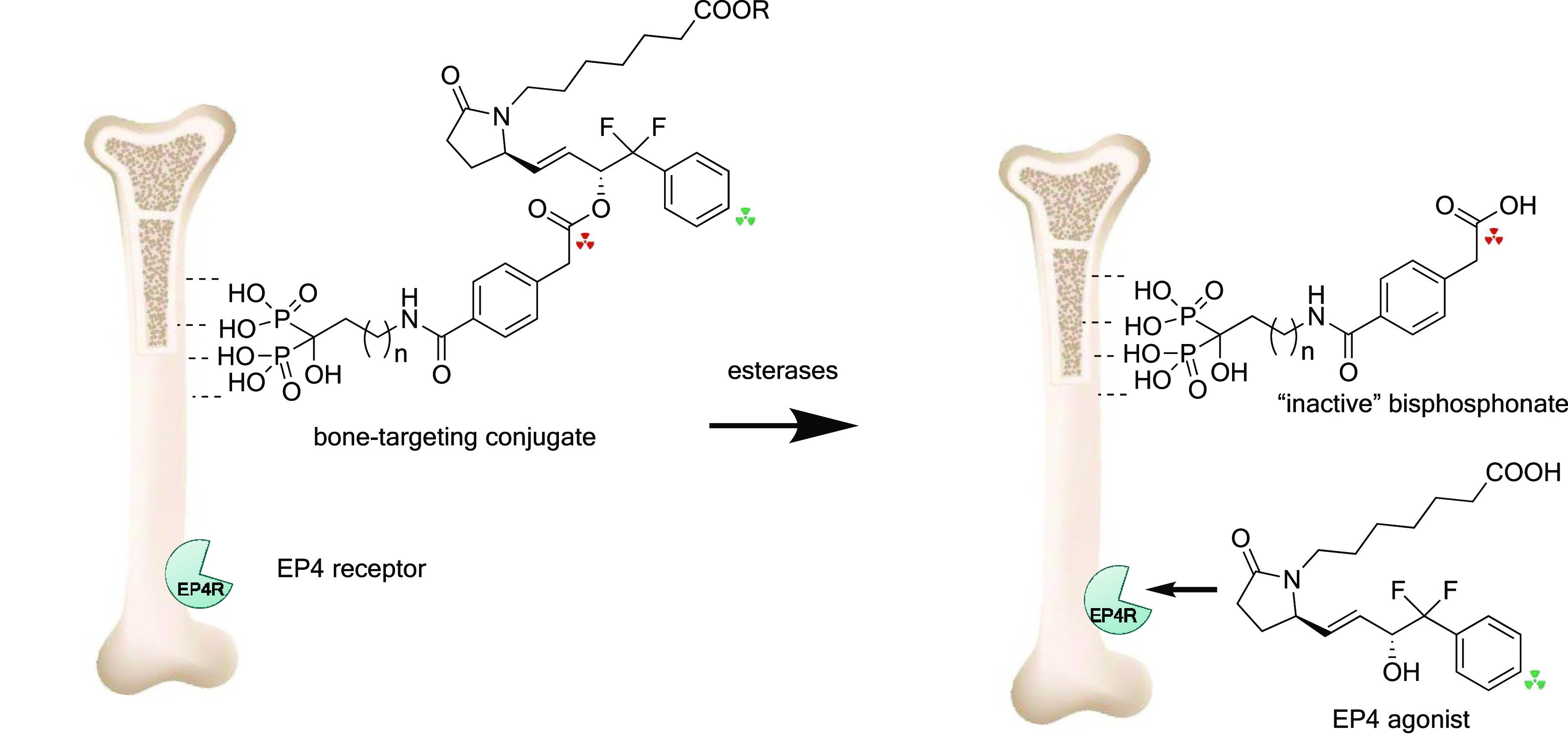
Conjugate prodrugs designed to target bone and liberate EP4 agonist and an inactive bisphosphonate which remains bound to bone. The red triangle triads indicate the 14C-labeled sites, and the green triangle triads indicate the 3H-labeled sites.
We therefore set out to prepare and evaluate several analogs of 3 where the carbamate group was replaced by an amide and the bisphosphonate chain length was varied. Our plan was to prepare compounds 4 (C3), 5 (C4), and 6 (C5) (Figure 4) incorporating the bisphosphonates alendronate, pamidronate, and neridronate and to evaluate these conjugates for chemical and blood plasma stability and degree of uptake into bones after intravenous administration. In addition, as a potentially higher efficiency EP4 agonist delivery conjugate, we prepared a conjugate where one alendronic acid was linked through the linker moiety, 3,5-bis-carboxymethylbenzoic acid, to two molecules of 2b to provide conjugate 7 (C6) with the EP4 agonist/bisphosphonate ratio of 2 (Figure 4).
Figure 4.

Target conjugates and their putative liberated bisphosphonate components.
We also wanted to evaluate the biological activity (anti-resorptive potential) of the corresponding bisphosphonate phenylacetic acid amide fragments (e.g., 8–11) (Figure 4) that would be expected to be liberated in vivo after enzymatic hydrolysis of the EP4-conjugate ester bond in 4–7. The mechanism of action of nitrogen-containing bisphosphonates (such as alendronate) involves the inhibition of farnesyl pyrophosphate synthase (FPPS); thus, suppression of the synthesis of isoprenoid lipids farnesyl–farnesyl pyrophosphate (FFPP) and geranyl–geranyl pyrophosphate (GGPP) metabolites is important in the post-translation prenylation of small Rho and Rac GTPases.15 Bisphosphonates used for the treatment of bone disorders inhibit squalene synthase and cholesterol biosynthesis15,16 and this is considered to be the basis of their antiosteoclast effects. Small GTPases are also integral to differentiation and function of neutrophils17 and Rac GTPases affect neutrophil function through the fMLP receptor.18 Nitrogen-containing alkyl bisphosphonates such as zoledronate19 inhibit neutrophil chemotaxis; thus, inhibition of fMLP-induced chemotaxis of mouse neutrophils can serve as a useful in vitro functional cellular assay to assess the potency of bisphosphonates. Thus, fragments 8–11 could be compared in this assay, with an active bisphosphonate as positive control, so as to demonstrate the anticipated lack of activity. Our plan was that a selected optimal compound and its bisphosphonate fragment would then be tested in more extensive efficacy models (i.e., the OVX rat model) to confirm both the expected anabolic and lack of anti-resorptive activity.
The main focus in this study was on the monovalent conjugates, so we prepared conjugates 4–6 with a radiolabel (tritium) in the EP4 agonist portion of the molecule (derived from [3H]-2b) to initially evaluate relative plasma stability, adherence to bone in vitro and in vivo, degree of uptake into bones, and tissue distribution after IV dosing. These data plus the activity (or lack of activity) of the putatively released bisphosphonate fragments then allowed selection of the optimal conjugate prodrug for further follow up.
We then selected conjugate 4, which achieved the greatest bone uptake, and prepared 4 in a doubly radiolabeled form so that the integrity of the conjugate could be tracked throughout the body and to confirm that while [3H]-2a was released from the bone over time, the bisphosphonate linker moiety was not hydrolyzed and remained bound to bones (Figure 3; tritium, indicated by green triangle triads). The labeling of the “linker” element with 14C (red triangle triads) would provide this information; thus, a radio-synthesis process was developed. The doubly labeled conjugate was also used to reconfirm the stability, biodistribution, elimination, and bone disposition of 4. We speculated that 4 would be largely eliminated via the liver into the intestines where it could potentially be hydrolyzed by putative gut enzymes to liberate free 2a that could then be reabsorbed into the bloodstream. This would potentially frustrate the selective delivery to bones. Therefore, we carried out a study where doubly radiolabeled 4 was administered orally to rats, and levels of labels in the GI tract, liver, and blood were monitored. Finally, the long-term stability of conjugate prodrug 4 was evaluated in both the solid state and in solution.
In compounds 3 and 4, the EP4 agonist component (derived from 2a) is present as the ethyl ester. The ethyl ester form, 2b, has been previously shown to be rapidly converted in blood plasma to form active free acid 2a(5) suggesting that this ethyl ester is a good substrate for esterases/lipases in the blood. Conjugate 3 has been shown to be very effective as an anabolic agent in vivo indicating that the active free acid form, 2a, was liberated in the bone compartment. We anticipated that this would also be true for conjugate prodrug 4.
Potential “Prebinding” of EP4-Bisphosphonate Conjugate Prodrugs to Bone or Bone Substitutes for Local Administration
An important aspect of the design of conjugate prodrugs such as 3 and 4 is their ability to bind strongly and essentially irreversibly to bone mineral via complexation of calcium phosphate with the bisphosphonate component. This has been demonstrated both in vivo and in vitro. We have used this property to monitor stability of the conjugates in solution and in blood plasma or serum and to provide a simple way to remove intact prodrugs from solution (by simply stirring with bone powder).5 Equally, however, this property can allow for preparation of bone, or biocompatible forms of calcium phosphate mineral, which incorporate the prodrug as an integral component. This “preloaded” material can be prepared with whatever degree of loading desired and then implanted at sites in the body where local stimulation of bone growth is desired. Such local administration could then provide effective treatments for a variety of indications such as in dental and jaw implants, joint fusions, and to stimulate fracture repair for example. Therefore, we evaluated the optimal conjugate, 4, for binding to bone and to biocompatible forms of calcium phosphate and to determine if such material could be formed into shapes or granules and if the binding was stable. It was important to show that once bound to calcium phosphate preparations the prodrug was chemically stable and survived intact during sterilization procedures that would be required before in vivo use. We also wanted to show that 2a could be completely liberated from the material through hydrolysis. The ability to successfully manufacture such material would potentially enable a wide variety of uses in orthopedics and bone repair.
Results and Discussion
Synthesis of Conjugates 4–6 and Corresponding Bisphosphonate Fragments
The synthesis of the desired conjugates and the respective fragments proved to be relatively straightforward (Scheme 1).
Scheme 1. Synthesis of Prodrug Conjugates 4–6 and Their Tritiated Analogs.
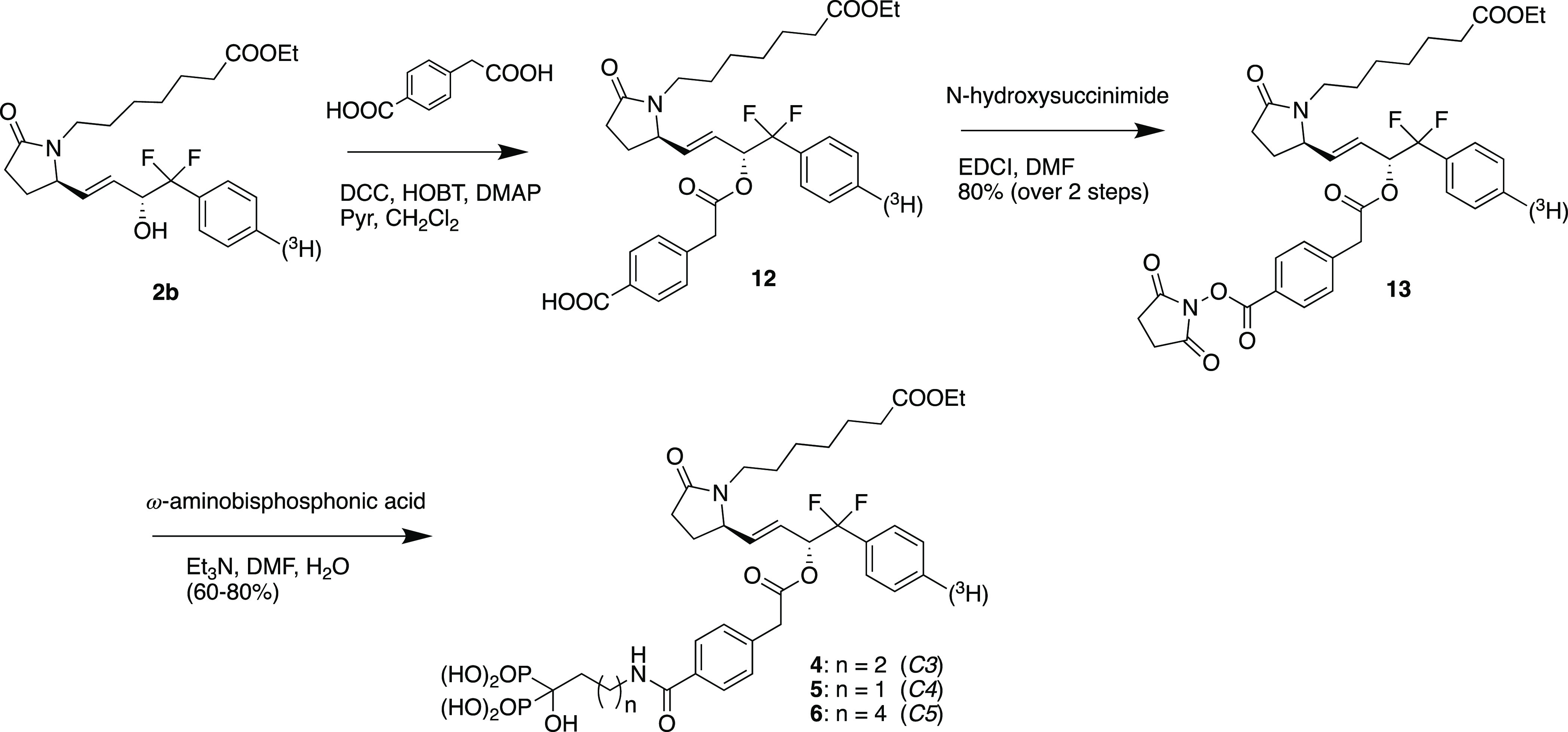
Esterification of the 15-hydroxyl function in the EP4 agonist ester 2b with 4-carboxyphenylacetic acid using DCC and HOBt proceeded efficiently and selectively to provide ester 12 which was then converted by reaction with N-hydroxysuccinimide and EDCI in DMF to provide activated ester 13, in 80% overall yield for the two steps. 13 was then reacted with excess alendronic acid (4 equiv) and triethyl amine (11.3 equiv) in aqueous DMF to provide crude conjugate 4 (designated as C3) which was purified by ion exchange chromatography followed by reverse phase chromatography to provide pure 4 in excellent yield (80%) as the disodium salt. Similar reaction of 13 with pamidronic acid gave conjugate 5 (designated as C4) (60% isolated yield), and reaction of 13 with neridronic acid gave conjugate 6 (designated as C5) in 60% yield.
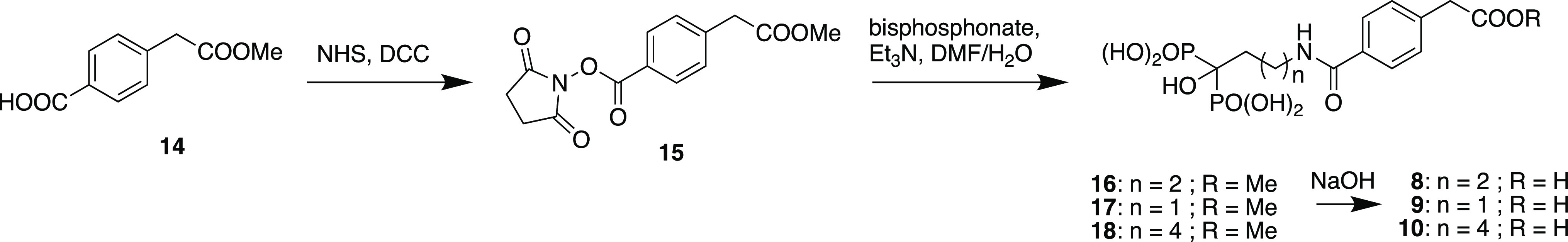
To prepare fragments 8–10, 4-carboxyphenylacetic acid was first selectively esterified with methanol using thionyl chloride,20 and resulting monoester 14 was converted to NHS-ester 15 and then reacted with alendronic acid, pamidronic acid, or neridronic acid to give corresponding monoesters 16–18 after ion-exchange and reverse-phase chromatography. These esters were then each hydrolyzed with sodium hydroxide, neutralized and purified by reverse phase chromatography to provide fragments 8–10 in 95, 71, and 58% yields, respectively, for the two-step procedures (Scheme 2).
Scheme 2. Synthesis of the Conjugate Fragments 8–10.
Synthesis of Conjugate 7
3,5-Dimethylbenzonitrile was brominated with NBS to give the bis-3,5-bis-bromomethylbenzonitrile (19) (28% yield) which was reacted with sodium cyanide and tetrabutylammonium bromide to give tricyano-compound 20 (59%) which was exhaustively hydrolyzed with sodium hydroxide to give tricarboxylic acid 21 (quantitative yield) after acidification. Selective esterification of the aliphatic carboxylic acid groups with 2b using DCC and DMAP gave adduct 22 which was converted directly to N-hydroxysuccinimide activated ester 23 in 55% overall yield. Reaction with alendronic acid triethylammonium salt and purification gave bis-EP4 agonist conjugate 7 (designated as C6) in 46% yield (Scheme 3).
Scheme 3. Synthesis of Conjugate 7 (C6).
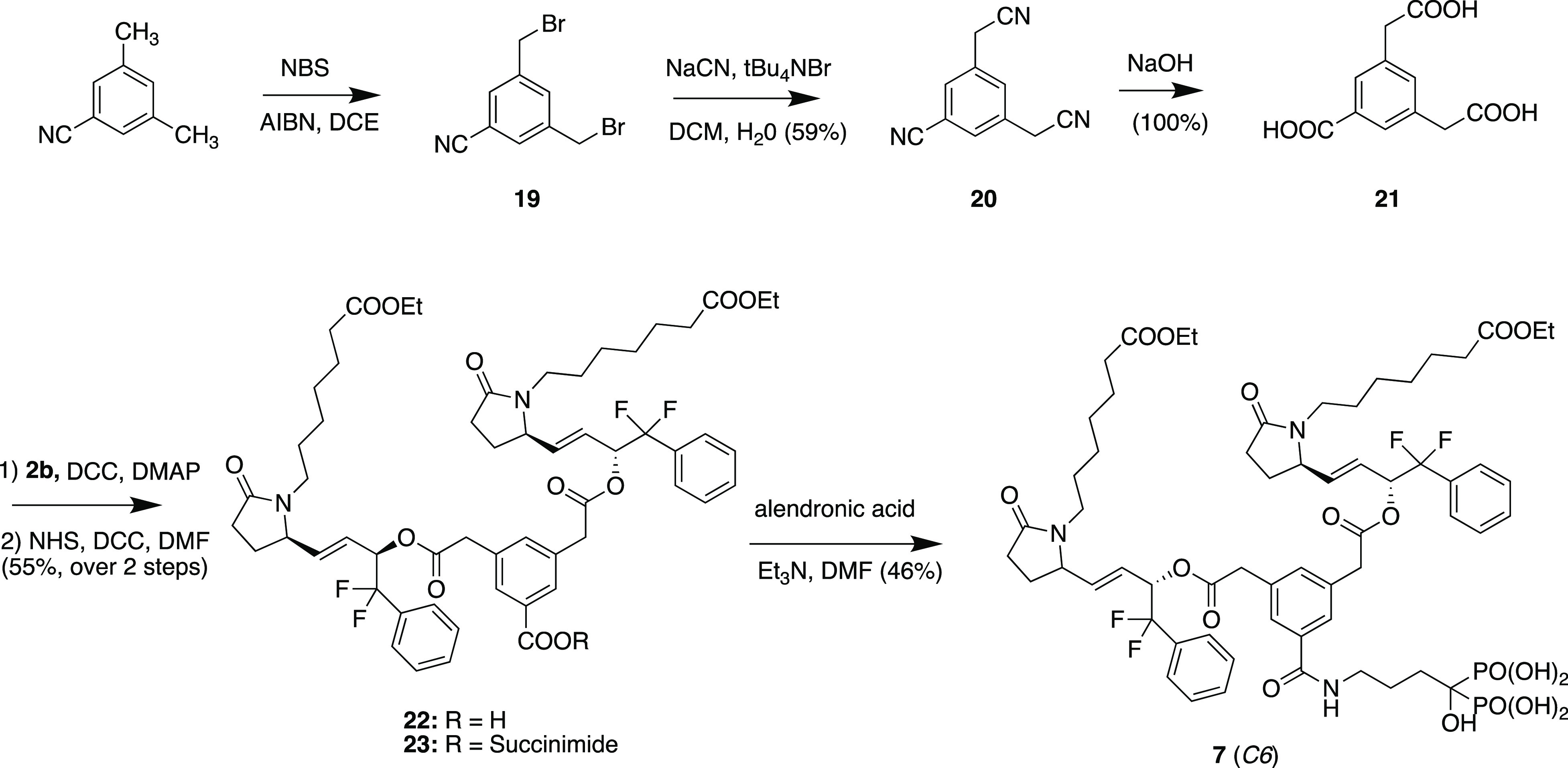
Synthesis of Bis-phenylacetic Acid Bisphosphonate Fragment 11
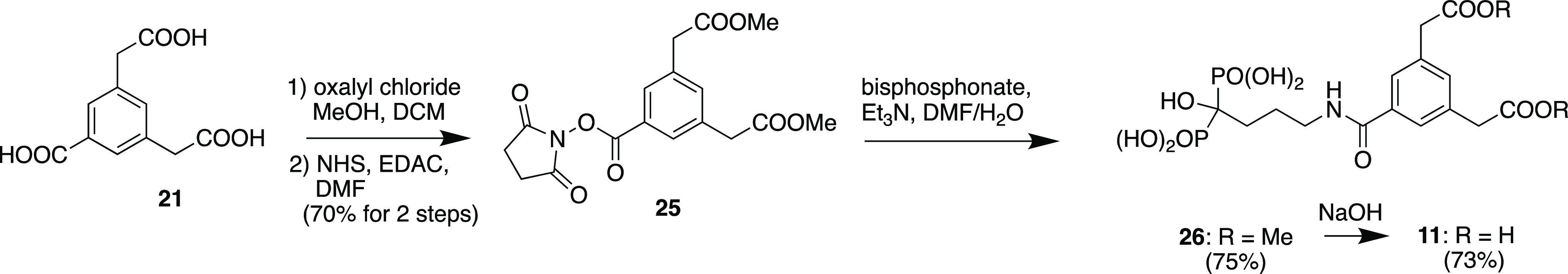
Tricarboxylic acid 21 was selectively esterified on the aliphatic carboxyl groups by reaction with oxalyl chloride and methanol to provide diester 24, and then the free benzoic acid was converted to activated ester 25 which was then reacted with alendronic acid (triethylammonium salt) followed by ion-exchange chromatography and then reverse-phase chromatography to provide bisphosphonate diester 26, which was then hydrolyzed with sodium hydroxide and purified (ion exchange and reverse phase chromatography) to provide desired bisphosphonate fragment 11 (Scheme 4).
Scheme 4. Synthesis of Conjugate Fragment 11.
Synthesis of Tritium-Labeled Conjugates 4–6
Conjugates [3H]-4, [3H]-5, and [3H]-6 radiolabeled with tritium on the EP4 agonist component were prepared essentially as in Scheme 1 but utilized corresponding 4′-[3H]-phenyl-substituted 2b available from a previous synthesis.21
Synthesis of [14C]-4
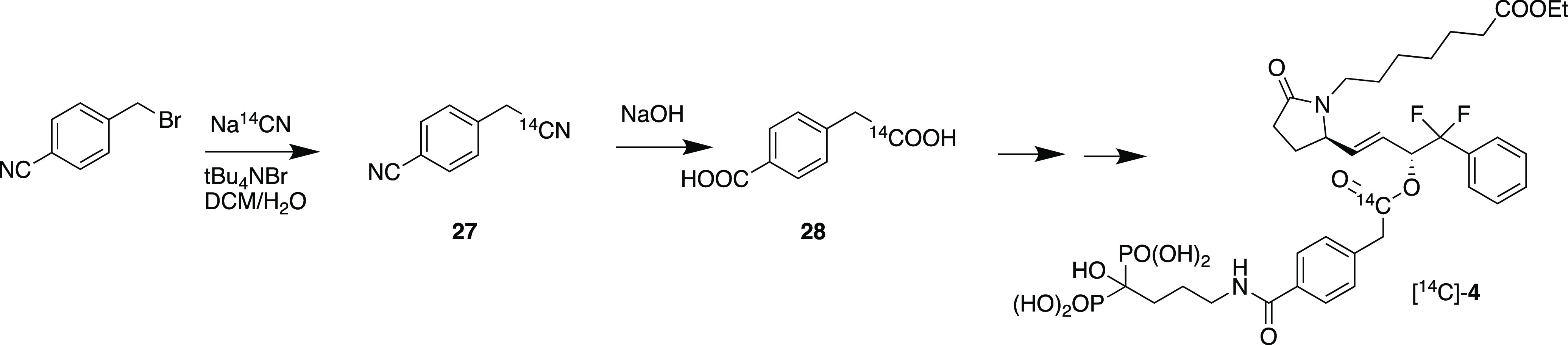
To effectively track the fate of 4 in the body as well as putative liberated fragment 7, we decided to label the linker 4-carboxyphenylacetic acid with 14C in the aliphatic carboxyl group (Scheme 5). Thus, 4-bromomethylbenzonitrile was reacted with sodium [14C]-cyanide in a two-phase reaction with tetra-n-butylammonium bromide to provide [14C]-4-cyanomethylbenzonitrile (27) in 76% yield. Hydrolysis of 27 with sodium hydroxide in a sealed tube gave 4-[14C]-(carboxymethyl)benzoic acid (28) (46% yield), which was then converted to [14C]-4 according to Scheme 1.
Scheme 5. Synthesis of [14C]-4.
Stability of Conjugates 4–6 in Blood Plasma
To evaluate the stability of the conjugates in blood, [3H]-4, [3H]-5, and [3H]-6 were each incubated at 37 °C in fresh rat plasma at a concentration of 100 μg/mL up to 24 h. At time 0 and various time intervals thereafter, aliquots were removed, and any free (hydrolyzed) EP4 agonist was quantified as tritium that remained in the solution after first removing intact conjugate by stirring with bone powder for 20 min. 4 showed about 10% increase in free radiolabel in the supernatant over 24 h (Table S1), while 5 and 6 were shown to be very stable in plasma with little release of free EP4 agonist 2a as indicated by <5% increase in the amount of free radioactivity in the supernatant over 24 h (Tables S2 and S3). In a second experiment, conjugate 4 was incubated under similar conditions in fresh human plasma and showed at most 5% liberation of free tritium over 24 h (data not shown).
In Vivo Uptake, Tissue Distribution, and Elimination of Conjugates 4–6 over 6 h
[3H]-4, [3H]-5, and [3H]-6 were each dosed intravenously (at 5 mg/kg) to rats, and blood samples were taken after 1, 2, 4, and 6 h. The animals were then euthanized, and tissues were examined for levels of radioactivity. In each case, after 1 h, most radioactivity was already eliminated from the blood (ca. 0.2–1% of administered dose remaining in blood at 1 h), and levels diminished rapidly thereafter with <0.2% remaining at 6 h (Figure 5, Table S4). Only traces of radioactivity were found in most tissues except for liver where 65–74% of the administered label was found, and in the bones the percent of the original label found was as follows: 20.3% (average, n = 2) with 4, 10.6 ± 2.1% with 5, and 9.5 ± 1.2% for 6 (Figure 6, Table S5). On the basis of these findings, we decided to proceed with follow up experiments using conjugate 4.
Figure 5.

Blood levels of tritium (±SEM) after intravenous dosing of [3H]-4, [3H]-5, and [3H]-6 (to 6 h) (average ± half differential for 4).
Figure 6.

Tissue levels of tritium (±SEM) after intravenous dosing of [3H]-4, [3H]-5, and [3H]-6 (after 6 h) (average ± half differential for 4).
In Vivo Uptake of [3H]-4 into Bones and Release of Tritium over 28 Days
To evaluate the rate of release of 2 from conjugate 4 once bound to bone in vivo, we first intravenously dosed singly radiolabeled [3H]-4 (at 5 mg/kg dose) to four groups of three rats. After 24 h and 7, 14, and 28 days, the rats were sacrificed, and blood, spleen, liver, and long bones were analyzed for radioactivity relative to the administered dose. Contrary to the initial finding, this time about 11.1% of the administered tritium label was found in bones after 24 h. The amount of label release diminished smoothly over the next month until only 1.4% remained at 28 days. This translated into a calculated elimination half-time from the bones of 7.1 days (Figure 7, Table S6). The elimination rate from the liver was moderate with a half-life of about 2.8 days (Table S6 and Figure S1).
Figure 7.

Release of tritium label from bones over 28 days after dosing with [3H]-4.
In Vivo Uptake and Release of 3H and 14C from [3H, 14C]-4 over 28 Days
To confirm the results of the single-label study with 4 and to evaluate the fate of the bisphosphonate component of the conjugate drug in the body, four groups of three rats each were dosed with doubly labeled [3H,14C]-4, dosed intravenously (at 5 mg/kg dose). For the first group of rats, serial blood samples were taken over 24 h. The animals were euthanized, and tissues were then examined for levels of radioactivity. Once again, only a small amount of the administered radioactivity was found in the blood (1.0 ± 0.2% 3H and 0.7 ± 0.2% 14C at 1 h), and clearance continued over 24 h where <0.2% the labels remained in the blood. Notably, levels of 14C and 3H radioactivity in the blood were similar and diminished at a similar rate (Figure 8, Table S7). Tissue distribution at 24 h showed the majority of both labels in the liver (57.0 ± 5.8% of 3H and 42.7 ± 5.0% of 14C) and bones (11.3 ± 0.3% of 3H and 10.9 ± 0.3% of 14C), with small amounts in the spleen (1.24 ± 0.14% of 3H and 0.91 ± 0.12% 14C) and very little found in kidneys (0.3 ± 0.02% of 3H and 0.15 ± 0.02% 14C) (Table S8). Other tissues (fat pad, brain, skeletal muscles, and heart) showed very low levels of radioactivity (≤0.01% for each label). The remaining groups of rats were sacrificed after 7, 14, or 28 days, and long bones, liver, and spleen were analyzed for radioactivity relative to the administered dose. In this study, unfortunately, bone samples from the 7 and 14 days groups were inadvertently contaminated with tritium; thus, tritium bone levels for these days could not be accurately analyzed. The carbon (14C) analysis however was not affected.
Figure 8.

Percentage of originally dosed radiolabel in blood after intravenous dosing of [3H]-4 and [14C]-4 at 5 mg/kg.
As noted above, at day 1, levels of 3H (11.3% of initial dose for 3H) were essentially as was seen in the second tritium experiment and the level of 14C (10.9% of initial dose) was very similar to the tritium level. 14C levels remained essentially unchanged at 11.7 ± 1.2% and 10.1 ± 1.3% of administered dose at 7 and 14 days after dosing, respectively. The level of 14C after 28 days was somewhat diminished at 8.6 ± 0.64% of the initial dose. In spite of the loss of tritium bone data from 7 and 14 days, the level of 3H in bones at 28 days postdosing was 1.6 ± 0.05% of the original dose (Figure 9; Table S9), again essentially the same as seen previously in the single-label experiment. Liver and spleen samples were also examined in this experiment, and although spleen levels were quite low, initially 1.2% of 3H and 0.9% of 14C administered dose after 1 day, they diminished only slowly thereafter to 0.34 and 0.25% of 3H and 14C initial dose respectively after 28 days. Liver levels of 3H and 14C diminished more rapidly and at a similar rate (calculated half-lives of 3.0 days, r2 = 0.966, and 4.1 days, r2 = 0.946; Figure 10; Table S9).
Figure 9.

Percentage of originally dosed radiolabel found in long bones after intravenous dosing of [3H]-4 and [14C]-4 at 5 mg/kg. Data for tritium levels at 7 and 14 days is from the previous experiment where [3H]-4 was dosed alone.
Figure 10.

Percentage of originally dosed radiolabel found in liver after intravenous dosing of [3H]-4 and [14C]-4 at 5 mg/kg.
Oral Dosing of [3H,14C]-4
Biodistribution studies (vide supra) with doubly radiolabeled [3H,14C]-4 showed rapid clearance of both labels from the bloodstream and into the liver and thus suggested that 4 remained unchanged wherein it could be excreted into the intestines largely intact. It was anticipated that the highly anionic nature of these prodrug conjugates would inhibit reabsorption, but we were concerned that if 4 reached the intestines intact then it might be hydrolyzed in the gut, liberating active 2a that might contribute to GI side effects. We therefore carried out a study where cohorts of rats were dosed orally with [3H,14C]-4, and after 1, 2, and 4 h, blood, liver, spleen, and sections of the GI tract were analyzed for radiolabel. Notably, only traces of label were found in the bloodstream, representing at most 0.07% of administered tritium and 0.02% of 14C after 1 h with less at 2 and 4 h, and even less was found in the liver and spleen. The vast majority of label was found in the GI tract, with the most in the stomach and jejunum after 1 h and the majority in the lower GI tract including the cecum and colon after 4 h. Notably, the ratio of 3H/14C remained largely constant and roughly equal to the dosing ratio throughout the GI tract, indicating little if any hydrolysis had taken place. Only the duodenum at 4 h indicated a higher ratio of 3H/14C than dosed which might suggest a small amount of hydrolysis (or decomposition) may have occurred in the upper GI tract. However, in these as well as other rat experiments with 4, no evidence of intestinal intolerance was observed.
Evaluation of Biological Activity of Conjugates 4–7 and Bisphosphonate Fragments 8–11 in a Neutrophil Chemotaxis Assay
EP4 agonist 2a, conjugates 4–7, and putative bisphosphonate fragments 8–11, liberated after release of active EP4 agonist 2, were evaluated for their effects on fMLP-stimulated chemotaxis of mouse neutrophils compared to alendronate and pamidronate as positive controls. The results of two experiments (Figure 11 and Tables S10 and S11) showed that only conjugate 5 had a partial but possibly significant effect on leukocyte migration. Notably, conjugates 4 did not have an effect at the doses tested, nor did the fragment 8 that would be derived from 4. Equally, agonist 2a had no significant effect.
Figure 11.

Neutrophil chemotaxis (nm/min) measured in a Zigmond chamber induced by fMLP (1 μM) and blank or with fMLP plus compound. Murine bone marrow neutrophils were pretreated at 37 °C for 30 min with compound (at 50 μM) or blank buffer in the case of fMLP alone. Each experiment is the average of measure of tracked movement of at least 30 cells over 15 min.
Stability of Conjugate 4
Conjugate 4 was obtained as a lyophilized amorphous powder after a final purification step of elution from a C-18 column with aqueous acetonitrile. Thus, the compound was obtained in its native state, and elemental analysis was consistent with 2 equiv of sodium (bis-sodium salt). Solid 4 was somewhat hygroscopic, but when stored in a desiccator, it was found to be very stable at room temperature (for more than 3 years as judged by periodic HPLC analysis). Conjugate 4 is freely soluble in water or PBS solution. The previous conjugate prodrug, 3 (C1), was found to be somewhat unstable in solution showing about 2–3% decomposition per day in solution at pH 7.4 and where HPLC analysis showed cleavage of the carbamate linkage. 4 in PBS solution at pH 7 showed no detectable hydrolysis of the amide bond. However, a slow hydrolysis of the 15-ester bond was observed at pH 7 liberating about 0.8% free 2b after 24 h.
Binding of 4 to Bone (Bio-Oss) and Forms of Calcium Phosphate
Binding of 4 to Bone
Initially, we evaluated the propensity for 4 to bind to granulated bovine bone (Bio-Oss). Solutions containing varying amounts of [3H]-4 in water were tumbled gently with Bio-Oss for 15–20 min, and then the supernatant was removed. The powder was washed with distilled water. The supernatant and aqueous washes were counted and showed very little tritium (<5%), indicating that most (>95%) of the radiolabel had been removed through binding to the bone powder. This rapid binding was employed to characterize the stability of 4 in aqueous solution and in plasma (vide supra). Repeated washing with water did not release any further radioactivity. After drying, the bone powder was treated with 2 M NaOH with stirring for about 1 h, and the supernatant solution was counted and shown to contain >95% of the original tritium label.
Binding of 4 to Brushite
Brushite is a form of calcium phosphate prepared by mixing β-tricalcium phosphate (β-TCP) with monocalcium phosphate hydrate (MCPM) with a small amount of water.22 When mixed, it forms a paste that over time sets to form a solid cement. The paste can be placed into a mold to provide brushite forms in whatever shapes desired. Brushite forms containing 4 could thus be obtained by using a solution of 4 in water in place of pure water during the preparation; thus, brushite containing 0.1–5% 4 by weight was prepared. Alternatively, 4 could first be adsorbed onto β-TCP powder by stirring a slurry of β-TCP with a solution of 4 for several hours followed by lyophilization to provide a fine anhydrous powder. This could then be mixed with MCPM and water to form brushite loaded with 4. Another option was to add a water solution of 4 to a preformed solid brushite and allow it to dry for a period of time (5 h). With each of these methods, the radiolabeled 4, once bound, could not be washed off with water.
To demonstrate the effective binding and stability of 4 in brushite and recovery of 2 from the material, we used [3H]-4 at 1% w/w to prepare brushite discs loaded with 4. After setting, the discs were converted to granules and dried overnight in a desiccator. The granules were then slurried with excess water, and only about 2.5% of the originally added radiolabel was found in the supernatant. After treatment of the remaining granules with 2 M NaOH for 1 h, 95% of the original radioactivity was found in the supernatant solution.
In a second experiment, a solution of unlabeled 4 was mixed with β-TCP and then lyophilized. This 4-β-TCP powder was mixed with MCPM and water and formed into brushite tablets which were dried several days under vacuum. Stirring these tablets or the powder obtained after crushing the tablets with water showed no liberation of 4 or 2a as judged by HPLC analysis of the supernatant. However, on shaking with 2 M NaOH, HPLC analysis showed that only 2a was liberated into the solution.
In a third experiment, an aqueous solution of 4 (and separately [3H]-4) was added dropwise to preformed brushite discs so as to saturate the top side of the disc but that all liquid was retained in the pellet. The discs were then dried in a vacuum desiccator for 5 h. Immersion of the discs into water showed no elution of 4 into the water, and counting radioactivity showed that <0.6% of the original radioactivity escaped into the supernatant. Hydrolysis of the discs was shown to liberate 2a both by radioactivity and by HPLC analysis of the basic supernatant solution. Finally, X-ray powder diffraction analysis of the brushite powders bearing 4 added by whatever method showed diffraction patterns identical to brushite alone (Figure S2).
Sterilization of 4 Bound to Brushite
To sterilize 4 once bound to brushite, discs bearing 1% w/w4 were autoclaved under standard conditions (20 min, 120 °C, 100% humidity, 15 psi). After cooling, the discs were suspended in water, and the supernatant showed no detectable 4 in solution. After the discs were crushed and stirred with water, a trace of 4 (<5%) could be detected by HPLC. After treating the powder with sodium hydroxide, the supernatant showed essentially complete recovery of 2a in solution. No indication of decomposition was observed. However, autoclaving brushite converts the mineral to a new solid form known as monetite,22 which has lost 1 mol of water. X-ray powder diffraction analysis of the 4-containing solid after autoclaving indicated that it had been converted to monetite (Figure S3).
An earlier series of bisphosphonate-based conjugate prodrugs represented by the conjugate 3 (C1) were designed to target bones and to slowly liberate both a bone-anabolic EP4 receptor agonist and a bone-resorption-inhibiting bisphosphonate. 3 showed moderate bone uptake after IV dosing (ca. 5%) and demonstrated efficacy in rat models of osteoporosis, but it proved to be somewhat unstable. Higher doses led to bone overgrowth in some skeletal areas. We felt that a similar but more chemically and metabolically stable prodrug which did not have anti-resorptive activity could be advantageous. Thus, a new series of prodrugs was designed and synthesized wherein the amino-bisphosphonate component of the prodrug was attached to the linker element via a stable amide bond rather than the less stable carbamate bond found in 3. A series of conjugate prodrugs 4–6 with varied chain lengths (n = 3, 2, and 5, respectively) in the bisphosphonate moiety were prepared. Another conjugate, 7, bearing two molecules of EP4 agonist on a single bone-targeting moiety was also prepared. 4–6 were found to be very stable in fresh rat blood plasma, and 4 was found to be similarly stable in fresh human plasma with <10% hydrolysis over 24 h incubation at 37 °C. Conjugates 4–6 were rapidly cleared from the bloodstream and effectively targeted bones after intravenous administration. Biodistribution studies on tritium-labeled 4–6 confirmed that the bulk of the conjugates were taken up into bones (10–20%) and liver (65–74%) within 6 h, while very little radioactivity was found in other tissue. Notably, only traces of radiolabels were found in the kidneys, which is a preferred route of elimination for many bisphosphonate drugs.23 Some bisphosphonates are deposited in the kidney and have been associated with kidney toxicity.24
In summary, on the basis of these preliminary experiments, conjugate 4 was selected for further follow-up studies to determine the rate of release of the active EP4 agonist in the bones. In a second experiment, [3H]-4 dosed intravenously to rats showed a significant uptake into bones (ca. 11.6% of the original dose after 24 h), somewhat less than found in the first experiment, and this level diminished exponentially over the next 28 days until only 1.4% remained. The elimination curve indicated a half-time for release of 7 days, a rate compatible with dosing once weekly.
While this result was gratifying, we wanted to ensure that the active EP4 agonist was effectively targeted to bones and not released in significant concentration in other tissues or blood which might negate the benefits of prodrug targeting. To define the integrity of 4 in vivo after dosing and to ensure that little or none of the active agonist was released into the bloodstream prior to binding to bones or after uptake into the liver, biodistribution studies were repeated with doubly radiolabeled 4, prepared with 14C incorporated into the bone-targeting moiety and 3H incorporated into the EP4 agonist moiety. This allowed determination of the fate of 4 in the blood and other tissues and in the liver and in the bones. Orally dosed 4 was shown to be not absorbed and was not significantly hydrolyzed in the GI tract. Intravenous dosing of [3H, 14C]-4 to rats confirmed that 4 was taken up into skeletal bones essentially intact and showed a degree of uptake that recapitulated the results from the earlier singly labeled study. The majority of both labels were found in the liver, and these labels decreased over 28 days at a similar rate, indicating that the conjugate was excreted intact (or metabolized completely as a unit). It was also notable that the amount of radiolabel found in the blood was very small and that both labels were found in roughly the same ratio as in the original dose and diminished at the same rate. This indicated that very little if any free EP4 agonist was circulating in the bloodstream and that bone targeting was efficient.
Both 3H and 14C labels were incorporated into bones (after 24 h) to the same extent (11.2% of dosed 3H and 10.9% of dosed 14C). Bone samples from cohorts sacrificed after 7, 14, and 28 days showed that the level of 14C remained essentially constant for 14 days, while the 3H label decreased steadily so that only 1.6% of the original label was left after 28 days. In contrast, the 14C level in bones was essentially constant up to 14 days, but by 28 days the 14C level was slightly, but significantly, diminished (8.6%) relative to that of day 1 (p = 0.01). The slow loss of 14C label may be attributed to ongoing bone turnover. EP4 agonists are known to stimulate bone turnover in rats25,26 and the rate of elimination of alendronate in rats has been estimated at about 200 days,27 so the small loss observed at 28 days may be due to dissolution of the bisphosphonate fragment from the bone surface. One premise of the study was that the residual bone targeting moiety would be inactive as an inhibitor of osteoclast activity (and thus not inhibit bone resorption), and in vitro studies on neutrophils showed that unlike active bisphosphonates, alendronate, and pamidronate, both 4 and its putatively liberated bisphosphonate component, 8, had no effect on fMLP-stimulated leukocyte chemokinesis, an effect considered to be mediated by the mechanism responsible for osteoclast inhibition. Equally, conjugate 6 and its liberated bisphosphonate compound, 10, also showed no effects on neutrophil function. Interestingly, conjugate 5 did appear to have some inhibitory activity.
The systemic in vivo bone effects of conjugate 4 (also known as C3) have been studied (and recently published)28 in curative OVX rat model of osteoporosis where 4 (dosed at 2.5 and 5 mg/kg/week) showed a dose response for bone anabolic activity and restored ovariectomy-induced bone loss to sham control levels while showing no observable anti-resorptive effects. These data confirm the predicted effects and benefits (at least in the rat) for compound 4 and have stimulated further development efforts. Conjugate 4 has been shown to be chemically stable as a solid sodium salt and thus may be suitable for development as a bone anabolic pharmaceutical agent.
Due to its propensity to bind quickly and avidly to calcium phosphate, 4 was shown to be suitable to be used to “preload” commercial bone powder (Bio-Oss) or various forms of calcium phosphate (brushite or monetite) that have been used as matrices to facilitate local bone repair and in bone implants. These conjugate-loaded materials were shown to be very stable and to be readily sterilized by autoclaving or gamma irradiation, a necessity to allow potential use in bone implant augmentation therapy. Some instability to ultraviolet irradiation indicated this would not be a suitable sterilization method and also suggested that materials containing 4 should be protected from light for longer term storage. The potential of 4 for local administration has been demonstrated in recently published studies where brushite and monetite preloaded with 4 have demonstrated significant efficacy to enhance implant integration in a rabbit calvarial implant model.29 Other recent studies have shown that Bio-Oss and monetite loaded with 4 or 7 can significantly speed and enhance bone repair in a rat mandible critical bone defect model.30
Experimental Section
Chemistry
General Information
1H and 13C NMR spectra were recorded with a Bruker Avance II 600 MHz spectrometer using a TCI cryoprobe, an Avance III 500 MHz spectrometer using a TXI inverse probe, or an Avance III 400 MHz spectrometer using a BBOF + ATM probe. NMR data processing was performed with MestReNova software (MestreLab Research, v 6.0.4–5850). The spectra were referenced to the corresponding solvent signals.31 LC-MS were recorded with an ESI ion source on an Agilent 6200 Time-of-Flight spectrometer coupled with Agilent 1200 series front-end. X-ray powder diffractions were recorded on a Bruker D8 advance X-ray diffractometer. Analytical thin-layer chromatography (TLC) was performed on aluminum plates precoated with silica gel 60F-254 as the adsorbent (EMD). The developed plates were air-dried, exposed to UV light and/or dipped in KMnO4 solution and heated. Flash chromatography was performed on a BioTage Isolera instrument using HP-silica cartridges from BioTage or SiliCycle Inc. Derivatized silica was obtained from SiliCycle Inc. Tetrahydrofuran (THF) was distilled from Na and benzophenone under nitrogen. Dichloromethane (DCM) was distilled from CaH2 under nitrogen. Pyridine was distilled from CaH2 under nitrogen. Other reagents and solvents were obtained from commercial vendors and used as received.
Purity was evaluated by HPLC. Solvent A contained 90% acetonitrile (ACN)/10% water/5 mM NH4OAc, and solvent B contained 90% water/10% acetonitrile (ACN)/5 mM NH4OAc. Method 1 used Halo C18 column (4.6 × 50 mm, 5 μm) with a gradient as follows: 0–0.5 min, 5% solvent B/95% solvent A; 0.5–3 min, 0–95% solvent B; 3–4.5 min, 95% solvent B/5% solvent A; 4.5–5 min, 95–5% solvent B; 5–6 min, 5% solvent B/95% solvent A. Method 2 used Halo ES-CN column (3.0 × 50 mm, 5 μm) with a gradient as follows: 0–0.5 min, 20% solvent B/80% solvent A; 0.5–1.5 min, 20–100% solvent B; 1.5–4 min, 100% solvent B; 4–4.2 min, 100–20% solvent B; 4.2–5 min, 20% solvent B/80% solvent A. Method 3 used Halo ES-CN column (3.0 × 50 mm, 5 μm) with a gradient as follows: 0–0.5 min, 5% solvent B/95% solvent A; 0.5–1.5 min, 5–100% solvent B; 1.5–4 min, 100% solvent B; 4–4.2 min, 100–5% solvent B; 4.2–5 min, 5% solvent B/95% solvent A.
Preparation of graphs and statistical analyses were performed using Prism software (GraphPad Prism 9.0.1).
Radioactive Analysis
Scintillation counting was conducted in a Beckman Coulter LS-6500 scintillation counter. The counting time was 1 min. Dual-label counting and chemiluminescence correction were enabled. Scintillant from Amersham Biosciences (cat. no. NBCS104) was used except for biological oxidizer samples, where special tritium and carbon-14 scintillants from the instrument manufacturer were used.
Tissue samples were analyzed in a Harvey OX-300 biological oxidizer using the 4 min program, and the resulting scintillation samples were counted as above.
2,5-Dioxopyrrolidin-1-yl 4-(2-(((R,E)-4-((R)-1-(7-Ethoxy-7-oxoheptyl)-5-oxopyrrolidin-2-yl)-1,1-difluoro-1-phenylbut-3-en-2-yl)oxy)-2-oxoethyl)benzoate (13)
To a solution of ester 2b (0.200 g, 0.47 mmol, 1 equiv) in dry CH2Cl2 (2.4 mL) were added 4-(carboxymethyl)benzoic acid (0.134 g, 0.75 mmol, 1.6 equiv), DMAP (0.001 g, 0.01 mmol, 0.02 equiv), pyridine (0.089 mL, 1.11 mmol, 2.4 equiv), and then DCC (0.150 g, 0.73 mmol, 1.5 equiv). The reaction mixture was allowed to stir under an argon atmosphere at room temperature for 2 h. The mixture was filtered, and the filter cake was washed with MTBE. The filtrate was washed with a solution of 0.5 M citric acid/water 1:1 (v/v). The aqueous layer was extracted three times with MTBE. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure in order to provide acid 12. HRMS m/z calcd for C32H36F2NO7 [M – H]− 584.2465. Found 584.2469. The residue was solubilized in DMF (10 mL). To this solution, N-hydroxysuccinimide (0.147 g, 1.28 mmol, 2.7 equiv) and N-(3-dimethylamino-propyl)-N′-ethylcarbodiimide hydrochloride (EDCI) (0.245 g, 1.28 mmol, 2.7 equiv) were added. The reaction mixture was stirred for 7 h at room temperature under an argon atmosphere. The mixture was diluted with EtOAc and water. The aqueous layer was extracted three times with EtOAc. The combined organic layers were washed four times with a saturated aqueous NaCl solution, dried over Na2SO4, and concentrated to dryness. The crude was then purified by flash chromatography (12 g BioTage HP Sil cartridge, 50–100% EtOAc/hexane gradient) to give NHS-ester 13 (0.364 g, 80%) as a colorless oil. 1H NMR (CDCl3, 500 MHz) δ 8.03 (d, 2H, J = 8.0 Hz), 7.45–7.32 (m, 5H), 7.27 (d, 2H, J = 8.0 Hz), 5.74–5.68 (m, 1H), 5.64–5.54 (m, 2H), 4.09 (q, 2H, J = 7.0 Hz), 4.03–3.98 (m, 1H), 3.72–3.63 (m, 2H), 3.46–3.40 (m, 1H), 2.93–2.82 (m, 4H), 2.65–2.58 (m, 1H), 2.38–2.25 (m, 4H), 2.19–2.12 (m, 1H), 1.65–1.56 (m, 3H), 1.42–1.20 (m, 9H). 13C NMR (CDCl3, 126 MHz) δ 174.7, 173.7, 169.3, 168.4, 161.5, 140.5, 138.1, 133.3 (t, JC–F= 31.7 Hz), 130.8, 130.6, 129.8, 128.4, 125.6 (t, JC–F= 7.6 Hz), 124.2, 123.7, 119.3 (t, JC–F= 299.0 Hz), 74.6 (t, JC–F= 40.8 Hz), 60.2, 59.6, 41.1, 40.4, 34.2, 29.8, 28.7, 27.0, 26.4, 25.7, 25.1, 24.8, 14.2. HRMS m/z calcd for C36H41F2N2O9 [M + H]+ 683.2775. Found 683.2796. HPLC purity: 100%, tR = 3.0 min (Method 1).
Sodium (4-(4-(2-(((R,E)-4-((R)-1-(7-Ethoxy-7-oxoheptyl)-5-oxopyrrolidin-2-yl)-1,1-difluoro-1-phenylbut-3-en-2-yl)oxy)-2-oxoethyl)benzamido)-1-hydroxybutane-1,1-diyl)bis(hydrogen phosphonate) (4)
A stock solution of alendronic acid triethylammonium salt (pH ∼10) was prepared by mixing alendronic acid (0.500 g, 2.01 mmol, 1 equiv), water (3 mL), DMF (5 mL), and triethylamine (0.8 mL, 5.74 mmol, 2.85 equiv). To a solution of NHS-ester 13 (0.100 g, 0.15 mmol, 1 equiv) in DMF (0.5 mL) was added the previously prepared stock solution of alendronic acid/Et3N (2.6 mL), representing 4 equiv (0.59 mmol) of alendronic acid and 11.3 equiv (1.69 mmol) of triethylamine. The reaction mixture was stirred at room temperature and monitored by HPLC. After 10 min of stirring, the reaction was complete. The reaction was quenched with 0.1% formic acid in water. The pH was adjusted to pH 6–7 with a 2% formic acid solution in water. The solution was then loaded onto an anion exchange column (1 g of Siliabond-TMA Acetate (Silicycle), loading: 0.94 mmol/g, packed in a SPE cartridge, activated by passing 0.1 M HCl/MeOH 1:1 (v/v), then 0.1% formic acid in water). It was sequentially eluted with 0.1% formic acid (3 column volumes (CV)), MeOH/0.1% formic acid 1:1 (v/v) (3 CV), MeOH (2 CV), and MeOH/0.1 M HCl (5 CV). This last acidic fraction was neutralized with 1 M NaOH and concentrated in vacuo to remove methanol. The remaining solution was loaded onto a 12 g C18 RP-chromatography column (activated by MeOH, then water) using as gradient the following: 1.5 CV of water, 12 CV of gradient 0–100% MeOH/H2O, 3 CV of 100% MeOH, and 3 CV of MeOH/H2O 1:1 (v/v). This provided, after lyophylization, 4 as a disodium salt (0.103 g, 80%) in the form of a white solid. 1H NMR (D2O, 600 MHz) δ 7.65 (d, 2H, J = 9.0 Hz), 7.48 (t, 1H, J = 7.2 Hz), 7.42–7.37 (m, 4H), 7.27 (d, 2H, J = 7.8 Hz), 5.77–5.68 (m, 2H), 5.19–5.10 (m, 1H), 4.06 (m, 3H), 3.80 (d, 1H, J = 15.0 Hz), 3.71 (d, 1H, J = 15.0 Hz), 3.37 (t, 2H, J = 7.2 Hz), 3.11–3.05 (m, 1H), 2.55–2.51 (m, 1H), 2.31–2.19 (m, 4H), 2.11–2.05 (m, 1H), 2.02–1.94 (m, 2H), 1.91–1.86 (m, 2H), 1.58–1.05 (m, 12H). 13C NMR (D2O, 151 MHz) δ 177.3, 176.7, 171.1, 169.4, 136.8, 132.4, 131.9 (t, JC–F= 25.7 Hz), 130.4, 129.2, 128.0, 127.0, 125.5 (t, JC–F= 6.0 Hz), 123.6, 119.4 (t, JC–F= 249 Hz), 73.8 (t, JC–F= 33.2 Hz), 73.4, 61.1, 60.3, 40.3, 40.3, 40.2, 33.4, 30.6, 29.4, 27.1, 25.5, 25.0, 23.8, 23.6, 23.5, 23.1, 12.9. HRMS m/z calcd for C36H49F2N2O13P2 [M + H]+ 817.2672. Found 817.2692. Elemental Analysis: Calcd for C36H46F2N2Na2O13P2: C, 47.27; H, 5.73; N, 3.06, F, 4.15. Found (2 determinations): C, 47.56, 47.62; H, 5.65, 5.64; N, 3.08,3.04, F, 4.08, 4.02. HPLC purity: 100%, tR = 2.0 min (Method 1).
Sodium (3-(4-(2-(((R,E)-4-((R)-1-(7-Ethoxy-7-oxoheptyl)-5-oxopyrrolidin-2-yl)-1,1-difluoro-1-phenylbut-3-en-2-yl)oxy)-2-oxoethyl)benzamido)-1-hydroxypropane-1,1-diyl)bis(hydrogen phosphonate) (5)
A stock solution of pamidronic acid triethylammonium salt (pH ∼10) was prepared by mixing pamidronic acid (0.500 g, 2.13 mmol, 1 equiv), water (3 mL), DMF (5 mL), and triethylamine (0.8 mL, 5.74 mmol, 2.70 equiv). To a solution of NHS-ester 13 (0.050 g, 0.07 mmol, 1 equiv) in DMF (0.5 mL), was added the previously prepared stock solution of pamidronic acid/Et3N (1.3 mL) representing 4 equiv (0.30 mmol) of pamidronic acid and 11 equiv (0.81 mmol) of triethylamine. The reaction mixture was stirred at room temperature for 45 min and then directly loaded onto a 12 g C18 RP-chromatography column (activated by MeOH, then water, gradient: 1.5 CV of water, 10 CV of gradient 0–100% MeOH/H2O, 3 CV of 100% MeOH, and 3 CV of MeOH/H2O 1:1 (v/v)) to give conjugate 5 as the triethylammonium salt. The latter was loaded onto a cation exchange column (0.32 g Siliabond-Tosic acid (Silicyle) 40–63 μm, 0.68 mmol/g) which was rinsed beforehand with 100% MeOH, 100% water, and 5% aqueous NaCl solution. The column was washed with MeOH/H2O 1:1 (v/v). The collected solution (pH 7) was concentrated in vacuo to remove methanol and loaded onto a 12 g C18 RP chromatography (activated by MeOH, water, and then gradient: 1.5 CV of water, 10 CV of gradient 0–100% MeOH/H2O, 3 CV of 100% MeOH, and 3 CV of MeOH/H2O 1:1 (v/v)). The latter fractions containing product were lyophilized to give 5 as a disodium salt (0.040 g, 60%) as a white solid. 1H NMR (D2O, 600 MHz) δ 7.75 (d, 2H, J = 7.8 Hz), 7.57–7.55 (m, 1H), 7.48–7.43 (m, 4H), 7.33 (d, 2H, J = 7.8 Hz), 5.84–5.80 (m, 1H), 5.77 (dd, 1H, J = 15.0, 6.0 Hz), 5.28 (dd, 1H, J = 15.0, 9.0 Hz), 4.15–4.11 (m, 3H), 3.86 (d, 1H, J = 15.0 Hz), 3.79–3.74 (m, 2H), 3.19–3.14 (m, 1H), 2.65–2.61 (m, 1H), 2.38–2.26 (m, 6H), 2.19–2.13 (m, 1H), 1.58–1.46 (m, 3H), 1.36–1.15 (m, 10H). 13C NMR (D2O, 151 MHz) δ 177.3, 176.6, 171.0, 168.9, 136.7, 136.0, 132.4, 131.9 (t, JC–F = 25.1 Hz), 130.4, 129.2, 128.0, 127.0, 125.4 (t, JC–F = 5.9 Hz), 123.6, 119.4 (t, JC–F = 248 Hz), 73.9 (t, JC–F = 32.6 Hz), 72.6 (t, JC–F = 132 Hz), 61.0, 60.3, 40.3, 40.2, 35.8 (t, JC–F = 7.9 Hz), 33.4, 32.4, 29.4, 27.2, 25.5, 25.0, 23.8, 23.6, 12.9. HRMS m/z calcd for C35H47F2N2O13P2 [M + H]+ 803.2514. Found 803.2516. HPLC purity: 100%, tR = 2.1 min (Method 1).
(Sodium (6-(4-(2-(((R,E)-4-((R)-1-(7-Ethoxy-7-oxoheptyl)-5-oxopyrrolidin-2-yl)-1,1-difluoro-1-phenylbut-3-en-2-yl)oxy)-2-oxoethyl)benzamido)-1-hydroxyhexane-1,1-diyl)bis(hydrogen phosphonate) (6)
A stock solution of neridronic acid triethylammonium salt (pH ∼10) was prepared by mixing neridronic acid (0.500 g, 1.80 mmol, 1 equiv), water (3 mL), DMF (5 mL), and triethylamine (0.8 mL, 5.74 mmol, 3.19 equiv). To a solution of NHS-ester 13 (0.050 g, 0.07 mmol, 1 equiv) in DMF (0.25 mL) was added the previously prepared stock solution of neridronic acid/Et3N (1.4 mL) representing 4 equiv (0.29 mmol) of pamidronic acid and 13 equiv (0.91 mmol) of triethylamine. The reaction mixture was stirred at room temperature for 20 min and then directly loaded onto a 12 g C18 RP-chromatography column (activated by MeOH, water, then gradient: 1.5 CV of water, 10 CV of gradient 0–100% MeOH/H2O, 3 CV of 100% MeOH, and 3 CV of MeOH/H2O 1:1 (v/v)) to give conjugate 6 as the triethylammonium salt. This latter fraction was loaded onto a cation exchange column (0.32 g Siliabond-Tosic acid (Silicyle) 40–63 μm, 0.68 mmol/g) which was rinsed beforehand with 100% MeOH, 100% water, and then 5% aqueous NaCl solution. The column was washed with MeOH/H2O 1:1 (v/v). The collected solution (pH 7) was concentrated in vacuo to remove methanol and loaded onto a 12 g C18 RP chromatography column (activated by MeOH and then water) with a gradient: 1.5 CV of water, 10 CV of gradient 0–100% MeOH/H2O, 3 CV of 100% MeOH, and 3 CV of MeOH/H2O 1:1 (v/v). The latter fractions containing product were lyophilized to provide 6 as the disodium salt (0.034 g, 55%) in the form of a white solid. 1H NMR (D2O, 500 MHz) δ 7.68 (d, 2H, J = 8.0 Hz), 7.55–7.52 (m, 1H), 7.48–7.42 (m, 4H), 7.33 (d, 2H, J = 8.0 Hz), 5.82–5.73 (m, 2H), 5.13–5.08 (m, 1H), 4.14–4.10 (m, 3H), 3.87 (d, 1H, J = 15.0 Hz), 3.76 (d, 1H, J = 15.0 Hz), 3.45–3.35 (m, 2H), 3.15–3.10 (m, 1H), 2.59–2.54 (m, 1H), 2.37–2.10 (m, 5H), 1.99–1.91 (m, 2H), 1.70–1.62 (m, 4H), 1.56–1.51 (m, 2H), 1.44–1.40 (m, 3H), 1.34–1.28 (m, 1H), 1.24–1.10 (m, 8H). 13C NMR (D2O, 151 MHz) δ 176.9, 175.8, 170.3, 168.6, 136.6, 136.5, 132.4, 132.2 (t, JC–F = 25.2 Hz), 130.3, 129.1, 128.0, 126.9, 125.3 (t, JC–F = 5.6 Hz), 123.6, 119.3 (t, JC–F = 248 Hz), 73.8 (t, JC–F = 33.0 Hz), 73.8 (t, JC–F = 134 Hz), 60.8, 60.2, 40.2, 40.0, 39.6, 33.4, 33.1, 29.4, 28.0, 27.4, 26.7, 25.7, 25.2, 23.9, 23.7, 22.8, 13.0. HRMS m/z calcd for C38H53F2N2O13P2 [M + H]+: 845.2985. Found: 845.2971. Purity: 100%, tR = 2.1 min (Method 1).
4-(2-Methoxy-2-oxoethyl)benzoic Acid (14)
To a solution of 4-(carboxymethyl)benzoic acid (1.00 g, 5.55 mmol, 1 equiv) in methanol (11 mL) was added thionyl chloride (0.020 mL, 0.28 mmol, 5 mol %). The reaction mixture was stirred overnight at room temperature. The solvent was evaporated under reduced pressure, and the residual material was taken up in MTBE and washed successively three times with a saturated aqueous NaHCO3 solution and once with water. The combined bicarbonate and aqueous extracts were acidified with 1 N HCl until precipitation of the monomethyl ester. The mixture was extracted three times with MTBE. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure to get 4-(2-methoxy-2-oxoethyl)benzoic acid (14) (0.985 g, 91%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 8.08 (d, 2H, J = 8.4 Hz), 7.40 (d, 2H, J = 8.4 Hz), 3.72 (s, 5H).20
2,5-Dioxopyrrolidin-1-yl 4-(2-methoxy-2-oxoethyl)benzoate (15)
To a solution of monoacid 14 (0.917 g, 4.72 mmol, 1 equiv) in DMF (107 mL) were added N-hydroxysuccinimide (1.47 g, 12.8 mmol, 2.7 equiv) and N-(3-dimethylamino-propyl)-N′-ethylcarbodiimide hydrochloride (2.44 g, 12.8 mmol, 2.7 equiv). The reaction mixture was stirred at room temperature for 3.5 h under an argon atmosphere. The colorless mixture was diluted with EtOAc and water. The solution was extracted three times with EtOAc. The combined organic layers were washed three times with water, dried over Na2SO4, and concentrated under reduced pressure to provide NHS-ester 15 (1.329 g, 97%) as a pale yellow oil. 1H NMR (CDCl3, 600 MHz) δ 8.10 (d, 2H, J = 8.1 Hz), 7.44 (d, 2H, J = 8.1 Hz), 3.72 (s, 2H), 3.71 (s, 3H), 2.90 (s, 4H). 13C NMR (CDCl3, 151 MHz) δ 169.9, 168.3, 160.8, 140.5, 130.0, 129.0, 123.2, 51.5, 40.4, 24.8; C14H14NO6 [M + H]+ 292.0816. Found 292.0824. C14H17N2O6 [M + NH4]+ 309.1081. Found 309.1084. Purity: 100%, tR = 2.4 min (Method 1).
Sodium 2-(4-((4-hydroxy-4,4 Bis(hydroxyoxidophosphoryl)butyl)carbamoyl)phenyl)-acetate (8)
A stock solution of alendronic acid triethylammonium salt (pH ∼10) was prepared by mixing alendronic acid (1.00 g, 4.02 mmol, 1 equiv), water (6 mL), DMF (10 mL), and triethylamine (1.6 mL, 11.5 mmol, 2.85 equiv). To a solution of NHS-ester 15 (0.200 g, 0.69 mmol, 1 equiv) in DMF (1 mL) was added the previously prepared stock solution of alendronic acid/Et3N (12.5 mL; alendronic acid: 2.86 mmol, 4 equiv; triethylamine: 8.17 mmol, 11.8 equiv). The reaction mixture was stirred at room temperature and monitored by HPLC. After 30 min of stirring, the reaction was complete. The reaction was quenched with 0.1% formic acid in water. The pH was adjusted to a pH of 6–7 with a 2% formic acid solution in water. The solution was then loaded onto an anion exchange column (6.2 g of SiliabondTMA Acetate (Silicycle), loading: 1.16 mmol/g, packed in a SPE cartridge, activated by passing 0.1 M HCl/MeOH 1:1 (v/v) then 0.1% formic acid in water). It was sequentially eluted with 0.1% formic acid (3 column volumes (CV)), MeOH/0.1% formic acid 1:1 (v/v) (3 CV), MeOH (2 CV), and MeOH/0.1 M HCl (5 CV). This last acidic fraction was neutralized with 1 M NaOH and concentrated in vacuo to remove methanol. The remaining solution was loaded onto a 12 g C18 RP-chromatography column (activated by MeOH, then water) using as gradient 1.5 CV of water, 12 CV of gradient 0–100% MeOH/H2O, 3CV of 100% MeOH, and 3 CV of MeOH/H2O 1:1 (v/v) in order to obtain ester 16. 1H NMR (D2O, 600 MHz) δ 7.77 (d, 2H, J = 8.4 Hz), 7.44 (d, 2H, J = 8.4 Hz), 3.85 (s, 2H), 3.74 (s, 3H), 3.45 (t, 2H, J = 6.0 Hz), 2.06–2.05 (m, 2H), 1.99–1.95 (m, 2H). 13C NMR (D2O, 151 MHz) δ 174.4, 170.2, 137.3, 132.4, 129.2, 127.0, 126.8, 52.2, 40.2, 39.7, 30.6, 23.1. C14H22NO10P2 [M + H]+ 426.0713. Found 426.0701.
To a solution of ester 16 in water (about 3 mL) was added a 1 M NaOH aqueous solution (2.7 mL, 2.74 mmol, 4 equiv). The reaction mixture was stirred overnight at room temperature; 1 M HCl was added to neutralize to pH 7. The mixture was then loaded onto a 25 g C18 RP-chromatography column (activated by MeOH, then water) using as gradient 1.5 CV of water, 12 CV of gradient 0–100% MeOH/H2O, 3 CV of 100% MeOH, and 3 CV of MeOH/H2O 1:1 (v/v) to yield, after lyophilization, 8 as the trisodium salt (0.310 g, 95%) in the form of a white solid. 1H NMR (D2O, 600 MHz) δ 7.74 (d, 2H, J = 8.1 Hz), 7.40 (d, 2H, J = 8.1 Hz), 3.62 (s, 2H), 3.44 (t, 2H, J = 6.7 Hz), 2.19–1.79 (m, 4H). 13C NMR (D2O, 151 MHz) δ 179.8, 170.4, 140.9, 131.5, 128.9, 126.8, 73.4 (t, JC–P = 132 Hz), 43.9, 40.3, 30.9, 23.2 (t, JC–P = 6.1 Hz). C13H20NO10P2 [M + H]+ 412.0557. Found 412.0568. Purity: 100% (Method 1).
Sodium 2-(4-((3-Hydroxy-3,3-bis(hydroxyoxidophosphoryl)propyl)carbamoyl)phenyl)-acetate (9)
A stock solution of pamidronic acid triethylammonium salt (pH ∼10) was prepared by mixing pamidronic acid (0.500 g, 2.13 mmol, 1 equiv), water (3 mL), DMF (5 mL), and triethylamine (0.8 mL, 5.74 mmol, 2.70 equiv). To a solution of NHS-ester 15 (0.050 g, 0.17 mmol, 1 equiv) in DMF (0.25 mL) was added the previously prepared stock solution of alendronic acid/Et3N (2.9 mL; alendronic acid: 0.70 mmol, 4 equiv; triethylamine: 2.00 mmol, 11.8 equiv). The reaction mixture was stirred at room temperature and monitored by HPLC. After 30 min of stirring, the reaction was complete. The reaction was quenched with 0.1% formic acid in water, and the pH was adjusted to a pH of 6–7 with a 2% formic acid solution in water. The solution was then loaded onto an anion exchange column (1.22 g of Si-TMA Acetate Silicycle, loading: 1.16 mmol/g, packed in a SPE cartridge, activated by passing 0.1 M HCl/MeOH 1:1 (v/v), then 0.1% formic acid in water). It was sequentially eluted with 0.1% formic acid (3 CV), MeOH/0.1% formic acid 1:1 (v/v) (3 CV), MeOH (2 CV), and MeOH/0.1 M HCl (5 CV). This last acidic fraction was neutralized with 1 M NaOH and concentrated in vacuo to remove methanol. The remaining solution was loaded onto a 12 g C18 RP-chromatography column (activated by MeOH, then water) using as gradient 1.5 CV of water, 12 CV of gradient 0–100% MeOH/H2O, 3 CV of 100% MeOH, and 3 CV of MeOH/H2O 1:1 (v/v) in order to provide ester 17. 1H NMR (D2O, 400 MHz) δ 7.75 (d, 2H, J = 8.1 Hz), 7.41 (d, 2H, J = 8.1 Hz), 3.82 (s, 2H), 3.74–3.67 (m, 5H), 2.41–2.21 (m, 2H). C13H20NO10P2 [M + H]+ 412.0557. Found 412.0566.
To a solution of 17 in water (1 mL) was added a 1 M NaOH aqueous solution (0.7 mL, 0.68 mmol, 4 equiv). The reaction mixture was stirred overnight at room temperature, and 1 N HCl was added to neutralize to pH 7. The mixture was then loaded onto a 4 g C18 RP-chromatography column (activated by MeOH, then water) using as gradient 1.5 CV of water, 12 CV of gradient 0–100% MeOH/H2O, 3 CV of 100% MeOH, and 3 CV of MeOH/H2O 1:1 (v/v) to yield, after lyophilization, 9 as the trisodium salt (0.056 g, 71%). 1H NMR (D2O, 600 MHz) δ 7.77 (d, 2H, J = 8.0 Hz), 7.41 (d, 2H, J = 8.0 Hz), 3.73 (t, 2H, J = 7.5 Hz), 3.62 (s, 2H), 2.35–2.16 (m, 2H). 13C NMR (D2O, 151 MHz) δ 179.8, 169.8, 140.9, 131.5, 128.9, 126.8, 72.6 (t, JC–P = 128 Hz), 43.9, 36.0 (t, JC–P = 8.1 Hz), 32.5. C12H18NO10P2 [M + H]+ 398.0400. Found 398.0407. Purity: 100% (Method 1).
Sodium 2-(4-((6-Hydroxy-6,6-Bis(hydroxyoxidophosphoryl)hexyl)carbamoyl)phenyl)-acetate (10)
A stock solution of neridronic acid triethylammonium salt (pH ∼10) was prepared by mixing neridronic acid (0.500 g, 1.80 mmol, 1 equiv), water (3 mL), DMF (5 mL), and triethylamine (0.8 mL, 5.74 mmol, 3.19 equiv). To a solution of NHS-ester 3 (0.10 g, 0.34 mmol, 1 equiv) in DMF (0.1 mL) was added the previously prepared stock solution of neridronic acid/Et3N (3.0 mL; neridronic acid: 0.29 mmol, 4 equiv; triethylamine: 0.91 mmol, 13 equiv). The reaction mixture was stirred at room temperature for 2 h. The reaction was quenched with 0.1% formic acid in water, and the pH was adjusted to pH 6–7 with a 2% formic acid solution in water. The solution was then loaded onto an anion exchange column (2 g of Si-TMA Acetate Silicycle, activated by passing 0.1 M HCl/MeOH 1:1 (v/v), then 0.1% formic acid in water). It was sequentially eluted with 0.1% formic acid (3 CV), MeOH/0.1% formic acid 1:1 (v/v) (2 CV), MeOH (2 CV), and MeOH/0.1 M HCl (2 CV). This last acidic fraction was neutralized with 1 M NaOH and lyophilized. The lyophilized powder was dissolved in water and loaded onto a 3 g C18 RP-chromatography column (activated by ACN, then water), using as gradient 2 CV of water, 4 CV of 20% ACN/H2O, 4 CV of 50% ACN/H2O, and 2 CV of 100% ACN (v/v) in order to provide ester 18 (110 mg, 73%). 1H NMR (500 MHz, D2O) δ 7.65 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 7.9 Hz, 2H), 3.74 (s, 2H), 3.68 (s, 3H), 3.34 (t, J = 7.2 Hz, 2H), 2.08–1.89 (m, 2H), 1.66–1.57 (m, 4H), 1.42–1.33 (m, 2H) ppm. 13C NMR (126 MHz, D2O) δ 174.69, 170.27, 137.81, 132.75, 129.70, 127.31, 73.51 (t, J = 142.0 Hz), 52.64, 40.14, 33.44, 28.17, 26.87, 22.99 ppm. 31P NMR (162 MHz, D2O) δ 19.63 ppm. C16H26NO10P2 [M + H]+ 454.1026. Found 454.0881.
To a solution of ester 18 in water (50 mg in 1 mL) was added a 1 M NaOH aqueous solution (pH was adjusted to pH 11–12). The reaction mixture was stirred for 8 h at room temperature, and 1 N HCl was added loaded onto an anion exchange column (2 g of Si-TMA Acetate Silicycle, activated by passing 0.1 M HCl/MeOH 1:1 (v/v), in water). It was sequentially eluted with 0.1 M HCl (2 CV), 50% ACN/0.1 M HCl (2 CV), and 100% ACN. The product was eluted in 50% ACN/0.1 M HCl, and it was neutralized with 1 M NaOH and lyophilized. The lyophilized powder was dissolved in water and loaded onto 4 g of C18 RP-chromatography column in order to provide, after lyophilization, fragment 10 (38 mg, 80%), eluted using water/ACN as follows: 4 CV of water, 4 CV of 50% ACN/H2O, and 2 CV of 100% ACN (v/v). 1H NMR (500 MHz,D2O) δ 7.70 (d, J = 8.2 Hz, 2H), 7.38 (d, J = 8.2 Hz, 2H), 3.60 (s, 2H), 3.41 (t, J = 7.0 Hz, 2H), 1.96–1.82 (m, 2H), 1.74–1.55 (m, 4H), 1.43–1.33 (m, J = 7.7 Hz, 2H) ppm. 13C NMR (126 MHz, D2O) δ 180.30, 170.89, 141.31, 132.11, 129.38, 127.22, 89.84, 44.35, 40.19, 35.99, 28.51, 27.52, 20.52 ppm. 31P NMR (162 MHz, D2O) δ 18.99 ppm. C15H24NO10P2 [M + H]+ 440.0870. Found 440.0922. HPLC purity: 100%, tR = 0.28 min (Method 1).
3,5-Bis(bromomethyl)benzonitrile (19)
To a solution of 3,5-dimethylbenzonitrile (1.00 g, 7.62 mmol, 1 equiv) in 1,2-dichloroethane (76 mL) were added N-bromosuccinimide (2.98 g, 16.8 mmol, 2.2 equiv) and azobis(isobutyronitrile) (AIBN) (0.250 g, 1.52 mmol, 0.2 equiv). The reaction mixture was stirred under an argon atmosphere for 9 h at 80 °C. H2O was added to the medium which was extracted three times with CH2Cl2. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by flash chromatography (25 g BioTage HP Sil cartridge, 2–15% EtOAc/hexane gradient, 20 CV) to yield 3,5-bis(bromomethyl)benzonitrile (19) (0.618 g, 28%) as white crystals. 1H NMR (CDCl3, 500 MHz) δ 7.64 (s, 1H), 7.61 (d, 2H, J = 1.2 Hz), 4.45 (s, 4H). HPLC purity: 99%, tR = 2.9 min (Method 1). Data were similar to those described in the literature.32
2,2′-(5-Cyano-1,3-phenylene)diacetonitrile (20)
To a solution the bis-bromomethyl compound 19 (0.581 g, 2.01 mmol, 1 equiv) in a mixture of CH2Cl2/H2O 1:1 (v/v) (30 mL) were added tetrabutylammonium bromide (0.648 g, 2.01 mmol, 1 equiv) and sodium cyanide (0.207 g, 4.22 mmol, 2.1 equiv). The phase-transfer reaction system was vigorously stirred overnight at room temperature. The aqueous phase was separated from the organic layer and extracted three times with DCM. The combined organic layers (included the one from the reaction mixture) were concentrated under reduced pressure. The crude was purified by flash chromatography (12 g BioTage HP Sil cartridge, 15–50% EtOAc/hexane gradient) to yield 2,2′-(5-cyano-1,3-phenylene)diacetonitrile (20) (0.215 g, 59%) as white crystals. 1H NMR (CDCl3, 400 MHz) δ 7.65–7.64 (m, 2H), 7.60–7.59 (m, 1H), 3.85 (s, 4H). 13C NMR (CDCl3, 101 MHz) δ 133.0, 132.0, 131.3, 117.2, 116.4, 114.5, 23.4. HRMS m/z calcd for C11H7N3Na [M + Na]+ 204.0532. Found 204.0542. HPLC purity: 100%, tR = 1.6 min (Method 1).
3,5-Bis(2-methoxy-2-oxoethyl)benzoic acid (21)
A mixture of 2,2′-(5-cyano-1,3-phenylene)diacetonitrile (20) (0.215 g, 1.19 mmol, 1 equiv) in an aqueous 2 M NaOH solution (15 mL, 30.0 mmol, 25 equiv) was heated to reflux for 4 h. Upon cooling, the solution was acidified to pH 2 using concentrated hydrochloric acid. The mixture was extracted three times with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure in order to yield 2,2′-(5-carboxy-1,3-phenylene)diacetic acid (21) (0.283 g, quant.) as a white solid. 1H NMR (acetone-d6, 400 MHz) δ 7.92 (d, 2H, J = 1.6 Hz), 7.52 (t, 1H, J = 1.6 Hz), 3.74 (s, 4H). 13C NMR (DMSO, 101 MHz) δ 172.5, 167.2, 135.5, 135.0, 130.8, 128.8, 21.1. HRMS m/z calcd for C11H9O6 [M + H]+ 237.0405. Found 204.0536.
Diethyl 7,7′-((5R,5′R)-((1E,1′E,3R,3′R)-((2,2′-(5-(((2,5-Dioxopyrrolidin-1-yl)oxy)carbonyl)-1,3-phenylene)bis(acetyl))bis(oxy))bis(4,4-difluoro-4-phenylbut-1-ene-3,1-diyl))bis(2-oxopyrrolidine-5,1-diyl))diheptanoate (23)
To a solution of 2b (0.356 g, 0.84 mmol, 2 equiv) and 21 (0.100 g, 0.42 mmol, 1 equiv) in distilled dichloromethane (2 mL) were successively added pyridine (0.159 mL, 1.97 mmol, 4.7 equiv), DMAP (0.002 g, 0.02 mmol, 0.04 equiv), and then DCC (0.260 g, 1.26 mmol, 3 equiv). The reaction mixture was stirred under nitrogen at room temperature for 3 h and then concentrated to dryness. The residue was solubilized with MTBE, and the mixture was filtered through Celite. The filter cake was washed with MTBE. The filtrate was washed with a solution of 0.5 M citric acid/water 1:1 (v/v). The aqueous layer was extracted twice with MTBE. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure in order to give 3,5-bis(2-(((R,E)-4-((R)-1-(7-ethoxy-7-oxoheptyl)-5-oxopyrrolidin-2-yl)-1,1-difluoro-1-phenylbut-3-en-2-yl)oxy)-2-oxoethyl)benzoic acid (22) as a colorless oil, used for the next step without any further purification.
Crude 22 (0.440 g, 0.42 mmol, 1 equiv) was dissolved in DMF (9 mL). To this solution were added N-hydroxysuccinimide (0.386 g, 3.36 mmol, 8 equiv) and N-(3-dimethylamino-propyl)-N′-ethylcarbodiimide hydrochloride (EDCI) (0.643 g, 3.36 mmol, 8 equiv). The reaction mixture was stirred overnight at room temperature under an argon atmosphere. The resulting red solution was diluted with water and EtOAc. The aqueous layer was extracted four times with EtOAc. The combined organic layers were washed four times with a saturated aqueous NaCl solution, dried over Na2SO4, and concentrated to dryness. The crude was then purified by flash chromatography (12 g BioTage HP Sil cartridge, 0–15% MeOH/CH2Cl2 gradient, 20 CV) followed by a second flash chromatography (12 g BioTage HP Sil cartridge, 20 CV of 90–100% AcOEt/hexane gradient, and then 20 CV of 1–15% MeOH/EtOAc gradient) to yield 23 (0.265 g, 55%) as a pale pink oil. 1H NMR (CDCl3, 400 MHz) δ 7.80 (s, 2H), 7.45–7.25 (m, 11H), 5.80–5.46 (m, 6H), 4.07 (q, 4H, J = 7.1 Hz), 4.00–3.95 (m, 2H), 3.73–3.55 (m, 4H), 3.46–3.30 (m, 2H), 2.88 (s, 4H), 2.58–2.51 (m, 2H), 2.33–2.21 (m, 8H), 2.17–2.05 (m, 2H), 1.65–1.52 (m, 6H), 1.40–1.16 (m, 18H). 13C NMR (CDCl3, 101 MHz) δ 174.7, 173.6, 169.2, 168.5, 161.2, 138.0, 136.7, 134.5, 133.2 (t, JC–F = 37.9 Hz), 130.5, 130.1, 128.3, 125.9, 125.5 (t, JC–F = 9.5 Hz), 123.7, 119.2 (t, JC–F = 373 Hz), 74.5 (t, JC–F = 47.1 Hz), 60.1, 59.6, 40.3, 40.3, 34.1, 29.7, 28.6, 26.9, 26.3, 25.6, 25.0, 24.7, 14.1. HRMS m/z calcd for C61H72F4N3O14 [M + H]+ 1146.4945. Found 1146.4918. HPLC purity: 97%, tR = 1.7 min (Method 2).
Sodium (4-(3,5-Bis(2-(((R,E)-4-((R)-1-(7-ethoxy-7-oxoheptyl)-5-oxopyrrolidin-2-yl)-1,1-difluoro-1-phenylbut-3-en-2-yl)oxy)-2-oxoethyl)benzamido)-1-hydroxybutane-1,1-diyl)bis(hydrogen phosphonate) (7)
A stock solution of alendronic acid triethylammonium salt (pH ∼10) was prepared by mixing alendronic acid (0.500 g, 2.01 mmol, 1 equiv), water (3 mL), DMF (5 mL), and triethylamine (0.8 mL, 5.74 mmol, 2.85 equiv). To a solution of 23 (0.025 g, 0.022 mmol, 1 equiv) in DMF (0.25 mL) was added the previously prepared stock solution of alendronic acid/Et3N (0.78 mL; alendronic acid: 0.18 mmol, 8 equiv; triethylamine: 0.51 mmol, 23 equiv). The reaction mixture was stirred at room temperature and monitored by HPLC. After 1.5 h, the solution was diluted 6- to 8-fold with 0.1% formic acid in water and finally, neutralized with a 2% formic acid solution to a neutral pH solution, which was loaded onto an anion exchange column (0.6 g of Si-TMA Acetate Silicycle, loading: 0.94 mmol/g, activated by passing MeOH, 0.1 M HCl/MeOH 1:1 (v/v), then 0.1% formic acid in water). It was sequentially eluted with 0.1% formic acid (5 CV), MeOH/0.1% formic acid 1:1 (v/v) (5 CV), MeOH (3 CV), and MeOH/0.1 M HCl (7 CV), and 1 M NaOH was added to the fractions of this last acid phase to increase the pH to 7. The mixture was concentrated under reduced pressure to remove methanol and get a smaller volume of solution which was loaded onto a 12 g C18 RP-chromatography column (activated by MeOH, then water) using as gradient 1.5 CV of water, 12 CV of gradient 0–100% MeOH/H2O, 3 CV of 100% MeOH, and 3 CV of MeOH/H2O 1:1 (v/v). One peak was observed, collected, and concentrated to get a few milliliters of a colorless solution which was frozen and lyophilized for 48 h in order to give 7 (0.013 g, 46%) as a white solid. 1H NMR (D2O, 500 MHz) δ 7.57 (s, 2H), 7.39–7.30 (m, 10H), 6.95 (s, 1H), 5.78–5.73 (m, 2H), 5.59 (dd, 2H, J = 14.7, 6.8 Hz), 5.48 (dd, 2H, J = 14.7, 9.0 Hz), 4.01 (q, 4H, J = 6.7 Hz), 3.92–3.91 (m, 2H), 3.67 (d, 2H, J = 15.5 Hz), 3.60 (d, 2H, J = 15.5 Hz), 3.37 (s, 2H), 3.10 (s, 2H), 2.49 (s, 2H), 2.29 (s, 4H), 2.20–2.18 (m, 4H), 2.04–1.92 (m, 6H), 1.54–1.39 (m, 6H), 1.27–1.01 (m, 18H). 13C NMR (D2O, 151 MHz) δ 176.7, 174.9, 169.8, 167.8, 137.6, 134.4, 133.6, 132.6 (t, JC–F = 27.2 Hz), 132.4, 130.2, 128.0, 126.8, 125.2, 123.5, 119.3 (t, JC–F = 247.6 Hz), 74.0 (t, JC–F = 31.7 Hz), 60.4, 60.2, 40.3, 40.1, 39.8, 33.4, 30.6, 29.3, 27.6, 25.8, 25.4, 24.0, 23.8, 23.2, 13.1). HRMS m/z calcd for C61H78F4N3O18P2 [M – H]− 1278.4697. Found 1278.4692. HPLC purity: 98%, tR = 2.5 min (Method 1).
3,5-Bis(2-methoxy-2-oxoethyl)benzoic Acid
To a solution of 2,2′-(5-carboxy-1,3-phenylene)diacetic acid (21) (0.18 g, 0.77 mmol, 1 equiv) in DCM (10 mL) were added oxalyl chloride (0.14 mL, 1.63 mmol, 2.1 equiv) and methanol (2 mL) at −78 °C, and the reaction was stirred for 10 min. The reaction mixture was then allowed to warm to room temperature and stirred for 20 min. Completion of the reaction was monitored using LC-MS and quenched with aqueous 1 M KHCO3 solution on ice/water bath. The bicarbonate extract was acidified with 1 N HCl until precipitation of the dimethyl ester. The mixture was extracted three times with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure and purified using silica gel chromatography and eluted with 70% EtOAc/hexane in order to get 3,5-bis(2-methoxy-2-oxoethyl)benzoic acid as a white solid (0.16 g, 77%). 1H NMR (500 MHz, acetone-d6) δ 7.89 (d, J = 1.7 Hz, 2H), 7.49 (t, J = 1.7 Hz, 1H), 3.76 (s, 4H), 3.66 (s, 6H) ppm. 13C NMR (126 MHz, acetone-d6) δ 171.96, 167.31, 136.18, 135.94, 135.79, 131.76, 130.19, 129.98, 52.24, 52.06, 40.77, 40.55 ppm.
Dimethyl 2,2′-(5-(((2,5-dioxopyrrolidin-1-yl)oxy)carbonyl)-1,3-phenylene)diacetate (25)
To a solution of 3,5-bis(2-methoxy-2-oxoethyl)benzoic acid (0.16 g, 0.6 mmol, 1 equiv) in DMF (10 mL) were added N-hydroxysuccinimide (0.187 g, 1.62 mmol, 2.7 equiv) and N-(3-dimethylamino-propyl)-N′-ethylcarbodiimide hydrochloride (311 mg, 1.62 mmol, 2.7 equiv). The reaction mixture was stirred at room temperature for 6 h under an argon atmosphere. The reaction mixture was quenched with extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with brine and water, dried over Na2SO4, and concentrated under reduced pressure in order to get a sticky white solid of NHS-ester (26) (0.2 g, 91%), which was used in the next step without further purification. 1H NMR (500 MHz, CDCl3) δ 7.96 (d, J = 1.7 Hz, 2H), 7.54 (t, J = 1.7 Hz, 1H), 3.71 (s, 6H), 3.69 (s, 4H), 2.91 (s, 4H) ppm. 13C NMR (126 MHz, CDCl3) δ 171.15, 169.24, 161.59, 137.10, 135.46, 130.38, 125.91, 52.45, 40.76, 25.82 ppm.
Sodium 2,2′-(5-((4-Hydroxy-4,4-diphosphonobutyl)carbamoyl)-1,3-phenylene)diacetic acid (11)
A stock solution of alendronic acid triethylammonium salt (pH ∼10) was prepared by mixing alendronic acid (68 mg, 0.27 mmol, 1 equiv), water (0.408 mL), DMF (0.680 mL), and triethylamine (0.108 mL, 0.78 mmol, 2.85 equiv). To a solution of NHS-ester 25 (0.010 g, 0.03 mmol, 1 equiv) in DMF (0.1 mL) was added the previously prepared stock solution of alendronic acid (1.196 mL). The reaction mixture was stirred at room temperature for 2 h. The reaction was quenched with 0.1% formic acid in water, and the pH was adjusted to pH 6–7 with a 2% formic acid solution in water. The solution was then loaded onto an anion exchange column (2 g of Si-TMA Acetate Silicycle, activated by passing 0.1 M HCl/MeOH 1:1 (v/v), then 0.1% formic acid in water). It was sequentially eluted with 0.1% formic acid (3 CV), MeOH/0.1% formic acid 1:1 (v/v) (3 CV), MeOH (2 CV), and MeOH/0.1 M HCl (5 CV). This last acidic fraction was neutralized with 1 M NaOH and lyophilized. The lyophilized powder was dissolved in water and loaded onto a 3 g C18 RP-chromatography column (activated by ACN, then water) in order to obtain bisphosphonate conjugate fragment ester 26 (11 mg, 75%), eluted using water/ACN as follows: 4 CV of water, 2 CV of 20% ACN/H2O, 4 CV of 50% ACN/H2O, and 2 CV of 100% ACN (v/v). 1H NMR (600 MHz, MeOD) δ 7.65 (d, J = 1.6 Hz, 2H), 7.38 (t, J = 1.6 Hz, 1H), 3.72 (s, 4H), 3.69 (s, 6H), 3.41 (t, J = 6.9 Hz, 2H), 2.16–2.07 (m, 2H), 2.05–1.98 (m, 2H) ppm. 13C NMR (151 MHz, MeOD) δ 172.06, 168.42, 134.97, 134.93, 133.18, 126.69, 72.71 (t, J = 143.5 Hz), 51.18, 40.20, 39.89, 31.02, 23.50 (t, J = 6.3 Hz) ppm. 31P NMR (243 MHz, MeOD) δ 20.30. C17H24NO12P2 [M – H]− 496.0779. Found 496.1039; C17H29N2O12P2 [M + NH4]+ 515.1190. Found 515.1231.
To a solution of ester 26 in water (11 mg, 1 mL) was added a 1 M NaOH aqueous solution (2.7 mL, 2.74 mmol, 4 equiv). The reaction mixture was stirred for 4 h at room temperature, and 1 N HCl was added to bring pH to 6-7 before loading onto an anion exchange column (2 g of Si-TMA Acetate Silicycle, activated by passing 0.1 M HCl/MeOH 1:1 (v/v), in water). It was sequentially eluted with 0.1 M HCl (2 CV), 50% ACN/0.1 M HCl (2 CV), and 100% ACN. The product was eluted in 50% ACN/0.1 M HCl, and it was neutralized with 1 M NaOH and lyophilized. The lyophilized powder was dissolved in water and loaded onto 4 g of C18 RP-chromatography column in order to provide 11 as white solid (8.3 mg, 73%), eluted using water/ACN as follows: 4 CV of water, 4 CV of 50% ACN/H2O, and 2 CV of 100% ACN (v/v). 1H NMR (600 MHz, D2O) δ 7.55 (d, J = 1.6 Hz, 1H), 7.37 (d, J = 1.7 Hz, 0H), 3.76 (s, 2H), 3.38 (t, J = 6.8 Hz, 1H), 2.05–1.95 (m, 1H), 1.92–1.82 (m, 2H) ppm. 13C NMR (151 MHz, D2O) δ 176.19, 170.39, 134.97, 134.71, 133.91, 126.98, 73.31 (t, J = 138.7 Hz), 40.34, 39.98, 30.83, 23.30 (t, J = 6.3 Hz). 31P NMR (243 MHz, D2O) δ 18.83. C15H25N2O12P2 [M + NH4]+ 487.0877. Found 487.0611. HPLC purity: 100%, tR = 0.213 min. (Method 1)
Synthesis of [3H]-4, [3H]-5, and [3H]-6
[3H]-4, [3H]-5, and [3H]-6 were synthesized on a ca. 10 mg scale in a sequence identical to that used for the unlabeled compounds but using tritium-labeled 2b ethyl 7-((R)-2-((R,E)-4,4-difluoro-3-hydroxy-4-(4-tritiophenyl)but-1-en-1-yl)-5-oxopyrrolidin-1-yl)heptanoate), available from a previous synthesis.21
Synthesis of [14C]-4
[14C]-4 was synthesized on a ca. 10 mg scale in a sequence essentially identical to that used to prepare unlabeled 4 but using 4-[14C]-carboxymethylbenzoic acid. 4-[14C]-Carboxymethylbenzoic acid was synthesized as follows: In a glove bag in a fume hood, tetra-n-butylammonium bromide (14 mg, 0.014 mmol) and 14C-sodium cynanide (ca. 1 mCi, estimated 4 mg, 0.085 mmol) were added to a 4 mL vial, and dichloromethane (1.5 mL) and water (1.5 mL) were added followed by 4-bromomethylbenzonitrile (50 mg, 0.256 mmol). The mixture was stirred vigorously overnight at room temperature and then the aqueous phase was separated from the organic layer and extracted twice with DCM. The combined organic layers were blown to dryness under a stream of nitrogen. The crude organics were chromatographed on a Pasteur pipet charged with silica eluting with 70% EtOAc/hexane. The fractions containing the desired product were re-chromatographed through a Pasteur pipet charged with silica and eluted with 70% EtOAc/hexane to yield pure 4-[14C]-cyanomethylphenylcyanide (3 mg, 0.74 mCi, 74% radiochemical yield) as white crystals. This material was diluted with unlabeled cyanomethylbenzonitrile to provide 10 mg that was treated with an aqueous 2 M NaOH (1 mL) in a 4 mL vial with stirring at 95 °C for 2 h. After cooling to room temperature, the reaction mixture was then acidified to pH 2 using concentrated HCl. After cooling to 4 °C overnight, a precipitate formed which was collected by centrifugation, washed, and dried to provide 4-[14C]-carboxymethylbenzoic acid (4.1 mg, 0.21 mCi). The combined aqueous layers were extracted twice with ethyl acetate, and the organic extract was evaporated under a stream of nitrogen to provide a further quantity of 4-[14C]-carboxymethylbenzoic acid as a white solid (6.4 mg, 0.35 mCi) (combined overall radiochemical yield, 57%). This latter sample was converted in the sequence identical to that used in the nonlabeled synthesis to provide [14C]-4 (0.12 mCi, radiochemical yield 34%), which gave a single peak by HPLC analysis with both UV and radiochemical detection (Method 1). In subsequent in vivo experiments, doubly labeled [3H, 14C]-4 was prepared by mixing the singly labeled compounds and diluting with unlabeled 4 as required.
Biology
All animal experiments were carried out in compliance with animal care guidelines and policies of the Canadian Council on Animal Care and under protocols approved by the Simon Fraser University Animal Care Committee.
Stability of Conjugates in Blood Plasma
The amide-linked conjugates were examined for their stability in rat and human blood plasma. Each [3H]-labeled conjugate, 4–6, was diluted with unlabeled conjugate and added to fresh rat plasma in order to get an activity of ca. 5 × 105 dpm and a concentration of 100 μg/mL. The mixture was then incubated at 37 °C for 24 h. Aliquots (10 μL) were removed after 0, 2, 4, 6, 7, and 24 h and 10-fold diluted with water in a 0.6 mL Eppendorf tube. Bovine bone powder (Bio-Oss powder, obtained as a generous gift from HansaMed Ltd.; 10 mg) was added and the mixture was gently stirred for 20 min. The sample was centrifuged, and 50 μL was then used determine the activity in the supernatant.
In Vivo Biodistribution Studies after Oral Dosing
Female Sprague–Dawley rats (Charles River) about 13 weeks old were acclimatized to the facility and operators and were randomly divided to groups of three. Rats were fasted overnight and then dosed by gavage in water (5 mL/kg) [3H, 14C]-4 (5 mg/kg; 1.4 μCi 3H and 2.65 μCi 14C; ratio 1.88 per rat). One cohort of 3 rats was terminated after 1 h. The others were fed, and then cohorts were terminated after 2, 3, and 4 h. The GI tracts, livers, spleens, blood, and long bones were collected and counted. Sections of the GI tract of about 2–3 cm length and 300 mg samples of stomachs, duodenums, jejumums, iliums, cecums, ampullae of colon, and colons were obtained and analyzed by oxidation and scintillation counting.
In Vivo Biodistribution and Elimination Experiments after Intravenous Dosing
Female Sprague–Dawley rats (Charles River) about 13 weeks old were acclimatized to the facility and operators and were randomly divided to groups of three. The dosing solutions were prepared by mixing labeled and unlabeled conjugate drugs in phosphate-buffered saline (PBS, pH 7.2). In all experiments, the final dosing level was 5 mg/kg (1–5 μCi radiolabel(s) per rat). The injection volume was 1 mL/kg body weight (ca. 200–300 μL). For initial experiments with 3H-labeled conjugates, blood samples were taken after 1, 2, 4, and 6 h, after which rats were euthanized, and tissues were harvested. Organs (other than bones) were weighed wet and portions (ca. 200–300 mg) analyzed for radioactivity in a biological oxidizer followed by scintillation counting. Data were corrected for the weight of the organs. For longer-term experiments, four groups of three rats were dosed as above (500 μL via tail vein) with radiolabeled conjugate drug (5 mg/kg; 1–5 μCi of radiolabel(s)), and cohorts were euthanized after 1, 7, 14, and 28 days. Tissues (bones, livers, and spleens) were collected, and radioactivity was analyzed in a biological oxidizer followed by scintillation counting.
Measurement of Bone-Bound Radioactivity
At specific time points, cohorts of three rats were euthanized by CO2 suffocation, and the long bones were harvested. Soft tissues from the bones were removed with a scalpel, and the bones were placed in a vacuum desiccator over Dririte for 1 day. The bones were then cut into sections and further dried until constant weight (<1 mg change over a 2 h period). Sections of the bones (200 ± 50 mg) were analyzed in a biological oxidizer followed by scintillation counting. Total bone radioactivity was calculated from counted dry weight assuming bones constituted 8% of the body weight.
Analysis of the Blood Samples
Blood samples (0.5 mL per time point per rat) for were taken at various times after dosing (1, 2, 4, and 6 h in preliminary experiments and for the doubly labeled 3H, 14C experiments after 24 h and 7, 14, and 28 days) and placed in heparinized microcentrifuge tubes. Blood samples from the 3H experiments (200 μL) were centrifuged at ∼5000 × g until plasma had separated. A 100 μL aliquot of the plasma was diluted with acetonitrile (ACN, 100 μL) and centrifuged again. Next, 50 μL of the supernatant was added to 15 mL of the scintillation mixture, and the radioactivity was counted.
Blood samples from the 3H and 14C experiments (0.5 mL) were frozen in liquid nitrogen, and the solid was divided into two portions of roughly equal size. Each portion was placed on a piece of quantitative filter paper (∼30 mg) and placed in the porcelain ladle. Samples were allowed to warm up to room temperature before being analyzed in a biological oxidizer. The readings from the two halves of the sample were corrected for the blank and summed to give the original radioactivity. The total blood radioactivity was determined assuming blood as 7% of body weight.
Neutrophil Chemotaxis Assay
Murine bone marrow neutrophils (106 cells/well) were pretreated with different drugs at 50 μM concentration for 30 min at 37 °C and then subjected to 5% BSA-coated microscope cover glass (22 × 40 mm2) for 10 min at 37 °C. The cover glass was inverted onto a Zigmond chamber. A 100 μL aliquot of HBSS media was added to the right chamber, and a 100 μL aliquot of HBSS media containing fMLP (10–6 M) was added to the left chamber. Time-lapse video microscopy was used to record neutrophil movements in the Zigmond chambers for 15 min (per frame/20 s). Captured images were analyzed using cell-tracking software (Retrac version 2.1.01 freeware).
Acknowledgments
The authors acknowledge the Canadian Institutes of Health Research (CIHR Team Grant No. 122069-2012), the Natural Sciences and Engineering Council of Canada (NSERC Discovery Grant RGPIN-2018-06275), Canada Foundation for Innovation, the British Columbia Government Leading Edge Endowment Fund, Simon Fraser University, and the University of Toronto for financial support. The authors also acknowledge the technical staff of Animal Care Services in the Animal Resources Facility, Simon Fraser University for animal experiments. The authors acknowledge Daniel Campbell for assistance in preparing original artwork.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00027.
Figures and tables, NMR spectra, HPLC tracings, and X-ray powder patterns (PDF)
Author Contributions
M.T. carried out the synthesis or the conjugates and the radiolabeled conjugates and the pharmacokinetic studies, stability studies, and formulations with natural and synthetic bone. G.C. helped with stability studies and radiosynthesis and formulation studies. C.S. and M.G. performed studies on the effects of conjugates and components on neutrophil function. R.Y. conceptualized the work and provided guidance. All authors contributed to writing and reviewing the manuscript.
The authors declare the following competing financial interest(s): R.Y. and M.G. hold equity positions in Mesentech, Inc., which has a license to right to the compounds described in this publication. R.Y., G.C., and M.T. are co-inventors on patents (WO2016IB53482 (June 12, 2016), published December 15, 2016, as WO16199111 A1; US-2018-0170951-A1 (June 21, 2018); US Patent No. 10,400,000 (September 3, 2019)) covering the intellectual property derived from this work.
Supplementary Material
References
- Gilsenan A.; Harding A.; Kellier-Steele N.; Harris D.; Midkiff K.; Andrews E. (2018) The Forteo Patient Registry linkage to multiple state cancer registries: study design and results from the first 8 years. Osteoporosis Int. 29, 2335–2343. 10.1007/s00198-018-4604-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machwate M.; Harada S.; Leu C. T.; Seedor G.; Labelle M.; Gallant M.; Hutchins S.; Lachance N.; Sawyer N.; Slipetz D.; Metters K. M.; Rodan S. B.; Young R.; Rodan G. A. (2001) Prostaglandin receptor EP(4) mediates the bone anabolic effects of PGE(2). Mol. Pharmacol. 60, 36–41. 10.1124/mol.60.1.36. [DOI] [PubMed] [Google Scholar]
- Billot X.; Chateauneuf A.; Chauret N.; Denis D.; Greig G.; Mathieu M. C.; Metters K. M.; Slipetz D. M.; Young R. N. (2003) Discovery of a potent and selective agonist of the prostaglandin EP4 receptor. Bioorg. Med. Chem. Lett. 13, 1129–1132. 10.1016/S0960-894X(03)00042-8. [DOI] [PubMed] [Google Scholar]
- Billot X., Young R. N., and Han Y. (2003) 1,5-Distributed pyrrolid-2-one derivatives for use as ep4 receptor agonists in the treatment of eye diseases such as glaucoma, WO2003103772A1.
- Arns S.; Gibe R.; Moreau A.; Monzur Morshed M.; Young R. N. (2012) Design and synthesis of novel bone-targeting dual-action pro-drugs for the treatment and reversal of osteoporosis. Bioorg. Med. Chem. 20, 2131–2140. 10.1016/j.bmc.2012.01.024. [DOI] [PubMed] [Google Scholar]
- Chen G.; Arns S.; Young R. N. (2015) Determination of the rat in vivo pharmacokinetic profile of a bone-targeting dual-action pro-drug for treatment of osteoporosis. Bioconjugate Chem. 26, 1095–1103. 10.1021/acs.bioconjchem.5b00160. [DOI] [PubMed] [Google Scholar]
- Liu C. C.; Hu S.; Chen G.; Georgiou J.; Arns S.; Kumar N. S.; Young R. N.; Grynpas M. D. (2015) Novel EP4 receptor agonist-bisphosphonate conjugate drug (C1) promotes bone formation and improves vertebral mechanical properties in the ovariectomized rat model of postmenopausal bone loss. J. Bone Miner. Res. 30, 670–680. 10.1002/jbmr.2382. [DOI] [PubMed] [Google Scholar]
- Hu S.; Liu C. C.; Chen G.; Willett T.; Young R. N.; Grynpas M. D. (2016) In vivo effects of two novel ALN-EP4a conjugate drugs on bone in the ovariectomized rat model for reversing postmenopausal bone loss. Osteoporosis Int. 27, 797–808. 10.1007/s00198-015-3284-x. [DOI] [PubMed] [Google Scholar]
- Migliorati C. A.; Brennan M. T.; Peterson D. E. (2019) (2019) Medication-Related Osteonecrosis of the Jaws. J. Natl. Cancer Inst. Monogr. 2019 (53), lgz009. 10.1093/jncimonographs/lgz009. [DOI] [PubMed] [Google Scholar]
- Widler L.; Jaeggi K. A.; Glatt M.; Müller K.; Bachmann R.; Bisping M.; Born A. R.; Cortesi R.; Guiglia G.; Jeker H.; Klein R.; Ramseier U.; Schmid J.; Schreiber G.; Seltenmeyer Y.; Green J. R. (2002) Highly potent geminal bisphosphonates. From pamidronate disodium (Aredia) to zoledronic acid (Zometa). J. Med. Chem. 45, 3721–3738. 10.1021/jm020819i. [DOI] [PubMed] [Google Scholar]
- Dempster D. W.; Bolognese M. A. (2006) Ibandronate: the evolution of a once-a-month oral therapy for postmenopausal osteoporosis. J. Clin Densitom 9, 58–65. 10.1016/j.jocd.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Vepsäläinen J. J. (2002) Bisphosphonate prodrugs. Curr. Med. Chem. 9, 1201–1208. 10.2174/0929867023369998. [DOI] [PubMed] [Google Scholar]
- Yewle J. N.; Puleo D. A.; Bachas L. G. (2011) Enhanced affinity bifunctional bisphosphonates for targeted delivery of therapeutic agents to bone. Bioconjugate Chem. 22, 2496–2506. 10.1021/bc2003132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y.; Ding H.; Qian L.; Yan X.; Yang C.; Xie Y. (2005) Synthesis and biological evaluation of novel bisphosphonates with dual activities on bone in vitro. Bioorg. Med. Chem. Lett. 15, 3267–3270. 10.1016/j.bmcl.2005.04.062. [DOI] [PubMed] [Google Scholar]
- Amin D.; Cornell S. A.; Gustafson S. K.; Needle S. J.; Ullrich J. W.; Bilder G. E.; Perrone M. H. (1992) Bisphosphonates used for the treatment of bone disorders inhibit squalene synthase and cholesterol biosynthesis. J. Lipid Res. 33, 1657–1663. 10.1016/S0022-2275(20)41388-4. [DOI] [PubMed] [Google Scholar]
- Reszka A. A.; Rodan G. A. (2003) Mechanism of action of bisphosphonates. Curr. Osteoporos Rep 1, 45–52. 10.1007/s11914-003-0008-5. [DOI] [PubMed] [Google Scholar]
- Sun C. X.; Downey G. P.; Zhu F.; Koh A. L.; Thang H.; Glogauer M. (2004) Rac1 is the small GTPase responsible for regulating the neutrophil chemotaxis compass. Blood 104, 3758–3765. 10.1182/blood-2004-03-0781. [DOI] [PubMed] [Google Scholar]
- Sun C. X.; Magalhães M. A.; Glogauer M. (2007) Rac1 and Rac2 differentially regulate actin free barbed end formation downstream of the fMLP receptor. J. Cell Biol. 179, 239–245. 10.1083/jcb.200705122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper J. W.; Forster C.; Sun C.; Peel S.; Glogauer M. (2012) Zoledronate and pamidronate depress neutrophil functions and survival in mice. Br. J. Pharmacol. 165, 532–539. 10.1111/j.1476-5381.2011.01592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton J. E., Fang J., Heyter D., Miller A. B., Navas F., Smalley T. L., Zuercher W. J., and Katamreddy S. R. (2005) Cycloalkylidene compounds as modulators of estrogen receptor, WO2005012220A2.
- Xie H.; Chen G.; Young R. N. (2017) Design, Synthesis, and Pharmacokinetics of a Bone-Targeting Dual-Action Prodrug for the Treatment of Osteoporosis. J. Med. Chem. 60, 7012–7028. 10.1021/acs.jmedchem.6b00951. [DOI] [PubMed] [Google Scholar]
- Tamimi F.; Le Nihouannen D.; Eimar H.; Sheikh Z.; Komarova S.; Barralet J. (2012) The effect of autoclaving on the physical and biological properties of dicalcium phosphate dihydrate bioceramics: brushite vs. monetite. Acta Biomater. 8, 3161–3169. 10.1016/j.actbio.2012.04.025. [DOI] [PubMed] [Google Scholar]
- Miller P. D. (2011) The kidney and bisphosphonates. Bone 49, 77–81. 10.1016/j.bone.2010.12.024. [DOI] [PubMed] [Google Scholar]
- Drake M. T.; Clarke B. L.; Khosla S. (2008) Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin. Proc. 83, 1032–1045. 10.4065/83.9.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jee W. S.; Ma Y. F. (1997) The in vivo anabolic actions of prostaglandins in bone. Bone 21, 297–304. 10.1016/S8756-3282(97)00147-6. [DOI] [PubMed] [Google Scholar]
- Tanaka M.; Sakai A.; Uchida S.; Tanaka S.; Nagashima M.; Katayama T.; Yamaguchi K.; Nakamura T. (2004) Prostaglandin E2 receptor (EP4) selective agonist (ONO-4819.CD) accelerates bone repair of femoral cortex after drill-hole injury associated with local upregulation of bone turnover in mature rats. Bone 34, 940–948. 10.1016/j.bone.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Lin J. H.; Duggan D. E.; Chen I. W.; Ellsworth R. L. (1991) Physiological disposition of alendronate, a potent anti-osteolytic bisphosphonate, in laboratory animals. Drug Metab. Dispos. 19, 926–932. [PubMed] [Google Scholar]
- Sheikh Z.; Chen G.; Al-Jaf F.; Thévenin M.; Banks K.; Glogauer M.; Young R. N.; Grynpas M. D. (2019) In Vivo Bone Effects of a Novel Bisphosphonate-EP4a Conjugate Drug (C3) for Reversing Osteoporotic Bone Loss in an Ovariectomized Rat Model. JBMR Plus 3, e10237. 10.1002/jbm4.10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh Z.; Chen G.; Thévenin M.; Young R. N.; Grynpas M. D.; Glogauer M. (2019) A Novel Anabolic Conjugate (C3) in the Matrix of Dicalcium Phosphate Onlay Block Grafts for Achieving Vertical Bone Augmentation: An Experimental Study on Rabbit Calvaria. Int. J. Oral Maxillofac Implants 34, e51–e63. 10.11607/jomi.7236. [DOI] [PubMed] [Google Scholar]
- Sheikh Z.; Abdallah M.-N.; Al-Jaf F.; Chen G.; Hamdan N.; Young R. N.; Grynpas M. D.; Glogauer M. (2020) Achieving enhanced bone regeneration using monetite granuals with bone anabolic conjugates (C3 and C6) in critical sized rat mandibular defects. J. Biomed. Mater. Res., Part B 108, 2670–2680. 10.1002/jbm.b.34598. [DOI] [PubMed] [Google Scholar]
- Gottlieb H. E.; Kotlyar V.; Nudelman A. (1997) NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 62, 7512–7515. 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- Easson M. W.; Fronczek F. R.; Jensen T. J.; Vicente M. G. (2008) Synthesis and in vitro properties of trimethylamine- and phosphonate-substituted carboranylporphyrins for application in BNCT. Bioorg. Med. Chem. 16, 3191–3208. 10.1016/j.bmc.2007.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.